

Abstract
The Japanese rhinoceros beetle Trypoxylus dichotomus is one of the largest beetle species in the world and is commonly used in traditional Chinese medicine. Ten subspecies of T. dichotomus and a related Trypoxylus species (T. kanamorii) have been described throughout Asia, but their taxonomic delimitations remain problematic. To clarify issues such as taxonomy, and the degree of genetic differentiation of Trypoxylus populations, we investigated the genetic structure, genetic variability, and phylogeography of 53 specimens of Trypoxylus species from 44 locations in five Asian countries (China, Japan, Korea, Thailand, and Myanmar). Using specific‐locus amplified fragment sequencing (SLAF‐seq) techniques, we developed 330,799 SLAFs over 114.16M reads, in turn yielding 46,939 high‐resolution single nucleotide polymorphisms (SNPs) for genotyping. Phylogenetic analysis of SNPs indicated the presence of three distinct genetic groups, suggesting that the various subspecies could be treated as three groups of populations. PCA and ADMIXTURE analysis also identified three genetic clusters (North, South, West), which corresponded to their locations, suggesting that geographic factors were important in maintaining within population homogeneity and between population divergence. Analyses of SNP data confirmed the monophyly of certain subspecies on islands, while other subspecies (e.g., T. d. septentrionalis) were found to be polyphyletic and nested in more than one lineage. AMOVA demonstrated high level of differentiation among populations/groups. Also, pairwise F ST values revealed high differentiation, particularly between South and West, as well as between North and South. Despite the differentiation, measurable gene flow was inferred between genetic clusters but at varying rates and directions. Our study demonstrated that SLAF‐seq derived markers outperformed 16S and COII sequences and provided improved resolution of the genetic differentiation of rhinoceros beetle populations from a large part of the species’ range.
Keywords: beetle taxonomy, genetic variation, island biogeography, phylogeography, population structure, SLAF‐seq
We estimate the evolution of morphological diversification as well as genetic differentiation, gene flow, and population genetic structure of Japanese rhinoceros beetle populations.

1. INTRODUCTION
The Japanese rhinoceros beetle, Trypoxylus dichotomus (Coleoptera, Scarabaeidae, Dynastinae) first described by Linnaeus (1771), is widely distributed in China, Japan, the Korean Peninsula, Vietnam, Myanmar, Laos, India, and Thailand (Adachi, 2017; Nagai, 2006, 2007; Satoru, 2014). It is typically found in broad‐leaved forests in tropical and subtropical mountainous habitats. The Japanese rhinoceros beetle has been important in Chinese traditional medicine for nearly 2000 years. Recent studies have found that extracts from the beetle have antihepatofibrotic, antineoplastic, and antibiotic effects (Chung et al., 2014; Kim et al., 2007; Miyanoshita et al., 1996; Ratcliffe et al., 2011; Sagisaka et al., 2001; Yoshikawa et al., 1999). A previously undescribed lectin purified from the larvae of Japanese rhinoceros beetles may assist in the suppression of human cervical cancer (HeLa), murine fibroblast (L929), and murine L1210 leukemic cells (Kui et al., 2000). Additional studies have found that Japanese rhinoceros beetles contain compounds with possible application as immunomodulators and tumor growth inhibitors (Hee et al., 2001; Ratcliffe et al., 2011; Umetsu et al., 1985, 1984). In addition to human medical applications, the bacterium Bacillus amyloliquefaciens KB3, isolated from feces of 3rd instar larvae of Japanese rhinoceros beetles, can be used as biocontrol against certain fungal plant pathogens (Nam et al., 2016). The beetle's high protein content gives it great potential as a food source when a suitable processing method is employed (Chung et al., 2013; Kim et al., 2016). In many East Asian countries, keeping Japanese rhinoceros beetles as pets has become a personal hobby and popular due to their large size and unique body shape.
Apart from the numerous applications, the Japanese rhinoceros beetle have received widespread attention owing mainly to their remarkable sexual dimorphism and individual variation, especially in the size and shape of the male's cephalic and pronotal horn (Buchalski et al., 2019; Hosoya & Araya, 2005; Ito et al., 2013; Morita et al., 2019; Ohde et al., 2018; Warren et al., 2013; Zinna et al., 2018). Male rhinoceros beetles have robust prominent cephalic horns for intraspecific competition over reproductive opportunities and in interspecific competition (like members of the Lucanidae family) over territory and resources. Horn shapes and sizes diverge greatly even among closely related species and sometimes between populations of beetles, suggesting differential selective pressures on horns among different lineages (Buchalski et al., 2019; Emlen et al., 2007, 2012; Hongo, 2003, 2007; Ito et al., 2013, Johns et al., 2014; Karino et al., 2005; Zinna et al., 2018). Studies on the genetic mechanism of horn development suggest that the genes underlying horn formation in rhinoceros beetles (Dynastinae) were the same genes recruited for the independent derivation of horns among the dung beetles (Scarabaeinae) (Ito et al., 2013; Ohde et al., 2018). As such rhinoceros beetles and related taxa are ideal for comparative studies of sexual selection, sexual dimorphism, and the evolution of morphological innovations.
T. dichotomus was initially described by Linnaeus (1771) from specimens collected in Southeast Asia (at the time referred to as Indiis). In total, 10 subspecies of T. dichotomus along with another Trypoxylus species (T. kanamorii Nagai, 2006) were proposed in later revisions (Figure 1). The subspecies include T. d. dichotomus (Linnaeus, 1771) in Southeastern China; T. d. inchachina (Kusui, 1976) in Japan (Kumejima Island); T. d. politus (Prell, 1934) in Northeastern India, Myanmar, Thailand, Laos, and Vietnam; T. d. septentrionalis (Kôno, 1931) in Japan, the Korean peninsula, and Northeastern China; T. d. tsunobosonis (Kôno, 1931) in Taiwan; T. d. takarai (Kusui, 1976) in Japan (Okinawa Island and Iheyajima Island); T. d. tsunobosonis Kôno in Taiwan; T. d. tsuchiyai (Nagai, 2006) in Japan (Kuchinoerabu‐jima Island); T. d. shizuae in Japan (Yakushima Island and Tanegashima Island); T. d. xizangensis (Li et al., 2015) in Tibet (China); T. d. shennongjii (Satoru, 2014) in China (Hubei Province) (Adachi, 2017; Kôno, 1931; Kusui, 1976; Li et al., 2015; Linnaeus, 1771; Nagai, 2006, 2007; Prell, 1934; Satoru, 2014); and the species T.kanamorii (Nagai, 2006) in Myanmar, India and China (Yunnan and Tibet). Species and subspecies division of Trypoxylus have been controversial, with traditional identifications mainly based on morphological characteristics, such as horn shape, body size and shape, lustrousness of elytra, and pubescence on the pygidium, pronotum, and elytra. Historically, delimitations among subspecies could be arduous because of limited divergence between the morphological characters used for subspecific identification. Later, Nagai (2006, 2007) proposed seven subspecies of T. dichotomus in Asia and illustrated differences among these subspecies based on morphological characteristics. Satoru (2014) and Adachi (2017) described additional T. dichotomus subspecies from Japan based on body color and pubescence on elytra. However, these studies have only employed a small number of diagnostic characters to separate the subspecies without placing the characters or taxa into a phylogenetic context to better understand character evolution within Trypoxylus. Taxonomy and distribution of T. dichotomus subspecies in Asia, especially in China, has also been challenging for many of the same reasons. As such, a molecular phylogenetic approach can provide much needed resolution in evaluating the monophyly of T. dichotomus subspecies that have been delimited with sometimes overlapping characters.
Figure 1.
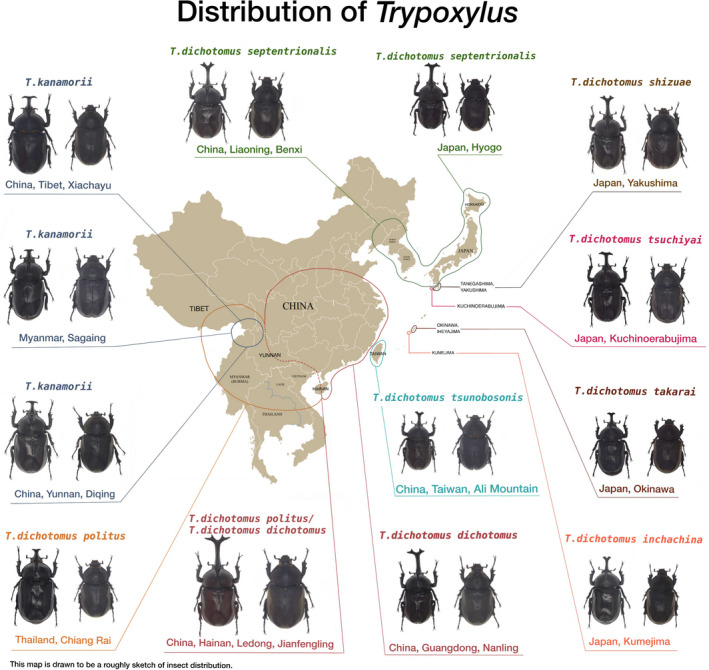
A map showing the distribution of Trypoxylus species in East Asia with illustrations of various subspecies
Understanding spatial distribution and species delineations is critical in clearly defining patterns of biodiversity and subsequently informing conservation decisions as well as providing lineage‐based comparisons for future studies, such as trait evolution and/or identifying novel medicinal compounds. The accurate inference of genetic clustering in natural beetle populations has important implications to understand the process and pattern of evolution. For instance, improved resolution of within‐species patterns of genetic diversity, divergence, and clustering can in turn improve estimates of population genetic processes, such as the resolution of spatial genetic differentiation among populations and increases accuracy and precision of gene flow rates estimates (Black & DuTeau, 1997; Zhu et al. 2018), which can be useful in identifying historic corridors of migration.
Numerous different molecular marker systems have been used to infer population structure and evolutionary patterns for a variety of insect species (e.g., Batista et al., 2016; Pascoal & Kilner, 2017). For instance, Pascoal and Kilner (2017) developed 14 polymorphic microsatellite markers and investigated levels of genetic differentiation in four populations of burying beetles (Nicrophorus vespilloides). With random amplified polymorphic DNA (RAPD) and mitochondrial DNA COII sequence analysis, homogeneity between small brown planthoppers from different geographic regions could be inferred (Xu et al., 2001; Wang et al. 2019). Single nucleotide polymorphisms (SNPs) from genome‐wide panels are known to improve the resolution of population genetic inferences such as linkage disequilibrium (LD) and population structure because of the abundant and generally even distribution across the genome over other markers, and the well‐understood models of evolution applied to nucleotide mutation analyses. The use of SNPs in studies dissecting complex traits is also attractive because they can be used to quantify heterozygosity in biallelic loci, they can be sequence‐tagged to ensure orthologous comparison of loci, and genotyping can be massively multiplexed to increase throughput and reduce cost (Blanco‐Bercial & Bucklin, 2016). High‐throughput next‐generation sequencing (NGS) technologies are now critical tools in developing SNP panels and genotyping subsequent individuals in studies of the ecology and evolution of traditionally poorly resolved cryptic groups. An example of a successful NGS method for large‐scale de novo SNP discovery and genotyping is SLAF‐seq (specific length amplified fragment sequencing) developed by Sun et al. (2013). The SLAF‐seq method has proven efficient and effective for SNP based population genetic analyses in numerous plant and animal studies (Su et al., 2017; Sun et al., 2013; Wang et al. 2019).
To better understand the evolution of morphological diversification as well as clarify genetic differentiation, gene flow, and population genetic structure of Japanese rhinoceros beetle populations, we conducted phylogenetic and population genetic analyses across 10 subspecies from a large portion of the species range in eastern Asia. DNA sequences from the mitochondrial cytochrome c oxidase subunit II (COII) and 16S ribosomal RNA (rRNA) genes were employed in reconstructing the phylogeny of Trypoxylus species and subspecies. SLAF‐seq derived SNPs were also used in population genetic and phylogenetic analyses for comparison to sequence‐based datasets and for improved resolution of inference.
