

Summary
Pancreatic ductal adenocarcinoma (PDA) is a deadly cancer characterized by complex metabolic adaptations that promote survival in a severely hypoxic and nutrient limited tumor microenvironment (TME). Modeling microenvironmental influences in cell culture has been challenging, and technical limitations have hampered the comprehensive study of tumor-specific metabolism in vivo. To systematically interrogate metabolic vulnerabilities in PDA, we employed parallel CRISPR-Cas9 screens using in vivo and in vitro systems. This work revealed striking overlap of in vivo metabolic dependencies with those in vitro. Moreover, we identified that intercellular nutrient sharing can mask dependencies in pooled screens, highlighting a limitation of this approach to study tumor metabolism. Furthermore, metabolic dependencies were similar between 2D and 3D culture, although 3D culture may better model vulnerabilities that influence certain oncogenic signaling pathways. Lastly, our work demonstrates the power of genetic screening approaches to define in vivo metabolic dependencies and pathways that may have therapeutic utility.
Graphical Abstract
Introduction
Pancreatic ductal adenocarcinoma (PDA) is the third leading cause of cancer death in the United States and is highly refractory to treatment (Siegel et al., 2018). One of the defining characteristics of PDA is a rewired cellular metabolism that facilitates growth in austere conditions (Biancur and Kimmelman, 2018). PDA are also exceptionally desmoplastic and lack functional vasculature creating a hypoxic and nutrient poor tumor microenvironment (TME) that necessitates alternative means of nutrient use and acquisition (Biancur and Kimmelman, 2018; Lyssiotis and Kimmelman, 2017). One mechanism by which PDA tumors maintain growth is through diverting fuel sources to support anabolic metabolism such as nucleotide synthesis (Ying et al., 2012). In addition to rewiring central carbon metabolism to provide metabolic plasticity (Biancur et al., 2017a), PDA cells also rely on high basal levels of nutrient scavenging by macropinocytosis and autophagy to meet energy demands (Davidson et al., 2017; Yang et al., 2011). The reliance of PDA on altered metabolic pathways has spawned great interest in discovering metabolic vulnerabilities and targeting them for therapeutic gain.
There is now emerging evidence that the metabolism of cancer cells grown in nutrient replete in vitro environments may not be entirely reflective of tumors and 2D culture may not faithfully unveil metabolic dependencies due to the artificial growth conditions. Physiological culture media may more accurately represent the nutrient availability of the TME, but it lacks environmental stimuli and stressors as well as the heterotypic cell-cell interactions that occur in tumors (Cantor et al., 2017). Furthermore, the precise nutrient content of most tumor types remains unknown. Thus, it is possible that in vitro culture may overestimate the contribution of certain metabolites or pathways to in vivo growth or conversely mask druggable metabolic vulnerabilities found in a tumor.
Nonetheless, development of robust methods to study the in vivo metabolism of PDA has been challenging. Techniques such as in vivo isotope tracing have been informative (Faubert et al., 2017; Hui et al., 2017), but assay limitations such as the inability to account for heterogeneous cell types within the TME, can make it difficult to ascertain the contribution of neoplastic cells to the metabolite profile. Additionally, intercellular nutrient sharing and scavenging will not be measured by bulk tumor measurements. To directly test the metabolic requirements of PDA, we developed a pooled genetic screening approach to determine the metabolic pathways that are indispensable for in vivo tumor growth. We compared these pathways to those required by in vitro cultured cells. These data systematically identify potential metabolic vulnerabilities of PDA tumors and also highlight the merits and limitations of performing metabolic screens in multiple in vitro culture systems. Using models in the context of both immune competent and immune compromised animals, we further investigate the role of multiple cellular compartments upon tumor metabolic perturbation. These data provide an invaluable resource for the investigation of the metabolic requirements of PDA tumors.
Results
Parallel in vivo and in vitro metabolic CRISPR Screens reveal striking similarity in metabolic dependencies
We created a custom library of lentiviral vectors encoding ~18,000 sgRNAs targeting ~3,000 mouse metabolic genes (Possemato et al., 2011) and performed CRISPR-Cas9 screens using a C57BL/6 (B6) mouse PDA line derived from a KrasG12D-driven autochthonous model (Figure 1A). Time-matched parallel screens were performed by in vitro passage or tumor implantation subcutaneously into the flanks of syngeneic hosts (Figure 1A, Figure S1A) and compared sgRNA abundance through next generation sequencing. Surprisingly, we found strong correlation of guide abundance when we compared in vivo to in vitro dropout (r = 0.845) (Figure 1B, FigureS1B). Indeed, analyzing the significantly depleted genes (FDR < 0.2) reveals that the standard in vitro conditions recapitulate approximately 86.5% of the in vivo metabolic dependencies (Figure 1C, Table S1). Interestingly, we observed that 38.1% of genes that score as in vitro dependencies fail to score in vivo. Thus, while standard tissue culture conditions can accurately model the overwhelming majority of tumor metabolic dependencies, in vitro metabolism may overestimate the contribution of certain genes and pathways to tumor cell growth. We also observed marked concordance between in vitro and in vivo dependencies at the pathway level. Pathways required for both in vitro and in vivo growth include those previously shown to be associated with PDA metabolism including glycolysis and nucleotide metabolism (Figure 1D) (Santana-Codina et al., 2018; Ying et al., 2012). While there is remarkable overlap between screens, we hypothesized that the genes and pathways that demonstrated selective in vivo dependency represent a novel class of tumor metabolic vulnerabilities that may have been masked by in vitro culture conditions.
Figure 1. Parallel CRISPR-Cas9 metabolism screens reveal metabolic dependencies in PDA.
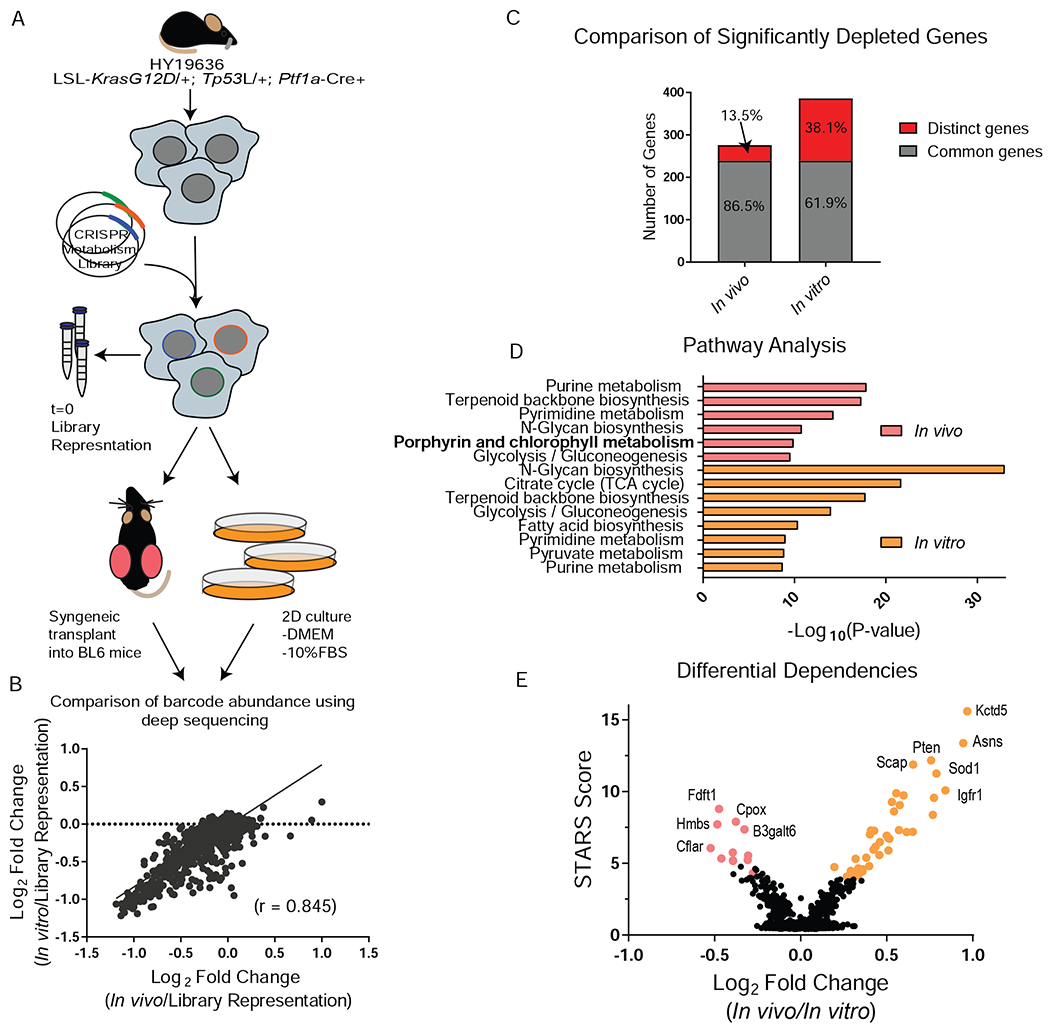
A, Schematic of parallel CRISPR/Cas9 screen design using a B6 KPC line. B, Gene score correlation between in vitro (passage 7, P7) and in vivo. C, Comparison of significantly depleted genes (FDR < 0.2) in the in vitro (P7) and in vivo screen. FDR is calculated using STARS algorithm. D, Pathway analysis of significantly depleted genes (FDR < 0.2) using Metaboanalyst. The –Log10 p-value of the most enriched pathways (FDR < 0.005) are represented. E, Differential dependencies are plotted by looking at the delta log fold change between in vivo and in vitro (P7) conditions. STARS score is plotted on the y axis and differentially required genes with an FDR < 0.02 are colored.
We first examined the possibility that selective in vivo dependencies were driven by the adaptive immune system. Tumors transduced with SpCas9 revealed no differences in tumor growth or T cell infiltration, suggesting SpCas9 itself was not contributing to an adaptive immune phenotype (Figure S1C–G). To assess the role of immune surveillance in metabolic dependencies, we performed the identical in vivo screen using immunodeficient athymic nude mice (Figure S1H). The nude and B6 screens showed strong concordance (r = 0.885), exemplifying the robustness of our in vivo screening platform. The majority of in vivo dependencies from the B6 screen validated in the nude mouse screen at an FDR<0.2 (Figure S1I, J).
Two of the top 3 most differentially dependent genes in the in vivo condition compared to the in vitro condition were members of the heme biosynthetic pathway [Hmbs (porphobilinogen deaminase), Cpox (coproporphyrinogen oxidase)] (Figure 1E, Table S1). Notably, pathway analysis highlighted heme biosynthesis as one of the major pathway differences between in vivo and in vitro metabolism (Figure 1D). Interestingly, Hmbs and Cpox did not score as differential dependencies between the immunocompetent and immunocompromised hosts, indicating the adaptive immune system was not contributing to the observed phenotype (FigureS1K).
Metabolic crosstalk in vitro can mask metabolic dependencies
Heme is a porphyrin ring containing a central iron ion and has diverse roles in cellular biology, including as a component of the oxygen-carrying protein hemoglobin and as a coenzyme for cytochromes and cyclooxygenases (Hamza and Dailey, 2012; Ponka, 1999; Tsiftsoglou et al., 2006). The biosynthesis of heme begins in the mitochondria, continues in the cytoplasm where porphyrin precursors are synthesized, and finishes in the mitochondria where iron is loaded onto the porphyrin ring to make heme (Figure 2A). Given the importance of heme in many cellular processes, it is surprising that loss of this pathway does not cause growth deficits in tissue culture. Consistent with the screen results, knockdown (KD) of the enzyme Hmbs with short-hairpin RNAs (shRNAs) caused no proliferation defect in 2D culture but significantly reduced in vivo tumor burden (FigureS2A–C). Polyclonal knockout (KO) using sgRNAs to Hmbs had no impact on 2D growth but significantly reduced tumor size in orthotopic pancreas tumors (Figure 2B, C, E), validating the importance of heme metabolism for PDA tumor growth. Interestingly, when polyclonal Hmbs KO cells were passaged greater than 10 days in culture and in vitro proliferation was re-assessed, a strong growth decrement was observed despite maintenance of similar levels of Hmbs KO in these late passaged cells (Figure 2D, E).These data indicate that over time, loss of Hmbs results in a striking growth inhibition.
Figure 2. Validation of heme biosynthetic screening hits.
