

SUMMARY
Many bacteria contain an RNA repair operon, encoding the RtcB RNA ligase and the RtcA RNA cyclase, that is regulated by the RtcR transcriptional activator. Although RtcR contains a divergent version of the CARF (CRISPR-associated Rossman fold) oligonucleotide-binding regulatory domain, both the specific signal that regulates operon expression and the substrates of the encoded enzymes are unknown. We report that tRNA fragments activate operon expression. Using a genetic screen in Salmonella enterica serovar Typhimurium, we find that the operon is expressed in the presence of mutations that cause tRNA fragments to accumulate. RtcA, which converts RNA phosphate ends to 2′, 3′-cyclic phosphate, is also required. Operon expression and tRNA fragment accumulation also occur upon DNA damage. The CARF domain binds 5′ tRNA fragments ending in cyclic phosphate, and RtcR oligomerizes upon binding these ligands, a prerequisite for operon activation. Our studies reveal a signaling pathway involving broken tRNAs and implicate the operon in tRNA repair.
Graphical Abstract
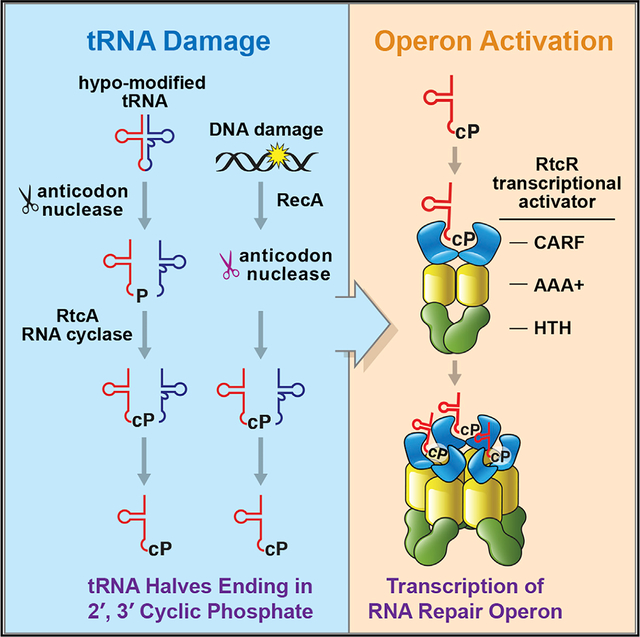
In Brief
Hughes et al. demonstrate that a bacterial RNA repair operon, containing the RtcB RNA ligase and the RtcA RNA cyclase, is regulated by binding of 5′ tRNA halves ending in 2′, 3′-cyclic phosphate to the RtcR transcriptional activator. These studies show how tRNA fragments can regulate bacterial gene expression.
INTRODUCTION
Most organisms contain repair systems that ligate RNA fragments generated by nuclease cleavage or removal of encoded intervening sequences. A major repair pathway involves RtcB, which joins pre-tRNA halves after intron excision in metazoans and archaea. In metazoans, RtcB also ligates mRNA encoding the XBP1 transcription factor as part of the unfolded protein response (Englert et al., 2011; Popow et al., 2011; Jurkin et al., 2014; Kosmaczewski et al., 2014). RtcB joins RNA 5′-OH ends to 2′, 3′-cyclic phosphate or 3′-phosphate RNA ends (Englert et al., 2011; Tanaka et al., 2011). RtcB is also present in bacteria, where its functions are less understood. The only reported substrate is 16S rRNA, since E. coli RtcB can re-ligate a 3′ fragment of 16S rRNA to the rRNA body after cleavage by a stress-induced endonuclease (Temmel et al., 2017). In vitro, E. coli RtcB can add a ppG cap to DNAs ending in 3′-phosphate (Das et al., 2013); however, the role of this activity in vivo is unclear.
In bacteria, RtcB is often expressed as part of a highly regulated “RNA repair” operon. In this operon, rtcB is adjacent to rtcA, which encodes an RNA cyclase that converts 3′-phosphate ends to 2′, 3′-cyclic phosphate (Genschik et al., 1998; Tanaka and Shuman, 2011). In some bacteria, including Salmonella enterica serovar Typhimurium (S. Typhimurium), the rtcBA operon also encodes orthologs of a human RNA-binding protein known as the Ro60 autoantigen and noncoding RNAs called Y RNAs (Chen et al., 2013; Das and Shuman, 2013; Burroughs and Aravind, 2016). The functions of bacterial Ro60 proteins (called Rsr for Ro sixty-related) and Y RNAs have only been studied in Deinococcus radiodurans, where Rsr is tethered by Y RNA to the 3′ to 5′ exoribonuclease polynucleotide phosphorylase (PNPase), specializing the nuclease for structured RNA decay (Chen et al., 2013). Transcription of the operon is often regulated by the enhancer binding protein RtcR, which is encoded adjacent to rtcBA and transcribed in the opposite direction (Genschik et al., 1998).
An impediment to studying the roles of the rtcBA operon has been a lack of information as to how the operon is regulated. RtcR is a member of the σ54-dependent enhancer binding protein family, which typically features an N-terminal signal-sensing ligand-binding domain fused to a C-terminal AAA+ ATPase domain that multimerizes and interacts with σ54 (Bush and Dixon, 2012). The N-terminal portion of RtcR contains a divergent form of the CARF (CRISPR-associated Rossman fold) domain (Makarova et al., 2014). Canonical CARF domains have been best characterized in type III CRISPR-Cas systems, where they are linked to effector domains such as ribonucleases. In these systems, binding of cyclic oligoadenylate (cOA) molecules to the CARF domain activates the adjacent effector (Kazlauskiene et al., 2017; Niewoehner et al., 2017). However, ligands that bind the RtcR domain have not been identified.
To determine how a rtcBA operon is activated, we performed a genetic screen to identify mutations that result in transcription of the S. Typhimurium rsr-yrlBA-rtcBA operon. We report that mutations that result in accumulation of tRNA fragments activate operon expression. Some mutations result in DNA damage, and we show that a feature of the DNA damage response in S. Typhimurium is the activation of one or more ribonucleases that cleave tRNAs in the anticodon loop, resulting in 5′ fragments ending in 2′, 3′-cyclic phosphate. Consistent with the hypothesis that RNAs ending in 2′, 3′-cyclic phosphate are important for operon activation, overexpression of RtcA increases operon expression. We show that the RtcR CARF domain binds 5′ tRNA fragments ending in 2′, 3′-cyclic phosphate and that RtcR forms oligomers upon ligand binding. Our studies identify a signaling pathway involving tRNA and implicate the operon in the repair of damaged tRNAs.
RESULTS
Identification of Mutations that Result in Operon Activation
To identify genes that, when mutated, result in activation of the rsr-yrlBA-rtcBA operon, we generated S. Typhimurium strains in which a lacZ reporter was inserted upstream of rsr under control of the rsr promoter (Figure 1A). We introduced the reporter into two different virulent strains, SL1344 and 14028s, because our early experiments revealed strain-specific differences in the extent to which the operon was repressed. Specifically, while β-galactosidase was fully repressed in the SL1344 Prsr-lacZ strain, we detected low levels of β-galactosidase in the 14028s Prsr-lacZ strain (Figures 1B and 1C). Expression of Prsr-lacZ was under control of the RtcR transcriptional activator, since β-galactosidase activity increased >2,500-fold in both strains when we expressed a constitutively active RtcR lacking part of the N-terminal ligand-binding domain (RtcRΔN; Genschik et al., 1998) (Figure 1C). Upon RtcRΔN expression, both Prsr-lacZ strains also appeared strongly blue when grown on X-gal-containing agar (Figure 1B).
Figure 1. Identification of Mutations that Activate the Operon.
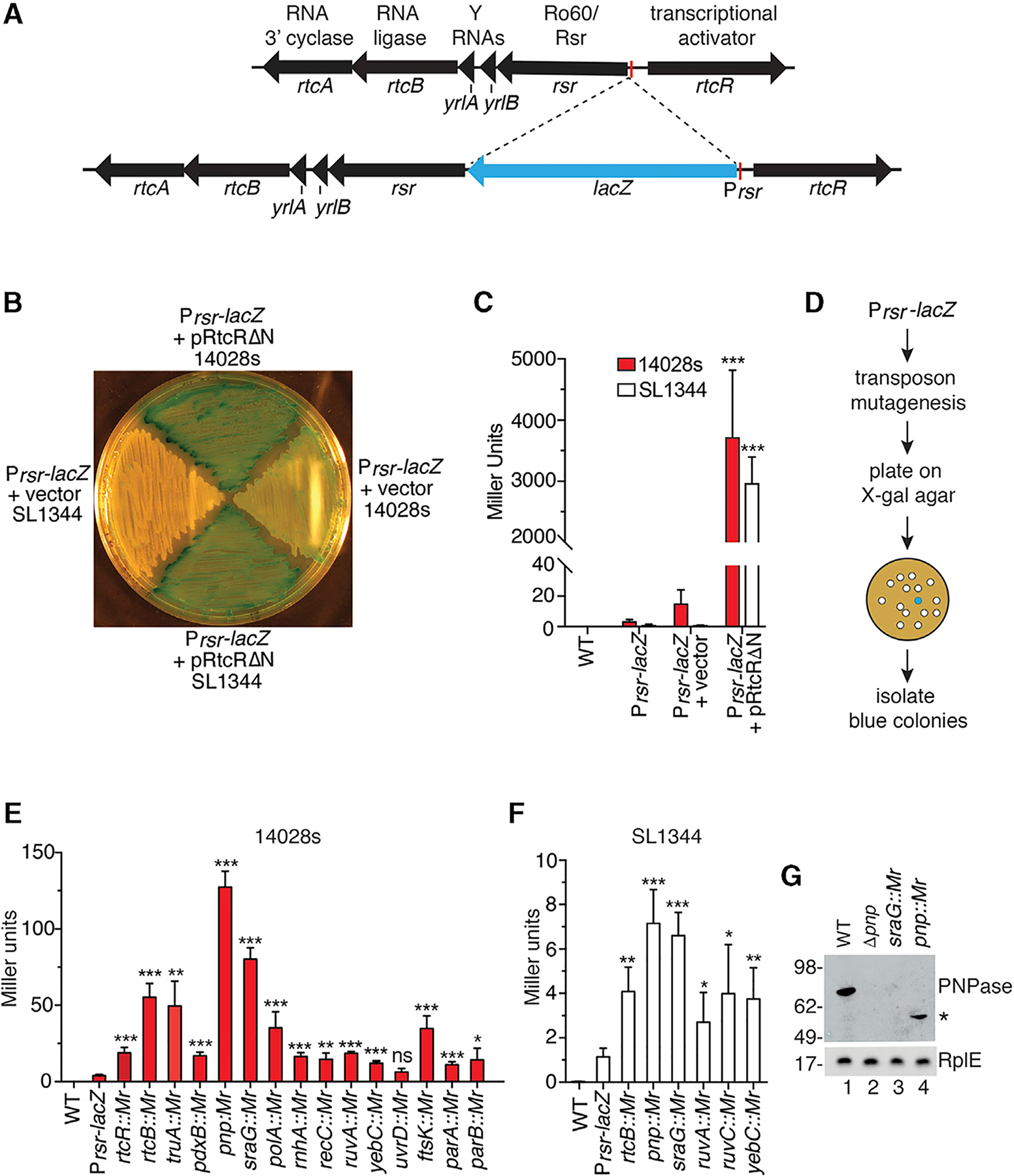
(A) Map of the operon and insertion of the lacZ reporter behind the Prsr promoter.
(B) β-Galactosidase activity was visualized by growing strains on X-gal-containing agar.
(C) After growth in liquid culture, β-galactosidase was measured in Prsr-lacZ strains carrying empty vector or pRtcRΔN.
(D) Screening strategy.
(E and F) β-Galactosidase activity was assayed in 14028s (E) and SL1344 (F) Prsr-lacZ strains carrying the indicated transposon insertions.
(G) Immunoblotting was performed on the indicated strains to detect PNPase. Asterisk, truncated form of PNPase.
Data in (C), (E), and (F) are mean (n = 3) ± SEM. p values were calculated with two-tailed unpaired t tests relative to Prsr-lacZ strains. *p < 0.05; **p < 0.01, and ***p < 0.001; ns, not significant. See also Figure S1.
We used a chloramphenicol-resistant derivative of pSAM, a mariner himar1C9 transposon delivery vector (Goodman et al., 2009), to create a library of transposon insertions in the Prsr-lacZ strains. Mutants that formed blue colonies on X-gal agar and exhibited at least a 50% increase in β-galactosidase activity compared to the parent strain were selected (Figure 1D). With these criteria, we obtained 45 mutants from 28,000 chloramphenicol-resistant 14028s Prsr-lacZ colonies and 26 mutants from 35,000 chloramphenicol-resistant SL1344 Prsr-lacZ colonies. Using ligation-mediated PCR, we mapped the transposon insertions to 28 distinct loci (Table S1). Six loci were identified in both strains, 17 were identified only in the 14028s strain and five only in the SL1344 strain. To confirm that the transposon insertion was responsible for the blue color, we used P22 phage transduction to transfer each transposon mutation into the parent Prsr-lacZ strain. Loci with at least two independent transposon insertions are shown in Figure S1, together with the insertion sites (red circles, 14028s; white circles, SL1344).
Mutations in Genes Involved in tRNA Metabolism and DNA Repair Activate the Operon
The genes we identified fell into three major functional categories. One group mapped within the rsr-yrlBA-rtcBA operon (Figure S1A). As expected, we recovered transposon insertions that, similar to the RtcRΔN mutation, truncate the RtcR N-terminal CARF domain to render the operon constitutively active (Genschik et al., 1998). As these mutations were only recovered in the 14028s strain, our screen may not have reached saturation. We also recovered transposon insertions from both strains within rtcB. One explanation is that the operon is normally expressed at low levels in both strains and that decreased RtcB ligase activity results, directly or indirectly, in increased levels of ligands that activate the operon.
A second category consisted of genes with roles in tRNA metabolism (Figure S1B). Insertions in truA, which encodes the pseudouridine synthase that modifies uridines at positions 38, 39, and 40 in the anticodon arm of some tRNAs (Cortese et al., 1974), activate the operon, as do insertions in pnp, which encodes the PNPase exoribonuclease. In E. coli, PNPase degrades aberrant pre-tRNAs (Li et al., 2002). We also obtained mutations in sraG, which encodes a small RNA that regulates pnp mRNA levels (Fontaine et al., 2016).
Surprisingly, the largest category consisted of genes involved in DNA replication, repair, and segregation (Figure S1C). These genes included polA, which encodes DNA polymerase I; rnhA, which encodes RNase H1, which cleaves the RNA strand of RNA-DNA hybrids that form during DNA replication and transcription (Hollis and Shaban, 2011); recC, which encodes a subunit of the RecBCD helicase-nuclease complex that functions in double-strand break repair (Dillingham and Kowalczykowski, 2008); ruvA and ruvC, whose products resolve Holliday junctions (Wyatt and West, 2014); and uvrD, which encodes a helicase involved in nucleotide excision repair (Kisker et al., 2013). We also identified mutations in yebC, a transcriptional regulator that contributes to E. coli survival after ionizing radiation (Byrne et al., 2014). Other mutations disrupted ftsK, which encodes a DNA translocase with roles in chromosome segregation (Kaimer and Graumann, 2011); and parA and parB, which encode proteins required for plasmid segregation (Gerdes et al., 2010). We also obtained transposon insertions in several genes that function in other processes (Figure S1D).