2. MATERIALS AND METHODS
2.1. Insect specimen collection
During the season of peak activity (from May to mid‐August) in the years 2017 to 2018, live Trypoxylus individuals were caught by light trap or collected on sap exudate (food source for beetles) of broad‐leaved trees. A total of 128 individual beetles from 44 locations in five different regions of Asia (19 provinces in China, seven locations in Japan, two locations in Korea, one location in Thailand, and one in Myanmar) were obtained in this study as well as outgroup taxa included two beetles of Xylotrupes mniszechi from Tibet (China) and Xyloscaptes davidis from Vietnam. 53 specimens of Trypoxylus belonging to two species (T. kanamorii and T. dichotomus) and nine subspecies from five regions of Asia were selected for SLAF‐seq analysis and phylogenetic analysis based on 16S and COII sequences (Table 1). Specimens from the two subspecies, T. d. shennongjii and T. d. xizangensis, could not be obtained. Immediately upon collection, beetles were preserved in absolute ethyl alcohol. Thereafter, collected specimens were mounted, morphological observations recorded, and digitally imaged by Nikon D7000 camera. Previous to these steps, the thoracic muscles were dissected for examination, preserved in absolute ethyl alcohol, and stored at −20°C until later use in DNA extractions. All examined voucher specimens were deposited in the School of Pharmaceutical Sciences, Sun Yat‐sen University (Guangzhou, China).
Table 1.
Sampling information of Trypoxylus specimens used in this investigation
| Cluster | Species | Location | Code | GenBank accession no. 16S | GenBank accession no. COII |
|---|---|---|---|---|---|
| North | T. d. septentrionalis | Tonghua, Jilin, China (JL) | T‐75 | MT080160 | MT103637 |
| T. d. septentrionalis | Dandong, Liaoning, China (LN1) | T‐50 | MT080148 | MT103651 | |
| T. d. septentrionalis | Benxi, Liaoning, China (LN2) | T‐52 | MT080164 | MT103632 | |
| T. d. septentrionalis | Gyungsangnum‐do, Korea (KO1) | T‐22 | MT080121 | MT103623 | |
| T. d. septentrionalis | Fukushima, Japan (FUK1) | T‐7 | MT080129 | MT103638 | |
| T..d septentrionalis | Fukushima, Japan (FUK2) | T‐8 | MT080132 | MT103631 | |
| T. d. septentrionalis | Kuriyama, Hokkaido, Japan (KUR1) | T‐6 | MT080141 | MT103640 | |
| T. d. septentrionalis | Kuriyama, Hokkaido, Japan (KUR2) | T‐5 | MT080157 | MT103643 | |
| T. d. septentrionalis | Hyogo, Japan (HYO) | T‐1 | MT080137 | MT103621 | |
| T. d. shizuae | Yakushima, Japan (YAK1) | T‐11 | MT080155 | — | |
| T. d. shizuae | Yakushima, Japan (YAK2) | T‐12 | MT080136 | MT103633 | |
| T. d. tsuchiyai | Kuchinoerabu‐jima, Japan (KUC1) | T‐13 | MT080133 | MT103608 | |
| T. d. tsuchiyai | Kuchinoerabu‐jima, Japan (KUC2) | T‐14 | MT080134 | MT103636 | |
| T. d. septentrionalis | Jeju Island, Korea (JEJ) | T‐25 | MT080124 | MT103613 | |
| T. d. dichotomus | Guiyang, Guizhou, China (GUI) | T‐38 | MT080119 | MT103634 | |
| West | T. d. dichotomus | Fuzhou, Jiangxi, China (JX1) | T‐31 | MT080131 | MT103647 |
| T. d. dichotomus | Xinyu, Jiangxi, China (JX2) | T‐34 | MT080135 | MT103652 | |
| T. d. dichotomus | Lishui, Zhejiang, China (ZJ1) | T‐76 | MT080126 | MT103641 | |
| T. d. dichotomus | Wuyishan, Fujian, China (FJ1) | T‐27 | MT080161 | MT103642 | |
| T. d. dichotomus | Nanling, Guangdong, China (GD) | T‐65 | MT080127 | MT103624 | |
| T. d. dichotomus | Zhumadian, Henan, China (HEN1) | T‐41 | MT080156 | MT103622 | |
| T. d. dichotomus | Xiangyang, Hubei, China (HB) | T‐49 | MT080114 | MT103630 | |
| T. d. dichotomus | Zhangzhou, Fujian, China (FJ2) | T‐28 | MT080159 | MT103639 | |
| T. d. dichotomus | Nanchang, Jiangxi, China(JX3) | T‐32 | MT080149 | MT103635 | |
| T. d. dichotomus | Meishan, Sichuan, China (SC1) | T‐57 | MT080163 | MT103609 | |
| T. d. dichotomus | Deyang, Sichuan, China (SC2) | T‐59 | MT080113 | MT103653 | |
| T. d. dichotomus | Ningshan, Shaanxi, China (SX) | T‐54 | MT080139 | MT103626 | |
| T. d. dichotomus | Chizhou, Anhui, China (AH) | T‐73 | MT080138 | MT103616 | |
| T. d. dichotomus | Suqian, Jiangsu, China (JS1) | T‐47 | MT080147 | MT103604 | |
| T. d. dichotomus | Yangzhou, Jiangsu, China (JS2) | T‐45 | MT080158 | MT103625 | |
| T. d. dichotomus | Huzhou, Zhejiang, China (ZJ2) | T‐78 | MT080125 | MT103629 | |
| T. d. dichotomus | Nanyang, Henan, China (HEN2) | T‐39 | MT080120 | MT103607 | |
| T. d. dichotomus | Jiujiang, Jiangxi, China (JX4) | T‐36 | MT080115 | MT103610 | |
| T. d. dichotomus | Suzhou, Jiangsu, China (JS2) | T‐43 | MT080146 | MT103649 | |
| T. d. dichotomus | Changde, Hunan, China (HN) | T‐70 | MT080128 | MT103612 | |
| T. d. dichotomus | Dayaoshan, Guangxi, China (GX) | T‐60 | MT080142 | MT103648 | |
| T. d. dichotomus | Wuzhishan, Hainan, China (HAN1) | T‐67 | MT080112 | MT103605 | |
| T. d. dichotomus | Jianfengling, Hainan, China(HAN2) | T‐69 | MT080130 | MT103614 | |
| T. kanamorii | Xiachayu, Tibet, China (TB) | T‐79 | MT080140 | MT103655 | |
| T. d. politus | Xishuangbanna, Yunnan, China (YN1) | T‐64 | MT080143 | MT103644 | |
| T. kanamorii | Diqing, Yunnan, China (YN2) | T‐62 | MT080144 | MT103645 | |
| T. d. politus | Wiang Papau, Thailand (THA1) | T‐17 | MT080118 | MT103618 | |
| T. d. politus | Wiang Papau, Thailand (THA2) | T‐18 | — | — | |
| T. kanamorii | Sagaing, Myanmar (MYA1) | T‐15 | MT080117 | MT103615 | |
| T. kanamorii | Sagaing, Myanmar (MYA2) | T‐16 | MT080145 | MT103646 | |
| South | T. d. septentrionalis | Gyungsang num‐do, Korea (KOR2) | T‐20 | MT080122 | MT103650 |
| T. d. tsunobosonis | Pingtung, Taiwan (TW1) | T‐81 | MT080150 | MT103627 | |
| T. d. tsunobosonis | Tengjhih, Taiwan (TW2) | T‐80 | MT080116 | MT103620 | |
| T. d. tsunobosonis | Taichung, Taiwan (TW3) | T‐82 | MT080151 | MT103619 | |
| T. d. takarai | Okinawa, Japan (OKI1) | T‐10 | MT080154 | MT103606 | |
| T. d. takarai | Okinawa, Japan (OKI2) | T‐9 | MT080110 | MT103602 | |
| T. d. inchachina | Kumejima Island, Japan (KUM1) | T‐3 | MT080152 | — | |
| T. d. inchachina | Kumejima Island, Japan (KUM2) | T‐4 | MT080153 | MT103611 | |
| Outgroup | Xylotrupes mniszechi | Chayu, Tibet, China (TB2) | T‐84 | MT080111 | MT103603 |
| Xyloscaptes davidis | Vietnam (VIET1) | T‐85 | MT080162 | MT103654 |
2.2. Morphological data
All specimens in our study were identified based on morphological characters published in previous studies (Adachi, 2017; Kôno, 1931; Kusui, 1976; Li et al., 2015; Nagai, 2006, 2007; Prell, 1934; Satoru, 2014). Some morphological characters, including body size, lustrousness, cuticle color, cephalic and pronotal horn size, pubescence on elytra, pronotum, head, and pygidium, have provided useful criteria for species identification (Table 2). In this study, the taxonomic revisions by Nagai (2006, 2007), Satoru (2014), and Adachi (2017) were followed as they provided the clearest subspecies descriptions. The genus Xylotrupes was treated as an outgroup in our comparisons and thought be a more distantly related to Trypoxylus than Xyloscaptes (also used as an outgroup) based on Dutrillaux et al. (2013). Digital images of diagnostic features were taken with a Nikon D7000 camera with AF‐S VR Micro‐Nikkor 105mm f/2.8G IF‐ED‐Nikon lenses and edited with Adobe Photoshop CS4, Microsoft Paint, and Autodesk Sketchbook.
Table 2.
Morphological features of Trypoxylus species in this investigation
| Species | Locations | Descriptions | Resource | Genetic group |
|---|---|---|---|---|
| T. kanamorii | Northwest of Myanmar; India, Tibet (China);Yunnan (China) |
♂: slender and dark brown to black body; strongly lustrous elytra; shorter horns of head and pronotum ♀: very slight pubescence on elytra compared to T. d. septentrionalis in Japan |
Nagai (2006, 2007) | West |
| T. d. dichotomus | Middle and South of China | Bigger body size and slender body shape; weak lustrous elytra compared to T. d. septentrionalis in Japan; lack of detailed description when first described as Scarabaeus by Linnaeus | Linnaeus (1771), Nagai (2007) | West |
| T. d. inchachina | Kumejima Island (Japan), Ryukyu Islands (Japan) |
♂: entirely black; very short forked cephalic horn with each apex weakly bifurcated and very short abruptly bifurcated pronotal horn; shorter legs; smaller body, strongly lustrous elytra; smaller cephalic horns compared to T. d. septentrionalis in Japan; slightly less lustrous than T. d. politus from Thailand; ♀: prothorax apical margin angulately sinuate at middle; head middle tubercle slightly lower than other outer ones; longitudinal hollow of prothorax shallow and not bifurcated near apical margin |
Kusui (1976), Nagai (2007) | South |
| T. d. politus | Myanmar, Thailand, Vietnam, Yunnan (China) and Laos | First described in Laos as having glossier body due to sparse pubescence and dark body color; shorter, wider body; ♂: strongly lustrous elytra compared to T. d. septentrionalis in Japan; ♂: most lustrous body among all subspecies | Nagai (2007), Prell (1934) | West |
| T. d. septentrionalis | Northeast of China; Korean Peninsula; Jeju Island; Japan (Honshu, Shikoku, Kyushu, Hokkaido, and some other small islands of Japan) | Robust body; ♂: weak lustrous elytra; sparse pubescence on pygidium | Kôno (1931), Nagai (2007) | North |
| T. d. shennongjii | Shennongjia, Hubei (China) | ♂: a large and magnificent V‐shaped cephalic horn, which has a small fork on the apex | Satoru (2014) | — |
| T. d. shizuae | Yakushima Island and Tanegashima Island (Japan) | Brighter reddish‐brown body, sometimes dark brown; robust body; slender front tibiae; ♂: shorter, convex pronotum; shorter cephalic and pronotal horn compared to T. d. septentrionalis; rounded pronotal lateral margin and curved gradually at the former point; fewer, shorter, and finer pubescence on metasternum, second abdominal sternite, and abdomen, ♀: faintly lustrous; otherwise similar to T. d. septentrionalis in Japan | Adachi (2017) | North |
| T. d. takarai | Mainland of Okinawa; Ryukyu Islands (Japan) | Smaller body size; ♂: smaller cephalic horn; darker body color; shorter pubescence on center of elytra and pronotum, therefore strongly lustrous compared to T. d. septentrionalis in Japan; ♀: middle tubercle of head lower or similar to outer ones in height; longitudinal hollow of prothorax not distinctly bifurcate near apical margin | Kusui (1976), Nagai (2007) | South |
| T. d. tsuchiyai | Kuchinoerabu‐jima Island (Japan) | Smaller and wider body than T. d. septentrionalis; naked or clothed with pubescence on pronotum; smoother and shining elytra; shorter and coarser pubescence on pygidium otherwise similar to T. d. septentrionalis in Japan | Nagai (2006, 2007) | North |
| T. d. tsunobosonis | Taiwan | ♂: thinner but longer pronotal horn compared to T. d. septentrionalis in Japan. | Kôno (1931), Nagai (2007) | South |
| T. d. xizangensis | Gamdo City, Tibet (China) | ♂: Thin horn; sharp‐pointed lateral horn; short and thick phallobase | Li et al. (2015) | — |
2.3. DNA extraction, PCR, and sequencing of 16S and COII
Genomic DNA was extracted from thoracic muscle tissue using the QIAGEN DNeasy Tissue Kit in accordance with manufacturer's instructions. For sequence‐based phylogenetic analysis, amplicons from cytochrome c oxidase II (COII) and 16S ribosomal RNA (16S rRNA) were used as they are common loci in barcoding. The 16S rRNA region was amplified by two primer sets (16sF/16sR and 16SB/16SA) (16sF: 5′‐CGCCTGTTTATCAAAAACAT‐3′; 16sR: 5′‐CTCCGGTTTGAACTCAGATCA‐3′), (16SA: 5′‐CGCCTGTTTAACAAAAACATGT‐3′; 16SB: 5′‐CCGGTTTGAACTCAGATCATGT‐3′), which were employed because they successfully amplified species in Lucanidae (Han et al., 2010; Rowland & Miller, 2012). The COII region was amplified by primers F‐lue and R‐lys (F‐lue: 5′‐TCTAATATGGCAGATTAGTGC‐3′; R‐lys: 5′‐GAGACCAGTACTTGCTTTCAGTCATC‐3′; Rowland & Miller, 2012). PCR reactions were performed in 25 μl volumes containing 1 μl DNA template, 2.5 μl forward primer (2 μM), 2.5 μl reverse primer (2 μM), 12.5 μl of 2× EsTaq Master Mix (Cwbio), and 6.5 μl ddH2O. The PCR thermocycle conditions were as follows: 94°C for 3 min; followed by 35 cycles at 95°C for 1 min, 40–50°C (depending on primer set) for 1 min, and 72°C for 1 min; and a final extension step of 72°C for 10 min. The PCR products were purified and visualized by gel electrophoresis on 1.5% agarose gel to confirm amplification. Sanger sequencing was performed at TSINGKE. All sequences are available from GenBank under accession numbers MT103602–MT103655 for COII and MT080110–MT080164 for 16S rRNA.