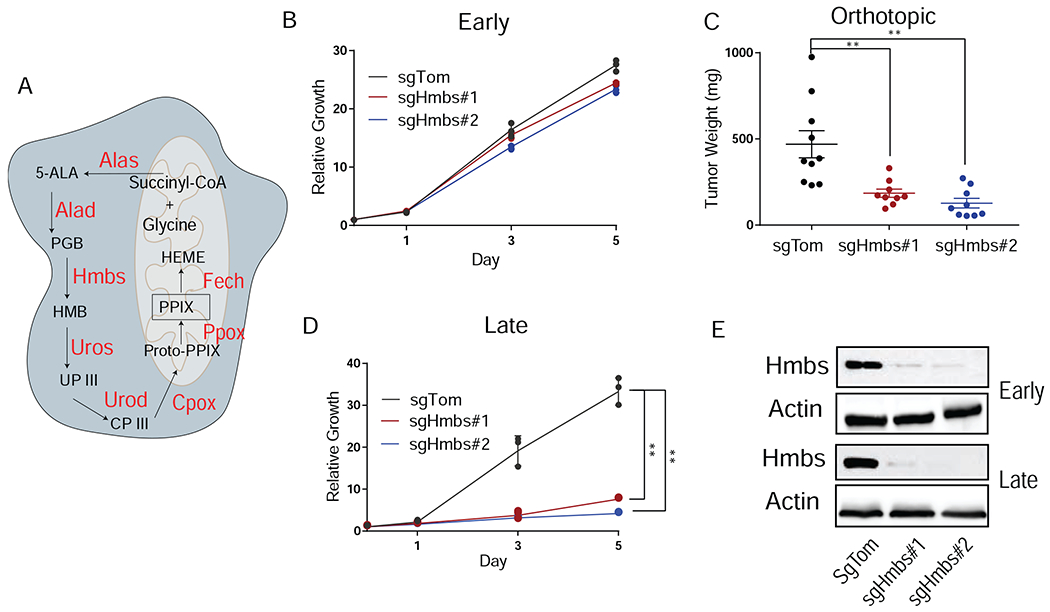
A, Schematic of heme biosynthesis. B, Relative proliferation rates of sgTom and sgHmbs cells performed immediately following transduction and selection (early passage) in 2D culture. Data are plotted as relative cell proliferation normalized to day 0 in arbitrary units. Error bars depict ± (s.d.) of three independent wells from a representative experiment. C, Tumor weight after orthotopic implantation of early passage sgTom (n = 10) and sgHmbs (KO#1 n = 9, KO#2 n = 9) cells into the pancreas of B6 mice harvested at endpoint. Error bars depict ± (s.e.m) of individual tumor weights. D, Relative proliferation rates of sgTom and sgHmbs cells that were cultured for greater than 10 days (late passage) after transduction and selection. Data are plotted as in B. Error bars depict ± s.d. of three independent wells from a representative experiment. E, Western blot of sgTom and sgHmbs cell lysates collected from early or late passage cells for Hmbs and Actin. For all panels, significance determined with an unpaired 2 tailed t-test. *P<0.05, **P<0.01, ***P<0.001, ns: non-significant, P>0.05.
To understand the cause of the Hmbs KO phenotype in the early versus late-passaged cells, we performed an in silico analysis of the in vitro screening data to determine which hemoproteins most potently impacted cell growth upon genetic ablation. This analysis revealed that heme-dependent mitochondrial enzymes were markedly more depleted in the screen than those targeting cytosolic heme-dependent enzymes, suggesting that the lack of heme is impacting mitochondrial function (FigureS2D). In line with these data, the decreased growth observed in late passage Hmbs KO cells was accompanied by a decrease in oxidative phosphorylation and a corresponding increase in glycolysis (Figure 3A, FigureS2E). These results stand in apparent contrast to the screening results since the in vitro screen was carried out over the course multiple weeks and no difference in sgRNA abundance was observed for Hmbs targeting sgRNAs.
Figure 3. Metabolic crosstalk masks the necessity for heme biosynthesis.
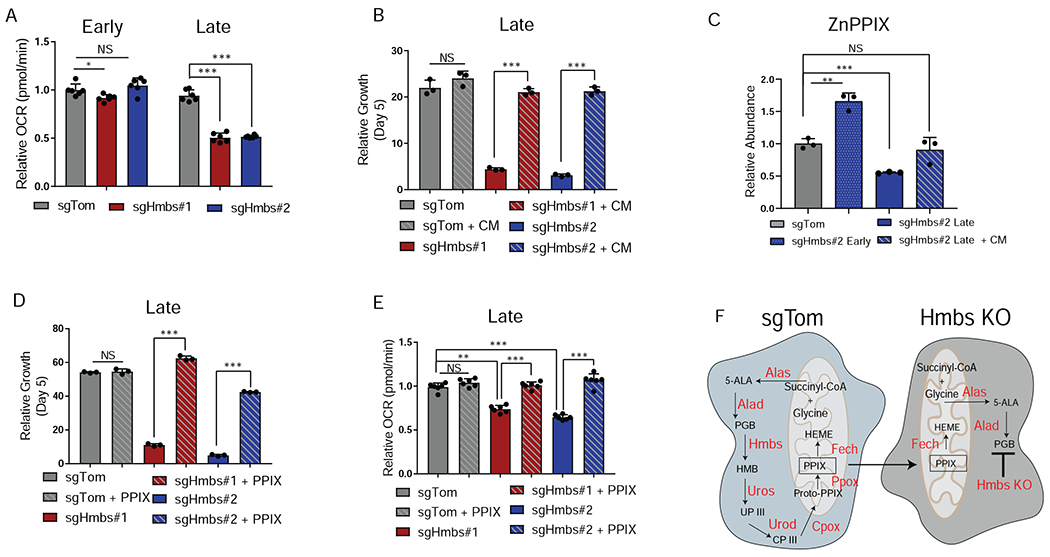
A, Oxygen consumption rates (OCR) of early passage (immediately after selection) or late passage (> 10 days in culture) cells. Data are plotted as relative OCR to sgTom early passage. Error bars depict ± s.d of 6 individual wells from a representative experiment. B, Relative growth of late passage sgTom and sgHmbs cells assessed at day 5 after seeding and grown in 2D culture. Cells were treated with either fresh media or conditioned media (CM) from sgTom cells. Error bars depict ± s.d. of three independent wells from a representative experiment. C, ZnPPIX abundance in sgTom cells, early passage sgHmbs cells, late passage sgHmbs cells, or late passage sgHmbs cells treated with CM in 2D culture. Data are plotted as relative abundance to sgTom cells. Error bars depict ± s.d. of three independent samples. D, Relative growth of late passage sgTom and late sgHmbs cells assessed at day 5 after seeding and grown in 2D culture. Cells were either treated DMSO or PPIX. Error bars depict ± s.d. of three independent wells from a representative experiment. E, Relative OCR of sgTom and late passage sgHmbs cells treated with either DMSO or PPIX. Data are plotted as relative OCR to DMSO treated sgTom samples. Error bars depict ± s.d of 6 individual wells from a representative experiment. F, Schematic depicting how cells proficient in heme biosynthesis (sgTom) can support cells with perturbed heme synthesis (Hmbs KO). For all panels, significance determined with an unpaired 2 tailed t-test. *P<0.05, **P<0.01, ***P<0.001, ns: non-significant, P>0.05.
We hypothesized that there may be a fundamental difference between loss of heme biosynthetic genes in a mixed sgRNA population versus the single guide setting and that cells in the mixed population screen were secreting metabolites which allowed cells with defective mitochondria to maintain proliferation. The addition of pyruvate and uridine (to support energy demands and nucleotide biosynthesis) in Hmbs KO cells only modestly restored growth, suggesting that this was not the reason that the cells are able to maintain their proliferative capacity (FigureS2F). We next posited that cells were able to restore mitochondrial activity by directly taking up heme itself or a heme biosynthetic intermediate secreted by neighboring cells proficient in heme biosynthesis. Strikingly, conditioned media (CM) from control cells applied to Hmbs KO cells completely restored growth and rescued oxygen consumption rates (Figure 3B, FigureS2G). To determine whether it was heme itself or a biosynthetic precursor that was responsible for the rescue, we used the ferrochetalase (Fech) inhibitor n-methyl protoporphyrin (NMPP), which inhibits the final step of heme biosynthesis (conversion of protoporphyrin IX (PPIX) into heme) (Shi and Ferreira, 2006). NMPP treatment blocked the rescue from the addition of CM in a dose dependent manner (FigureS2H). These data demonstrate that cells capable of synthesizing heme are exporting a biosynthetic precursor of heme that can rescue heme deficiency in Hmbs KO cells. To determine the identity of the secreted metabolite we performed a porphyrin analysis on Hmbs KO cells with or without the addition of CM. This analysis revealed the ZnPPIX (as a surrogate measurement for PPIX) (Nelson et al., 1998; Sachar et al., 2016) levels are elevated in early passage KO cells, but significantly reduced in late passage Hmbs KO cells. The addition of CM to late passage cells is able to rescue ZnPPIX abundance to wildtype levels. (Figure 3C). Interestingly, intermediate porphyrins failed to increase upon treatment with CM, suggesting that this rescue was specifically from PPIX itself (Figure S2I). Furthermore, the overall decrease in intermediate porphyrins suggests that this phenotype is driven by the lack of heme and not the accumulation of heme precursors to toxic levels. PPIX is known to be secreted in mammalian cells (Kitajima et al., 2019; Kobuchi et al., 2012; Ogino et al., 2011), and treatment of Hmbs KO cells with PPIX was capable of completely restoring growth and oxygen consumption rates (Figure 3D, E). We knocked out Hmbs in 2 additional independent KPC cell lines and saw a similar pattern, with later timepoints exhibiting a more profound growth suppression that was capable of being rescued by addition of CM or PPIX (Figure S2J, K). While MPDAC4 cells closely mimicked HY19636 growth rates when assessed during early passage, the HY15449 cells exhibited a more significant early growth inhibition, suggesting metabolic heterogeneity and different efficiencies of conserving or recycling heme. Indeed, in HY19636 cells there appears to be a mechanism of heme conservation or recycling as ZnPPIX levels undergo a transient increase followed by a precipitous drop upon Hmbs KO. Taken together, these data reveal a limitation of pooled 2D in vitro screens where metabolic crosstalk between cancer cells can mask metabolic dependencies (Figure 3F).
In the context of screening where Hmbs KO cells were injected in a pool with cells proficient in heme biosynthesis, Hmbs scored as a dependency suggesting that the wild-type cells capable of synthesizing heme were unable to rescue the Hmbs KO cells in vivo (Figure 1E, FigureS1K). This suggested that metabolic crosstalk in a tumor is distinct from metabolic crosstalk that occurs in 2D culture, perhaps due to lower nutrient abundance and the more complex microenvironment of a tumor. In line with this hypothesis, CM from control cells cultured in physiologic glucose could no longer rescue Hmbs KO cells to the same extent as CM from cells cultured in full DMEM (FigureS2L). Furthermore, the enzyme that is responsible for heme degradation, heme oxygenase (Hmox1), is transcriptionally increased in tumors (FigureS2M). This is likely due to the lower oxygen tensions and increased reactive oxygen species (ROS) found in the TME which have been shown to drive expression of Hmox1 (Dunn et al., 2014; Lyssiotis and Kimmelman, 2017). We speculate that less secretion of PPIX from cells proficient in heme synthesis due to lower nutrient abundance coupled with higher Hmox1 expression and thus more heme degradation leads to increased reliance on heme biosynthesis in tumors.
3D culture system better models tumor metabolism-cell signaling axis
Tumor cells grown in 3D culture typically demonstrate slower proliferation rates and nutrient and oxygen restriction at their core, which may be more akin to in vivo growth (Langhans, 2018). Moreover, the matrix in 3D culture may act to limit nutrient exchange. To investigate the hypothesis that cells grown in a 3D matrix may better recapitulate metabolite exchange found in the TME, we performed the same screen under 3D culture conditions (Figure 4A, FigureS3A). To enable a comparison with the 2D results, we utilized the same media (DMEM, 10% FBS) with the addition of 5% Matrigel. Since 3D cells proliferate significantly slower (FigureS3B), we sought to evaluate 3D and 2D screens at more comparable population doublings and compared sgRNA abundance in 3D at 20 days post-induction with the abundance in the 2D screen after 3 passages (P3, 9 days). Overall, the 3D screen yielded similar results as the 2D screen (r = 0.759) (FigureS3C). Genes involved in heme biosynthesis also failed to score in the 3D screen suggesting that crosstalk between wild type cells and those deficient in heme biosynthesis still exists when cells are grown in a 3D matrix in full media (FigureS3D). Similarly, single-gene knockout experiments also demonstrated that Hmbs is still required for growth in 3D (FigureS3E).
Figure 4. Three-dimensional culture reveals differential dependency on Fdft1.
