

Abstract
Senescence is the consequence of a signaling mechanism activated in stressed cells to prevent proliferation of cells with damage. Senescent cells (Sncs) often develop a senescence-associated secretory phenotype to prompt immune clearance, which drives chronic sterile inflammation and plays a causal role in aging and age-related diseases. Sncs accumulate with age and at anatomical sites of disease. Thus, they are regarded as a logical therapeutic target. Senotherapeutics are a new class of drugs that selectively kill Sncs (senolytics) or suppress their disease-causing phenotypes (senomorphics/senostatics). Since 2015, several senolytics went from identification to clinical trial. Preclinical data indicate that senolytics alleviate disease in numerous organs, improve physical function and resilience, and suppress all causes of mortality, even if administered to the aged. Here, we review the evidence that Sncs drive aging and disease, the approaches to identify and optimize senotherapeutics, and the current status of preclinical and clinical testing of senolytics.
Keywords: senescence, senolytics, senomorphics, senescence-associated secretory phenotype, aging
1. INTRODUCTION
1.1. Geroscience Hypothesis
Old age is the greatest risk factor for the chronic diseases responsible for the majority of morbidity, mortality, and health-care costs worldwide (1, 2). These chronic diseases include atherosclerosis and other cardiovascular diseases, strokes, dementias, most cancers, diabetes, renal failure, chronic lung diseases, osteoporosis, arthritis, blindness, and neurodegenerative diseases. Aging also predis-poses people to geriatric syndromes and decreased physical resilience. The geriatric syndromes include frailty, sarcopenia, immobility, falling, depression, mild cognitive impairment, incontinence, metabolic syndrome, and weight loss. Decreased physical resilience can be reflected in delayed recovery following myocardial infarction, stroke, injuries, infection, chemotherapy, surgery, or fractures and in impaired response to immunization. Combinations of these conditions often occur in older individuals, with the frequency of multimorbidity (three or more conditions occurring simultaneously) increasing exponentially in the third tertile of life (3). The presence of multimorbidity confounds the potential gains from curing any single age-related disorder (4).
The recognition that old age is the greatest risk factor for most chronic diseases led to the geroscience hypothesis, which posits that therapeutically targeting fundamental aging mechanisms will successfully impact multiple chronic diseases because these diseases all share the same underlying mechanism (1). There is a tremendous potential health and economic benefit to targeting fundamental aging mechanisms as opposed to treating each disease separately (2). Indeed, by one estimate, a 2% delay in the progression of aging processes, compared with doing nothing or curing cancer or heart disease, would lead to 10 million more healthy, rather than disabled, elderly people in the United States and a savings of $7.1 trillion over the next 40 years.
1.2. Cellular Senescence
Recent studies of animal models have established the veracity of the geroscience hypothesis (1). One fundamental aging mechanism that underlies numerous age-related diseases is cellular senescence (5). Senescence is a cell fate that typically includes irreversible loss of proliferative potential, resistance to cell death, and increased metabolic activity (Figure 1). Persistent mitogenic signals, telomere shortening, DNA damage, high glucose, increased reactive oxygen species, or protein aggregation activates the p53/p21CIP1 and p16INK4a/Rb tumor suppressor pathways to initiate senescence (6) (Figure 2). Downstream of these signals, the senescence response is amplified by several signal transducers, including ATM (ataxia telangiectasia mutated; a kinase that mobilizes the cellular response to DNA double-strand breaks), IKK/NF-κB [an IKK complex that activates the transcription factor nuclear factor κB (NF-κB) in response to cellular stress], JAK/STAT (a signaling pathway that drives transcription of genes regulating cell fate), GATA4 (a transcription factor that regulates development and cellular differentiation), and mTOR (mammalian target of rapamycin; a kinase that regulates the cellular response to nutrient sensing). A deeply senescent cell (Snc) can acquire many characteristics, including increased cell size and protein content; expression of antiapoptotic proteins; increased lysosomal hydrolase activity [senescence-associated β-galactosidase (SA-βgal)]; and evidence of DNA damage including γH2AX foci, telomere-associated foci (TAFs), and decondensation of pericentromeric satellite heterochromatin [senescence-associated heterochromatin foci (SAHFs)]. Sncs are often metabolically very active with a robust secretome [senescence-associated secretory phenotype (SASP)] (7). SASP factors include proinflammatory interleukins, chemokines, growth factors, proteases, receptors, enzymes that modify the extracellular matrix, stem cell/progenitor toxins, hemostatic factors, reactive metabolites, bioactive lipids (e.g., bradykines, ceramides, and prostanoids), microRNAs, noncoding nucleotides, and extracellular vesicles (Figure 2). Evidence suggests that the role of the SASP is to entice immune clearance. However, with age, an increasing dysfunctional immune system contributes to an accumulation of Sncs. Even a relatively low number of Sncs are sufficient to cause a chronic, sterile inflammation that disrupts tissue homeostasis and reduces resilience to stress, termed inflammaging (Figure 1). The SASP also has negative effects on stem cell function, leading to impaired tissue regeneration (Figure 1). Sncs are found at anatomical sites of many diseases (see the list in the Supplemental Appendix) and play a causal role in pathology.