2.4. Phylogenetic analysis
16S and COII sequences were edited using Seqman v. 7.1.0 in the DNASTAR Lasergene core suite software (DNASTAR Inc., Madison, WI, USA), and aligned using MAFFT v.6 (Katoh & Standley, 2013). Ambiguously aligned sequences were excluded from the analysis.
Maximum parsimony (MP) analysis of concatenated 16S and COII alignment was performed in PAUP v.4.0b10 (Swofford, 2003). Gaps were treated as missing data, and all characters were equally weighted. Trees were inferred using the heuristic search option with TBR branch swapping. The robustness of the most parsimonious trees was evaluated by 1,000 bootstrap replications (Felsenstein, 1985). Other calculated measures were the tree length (TL), consistency index (CI), retention index (RI), and rescaled consistency (RC).
Bayesian analyses (BI) were performed using MrBayes v.3.1.2 with Markov chain Monte Carlo (MCMC) and Bayesian posterior probabilities (Ronquist & Huelsenbeck, 2003). Default parameters were selected, and the evolutionary model was set to the GTR + I + G model, which was the best model predicted for the concatenated 16S and COII alignment by MrModeltest v. 2.3 (Nylander, 2004). Simultaneous Markov chains were computed for 1,000,000 generations, and the trees were sampled every 100th generation (You et al., 2019).
Maximum likelihood analysis (ML) was performed by RAxMl v. 1.3 (Stamatakis, 2006). The bootstrapping was carried out with 1,000 replicates using the GTR + I + G model. Xylotrupes mniszechi and Xyloscaptes davidis were selected as outgroup taxa.
2.5. SLAF library construction and high‐throughput sequencing
For SLAF‐seq analysis, DNA concentration was quantified using a NanoDrop‐2000 spectrophotometer, and all DNA samples were diluted to 50 ng/μl. SLAF library construction was carried out following Sun et al. (2013) with minor modifications. To obtain evenly distributed SLAF tags and to avoid repetitive SLAF tags for maximum SLAF‐seq efficiency, simulated restriction enzyme digestion was carried out in silico. Genomic DNA was digested using RsaI‐HaeIII restriction enzyme, and the reference genome of Dendroctonus ponderosae (https://www.ncbi.nlm.nih.gov/genome/?term=Dendroctonus+ponderosae) was used to predict enzyme digestion. DNA fragments of 264–364 bp were selected as SLAFs and prepared for paired‐end sequencing on the Illumina High‐Seq 2,500 sequencing platform (Illumina, Inc.) at Biomarker Technologies Corporation.
2.6. SLAF‐seq data grouping and genotyping
Raw pair‐end reads were clustered based on sequence similarity. Sequences with over 90% identity were grouped in one SLAF tag, SLAFs with low‐depth coverage were filtered out (Huang et al., 2016; Sun et al., 2013; Wang et al. 2019). Only groups with higher depth and four tags or fewer were identified as high‐quality SLAFs with SLAFs possessing two, three, or four tags identified as polymorphic. In this study, depth was 17.63× on average, and a total of 1,374,985 high‐quality unique SLAF tags were obtained with 330,799 of those tags considered polymorphic.
Development of SNP markers was based on reference sequence with very high depth in each SLAF tag. SAMtools and GATK were used for mapping and SNP calling (Li et al., 2009; McKenna et al., 2010; Wang et al. 2019). A total of 46,939 SNPs with minor allele frequencies (MAF) of ≥0.05 and an integrity score of ≥80% were employed in downstream analyses.
2.7. SNP Data analysis
2.7.1. Population genetics analyses
Phylogenetic trees based on a polymorphic SNP matrix (53 individuals × 46,939 SNPs, also used in all subsequent analyses unless noted otherwise) were constructed using the neighbor joining (NJ) method employed in MEGA X with p‐distance and pairwise deletion option parameters (Kumar et al., 2018; Saitou & Nei, 1987; Tamura et al., 2011). Bootstrap tests with 1,000 replicates were applied to assess branch support in the final consensus trees. Bayesian Inference (BI) was performed using MrBayes 3.1.2 (Ronquist & Huelsenbeck, 2003) with Markov chain Monte Carlo (MCMC) and Bayesian posterior probabilities to assess branch support. Default parameters were selected, using a GTR (general time reversible) substitution model with gamma‐distributed rate variation across sites and a proportion of invariable sites (Ronquist & Huelsenbeck, 2003). Simultaneous Markov chains were run for 1,000,000 generations, and with sampling every 100th generation (You et al., 2019).
2.7.2. Population structure
Population structure and genetic clustering of the 53 specimens was inferred using ADMIXTURE (Alexander et al., 2009), which does so through running population expansion models as well as inferring evolutionary and ecological processes among populations. ADMIXTURE is a model‐based clustering method used to analyze the association of individuals from multilocus SNP genotypes and infer clusters to test for population admixture. A range of cluster values (K) from 1 to 10 was assessed, with each run set to allow admixture and with a correlated allele frequency model, using the first 100,000 MCMC generations as burn‐in followed by 900,000 MCMC generations. The clustering results were cross‐verified, and the optimal number of clusters (K) was determined according to the lowest value of the cross‐validation error rate between cluster values (Debnath, 2014; Wang et al. 2019). A nonmodel‐based principal components analysis (PCA) was performed with EIGENSOFT v. 6.0, to further assess clustering of genotypes (Price et al., 2006).
2.8. Genetic diversity
2.8.1. Polymorphism analysis
Power Marker (Liu and Muse, 2005) was used to calculate measures of genetic diversity: observed and expected allele number, observed and expected heterozygosity, Nei's diversity index, Shannon–Weiner index, and polymorphism information content. Observed (H o) and expected heterozygosity (H e) were defined as the probability that two randomly chosen alleles from the population are different (Nei, 1978). The polymorphism information content (PIC) value is commonly used as an estimate of the probability of finding polymorphism between two random samples (Shete et al., 2000).
2.8.2. Genetic differentiation and gene flow between genetic clusters
Genetic differentiation was calculated using an analysis of molecular variance (AMOVA) as applied in ARLEQUIN v. 3.5.1.2, which is a method of quantifying at what level (individual, population, or total) genetic variation is greatest (Excoffier & Lischer, 2010; Peakall & Smouse, 2005; Tsui et al., 2014, 2012). The divergence index and F‐statistics (F ST) were used to measure the level of genetic differentiation among T. dichotomus populations based on genetic polymorphism in the SNP data (Hudson et al., 1992), and F ST was calculated via the PopGen 32 package in BIOPERL (Yeh et al., 1997) using a 100‐kb sliding windows in 10‐kb steps. Pairwise F ST values were evaluated using a randomization test with 1,000 iterations using ARLEQUIN v. 3.5.1.2 (Excoffier & Lischer, 2010; Tsui et al., 2014, 2012).
The direction and rate of migration between genetic clusters (derived from the results of ADMIXTURE) were inferred using MIGRATE‐N v. 3.6.4 (Beerli & Palczewski, 2010), which uses an expansion of the coalescent theory to estimate migration rates. Four possible models for migration between each genetic cluster were evaluated (Yang et al., 2018). The number of recorded steps in the chain was set to 500,000, and the models were run three separate times to confirm convergence of parameter estimates. The marginal likelihoods of all models were compared to infer the direction and rate of gene flow, and only the results of the run that yielded the highest Bezier approximation score (1b) value were presented (Han et al., 2016; Tsui et al., 2014, 2012; Yang et al., 2018).
3. RESULTS
3.1. Morphology
External morphology of all Trypoxylus subspecies were observed and recorded (Table 3). Morphological diagnostic features for delimiting taxa were compared by taxa and geographic origin to assess the degree of overlap or separation of each character. Based on morphological criteria, these Trypoxylus samples included four T. kanamorii from Yunnan (China), Tibet (China), Sagaing (Myanmar); 24 T. d. dichotomus from Southern China; 11 T. d. septentrionalis from Northeastern China, Korean Peninsula, Jeju Island (Korea), Hokkaido (Japan), Hyogo (Japan), and Fukushima (Japan); three T. d. politus from Yunnan (China) and Wiang Papau (Thailand); two T. d. shizuae from Yakushima (Japan); three T. d. tsunobosonis from Taiwan; two T. d. takarai from Okinawa (Japan); and two T. d. tsuchiyai from Kuchinoerabu‐jima (Japan).
Table 3.