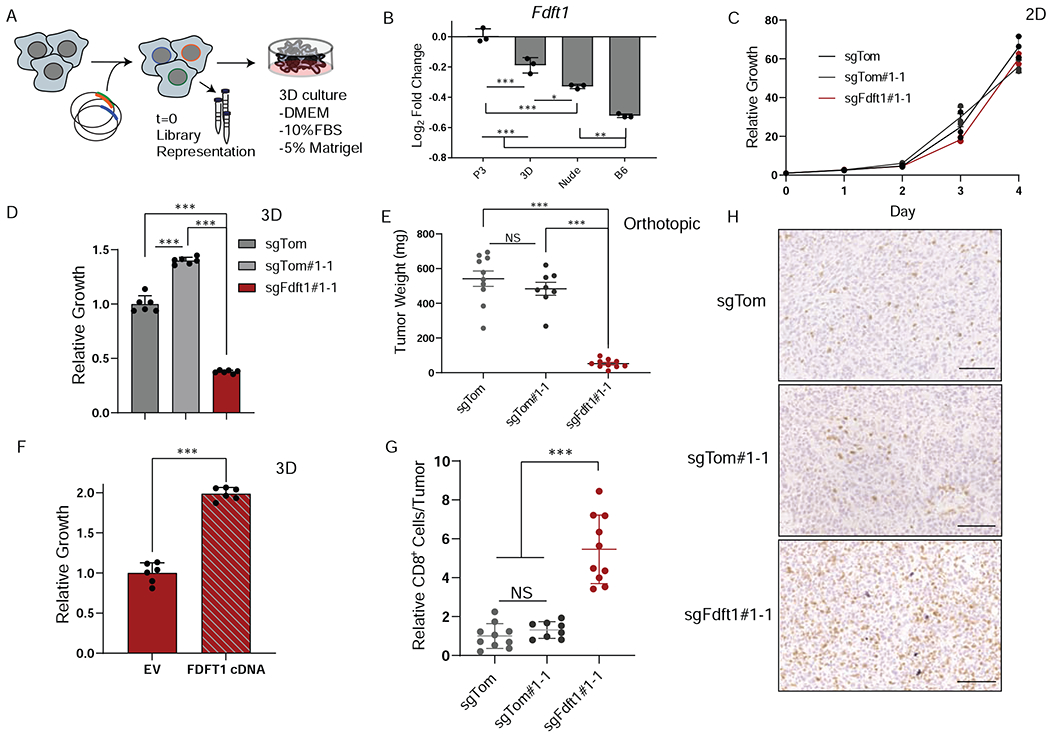
A, Schematic depicting experimental design for 3D metabolism screen. B, Log fold change of Fdft1 is plotted for P3, 3D, Nude, and B6 experimental conditions. Error bars depict ± s.d. of experimental condition (screens performed in triplicate). C, Relative proliferation rates of sgTom (polyclonal), sgTom#1-1 (clone), and sgFdft1#1-1 (clone) cells grown in 2D culture. Data are plotted as relative cell proliferation normalized to day 0 in arbitrary units. Error bars depict ± s.d. of three independent wells from a representative experiment.. D, Relative growth of sgTom (polyclonal), sgTom#1-1 (clone), and sgFdft1#1-1 (clone) cells grown in 3D culture and assessed by cell titer-glo (CTG). Data are plotted as relative luminescent signal to sgTom. Error bars depict ± s.d. of 6 independent wells from a representative experiment. E, Tumor weight after orthotopic implantation of sgTom (polyclonal) (n = 10), sgTom#1-1 (clone) (n = 8), and sgFdft1#1-1 (clone) (n = 10) cells into the pancreata of B6 mice harvested at endpoint. Error bars depict ± s.e.m of individual tumor weights. F, Relative growth of sgFdft1#1-1+ empty vector (EV) and sgFdft1#1-1 + human FDFT1 cDNA (FDFT1 cDNA) cells grown in 3D and assessed by cell titer-glo (CTG). Data are plotted as relative luminescent signal to sgFdft1#1-1 + EV. Error bars depict ± s.d. of 5 independent wells from a representative experiment G, Quantification of CD8a positive cells in either sgTom (polyclonal), sgTom#1-1 (clone), and sgFdft1#1-1 (clone) orthotopic tumors injected into the pancreata of B6 mice. Error bars depict ± s.d. of 10 independent tumors (sgTom), 8 independent tumors (sgTom#1-1) and 10 independent tumors (sgFdft1#1-1). F, Representative images of IHC for CD8a cells performed on sgTom (polyclonal), (n = 10), sgTom#1-1 (clone) (n = 8), and sgFdft1#1-1 (clone) (n = 10) orthotopic tumors. Scale bar represents 100μm. For all panels, significance determined with an unpaired 2 tailed t-test. *P<0.05, **P<0.01, ***P<0.001, ns: non-significant, P>0.05.
While our data revealed that many metabolic dependencies in 3D growth are similar to those in 2D culture, we also observed that 3D systems are better at modeling certain aspects of tumor metabolism. For instance, we observed differential dependency of the gene squalene synthase (Fdft1) for 2D, 3D, and in vivo cancer growth (Figure 4B). Fdft1 encodes an enzyme in the mevalonate pathway that catalyzes the synthesis of squalene from farnesyl pyrophosphate and is the first committed step to cholesterol synthesis (Tansey and Shechter, 2000). Fdft1 appears to be dispensable for 2D growth but is required for both in vivo and 3D proliferation. We generated clonal Fdft1 KO cells to assess growth kinetics in a pure population. These cells had no growth decrement in 2D culture but exhibited profound growth inhibition in 3D culture and upon orthotopic transplantation (Figure 4C–E, FigureS3F). Expectedly, overexpression of human FDFT1 cDNA was able to significantly rescue 3D growth (Figure 4F, FigureS3G). Knockout of Fdft1 in a polyclonal population also had no effect on proliferation in 2D culture but resulted in a growth disadvantage in both 3D culture and in orthotopic transplantation models (Figure S3H–K). The magnitude of the effect in polyclonal KO cells is not as striking as in the clonal KO, likely due to the outgrowth of wildtype cells within the polyclonal populations. Finally, KD of Fdft1 using shRNAs also resulted in no effect in 2D culture but revealed a significant anti-proliferative effect when cells were injected into either nude or B6 hosts (FigureS3L–M). Interestingly, Fdft1 KD cells grew slower in immunocompetent B6 mice than in the immunocompromised nude mice, suggesting that loss of Fdft1 activity may increase sensitivity to the adaptive immune system (FigureS3O). Indeed, when we examined clonal Fdft1 KO tumors by IHC we saw a marked increase in infiltrating CD8+ T-cells (Figure 4G, H). These data support that disruption of Fdft1 acts in both a cell autonomous and non-cell autonomous fashion by decreasing proliferation, but also rendering cells sensitive to immune surveillance.
Fdft1 loss disrupts cell signaling in PDA
Given that Fdft1 activity is required for cholesterol production, we hypothesized that tumors and 3D cultured cells rely more strongly on de novo cholesterol synthesis. In silico analysis of the screening data, however, revealed that no other cholesterol biosynthesis genes scored as a dependency in vivo (Figure S4A). Furthermore, addition of a lipid mix (containing exogenous cholesterol) failed to rescue growth of Fdft1 KO cells in 3D culture (FigureS4B). We then performed RNAseq, comparing control cells to Fdft1 KO in 3D culture (Table S2). We observed that Fdft1 KO cells grown in 3D upregulated genes involved in detoxification of cellular reactive oxygen species (ROS) (FigureS4C). Recent reports have highlighted that squalene has lipid antioxidant properties and may be responsible for quenching ROS in cellular membranes (Finotti et al., 2000; Garcia-Bermudez et al., 2019). To investigate if elevated lipid ROS may account for decreased growth of Fdft1 KO cells in 3D culture, we treated cells with the lipid antioxidant tocopherol acetate (Vit. E) which failed to restore growth in Fdft1 KO cells (FigureS4D). Furthermore, control and Fdft1 KO cells had similar levels of lipid ROS (FigureS4E). These data suggest that redox imbalance is not the root cause of the observed phenotype.
Further analysis of the RNAseq data revealed that Fdft1 KO in 3D culture was also associated with a decreased growth factor receptor signaling signature (Figure S4F, Table S2). Consistent with this, Kras scores as more essential in the 3D versus the 2D (P3) screen (FigureS4G). Given recent reports highlighting enhanced sensitivity to novel KRAS inhibitors for cancer cells cultured in 3D compared with 2D (Janes et al., 2018), we hypothesized that squalene depletion in cells may disrupt RTK activation and impair downstream signaling. To test this, we performed an RTK activation array to look globally at RTK signaling in Fdft1 KO cells which revealed that Fdft1 KO cells grown in 3D have significantly decreased levels of phosphorylated Erbb3 (FigureS4H, I). To understand which pathways could be affected by reduced Erbb3 signaling, we assessed the canonical cancer associated signaling pathways Ras-Mapk and Pi3k-Akt-mTor. Comparing Fdft1 KO to control cells revealed that Kras activity and Erk phosphorylation were unperturbed, suggesting that the decrease in Erbb3 phosphorylation was not related to the Ras-Mapk signaling cascade (FigureS4J). These data also support that notion that loss of Fdft1 does not affect Ras farnesylation by disruption of upstream farnesyl precursors. We then assessed Pi3k-Akt-mTor signaling axis in clonal KO cells. Indeed, p-Akt and p-S6 levels were decreased in clonal Fdft1 KO cells (Figure 5A), and polyclonal Fdft1 KO cells showed a similar decrease in Akt phosphorylation (Figure S4K). These data validated the results from the RTK array, revealing that both p-Erbb3 and total levels of Erbb3 were decreased upon loss of Fdft1 despite cells maintaining similar levels of Erbb3 mRNA (Figure S4L). Furthermore, we were able to restore the signaling changes in Fdft1 KO clones by expressing an FDFT1 cDNA (Figure 5A). Similar results were obtained in an independent KPC cell line (Figure S4M–O). To understand if the cell signaling phenotype was driven by decreased Erbb3 protein and/or activity, we knocked out Erbb3. Surprisingly, Erbb3 KO resulted in no growth inhibition in 3D culture and failed to recapitulate decreased Akt phosphorylation (Figure S4P, Q). We hypothesized that a reduction in Akt activation due to Fdft1 loss impacted Erbb3 protein levels. Indeed, it has been shown that Akt phosphorylation is able to regulate genes involved in Erbb3 regulation at the protein level (Gaborit et al., 2016; Panner et al., 2010; Wu et al., 2004). To understand if decreased Akt signaling was leading to loss of Erbb3 protein in our cells, we treated cells with an AKT inhibitor (AKTi). Treatment with the inhibitor phenocopied Fdft1 KO, showing decreased Akt phosphorylation, loss of Erbb3 protein (Figure 5B), and a marked growth disadvantage (Figure 5C). We next evaluated the FDFT1 inhibitor TAK-475 in our cell culture models. TAK-475 is a potent inhibitor of FDFT1 developed to treat hypercholesterolemia and is reported to have lipid lowering effects in multiple animal models (Nishimoto et al., 2003). Treating HY19636 cells grown in 3D with TAK-475 reveals a similar signaling phenotype to Fdft1 KO cells, with decreased Akt activation and decreased Erbb3 protein and phosphorylation levels (Figure 5D). In the acute treated setting of AKTi and TAK-475 there are minimal changes to p-S6 perhaps suggesting that chronic loss of Fdft1 activity results in decreased signaling to S6. Consistent with screening data, the inhibitor caused marked growth inhibition in 3D culture but only a modest growth phenotype in 2D culture (Figure 5E). While control cells exhibited growth delay, Fdft1 KO clones experienced no additive growth inhibition upon TAK-475 treatment, suggesting the inhibitor was on-target at the concentrations used in culture (Figure 5E). Taken together, these data support that Fdft1 potentiates Akt signaling and is critical for PDA cancer cell growth in 3D conditions.
Figure 5. Fdft1 potentiates Akt signaling in PDA cells.