Figure 1.
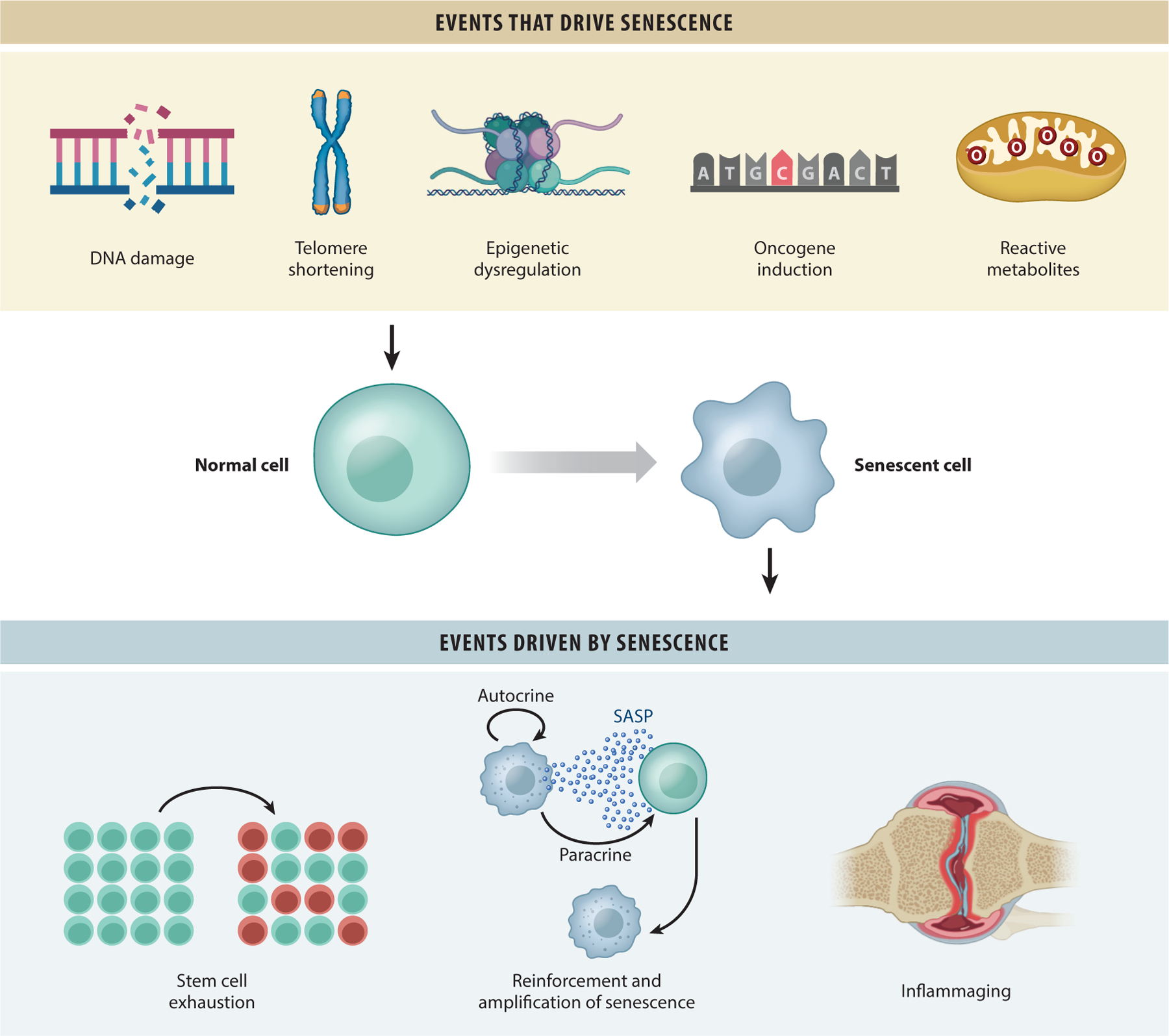
Events that drive cellular senescence and events driven by senescence. Senescence can be driven by different types of cellular stress, including genotoxicity, telomere shortening, epigenetic dysregulation, oncogene activation, mitochondrial dysfunction, and metabolic and oxidative stress. This leads to signaling events that result in senescence. Senescent cells have a robust SASP, which can reinforce and spread senescence, locally and systemically, inhibiting stem cell function and disrupting tissue homeostasis while increasing sterile inflammation, termed inflammaging. Figure adapted, with permission, from the original figure by Dr. Rajesh Vyas. Abbreviation: SASP, senescence-associated secretory phenotype.
Figure 2.