Additional morphological descriptions of Trypoxylus specimens in different regions
| Specimens | Features | ♂ | ♀ |
|---|---|---|---|
| Hainan Island (China) | Cuticle coloration is dark wine red in most specimens. Wider, robust body. Morphology and distribution are similar to T. d. politus in Vietnam | Elytra and pronotum highly lustrous in males due to sparse pubescence. Long robust cephalic horn in large males. large males commonly come to light trap in tropical rain forests but rarely found on trees by collectors | |
| Taiwan | Commonly identified to be T. d. tsunobosonis where tsunobosonis implies their thin pronotal horn in Latin. Cuticle coloration ranges from dark red to dark orange. Thinner pronotal horn compared to T. d. septentrionalis in Japan. Slightly smaller on average than T. d. dichotomus from mainland China. Large body size individuals are common, commonly and easily found on broad‐leaved trees across Taiwan thereby light trap is not needed | ||
| Mainland Okinawa (Japan) | Commonly identified to be T. d. takarai. Dark red cuticle coloration, smaller body size, smaller cephalic and pronotal horn size, shorter pubescence on center of elytra and pronotum therefore strongly lustrous compared to T. d. septentrionalis in Japan. similar to T. d. inchachina from Kumejima (Japan). Slightly thinner pronotal horn than T. d. inchachina in most individuals | ||
| Kumejima (Japan) | Commonly identified to be T. d. inchachina. Dark red cuticle coloration, smaller body size, strongly lustrous elytra, smaller cephalic horns compared to T. d. septentrionalis in Japan. Slightly more lustrous than T. d. takarai and less lustrous than T. d. politus from Thailand. Slightly shorter and thicker pronotal horn than T. d. takarai in most individuals. Unlike T. d. septentrionalis from Japan, population on Kumejima are collected in deep jungles that are guarded by military force which is out of most local collectors’ collecting range | ||
| Kuchinoerabu‐jima (Japan) | Commonly identified to be T. d. tsuchiyai. Smaller and wider body, naked or clothed with pubescence on pronotum, shorter and coarser pubescence on pygidium compared to T. d. septentrionalis in Japan. Smaller average body size, smoother and shining lustrous elytra otherwise similar to T. d. septentrionalis in Japan. T. d. shizuae, T. d. tsuchiyai both lined similar to T. d. septentrionalis in morphology, genetics, and geology, these two subspecies were established mostly evidential to their island distribution and slight differences in their morphological characteristics | ||
| Yakushima (Japan) | Commonly identified to be T. d. shizuae. Brighter reddish‐brown, sometimes dark brown cuticle coloration, robust body, slender front tibiae, elytra strongly convex, shorter pronotum, rounded pronotal lateral margin and curved gradually at the former point | Shorter cephalic horn compared to T. d. septentrionalis | Faintly lustrous, fewer, shorter and finer pubescence on metasternum, second abdominal sternite, and abdomen compared to T. d. septentrionalis in Japan but very similar. T. d. shizuae, T. d. tsuchiyai both lined similar to T. d. septentrionalis in morphology, genetics, and geology, these two subspecies were established mostly evidential to their island distribution and slight differences in their morphological characteristics |
| Honshu (Japan), Shikoku (Japan), Kyushu (Japan), and Hokkaido (Japan) | Commonly identified to be T. d. septentrionalis. Cuticle coloration ranges from bright red to dark red, large body size is commonly observed, commonly captured on broad‐leaved trees and by light trap, robust body, weak lustrous elytra, sparse pubescence on pygidium. Population in Hokkaido was introduced by human in recent years due to pet importation, and the population is still growing in Hokkaido every year, imagines collected by light trap or on host trees, sometimes collecting larvae by chopping logs at timber piles near forests | ||
| Korea, Liaoning (China), Jilin (China) | Commonly identified to be T. d. septentrionalis. Cuticle coloration ranges from dark red to black, darker than T. d. septentrionalis in Japan. Large body size is commonly observed, commonly captured on broad‐leaved trees and by light trap, sometimes collecting larvae by chopping logs at timber piles near forests, robust body, weak lustrous elytra, sparse pubescence on pygidium | ||
| Zhangzhou, Fujian (China) | Commonly identified to be T. d. dichotomus. All three male specimens examined have similar small size, thinner pronotal horn, and dark body color. Genitals and geology are also close to T. d. tsunobosonis in Taiwan, yet genetic data clusters it together with the rest of T. d. dichotomus in China | ||
| Mt. Tianmushan, Linan, Zhejiang (China) | Commonly identified to be T. d. dichotomus. It should be noted that specimens caught on Mt. Tianmushan are significantly smaller in average body size compared with other specimens across China | ||
| Southeast of mainland China | Commonly identified to be T. d. dichotomus. Cuticle coloration ranging from dark red to bright red, commonly bigger body size, slender body shape, weak lustrous elytra compared to T. d. septentrionalis in Japan | ||
| Sichuan (China) and Shaanxi (China) | Commonly identified to be T. d. dichotomus. Large body size | Cuticle coloration is darker, ranging from black to dark red, robust cephalic horn | |
| Guangxi (China) | Cuticle coloration is bright wine red. | Highly lustrous elytra and pronotum in males, similar to T. d. politus from Vietnam in morphology and geology. Lustrousness greater than specimens collected in Hainan but color is more reddish | |
| Tibet (China), Diqing Yunnan (China), and Sagaing (Myanmar) | Commonly identified to be T. kanamorii. Smaller body size, cuticle color black, slender body | Strongly lustrous elytra, shorter horns of head and pronotum, very slight pubescence on elytra in female, commonly found in higher elevation than other T. dichotomus subspecies. Its morphological difference is likely to be an adaptation to its higher elevation distribution habitats, male individuals are very similar to T. d. politus in Thailand | |
| Xishuangbanna (Yunnan) and Thailand | Commonly identified to be T. d. politus where politus describes their lustrous elytra and pronotum in Latin, medium to large body size, dark cuticle color ranging from dark red to commonly seen black | Most lustrous elytra and pronotum among all subspecies, shorter and wider body, commonly imported to China, Taiwan, Japan, and Korea as pets due to the fact that T. d. politus imagines do not have stinky smells like T. d. dichotomus from China and T. d. septentrionalis from Japan, usually collected by light trap | |
| Vietnam and Laos | Commonly identified to be T. d. politus. Large body size, cuticle color ranging from dark red to black | Lustrous elytra and pronotum | |
| Dibang (India) | Commonly identified close to T. kanamorii or T. d. politus. Lighter culicle color (dark reddish‐brown to black) than T. kanamorri from Myanmar, small pronotal horn and cephalic horn, small body size; rounded body shape | Lustrous elytra and pronotum, sparse pubescence on pronotum and elytra in female, commonly collected by light trap at altitude above 2,000 m, distribution close to Chayu, Tibet, China |
The coloration of cuticle ranges from black to red; body length, including horn, ranges from 3 cm to over 9 cm, and all other external morphological characters had similarly broad and sometimes overlapping values across regions. Similarly, none of the analyzed morphological characters (commonly used in the Trypoxylus taxonomy) was individually proved to be unambiguously species‐ or subspecies‐specific. As such, additional morphological characters should be used in the further taxonomic study of Trypoxylus. For example, male genitalia have been successfully employed to delimit numerous Coleopteran species.
3.2. Phylogenetic analysis based on 16S and COII
To assess the monophyly of the Trypoxylus subspecies, a phylogenetic analysis was performed based on combined 16S and COII sequence dataset (Figure 2). The alignment included 51 Trypoxylus ingroup sequences that when aligned were 1,239 characters in length including gaps, with 1,042 characters monomorphic, another 113 variable characters were parsimony‐uninformative, and 84 characters were variable and parsimony‐informative. MP analyses generated 12 most parsimonious trees of equal length with the following parameters: tree length (TL) = 274; consistency index (CI) = 0.818; retention index (RI) = 0.839; and rescaled consistency index (RC) = 0.686. ML and BI trees were similar in topology to the MP tree.
Figure 2.
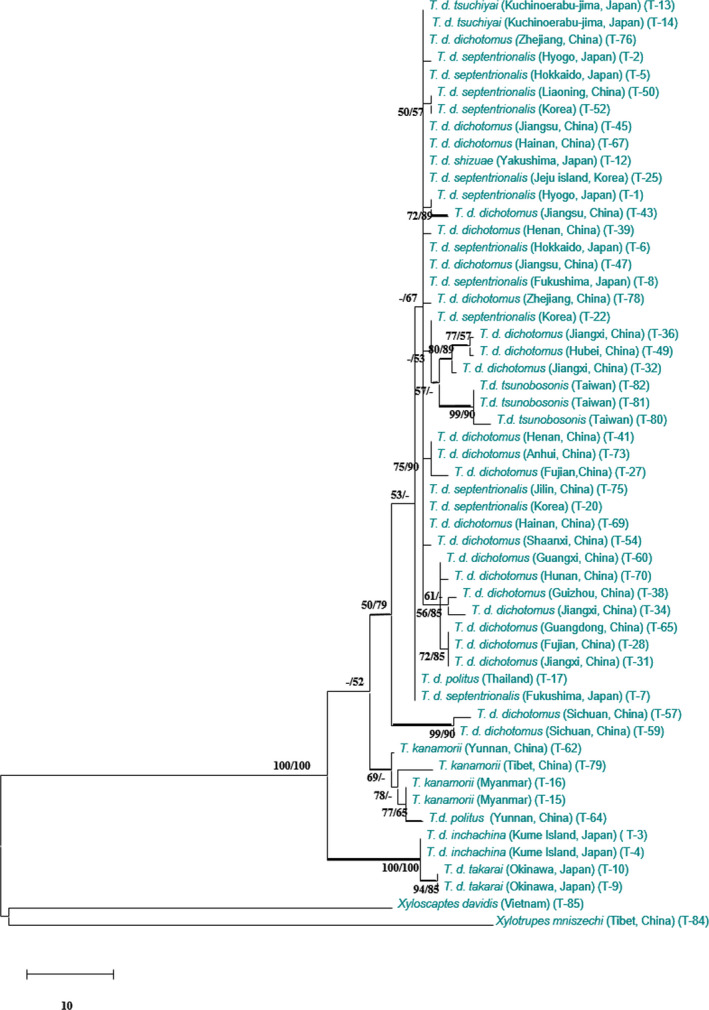
Phylogram of Trypoxylus species based on 16S rRNA and COII gene. MP and ML bootstrap support values above 50% are shown at the first and second position. Thickened branches represent posterior probabilities above 0.95 from BI
The phylogenetic tree based on 16S and COII sequence dataset did not clearly resolve the relationships among the Trypoxylus subspecies (Figure 2), as representatives of T. d. dichotomus and T. d. septentrionalis from Korea and different parts of China formed a clade with weak branch support. However, the 16S and COII data supported the sister relationship between T. d. takarai (Okinawa T‐9, T‐10) and T. d. inchachina (Kumejima T‐3, T‐4; branch support for the two subspecies MP/ML/BI = 100/100/1) in Japan, as well as a clade of T. kanamorii and T. d. politus from Myanmar and China.
3.3. SLAF sequencing and SNP discovery
Using the genome of mountain pine beetles D. ponderosae (Coleoptera: Scolytidae) as a reference, the restriction fragment length of RsaI‐HaeIII, ranging from 264 to 364 bp, was used for defining SLAF tags in our study taxa. Polymorphism analysis of the SLAF markers was summarized in Table 4. Fifty‐three DNA libraries were sequenced using the SLAF‐seq technique, which generated 114.16 Mb of data, ranging from 816,730 to 9,167,064 reads for each library. In all libraries, 92.20% of bases had high‐quality Q30 values. The Q30 data of tested sequences were higher, indicating that the base error rate was very low, and thereby the sequencing results were reliable. Major characteristics of the 53 SLAF‐seq libraries were summarized in Table 4. The average guanine–cytosine content was 35.28%, with each library ranging from 36.24% to 53.77%. The average depth of sequencing obtained was 17.63, ranging from 6.09 (in T‐67) to 109.26 (in T‐25) for each DNA library. Additionally, the total number of SLAF tags was 1,374,985. The highest number of SLAFs by individual was 245,614 in library T‐80, whereas the lowest one was 13,864 in library T‐22, whose depth was 59.12. Out of all tags identified, 330,799 were polymorphic SLAF markers.
Table 4.
Polymorphism analysis results of the SLAF markers of Trypoxylus
| Feature | Value |
|---|---|
| No. of reads | 114.16 Mb |
| Average Q30 percentage | 92.20% |
| Average GC percentage | 37.42% |
| Average depth in individuals | 17.63 |
| Total number of SLAFs | 1,374,985 |
| No. of polymorphic SLAFs | 330,799 |
| No. of SNPs | 2,127,917 |
| No. of SNPs (MAF > 0.5) | 46,939 |
SNP markers were developed based on high depth mapped to the reference genome in each SLAF tag. We identified 2,127,917 SNPs, with an integrity score between 30.16% and 53.93%. To avoid bias in the estimation of the baseline differentiation and to reduce sequencing and PCR error from the SNPs dataset, SNPs with a minor allele frequency (MAF > 0.05) on average across sampling locations were retained and used for analyses. It has been shown that very low‐frequency SNPs (MAF < 0.05) created biases in quantifying genetic connectivity and should therefore be excluded when inferring demographic processes (Roesti et al., 2012). With these criteria, a total of 46,939 high‐integrity SNPs with an MAF > 0.05 were identified among 53 individuals. There was a broad difference in SNP heterozygosity among different samples, with the number of heterozygous SNPs before filtering ranging from 21,157 in T‐20 to 1,188,339 in T‐80. The filtered SNP dataset provided sufficient information to detect the genetic structure and genetic diversity of Trypoxylus taxa.
3.4. Phylogenetic analysis and genetic structure based on SLAF‐seq derived SNPs
Phylogenetic analyses were conducted to determine the relationships among 53 specimens of Trypoxylus subspecies in different regions of Asia (Figure 3). Three well‐supported genetic clusters (North, South, West) were recognized. Genetic clusters North contained individuals from Northeast China (Jilin, Liaoning), mainland of Japan (Fukushima, Kuriyama, Hyogo, Yakushima, Kuchinoerabu‐jima), Korean Peninsula and Jeju Island of Korea; West was made up of individuals from Central and Southern China, including Tibet, Hainan Island, Thailand, and Myanmar, with the remaining individuals from Taiwan (China), the islands of Okinawa and Kumejima (Japan) placed in the South genetic cluster.
Figure 3.