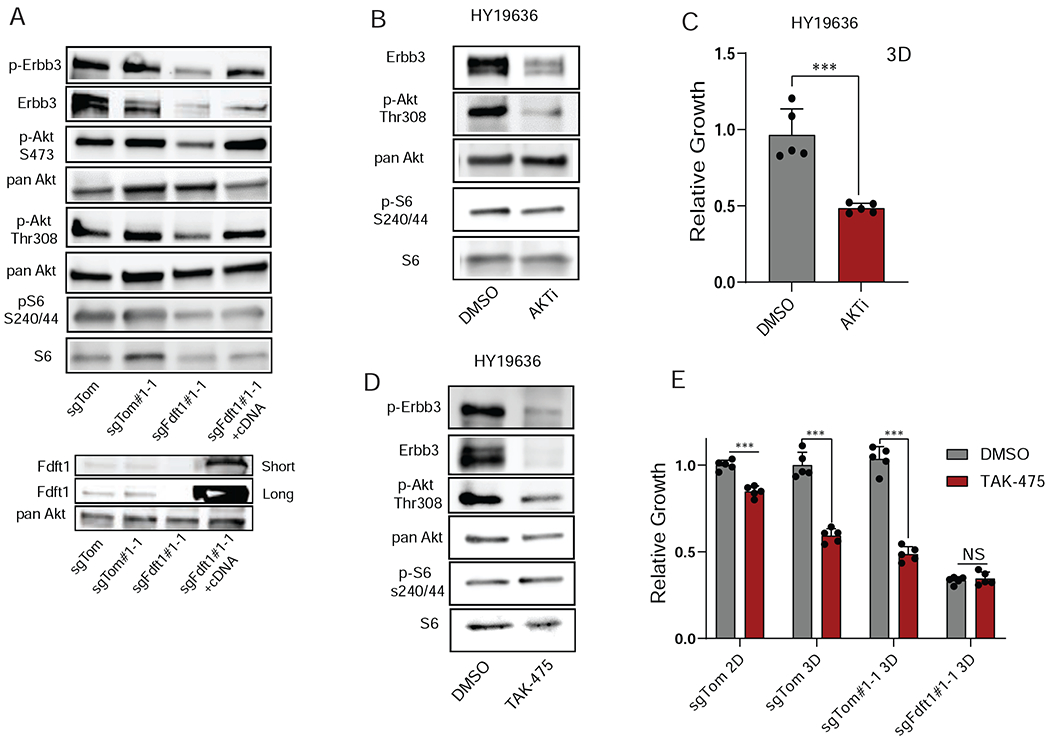
A, Western blot of sgTom (polyclonal), sgTom#1-1 (clone), sgFdft1#1-1 (clone), sgFdft1#1-1 + FDFT1 cDNA (rescue clone) cell lysates for p-Erbb3, Erbb3, p-Akt S473, pan Akt, p-Akt Thr 308, pS6 S240/44, S6, and Fdft1 in 3D conditions. B, Western blot of HY19636 cells treated with either DMSO or 1μM AKTi (MK-2206) in 3D conditions. Cell lysates were probed for Erbb3, p-Akt Thr308, pan Akt, p-S6 S240/44, and S6. C, Relative growth of HY19636 cells treated with either DmSo or 1μM AKTi (MK-2206). Growth was assessed by cell titer-glo (CTG). Data are plotted as relative luminescent signal to DMSO. Error bars depict ± s.d. of 5 independent wells from a representative experiment. D, Western blotting of HY19636 cells treated with either DMSO or 5μM TAK-475 in 3D conditions. Cell lysates were probed for p-Erbb3, Erbb3, p-Akt Thr 308, pan Akt, p-S6 S240/44, and S6. E, Relative growth of sgTom (polyclonal), sgTom#1-1 (clone), sgFdft1#1-1 (clone) cells treated with either DMSO or TAK-475 in 2D (sgTom) and 3D (sgTom, sgTom#1-1, sgFdft1#1-1) culture. Data are plotted as relative absorbance or luminescent signal to sgTom for 2D and 3D conditions respectively. Error bars depict ± s.d.of 3 (2D) or 5 (3D) independent wells from a representative experiment. For all panels, significance determined with an unpaired 2 tailed t-test. *P<0.05, **P<0.01, ***P<0.001, ns: non-significant, P>0.05.
Fdft1 is a targetable metabolic liability for PDA tumors
Our data suggest that knockout of Fdft1 impedes proliferation of PDA tumors through inhibition of Pi3k/Akt signaling and elicits an immune response, therefore credentialing Fdft1 as a prospective therapeutic target in PDA. We evaluated the efficacy and tolerability of Fdft1 inhibition using TAK-475 in a B6 syngeneic subcutaneous transplant model of PDA (Figure 6A). Tumor-bearing mice treated with TAK-475 showed significantly reduced tumor growth (Figure 6B, C). Furthermore, TAK-475 treated tumors displayed increased infiltration of CD8+ T-cells (Figure 6D, E), consistent with previous findings (Figure 4G, H, Figure S3O). Analysis of treated tumors also revealed increased apoptotic death (Figure 6F, G). To determine if the inhibitor was working as expected in tumors, we checked for canonical transcriptional changes that should occur upon loss of Fdft1. Indeed, it has been shown that Fdft1 inhibition activates transcription factor Srebp2 resulting the in the transcriptional upregulation of mevalonate pathway genes (Pai et al., 1998; Weber et al., 2004). We verified that this was happening in our Fdft1 KO cells and tumors by examining RNAseq data for Srebp2 target genes. Srebp2 targets were robustly increased in Fdft1 KO cells (Figure S5A). We then performed qPCR on TAK-475 treated cells and tumors for a subset of these targets and revealed a similar transcriptional increase in Fdft1 KO cells and tumors suggesting the drug was on-target in both in vitro and in vivo settings (Figure S5B). These data highlight the single agent anticancer activity of TAK-475 in PDA tumors. FDFT1 KO also reduced the 3D growth of multiple human PDA cell lines (Figure 6H, Figure S5C, D). Furthermore, we were able to show a differential dependency on FDFT1 between 2D and 3D growth as we had demonstrated in our screening data with Patu-8902 (Figure S5E). Indeed, FDFT1 KO in Patu-8902 cells also resulted in similar signaling changes to the KPC mouse cell lines suggesting that the mechanism for growth inhibition upon loss of FDFT1 is conserved across species (Figure 6I). Consistently, we observe marked decreases in phosphorylation of both Akt Thr308 and Akt S473 sites across all the cell lines tested. However, the differences in S6 phosphorylation in the various cell lines tested are perhaps suggestive of cell line dependent differences in Akt signaling to mTor. Consistent with the importance of FDFT1 in human PDA cell lines, PDA patient tumors that express high levels of FDFT1 showed significantly poorer overall survival (Figure 6J). Interestingly, bulk RNA sequencing from patient tumor datasets revealed that FDFT1 low tumors had greater overall immune infiltrate compared to FDFT1 high expressing tumors (FigureS5F). These data suggest that FDFT1 inhibition may be a tractable therapeutic opportunity for treating PDA.
Figure 6. Fdft1 inhibition decreases PDA tumor growth.
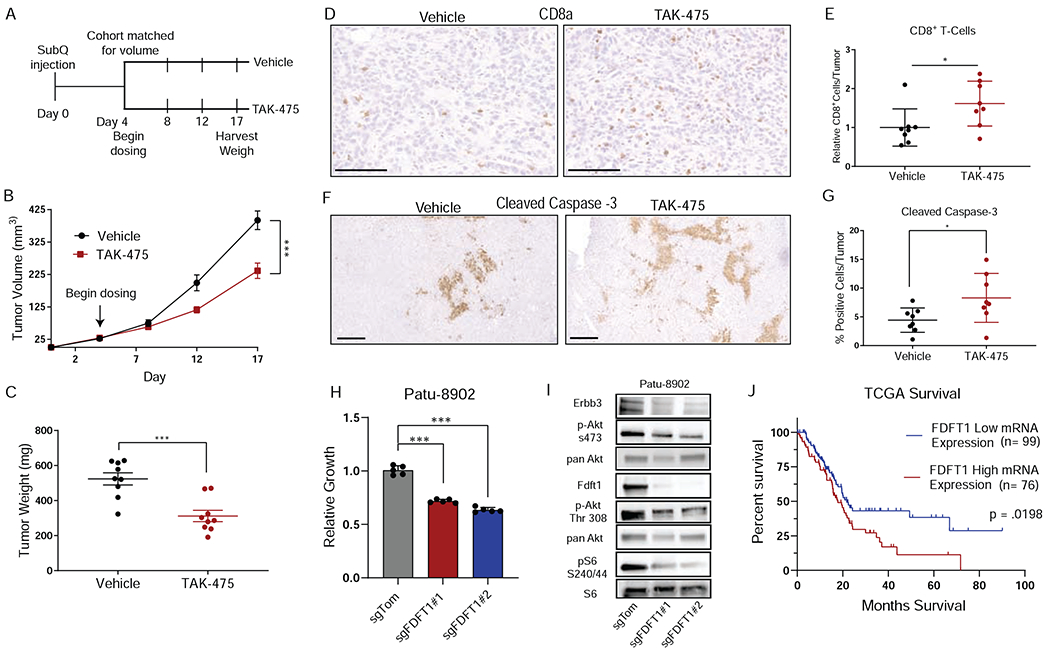
A, Schematic depicting experimental design for treating mice with Fdft1 inhibitor TAK-475. B, Tumor volume of either vehicle treated (n = 9) or TAK-475 treated (n = 9) mice at the indicated time points. Error bars depict ± s.e.m of individually measured tumors. C, Tumor weight harvested at endpoint of panel B (day 17) of either vehicle or TAK-475 treated tumors. Error bars depict ± s.e.m of individually weighed tumors. D, Representative images of Immunohistochemistry (IHC) performed on vehicle treated (n = 8) and TAK-475 treated (n = 8) tumors for intratumoral CD8a positive cells. Scale bar represents 100μm. E, Quantification of positive CD8a positive cells in either vehicle or TAK-475 treated tumors as in panel d. Error bars depict ± s.d. of 8 independent tumors. F, Representative images of IHC performed on vehicle (n = 8) and TAK-475 (n = 8) treated tumors for cleaved caspase-3 positive cells. Scale bar represents 500μm. G, Quantification of positive cleaved caspase-3 positive cells in either vehicle or TAK-475 treated as in panel d. Error bars depict ±s.d. of 8 independent tumors. H, Relative growth of human PDA cells Patu-8902 transduced with either sgTom or sgFdft1 and grown in 3D culture and assessed by cell titer-glo (CTG). Data are plotted as relative luminescent signal to sgTom. Error bars depict ± s.d. of 5 independent wells from a representative experiment. I, Western blot of sgTom, sgFdft1#1, and sgFdft1#2 in Patu-8902cell lysates for p-Erbb3, Erbb3, p-Akt S473, pan Akt, p-Akt Thr 308, pS6 S240/44, S6, and Fdft1 in 3D conditions. J, Survival plot of human pancreatic cancer cases from the cancer genome atlas (TCGA). Tumors were stratified by mRNA expression of FDFT1 into either high (top 50% of expression, n = 76) or low expressing (bottom 50% of expression, n = 99) tumors. P-value is calculated by Log-rank (Mantel-Cox) test. For all panels (except panel J), significance determined with an unpaired 2 tailed t-test. *P<0.05, **P<0.01, ***P<0.001, ns: non-significant, P>0.05.
Discussion
The ability to interrogate tumor cell metabolism and discover metabolic bottlenecks in vivo has proven difficult with conventional techniques. Here, we used parallel loss-of-function screens in vitro and in vivo to systematically define key metabolic vulnerabilities and pathways that are essential for the tumor growth of murine PDA. Our data demonstrates that a majority of tumor metabolic vulnerabilities are well represented in 2D in vitro culture. As with all systems, however, there are limitations that need to be taken into consideration. We demonstrate that pooled metabolic screens performed in standard 2D in vitro culture can mask metabolic vulnerabilities by presenting the metabolic crosstalk of heme biosynthetic intermediates as an example. Additionally, 2D culture appears to overestimate the contribution of certain metabolites and pathways to cancer cell growth. Our screens also reveal that media formulation and growth conditions can play an important role in defining the metabolic dependencies in vitro. Accordingly, the enzyme Asns (Asparagine Synthetase) scores as a strong dependency in vitro but fails to score as a dependency in vivo. We speculate that the reason for this is that mouse plasma contains approximately 100 μM asparagine while it is absent in DMEM (Sullivan et al., 2019). Asparagine is a nonessential amino acid and cellular demand can be met through synthesis via the gene Asns; however, when this gene is lost, cells are forced to rely on exogenous sources of asparagine, which is not present in DMEM (Krall et al., 2016). Furthermore, cells grown in vitro appear to rely on the fatty acid synthesis pathway whereas this pathway does not score in vivo. One hypothesis for this discordance is that the fatty acid content in the TME is higher than in current media formulations allowing cells to meet requirements through exogenous uptake. Alternatively, it is possible that the rapid doubling times that occur in vitro create a reliance on fatty acid synthesis for synthesizing new cell membranes. Indeed, more work needs to be done to explain these differential dependencies. In conclusion, while standard 2D culture conditions are a mainstay for mechanistic investigation of tumor cell metabolism, there is potential room for improvement through multiple considerations such as rational media design.
Additionally, we demonstrated the benefits of 3D culture in studying metabolism. While 3D culture appears to suffer from the same limitations as 2D culture with regards to nutrient crosstalk and media composition, it does appear to better model important aspects of metabolism-cell signaling axis. For example, our data reveal that one consequence of inhibiting the metabolic enzyme Fdft1 is impaired Pi3k-Akt-mTor signaling. This finding was especially intriguing to us given reports highlighting differential sensitivity to Kras inhibitors and genetic KO between 2D and 3D cultured cells (Han et al., 2020; Janes et al., 2018). The growth disadvantage upon Fdft1 inhibition is only evident in 3D culture and tumors, further supporting the claims that there are different signaling requirements between 2D and 3D cultures. Interestingly, our data suggests that perturbation of this signaling cascade is likely downstream of RTKs since KO of Erbb3 failed to recapitulate any of the biochemical or growth phenotypes observed in Fdft1 KO. Similarly, RTK array data showed a loss of Erbb3 is accompanied by a modest but global upregulation of other RTKs likely as a compensatory mechanism. While the cells can compensate for loss of Erbb3 (likely through upregulation of other RTKs) they are unable to fully restore downstream signaling upon Fdft1 KO. Importantly, these data reveal a critical insight into studying metabolic pathways especially with regards to the interconnection between metabolism and cell signaling where 3D culture better represents the in vivo scenario.