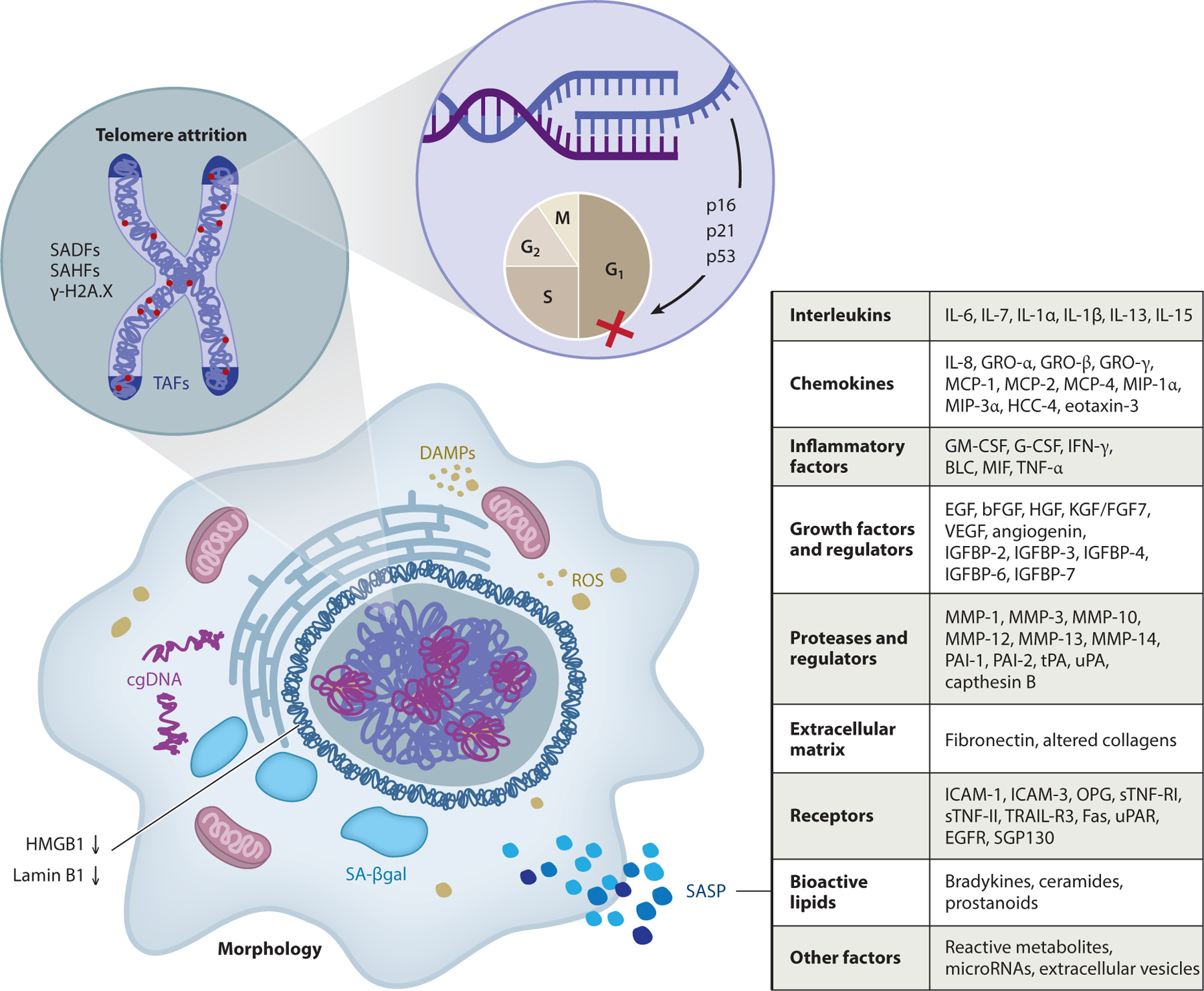
Characteristics of a Snc that can be exploited as biomarkers to detect and quantify senescence. Cells undergoing senescence due to damage and stress (see Figure 1) have activated signaling pathways, including the DNA damage response/ATM, GATA-4, IKK/NF-κB, JAK/STAT, and mTOR signaling pathways. There is also upregulation of the cell cycle inhibitors p53, p16INK4a, and p21CIP1. In addition, Sncs can show evidence of telomere shortening or damage, called TAFs; damage elsewhere in the genome, called SADFs; and epigenetic changes, called SAHFs. Sncs also have decreased levels of Lamin B1 and increased SA-βgal activity. Many Sncs have a SASP composed of chemokines, inflammatory factors and interleukins, growth factors and regulators, extracellular matrix components, soluble receptors, proteases and regulators, reactive metabolites, bioactive lipids, microRNAs, and extracellular vesicles, and early in senescence they secrete HMBG1, a key DAMP (an endogenous molecule that activates the innate immune system), which amplifies the SASP. There is no Snc-specific marker and not all Sncs express the same markers, especially in terms of the SASP. Figure adapted, with permission, from the original figure by Matthew Moore. Abbreviations: cgDNA, cytoplasmic genomic DNA; DAMP, danger-associated molecular pattern; ROS, reactive oxygen species; SA-βgal, senescence-associated β-galactosidase; SADFs, senescence-associated DNA damage foci; SAHFs, senescence-associated heterochromatin foci; SASP, senescence-associated secretory phenotype; Snc, senescent cell; TAFs, telomere-associated foci.