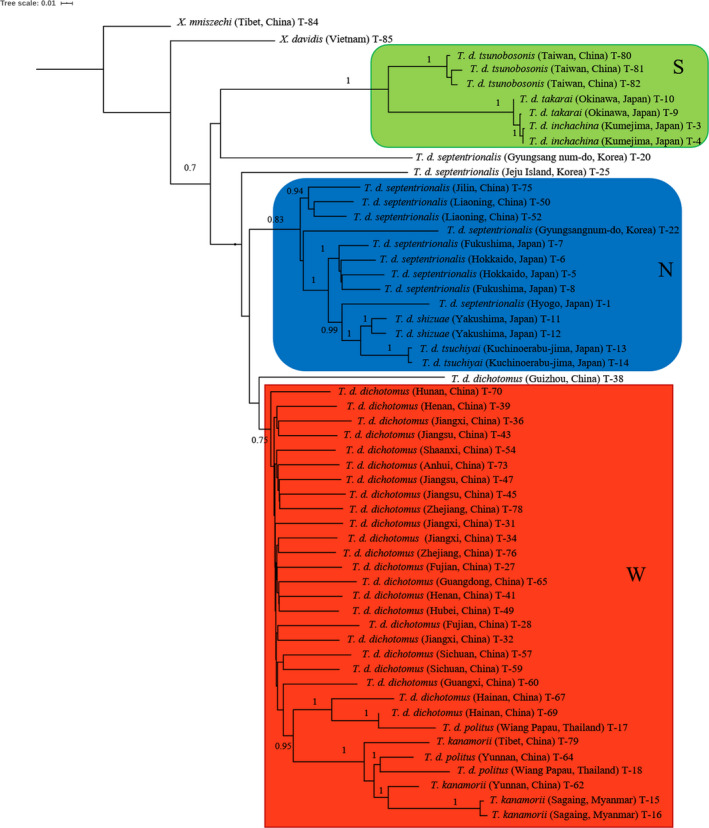
Phylogenetic tree of Trypoxylus subspecies based on the analysis of 46,939 SNPs developed from SLAF. Three major groups were colored
There were discrepancies between the phylogenetic groups and subspecies established with traditional morphological studies; some subspecies were monophyletic, but some were not. Two subspecies in South were monophyletic; T. d. inchachina from Kumejima, Japan, and T. d. takarai from Okinawa, Japan, were closely related to each other, and together, they formed a sister relationship to T. d. tsunobosonis sampled from different locations in Taiwan (Figure 3).
Within group North, T. d. shizuae from Yakushima and T. d. tsuchiyai from Kuchinoerabu‐jima in Japan were in a sister relationship; however, they were more closely related to T. d. septentrionalis population in Japan than other T. d. septentrionalis population from Korea and Northeastern China. Three samples of T. d. septentrionalis from Northeastern China were closely related to each other. In group West, T. kanamorii from Myanmar, Tibet (China) and Yunnan (China) were clustered together with T. d. politus and T. d. dichotomus from Thailand, Yunnan (China), Hainan (China). These samples also clustered with T. d. dichotomus from Sichuan and Guangxi in China. The rest of samples from Eastern China formed a paraphyletic clade. Two T. d. septentrionalis (T‐20, T‐25) from Korea, and one T. d. dichotomus (T‐38) did not cluster with these three genetic clusters and may represent hybrid or relictual lineages.
PCA also separated specimens into three major clusters (Figure 4a,b), which was similar to the phylogenetic analysis based on identified SNPs. PCA spatially placed the group South on the left bottom quadrant of the first PCA axis (account for 22.14% of variation), while the other two groups clustered within the right quadrant of the first PCA axis (Figure 4b). Considerable divergence was also illustrated within group South, in which samples from Taiwan and southern Japan (Okinawa and Kumejima) could be further divided into two well‐supported clades. The North population was slightly differentiated from the West population along the third PCA axis (7.89% of the variation; however, they were apparently separated along the second PCA axis (10.4% of the variation).
Figure 4.
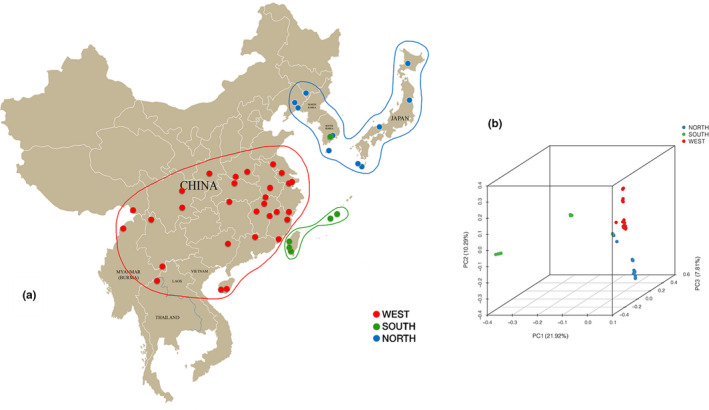
Genetic structure of Trypoxylus species in East Asia. (a) Sampling locations and the Admixture result of three populations. (refer to Table 2 for population abbreviations). (b) PCA analysis of Trypoxylus species depicting the genetic structure
ADMIXTURE analysis generated similar genetic clustering patterns to those based on phylogenetic analysis of SNPs and PCA. The estimated membership fractions of 53 specimens for different values of K were assessed across a range from 1 to 10, with the optimum cluster value of 3 (K = 3) determined using the cross‐validation error rate (Figure 5), which indicated that the sampled specimens could be categorized into three genetic groups. Group North contained 14 specimens, which were from Northeastern China (Jilin, Liaoning), mainland of Japan (Fukushima, Kuriyama, Hyogo, Yakushima, Kuchinoerabu‐jima), Jeju Island (South Korea) and the Korean Peninsula. Group West contained 32 specimens, most of which were from different provinces in China, including central China, Southwestern China, Southeastern China, Tibet, and Hainan Island, while the remainder were from Thailand and Myanmar. Group South contained 7 specimens, 3 of which were from Taiwan, and the rest were from Okinawa, and Kumejima of Japan. Two specimens (T‐25, T‐20) appeared to have admixed origin.
Figure 5.
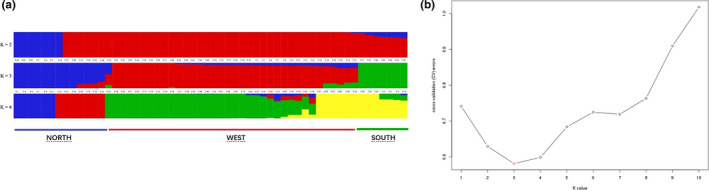
Genetic structure of Trypoxylus inferred by ADMIXTURE (K = 2 to 4). Population codes are given in Table 2
3.5. Genetic diversity among populations
The genetic diversity of the three genetic clusters inferred by genome‐wide SNP data is presented in Table 5. Expected heterozygosity in the South genetic cluster was the highest (H e = 0.338), followed by genetic cluster North (H e = 0.356) and lowest in the West genetic cluster (H e = 0.29). However, observed heterozygosity (South = 0.097, North = 0.197, and West = 0.177) was lower than expected in all genetic clusters suggesting past genetic bottlenecks or some other demographic or selective process is common in Trypoxylus beetles. The mean PIC also revealed that group South was highly polymorphic (0.281), whereas group West exhibited the lowest PIC (0.240).
Table 5.
Genetic diversity metrics from genetic clusters determined in ADMIXTURE
| Population | Observed allele number | Expected Allele number | Observed heterozygous number (H o) | Expected heterozygous number (H e) | Nei's diversity index | Shannon Wiener index | Polymorphism information content (PIC) |
|---|---|---|---|---|---|---|---|
| North | 2 | 1.567 | 0.197 | 0.338 | 0.352 | 0.511 | 0.273 |
| South | 2 | 1.638 | 0.097 | 0.356 | 0.384 | 0.528 | 0.281 |
| West | 2 | 1.457 | 0.177 | 0.290 | 0.295 | 0.455 | 0.240 |
| Total | 2 | 1.392 | 0.111 | 0.257 | 0.260 | 0.414 | 0.216 |
3.6. Population divergence and gene flow
Analysis of molecular variance (AMOVA) indicated that 35.89% of the total variation attributed to variation among populations, while 28.29% and 35.82% were attributed to among individuals within populations and within individual, respectively (Table 6).
Table 6.
Analysis of molecular variance (AMOVA) for T. dichotomus based on three major populations inferred from SNPs data and ADMIXTURE analysis
| Source of variation | df | SS | Variance | % | p‐values |
|---|---|---|---|---|---|
| Among populations | 2 | 145,731.77 | 2,607.16 | 35.89 | <.001 |
| Among individuals within populations | 50 | 292,407.42 | 2,054.62 | 28.29 | <.001 |
| Within individuals | 53 | 121,563.00 | 2,602.30 | 35.82 | <.001 |
| Total | 105 | 559,702.19 | 7,264.07 |
Degree of freedom (df), sum of squares (SS), variance components (Variance), percentage of total variation (%) contributed by populations.
Pairwise F ST revealed differing levels of divergence between North, West, and South genetic clusters. The highest differentiation was between genetic clusters North and South (F ST = 0.345), while the least differentiation was between West and North (F ST = 0.094) (Table 7), which was corroborated with the results based on SNPs.
Table 7.
Pairwise F ST among the three populations inferred from ADMIXTURE analysis
| South | West | North | |
|---|---|---|---|
| South | 0.174 | 0.345 | |
| West | 0.094 |
In addition, the gene flow and migration rates among populations were estimated using MIGRATE‐N. Based on the Bezier approximation scores in model selection, the following gene flow rates and directionality were inferred (Table 8). Bidirectional gene flow between North and West as well as between South and West genetic clusters was inferred. The rate of bidirectional gene flow was similar between these genetic clusters, but the rates were different when compared to different genetic clusters as the average rate of gene flow (N m) between North and West genetic clusters was 3.70 and 2.13 between South and West genetic clusters. Gene flow was inferred to be unidirectional from the South to North genetic cluster at a rate of N m 0.94 (<1), and the genetic differentiation (F ST value = 0.345) between these genetic clusters was also inferred to be the highest among the clusters we compared. As such, a strong barrier to gene flow was inferred between the two population groups (Figure 6).
Table 8.
The estimates of migration rate among three populations using Migrate‐N
| Group | Model | Bezier approximation score (1b) |
|---|---|---|
| NORTH‐SOUTH | Mo1 | −60,030.15 |
| Mo2 | −59,873.85 | |
| Mo3 | −61,398.83 | |
| Mo4 | −60,560.66 | |
| NORTHWEST | Mo1 | −138,945.00 |
| Mo2 | −141,897.74 | |
| Mo3 | −139,602.63 | |
| Mo4 | −141,312.02 | |
| SOUTHWEST | Mo1 | −120,988.34 |
| Mo2 | −124,061.54 | |
| Mo3 | −122,687.21 | |
| Mo4 | −123,467.90 |
Figure 6.
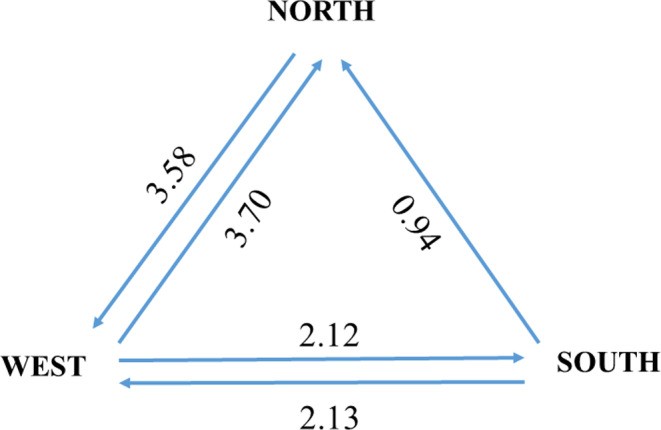
Migration rate estimates among three population groups. The arrow represented the direction of geneflow
4. DISCUSSION
4.1. Population structure and biogeography of Trypoxylus
Resolving patterns of intraspecific variation such as genetic diversity, populations genetic structure, and gene flow within and among populations are essential to understand historic processes such as migration, isolation, and adaptive radiation (Mendelson & Shaw, 2005). Furthermore, patterns of genetic partitioning and divergence when compared to abiotic and biotic landscape features can be an effective method in finding correlations and from there testing hypotheses regarding processes that may have shaped the observed patterns. We investigated the population structure of T. dichotomus and related species using high density SNP markers covering the whole genome, as well as sequences from 16S rRNA and COII. Phylogenetic analysis using the SNP dataset, in addition to ADMIXTURE and PCA analyses, revealed the presence of three genetic groups in the Trypoxylus subspecies/populations.