Our findings suggest that FDFT1 has both cell autonomous and non-cell autonomous effects on PDA tumors likely through impaired PI3K/AKT signaling. Although PDA tumors are driven by KRAS signaling, the PI3K/AKT signaling pathway is highly activated and associated with poor prognosis (Collisson et al., 2012; Ying et al., 2012). Aberrant activation of this pathway is associated with changes in cancer cell metabolism, increased cell cycling, and decreased apoptosis (Murthy et al., 2018). Our data suggest that TAK-475 may be an alternative and effective means of targeting one of the critical signaling axes in PDA. While there has been great interest in targeting PI3K signaling in solid tumors, the results have been overall disappointing. There are multiple reasons for the lackluster results of PI3K inhibitors including poor bioavailability and toxicity as well as mechanisms of resistance. Here we show significant single agent activity of the FDFT1 inhibitor TAK-475. One caveat of high dose TAK-475 treatment is that it caused mild liver damage in a small fraction of patients being treated for hypercholesterolemia (Stein et al., 2011). While this should not preclude investigating the therapeutic efficacy of TAK-475 in an oncological setting, liver damage would need to be carefully assessed during clinical trials especially in the context of combinatorial treatments with other agents with potential hepatotoxicity. Moreover, we have also shown that Fdft1 inhibition leads to an accumulation of T-cells in the TME. We postulate that this may be due to increased immune surveillance as it has been reported that PI3K inhibition in pancreatic cancer leads to upregulation of MHC-I and co-stimulatory molecules on the cancer cell surface (Sivaram et al., 2019). Thus, we speculate that TAK-475 may synergize with other immunotherapy approaches.
Overall, these data demonstrate the power of loss-of-function screens to identify metabolic dependencies across in vitro and in vivo contexts in a robust and efficient manner. Importantly, we have demonstrated the merits and limitations of performing metabolic screens in 2D and 3D in vitro systems compared to tumor transplant models. A major concern for in vivo screens is achieving high representation to account for tumor initiating capacity and growth rates of tumor cells in vivo. For these reasons, comparing cells grown in vitro to tumors can present challenges and to account for these variables we collected multiple time points in the 2D in vitro screens. Indeed, the high concordance between the nude, B6, and in vitro screens (at multiple time points) exemplifies the robustness of our screening platforms. We also highlighted a caveat of the in vitro screens where metabolite transfer between mixed populations of cells can mask metabolic dependencies. While the in vivo screens are also a mixed population, the complex TME does not appear to facilitate metabolic crosstalk to the same degree. Thus, it is possible that there are dependencies that are masked by some level of metabolite transfer in vivo. The hits that do arise, however, are likely to be robust therapeutic targets as they are not readily rescued by uninhibited neighboring cells. Another potential caveat is that this screen was performed in one genetic context (Kras mutant; p53 loss). Future studies utilizing this approach can be applied to other genetic variants of PDA (LKB1, CDKN2A, SMAD4 mutants) to create metabolic landscapes of the disease and to elucidate mutation-specific metabolic vulnerabilities. Ultimately, understanding the essential metabolic pathways of PDA may provide novel therapeutic strategies and improve patient care.
STAR ★ Methods
Resource Availability
Lead Contact
Further information and requests for reagents may be directed to, and will be fulfilled by the corresponding author Alec C. Kimmelman (alec.kimmelman@nyulangone.org).
Material availability
The unique reagents generated in this study are available from the Lead Contact Alec C. Kimmelman (alec.kimmelman@nyulangone.org).
Data and code availability
The accession number for the CRISPR screening and RNA sequencing is GSE158787 and GSE158657 respectively. All codes in this study are available on Github (Biancur-CellMetabolism-2020).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
MiaPaCa2 cell line was obtained from the American Type Culture Collection. PaTu-8902 was obtained from the German Collection of Microorganisms and Cell Cultures. Primary mouse PDAC line (HY19636) was generated from tumours from B6 genetically engineered mice (LSL-KrasG12D; p53 L/+, Ptf1a-Cre+) as previously described (Bardeesy et al., 2006). Control and knockout clones were generated by limiting dilution and clonal expansion. Cell lines were authenticated by periodic fingerprinting as well as visual inspection and carefully maintained in a centralized cell bank. All cell lines were tested routinely, and before all screening efforts, for mycoplasma contamination by PCR. Cell lines were cultured in DMEM (Corning 10-017-CV) with 10% fetal bovine serum (FBS) (Atlanta Biologicals S11550H), and 1% Penicillin/Streptomycin (Thermo, 15140122) except for 3D growth assays where cells were grown in DMEM with 10%FBS 1% Penicillin/Streptomycin and 5% growth factor reduced Matrigel (Corning, 354230). All cells were maintained at 37°C in a humidified atmosphere of 95% air and 5% CO2. The Key Resource Table contains the complete list of cell lines.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-Flag | Cell Signaling | 8146T |
| Rabbit anti-HMBS | Abcam | ab129092 |
| Rabbit anti-FDFT1 | Proteintech | 13128-1-AP |
| Rabbit anti-S6 | Cell Signaling | 2217 |
| Rabbit anti-S6 phospho S240/44 | Cell Signaling | 2215 |
| Rabbit anti-AKT | Cell Signaling | 9272 |
| Rabbit anti-AKT phospho Thr308 | Cell Signaling | 9275 |
| Rabbit anti-AKT phospho S473 | Cell Signaling | 9271 |
| Mouse ant-ERK II | Santa Cruz | Sc-1647 |
| Rabbit anti-MAPK phospho 44/42 | Cell Signaling | 9102 |
| Rabbit anti-ERBB3 | Cell Signaling | 12708 |
| Rabbit anti-ERBB# phospho Tyr1289 | Cell Signaling | 4791 |
| Mouse anti-Actin | Sigma | A2228 |
| Rat anti-Mouse CD45 | BD Biosciences | 564279 Clone 30-F-11 |
| Rat anti-Mouse CD3 | Biolegend | 100241 Clone 17a2 |
| Rat anti-Mouse CD8 | Biolegend | 100730, Clone AF700 |
| Rat anti-Mouse CD4 | Biolegend | 100451, Clone BV605 |
| Anti-Mouse IgG, HRP linked | Cell Signaling | 7076 |
| Anti-Rabbit IgG HRP linked | Cell Signaling | 7074 |
| Rabbit anti-Cleaved caspase-3 | Cell Signlaing | 9664 |
| Rat anti-CD8a | eBioscience | 14-0808-82 |
| Bacterial and Virus Strains | ||
| N/A | ||
| Biological Samples | ||
| N/A | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Crystal Violet | Sigma | C0755 |
| PPIX | Santa Cruz | 553-12-8 |
| NMPP | Cayman | 20846 |
| Pyruvate | Sigma | S8636 |
| Uridine | Sigma | U3750 |
| TAK-475 | Sigma | SML2168 |
| Lipid Mix | Sigma | LO288 |
| Vitamin E (tocopherol acetate) | Sigma | T3376 |
| AKTi (MK-2206) | Cayman | 11593 |
| Cell Titer Glo | Promega | G7570 |
| growth factor-reduced Matrigel | Fisher Scientific | CB-40230C |
| SYBR Green Supermix | Bio-Rad | 1725275 |
| T4 RNA ligase 2, truncated | NEB | M0242S |
| T4 polynucleotide kinase | NEB | M0201S |
| TRizol | Thermo | 15596026 |
| Liperfluo | Dojindo | SKU:L248 |
| Collagenase | Gibco | 17018029 |
| DNAse I | Roche | 4716728001 |
| Antibiotic-Antimycotic | Thermo | 15240062 |
| Zombie Aqua | Biolegend | 423101 |
| Mouse BD Fc Block | Biolegend | 553141 |
| L-Lysine | Sigma | 56-87-1 |
| Paraformaldehyde | Ted Pella | 18505 |
| Elite ABC Kit | Vector | PK-6100 |
| DAB | Vector | SK-4100 |
| Methylcellulose | Sigma | M0512 |
| Critical Commercial Assays | ||
| Phospho-RTK Array | R&D System | ARY014 |
| PureLink RNA mini kit | ThermoFisher Scientific | 12183025 |
| Superscript Vilo IV | ThermoFisher Scientific | 11766050 |
| Active Ras detection kit | Cell Signaling | 8821 |
| Deposited Data | ||
| CRISPR NGS from various screens | This study | GSE158787 |
| RNA sequencing | This study | GSE158657 |
| Experimental Models: Cell Lines | ||
| MIAPACA2 | ATCC | CRL-1420 |
| PATU-8902 | DSMZ | ACC 179 |
| 293T | ATCC | CRL-3216 |
| HY19636 | Haoqiang Ying | |
| HY15449 | Haoqiang Ying | |
| MPDAC4 | Biancur et al. 2017 | |
| Experimental Models: Organisms/Strains | ||
| Athymic nude (CrTac:NCr-Foxn1nu) | Taconic | NCRNU-F |
| C57BL/6 | Taconic | B6NTac |
| Oligonucleotides | ||
| qPCR Primers | ||
| Hmbs (mouse) Forward 5’-ATGAGGGTGATTCGAGTGGG-3’ | MGH Primer Bank | |
| Hmbs (mouse) Reverse 5’-TTGTCTCCCGTGGTGGACATA-3’ | MGH Primer Bank | |
| Fdft1 (mouse) Forward 5’-AGAGTGGCGGTTCACTGAGA-3’ | MGH Primer Bank | |
| Fdft1 (mouse) Reverse 5’-GAGAAAGGCCAATTCCCACCA-3’ | MGH Primer Bank | |
| Hmgcs (mouse) Forward 5’-AACTGGTGCAGAAATCTCTAGC-3’ | MGH Primer Bank | |
| Hmgcs (mouse) Revese 5’-GGTTGAATAGCTCAGAACTAGCC-3’ | MGH Primer Bank | |
| Sqle (mouse) Forward 5’-ATAAGAAATGCGGGGATGTCAC-3’ | MGH Primer Bank | |
| Sqle (mouse) Reverse 5’-ATATCCGAGAAGGCAGCGAAC-3’ | MGH Primer Bank | |
| Mvd (mouse) Forward 5’-ATGGCCTCAGAAAAGCCTCAG-3’ | MGH Primer Bank | |
| Mvd (mouse) Reverse 5’-TGGTCGTTTTTAGCTGGTCCT-3’ | MGH Primer Bank | |
| 18S Forward 5’-GTAACCCGTTGAACCCCATT-3’ | MGH Primer Bank | |
| 18S Reverse 5’-CCATCCAATCGGTAGTAGCG-3’ | MGH Primer Bank | |
| shRNA | ||
| shHmbs#1 (mouse) CCATACTACCTCCTGGCTTTA | Sigma | TRCN0000324954 |
| shHmbs#2 (mouse) CCCTGGCATACAGTTTGAAAT | Sigma | TRCN0000075689 |
| shFdft1#1 (mouse) CAGTGCTTGAATGAACTCATA | Sigma | TRCN0000099192 |
| shFdft1#2 (mouse) TCTATAACCTGCTGCGATTCC | Sigma | TRCN0000319438 |
| sgRNA | ||
| sgHmbs#1 (mouse) CAATACTCTTGAATTCCAGG | This study | |
| sgHmbs#2 (mouse) CCTGGTCGTTCACTCCCTGA | This study | |
| sgFdft1#1 (mouse) CCAGAGAGCCCAGTACCGTG | This study | |
| sgFdft1#2 (mouse) GGATGACATGAGCATCAGTG | This study | |
| sgFDFT1#1 (human) GAAGGATCGCCAGGTGCTGG | Park et al. 2016 | |
| sgFDFT1#1 (human) GGTGCTGGAGGACTTCCCAA | Park et al. 2016 | |
| sgErbb3#1 (mouse) GAACCAGAGCAACCTTGGGG | Wang et al. 2017 | |
| sgErbb3#2 (mouse) GGGTTCCCCGGACCACTCGG | Wang et al. 2017 | |
| sgErbb3#3 (mouse) AAGATGTGTGAGCCTTGCAG | Wang et al. 2017 | |
| Recombinant DNA | ||
| psPAX2 | Addgene | 12260 |
| pMD2.G | Addgene | 12259 |
| pLenticrisprV2 | Addgene | 52961 |
| pDONR 221 | Thermo | 12536017 |
| pLenti CMV Blast DEST | Addgene | 12260 |
| FDFT1 cDNA | Genscript | OHu14273 |
| Software and Algorithms | ||
| GraphPad Prism 7 | Version 7.05.234 | |
| STARS algorithm | Version 1.3 | |
| DESeq | Version 1.24.0 | |
| R | Version 3.6.1 | |
| Python | Version 3.8 | |
| Other | ||
| 96 well Ultra-low attachment plates | Corning | CLS3474 |
| 6 well Ultra-low attachment plates | Corning | CLS3261 |
Mouse subcutaneous and orthotopic xenografts
For the in vivo screen, female athymic nude (Taconic, NCrNU) and B6 (Taconic, C57BL/6) mice were injected with 1×10^6 cells in HBSS per flank (2 flank injections per animal). Mouse experiments were performed at 1000× library representation in triplicate accounting for the tumor initiating capacity of HY19636 cells. Mouse tumors were grown over the course of 21 days (B6) and 17 days (nude) where tumors were harvested and pooled for sequencing. Mouse orthotopic experiments were performed as previously described (Sousa et al., 2016). Briefly, mice were anaesthetized by intra-peritoneal injection of ketamine (120mg/kg) and xylazine (10mg/kg). A small incision was made on the upper left quadrant of the abdomen and the pancreas externalized. Cells were suspended in a 1:1 solution of HBSS:Growth factor-reduced Matrigel (Corning, 354230) and injected into the pancreas at a concentration of 20,000 cells per injection in 20ul using insulin syringes (29-gauge needle, BD 324702). Mice were treated with buprenex every 12 hours after surgery for 48 hours. For subcutaneous mouse experiments, cells were suspended in a 1:1 solution of HBSS:Growth factor-reduced Matrigel and injected into the flanks of either athymic nude or B6 mice at a concentration of 100,000 cells per flank. For TAK-475 (Sigma, SML2168) treatment studies, mice were inoculated with cells and when tumors were measurable, mice were matched for tumor size and split into groups to receive vehicle or TAK-475. The average starting volume of groups (vehicle and TAK-475) was approximately 25mm3. Mice were treated with 50mg/kg/day of TAK-475 by oral gavage in a 0.5% methylcellulose suspension (Sigma, M0512). Tumors were measured manually with calipers and volume was estimated with the equation (L × W2/2). For all experiments (treatment, shRNA or sgRNA) tumors were harvested and weighed at endpoint.