Evidence that Sncs play a causal role in aging first came from transgenic mice, in which cells that express p16Ink4a, a cyclin-dependent kinase inhibitor that arrests the cell cycle and is commonly upregulated in Sncs, can be selectively killed. Ablating p16+ cells in these mice suppresses age-related disease, extending the health span of two independent strains of transgenic mice: INK-ATTAC and p16-3MR (see Supplemental Figure 1 for a description of the models) (8–10). Clearing Sncs from aged INK-ATTAC mice improves age-related changes in metabolic function (11), and chronic clearance of p16-expressing cells in adult mice extends their median life span (9). Clearance of Sncs in INK-ATTAC or p16-3MR mice restores vascular reactivity (12), stabilizes atherosclerotic plaques (13), improves pulmonary function (14), alleviates osteoarthritis (10), improves fatty liver disease (15), improves lung function in a pulmonary fibrosis model (14), and prevents age-related bone loss (16). Conversely, transplantation of as few as 1 million Sncs into a mouse is sufficient to drive frailty and age-related disease (17). Thus, Sncs appear to play a major role in driving life-limiting age-related diseases.
1.3. Senotherapeutics
A key study demonstrating that caloric restriction, which increases health span and life span, decreases the accumulation of Sncs (18) initiated the search in the early 2000s for pharmacologic approaches to kill Sncs (well before the transgenic p16+-cell ablating models were developed). Originally, this was done with bioinformatics, based on the expression profile of Sncs versus non-Sncs (19). This approach revealed that Sncs express prosurvival proteins that block apoptosis of Sncs. Inhibitors of these prosurvival proteins, often using repurposed cancer chemotherapeutics, kill Sncs (senolysis), leading to the creation of the first senolytic drugs (19). Subsequently, additional senolytics were identified through drug screens (20–22). Note that Sncs are not always pathological. They play important roles in, for example, wound healing and parturition (23). Another strategy for therapeutically targeting Sncs is to attempt to dampen their disease-causing phenotypes, which is termed senomorphic or senostatic therapy (20). Senomorphics are aimed largely at suppressing the SASP and thereby inflammation. However, this is not necessarily a safer approach, as the SASP is also critical for wound healing (23). Importantly, no senotherapeutic approach should interfere with pathways controlling the cell cycle or the mechanisms driving senescence, as this could promote cancer. Senolytics and senomorphics do not interfere with Snc generation, allowing Sncs to form as needed to arrest damaged cells.
Although not the focus of this review, several senomorphic/senostatic drugs have potential clinical applications. Rapamycin, an immunosuppressant, decreases the SASP and maintains cell cycle arrest but does not kill Sncs (24). IKK/NF-κB and JAK inhibitors also suppress the SASP (11). In addition, metformin, a drug widely used to treat type 2 diabetes, limits NF-κB activation (25), thereby reducing the SASP. All these drugs extend health span in mice, illustrating the utility of senomorphic approaches.
Table 1 lists the senolytics identified to date. Several senolytics target prosurvival pathways, including the BCL-2/BCL-xL, p53/p21, and PI3K/AKT, and the antiapoptotic pathways, including serpins. Pharmacologic inhibitors of these proteins, similar to inhibition by small interfering RNA (siRNA), cause death of murine and human Sncs (19). Drugs that target these pathways also have senolytic activity in vivo. The first demonstration of senolysis in vivo employed the combination of dasatinib (a kinase inhibitor of BCR-ABL, SRC family, c-KIT, and ephrin receptors that is used to treat leukemias) and quercetin (an inhibitor of PI3Ks and serpins) (19). Dasatinib plus quercetin (D+Q) extends health span and life span in aged mice (17), improving cardiovascular function and treadmill endurance, while decreasing frailty, neurologic dysfunction, and bone loss in mice (19). D+Q also reduces aortic calcification of atherosclerotic ApoE−/− mice (12), improves lung function in a murine model of idiopathic pulmonary fibrosis (14), reduces hepatic steatosis (15), and prevents age-related bone loss (16). Fisetin, a quercetin-related natural flavonoid, has both senolytic and senomorphic activity depending on the cell type (21, 22). Fisetin extends the health span and life span of mice and reverses tissue damage when administered to aged animals (26). Fisetin reduces senescence in aged nonhuman primates and human tissue explants (26).
Table 1.