To compare the extent to which the various mutations increased Prsr-lacZ expression, we performed β-galactosidase assays. We focused on the three major categories (Figures S1A–S1C) and assayed cells between the mid-logarithmic and early stationary phases of growth. For all genes that were sites of transposon insertion in both strains (rtcB, pnp, sraG, ruvA, and yebC), β-galactosidase levels were >10-fold higher in the 14028s strain (Figures 1E and 1F). Due to the stronger induction, subsequent experiments were performed in the 14028s strain.
As the sraG RNA downregulates pnp mRNA (Fontaine et al., 2016), it was surprising that mutations in both pnp and sraG increased operon activation. Since sraG overlaps the pnp promoter, and our mutations are within or near this promoter, we examined PNPase levels by immunoblotting. PNPase was undetectable in sraG mutants (Figure 1G), indicating the transposon insertions abrogate PNPase synthesis.
To confirm that loss of function of the affected genes was responsible for operon activation, we generated strains lacking open reading frames (ORFs) that were sites of transposon insertion. We monitored operon activation using western blotting to detect expression of Rsr fused at the N terminus to three copies of the FLAG epitope. When strains containing deletions in truA, pnp, ruvA, or rnhA were grown to mid-logarithmic phase, FLAG3-Rsr increased compared to wild-type cells (Figure 2A). Although the low levels of FLAG3-Rsr in ΔrtcB strains were not significantly different from wild-type strains, strains lacking pnp and either truA or rtcB contained higher levels of FLAG3-Rsr than either deletion alone, revealing that deletions in PNPase, TruA, and RtcB act additively.
Figure 2. tRNA Fragments Accumulate in Some Mutant Strains.
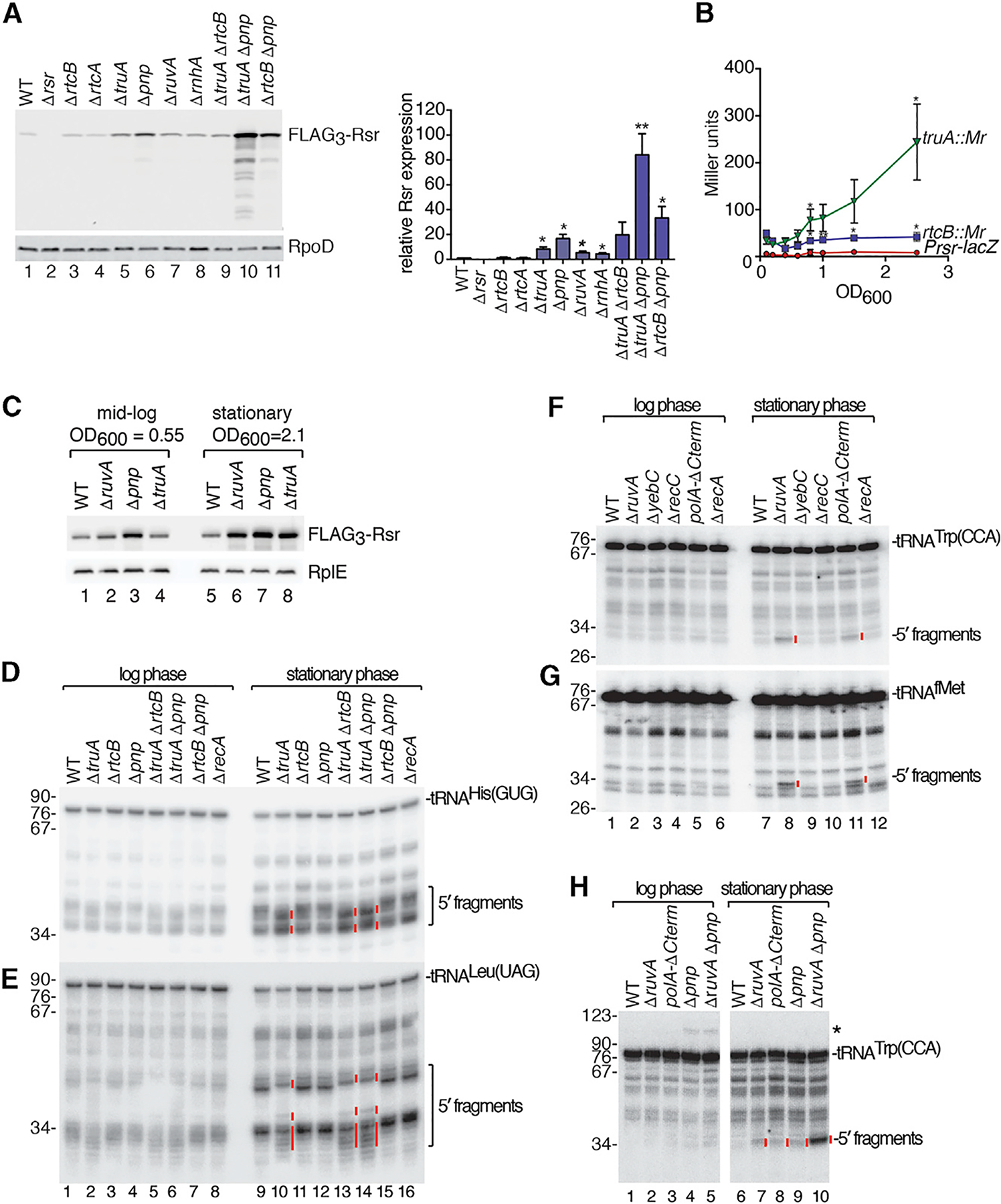
(A) 14028s FLAG3-rsr strains carrying the indicated deletions were subjected to immunoblotting to detect FLAG3-Rsr. RpoD, loading control. Right, quantitation (n = 3). p values are relative to the wild-type strain.
(B) β-Galactosidase activity was assayed in the indicated Prsr-lacZ strains as a function of growth. p values are relative to the Prsr-lacZ strain.
(C) Lysates of the indicated FLAG3-rsr strains were subjected to immunoblotting to detect FLAG3-Rsr. RplE, loading control.
(D and E) RNA from the indicated strains was subjected to northern blotting to detect 5′ halves of tRNAHis(GUG) (D) and tRNALeu(UAG) (E).
(F and G) RNA from the indicated strains was subjected to northern blotting to detect 5′ halves of tRNATrp(CCA) (F) and tRNAfMet (G).
(H) RNA from the indicated strains was probed to detect 5′ halves of tRNATrp(CCA). Asterisk denotes a tRNA precursor.
Data in (A) and (B) are mean (n = 3) ± SEM. p values were calculated with two-tailed unpaired t tests. *p < 0.05 and **p < 0.01.
In (D)–(H), red lines denote fragments unique to mutant strains in stationary phase. See also Figure S2.
Since expression of the E. coli rtcBA operon increases in stationary phase (Temmel et al., 2017), we examined expression of the S. Typhimurium operon as a function of growth. Although Prsr-lacZ expression did not change in the parental strain, expression increased more than 5-fold in the truA transposon mutant (truA::Mr) between mid-log and stationary phase (Figure 2B). Western blotting revealed that although the levels of FLAG3-Rsr in wild-type cells increased less than 1.5-fold, FLAG3-Rsr increased 2.8-fold in ΔruvA strains, 2-fold in Δpnp strains, and 5-fold in ΔtruA strains in stationary phase relative to mid-log phase (Figure 2C). Thus, mutations that disrupt certain genes involved in tRNA metabolism and DNA repair activate expression of the rsr-yrlBA-rtcBA operon and the effects are more severe in stationary phase.
Distinct tRNA Fragments Accumulate in Mutant Strains
Since one category of mutations that resulted in operon activation were predicted to affect tRNA metabolism, we examined tRNA levels in the mutant strains. We examined truA, since pseudouridines in tRNA anticodon arms contribute to structural stability (Arnez and Steitz, 1994); rtcB, since RtcB ligates eukaryotic and archaeal tRNAs after intron excision; and pnp, which degrades aberrant pre-tRNAs (Li et al., 2002). We grew strains lacking each gene to mid-log or stationary phase, isolated RNA, and performed northern blotting to detect two tRNAs that are TruA substrates, tRNAHis(GUG) and tRNALeu(UAG). Several 5′ (Figures 2D and 2E) and 3′ (Figures S2A and S2B) fragments of these tRNAs were detected in wild-type and mutant strains during logarithmic growth and increased in cells in stationary phase.
Notably, some fragments in ΔtruA strains were altered in mobility and/or levels compared to wild-type strains. For both tRNAHis(GUG) and tRNALeu(UAG), some 5′ and 3′ fragments were reduced in ΔtruA strains, while others became prominent (Figures 2D, 2E, S2A, and S2B, lanes 2 and 10, red lines). Possibly, the decreased stability of these tRNAs in ΔtruA strains renders them susceptible to cleavage at distinct sites. Fragments of tRNAs that are not TruA substrates, such as tRNATrp(CCA), were unchanged in ΔtruA strains (Figure S2C).
To determine if accumulation of specific tRNA fragments was a common feature of our mutants, we examined strains carrying deletions in genes important for DNA replication and repair. Remarkably, we detected discrete tRNA fragments in some strains. This was most apparent in stationary phase, where we detected 5′ fragments of tRNATrp(CCA) and tRNAfMet in strains that lacked ruvA or were deleted for the 3′ end of the essential polA gene (polAΔC) (Figures 2F and 2G). These fragments, as well as 5′ and 3′ fragments of other tRNAs, such as tRNACys(GCA), increased further in ΔruvA cells that also lacked pnp (Figures 2H and S2E–S2G), indicating that PNPase may degrade the fragments. Although we did not detect specific fragments in ΔyebC or ΔrecC strains, low levels of these fragments may be obscured by the background of nonspecific fragments that we show later to be irrelevant for operon activation.
Accumulation of tRNA Fragments and Operon Induction Occur through at Least Two Pathways, One of Which Requires RecA
Many of the identified genes, such as ruvA, polA, ruvC, yebC, uvrD and ftsK, were also isolated in a screen for E. coli mutations that result in expression of the SOS regulon, a gene network induced upon DNA damage (O’Reilly and Kreuzer, 2004). To determine if the SOS response was required for tRNA fragment accumulation or activation of the rsr-yrlBA-rtcBA operon, we examined strains lacking RecA, since expression of the SOS regulon initiates when activated RecA assists cleavage of the LexA repressor. Notably, the tRNATrp(CCA) fragments that accumulated in ΔruvA cells were strongly reduced in ΔruvA ΔrecA cells (Figure 3A). Western blotting revealed that although the operon was activated in ΔruvA stationary phase cells, it was expressed similarly to wild-type cells in ΔruvA ΔrecA cells (Figure 3B).
Figure 3. Accumulation of tRNA Fragments and Operon Activation Occur via Two Path- ways.
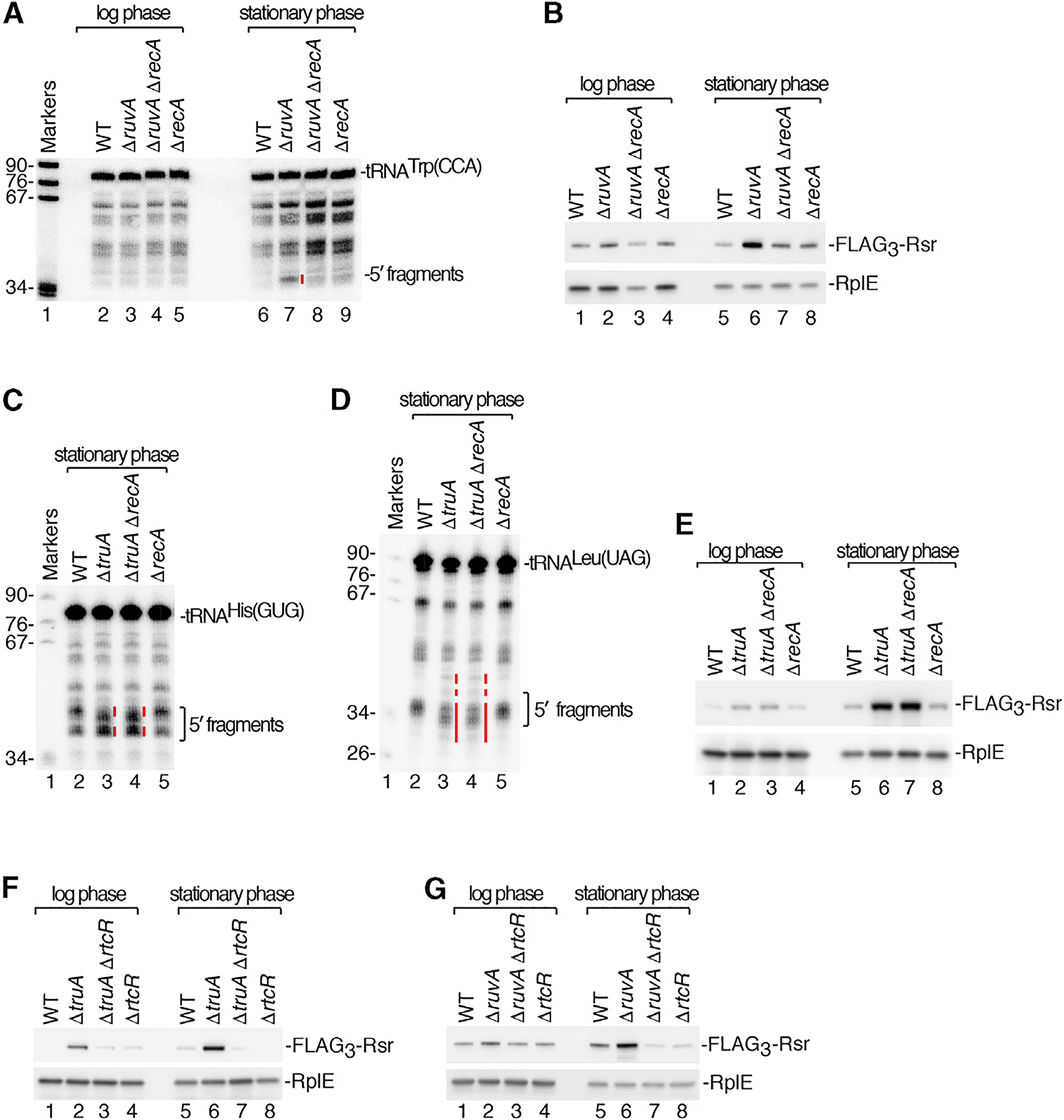
(A) RNA from strains grown to mid-log or stationary phase was subjected to northern blotting to detect tRNATrp(CCA) 5′ halves. Lane 1, size markers (nt).
(B) Immunoblots were performed on lysates from the indicated FLAG3-rsr strains to detect FLAG3-Rsr. RplE, loading control.
(C and D). RNA from the indicated strains grown to stationary phase was subjected to northern blotting to detect 5′ halves of tRNAHis(GUG) (C) and tRNA-Leu(UAG) (D).
(E–G) Lysates from the indicated strains were immunoblotted to detect FLAG3-Rsr. RplE, loading control.
In (A), (C), and (D), red lines denote fragments differing in mobility or levels in mutant strains.