All animal studies were approved by the NYULMC IACUC (Institutional Animal Care and Use Committee, Protocol #161113 and 170204).
METHOD DETAILS
CRISPR Metabolism Screen
We generated a mouse metabolic library of optimized sgRNA’s and controls targeting 2,918 metabolic genes. The metabolic gene list was generated by compiling lists from previous human metabolic screens and mapping these genes to the mouse homologues with the addition of some manually curated genes. All sgRNA’s were developed using established algorithms to optimize on-target and minimize off-target genome editing High titer lentivirus was generated from the mouse metabolism library and cells infected at a multiplicity of integration (MOI) of 0.3. HY19636 cells were infected with previously tittered virus with 8ug/ml of polybrene (Sigma, TR-1003) by spin infection (2 hours, 30°C, 2000 RPM). After cells have settled on plates (1-2 hours), media was changed and cells allowed to grow overnight. The following day cells were trypsinized and plated in puromycin containing media and selected for 2 days (a timepoint where uninfected control cells were dead). Immediately after selection cells were split for the following experiments: cells were collected to assess library representation at time = 0, passaged in vitro in 2D passaged in vitro in 3D, or implanted subcutaneously in the flanks of nude or B6 mice. Cells were collected at 1000× library coverage and in triplicate. Pellets were frozen until DNA extraction was performed. For 2D culture, 1000x library representation was passaged in triplicate in DMEM+10%FBS+1%P/S every 3 days for either 3 passages (P3/9 days) or 7 passages (P7/21 days). For 3D culture, 1000x library representation was passaged in DMEM+10%FBS+1%P/S+ 5% growth factor reduced matrigel (BD Biosciences). Cells were passaged once at day 10 and collected at day 20. For tumors, both athymic nude (Taconic, NCrNU) and B6 (Taconic, C57BL/6), mice were injected with 1×10^6 cells in HBSS per flank (2 flank injections per animal). Mouse experiments were performed at 1000x library representation in triplicate accounting for the tumor initiating capacity of HY19636 cells. Mouse tumors were grown over the course of 21 days (B6) and 17 days (nude) where tumors were harvested and pooled for sequencing.
CRISPR sgRNA Sequencing
Cell pellets from mouse tumors was created by pooling tumors from cages detailed below. Animals were sacrificed and tumors harvested and weighed, being careful to only include tumor and avoiding skin, muscle, and peritoneum. Both tumors from one mouse were combined into one 50mL conical tube. DMEM with 2%FBS was added to the tumors so that tumors will be resuspended at a concentration of 0.1mg/mL. Tumors were then carefully and thoroughly homogenized with scissors on ice trying to get as close to a single cell suspension as possible. Once tumors were homogenized, 1.6mL of tumor homogenate from each mouse was added to a 15mL conical tube, pooling all the tumors from a single cage. This was then spun down, supernatant aspirated, and tumor cell pellet frozen until DNA extraction. DNA from cell/tumor pellets was extracted using the Qiagen Blood Maxi Kit as per manufacturer’s instructions. PCR was used to amplify the sgRNA region followed by sequencing on an Illumina HiSeq to determine sgRNA abundance.
Data Normalization
Reads per million (RPM) values were calculated for each sample by dividing sgRNA count values for a given sample by the total counts for that sample. These values were then multiplied by 106 , to obtain the RPM value for the given sgRNA. The log2(RPM + 1) value was then calculated for each sgRNA. Following this procedure, replicates were then collapsed down by taking the median value. The log fold change value (LFC) was then calculated by subtracting the corresponding early time-point of infection, referred to as the Full Library Representation. For each sample, the median of the individual sgRNAs from the negative control genes was then subtracted from each of the LFC values. Finally, each LFC value was divided by the magnitude of the median of the LFC values of the individual sgRNAs for the positive control genes.
Data Quality Control
Distributions of the log2(RPM+1) values were generated to look at relative drop out of total guide counts relative to condition and empirical cumulative distribution functions, ECDF, were used to ensure that all guides were present in the initial library. ECDFs were created using the ECDF function in the base R stats package (R version 3.6.1).
STARS Analysis
The STARS algorithm version 1.3 was used to provide p-value, FDR, and q-value estimations for the gene LFC scores (Doench et al., 2014). The Normalized LFC data at the sgRNA level, as described in Data Normalization, was provided. The null distribution was created with a threshold of 60, 1000 iterations, and excluding the first ranked perturbation. STARS was then subsequently run with a threshold of 0.6 in both the positive and negative directions on the normalized log fold change data. The positive and negative runs for a given condition were combined by assigning STARS scores from the negative direction a negative sign and then merging the lists. Conflicts in this merge were resolved by either: taking the corresponding STARS score and statistics associated with the higher p-value if both were insignificant or taking STARS score and statistics associated with the statistically significant result of a p-value less than 0.05 and an FDR of less than 0.25.
Comparison of Significantly Enriched Genes and Gene Sets
Significantly depleted genes were determined by taking all genes that dropped out of the screen with an FDR of <0.2 as determined by STARS analysis of datasets. For figure 1c, the number of significant genes is plotted on the y-axis. For the in vivo (B6) analysis, there were a total of 276 significantly depleted genes. For the in vitro (P7) portion of the screen, there were 386 significantly depleted genes. Comparing these genes, there were 239 genes in common between the two screens. The percentage of genes that were common between the two conditions (in vivo B6) and (in vitro P7) were plotted on the graph as common genes. The genes that only scored in vivo or in vitro were plotted as distinct genes.
Metaboanalyst was performed on significantly enriched genes (FDR < 0.2 as determined by STARS analysis of datasets). Analysis was performed using joint pathway analysis by inputting official gene symbols into the software using KEGG (prior to October, 2019) and all pathways option (gene only). Enrichment analysis used the hypergeometric test and topology used degree centrality as parameters. Metaboanalyst pathway readout of the –Log10 p-values were plotted for all pathways with an FDR of < 0.005.
Differential dependencies (Figure 1e) between the in vivo (B6) and in vitro (P7) were determined by looking at the delta log2 fold change of in vivo – in vitro plotted on the x-axis. STARS score as determined by STARS analysis of the datasets is plotted on the y-axis. Genes with an FDR <0.02 are colored in pink as a selective in vivo dependency or in yellow as a selective in vitro dependency.
shRNA, sgRNA and Cloning
All shRNA’s were purchased as glycerol stocks (pLKO.1 puro) from the Sigma MISSION TRC shRNA library. Mouse sgRNA sequences were taken from the mouse metabolism CRISPR library (except for sgErbb3 which was taken from previously published CRISPR library (Wang et al., 2017). Human sgRNA sequences were taken from previously published CRISPR library (Park et al., 2017). All sgRNA’s were cloned into pLentiCRISPRv2 (Addgene, plasmid #52961) and sequence verified prior to viral transduction.
The generation of lentivirus was performed by transfecting 293T cells with lentiviral vector (pLKO.1, plentiCRISPRv2) and packaging plasmids psPAX2 (Addgene, plasmid #12260) and pMD2.G (Addgene, plasmid #12259) at a 0.5:0.25:0.25 ratio with polyethylenimine (PEI). Viral supernatant was collected 48hr after transfection. Filtered viral supernatant and polybrene (8ug/mL) was used to infect target cells. Cells were then subject to antibiotic selection until control plates (uninfected cells) were dead.
FDFT1 rescue was performed by cloning human FDFT1 cDNA (Genscript OHu14273) into pLenti CMV Blast DEST vector (Addgene plasmid #17451) using gateway cloning. Sequences for sgRNA and shRNA are as follows:
| shRNA | Sequence | TRCN # |
|---|---|---|
| shHmbs#1 (mouse) | CCATACTACCTCCTGGCTTTA | (TRCN0000324954) |
| shHmbs#2 (mouse) | CCCTGGCATACAGTTTGAAAT | (TRCN0000075689) |
| shFdft1#1 (mouse) | CAGTGCTTGAATGAACTCATA | (TRCN0000099192) |
| shFdft1#2 (mouse) | TCTATAACCTGCTGCGATTCC | (TRCN0000319438) |
| sgRNA | Sequence | |
| sgHmbs#1 (mouse) | CAATACTCTTGAATTCCAGG | |
| sgHmbs#2 (mouse) | CCTGGTCGTTCACTCCCTGA | |
| sgFdft1#1 (mouse) | CCAGAGAGCCCAGTACCGTG | |
| sgFdft1#2 (mouse) | GGATGACATGAGCATCAGTG | |
| sgFDFT1#1 (human) | GAAGGATCGCCAGGTGCTGG | |
| sgFDFT1#1 (human) | GGTGCTGGAGGACTTCCCAA | |
| sgErbb3#1 (mouse) | GAACCAGAGCAACCTTGGGG | |
| sgErbb3#2 (mouse) | GGGTTCCCCGGACCACTCGG | |
| sgErbb3#3 (mouse) | AAGATGTGTGAGCCTTGCAG |
Cell Proliferation Assays
For 2D growth assays, HY19636 cells were plated in 24-well plates at 2,500 cells per well in 0.5 mL of media. Conditioned media (Cm) for Hmbs KO rescue experiments was generated by plating 2 million sgTom cells in 10cm plates and conditioning 10ml of media for 24-36 hours. Rescue experiments were performed by adding 0.5mL of .45um filtered CM or PPIX to 24-well growth curves the day after plating. PPIX (Santa Cruz, 553-12-8) stocks were generated by resuspending 10mg/mL of PPIX in DMSO. This was diluted 1:6,000 in media, vortexed, and filtered through 0.45uM filter. For the n-methyl-protoporphyrin (NMPP) (Cayman, 20846) treatment of cells, NMPP was added with 0.5 mL conditioned media the day after plating at the indicated concentrations. Pyruvate (Sigma, S8636) and uridine (Sigma, U3750) treatment of Hmbs KO cells was performed by adding 1mM pyruvate and 50ug/mL uridine to cells in 0.5mL of media the day after plating. For sgHmbs rescue with WT cells cultured in varying concentrations of glucose (5mM and 1mM), WT cells were grown for 24-36 hours in low glucose media. Media was collected, filtered and added to the wells of HMBs KO cells. The final concentration of glucose in the wells was adjusted to 25mM. Growth was assessed by crystal violet. For 2D FDFT1 KO in Patu-8902, cells were grown for 5 days and compared to cells grown for 5 days in 3D culture. Relative growth was determined by normalizing perturbations to sgTom control in 2D and 3D respectively. For 2D FDFTi treatment in mouse cells were plated overnight and treated the following day treated with 5uM of TAK-475 (Sigma, SML2168) and collected 72 hours later (to compare to 72 hour 3D treated samples). For all other experiments, cells were collected at the indicated time points. Cells in 2D were fixed in 10% formalin and stained with 0.1% crystal violet. Dye was extracted with 10% acetic acid and the relative proliferation was determined by OD at 595 nm by normalizing to day 0 absorbance. For 3D growth assays, mouse cells with the indicated perturbations were plated in 96 well ultra-low attachment plates (Corning, CLS3474) at a concentration of 2,500 cells per well. Cells were plated into media containing lipid mix (Sigma, LO288) or vitamin E (Sigma, T3376). For 3D treatments, cells were plated overnight and drug added the following day and growth assessed at 72 hours. For TAK-475 (Sigma, SML2168) cells were treated with 5uM. For AKTi (MK-2206 Cayman 11593) cells were treated with 1uM. At the indicated time points, cells confluence was determined by cell-titer glo (CTG) reagent (Promega, G7570). Briefly, 100ul of CTG was added to each well of the 96 well plate. Plate was incubated on shaker for 10 minutes and the CTG-cell culture solution transferred to white walled clear bottom pate (Corning, 3610). The luminescence intensity was determined by plate reader (BMG Labtech). For cell growth fold change in supplementary figure 3b, 200,000 HY19636 cells were plated into either 6cm dishes with DMEM or ultra-low attachment 6cm dishes (Corning, CLS3261) with DMEM and 5% matrigel. Cells were grown for 48 hours, trypsinized, and counted.