Senotherapeutics reported to date and methods of discovery
| First-generation senolytics: hypothesis-driven, mechanism-based discovery | |
|---|---|
| Agent | Reference(s) |
| Dasatinib | 19 |
| Quercetin | 19 |
| Fisetin | 22, 26 |
| Luteolin | 26 |
| Curcumin | 26 |
| Curcumin analog EF24 | 85 |
| Navitoclax (ABT263) | 27, 28 |
| A1331852 | 22 |
| A1155463 | 22 |
| Geldanamycin, tanespimycin, alvespimycin, and other HSP90 inhibitors | 20 |
| Piperlongumine | 86 |
| FOXO4-related peptide | 29 |
| Nutlin3a [although Nutlin3a can also cause senescence (87)] | 88 |
| Cardiac glycosides such as ouabain, proscillaridin A, and digoxin | 89, 90 |
| Aspirin | 91 |
| Second-generation senolytics: traditional and other drug discovery methods | |
| Method | Reference(s) |
| High-throughput compound library screens | 92 |
| Vaccines | 93, 94 |
| Toxin-loaded nanoparticles preferentially lysed by Sncs | 95 |
| Immunomodulators | 96 |
Screening a small library of autophagy regulators revealed HSP90 (heat shock protein 90) inhibitors as potent senolytics (20). The HSP90 inhibitor 17-DMAG extends the health span of progeroid mice (20). On the basis of our discovery that BCL-xL siRNA selectively eliminates human senescent endothelial cells (19), we and others demonstrated that inhibitors of BCL-2 family members, including navitoclax (ABT263), ABT737, A1331852, and A1155463, are senolytic in some but not all human cell types (27). Treating mice with navitoclax reduces Snc burden, alleviates radiation-induced hematopoietic stem cell dysfunction (28), and improves osteoarthritis (10). In addition, a peptide that blocks the interaction of p53 with FOXO4 (a forkhead transcription factor family member that binds p53 to induce cellular senescence) induces apoptosis of Sncs in vitro and in vivo, improving fitness, hair growth, and renal function in progeroid and aged mice (29). These preclinical studies of numerous different drugs targeting Sncs via a variety of mechanisms illustrate the tremendous potential of senotherapeutics. The success is particularly remarkable because the field is young and little attention has yet been paid to chemically optimizing the therapeutics or their methods of delivery to enhance safety and efficacy.
2. MEASURING SENESCENCE
One of the key challenges in senescence research is the lack of biomarkers that unambiguously discriminate Sncs from non-Sncs, particularly in vivo. Such biomarkers are of course essential for proving efficacy of senolytics. They would also be valuable for determining who is most likely to benefit from senotherapy. Currently, it is accepted that there is not one single biomarker of Sncs (Figure 2) and that only through the simultaneous measure of multiple end points can Sncs be identified accurately (30).
2.1. Multiple Markers of Senescence
Sncs are heterogeneous for several reasons. First, senescence is a dynamic, not static, process. Thus, it is reasonable to assume that within a tissue both early and late Sncs coexist and have distinct phenotypes. Second, Sncs have different expression profiles based on their cell lineage as well as the type of stress that triggered senescence (31). Finally, Sncs play different roles depending on their physiological context. For example, Sncs are critical during development, can suppress tumor growth, and assist with tissue repair (23, 32), but they also drive pathology, including cancer (9, 17).
Currently, the most widely used markers of cellular senescence include SA-βgal, which is measured at a higher pH than normal; the expression of cell cycle kinase inhibitors p16INK4a and p21CIP1; the absence of proliferation markers such as Ki67, markers of DNA damage such as p53, p-p53, γH2AX foci, TAFs, and SAHFs; the nuclear exclusion of HMGB1; the loss of Lamin B1 expression; the presence of senescence-associated distension of satellite DNA (SADS); and the expression of SASP components such as interleukin-1β (IL-1β), IL-6, monocyte chemoattractant protein 1 (MCP-1), plasminogen activator inhibitor 1 (PAI-1), and certain matrix metallo-proteinases (30). Obviously, none of these markers are unique to Sncs. For instance, increased SA-βgal activity occurs in confluent and immortalized cells in culture. Increased SA-βgal activity and p16INK4a expression occur during macrophage activation (33). Adding to this complexity, senescence markers are found in postmitotic, terminally differentiated cells such as neurons, cardiomyocytes, osteoclasts and osteoblasts, and adipocytes, seemingly contradictory to the definition of senescence as a permanent cell cycle arrest. Senolytics do clear postmitotic cells with senescent features, resulting in beneficial health outcomes (16, 34, 35) and reinforcing Sncs’ role in causing disease.
To date, most in vivo studies report the measure of senescence markers in tissue homogenates, with fewer studies using histochemical and immunohistochemical approaches in tissue sections. Multiple markers of senescence are measured because they lack specificity. What these methods lack is the ability to colocalize several biomarkers of senescence in a single cell. New approaches that permit the simultaneous measure of multiple senescence markers [e.g., cytometry by time of flight (CyTOF) (35) or single-cell omics such as single-cell RNA sequencing (36)] are needed.