In contrast, accumulation of tRNA fragments in ΔtruA cells was unaffected by recA deletion (Figures 3C and 3D), supporting the hypothesis that tRNA breakage occurs in these cells due to lack of pseudouridine in some anticodon stems. Activation of the rsr-yrlBA-rtcBA operon in ΔtruA strains was also unaffected by recA deletion (Figure 3E). Our results support a model in which tRNA fragments can be generated through at least two pathways, one involving tRNA fragility and a second requiring the RecA component of the SOS response. In this model, accumulation of tRNA fragments may result, directly or indirectly, in production of a ligand that binds the RtcR CARF domain to activate the operon. Consistent with this hypothesis, operon activation in both ΔtruA and ΔruvA cells requires RtcR (Figures 3F and 3G).
DNA Damaging Agents Cause tRNA Cleavage and Operon Activation
To obtain further evidence that tRNA fragment accumulation was linked to operon activation, we asked if treatment with DNA damaging agents results in tRNA cleavage. Upon treatment with the interstrand crosslinker mitomycin C (MMC), which leads to operon activation (Kurasz et al., 2018), FLAG3-Rsr expression was detected within one hour and peaked by 2 h (Figure 4A). Cleavage of the LexA repressor occurred with similar kinetics. Operon induction required RecA and LexA cleavage, as FLAG3-Rsr was not detected in ΔrecA strains or strains carrying the uncleavable lexA3 allele (Little and Harper, 1979) (Figures 4A and S3A). (Deletions of lexA are lethal, because S. Typhimurium contains LexA-regulated prophages; Bunny et al., 2002; Lemire et al., 2011.) Similar to ΔruvA and ΔtruA strains, operon expression required RtcR (Figure S3B).
Figure 4. DNA Damaging Agents Result in tRNA Cleavage and Operon Activation.
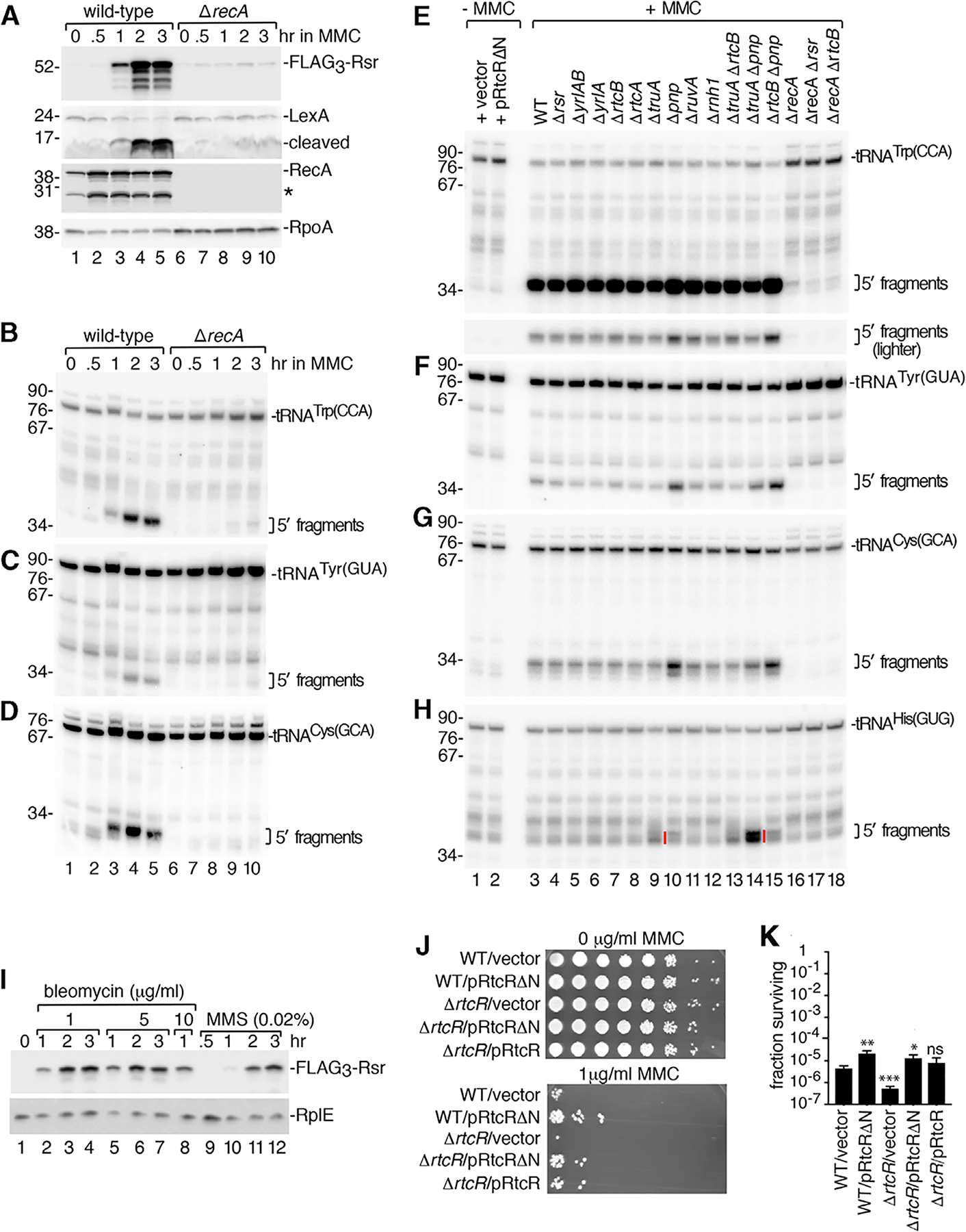
(A) After growing wild-type and ΔrecA FLAG3-rsr strains in MMC for the indicated times, FLAG3-Rsr, LexA, RecA, and RpoA (loading control) were detected on immunoblots. Asterisk, RecA fragment. (B–D) After growing wild-type and ΔrecA strains in MMC, northern analysis was performed to detect 5′ halves of the indicated tRNAs.
(E–H) RNA from strains grown in MMC was subjected to northern analysis to detect 5′ halves of the indicated tRNAs (lanes 3–18). Lanes 1 and 2, RNA from wild-type cells carrying empty vector or pRtcRΔN, respectively. In (E), the bottom panel is a lighter view of the 5′ halves. In (H), red lines denote fragments differing in mobility in ΔtruA and ΔtruA Δpnp strains.
(I) After treating FLAG3-rsr strains with bleomycin or MMS for 0.5, 1, 2, or 3 h, lysates were subjected to immunoblotting to detect FLAG3-Rsr and RplE.
(J) After growth of the indicated strains in 0 (top) and 1 μg/mL MMC (bottom panel) for 2 h, serial 10-fold dilutions were spotted on Luria-Bertani broth (LB) agar and grown at 37°C.
(K) Aliquots of the cells in (J) were plated on LB agar and colonies counted to determine the fraction of surviving cells. Data are mean (n = 3) ± SEM. p values were calculated with two-tailed unpaired t tests. *p < 0.05, **p < 0.01, and ***p < 0.001; ns, not significant.
See also Figure S3.
Upon MMC treatment, many tRNAs, including tRNATrp(CCA), tRNATyr(GUA), and tRNACys(GCA), underwent cleavage, as both 5′ (Figures 4B–4H) and 3′ (Figures S3D and S3E) fragments accumulated. Fragments were detected within 1 h of adding MMC but were absent or reduced in ΔrecA and lexA3 strains (Figures 4B–4D and S3C–S3E). Levels of some full-length tRNAs, such as tRNATrp(CCA), decreased concomitantly (Figures 4E and S3D), indicating the fragments derive from cleavage of mature tRNAs rather than reduced decay of preexisting fragments. Although the operon-encoded YrlA RNA contains a tRNA-like domain (Chen et al., 2014), fragments of this RNA were not detected (Figure S3F), indicating it is not a substrate for the RecA-dependent nuclease. Operon activation and tRNA fragment accumulation also occurred upon treatment with other DNA damaging agents, such as bleomycin, which cleaves single- and double-stranded DNA (Bolzán and Bianchi, 2018), and methyl methanesulfonate (MMS), which methylates DNA (Wyatt and Pittman, 2006) (Figures 4I, S3G, and S3H). Together, our data indicate that a tRNA anticodon nuclease is activated as part of the S. Typhimurium SOS response.
Inspection of the tRNA fragments that accumulated upon MMC treatment revealed some differences between the mutant strains. Most 5′ and 3′ fragments increased in cells lacking PNPase (Figures 4E–4G and S3E, lane 10). For some tRNAs that are truA substrates, the fragments that accumulated were altered in mobility in ΔtruA strains compared to wild-type cells and increased further in ΔtruA Δpnp strains (Figure 4H, lanes 9 and 14, red lines), implicating PNPase in their degradation. Fragments of tRNATrp(CCA) and tRNATyr(GUA) were slightly increased in ΔrtcB Δpnp strains compared to Δpnp strains (Figures 4E, 4F, and S3D, lanes 10 and 15), supporting a possible role for RtcB in repairing broken tRNAs.
To further assess the link between tRNA fragments and operon activation, we examined the E. coli rtcBA operon, which is regulated by RtcR. Overexpression of two toxins that cleave tRNAs, but not MMC incubation, activates this operon (Engl et al., 2016; Kurasz et al., 2018). If tRNA fragments are important for activation, then failure of the E. coli operon to be expressed in the presence of MMC may be due to the absence of these fragments. Consistent with our model, tRNA fragments did not accumulate in E. coli during growth in MMC (Figures S3I and S3J). Western blotting to detect RtcB confirmed that the E. coli operon was not induced (Figure S3K).
Together, our results reveal that RecA-dependent tRNA cleavage and rsr-yrlBA-rtcBA operon activation occur during conditions that induce the SOS response in S. Typhimurium. Since tRNA fragments do not accumulate when the operon is activated by overexpressing constitutively active RtcRΔN (Figures 4E–4H, S3D, and S3E, lane 2), operon activation in itself does not cause fragment accumulation. Instead, this result, together with our data that the operon is activated in ΔtruA strains in the absence of DNA damage, supports a model in which tRNA cleavage results in a ligand that activates the operon.
We determined whether expression of the rsr-yrlBA-rtcBA operon is important for survival after MMC exposure. Wild-type strains expressing constitutively active RtcR (RtcRΔN) were more resistant to 1 μg/mL MMC than the same cells carrying an empty vector, while strains lacking RtcR (ΔrtcR) were less resistant than wild-type cells (Figure 4J). Quantitation revealed that wild-type strains overexpressing RtcR were 4.8-fold more resistant than cells carrying an empty vector, while ΔrtcR cells were 9.4-fold less resistant than wild-type cells (Figure 4K). The decreased resistance of ΔrtcR strains could be complemented by expressing either RtcRΔN or wild-type RtcR on a plasmid. Thus, operon activation confers a growth advantage in MMC.
Most tRNA Cleavage Occurs in the Anticodon Loop, Leaving a Cyclic Phosphate End
To understand how tRNA cleavage could result in a ligand, we characterized the fragments that accumulate upon DNA damage. Since endonucleases can leave 2′, 3′-cyclic phosphate, 3′-phosphate, or 3′-OH at the 3′ end of the 5′ fragment, we tested if pre-treatment with T4 polynucleotide kinase (PNK), which converts 2′, 3′-cyclic phosphate and 3′-phosphate to 3′-OH, was needed for T4 RNA ligase (which requires 3′-OH) to ligate a 5′-phosphate-containing oligonucleotide to the 5′ fragments (Figure S4A). Comparison of RNA from wild-type and Δpnp strains, followed by northern blotting to detect specific tRNAs, revealed that ligation to the 5′ fragments of all examined tRNAs increased in the presence of PNK (Figures S4B–S4E).To distinguish between 3′-phosphate and cyclic phosphate, we asked if treatment with calf intestinal phosphatase (CIP) (which removes 3′-phosphate, but not 2′, 3′-cyclic phosphate) could substitute for PNK or if treatment with acid (which opens up cyclic phosphate) was also required. Maximal ligation of the oligonucleotide to the 5′ tRNA fragments occurred in the presence of acid and CIP, indicating some fragments end in cyclic phosphate (Figures S4G–S4J). Thus, one or more metal-independent endoribonucleases (which leave 2′, 3′-cyclic phosphate) (Yang, 2011) contribute to cleavage. As some ligation occurred with CIP alone, 5′ fragments ending in 3′ phosphate were also present (Figures S4G–S4J). These species could derive from fragments ending in 2′, 3′-cyclic phosphate or from cleavage by additional nucleases.
Using 3′ rapid amplification of cDNA ends, we determined that cleavage occurred within the anticodon loops. For tRNATrp(CCA), tRNATyr(GUA), tRNACys(GCA), and tRNAPhe(GAA), most 5′ fragments terminated at position 36, the last nucleotide of the anticodon, with the apparent cleavage site between two adenines (Figure S4K, arrowheads; Table S2). For tRNAfMet, most cleavage occurred after position 37, between two adenines (Figure S4K; Table S2). For tRNALeu(UAG), cleavage was heterogenous, with fragments ending at multiple sites (Figure S4K, arrows; Table S2). Slightly shorter 5′ fragments of most tRNAs were also recovered, which may represent exonucleolytic nibbling and/or other cleavages (arrows).
We tested if the abundant metal-independent endonuclease RNase I was important for cleavage. In strains lacking RNase I (Δrna), we observed striking decreases in all nonspecific fragments (Figure 5A, lanes 10–17). However, the specific tRNATrp(CCA) and tRNALeu(UAG) fragments that accumulate in ΔruvA strains were unchanged in ΔruvA Δrna strains (Figures 5A and 5B, lanes 6 and 14). Although we could not detect specific tRNACys(GCA) or tRNATyr(GUA) fragments in Δpnp and ΔruvA strains due to the background of nonspecific fragments, specific fragments were evident in Δpnp Δrna and ΔruvA Δrna strains (Figures 5C and S5A, lanes 13 and 14). The RecA-dependent tRNA fragments detected in MMC were also unaffected (Figures 5D–5F and S5B, lanes 14–17). As expected if these fragments are important for formation of the ligand that binds RtcR, operon induction was unaffected in Δrna strains (Figure 5G).
Figure 5. RtcA Is Important for Operon Activation.