The genetic partitioning of individuals is clearly related to geographic distribution. For instance, the South genetic cluster is made up of individuals found on the islands of Taiwan, Okinawa, and Kumijima. The phylogenetic branching pattern within this genetic cluster is consistent with the pattern expected for an island radiation wherein individuals from the same island cluster together and the distance of islands from one another is reflected in branching order (i.e., ancestors from islands nearby to each other coalesce before coalescing to ancestors from more distant islands) (MacArthur & Wilson, 1967; Wilson, 1959; Zimmerman, 1970). From the branching pattern, it appears that Taiwan was colonized first, and then Okinawa followed by Kumejima. The subspecific designations in this genetic cluster/clade follow the pattern of geographic/island distribution suggesting that morphological traits associated with subspecific designations may also be involved with adaptive traits which evolved after island colonization (e.g., Darwin's finches). The T‐20 individual, which is a member of the South genetic cluster, does not follow the island model as it was close to the individuals in the North cluster on the Korean peninsula. Possible explanation for the presence of T‐20 on the Korean peninsula could be a recent dispersal from one of the islands or T‐20 may represent an ancient or admixed genotype. The North and West genetic clusters are made up of individuals that are geographically adjacent and from a similar set of subspecific taxa. Branching patterns within these clusters further resolve the pattern of genetic partitioning correlating with geography. For instance, individuals (T. kanamorii, T. d. dichotomus, and T. d. politus) from the southern part of West genetic cluster form a well‐supported clade within the West genetic cluster suggesting a recent radiation with subsequent isolation. In general, the partitioning of individuals into North and South genetic clusters follows previous observations for Trypoxylus populations).
Historic and ongoing gene flow reduces differentiation between populations (Ellstrand & Elam, 1993). The high differentiation and relatively low level of migration from genetic cluster South to North and West indicate barriers to gene flow. The geographic distance from Taiwan to mainland China (closest geographic distance between the South and West genetic clusters) is only about 130 km, while the distance from Taiwan to Okinawa is about 700 km. Given that individuals on Taiwan are more closely related to individuals on Okinawa than they are to individuals on mainland China, suggests that dispersal from the mainland to islands is rare and may be more strongly influenced by ocean currents (Karl, 1999) than geographic proximity. While island dispersal has occurred in both North (Japan and associated islands and Jeju, Korea) and West (Hainan island, China) genetic clusters, these dispersal events are inferred to be more recent and/or include ongoing migration as individuals on these islands did not resolved in separate genetic clusters. However, branching patterns in the phylogenetic analysis (Figure 3) suggests subclustering of island lineages within North and West genetic clusters. Additionally, gene flow between North and West genetic clusters is more frequent, possibly because of fewer geographic barriers.
In all genetic clusters, the observed heterozygosity was lower than expected heterozygosity suggesting that demographic and/or behavioral processes might be decreasing genetic diversity in Trypoxylus populations. It is well known that male Trypoxylus beetles compete against one another for access to females (Hongo, 2003, 2007). These types of competitive behaviors have been associated with increases in heterozygosity in other animal species (e.g., Bensch et al., 2006; Seddon et al., 2004). However, founder effects associated with colonizing new territory have been shown to strongly decrease observed heterozygosity in animal populations (e.g., Kekkonen et al., 2012; Keller & Waller, 2002). The greatest difference between observed and expected heterozygosity was found in the South genetic cluster which is consistent with the inferred founder effects associated with island dispersal. More work is needed to elucidate what factors are contributing to the low observed heterozygosity found in this study.
Despite the geographic isolation of various subspecies/population, some individuals were inferred to be of admixed origins (Figures 3 and 4). Given the popularity of keeping Trypoxylus beetles as pets and importance in medicine, they have been translocated throughout Asia. Beetles kept as pets or used in breeding are often released back into the wild (Nagai, 2007). Through such actions, beetles from divergent lineages may be brought into contact and reproduce. Translocation and release into the wild may explain the presence of a South genetic cluster genotype (T‐20) found in the geographic range of North genetic cluster individuals. Translocation and release into the wild can have detrimental effects on wild locally adapted populations through outbreeding depression (Frankham, 2010) and should be studied further especially among isolated island populations. T. d. septentrionalis samples from Korea were quite divergent as isolates from Jeju Island (T‐25) and Gyungsang num‐do (T‐20) did not cluster with beetle populations from Northeastern China and Japan. They could represent different independent lineages as a result of historic separation or recent geographic isolation.
4.2. Taxonomic implications
Ten recognized subspecies of T. dichotomus and closely allied species T. kanamorii have been described from different parts of Asia, including the countries China, Japan, Korea, Myanmar, Thailand, Laos, and India (Adachi, 2017; Kôno, 1931; Kusui, 1976; Li et al., 2015; Linné, 1771; Nagai, 2006, 2007; Prell, 1934; Satoru, 2014). Taxonomic boundaries among these subspecies were not clearly defined in the past. We attempted to resolve the phylogenetic relationships among Trypoxylus subspecies based on 16S and COII sequences, which have provided phylogenetic resolution for delineating Coleoptera genera in past studies (Lee et al., 2015). However, 16S and COII did not resolve Trypoxylus subspecies delineations with high support.
Using broadly distributed SNP markers, subspecies were resolved in three well‐supported genetic clusters that generally corresponded to geographic origins. Although T. dichotomus and related species share similar morphological features (body size, lustrousness, cuticle color, and distribution of pubescence; Table 2), none of the analyzed morphological characters taken individually and commonly used in the traditional taxonomy of T. dichotomus proved to be unambiguously species‐ or subspecies‐specific. The overlap of some characters in Trypoxylus may be the result of phenotypic plasticity, not previous observed in taxonomic descriptions, resulting from the interactions between genetic, environmental conditions (e.g., temperature or latitude), and/or biotic factors seen in other species (Davis et al., 2008; Gross et al., 2004). However, according to Buchalski et al. (2019), T. dichotomus populations in Kyoto and Hokkaido (in central and Northern Japan, corresponding to North group in this study) had significantly longer horns for a given body size, than those specimens in Taiwan and Yakushima (largely corresponding to group South) in this study.
Several subspecies circumscribed by morphological features were supported by the molecular data in this study. The best example was T. d. tsuchiyai and T. d. shizuae from Japan that were resolved as monophyletic with high branch support (PP = 1). Both subspecies were similar to T. d. septentrionalis. However, T. d. tsuchiyai was smaller and wider in both sexes, with pronotum and middle frontal areas of elytra clothed with pubescence, and the female individuals had shorter and coarser pubescence on pygidium comparing to T. d. septentrionalis. T. d. shizuae lacked an obvious presence absence morphological trait but was different in body size ratio to T. d. septentrionalis among individuals examined for this study. Based on our results, T. d. tsuchiyai and T. d. shizuae should be retained as subspecies and considered for elevation to species following further study including genitalic dissections. T. d. septentrionalis is paraphyletic with the inclusion of T. d. tsuchiyai and T. d. shizuae. However, the resolution of several well‐supported clades within the North genetic cluster suggests that T. d. septentrionalis should be reexamined and possible refined to account for these well‐supported clades.
Both T. d. inchachina and T. d. takarai have been reported from the Okinawan Islands (Japan), and together, they formed a monophyletic clade with small genetic distance. T. d. takarai has been separated from T. d. inchachina as being a smaller and black bodied beetle, with shorter pronotal horns on male individuals. However, these two subspecies were found to be very similar to each other in morphological traits (Nagai, 2007). Compared with T. d. septentrionalis, they were both smaller in body size and possessed smaller cephalic/pronotal horns and were darker in body color in male individuals. Differences between these two subspecies were indistinguishable in smaller individuals. T. d. inchachina and T. d. takarai may be considered as independent lineages possibly because of allopatry. From Taiwan, T. d. tsunobosonis resolved as monophyletic and should be retained as subspecies and studied further for possible elevation to species. T. d. tsunobosonis had thinner pronotal horn in male individuals than T. d. septentrionalis, which was similar to T. d. takarai in male genitalia (Nagai, 2007).
The West genetic cluster comprised of 23 specimens of T. d. dichotomus, which was resolved as paraphyletic basal to T. kanamorii and T. d. politus. T. d. politus nested within. T. d. dichotomus is separated from T. d. septentrionalis by having a longer, thinner, and less glossy body as well as a longer thicker cephalic horn. However, specimens from Southern China and Central China did not show any significant difference in morphology. Within the paraphyletic grade of T. d. dichotomus, a well‐supported clade containing the subspecies T. d. politus. and the species T. kanamorii was resolved. Morphologically, T. kanamorii is very similar to T. d. politus, but it can be distinguished from the later in having a slender and black body, parallel‐sided elytra, shorter horns on head and pronotum, and slight pubescence on elytra (Nagai, 2006). The clade containing T. kanamorii is paraphyletic with respect to T. d. politus. As such the designation of “species” does not appear to be valid for T. kanamorii. Furthermore, if strictly applying a phylogenetic approach to taxonomic designations (de Queiroz & Gauthier, 1990), the recognition of clades (like that containing T. kanamorii) that result in the creation of basal paraphyletic grades does not adhere to the goal of species delineations recognizing monophyletic groupings.
5. CONCLUSION
Our study demonstrated that SLAF‐seq derived markers outperformed 16S and COII sequences in beetle taxonomy and provided improved resolution of the genetic differentiation of rhinoceros beetle populations from a large part of the species’ range. Phylogenetic analysis of SNPs indicated the presence of three distinct genetic groups, suggesting that the various subspecies fall into three distinct and well‐supported lineages. PCA and ADMIXTURE analysis also identified three genetic clusters (North, South, West), which corresponded to their origin, suggesting that geographic factors were important in maintaining within population homogeneity and between population divergence. Further study including more detailed morphological work, increased taxon sampling, and taxonomic circumscription is needed to improve the current systematics of Trypoxylus beetles. The current study provides an important dataset, analyses, and method for genotyping (SLAF‐seq) that can be used and built upon to answer unresolved questions of systematics, character evolution, chemical and immunological chemistry, and phylogeography in Trypoxylus and related taxa.
CONFLICT OF INTEREST
All authors declare that they have no competing interests.
AUTHOR CONTRIBUTION
Huan Yang: Data curation (equal); Formal analysis (equal); Writing‐original draft (equal). Chongjuan You: Data curation (equal); Funding acquisition (lead); Methodology (equal); Writing‐original draft (equal). Clement Tsui: Investigation (equal); Methodology (equal); Writing‐review & editing (equal). Luke Tembrock: Writing‐review & editing (equal). Zhiqiang Wu: Writing‐review & editing (equal). Depo Yang: Supervision (equal).
ACKNOWLEDGMENTS
We are grateful to Masashi Sakamoto, Chang Chin Chen, Qiao Zhi Yang, Ching Lin Chu, Do Young Kim, Sang Il Kim, Yuan Yuan Zhu, Sai Huang, Rui Yang, Yuan Dong Zheng, Yi Zhou Liu, Hong Liang Shi, Jun Qiang Xu, Meng Ya Ni, Yang Zhang, Yang He, Zhi Hao Qi, Xiao Dong Yang, Yi Xin Su, Tong Ye, Zhi Chao Zhang, Cheng Hui Zhan, Ming Qiang Xu, Jun Qi, Jia Qi Wang, Zhao Yang Tang, Ryan Yu, Ren Zhi Zhang, Hao Huang, Andrew Zhu, Ming Yu Ma, Cheng Ming Xiao, Ze Nan Zhang, Jian Rong Chen, Ming Jin, Cheng Bin Wang, Jian Hao, Song Yang, Ming Fei Hou, and Yao Qin Hong, who are amateur entomologists from China, Korea, Canada, USA, and Japan for providing their valuable specimens and valuable advice. This work was supported through grants to CJY by the Fundamental Research Funds for the Central Universities, China (Grant No. 2018ZY23).
Yang H, You CJ, Tsui CKM, Tembrock LR, Wu ZQ, Yang DP. Phylogeny and biogeography of the Japanese rhinoceros beetle, Trypoxylus dichotomus (Coleoptera: Scarabaeidae) based on SNP markers. Ecol Evol.2021;11:153–173. 10.1002/ece3.6982
Funding information
Fundamental Research Funds for the Central Universities, China (Grant No. 2018ZY23).
DATA AVAILABILITY STATEMENT
Data have been deposited at the Dryad Digital Repository: https://doi.org/10.5061/dryad.qv9s4mwcz.