Immunohistochemistry
Tumors were fixed in 10% formalin overnight and embedded in paraffin. Hematoxylin and eosin and immunohistochemical analysis was performed as described (Biancur et al., 2017b). In brief, slides were deparaffinized in xylene and rehydrated sequentially in ethanol. Antibody retrieval was performed by incubating slides in sodium citrate buffer (pH6) in a pressure cooker for 15 min. Slides were quenched in hydrogen peroxide (3%) and blocked in normal serum for 1 hr at room temperature. Primary antibody was applied overnight at 4 °C then developed using vectastain Elite ABC kit (Vector labs PK-6100) and DAB (Vector labs SK-4100). Slides were counterstained with hematoxylin (Vector Labs H-3401). Cleaved Caspase-3 (CST, 9664) at 1:200; CD8a (eBioscience, 14-0808-82) 1:100. Quantification of IHC was performed using imageJ. For cleaved caspase-3 staining, the percentage of total tumor area staining positively for cleaved caspase-3 was assessed. For CD8 staining, the percentage of CD8 positive cells was calculated and plotted relative to vehicle or sgTom tumors.
Immunoblotting
For immunoblotting, whole cell lysates were generated in modified radioimmunoprecipitation (RIPA) buffer (50mM Tris-HCl pH 8.0, 150mM NaCl, 2mM EDTA, 1% NP-40, and 0.1% SDS, without sodium deoxycholate supplemented with protease (ThermoFisher Scientific) and phosphatase (10mM NaF, 1mM Na3VO4, 10mM β-glycerophosphate, and 10mM sodium pyrophosphate) inhibitors. Protein lysates (25ug) were loaded into 4-20% gradient gels (BioRad, 4561096) and transferred to nitrocellulose (Thermo, 88018) or PVDF (BioRad 1620177) membranes with 1X transfer buffer (Tris-glycine) and 10% methanol. Membranes were blocked with 5% bovine serum albumin (Sigma, A2058) for 1hr at room temperature before incubation overnight at 4 °C with primary antibody. The following day membranes were washed in TBST and incubated for 1hr with appropriate horseradish peroxidase-conjugated secondary antibody (1:5,000) for 1hr. Membrane were stripped with mild stripping buffer (15g glycine, 1g SDS, 10ml Tween20, pH 2.2, for 1L of stripping buffer). Membranes were visualized by chemiluminescence (Bio-rad, 1705061) using a ChemiDoc (Bio-Rad). For signaling studies cells were plated into ultra-low attachment 6 well plates (Corning 3471) and collected at 48 or 72 hours. For signaling studies with inhibitors cells were plated and treated with inhibitor the follow day and collected at 48 or 72 hours (TAK-475 (Sigma, SML2168), MK-2206 Cayman 11593). Flag (CST 8146T) 1:1000; HMBS (Abcam, ab129092) 1:1000; FDFT1 (Proteintech, 13128-1-AP) 1:1000; S6 Ribosomal Protein (CST, 2217) 1:1000; Phospho-S6 Ribosomal Protein (Ser240/244) (CST, 2215) 1:1000; AKT (CST, 9272) 1:1000; Phospho-AKT (Thr308) (CST, 9275) 1:500; Phospho-AKT (S473) (CST 9271) 1:500; ERK II (Santa Cruz, sc-1647); 1:1000; MAPK p44/42 (ERK1/2) (CST, 9102) 1:1000; Her3/Erbb3 (CST, 12708) 1:500; Akt (pan) (40D4) (CST 2920) 1:1000); Phospho-Her3/Erbb3 (Tyr 1289) (CST, 4791) 1:500; Actin (Sigma, A2228) 1:2000;
Porphyrin Analysis
Porphyrin profiling was performed at the Iron and Heme Core facility at the University of Utah. Briefly, porphyrin levels in control cells (sgTom) or KO cells (sgHmbs#2) were assessed basally or after 4hr supplementation with conditioned media (1:1) which was prepared as described above. Porphyrins and heme were separated on a reverse-phase C18 column, 2.1× 100 mm BEH, and visualized with the tandem high sensitivity photodiode array and fluorescence detectors of the UPLC.
Seahorse Analyzer
Oxygen consumption rate (OCR) was measured with an XF96 analyzer (Seahorse Bioscience). Briefly, cells (10,000-12,000) were seeded in DMEM+10%FBS+1%P/S and allowed to adhere overnight. The following day media was exchanged and cells were incubated in DMEM (Sigma, D5030) supplemented with 10mM glucose and 2mM glutamine (no sodium bicarbonate, phenol red, or FBS) for 1hr at 37°C non-CO2 incubator and the assay run. For conditioned media rescue experiments, cells were plated 1:1 mixture of DMEM+10%FBS+1%P/S with CM or PPIX overnight. The following morning fresh CM or PPIX media was added 1:1 into plates and grown for an additional 4 hours. After 4 hours, media was exchanged with DMEM containing 10mM glucose and 2mM glutamine (with no sodium bicarbonate, phenol red, FBS) for 1hr at 37°C non-CO2 incubator and the assay run. Injection ports for mitochondrial stress test were loaded to achieve a final concentration of oligomycin A (2μM), FCCP (0.5μM), rotenone (1μM) and antimycin A (1μM) in the plate. All seahorse experiments were normalized to protein content of each well using DC protein assay kit (Bio-Rad, 5000111).
RNA sequencing
RNA isolation was performed on 2D cells, 3D cells, and tumors using TRizol (Thermo, 15596026) and PureLink RNA Midi Kit (Thermo, 12183025) according to the manufacturer’s instructions. Bulk RNA-seq analysis was performed by Novogene using DESeq2(Love et al., 2014). Gene set enrichment analysis (GSEA) was performed by inputting genes and associated log fold change into GSEA software and running gene ontology (GO) and Kyoto encyclopedia of genes and genomes (KEGG) analysis packages.
Quantitative reverse-transcription PCR
Total RNA was extracted from cell pellets in 2D, 3D, or tumors using TRIzol (Thermo, 15596026) and PureLink RNA Mini Kit (Thermo, 12183025). Reverse transcription was performed using Superscript Vilo IV (Thermo, 11766050) with oligo-dT primers. Quantitative PCR was performed with SYBR Green Supermix (Bio-rad, 1725274) on the CFX96 real-time PCR machine (Bio-Rad). The quantity of mRNA was calculated using ΔCT method normalizing to 18S. Primer Sequences are as follows:
| Gene Name | Sequence |
|---|---|
| Hmbs (mouse) Forward | 5’-ATGAGGGTGATTCGAGTGGG-3’ |
| Hmbs (mouse) Reverse | 5’-TTGTCTCCCGTGGTGGACATA-3’ |
| Fdft1 (mouse) Forward | 5’-AGAGTGGCGGTTCACTGAGA-3’ |
| Fdft1 (mouse) Reverse | 5’-GAGAAAGGCCAATTCCCACCA-3’ |
| Hmgcs (mouse) Forward | 5’-AACTGGTGCAGAAATCTCTAGC-3’ |
| Hmgcs (mouse) Reverse | 5’-GGTTGAATAGCTCAGAACTAGCC-3’ |
| Sqle (mouse) Forward | 5’-ATAAGAAATGCGGGGATGTCAC-3’ |
| Sqle (mouse) Reverse | 5’-ATATCCGAGAAGGCAGCGAAC-3’ |
| Mvd (mouse) Forward | 5’-ATGGCCTCAGAAAAGCCTCAG-3’ |
| Mvd (mouse) Reverse | 5’-TGGTCGTTTTTAGCTGGTCCT-3’ |
| 18S Forward | 5’-GTAACCCGTTGAACCCCATT-3’ |
| 18S Reverse | 5’-CCATCCAATCGGTAGTAGCG-3’ |
Flow cytometry
For the analysis of in vitro cultured cells, single cell suspensions were prepared, washed with FCM buffer (HBSS containing 1% FBS, 1 mM EDTA, and 10 mM HEPES) and stained with Liperfluo (Dojindo) for 15 minutes at 37°C in the dark. Dead cells were depleted by DAPI staining. For the immunophenotyping of orthotopic tumours, tissues were mechanically minced with scissors, and then digested in DMEM containing 1 mg/mL Collagenase IV (Gibco), 100 μg/mL DNase I (Roche), 1% FBS, 10 mM HEPES, 2% Antibiotic-Antimycotic (ThermoFisher Scientific) for 40 min at 37°C in the dark with gentle agitation every 10 min. Digested tissues were then washed twice in DMEM containing 10% FBS, filtered through a 40-μm nylon mesh strainer (Corning). Cells were suspended in ACK Lysing Buffer (ThermoFisher Scientific), incubated for 10 min at 4°C in the dark to remove red blood cells. Cells were washed twice in FCM buffer and counted. Cells were stained with Zombie Aqua Fixable Viability Kit (BioLegend) and blocked with anti-mouse CD16/CD32 antibody (Mouse BD Fc Block™, clone 2.4G2, BD Bioscience). One million cells were incubated with appropriate antibodies diluted in FCM buffer at 4°C in the dark for 40 min. Cells were then washed twice with FCM buffer and analyzed or further fixed in phosphate-buffered saline (PBS) containing 0.1 M L-Lysine (Sigma) and 2% paraformaldehyde (Ted Pella) at 4°C in the dark overnight. Cells were analyzed on a BD LSR Fortessa or a BD LSR-II UV and analyzed by FlowJo software (FlowJo, LLC, version 10.4). Dead cells were excluded by zombie staining and singlets were analyzed using the following definitions: CD3+ cells are CD45+ and CD3+ ; CD8+ cells are CD45+, CD3+,and CD8+ ; CD4+ cells are defined as CD45+, CD3+,and CD4+.. Antibodies are as follows: CD45 (BD Biosciences, 564279, Clone 30-F-11, 1:100 dilution), CD3 (Biolegend, 100241, Clone 17a2, 1:100 dilution), CD8 (Biolegend, 100730, Clone AF700, dilution 1:100), CD4 (Biolegend, 100451, Clone BV605, 1:100 dilution). The gating strategy is highlighted in Figure S6.
RTK Array
Analysis of global RTK activation was performed using Proteome Profiler Mouse Phospho-RTK Array Kit (R&D systems, ARY014). Assay was carried out according to manufacturer instructions with 300ug of protein used for each membrane.
Active Ras Detection
Active Ras detection was performed using Active Ras Detection Kit (Cell Signaling Technologies, #8821). Assay was performed according to manufacturer instructions with 300ug of cell lysate.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using Prism (GraphPad 7.0). Results are expressed as mean +/− standard deviation unless otherwise indicated. Unpaired, two tailed Student’s t-tests were performed when comparing two groups to each other. Log-rank Mantel-Cox tests were performed when conducting survival curve statistical analysis. Sample sizes and statistical tests for each experiment are denoted in the figure legends. Immunoblots were performed at least 3 times. All in vitro growth data and seahorse was performed at least 3 times. Mouse experiments represent an independent experiment with tumor biological replicates noted. Immunohistochemical images show samples from a single mouse of each group. For all significance analyses, groups were considered significantly different if p-value <0.05.
Supplementary Material
In this issue of Cell Metabolism, Biancur et al. use CRISPR/Cas9 screening to map the metabolic dependencies of pancreatic cancer in vitro and in vivo. These data provide insight into the merits and limitations of multiple model systems for studying cancer metabolism and highlight tractable metabolic targets in pancreatic tumors.