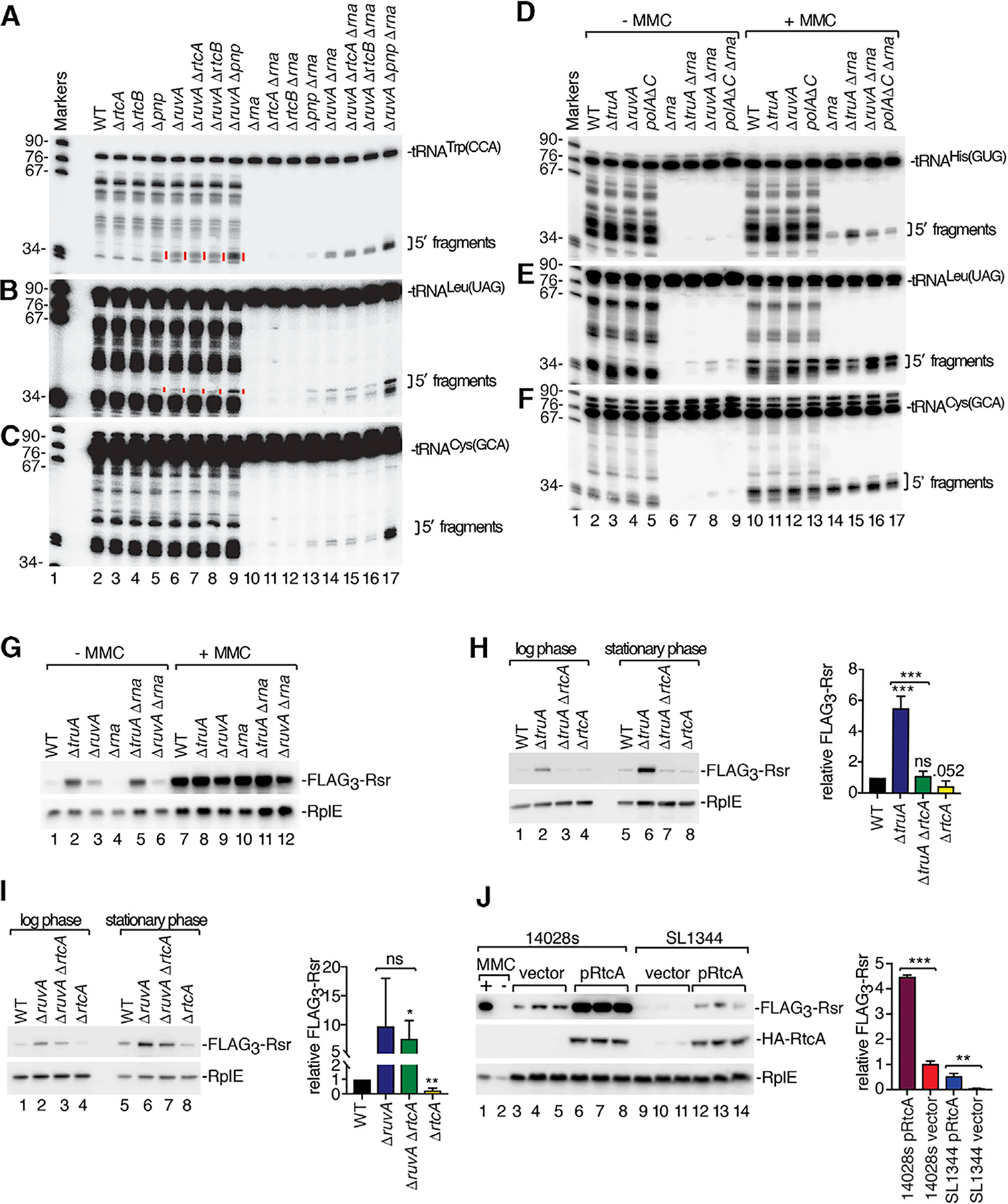
(A–C) RNA from the indicated strains was subjected to northern blotting to detect 5′ halves of tRNATrp(CCA) (A), tRNALeu(UAG) (B), and tRNACys(GCA)
(C). In (A) and (B), specific fragments that become evident in Δrna strains are denoted by red lines in strains containing RNase I (lanes 5–8).
(D–F) After growing the indicated strains without or with MMC, RNA was subjected to northern blotting to detect 5′ halves of tRNAHis(GUG) (D), tRNALeu(UAG) (E) and tRNACys(GCA) (F).
(G) After growing the indicated FLAG3-rsr strains without or with MMC, lysates were subjected to immunoblotting to detect FLAG3-Rsr and RplE.
(H and I) Lysates of FLAG3-rsr strains carrying the indicated deletions were immunoblotted to detect FLAG3-Rsr. RplE, loading control. Right, quantitation (n = 3).
(J). Lysates of 14028s and SL1344 FLAG3-rsr strains carrying empty vector (lanes 3–5 and 9–11) or pHA-RtcA expressing RtcA fused to a hemagglutinin (HA) epitope tag (lane 6–8 and 12–14) were subjected to immunoblotting to detect FLAG3-Rsr, HA-RtcA, and RplE. Right, quantitation (n = 3). Data in (H), (I), and (J) are mean (n = 3) ± SEM. p values were calculated with two-tailed unpaired t tests. *p < 0.05, **p < 0.01, and ***p < 0.001; ns, not significant. See also Figures S4 and S5 and Table S2.
Despite much effort, we were unable to identify the endonuclease responsible for the RecA-dependent tRNA cleavage. We examined strains lacking 22 other toxins and potential nucleases, including three metal-independent nucleases encoded adjacent to consensus LexA sites. These are YafQ, the toxin component of an antitoxin:toxin cassette encoded downstream of the rsr-yrlBA-rtcBA operon (Figure S5C); HigB, which is encoded with its HigA antitoxin downstream of a LexA-regulated ORF; and HigB2, which is encoded on the opposite strand such that HigB and HigB2 may be controlled through the same palindromic LexA site (Figure S5D). All three toxins are part of the RelE family, which bind ribosomes and cleave translating mRNAs (Harms et al., 2018). Northern and western blotting of ΔruvA strains carrying deletions of all three toxins revealed that they were not required for accumulation of tRNATrp(CCA) fragments or operon activation (Figures S5E and S5F). We tested strains deleted for 19 other toxins and potential nucleases, with similar negative results (Figures S5G–S5J). Since RNA decay pathways are often redundant (Houseley and Tollervey, 2009), multiple endonucleases may carry out tRNA cleavage.
RtcA Is Important for Induction of the rsr-yrlBA-rtcBA Operon
To test if 2′, 3′-cyclic phosphate RNA ends were important for operon activation, we examined the role of RtcA, which converts 3′-phosphate ends to 2′, 3′-cyclic phosphate (Genschik et al., 1998). Operon activation decreased 4.8-fold in ΔtruA ΔrtcA cells compared to ΔtruA cells and was similar to wild-type cells (Figure 5H). Thus, the lower stability of some tRNA anticodon stems in ΔtruA strains may make them susceptible to nucleases that leave 3′-phosphate ends that are subsequently converted by RtcA to 2′, 3′-cyclic phosphate. In contrast, operon activation was not significantly different between ΔruvA and ΔruvA ΔrtcA strains (Figure 5I), supporting the idea that tRNA halves that accumulate in ΔruvA strains derive from cleavage by a metal-independent endonuclease.
We also found that the low level of expression of the rsr-yrlBA-rtcBA operon in wild-type 14028s strains in stationary phase was reduced in ΔrtcA strains (Figures 5H and 5I). This suggested that the RtcA levels in 14028s, but not SL1344 strains, might be sufficient to convert broken tRNA ends to 2′, 3′-cyclic phosphates as part of a feedback loop resulting in low levels of operon expression. To test if increased RtcA levels were sufficient for operon activation, we expressed RtcA under control of the arabinose-inducible promoter in the SL1344 and 14028s strains. We detected RtcA in both strains in the absence of arabinose due to the leakiness of this promoter (Figure 5J). Importantly, operon expression increased, as Rsr levels were 4.5-fold higher in 14028s strains and 11-fold higher in SL1344 strains (Figure 5J) compared to the same cells carrying empty vector. We conclude that RtcA expression is sufficient to activate the rsr-yrlBA-rtcBA operon. As expression of the operon in the SL1344 strain did not reach that of 14028s, additional differences between these strains contribute to operon activation.
The RtcR CARF Domain Binds tRNA Fragments with Cyclic Phosphate Ends
A model that accommodates all our data is that 5′ tRNA fragments ending in 2′, 3′-cyclic phosphate are the ligands that bind the CARF domain of RtcR to activate operon transcription. However, since many tRNAs undergo cleavage during growth in MMC, it was unclear which 5′ fragments could be responsible. To reduce the candidates, we used northern blotting to identify the 5′ tRNA fragments that accumulate in ΔtruA strains, since only some tRNAs are TruA substrates. To assist visualization, we analyzed strains that also lacked RNase I. Of the tRNAs assayed (tRNAGln(CUG), tRNAHis(GUG), tRNALeu(UAG), tRNALeu(UAA), tRNALeu(CAA), tRNACys(GCA), tRNATyr(GUA), and tRNAPhe(GAA)), the most prominent fragments were from tRNALeu(UAG) (Figure 5E, lane 7). These fragments increased when PNPase was also deleted (Figure 6A), consistent with the enhanced operon activation in ΔtruA Δpnp strains (Figure 2A). The fragments ended in cyclic phosphate, as ligation of an oligonucleotide to most tRNALeu(UAG) 5′ fragments required treatment with acid and CIP (Figure S6A). Thus, tRNALeu(UAG) 5′ fragments were good candidates for a ligand that activates the operon.
Figure 6. RtcR Oligomerizes on Binding tRNA Fragments Ending in Cyclic Phosphate.
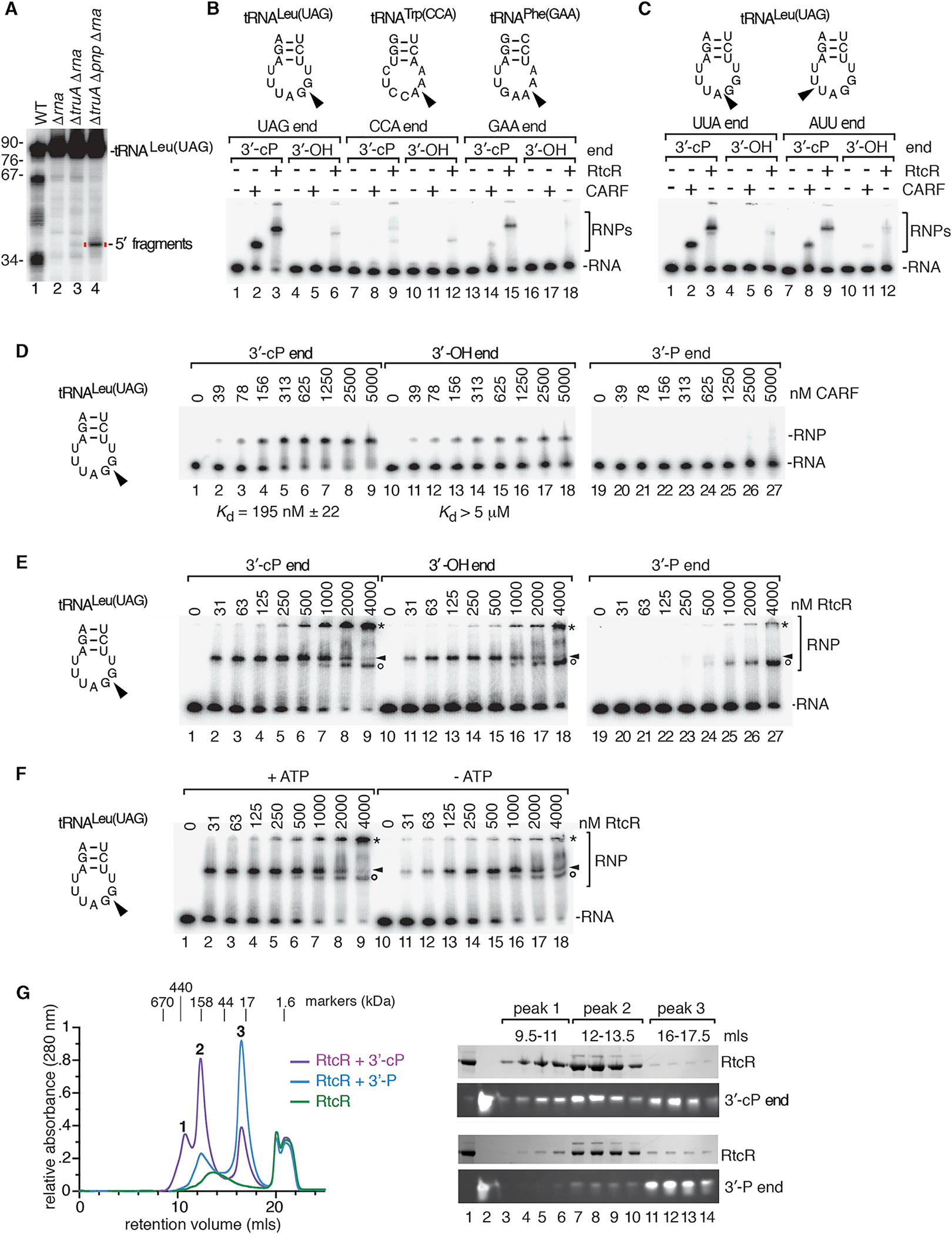
(A) RNA from the indicated strains was subjected to northern blotting to detect tRNALeu(UAG) 5′ halves.
(B) 32P-labeled 5′ tRNA halves (1 nM) ending after the anticodon in 2′, 3′-cyclic phosphate (lanes 1–3, 7–9, and 13–15) or 3′-OH (lanes 4–6, 10–12, and 16–18) were incubated with no protein, 0.5 μM CARF domain, or 0.5 μM RtcR. Reactions contained 1 mM ATP. RNA-protein complexes (RNPs) were separated from naked RNA in native gels.
(C) Similar to (B), except that 5′ tRNALeu(UAG) halves ended at the indicated positions.
(D and E) 5′ tRNALeu(UAG) halves ending after the G of the anticodon were incubated with the indicated concentrations of CARF domain (D) or RtcR (E). RNAs ended in 2′, 3′-cyclic phosphate (lanes 1–9), 3′-OH (lines 10–18), or 3′-phosphate (lanes 19–27). In (E), arrowheads denote the first complexes formed, while asterisks denote complexes that could represent oligomers. Circles, complexes that form on all three RNAs at the highest RtcR concentrations. In (D) and (E), the samples were fractionated in two gels. Binding reactions in (E) contained 1 mM ATP.
(F) tRNALeu(UAG) halves ending in cyclic phosphate were mixed with the indicated concentrations of RtcR with or without 1 mM ATP. Complexes are designated with arrowheads, asterisks, and circles as in (E).
(G) Size exclusion chromatography was performed on RtcR (48 μM) alone or bound to 48 μM 5′ tRNALeu(UAG) halves ending in 3′-phosphate or 2′, 3′-cyclic phosphate. Left, overlay of chromatograms. Right, proteins and RNA in the indicated peaks were fractionated in SDS-PAGE and denaturing polyacrylamide gels, respectively. The complex eluting at 20 mL is ATP from the binding reaction.
See also Figure S6.
We tested whether 5′ fragments of tRNALeu(UAG) or other tRNAs could bind the CARF domain. We expressed both the CARF domain and RtcR in E. coli (Figures S6B and S6C) and performed electrophoretic mobility shift assays (EMSAs) with the purified proteins. Initial studies in which we incubated 5′ tRNA halves derived from tRNALeu(UAG), tRNATrp(CCA), and tRNAPhe(GAA) ending in 2′, 3′-cyclic phosphate or 3′-OH revealed that both the isolated CARF domain and RtcR bound most efficiently to 5′ halves of tRNALeu(UAG) (Figure 6B). We also tested tRNALeu(UAG) 5′ halves ending at other nucleotides in the anticodon loop. Although all tested 5′ halves ending in cyclic phosphate bound both the CARF domain and RtcR, fragments that ended after the G of the UAG were most efficiently bound (Figures 6B and 6C). Quantitative assays revealed that the amount of CARF domain required to shift 50% of these tRNALeu(UAG) halves ending in cyclic phosphate was ~200 nM, while more than 5 μM was required to shift 50% of the same RNAs ending in in 3′-OH (Figure 6D). Specific binding to the CARF domain was not detected when the fragments ended in 3′-phosphate.