REFERENCES
- Adachi, N. (2017). A new subspecies of Trypoxylus dichotomus (Linnaeus, 1771) (Coleoptera, Scarabaeidae, Dynastinae) from Yakushima Island and Tanegashima Island, Kagoshima Prefecture, Japan. Kogane, Tokyo, 20, 11–16. [Google Scholar]
- Alexander, D. H. , Novembre, J. , & Lange, K. (2009). Fast model‐based estimation of ancestry in unrelated individuals. Genome Research, 19, 1655–1664. 10.1101/gr.094052.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batista, P. D. , Janes, J. K. , Boone, C. K. , Murray, B. W. , & Sperling, F. A. (2016). Adaptive and neutral markers both show continent‐wide population structure of mountain pine beetle (Dendroctonus ponderosae). Ecology and Evolution, 6, 6292–6300. 10.1002/ece3.2367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beerli, P. , & Palczewski, M. (2010). Unified framework to evaluate panmixia and migration direction among multiple sampling locations. Genetics, 185, 313–326. 10.1534/genetics.109.112532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensch, S. , Andrén, H. , Hansson, B. , Pedersen, H. C. , Sand, H. , Sejberg, D. , Wabakken, P. , Åkesson, M. , & Liberg, O. (2006). Selection for heterozygosity gives hope to a wild population of inbred wolves. PLoS One, 1(1), e72 10.1371/journal.pone.0000072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black, W. C. , & DuTeau, N. M. (1997). In The molecular biology of insect disease vectors. Springer. [Google Scholar]
- Blanco‐Bercial, L. , & Bucklin, A. (2016). New view of population genetics of zooplankton: RAD‐seq analysis reveals population structure of the North Atlantic planktonic copepod Centropages typicus . Molecular Ecology, 25, 1566–1580. 10.1111/mec.13581 [DOI] [PubMed] [Google Scholar]
- Buchalski, B. , Gutierrez, E. , Emlen, D. , Lavine, L. , & Swanson, B. (2019). Variation in an extreme weapon: Horn performance differences across rhinoceros beetle (Trypoxylus dichotomus) populations. Insects, 10, 346 10.3390/insects10100346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung, M. Y. , Kwon, E. Y. , Hwang, J. S. , Goo, T. W. , & Yun, E. Y. (2013). Establishment of food processing methods for larvae of Allomyrina dichotoma, Korean Horn Beetle. Life Science Journal, 23, 426–431. 10.5352/jls.2013.23.3.426 [DOI] [Google Scholar]
- Chung, M. Y. , Yoon, Y. I. , Hwang, J. S. , Goo, T. W. , & Yun, E. Y. (2014). Anti‐obesity effect of Allomyrina dichotoma (Arthropoda: Insecta) larvae ethanol extract on 3T3‐L1 adipocyte differentiation. Entomological Research, 44, 9–16. 10.1111/1748-5967.12044 [DOI] [Google Scholar]
- Davis, A. L. V. , Brink, D. J. , Scholtz, C. H. , Prinsloo, L. C. , & Deschodt, C. H. (2008). Functional implications of temperature‐correlated colour polymorphism in an iridescent, scarabaeine dung beetle. Ecological Entomology, 33, 771–779. 10.1111/j.1365-2311.2008.01033.x [DOI] [Google Scholar]
- De Queiroz, K. , & Gauthier, J. (1990). Phylogeny as a central principle in taxonomy: Phylogenetic definitions of taxon names. Systematic Zoology, 39, 307–322. 10.2307/2992353 [DOI] [Google Scholar]
- Debnath, S. C. (2014). Structured diversity using EST‐PCR and EST‐SSR markers in a set of wild blueberry clones and cultivars. Biochemical Systematics and Ecology, 54, 337–347. 10.1016/j.bse.2014.03.018 [DOI] [Google Scholar]
- Dutrillaux, A.‐M. , Mamuris, Z. , & Dutrilllaux, B. (2013). Chromosome analyses challenge the taxonomic position of Augosoma centaurus Fabricius, 1775 (Coleoptera: Scarabaeidae: Dynastinae) and the separation of Dynastini and Oryctini. Zoosystema, 35, 537–549. 10.5252/z2013n4a7 [DOI] [Google Scholar]
- Ellstrand, C. N. , & Elam, R. D. (1993). Population genetic consequences of small population size: Implication for plant conservation. Annual Review of Ecology and Systematics, 24, 217–242. 10.1146/annurev.es.24.110193.001245 [DOI] [Google Scholar]
- Emlen, D. , Lavine, L. , & Ewen‐Campen, B. (2007). On the origin and evolutionary diversification of beetle horns. Proceedings of the National Academy of Sciences, 104, 8661–8668. 10.1073/pnas.0701209104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emlen, D. , Warren, I. , Johns, A. , Dworkin, I. , & Lavine, L. (2012). A mechanism of extreme growth and reliable signaling in sexually selected ornaments and weapons. Science, 337, 860–864. 10.1126/science.1224286 [DOI] [PubMed] [Google Scholar]
- Excoffier, L. , & Lischer, H. E. L. (2010). Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Resources, 10, 564–567. 10.1111/j.1755-0998.2010.02847.x [DOI] [PubMed] [Google Scholar]
- Felsenstein, J. (1985). The American Naturalist. Phylogenies and the Comparative Method, 125, 1–15. [Google Scholar]
- Frankham, R. (2010). Challenges and opportunities of genetic approaches to biological conservation. Biological Conservation, 143, 1919–1927. 10.1016/j.biocon.2010.05.011 [DOI] [Google Scholar]
- Gross, J. , Schmolz, E. , & Hilker, M. (2004). Thermal adaptations of the leaf beetle Chrysomela lapponica (Coleoptera: Chrysomelidae) to different climes of Central and Northern Europe. Enviroment Entomology, 33, 799–806. 10.1603/0046-225x-33.4.799 [DOI] [Google Scholar]
- Han, T. M. , Jeong, J. C. , Kang, T. H. , Lee, Y. B. , & Park, H. C. (2010). Phylogenetic relationships of Dorcus koreanus Jang and Kawai, 2008 (Coleoptera, Lucanidae): Species or subspecies? Zoological Science, 27, 362–368. 10.2108/zsj.27.362 [DOI] [PubMed] [Google Scholar]
- Han, Y. , Zhao, X. , Liu, D. , Li, Y. , Lightfoot, D. A. , Yang, Z. , Zhao, L. , Zhou, G. , Wang, Z. , Huang, L. , Zhang, Z. , Qiu, L. , Zheng, H. , & Li, W. (2016). Domestication footprints anchor genomic regions of agronomic importance in soybeans. New Phytologist, 209, 871–884. 10.1111/nph.13626 [DOI] [PubMed] [Google Scholar]
- Hee, J. K. , Yeun, J. M. , & Ho, L. S. (2001). Lectin from the larvae of Allomyrina dichotoma as immunomodulator and antitumor agent. Autumn Conferences, 2, 264–265. [Google Scholar]
- Hongo, Y. (2003). Appraising behaviour during male‐male interaction in the Japanese horned beetle Trypoxylus dichotomus septentrionalis (Kôno). Behaviour, 140, 501–517. 10.1163/156853903322127959 [DOI] [Google Scholar]
- Hongo, Y. (2007). Evolution of male dimorphic allometry in a population of the Japanese horned beetle Trypoxylus dichotomus septentrionalis . Behavioral Ecology and Sociobiology, 62, 245–253. 10.1007/s00265-007-0459-2 [DOI] [Google Scholar]
- Hosoya, T. , & Araya, K. (2005). Phylogeny of Japanese stag beetles (Coleoptera: Lucanidae) inferred from 16S mtrRNA gene sequences, with reference to the evolution of sexual dimorphism of mandibles. Zoological Science, 22, 1305–1319. 10.2108/zsj.22.1305 [DOI] [PubMed] [Google Scholar]
- Huang, H. R. , Wu, W. , Zhang, J. X. , Wang, L. J. , Yuan, Y. M. , & Ge, X. J. (2016). A genetic delineation of Patchouli (Pogostemon cablin) revealed by specific‐locus amplified fragment sequencing. Journal of Systemaatics and Evolution, 54, 491–501. 10.1111/jse.12195 [DOI] [Google Scholar]
- Hudson, R. R. , Boos, D. D. , & Kaplan, N. L. (1992). A statistical test for detecting geographic subdivision. Molecular Biology and Evolution, 9, 138–151. 10.1093/oxfordjournals.molbev.a040703 [DOI] [PubMed] [Google Scholar]
- Ito, Y. , Harigai, A. , Nakata, M. , Hosoya, T. , Araya, K. , Oba, Y. , Ito, A. , Ohde, T. , Yaginuma, T. , & Niimi, T. (2013). The role of doublesex in the evolution of exaggerated horns in the Japanese rhinoceros beetle. EMBO Reports, 14, 561–567. 10.1038/embor.2013.50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johns, A. , Gotoh, H. , McCullough, E. , Emlen, D. , & Lavine, L. (2014). Heightened condition‐ dependent growth of sexually selected weapons in the rhinoceros beetle, Trypoxylus dichotomus (Coleoptera: Scarabaeidae). Integrative and Comparative Biology, 54, 614–621. 10.1093/icb/icu041 [DOI] [PubMed] [Google Scholar]
- Karino, K. , Niiyama, H. , & Chiba, M. (2005). Horn length is the determining factor in the outcomes of escalated fights among male Japanese horned beetles, Allomyrina dichotoma L. (Coleoptera: Scarabaeidae). Journal of Insect Behavior, 18, 805–815. 10.1007/s10905-005-8741-5 [DOI] [Google Scholar]
- Karl, D. M. (1999). A sea of change: Biogeochemical variability in the North Pacific Subtropical Gyre. Ecosystems, 2, 181–214. 10.1007/s100219900068 [DOI] [Google Scholar]
- Katoh, K. , & Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Molecular Biology and Evolution, 30, 772–780. 10.1093/molbev/mst010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kekkonen, J. , Wikström, M. , & Brommer, J. E. (2012). Heterozygosity in an isolated population of a large mammal founded by four individuals is predicted by an individual‐based genetic model. PLoS One, 7, e43482 10.1371/journal.pone.0043482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller, L. F. , & Waller, D. M. (2002). Inbreeding effects in wild populations. Trends in Ecology and Evolution, 17, 230–241. 10.1016/s0169-5347(02)02489-8 [DOI] [Google Scholar]
- Kim, D. S. , Huh, J. , You, G. C. , & Chae, S. C. (2007). Allomyrina dichotoma larvae extracts protect streptozotocin‐induced oxidative cytotoxicity. Journal of Environmental Toxicology, 22, 349–355. [Google Scholar]
- Kim, J. , Yun, E. Y. , Park, S. W. , Goo, T. W. , & Seo, M. (2016). Allomyrina dichotoma larvae regulate food intake and body weight in high fat diet‐induced obese mice through mTOR and Mapk signaling pathways. Nutrients, 8, 100 10.3390/nu8020100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kôno, H. (1931). Die Trypoxylus‐Arten aus Japan und Formosa (Col. Scarabaeidae). Insecta Matsumurana, 5, 159–160. [Google Scholar]
- Kui, Z. , Chen, J. F. , & Cai, Y. X. (2000). Cultivation of medicinal insect of Japanese rhinoceros beetles. Zhong Cao Yao, 31, 633–634. [Google Scholar]
- Kumar, S. , Stecher, G. , Li, M. , Knyaz, C. , & Tamura, K. (2018). MEGA X: Molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution, 35, 1547–1549. 10.1093/molbev/msy096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusui, Y. (1976). Notes on the Allomyrina dichotoma (Linne) from Okinawa Islands (Coleoptera, Scarabaeidae). Entomological Review of Japan, 29, 51–54. [Google Scholar]
- Lee, G.‐E. , Han, T. , Jeong, J. , Kim, S.‐H. , Park, I. G. , & Park, H. (2015). Molecular phylogeny of the genus Dicronocephalus (Coleoptera, Scarabaeidae, Cetoniinae) based on mtCOI and 16S rRNA genes. ZooKeys, 501, 63–87. 10.3897/zookeys.501.8658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , Handsaker, B. , Wysoker, A. , Fennell, T. , Ruan, J. , Homer, N. , Marth, G. , Abecasis, G. , & Durbin, R. (2009). The sequence alignment/map format and SAMtools. Bioinformatics, 25, 2078–2079. 10.1093/bioinformatics/btp352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, J. K. , Gao, M. X. , & Zhang, X. P. (2015). Color illustrations of soil beetles in Northeast China: Scarabaeoidea 103–104. (Harbin Cartographic Publishing House). [Google Scholar]
- Linnaeus, C. (1771). Mantissa Plantarum altera generum editions VI. et specierum edition II. Laurentii Salvii, Holamiae.
- Linné, C. (1771). Mantissa Plantarum altera generum editions VI. et specierum edition II. 144–588 (Laurentii Salvii).