Highlights:
CRISPR screens reveal metabolic dependencies in PDA tumors in vitro and in vivo
Screen reveals targetable metabolic liabilities and limitations of model systems
The majority of tumor metabolic dependencies are recapitulated in 2D culture
3D culture better recapitulates aspects of tumor metabolism-cell signaling axis
Acknowledgements
We thank members of the Kimmelman and Aguirre labs for technical assistance and thoughtful suggestions and Haoqiang Ying for providing the B6 KPC cells. This work was supported by National Cancer Institute Grants R01CA157490, R01CA188048, P01CA117969, R35CA232124; P30CA016087 and the Lustgarten Foundation, and SU2C to A.C.K. A.J.A. is funded by the Lustgarten Foundation, Dana-Farber Cancer Institute Hale Center for Pancreatic Cancer Research, the Doris Duke Charitable Foundation, Pancreatic Cancer Action Network, NIH-NCI K08 CA218420-02, P50 CA127003, and U01 CA224146. Heme/porphyrin/metal analysis was performed at the University of Utah, supported in part by a grant from the NIH National Institute of Diabetes and Digestive and Kidney Diseases, Grant number U54DK110858 D.E.B. is supported by a Ruth L. Kirschstein Institutional National Research Service Award, T32 CA009161 (Levy), and the NCI Predoctoral to Postdoctoral Fellow Transition Award (F99/K00) F99 CA245822. K.Y was supported by the Uehara Memorial Foundation Research Fellowship. J.E.N. is supported by a Damon Runyon Cancer Research Foundation fellowship award (DRG: 2384-19). R.S.B. is a Merck Fellow of the Damon Runyon Cancer Research Foundation (DRG-2348-18). We thank the NYU Langone Experimental Pathology Laboratory, Flow Cytometry Core, and Genome Technology Center, each supported in part by the CCSG P30CA016087 at the Perlmutter Cancer Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interests
A.C.K. has financial interests in Vescor Therapeutics, LLC. A.C.K. is an inventor on patents pertaining to KRAS regulated metabolic pathways, redox control pathways in pancreatic cancer, targeting GOT1 as a therapeutic approach, and the autophagic control of iron metabolism. A.C.K. is on the SAB of Rafael/Cornerstone Pharmaceuticals. A.C.K. is a consultant for Deciphera. R.TM. consults for Bristol-Myers Squibb. The other authors declare no competing interests. A.J.A. has consulted for Oncorus, Inc., Arrakis Therapeutics, and Merck & Co., Inc, and has research funding from Mirati Therapeutics and Deerfield, Inc. that are unrelated to this project.
Supplementary Table and Information
Supplementary Table 1 (related to Figure 1). Analyzed CRISPR screening data
Supplementary Table 2 (related to Figure S4 and Figure 5). Analyzed RNA sequencing for WT and Fdft1 KO cells.
References
- Bardeesy N, Aguirre AJ, Chu GC, Cheng KH, Lopez LV, Hezel AF, Feng B, Brennan C, Weissleder R, Mahmood U, et al. (2006). Both p16(Ink4a) and the p19(Arf)-p53 pathway constrain progression of pancreatic adenocarcinoma in the mouse. Proceedings of the National Academy of Sciences of the United States of America 103, 5947–5952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biancur DE, and Kimmelman AC (2018). The plasticity of pancreatic cancer metabolism in tumor progression and therapeutic resistance. Biochimica et biophysica acta. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biancur DE, Paulo JA, Małachowska B, Del Rey MQ, Sousa CM, Wang X, Sohn AS, Chu GC, Gygi SP, and Harper JW (2017a). Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nature communications 8, 15965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biancur DE, Paulo JA, Malachowska B, Quiles Del Rey M, Sousa CM, Wang X, Sohn ASW, Chu GC, Gygi SP, Harper JW, et al. (2017b). Compensatory metabolic networks in pancreatic cancers upon perturbation of glutamine metabolism. Nature communications 8, 15965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantor JR, Abu-Remaileh M, Kanarek N, Freinkman E, Gao X, Louissaint A Jr., Lewis CA, and Sabatini DM (2017). Physiologic Medium Rewires Cellular Metabolism and Reveals Uric Acid as an Endogenous Inhibitor of UMP Synthase. Cell 169, 258–272.e217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collisson EA, Trejo CL, Silva JM, Gu S, Korkola JE, Heiser LM, Charles R-P, Rabinovich BA, Hann B, Dankort D et al. (2012). A central role for RAF➔MEK➔ERK signaling in the genesis of pancreatic ductal adenocarcinoma. Cancer discovery 2, 685–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davidson SM, Jonas O, Keibler MA, Hou HW, Luengo A, Mayers JR, Wyckoff J, Del Rosario AM, Whitman M, Chin CR, et al. (2017). Direct evidence for cancer-cell-autonomous extracellular protein catabolism in pancreatic tumors. Nature medicine 23, 235–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench JG, Hartenian E, Graham DB, Tothova Z, Hegde M, Smith I, Sullender M, Ebert BL, Xavier RJ, and Root DE (2014). Rational design of highly active sgRNAs for CRISPR-Cas9-mediated gene inactivation. Nature biotechnology 32, 1262–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn LL, Midwinter RG, Ni J, Hamid HA, Parish CR, and Stocker R (2014). New insights into intracellular locations and functions of heme oxygenase-1. Antioxidants & redox signaling 20, 1723–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, Yang C, Do QN, Doucette S, Burguete D, et al. (2017). Lactate Metabolism in Human Lung Tumors. Cell 171, 358–371.e359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finotti E, D’Ambrosio M, Paoletti F, Vivanti V, and Quaglia G (2000). Synergistic effects of alpha-tocopherol, beta-sitosterol and squalene on antioxidant activity assayed by crocin bleaching method. Die Nahrung 44, 373–374. [DOI] [PubMed] [Google Scholar]
- Gaborit N, Lindzen M, and Yarden Y (2016). Emerging anti-cancer antibodies and combination therapies targeting HER3/ERBB3. Human vaccines & immunotherapeutics 12, 576–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Bermudez J, Baudrier L, Bayraktar EC, Shen Y, La K, Guarecuco R, Yucel B, Fiore D, Tavora B, Freinkman E, et al. (2019). Squalene accumulation in cholesterol auxotrophic lymphomas prevents oxidative cell death. Nature 567, 118–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamza I, and Dailey HA (2012). One ring to rule them all: trafficking of heme and heme synthesis intermediates in the metazoans. Biochimica et biophysica acta 1823, 1617–1632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han K, Pierce SE, Li A, Spees K, Anderson GR, Seoane JA, Lo YH, Dubreuil M, Olivas M, Kamber RA, et al. (2020). CRISPR screens in cancer spheroids identify 3D growth-specific vulnerabilities. Nature 580, 136–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, Esparza LA, Reya T, Le Z, Yanxiang Guo J, et al. (2017). Glucose feeds the TCA cycle via circulating lactate. Nature 551, 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, Chen Y, Babbar A, Firdaus SJ, Darjania L, et al. (2018). Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 172, 578–589.e517. [DOI] [PubMed] [Google Scholar]
- Kitajima Y, Ishii T, Kohda T, Ishizuka M, Yamazaki K, Nishimura Y, Tanaka T, Dan S, and Nakajima M (2019). Mechanistic study of PpIX accumulation using the JFCR39 cell panel revealed a role for dynamin 2-mediated exocytosis. Scientific reports 9, 8666–8666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobuchi H, Moriya K, Ogino T, Fujita H, Inoue K, Shuin T, Yasuda T, Utsumi K, and Utsumi T (2012). Mitochondrial localization of ABC transporter ABCG2 and its function in 5-aminolevulinic acid-mediated protoporphyrin IX accumulation. PLoS One 7, e50082–e50082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krall AS, Xu S, Graeber TG, Braas D, and Christofk HR (2016). Asparagine promotes cancer cell proliferation through use as an amino acid exchange factor. Nature communications 7, 11457–11457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langhans SA (2018). Three-Dimensional in Vitro Cell Culture Models in Drug Discovery and Drug Repositioning. Front Pharmacol 9, 6–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyssiotis CA, and Kimmelman AC (2017). Metabolic Interactions in the Tumor Microenvironment. Trends in cell biology 27, 863–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy D, Attri KS, and Singh PK (2018). Phosphoinositide 3-Kinase Signaling Pathway in Pancreatic Ductal Adenocarcinoma Progression, Pathogenesis, and Therapeutics. Front Physiol 9, 335–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson JC, Westwood M, Allen KR, Newton KE, and Barth JH (1998). The ratio of erythrocyte zinc-protoporphyrin to protoporphyrin IX in disease and its significance in the mechanism of lead toxicity on haem synthesis. Annals of clinical biochemistry 35 (Pt 3), 422–426. [DOI] [PubMed] [Google Scholar]
- Nishimoto T, Amano Y, Tozawa R, Ishikawa E, Imura Y, Yukimasa H, and Sugiyama Y (2003). Lipid-lowering properties of TAK-475, a squalene synthase inhibitor, in vivo and in vitro. Br J Pharmacol 139, 911–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogino T, Kobuchi H, Munetomo K, Fujita H, Yamamoto M, Utsumi T, Inoue K, Shuin T, Sasaki J, Inoue M, et al. (2011). Serum-dependent export of protoporphyrin IX by ATP-binding cassette transporter G2 in T24 cells. Molecular and cellular biochemistry 358, 297–307. [DOI] [PubMed] [Google Scholar]
- Pai JT, Guryev O, Brown MS, and Goldstein JL (1998). Differential stimulation of cholesterol and unsaturated fatty acid biosynthesis in cells expressing individual nuclear sterol regulatory element-binding proteins. The Journal of biological chemistry 273, 26138–26148. [DOI] [PubMed] [Google Scholar]
- Panner A, Crane CA, Weng C, Feletti A, Fang S, Parsa AT, and Pieper RO (2010). Ubiquitin-specific protease 8 links the PTEN-Akt-AIP4 pathway to the control of FLIPS stability and TRAIL sensitivity in glioblastoma multiforme. Cancer research 70, 5046–5053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park RJ, Wang T, Koundakjian D, Hultquist JF, Lamothe-Molina P, Monel B, Schumann K, Yu H, Krupzcak KM, Garcia-Beltran W, et al. (2017). A genome-wide CRISPR screen identifies a restricted set of HIV host dependency factors. Nature genetics 49, 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponka P (1999). Cell biology of heme. The American journal of the medical sciences 318, 241–256. [DOI] [PubMed] [Google Scholar]
- Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, Sethumadhavan S, Woo HK, Jang HG, Jha AK, et al. (2011). Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476, 346–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachar M, Anderson KE, and Ma X (2016). Protoporphyrin IX: the Good, the Bad, and the Ugly. J Pharmacol Exp Ther 356, 267–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santana-Codina N, Roeth AA, Zhang Y, Yang A, Mashadova O, Asara JM, Wang X, Bronson RT, Lyssiotis CA, Ying H, et al. (2018). Oncogenic KRAS supports pancreatic cancer through regulation of nucleotide synthesis. Nature communications 9, 4945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Z, and Ferreira GC (2006). Modulation of inhibition of ferrochelatase by N-methylprotoporphyrin. Biochem J 399, 21–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, and Jemal A (2018). Cancer statistics, 2018. CA: a cancer journal for clinicians 68, 7–30. [DOI] [PubMed] [Google Scholar]
- Sivaram N, McLaughlin PA, Han HV, Petrenko O, Jiang YP, Ballou LM, Pham K, Liu C, van der Velden AW, and Lin RZ (2019). Tumor-intrinsic PIK3CA represses tumor immunogenecity in a model of pancreatic cancer. The Journal of clinical investigation 129, 3264–3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, Kremer D, Hwang RF, Witkiewicz AK, Ying H, et al. (2016). Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536, 479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein EA, Bays H, O’Brien D, Pedicano J, Piper E, and Spezzi A (2011). Lapaquistat acetate: development of a squalene synthase inhibitor for the treatment of hypercholesterolemia. Circulation 123, 1974–1985. [DOI] [PubMed] [Google Scholar]
- Sullivan MR, Danai LV, Lewis CA, Chan SH, Gui DY, Kunchok T, Dennstedt EA, Vander Heiden MG, and Muir A (2019). Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. eLife 8, e44235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tansey TR, and Shechter I (2000). Structure and regulation of mammalian squalene synthase. Biochimica et biophysica acta 1529, 49–62. [DOI] [PubMed] [Google Scholar]
- Tsiftsoglou AS, Tsamadou AI, and Papadopoulou LC (2006). Heme as key regulator of major mammalian cellular functions: molecular, cellular, and pharmacological aspects. Pharmacology & therapeutics 111, 327–345. [DOI] [PubMed] [Google Scholar]
- Wang T, Yu H, Hughes NW, Liu B, Kendirli A, Klein K, Chen WW, Lander ES, and Sabatini DM (2017). Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions with Oncogenic Ras. Cell 168, 890–903.e815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weber L-W, Boll M, and Stampfl A (2004). Maintaining cholesterol homeostasis: Sterol regulatory element-binding proteins. World journal of gastroenterology : WJG 10, 3081–3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu X, Yen L, Irwin L, Sweeney C, and Carraway KL 3rd (2004). Stabilization of the E3 ubiquitin ligase Nrdp1 by the deubiquitinating enzyme USP8. Molecular and cellular biology 24, 7748–7757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S, Wang X, Contino G, Liesa M, Sahin E, Ying H, Bause A, Li Y, Stommel JM, Dell’antonio G, et al. (2011). Pancreatic cancers require autophagy for tumor growth. Genes & development 25, 717–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying H, Kimmelman AC, Lyssiotis CA, Hua S, Chu GC, Fletcher-Sananikone E, Locasale JW, Son J, Zhang H, Coloff JL, et al. (2012). Oncogenic Kras maintains pancreatic tumors through regulation of anabolic glucose metabolism. Cell 149, 656–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The accession number for the CRISPR screening and RNA sequencing is GSE158787 and GSE158657 respectively. All codes in this study are available on Github (Biancur-CellMetabolism-2020).