We also examined binding to full-length RtcR, in which the CARF domain is followed by AAA+ (ATPases associated with multiple cellular activities) and helix-turn-helix DNA-binding domains. Based on other bacterial enhancer proteins, RtcR is expected to be a dimer in its unliganded form. In the absence of ligand, the CARF domain negatively regulates transcription, most likely by preventing the AAA+ domain from oligomerizing (Genschik et al., 1998; Bush and Dixon, 2012). The amount of RtcR required to shift 50% of the tRNALeu(UAG) fragments ending in cyclic phosphate was slightly less than that of the isolated CARF domain (~125 nM; Figure 6E, arrowheads). Beginning at 125 nM RtcR, we also detected a larger complex. This complex, which could represent oligomerization, eventually became the predominant species (Figure 6E, asterisk). Formation of both complexes, but not the complex formed with the isolated CARF domain, was enhanced when ATP was present, suggesting conformational changes mediated by the AAA+ domain contribute to their formation (Figures 6F and S6D). As both RtcR-containing complexes were reduced in levels when the fragments ended in 3′-OH and were barely detectable when the fragments ended in 3′-phosphate, tRNA 5′ halves ending in cyclic phosphate are the preferred ligand for their formation. At the highest protein concentrations, a third complex that migrated slightly faster than the initial complex was detected (Figure 6E, circles). As formation of this complex was not dependent on the presence of either ATP or a cyclic phosphate RNA end, this complex may represent nonspecific interactions of the RNA fragment with RtcR.
To further characterize these complexes, we performed size exclusion chromatography and analyzed the composition of the peaks using gel electrophoresis. In these experiments, we mixed RtcR with equimolar amounts of 5′ tRNALeu(UAG) halves to maximize complex formation. When the tRNA halves ended in cyclic phosphate, two RtcR/tRNA complexes formed that migrated at ~400 and 160 kDa, consistent with hexameric (358 kDa) and dimeric (119 kDa) forms of RtcR bound to three and one tRNA halves, respectively (12 kDa each). A third peak consisted of unbound tRNA halves (Figure 6G). Notably, when the tRNA halves terminated in 3′-phosphate, the 400-kDa complex was not detected, the 160-kDa complex was reduced, and the peak corresponding to unbound RNA increased (Figure 6G). We conclude that RtcR forms oligomers upon binding tRNA fragments ending in cyclic phosphate.
DISCUSSION
Although rtcBA and rsr-yrlBA-rtcBA RNA repair operons are widespread in bacteria, the signals that activate the operons have been obscure. We showed that tRNA 5′ fragments ending in 2′, 3′-cyclic phosphate bind the RtcR CARF domain. These tRNA fragments accumulate in the presence of mutations in genes that maintain tRNA homeostasis and upon activation of a LexA-regulated endoribonuclease. Since operon expression is important for S. Typhimurium survival after MMC exposure, our experiments uncover a signaling pathway involving tRNA and implicate rsr-yrlBA-rtcBA operon components in the repair of nucleic acid damage.
A Signaling Pathway Involving tRNA
Our data support a model in which 5′ tRNA fragments ending in 2′, 3′-cyclic phosphate bind the RtcR CARF domain, triggering oligomerization of the AAA+ domain. These fragments accumulate in ΔtruA strains, which contain hypomodified tRNAs with unstable anticodon arms (Figure 7A), in strains carrying mutations that result in DNA damage and in wild-type cells treated with DNA damaging agents (Figure 7B). These tRNA fragments also accumulate in strains lacking PNPase (Figure 7C). Binding of the fragments to RtcR triggers oligomerization, resulting in operon transcription (Figure 7D). Our proposal that 5′ tRNA fragments ending in 2′, 3′-cyclic phosphate are critical ligands is consistent with data that the E. coli operon is activated by ectopic expression of colicin D and S. Typhimurium LT2 VapC (Engl et al., 2016), toxins that cleave the anticodon loops of tRNAArg and tRNAMet, respectively (Masaki and Ogawa, 2002; Winther and Gerdes, 2011).
Figure 7. Model for Operon Activation.
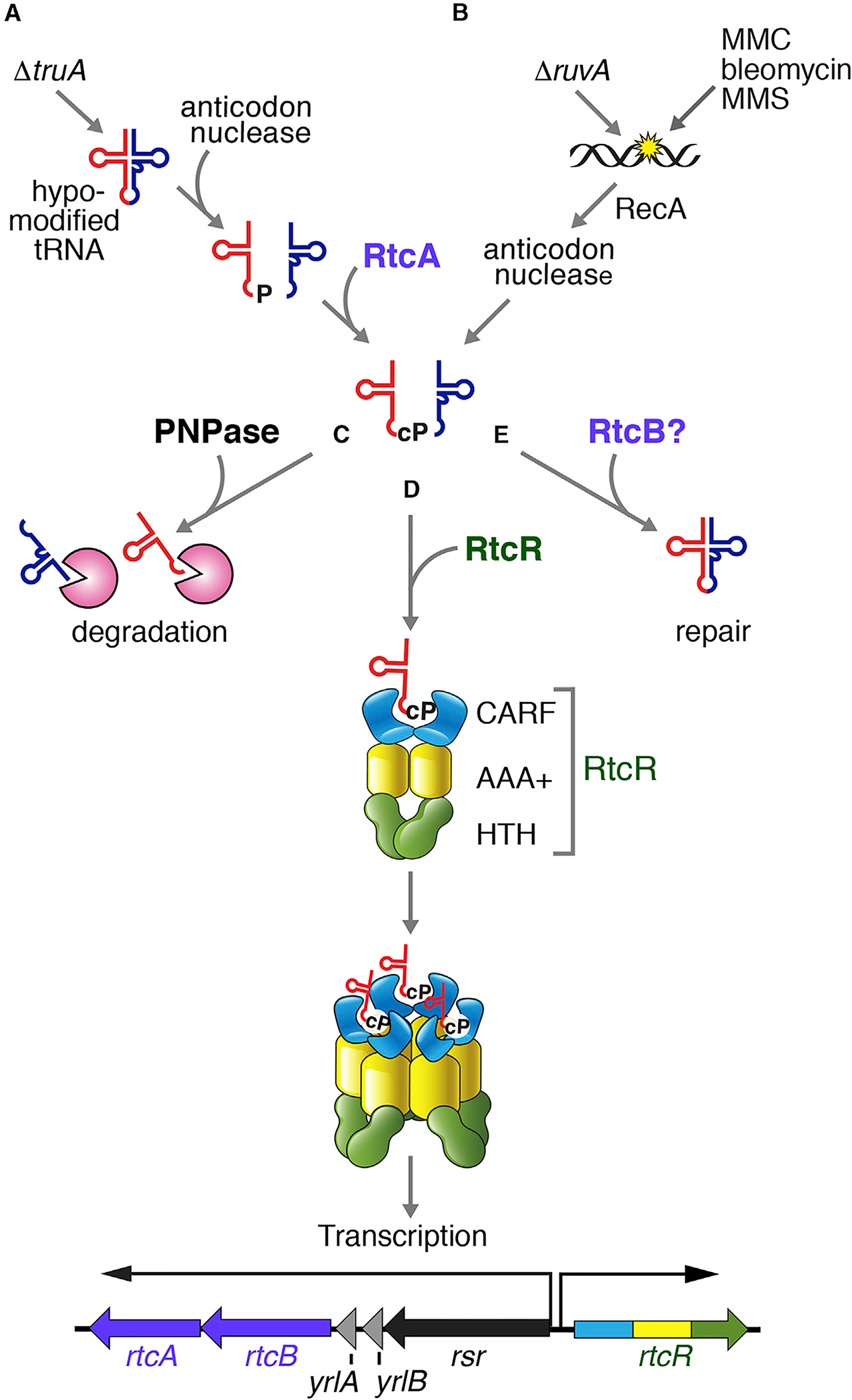
(A) In ΔtruA strains, hypomodified tRNAs are susceptible to nuclease, leading to tRNA fragment accumulation. RtcA converts the ends of the 5′ fragments to 2′, 3′-cyclic phosphate (cP).
(B) In ΔruvA strains and after treatment with DNA damaging agents, a RecA-regulated endonuclease cleaves tRNAs, resulting in 5′ halves ending in cyclic phosphate.
(C–E) We propose that 5′ tRNA halves ending in cyclic phosphate can be degraded by PNPase (C); bind the CARF domain of a RtcR dimer, resulting in oligomerization and operon activation (D); and may also be repaired by RtcB (E).
Although canonical CARF domains, such as those of Csm6 and Csx1, dimerize to form a symmetric binding pocket for cOA (Jia et al., 2019; Molina et al., 2019; Garcia-Doval et al., 2020), structural analyses will be required to determine how the divergent RtcR CARF domain recognizes asymmetric tRNA fragments ending in cyclic phosphate. As the affinity of the RtcR CARF domain for the in-vitro-synthesized tRNA halves used in our assays is lower than the 0.5–5 nM affinity of canonical CARF domains for cOA ligands (Kazlauskiene et al., 2017; Niewoehner et al., 2017), other proteins and/or tRNA modifications, which are often present in the vicinity of the anticodon, may contribute to recognition. It is also possible that another tRNA 5′ half may be a better ligand than the tRNALeu(UAG) fragment used in our studies or that tRNA 3′ fragments contribute.
If binding of 5′ tRNA fragments ending in 2′, 3′-cyclic phosphate activates the operon, how is signaling regulated? For CARF domains activated by cOA, signaling is terminated by ring nuclease activity that is encoded within the CARF domain (Athukoralage et al., 2019; Jia et al., 2019) or supplied by standalone nucleases (Athukoralage et al., 2018). One possibility is that RtcA and RtcB function as part of a feedback loop to ligate cleaved tRNAs and terminate operon expression. PNPase may contribute to reducing operon expression by degrading tRNA fragments (Figure 7C). However, since RNAs ending in 3′-phosphate or 2′, 3′-cyclic phosphate are not PNPase substrates (Singer, 1958), other enzymes must first convert these ends to 3′-OH.
It is curious that tRNA cleavage occurs upon DNA damage in S. Typhimurium. Since S. Typhimurium contains LexA-regulated prophages (Lemire et al., 2011), prophage-encoded endonucleases may carry out cleavage. In this case, the decreased levels of specific tRNAs may be peripheral to the DNA damage response, instead allowing the phage to reduce host protein synthesis or favor translation of specific proteins. It is also possible that tRNA cleavage, by decreasing protein synthesis and slowing division, allows more time for DNA repair. Given the growing evidence that RNA can template and otherwise enhance DNA repair (Michelini et al., 2018), another possibility is that the tRNA halves play a role in restoring genome integrity.
Functions of the rsr-yrlBA-rtcBA Operon
Our findings that (1) the operon is activated by tRNA cleavage, (2) RtcA is required for operon activation in ΔtruA cells, and (3) some tRNA fragments are present at slightly higher levels in ΔrtcB Δpnp cells than Δpnp cells support a role for RtcA and RtcB in tRNA repair (Figure 7E). Such a role would be consistent with the finding that RtcB seals anticodon loops after excision of intervening sequences in eukaryotes and archaea (Englert et al., 2011; Popow et al., 2011; Kosmaczewski et al., 2014). It would also be consistent with our observation that mutations in RtcB trigger operon expression, since tRNA halves might be expected to accumulate in ΔrtcB cells. Although we did not detect accumulation of tRNA halves in ΔrtcB cells, we did not examine the full complement of tRNAs. Our identification of conditions that activate the operon should allow us to determine the diversity of RtcA and RtcB substrates.
Although our studies did not address the roles of Rsr and Y RNAs, several possibilities can be envisioned. Since D. radiodurans Rsr and Y RNA assist PNPase in degrading structured RNA (Chen et al., 2013), they could potentially function in tRNA decay. An alternative, but not exclusive, possibility is that the tRNA-like domain of YrlA RNA, which is not a substrate for the anticodon nuclease (Figure S3F), acts as a competitive inhibitor of this nuclease.
Finally, we note that RtcB, Rsr, and Y RNAs could potentially function in DNA repair. E. coli RtcB can ligate DNA 3′-PO4 ends to DNA 5′-OH ends and can add a ppG cap to DNAs ending in 3′-phosphate, a modification that allows priming by DNA polymerase (Das et al., 2013, 2014). D. radiodurans Rsr contributes to survival after UV irradiation and Rsr and Y RNAs are among the most upregulated genes during recovery of D. radiodurans from DNA damage (Chen et al., 2000; Tanaka et al., 2004). Since Rsr and Y RNAs are encoded adjacent to RtcB in diverse bacteria, it will be interesting to determine if activation in response to DNA damage is a conserved feature of rsr-yrlBA-rtcBA operons.
STAR★METHODS
Detailed methods are provided in the online version of this paper and include the following:
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Sandra Wolin (sandra.wolin@nih.gov).
Materials Availability
Bacterial strains and plasmids are available on request.
Data and Code Availability
The accession number for the RNA sequencing data reported in this paper is Gene Expression Omnibus: GSE153782.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All bacterial strains used in this study are listed in Table S3. Two virulent S. Typhimurium strains were used: SL1344 (Hoiseth and Stocker, 1981) (gift of Jorge Galan, Yale School of Medicine) and 14028s (Jarvik et al., 2010) (gift of Eduardo Groisman, Yale School of Medicine). Mutant strains were created using allelic exchange (Kaniga et al., 1994) using the donor strain E. coli β-2163Δnic35 and R6K-based conjugative transfer of suicide vectors (Demarre et al., 2005). To generate deletion mutants, DNA fragments 5′ and 3′ to the ORF were amplified from genomic DNA using primers p1 and p2r, and p3 and p4r, respectively (Table S3), which were joined with overlap PCR. To generate the RtcR mutant, which encodes a truncated protein containing 185 amino acids at the N terminus, DNA fragments were amplified from genomic DNA using 5′-ATCCTTCATCATCCCGCGTTCG-3′ and 5′-GTATTAAAACGCACGCTGTCGCCTGCCGGACTTCAGGAAGTTGAG-3′, and 5′-GGCGACAGCGTGCGTTTTAATAC-3′ and 5′-GATCTTAACGATCTGGCGGAACAG-3′ and joined by overlap PCR. DNAs were cloned into the SmaI site of pSB890 and transformed into donor strain E. coli β-2163Δnic35, which was used to conjugate recipient strains. P22 phage transductants were constructed as described (Davis et al., 1980). All strains were grown at 37° in Luria-Bertani broth (LB; 10 g/L tryptone, 10 g/L NaCl, 5 g/L yeast extract) with appropriate antibiotic unless otherwise stated. Chloramphenicol was used at 15 μg/ml, carbenicillin was used at 100 μg/ml, tetracycline was used at 12–20 μg/ml, and diaminopimelic acid (DAP) was used at 50 μg/ml. For treatment with DNA damaging agents, cells were grown in LB to OD600 = 0.9 and incubated with 3 μM (1 μg/ml) MMC (Sigma-Aldrich), 0.02% methyl methanesulfonate (MMS) (Sigma) or 1 or 5 μg/ml bleomycin for 2 h unless otherwise stated. Bacteria were collected and resuspended in RNAprotect Bacteria reagent (QIAGEN). Afterward, bacteria were centrifuged and RNA extracted using hot acid phenol (Chen et al., 2007). E. coli strain MG1655 was a gift of Donald Court (National Cancer Institute, Frederick, MD, USA).