3. EVIDENCE THAT SENESCENCE DRIVES AGING AND AGE-RELATED DISEASES
3.1. Adipose Tissue
Adipose tissue is the largest organ in many people; more than 25% of those people age 65 or older are obese. With aging and obesity, adipose tissue undergoes extensive changes in distribution (subcutaneous versus visceral), cell composition, cell size, responsiveness to insulin and other hormones, differentiation capacity, and extent of inflammation (37). With aging, visceral fat often expands, whereas subcutaneous fat undergoes lipoatrophy. Adipose tissue becomes a major contributor to insulin resistance in old age due to changes in immune cell populations, including a shift of macrophages toward a proinflammatory phenotype and a loss of regulatory T cells, and increased release of inflammatory cytokines, including tumor necrosis factor-α (TNF-α) and osteopontin (38). Similarly, inflammation of adipose tissue is implicated in the genesis of insulin resistance and diabetes in obesity. Both aging and obesity are associated with the impaired capacity of adipocyte progenitors to replicate and differentiate into fully functional, insulin-responsive adipocytes (37). This decreased adipogenic potential impairs the ability of adipose tissue to expand when challenged with excess nutrients. This leads to adipocyte hypertrophy, which drives inflammation, lipolysis, and systemic insulin resistance. As a result, Sncs accumulate abundantly in adipose tissue of aging and obese humans and mice. This accumulation is associated with increased activity of SA-βgal and expression of PAI-1, p53, and p16INK4a in adipose tissue and contributes to reduced adipogenesis. This in turn leads to increased accumulation of ectopic lipids in tissues such as liver, muscle, and brain (15), which impairs tissue homeostasis and exacerbates insulin resistance.
Senescence of preadipocytes is a major negative regulator of adipogenesis through both cellautonomous and paracrine mechanisms, as demonstrated in coculture experiments (11). Sncs can directly cause insulin resistance through release of SASP factors such as IL-6 and TNF-α (39, 40). Sncs contribute to chemoattraction of immune cells to visceral adipose tissue in obesity, further contributing to insulin resistance and risk of diabetes (41). Activin A, a transforming growth factor-β (TGF-β) family member that is a component of the senescent adipocyte progenitor SASP (11), is increased in obese mice and correlates with levels of p16Ink4a. Activin A impedes expression of the insulin-sensitizing adipogenic transcription factors peroxisome proliferator-activated receptor γ (PPARγ) and CCAAT-enhancer-binding protein α (C/EBPα), thereby contributing to failed adipogenesis, lipotoxicity, and insulin resistance (11). p53 activation, as occurs in senescence, also inhibits adipocyte differentiation, blunts insulin-induced glucose transport, and increases lipolysis in adipocytes, further contributing to both inflammation and insulin resistance. As with aging, p53 is increased in adipose tissue in type 2 diabetes, and genetic activation of p53 in adipose tissue in animal models leads to systemic insulin resistance. Transplanting senescent adipocyte precursors into young animals induces an aging-like phenotype with accelerated onset of multiple age-related diseases and premature death (17).
Adipose tissue from mice treated with D+Q have significantly decreased p16Ink4a-, centromere binding protein (Cenbp)-, and p21Cip1-expressing Sncs (42). Adipose progenitor cells are the main cell type targeted by senolytics in fat. Chronic, intermittent D+Q treatment alleviates metabolic dysfunction in genetically or diet-induced obese mice or mice transplanted with Snc preadipocytes (17, 42). Senolytics reduce adipocyte hypertrophy, increase the ratio of subcutaneous to visceral adipose tissue, decrease HbA1c, improve glucose tolerance, enhance insulin sensitivity, lower circulating inflammatory mediators, and promote adipogenesis in obese mice (34). Elimination of Sncs also prevents migration of transplanted monocytes into intra-abdominal adipose tissue and reduces the number of macrophages in this tissue. In addition, microalbuminuria is decreased and renal podocyte and cardiac diastolic function are improved with senolytic therapy. Thus, the accumulation of Sncs in adipose tissue appears to be a causal factor in ageand obesity-related inflammation and metabolic derangements.
3.2. Bone
Osteoporosis is a skeletal fragility syndrome of old age that affects approximately 10 million people in the United States, and more than 43 million have osteopenia (43). If left unchecked, the 2 million osteoporotic fractures that occur annually could exceed 3 million in 2025, with health-care costs rising from $16.9 to $25.3 billion per year (44). Thus, there is a crucial need to define the key mechanisms responsible for age-related bone loss in order to identify more effective therapeutic approaches for preventing and treating osteoporosis.
p16Ink4a is significantly increased in osteoprogenitors, osteoblasts, and osteocytes from old mice compared with those from young mice (45). p21Cip1 is significantly increased predominantly in osteocytes (45). The percentage of senescent osteocytes in bone cortices in old mice is approximately sixfold higher than that in young mice, as measured by SADS. Expression of SASP factors, as measured by quantitative polymerase chain reaction (qPCR), is significantly increased in osteocytes but not in osteoblast progenitors or osteoblasts (45). The same SASP factors are upregulated in bone marrow–derived myeloid cells but not in lymphocytes from old mice relative to those from young mice (45). These findings suggest that within the bone microenvironment, senescent osteocytes and senescent myeloid cells may be the key producers of the SASP.
Genetic clearance of p16-expressing Sncs from old INK-ATTAC mice results in a 46% reduction in senescent osteocytes, as assessed by measuring SADS, and significantly improves trabecular bone, thicker cortices, and bone strength in the spine and femur (16). Thus, Sncs increase with age in bones of mice and contribute to osteoporosis, suggesting that senolytics might prove useful to treat this disease and prevent the risk of fractures in the elderly.