- Liu, K. , & Muse, S. V. (2005). Power marker: An integrated analysis environment for genetic marker analysis. Bioinformatics, 21, 2128–2129. 10.1093/bioinformatics/bti282 [DOI] [PubMed] [Google Scholar]
- MacArthur, R. H. , & Wilson, E. O. (1967). The theory of Island biogeography. (Princeton University Press). [Google Scholar]
- McKenna, A. , Hanna, M. , Banks, E. , Sivachenko, A. , Cibulskis, K. , Kernytsky, A. , Garimella, K. , Altshuler, D. , Gabriel, S. , Daly, M. , & DePristo, M. A. (2010). The Genome Analysis Toolkit: A map reduce framework for analyzing next‐generation DNA sequencing data. Genome Research, 20, 1297–1303. 10.1101/323535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelson, T. , & Shaw, K. (2005). Sexual behaviour: rapid speciation in an arthropod. Nature, 433, 375–376. 10.1038/433375a [DOI] [PubMed] [Google Scholar]
- Miyanoshita, A. , Hara, S. , Sugiyama, M. , Asaoka, A. I. , Taniai, K. , Yukuhiro, F. , & Yamakawa, M. (1996). Isolation and characterization of a new member of the insect defensin family from a beetle, Allomyrina dichotoma . Biochemical and Biophysical Research Communications, 220, 526–531. 10.1006/bbrc.1996.0438 [DOI] [PubMed] [Google Scholar]
- Morita, S. , Ando, T. , Maeno, A. , Mizutani, T. , Mase, M. , Shigenobu, S. , & Niimi, T. (2019). Precise staging of beetle horn formation in Trypoxylus dichotomus reveals the pleiotropic roles of doublesex depending on the spatiotemporal developmental contexts. PLoS Genetics, 15, e1008063 10.1371/journal.pgen.1008063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai, S. (2006). A new species and new subspecies of the genus Trypoxylus from Asia and a new subspecies of the genus Beckium from New Guinea (Coleoptera, Scarabaeidae, Dynastinae). Gekkan‐Mushi, 428, 13–17. [Google Scholar]
- Nagai, S. (2007). Nihon no Kabutomushi Daizukan. BE‐KUWA, 22, 8–29. [Google Scholar]
- Nam, H. S. , Yang, H. J. , Oh, B. J. , Anderson, A. J. , & Kim, Y. C. (2016). Biological control potential of Bacillus amyloliquefaciens KB3 isolated from the feces of Allomyrina dichotoma larvae. Plant Pathology Journal, 32, 273–280. 10.5423/ppj.nt.12.2015.0274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei, M. (1978). Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics, 89, 583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nylander, J. A. A. (2004). MrModeltest. Program distributed by the Author (vol. 2). Evolutionary Biology Centre, Uppsala University. [Google Scholar]
- Ohde, T. , Morita, S. , Shigenobu, S. , Morita, J. , Mizutani, T. , Gotoh, H. , Zinna, R. A. , Nakata, M. , Ito, Y. , Wada, K. , Kitano, Y. , Yuzaki, K. , Toga, K. , Mase, M. , Kadota, K. , Rushe, J. , Lavine, L. C. , Emlen, D. J. , & Niimi, T. (2018). Rhinoceros beetle horn development reveals deep parallels with dung beetles. PLoS Genetics, 14, e1007651 10.1371/journal.pgen.1007651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pascoal, S. , & Kilner, R. M. (2017). Development and application of 14 microsatellite markers in the burying beetle Nicrophorus vespilloides reveals population genetic differentiation at local spatial scales. PeerJ, 5, e3278 10.7717/peerj.3278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peakall, R. , & Smouse, P. E. (2005). GenAlEx 6: Genetic analysis in Excel. Population genetic software for teaching and research. Molecular Ecology Notes, 6, 288–295. 10.1093/bioinformatics/bts460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prell, H. (1934). Beitrage zur Kenntnis der Dynastinen (XII). Beschreibungen Und Bemerkungen. Entomologische Blatter, 30, 54–60. [Google Scholar]
- Price, A. L. , Patterson, N. J. , Plenge, R. M. , Weinblatt, M. E. , Shadick, N. A. , & Reich, D. (2006). Principal components analysis corrects for stratification in genome‐wide association studies. Nature Genetics, 38, 904–909. 10.1038/ng1847 [DOI] [PubMed] [Google Scholar]
- Ratcliffe, N. A. , Mello, C. B. , Garcia, E. S. , Butt, T. M. , & Azambuja, P. (2011). Insect natural products and processes: New treatments for human disease. Insect Biochemistry and Molecular Biology, 41, 747–769. 10.1016/j.ibmb.2011.05.007 [DOI] [PubMed] [Google Scholar]
- Roesti, M. , Salzburger, W. , & Berner, D. (2012). Uninformative polymorphisms bias genome scans for signatures of selection. BMC Evolutionary Biology, 12, 94 10.1186/1471-2148-12-94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronquist, F. , & Huelsenbeck, J. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics, 19, 1572–1574. 10.1093/bioinformatics/btg180 [DOI] [PubMed] [Google Scholar]
- Rowland, J. M. , & Miller, K. B. (2012). Phylogeny and systematics of the giant rhinoceros beetles (Scarabaeidae: Dynastini). Insecta Mundi, 263, 1–15. [Google Scholar]
- Sagisaka, A. , Miyanoshita, A. , Ishibashi, J. , & Yamakawa, M. (2001). Purification, characterization and gene expression of a glycine and proline‐rich antibacterial protein family from larvae of a beetle. Allomyrina Dichotoma. Insect Molecular Biology, 10, 293–302. 10.1046/j.0962-1075.2001.00261.x [DOI] [PubMed] [Google Scholar]
- Saitou, N. , & Nei, M. (1987). The neighbor‐joining method: A new method for reconstructing phylogenetic trees. Molecular Biology and Evolution, 4, 406–425. 10.1093/oxfordjournals.molbev.a040454 [DOI] [PubMed] [Google Scholar]
- Satoru, T. (2014). A new subspecies of Trypoxylus dichotomus (Coleoptera: Scarabaeidae: Dynastinae) from China. India Journal of Entomology, 76, 149–151. [Google Scholar]
- Seddon, N. , Amos, W. , Mulder, R. A. , & Tobias, J. A. (2004). Male heterozygosity predicts territory size, song structure and reproductive success in a cooperatively breeding bird. Proceedings of the Royal Society of London Series B‐Biological Sciences, 271, 1823–1829. 10.1098/rspb.2004.2805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shete, S. , Tiwari, H. , & Elston, R. C. (2000). On estimating the heterozygosity and polymorphism information content value. Theoretical Population Biology, 57, 265–271. 10.1006/tpbi.2000.1452 [DOI] [PubMed] [Google Scholar]
- Stamatakis, A. (2006). RAxML‐VI‐HPC: Maximum likelihood‐based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics, 22, 2688–2690. 10.1093/bioinformatics/btl446 [DOI] [PubMed] [Google Scholar]
- Su, W. , Wang, L. , Lei, J. , Chai, S. , Liu, Y. I. , Yang, Y. , Yang, X. , & Jiao, C. (2017). Genome‐wide assessment of population structure and genetic diversity and development of a core germplasm set for sweet potato based on specific length amplified fragment (SLAF) sequencing. PLoS One, 12, e0172066 10.1371/journal.pone.0172066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, X. , Liu, D. , Zhang, X. , Li, W. , Liu, H. , Hong, W. , Jiang, C. , Guan, N. , Ma, C. , Zeng, H. , Xu, C. , Song, J. , Huang, L. , Wang, C. , Shi, J. , Wang, R. , Zheng, X. , Lu, C. , Wang, X. , & Zheng, H. (2013). SLAF‐seq: An efficient method of large‐scale de novo SNP discovery and genotyping using high‐throughput sequencing. PLoS One, 8, e58700 10.1371/journal.pone.0058700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swofford, D. L. (2003). PAUP*: Phylogenetic Analysis Using Parsimony, * and Other Methods. Version 4.0b10. (Sunderland). [Google Scholar]
- Tamura, K. , Peterson, D. , Peterson, N. , Stecher, G. , Nei, M. , & Kumar, S. (2011). MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution, 28, 2731–2739. 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui, C.‐M. , Farfan, L. , Roe, A. D. , Rice, A. V. , Cooke, J. E. K. , El‐Kassaby, Y. A. , & Hamelin, R. C. (2014). Population structure of mountain pine beetle symbiont Leptographium longiclavatum and the implication on the multipartite beetle‐fungi relationships. PLoS ONE, 9 10.1371/journal.pone.0105455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsui, C. K. M. , Roe, A. D. , El‐Kassaby, Y. A. , Rice, A. V. , & Hamelin, R. C. (2012). Population structure and migration pattern of a conifer pathogen, Grosmannia clavigera, as influenced by its symbiont, the mountain pine beetle. Molecular Ecology, 21, 71–86. 10.1111/j.1365-294x.2011.05366.x [DOI] [PubMed] [Google Scholar]
- Umetsu, K. , Ikeda, N. , Kashimura, S. , & Suzuki, T. (1985). Affinity chromatography of human serum proteins using immobilized lectin from Allomyrina dichotoma . Biochemical. International, 10, 549–552. [PubMed] [Google Scholar]
- Umetsu, K. , Kosaka, S. , & Suzuki, T. (1984). Purification and characterization of a lectin from the beetle, Allomyrina Dichotoma. Biochemcal Journal, 95, 239–245. 10.1093/oxfordjournals.jbchem.a134590 [DOI] [PubMed] [Google Scholar]
- Wang, X. , Lu, B. , Shao, L. , Yu, F. , Zheng, W. , & Sun, F. (2019). Genetic variation and population genetic structure of Laodelphax striatellus via genome‐wide single nucleotide polymorphisms from specific‐locus amplified fragment‐sequencing. Journal of Applied Entomology, 143, 315–327. 10.1111/jen.12597 [DOI] [Google Scholar]
- Warren, I. A. , Gotoh, H. , Dworkin, I. M. , Emlen, D. J. , & Lavine, L. C. (2013). A general mechanism for conditional expression of exaggerated sexually‐selected traits. BioEssays, 35, 889–899. 10.1002/bies.201300031 [DOI] [PubMed] [Google Scholar]
- Wilson, E. O. (1959). Adaptive shift and dispersal in a tropical island fauna. Evolution, 13, 122–144. 10.2307/2405948 [DOI] [Google Scholar]
- Xu, J. , Zhao, Y. , Wu, A. Z. , & Pan, C. G. (2001). RAPD analysis for brown planthopper groups from different areas. Journal of Shanghai Jiaotong University, 9, 20–23. [Google Scholar]
- Yang, J. B. , Dong, Y. R. , Wong, K. M. , Gu, Z. J. , Yang, H. Q. , & Li, D. Z. (2018). Genetic structure and differentiation in Dendrocalamus sinicus (Poaceae: Bambusoideae) populations provide insight into evolutionary history and speciation of woody bamboos. Science Report, 8 10.1038/s41598-018-35269-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh, F. C. , Yang, R. C. , Boyle, J. , Ye, Z. H. , & Mao, J. X. (1997). POPGENE, the user‐friendly shareware for population genetic analysis. Edmonton, Canada: Molecular Biology and Biotechnology Center, University of Alberta. [Google Scholar]
- Yoshikawa, K. , Umetsu, K. , Shinzawa, H. , Yuasa, I. , Maruyama, K. , Ohkura, T. , Yamashita, K. , & Suzuki, T. (1999). Determination of carbohydrate‐deficient transferring separated by lectin affinity chromatography for detecting chronic alcohol abuse. FEBS Letters, 458, 112–116. 10.1016/s0014-5793(99)01137-0 [DOI] [PubMed] [Google Scholar]
- You, C. J. , Yang, L. J. , & Tian, C. M. (2019). Resolving the phylogenetic position of Caeoma spp. that infect rhododendron and Chrysomyxa from china. Mycological Progress, 18, 1285–1299. 10.1007/s11557-019-01524-z [DOI] [Google Scholar]
- Zhu, W. C. , Sun, J. T. , Dai, J. , Huang, J. R. , Chen, L. , & Hong, X. Y. (2018). New microsatellites revealed strong gene flow among populations of a new outbreak pest, Athetis lepigone (Möschler). Bulletin Entomological Research, 108, 636–644. 10.1017/s000748531700116x [DOI] [PubMed] [Google Scholar]
- Zimmerman, E. C. (1970). Adaptive radiation in Hawaii with special reference to insects. Biotropica, 2, 32–38. 10.2307/2989786 [DOI] [Google Scholar]
- Zinna, R. , Emlen, D. , Lavine, L. C. , Johns, A. , Gotoh, H. , Niimi, T. , & Dworkin, I. (2018). Sexual dimorphism and heightened conditional expression in a sexually selected weapon in the Asian rhinoceros beetle. Molecular Ecology, 27, 5049–5072. 10.1111/mec.14907 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data have been deposited at the Dryad Digital Repository: https://doi.org/10.5061/dryad.qv9s4mwcz.