METHOD DETAILS
Transposon mutagenesis
Plasmid pSRS_CM1 (Shames et al., 2017), also called pSB4807 (Fowler and Galan, 2018), a chloramphenicol-resistant derivative of pSAM_Bt (Goodman et al., 2009), was a gift of C. Fowler and J. Galan. This plasmid was transferred into donor strain E. coli β-2163Δnic35 (Demarre et al., 2005) and propagated at 37°C in LB containing DAP and carbenicillin. Conjugative-based transposon mutagenesis of recipient S. Typhimurium strains was performed by mixing donor and recipient strains in a 3:1 ratio. Bacterial conjugations were performed at room temperature for 24 h and transconjugants selected by plating on LB agar plates containing 15 mg/ml chloramphenicol and 40 mg/ml X-gal (MP Biomedicals) to identify blue colonies. Approximately 63,000 colonies were screened (28,000 14028s and 35,000 SL1344) and blue colonies purified by streaking to single colonies on X-gal/chloramphenicol plates. Genomic DNA was isolated with DNAzol (Thermo Fisher), partially digested with Sau3AI and ligated to a DNA adaptor digested with BamHI. Ligation-mediated PCR (Mueller and Wold, 1989), using primers that anneal to adaptor and transposon sequences, was used to amplify sites of transposon insertion from genomic DNA.
β-Galactosidase assays
All strains were grown at 37°C in LB with the appropriate antibiotic. Expression of rtcR and rtcRΔN was induced from pBAD24 with 0% and 0.1% arabinose respectively. (Some product is produced in 0% arabinose because the promoter is leaky). β-galactosidase activity was measured as described (Miller, 1972) from cells grown to OD600 between 0.320 and 1.160 (with most strains kept below 1.0) unless otherwise stated. Briefly, after cultures were incubated on ice for 20 minutes to stop growth, cells were pelleted and resuspended in Z buffer (60 mM Na2HPO4.7H2O, 40 mM NaH2PO4.H2O, 10 mM KCl, 1 mM MgSO4, 50 mM 2-mercaptoethanol, adjusted to pH 7.0). Cells were diluted in Z buffer to 1 mL and permeabilized by adding 100 μl chloroform and 50 μl 0.1% sodium dodecyl sulfate and vortexing. After incubating at 28°C for 5 minutes, 200 μl 2-nitrophenyl-β-D-galactopyranoside (Sigma-Aldrich) was added. After yellow color developed, the reaction was stopped by adding 500 μl 1 M Na2CO3 and 1 mL transferred to a micro-centrifuge tube and the tube sedimented at 16,000 × g for 5 minutes. After measuring the OD420 and OD550 of the supernatant, units of activity were calculated according to the equation Miller units = 1000 × [(OD420 − 1.75 × OD550)] / [reaction time (minutes) × volume of culture (ml) × OD600 of original culture].
Construction of expression plasmids
The pRtcRΔN plasmid for overexpressing N-terminally truncated RtcR was described (Chen et al., 2013). To construct pRtcR, full-length RtcR was amplified from genomic DNA using 5′-GGAATTCATGCGAAAAACGGTGGCCTTTG-3′and 5′-GCTCTAGATTAATTCTGTAAAACGTCCCACGTCAG-3′, digested with EcoRI and XbaI and cloned into pBAD24. To construct pHA-RtcA (in which the HA epitope is fused to RtcA), full-length RtcA was amplified from genomic DNA with 5′-GGAATTCATGTACCCATACGATGTTCCAGATTACGCTATGGCAAGGATCATCGCGCTG-3′ and 5′-GCTCTAGATTAGTCGCTTACCCGGACAAGATAGC-3′, digested with EcoRI and XbaI and cloned into pBAD24.
Immunoblotting
Bacterial pellets were resuspended in 1X SDS sample buffer (50 mM Tris-HCl pH 6.8, 2% SDS, 10% glycerol, 100 mM DTT, 12.5 mM EDTA, 0.02% bromophenol blue). Cells were lysed by boiling for 10 min. After centrifuging at 16,000 × g, cleared lysates were fractionated in SDS-polyacrylamide gels and transferred to nitrocellulose. Membranes were blocked overnight in 5% milk in TBS-T (20 mM Tris-HCl pH 7.6, 137 mM NaCl, 0.1% Tween 20) overnight at 4°C, washed twice with TBS-T at room temperature, and incubated with primary antibody diluted in 5% milk in TBS-T at room temperature for 1 hour. Membranes were washed in TBS-T before incubation with HRP-coupled secondary antibody diluted in 5% milk in TBS-T for 1 hour at room temperature followed by washing with TBS-T. HRP was detected by enhanced chemiluminescence (ECL) using the Pierce ECL Western Blotting Substrate (ThermoFisher). Antibodies used for immunoblotting are listed in the Key Resources Table. In Figure S3K, the anti-LexA antibody was purchased from Abcam, while in Figure 4A, the anti-LexA antibody was a gift of Dr. I. Narumi (Toyo University).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Monoclonal ANTI-FLAG M2 antibody produced in mouse | Sigma-Aldrich | Cat#F1804; RRID: AB_262044 |
| HRP anti-E coli RNA polymerase Sigma 70 | BioLegend | Cat#663205; RRID: AB_2629596 |
| Rabbit anti-E coli RecA | Narumi et al., 2001 | N/A |
| Rabbit anti-E coli LexA | gifts of I. Narumi, Toyo University, Itakura, Gunma Japan | N/A |
| Rabbit anti-E coli RtcB | Temmel et al., 2017 | N/A |
| HA Tag Monoclonal Antibody, HRP | Thermo Fisher Scientific | Cat#26183; RRID: AB_2533056 |
| Rabbit anti-E coli RplE | gift of J. Zengel, University of Maryland, Baltimore County, MD | N/A |
| Rabbit anti-E coli LexA | Abcam | Cat#ab174384; RRID: N/A |
| Rabbit IgG HRP Linked Whole Ab | Sigma-Aldrich | Cat#GENA934; RRID: AB_2722659 |
| Goat anti-Mouse IgG Fc Cross-Adsorbed Secondary Antibody, HRP | Thermo Fisher Scientific | Cat#31439; RRID: AB_228292 |
| Bacterial and Virus Strains | ||
| List of Strains Used in This Study | This paper | See Table S3 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 2,6-Diaminopimelic acid | Sigma-Aldrich | Cat#D1377; CAS: 583-93-7 |
| Mitomycin C | Sigma-Aldrich | Cat#M4287; CAS: 50-07-7 |
| Methyl methanesulfonate | Sigma-Aldrich | Cat#129925; CAS: 66-27-3 |
| Bleomycin | Sigma-Aldrich | Cat#B8416; CAS: 9041-93-4 |
| Phenol solution (acid) | Sigma-Aldrich | Cat#P4682; CAS: 108-95-2 |
| X-gal | MP Biomedicals | Cat#114063102; CAS: 7240-90-6 |
| DNAzol | Thermo Fisher Scientific | Cat#10503027 |
| RNAprotect Bacteria Reagent | QIAGEN | Cat#76506 |
| Turbo DNase I | Thermo Fisher Scientific | Cat#AM2238 |
| Calf intestinal alkaline phosphatase | Sigma-Aldrich | Cat#11097075001 |
| T4 polynucleotide kinase | New England Biolabs | Cat#M0201S |
| T4 RNA Ligase 2, truncated KQ | New England Biolabs | Cat#M0373S |
| Polyethylene glycol 8000 | Sigma-Aldrich | Cat#1546605; CAS: 25322-68-2 |
| RNaseOUT | Thermo Fisher Scientific | Cat#10777019 |
| Superscript III Reverse Transcriptase | Thermo Fisher Scientific | Cat#18080093 |
| Phusion DNA polymerase | New England Biolabs | Cat#M0530S |
| Factor Xa Protease | New England Biolabs | Cat#P8010S |
| ATP, [γ-32P] | PerkinElmer | Cat#BLU002Z250UC |
| Chloramphenicol | Sigma-Aldrich | Cat#C0378; CAS 56-75-7 |
| Carbenicillin | Sigma-Aldrich | Cat#C1389; CAS 4800-94-6 |
| Tetracycline | Sigma-Aldrich | Cat#T3383; CAS 64-75-5 |
| 2-Mercaptoethanol | Sigma-Aldrich | Cat#M6250; CAS 60-24-2 |
| 2-Nitrophenyl-β-D-galactopyranoside | Sigma-Aldrich | Cat#N1127; CAS 369-07-3 |
| Pierce ECL Western Blotting Substrate | Thermo Fisher Scientific | Cat#32106 |
| SYBR Gold Nucleic Acid Gel Stain | Thermo Fisher Scientific | Cat#S11494 |
| Critical Commercial Assays | ||
| RNA Clean and Concentrator-5 | Zymo Research | Cat#R1013 |
| Zero Blunt PCR cloning kit | Thermo Fisher Scientific | Cat#K270020 |
| TruSeq RNA Library Prep Kit v2 | Illumina | Cat#RS-122–2001 |
| Bacteria Ribo-Zero Magnetic Kit | Illumina | Cat#MRZB12424 |
| Deposited Data | ||
| Raw and Analyzed Data | This paper | GEO: GSE153782 |
| Oligonucleotides | ||
| List of Oligonucleotides Used in This Study. | This paper | See Table S3 |
| Recombinant DNA | ||
| List of Plasmids used in this study. | This paper | See Table S3 |
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.net/Welcome |
| GraphPad Prism 8.0 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Pfam | El-Gebali etal., 2019 | N/A |
| CDD | Yang et al., 2020 | N/A |
| HMMSCAN (HMMER3 package) | Potter et al., 2018 | https://www.ebi.ac.uk/Tools/pfa/hmmer3_hmmscan/ |
| PSI-BLAST | Altschul and Koonin, 1998 | https://blast.ncbi.nlm.nih.gov/Blast.cgi?CMD=Web&PAGE=Proteins&PROGRAM=blastp&RUN_PSIBLAST=on |
| JACKHMMER | Johnson et al., 2010 | https://www.ebi.ac.uk/Tools/hmmer/search/jackhmmer |
| HHPRed | Zimmermann et al., 2018 | https://toolkit.tuebingen.mpg.de/tools/hhpred |
| Kalign program | Lassmann, 2019 | N/A |
| Pymol | Schrödinger | https://pymol.org/2/ |
| Illumina basecalling RTA 1.18.66.3 | Illumina | N/A |
| Trimmomatic version 0.30 software | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| Bowtie2 version 2.3.3. | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Cuffdiff | Trapnell et al., 2013 | http://cole-trapnell-lab.github.io/cufflinks/cuffdiff/ |
| Other | ||
| Amersham Hybond-N | Cytiva | Cat#RPN303N |
| Ni-NTA Agarose | Thermo Fisher Scientific | Cat#R90101 |
| Superdex 200 Increase 10/300 Column | Cytiva | Cat#28990944 |
| Amicon Ultra-15 Centrifugal Filter Units | Millipore | Cat#UFC901024 |
| Whatman Optitran Nitrocellulose Blotting Membrane | Cytiva | Cat# 10439196 |
RNA analyses and Northern blotting
Total RNA was extracted from bacterial pellets treated with RNAprotect Bacteria Reagent (QIAGEN) using hot acid phenol. Pellets (3 O.D. units) were resuspended in 400 μL of 10 mM Tris-HCl (pH 7.5), 10 mM EDTA, 0.5% SDS and 400 μL of acid phenol (pH 5) and incubated at 65°C for 30 minutes with occasional vortexing. The sample was then cooled briefly on ice, sedimented at 25°C for 10 minutes at 16,000 × g and the supernatant removed to a new microcentrofuge tube. After extraction with phenol:chloroform: isoamyl alcohol (50:49:1), RNA was precipitated with 2.5 volumes ethanol in 40 μL 3M NaOAc.
For 3′ end analyses, 10 μg of RNA was incubated in 5 μl 10X Turbo DNase I buffer (Ambion) and 2 U Turbo DNase I (Ambion) at 37°C for 30 minutes followed by purification using RNA Clean and Concentrator-5 (Zymo Research Corporation). To remove 3′ phosphates, RNA was first incubated with 10 mM HCl (Sigma) on ice for 4 hours, followed by incubation with 5 U of CIP (Roche), or 10 U T4 PNK (New England Biolabs) according to the manufacturers’ instructions and purified using RNA Clean and Concentrator-5 kit (Zymo Research Corporation). Afterward, RNA (1.5 μg) was ligated overnight at 16°C to 750 ng of adenylated adaptor (Table S3) using T4 RNA Ligase 2, truncated KQ (New England Biolabs) in the presence of 25% PEG 8000 and RNaseOUT (Thermo Fisher). Ligated RNAs were extracted with phenol:chloroform:isoamyl alcohol (50:49:1), precipitated with ethanol, and resuspended in 10 μl water. Half the sample was used for Northern analyses, while the other half was subjected to reverse transcription using Superscript III (Thermo Fisher) and a primer complementary to the adenylated adaptor (Table S3). After reverse transcription, cDNA was amplified using Phusion DNA polymerase (New England Biolabs), a tRNA-specific forward primer (Table S3), and the RT-adaptor reverse primer. Gel purified DNAs were cloned using Zero Blunt PCR cloning kit (Thermo Fisher) according to manufacturer instructions. At least 9 clones were sequenced for each sample. For Northern analyses, RNA was separated in 5% or 8% polyacrylamide/8.3 M urea gels and transferred to Hybond N (Cytiva) in 0.5X TBE at 150 mA for 16 h. Blots were hybridized with [32P]-labeled oligonucleotides as described (Tarn et al., 1995). Oligonucleotide probes are listed in Table S3. Radioactive signals were detected using a Typhoon FLA 7000 Phosphorimager (GE Healthcare).
RNA sequencing and analysis
Salmonella RNA was isolated from 14028s cells treated with or without MMC for two hours using hot acid phenol. The RNA library was prepared by the National Cancer Institute Sequencing Facility using the TruSeq RNA Library Prep Kit v2 (Illumina) following ribosomal RNA removal using the Ribo-Zero Magnetic Kit for bacteria (Illumina). Sequencing was performed on a NextSeq 500 (Illumina) with 75 bp paired end reads. After trimming adapters and low quality sequence using Trimmomatic version 0.30 software, reads were aligned to the S. Typhimirium (14028s) genome using Bowtie2 software version 2.3.2. Mapped BAM files were used as input for Cuffdiff to determine differential gene expression between MMC-treated and control untreated samples.