3.3. Skeletal Muscle
Humans achieve peak skeletal muscle mass and strength in midlife and then experience a progressive decline of up to 50% by the ninth decade. This reduction in skeletal muscle mass due to aging is termed sarcopenia, from the Greek sarx (flesh) and penia (poverty). Sarcopenia is evident in up to 25% of community-dwelling individuals (46) and reflects a reduction in the cross-sectional area and the number of muscle fibers and the accumulation of fat and connective tissue. As recently reviewed (47), sarcopenia leads to age-related functional limitations (e.g., difficulty walking and climbing stairs), falls, disability, loss of independence, and even death. Sarcopenia is also a major contributor to the loss of physical resilience with advancing age. Older persons with low muscle mass experience higher rates of surgical complications and infections (48), delayed recovery (49), chemotherapy toxicity (50), and disease-specific and all-cause mortality (51). Unfortunately, the consequences of age-related muscle loss are far clearer than the causes. As a result, there are currently no approved pharmacologic therapies.
Several lines of evidence suggest that cellular senescence may contribute to skeletal muscle aging. Transplantation of senescent syngeneic preadipocytes or autologous ear fibroblasts into young adult mice impairs grip strength and measures of physical function, including walking speed and hanging endurance (17). Correspondingly, systemic elimination of p16-expressing cells in INK-ATTAC mice, or elimination of Sncs through administration of senolytic drugs, modestly improves measures of physical function in older mice. Additional research is needed to determine the extent to which the detrimental effects of Sncs on parameters of muscle performance and physical function, or the salutary effects of their removal, are caused by alterations in skeletal muscle aging (i.e., atrophy, fibrosis, denervation, vascularization, or cellular composition). Indeed, reductions in Snc burden in tissues other than skeletal muscle may influence systemic health and in turn integrative measures of function. This concept is supported by a study of aged mice demonstrating that pharmacologic inhibition of the JAK pathway, which regulates the SASP in senescent preadipocytes and endothelial cells in vitro, suppresses markers of both adipose tissue and systemic inflammation and improved parameters of muscle strength, endurance, and coordination (39). Correspondingly, in older humans, the number of p16INK4a-expressing cells in thigh adipose tissue is negatively associated with performance-based measures of strength and walking ability (52).
Whether cell populations within the skeletal muscle are prone to becoming senescent remains to be determined. Skeletal muscle is a complex tissue composed of postmitotic multinucleated fibers and several functionally diverse mononuclear cell types. Satellite cells are the quintessential skeletal muscle stem cell. Satellite cells derived from geriatric mice (28–32 months of age) exhibit elevated expression of p16Ink4a (53). Following skeletal muscle injury in young mice, transplanted satellite cells from geriatric mice are unable to become activated and proliferate. Targeted repression of p16Ink4a, however, restores satellite cell activation in vitro and augments stem cell engraftment and self-renewal in injured tissue in vivo. The longer-term effects of p16Ink4a-expressing cells on sarcopenia, muscle fibrosis, fat infiltration, and measures of muscle performance have not been carefully investigated. In part, this is a challenge because sarcopenia is a relatively late phenomenon in mice, whereas it is progressive in humans beyond the fourth decade of life. As emphasized, aged muscle is compositionally heterogeneous, and senescence of resident cell populations beyond satellite cells, including fibroadipogenic progenitors, endothelial cells, and immune cells, may also contribute to muscle aging. Indeed, key SASP components (cytokines, growth factors, and extracellular matrix proteins and remodelers) are produced by fibroadipogenic progenitors, endothelial cells, and resident immune cells in skeletal muscle, as well as from invading monocytes that differentiate into macrophages (54). Dysregulation of the niche has been implicated in impaired satellite cell proliferation and differentiation, increased fibrosis, and fat accumulation, at least in the context of muscle regeneration. Consequently, there is an unmet need to identify and comprehensively phenotype the cell populations within aged muscle that are prone to becoming senescent and to determine the extent to which those Sncs mechanistically contribute to muscle loss and dysfunction.
3.4. Brain
Aging is a major risk factor for the development of many neurodegenerative diseases, including Alzheimer’s disease (AD) and Parkinson’s disease (PD). In 2010, there were an estimated 4.7 million individuals age 65 or older in the United States with AD and this number is projected to increase to 13.8 million (with 7 million of these individuals aged 85 years or older) in 2050 (55). The financial costs of treating dementia are substantially higher than the costs of treating other age-related diseases (56). Furthermore, most clinical trials have been unsuccessful, which suggests that new treatment options are desperately needed.
Recent studies demonstrated that Sncs accumulate with age in the mouse brain, as measured by expression of TAFs and p21Cip1. Neurons microdissected from postmortem human brain with AD exhibit increased expression of senescence markers (57). Importantly, clearance of Sncs either genetically or pharmacologically in mice leads to functional improvements in models of neurodegenerative diseases such as PD and AD (57–59).