Genome screening for candidate tRNA endoribonucleases
All proteins encoded by the genomes of S. Typhimurium SL1344 and 14028s strains were screened against a library of profiles constructed from Pfam (El-Gebali et al., 2019), CDD (Yang et al., 2020) and a custom database of profiles of RNase domains from diverse biological conflict systems using the RPSBLAST and HMMSCAN program (HMMER3 package). Statistically significant hits (e < 10−3) were checked for recovery of known and predicted endoribonuclease domains and these were set aside as potential candidates; for example, proteins containing domains that were members of the BECR fold (Zhang et al., 2014; Iyer et al., 2017), PIN domain superfamily (Matelska et al., 2017), SNase fold (Ponting, 1997), HEPN domain (Anantharaman et al., 2013), potential RNA endonucleases of the RNase H fold (Majorek et al., 2014) and metallo-beta-lactamase fold (Aravind, 1999), the RNase T2 domain (Watanabe et al., 1995), the RNaseE/G superfamily (Callaghan et al., 2005), and the SymE domain (Kawano et al., 2007) were considered valid hits. Proteins encoded by the two S. Typhimurium genomes were also subject to iterative sequence profile searches using PSI-BLAST (Altschul and Koonin, 1998) and JACKHMMER (Johnson et al., 2010) and profile-profile searches using the HHPRed program (Zimmermann et al., 2018) in a further effort to unify them with known domains. Successful unification with any of the known RNase domains led to their inclusion in the candidate list. Genome contexts of all candidates were then isolated and screened for 1) links to known translation or ribosome assembly factors and 2) inclusion in biological conflict systems. Active site conservation patterns of each candidate were examined for potential loss of catalytic activity using multiple alignments [constructed with the Kalign program (Lassmann, 2019) and, if required, examination of homologous structures using Pymol (https://pymol.org/2/)]. Predicted inactive representatives of the above RNase domains were removed from further consideration. We arrived at a list of 40 potential RNases. To prioritize, whole transcriptome sequencing of rRNA-depleted total RNA from wild-type cells treated with or without MMC was performed. Potential RNases that were expressed in the presence of MMC were prioritized for deletion.
Survival assays
Overnight cultures containing 100 μg/ml carbenicillin were diluted to OD600 = 0.05 in LB containing 0.1% glucose, which reduces expression of genes controlled by the PBAD promoter (Guzman et al., 1995). After 2.5 h at 37°, bacteria were diluted to OD600 = 0.2 in LB containing 0.1% glucose and 1 μg/ml MMC. After 2 additional h at 37°, bacteria were diluted and spotted or spread on LB agar plates. Colonies from both MMC- and mock-treated bacteria were counted to determine the fraction of surviving cells.
MazEF purification and MazF activation
To overexpress MazEF in E. coli, double-stranded DNA encoding the 41 C-terminal coding residues of the MazE antitoxin, a Factor Xa cleavage site and the coding sequence of the MazF toxin as a fusion protein (Park et al., 2012) was synthesized (gBlocks Gene Fragments, Integrated DNA Technologies), digested with NdeI and XhoI and inserted into pET28b (EMD Biosciences). After transforming the recombinant plasmid into E. coli BL21(DE3), cells were grown to OD600 = 0.8, induced with 0.5 mM IPTG at 25°C for 4 h and harvested by centrifugation. After resuspending in 20 mM Tris-HCl pH 7.5, 300 mM NaCl, 20 mM imidazole, cells were lysed by passing through a French Press at 8000 psi three times. After sedimenting the lysate at 40,000 rpm in a Beckman Type 50.2 Ti rotor, the supernatant was passed through a Ni-NTA agarose column (Thermo Fisher), washed with 20 mM Tris-HCl pH 7.5, 300 mM NaCl, 30 mM imidazole until the OD600 of the flowthrough was below 0.01. His-tagged MazEF was eluted with 20 mM Tris-HCl pH 7.5, 300 mM NaCl, 250 mM imidazole. Further purification was by size exclusion chromatography using a Superdex 200 Increase 10/300 column (GE Healthcare Life Sciences) in 20 mM Tris-HCl pH 7.5, 150 mM NaCl, 10% glycerol. Fractions containing MazEF were pooled, concentrated using Amicon Ultra-15 Centrifugal Filter Units (Millipore), aliquoted and frozen in liquid nitrogen. MazF was activated by incubating 1 mg MazEF with 10 μg protease Factor Xa (NEB) in 10 mM Tris-HCl pH 8.0, 1 mM DTT, 2 mM CaCl2 at 37°C for 3 h (Rouillon et al., 2019).
Generation of 5′ tRNA halves for binding studies
RNA oligonucleotides corresponding to 5′ tRNA halves containing 3′-OH or 3′-phosphate ends were purchased from Integrated DNA Technologies. To generate RNAs containing 2′, 3′-cyclic phosphate, activated MazF was incubated with RNA oligonucleotides containing a MazF cleavage site ACACUG at the 3′ end (Rouillon et al., 2019). For EMSAs, RNAs were were labeled at the 5′ end using [γ-32P]-ATP and T4 PNK. After incubating at 70°C for 20 min to inactivate T4 PNK, activated MazF was added to 10 μg/μl and incubated for 2 h at 37°C to generate RNAs ending in 2′, 3′-cyclic phosphate. To remove the cyclic phosphate and generate a labeled RNA with 3′-OH, the reaction was incubated with T4 PNK (Amitsur et al., 1987). To generate labeled RNA ending with 3′-phosphate, the desired sequence was synthesized with an additional uridylate, labeled at the 5′ end with [γ-32P]-ATP and T4 PNK, and incubated in 100 μl 1 M DL-lysine-Cl and 0.025 M NaIO4 (pH 8.3) at 45°C for 2.5 h (Neu and Heppel, 1964). Afterward, the RNA was extracted with phenol:chloroform:isoamyl alcohol (50:49:1) and precipitated with 2.5 volumes ethanol. All labeled RNAs were purified from 15% polyacrylamide, 8.3M urea gels before use.
Purification of full-length RtcR and the CARF domain
Sequences encoding full-length S. Typhimurium RtcR and the CARF domain (amino acids 1–188) were amplified from genomic DNA using primers 5′-CGGGATCCGATGCGAAAAACGGTGGCCTTTG-3′ and 5′-CCGCTCGAGTTAATTCTGTAAAACGTCCCACGTCAG-3′, and primers 5′-CGGGATCCGATGCGAAAAACGGTGGCCTTTG-3′ and 5′-CCGCTCGAGTTAGGTTGCAAT GCCGGACTTCAG-3′, respectively, digested with BamHI and XhoI and cloned into the same sites of pRSFDuet-1 (Novagen). The resulting plasmids were transformed into E. coli BL21(DE3). To express recombinant protein, cells were cultured in LB containing 20 mM Tris-HCl pH 7.5 and 50 μg/ml kanamycin to OD600 = 0.7, 0.1mM IPTG was added and the culture incubated at 16°C for 20 h. His-tagged proteins were purified as described above, except that the lysis buffer was 50 mM Tris-HCl pH 7.5, 500 mM NaCl, 10% glycerol, 20 mM imidazole, the wash buffer was 50 mM Tris-HCl pH 7.5, 500 mM NaCl, 10% glycerol, 30 mM imidazole, the elution buffer was 50 mM Tris-HCl pH 7.5, 500 mM NaCl, 10% glycerol, 250 mM imidazole and the buffer used in gel filtration was 20 mM Tris-HCl pH 7.5, 250 mM NaCl, and 10% glycerol.
RNA binding assays
32P-labeled RNAs in binding buffer (20 mM Tris-HCl pH 7.5, 50 mM NaCl, 1 mM MgCl2, 0.05% Tween 20) were refolded by heating to 95°C for 2 min, frozen on dry ice and thawed on ice. Refolded RNAs were mixed with RtcR or the CARF domain in binding buffer in 5 μl total volume, incubated at 4°C for 30 min and at room temperature for 30 min. For RtcR, the reaction included 1 mM ATP except where stated. Reactions were fractionated in 6% polyacrylamide (80:1 acrylamide:bisacrylamide)/5% glycerol gels for CARF domain EMSAs and 4% polyacrylamide (80:1 acrylamide:bisacrylamide)/2.5% glycerol gels for RtcR EMSAs. Gels were run at 4°C, 5 V/cm for 20 min, then 10 V/cm in 0.5xTBE (50 mM Tris, 45 mM boric acid, 1.25 mM EDTA) until the bromophenol blue dye migrated 4 cm. The gels were dried and scanned using a Typhoon FLA 7000 Phosphorimager (GE Healthcare Life Sciences). Fractions of bound RNA were quantitated using ImageJ (NIH). Results were analyzed with GraphPad Prism 8 and fitted by nonlinear regression using the equation for one site specific binding: Y = Bmax*X/(Kd + X), where Y is the fraction of bound RNA, Bmax is the maximum specific binding, X is the protein concentration, and Kd is the equilibrium binding constant.
Size exclusion chromatography
Size exclusion chromatography was performed on an ÄKTA Pure 25 using a Superdex 200 Increase 10/300 column, which was equilibrated and run in 20 mM Tris-HCl pH 7.5, 100 mM NaCl, 1 mM MgCl2, 2% glycerol, 0.5% Tween 20, 1 mM ATP, with a flow rate at 0.25 ml/min. Next, 48 μM of RtcR and 48 μM of the refolded RNA were mixed in gel filtration buffer. After incubating 30 min on ice and at room temperature for 30 min to allow complex formation, 100 μl of the sample was injected onto the column. Elution volumes were monitored by measuring the absorbance at 280 nm. Proteins in peak fractions were analyzed using 4%–12% SDS-PAGE and staining with Coomassie blue, while RNAs was detected by fractionation in 8% polyacrylamide/8.3 M urea gels, followed by staining with SYBR Gold Nucleic Acid Gel Stain (Thermo Fisher).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis
All results are presented as the mean (n = 3) of three biological replicates ± SEM. GraphPad Prism v8 was used for statistical analysis. P values were calculated using two-tailed unpaired t tests. *, p < 0.05; **, p < 0.01, ***, p < 0.001 indicate significant differences between samples; ns indicates not significant.
Supplementary Material
Highlights.
An RNA repair operon is activated by mutations that cause tRNA halves to accumulate
Operon expression and accumulation of tRNA halves occur upon DNA damage
The 5′ tRNA halves that accumulate end in 2′, 3′-cyclic phosphate
The RtcR transcriptional activator oligomerizes upon binding these 5′ tRNA halves
ACKNOWLEDGMENTS
We thank Casey Fowler and Jorge Galan for the SL1344 strain, the pSRS_CM1/pSB4807 plasmid, and advice on the screen; Lynn Thomason and Donald Court for E. coli strains and advice; Eduardo Groisman, Issay Narumi, Isabella Moll, and Janice Zengel for other strains and antibodies; Eugene Koonin and Kira Makarova for help with identifying endonucleases; and Stephanie Shames, Eugene Valkov, Soyeong Sim, and Marco Boccitto for other helpful advice. We also thank Donald Court, Susan Gottesman, Traci Hall, Gisela Storz, Lynn Thomason, and members of the Wolin laboratory for comments on the manuscript. This work was supported in part by a National Science Foundation Graduate Research Fellowship (K.J.H.); National Institutes of Health grant R01 GM073863 (S.L.W.) and training grant T32 GM007223 (K.J.H.); and the Intramural Research Program, Center for Cancer Research, National Cancer Institute, National Institutes of Health (K.J.H., X.C., and S.L.W.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108527.
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPORTING CITATIONS
The following references appear in the supplemental information: Blakely et al. (1993); Padan et al. (2005); Shiomi et al. (2008).
REFERENCES
- Altschul SF, and Koonin EV (1998). Iterated profile searches with PSI-BLAST: a tool for discovery in protein databases. Trends Biochem. Sci 23, 444–447. [DOI] [PubMed] [Google Scholar]
- Amitsur M, Levitz R, and Kaufmann G (1987). Bacteriophage T4 anticodon nuclease, polynucleotide kinase and RNA ligase reprocess the host lysine tRNA. EMBO J 6, 2499–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantharaman V, Makarova KS, Burroughs AM, Koonin EV, and Aravind L (2013). Comprehensive analysis of the HEPN superfamily: identification of novel roles in intra-genomic conflicts, defense, pathogenesis and RNA processing. Biol. Direct 8, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravind L (1999). An evolutionary classification of the metallo-beta-lactamase fold proteins. In Silico Biol 1, 69–91. [PubMed] [Google Scholar]
- Arnez JG, and Steitz TA (1994). Crystal structure of unmodified tRNA(Gln) complexed with glutaminyl-tRNA synthetase and ATP suggests a possible role for pseudo-uridines in stabilization of RNA structure. Biochemistry 33, 7560–7567. [DOI] [PubMed] [Google Scholar]
- Athukoralage JS, Rouillon C, Graham S, Grüschow S, and White MF (2018). Ring nucleases deactivate type III CRISPR ribonucleases by degrading cyclic oligoadenylate. Nature 562, 277–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Athukoralage JS, Graham S, Grüschow S, Rouillon C, and White MF (2019). A type III CRISPR ancillary ribonuclease degrades its cyclic oligoadenylate activator. J. Mol. Biol 431, 2894–2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blakely G, May G, McCulloch R, Arciszewska LK, Burke M, Lovett ST, and Sherratt DJ (1993). Two related recombinases are required for site-specific recombination at dif and cer in E. coli K12. Cell 75, 351–361. [DOI] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolzán AD, and Bianchi MS (2018). DNA and chromosome damage induced by bleomycin in mammalian cells: An update. Mutat. Res 775, 51–62. [DOI] [PubMed] [Google Scholar]
- Bunny K, Liu J, and Roth J (2002). Phenotypes of lexA mutations in Salmonella enterica: evidence for a lethal lexA null phenotype due to the Fels-2 prophage. J. Bacteriol 184, 6235–6249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burroughs AM, and Aravind L (2016). RNA damage in biological conflicts and the diversity of responding RNA repair systems. Nucleic Acids Res 44, 8525–8555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush M, and Dixon R (2012). The role of bacterial enhancer binding proteins as specialized activators of σ54-dependent transcription. Microbiol. Mol. Biol. Rev 76, 497–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byrne RT, Chen SH, Wood EA, Cabot EL, and Cox MM (2014). Escherichia coli genes and pathways involved in surviving extreme exposure to ionizing radiation. J. Bacteriol 196, 3534–3545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaghan AJ, Marcaida MJ, Stead JA, McDowall KJ, Scott WG, and Luisi BF (2005). Structure of Escherichia coli RNase E catalytic domain and implications for RNA turnover. Nature 437, 1187–1191. [DOI] [PubMed] [Google Scholar]
- Chen X, Quinn AM, and Wolin SL (2000). Ro ribonucleoproteins contribute to the resistance of Deinococcus radiodurans to ultraviolet irradiation. Genes Dev 14, 777–782. [PMC free article] [PubMed] [Google Scholar]
- Chen X, Wurtmann EJ, Van Batavia J, Zybailov B, Washburn MP, and Wolin SL (2007). An ortholog of the Ro autoantigen functions in 23S rRNA maturation in D. radiodurans. Genes Dev 21, 1328–1339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Taylor DW, Fowler CC, Galan JE, Wang HW, and Wolin SL (2013). An RNA degradation machine sculpted by Ro autoantigen and noncoding RNA. Cell 153, 166–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Sim S, Wurtmann EJ, Feke A, and Wolin SL (2014). Bacterial noncoding Y RNAs are widespread and mimic tRNAs. RNA 20, 1715–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortese R, Kammen HO, Spengler SJ, and Ames BN (1974). Biosynthesis of pseudouridine in transfer ribonucleic acid. J. Biol. Chem 249, 1103–1108. [PubMed] [Google Scholar]
- Das U, and Shuman S (2013). 2′-Phosphate cyclase activity of RtcA: a potential rationale for the operon organization of RtcA with an RNA repair ligase RtcB in Escherichia coli and other bacterial taxa. RNA 19, 1355–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das U, Chakravarty AK, Remus BS, and Shuman S (2013). Rewriting the rules for end joining via enzymatic splicing of DNA 3′-PO4 and 5′-OH ends. Proc. Natl. Acad. Sci. USA 110, 20437–20442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das U, Chauleau M, Ordonez H, and Shuman S (2014). Impact of DNA3’pp5’G capping on repair reactions at DNA 3′ ends. Proc. Natl. Acad. Sci. USA 111, 11317–11322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RW, Botstein D, and Roth JR (1980). Advanced Bacterial Genetics: A Manual for Genetic Engineering (Cold Spring Harbor Laboratory Press; ). [Google Scholar]
- Demarre G, Guérout AM, Matsumoto-Mashimo C, Rowe-Magnus DA, Marlière P, and Mazel D (2005). A new family of mobilizable suicide plasmids based on broad host range R388 plasmid (IncW) and RP4 plasmid (IncPalpha) conjugative machineries and their cognate Escherichia coli host strains. Res. Microbiol 156, 245–255. [DOI] [PubMed] [Google Scholar]
- Dillingham MS, and Kowalczykowski SC (2008). RecBCD enzyme and the repair of double-stranded DNA breaks. Microbiol. Mol. Biol. Rev 72, 642–671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Gebali S, Mistry J, Bateman A, Eddy SR, Luciani A, Potter SC, Qureshi M, Richardson LJ, Salazar GA, Smart A, et al. (2019). The Pfam protein families database in 2019. Nucleic Acids Res 47 (D1), D427–D432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engl C, Schaefer J, Kotta-Loizou I, and Buck M (2016). Cellular and molecular phenotypes depending upon the RNA repair system RtcAB of Escherichia coli. Nucleic Acids Res 44, 9933–9941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englert M, Sheppard K, Aslanian A, Yates JR 3rd, and Söll D (2011). Archaeal 3′-phosphate RNA splicing ligase characterization identifies the missing component in tRNA maturation. Proc. Natl. Acad. Sci. USA 108, 1290–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaine F, Gasiorowski E, Gracia C, Ballouche M, Caillet J, Marchais A, and Hajnsdorf E (2016). The small RNA SraG participates in PNPase homeostasis. RNA 22, 1560–1573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler CC, and Galan JE (2018). Decoding a Salmonella Typhi regulatory network that controls typhoid toxin expression within human cells. Cell Host Microbe 23, 65–76. e66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Doval C, Schwede F, Berk C, Rostøl JT, Niewoehner O, Tejero O, Hall J, Marraffini LA, and Jinek M (2020). Activation and self-inactivation mechanisms of the cyclic oligoadenylate-dependent CRISPR ribonuclease Csm6. Nat. Commun 11, 1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genschik P, Drabikowski K, and Filipowicz W (1998). Characterization of the Escherichia coli RNA 3′-terminal phosphate cyclase and its sigma54-regulated operon. J. Biol. Chem 273, 25516–25526. [DOI] [PubMed] [Google Scholar]
- Gerdes K, Howard M, and Szardenings F (2010). Pushing and pulling in prokaryotic DNA segregation. Cell 141, 927–942. [DOI] [PubMed] [Google Scholar]
- Goodman AL, McNulty NP, Zhao Y, Leip D, Mitra RD, Lozupone CA, Knight R, and Gordon JI (2009). Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe 6, 279–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman LM, Belin D, Carson MJ, and Beckwith J (1995). Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol 177, 4121–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harms A, Brodersen DE, Mitarai N, and Gerdes K (2018). Toxins, targets, and triggers: an overview of toxin-antitoxin biology. Mol. Cell 70, 768–784. [DOI] [PubMed] [Google Scholar]
- Hoiseth SK, and Stocker BA (1981). Aromatic-dependent Salmonella typhimurium are non-virulent and effective as live vaccines. Nature 291, 238–239. [DOI] [PubMed] [Google Scholar]
- Hollis T, and Shaban NM (2011). Structure and function of RNase H enzymes. Nucleic Acids Mol. Biol 26, 299–317. [Google Scholar]
- Houseley J, and Tollervey D (2009). The many pathways of RNA degradation. Cell 136, 763–776. [DOI] [PubMed] [Google Scholar]
- Iyer LM, Burroughs AM, Anand S, de Souza RF, and Aravind L (2017). Polyvalent proteins, a pervasive theme in the intergenomic biological conflicts of bacteriophages and conjugative elements. J. Bacteriol 199, e00245–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarvik T, Smillie C, Groisman EA, and Ochman H (2010). Short-term signatures of evolutionary change in the Salmonella enterica serovar Typhimurium 14028 genome. J. Bacteriol 192, 560–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia N, Jones R, Yang G, Ouerfelli O, and Patel DJ (2019). CRISPR-Cas III-A Csm6 CARF domain is a ring nuclease triggering stepwise cA4 cleavage with ApA > p formation terminating RNase activity. Mol. Cell 75, 944–956. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LS, Eddy SR, and Portugaly E (2010). Hidden Markov model speed heuristic and iterative HMM search procedure. BMC Bioinformatics 11, 431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurkin J, Henkel T, Nielsen AF, Minnich M, Popow J, Kaufmann T, Heindl K, Hoffmann T, Busslinger M, and Martinez J (2014). The mammalian tRNA ligase complex mediates splicing of XBP1 mRNA and controls antibody secretion in plasma cells. EMBO J 33, 2922–2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaimer C, and Graumann PL (2011). Players between the worlds: multifunctional DNA translocases. Curr. Opin. Microbiol 14, 719–725. [DOI] [PubMed] [Google Scholar]
- Kaniga K, Bossio JC, and Galán JE (1994). The Salmonella typhimurium invasion genes invF and invG encode homologues of the AraC and PulD family of proteins. Mol. Microbiol 13, 555–568. [DOI] [PubMed] [Google Scholar]
- Kawano M, Aravind L, and Storz G (2007). An antisense RNA controls synthesis of an SOS-induced toxin evolved from an antitoxin. Mol. Microbiol 64, 738–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazlauskiene M, Kostiuk G, Venclovas Č, Tamulaitis G, and Siksnys V (2017). A cyclic oligonucleotide signaling pathway in type III CRISPR-Cas systems. Science 357, 605–609. [DOI] [PubMed] [Google Scholar]
- Kisker C, Kuper J, and Van Houten B (2013). Prokaryotic nucleotide excision repair. Cold Spring Harb. Perspect. Biol 5, a012591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosmaczewski SG, Edwards TJ, Han SM, Eckwahl MJ, Meyer BI, Peach S, Hesselberth JR, Wolin SL, and Hammarlund M (2014). The RtcB RNA ligase is an essential component of the metazoan unfolded protein response. EMBO Rep 15, 1278–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurasz JE, Hartman CE, Samuels DJ, Mohanty BK, Deleveaux A, Mrázek J, and Karls AC (2018). Genotoxic, metabolic, and oxidative stresses regulate the RNA repair operon of Salmonella enterica serovar Typhimurium. J. Bacteriol 200, e00476–e00418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassmann T (2019). Kalign 3: multiple sequence alignment of large data sets. Bioinformatics 36, 1928–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemire S, Figueroa-Bossi N, and Bossi L (2011). Bacteriophage crosstalk: coordination of prophage induction by trans-acting antirepressors. PLoS Genet 7, e1002149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Reimers S, Pandit S, and Deutscher MP (2002). RNA quality control: degradation of defective transfer RNA. EMBO J 21, 1132–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little JW, and Harper JE (1979). Identification of the lexA gene product of Escherichia coli K-12. Proc. Natl. Acad. Sci. USA 76, 6147–6151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majorek KA, Dunin-Horkawicz S, Steczkiewicz K, Muszewska A, Nowotny M, Ginalski K, and Bujnicki JM (2014). The RNase H-like superfamily: new members, comparative structural analysis and evolutionary classification. Nucleic Acids Res 42, 4160–4179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Anantharaman V, Grishin NV, Koonin EV, and Aravind L (2014). CARF and WYL domains: ligand-binding regulators of prokaryotic defense systems. Front. Genet 5, 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masaki H, and Ogawa T (2002). The modes of action of colicins E5 and D, and related cytotoxic tRNases. Biochimie 84, 433–438. [DOI] [PubMed] [Google Scholar]
- Matelska D, Steczkiewicz K, and Ginalski K (2017). Comprehensive classification of the PIN domain-like superfamily. Nucleic Acids Res 45, 6995–7020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelini F, Jalihal AP, Francia S, Meers C, Neeb ZT, Rossiello F, Gioia U, Aguado J, Jones-Weinert C, Luke B, et al. (2018). From “cellular” RNA to “smart” RNA: multiple roles of RNA in genome stability and beyond. Chem. Rev 118, 4365–4403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JH (1972). Experiments in Molecular Genetixs (Cold Spring Harbor Laboratory Press; ). [Google Scholar]
- Molina R, Stella S, Feng M, Sofos N, Jauniskis V, Pozdnyakova I, López-Méndez B, She Q, and Montoya G (2019). Structure of Csx1-cOA4 complex reveals the basis of RNA decay in Type III-B CRISPR-Cas. Nat. Commun 10, 4302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller PR, and Wold B (1989). In vivo footprinting of a muscle specific enhancer by ligation mediated PCR. Science 246, 780–786. [DOI] [PubMed] [Google Scholar]
- Narumi I, Satoh K, Kikuchi M, Funayama T, Yanagisawa T, Kobayashi Y, Watanabe H, and Yamamoto K (2001). The LexA protein from Deinococcus radiodurans is not involved in RecA induction following gamma irradiation. J. Bacteriol 183, 6951–6956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neu HC, and Heppel LA (1964). Nucleotide sequence analysis of polyribo-nucleotides by means of periodate oxidation followed by cleavage with an amine. J. Biol. Chem 239, 2927–2934. [PubMed] [Google Scholar]
- Niewoehner O, Garcia-Doval C, Rostøl JT, Berk C, Schwede F, Bigler L, Hall J, Marraffini LA, and Jinek M (2017). Type III CRISPR-Cas systems produce cyclic oligoadenylate second messengers. Nature 548, 543–548. [DOI] [PubMed] [Google Scholar]
- O’Reilly EK, and Kreuzer KN (2004). Isolation of SOS constitutive mutants of Escherichia coli. J. Bacteriol 186, 7149–7160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padan E, Bibi E, Ito M, and Krulwich TA (2005). Alkaline pH homeostasis in bacteria: new insights. Biochim. Biophys. Acta 1717, 67–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JH, Yamaguchi Y, and Inouye M (2012). Intramolecular regulation of the sequence-specific mRNA interferase activity of MazF fused to a MazE fragment with a linker cleavable by specific proteases. Appl. Environ. Microbiol 78, 3794–3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponting CP (1997). P100, a transcriptional coactivator, is a human homologue of staphylococcal nuclease. Protein Sci 6, 459–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popow J, Englert M, Weitzer S, Schleiffer A, Mierzwa B, Mechtler K, Trowitzsch S, Will CL, Lührmann R, Söll D, and Martinez J (2011). HSPC117 is the essential subunit of a human tRNA splicing ligase complex. Science 331, 760–764. [DOI] [PubMed] [Google Scholar]
- Potter SC, Luciani A, Eddy SR, Park Y, Lopez R, and Finn RD (2018). HMMER web server: 2018 update. Nucleic Acids Res 46 (W1), W200–W204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouillon C, Athukoralage JS, Graham S, Grüschow S, and White MF (2019). Investigation of the cyclic oligoadenylate signaling pathway of type III CRISPR systems. Methods Enzymol 616, 191–218. [DOI] [PubMed] [Google Scholar]
- Shames SR, Liu L, Havey JC, Schofield WB, Goodman AL, and Roy CR (2017). Multiple Legionella pneumophila effector virulence phenotypes revealed through high-throughput analysis of targeted mutant libraries. Proc. Natl. Acad. Sci. USA 114, E10446–E10454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiomi D, Sakai M, and Niki H (2008). Determination of bacterial rod shape by a novel cytoskeletal membrane protein. EMBO J 27, 3081–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer MF (1958). Phosphorolysis of oligoribonucleotides by polynucleotide phosphorylase. J. Biol. Chem 232, 211–228. [PubMed] [Google Scholar]
- Tanaka N, and Shuman S (2011). RtcB is the RNA ligase component of an Escherichia coli RNA repair operon. J. Biol. Chem 286, 7727–7731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka M, Earl AM, Howell HA, Park MJ, Eisen JA, Peterson SN, and Battista JR (2004). Analysis of Deinococcus radiodurans’s transcriptional response to ionizing radiation and desiccation reveals novel proteins that contribute to extreme radioresistance. Genetics 168, 21–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka N, Chakravarty AK, Maughan B, and Shuman S (2011). Novel mechanism of RNA repair by RtcB via sequential 2′,3′-cyclic phosphodies-terase and 3′-phosphate/5′-hydroxyl ligation reactions. J. Biol. Chem 286, 43134–43143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarn W-Y, Yario TA, and Steitz JA (1995). U12 snRNA in vertebrates: Evolutionary conservation of 5’ sequences implicated in splicing of pre-mRNAs containing a minor class of introns. RNA 1, 644–656. [PMC free article] [PubMed] [Google Scholar]
- Temmel H, Müller C, Sauert M, Vesper O, Reiss A, Popow J, Martinez J, and Moll I (2017). The RNA ligase RtcB reverses MazF-induced ribosome heterogeneity in Escherichia coli. Nucleic Acids Res 45, 4708–4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, and Pachter L (2013). Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol 31, 46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe H, Fauzi H, Iwama M, Onda T, Ohgi K, and Irie M (1995). Base non-specific acid ribonuclease from Irpex lacteus, primary structure and phylogenetic relationships in RNase T2 family enzyme. Biosci. Biotechnol. Biochem 59, 2097–2103. [DOI] [PubMed] [Google Scholar]
- Winther KS, and Gerdes K (2011). Enteric virulence associated protein VapC inhibits translation by cleavage of initiator tRNA. Proc. Natl. Acad. Sci. USA 108, 7403–7407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt MD, and Pittman DL (2006). Methylating agents and DNA repair responses: methylated bases and sources of strand breaks. Chem. Res. Toxicol 19, 1580–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyatt HD, and West SC (2014). Holliday junction resolvases. Cold Spring Harb. Perspect. Biol 6, a023192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W (2011). Nucleases: diversity of structure, function and mechanism. Q. Rev. Biophys 44, 1–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Derbyshire MK, Yamashita RA, and Marchler-Bauer A (2020). NCBI’s conserved domain database and tools for protein domain analysis. Curr. Protoc. Bioinformatics 69, e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Iyer LM, Burroughs AM, and Aravind L (2014). Resilience of biochemical activity in protein domains in the face of structural divergence. Curr. Opin. Struct. Biol 26, 92–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann L, Stephens A, Nam SZ, Rau D, Kübler J, Lozajic M, Gabler F, Söding J, Lupas AN, and Alva V (2018). A completely reimplemented mpi bioinformatics toolkit with a new HHpred server at its core. J. Mol. Biol 430, 2237–2243. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the RNA sequencing data reported in this paper is Gene Expression Omnibus: GSE153782.