An emerging concept is that obesity may accelerate the onset of age-related diseases and therefore may pose a major risk for early onset of neurodegenerative diseases such as AD. Diet-induced obesity contributes to early memory impairment and amyloid-β or tau neuropathology in mouse models of AD (60). Obesity accelerates the accumulation of Sncs in multiple tissues (15), including the brain. There is also a causal link between senescence and neuropsychiatric disorders, such as anxiety, in the context of obesity (34). Sncs accumulate in glial cells and neurons in different brain regions of both obese and aged mice. Importantly, clearance of Sncs, by either genetic or pharmacologic approaches, restores neurogenesis and significantly decreases obesity-induced anxiety-like behavior. Senescence has been observed in neurons, astrocytes, microglia, oligodendrocytes, and oligodendrocyte progenitor cells (57–59). However, the relative contribution of each cell type to neurodegenerative diseases is not currently known.
3.5. Cardiovascular System
While the size of the population over the age of 65 has dramatically increased in recent years, our understanding of biological contributors to age-related dysfunction of the systemic vasculature has not kept pace. Aging is associated with progressive increases in atherosclerotic burden, vascular stiffening and fibrosis, vascular calcification, and losses in organ capillary density. Changes in conduit and peripheral vascular structure and function are strongly predictive of end-organ dysfunction, morbidity, and mortality. Furthermore, development of truly innovative classes of drugs (i.e., drugs that do not target risk factors such as blood pressure or blood lipids) has remained stagnant for over a decade. Identification of novel mechanisms contributing to age-related cardiovascular disease could result in transformative treatments to reduce cardiovascular morbidity and mortality with aging and improve multiorgan system function with aging, thereby reducing health-care costs and improving quality of life in elderly patients. As cardiovascular disease continues to be the leading cause of death in the United States, development of new and transformative therapeutic strategies to prevent age-associated cardiovascular dysfunction is critical.
Emerging data suggest the increased Snc burden influences age-associated cardiovascular dysfunction via three major mechanisms: (a) by augmenting known risk factors for cardiovascular disease, including systemic inflammation, type 2 diabetes, and subsequent hyperglycemia (all of which are strong predictors of cardiovascular morbidity); (b) by altering local paracrine signaling from senescent endothelial and inflammatory cells in cardiovascular microenvironments, including reduced nitric oxide bioavailability and impaired endothelial function (which are strong predictors of all-cause mortality) (61); and (c) by promoting redifferentiation of local cells to osteoblast-like or synthetic cell phenotypes (62), both of which favor matrix disruption and excess/alternate matrix production and are mechanistic contributors to vascular stiffening, hypertension, and resultant end-organ failure. Interventions that reduce Snc burden can dramatically attenuate arterial calcification, improve endothelial function and nitric oxide signaling (12), and improve survival after major cardiovascular insults, which suggests that Sncs play an important role in cardiovascular morbidity and mortality.
3.6. Liver
Nonalcoholic fatty liver disease (NAFLD) is characterized by excess hepatic fat (steatosis) in individuals who drink little or no alcohol and is more prevalent in older and obese populations. NAFLD encompasses steatosis, nonalcoholic steatohepatitis, advanced fibrosis, cirrhosis, and hepatocellular carcinoma. Cellular senescence is implicated in the progression of liver disease. Telomere dysfunction, a major driver of hepatocyte senescence, impairs liver regeneration and induces liver cirrhosis in mice (63). Furthermore, chronic inflammation driven by the SASP spreads hepatocyte senescence, contributing to liver fibrosis (64) and hepatocellular carcinoma (65). Increased hepatocyte Snc burden is observed during aging and obesity in mice (15) and in chronic parenchymal liver disorders in humans such as chronic viral hepatitis (66), alcohol-related liver disease (65), NAFLD (15, 67), and genetic hemochromatosis (68). Induction of senescence in hepatocytes results in fat accumulation in vitro and in vivo (15). Elimination of Sncs by genetic ablation of p16Ink4a-expressing cells or by the senolytic drug cocktail D+Q reduces hepatic steatosis. These studies suggest that elimination of Sncs may be a therapeutic strategy to alleviate NAFLD.
4. APPROACHES TO IDENTIFY SENOLYTICS
Senolytics were initially discovered through a hypothesis-driven, bioinformatics-informed approach and subsequently by screening of drug libraries.
4.1. Hypothesis-Driven, Mechanism-Based Approach
Traditional approaches were not initially successful for developing senolytics, so hypothesis-driven discovery was implemented to discover the first drugs. We asked how Sncs expressing a SASP can survive, despite their own proapoptotic and damaging metabolic milieu. We hypothesized that (a) Sncs resist apoptotic stimuli, implying existence of prosurvival/antiapoptotic defenses against their own SASP and harsh metabolic internal state, and that (b) in some respects, including apoptosis resistance and metabolic shifts, Sncs are like cancer cells that do not divide. From a bioinformatic analysis of proteomic and transcriptomic data from Sncs versus non-Sncs, we identified several senescence cell antiapoptotic pathways (SCAPs) (19), including ephrins/dependence receptors; PI3Kδ/AKT/metabolic; BCL-2/BCL-xL/BCL-W; p53/FOXO4a/p21/serpin [plasminogen activator inhibitors 1 and 2 (PAI-1, PAI-2)]; hypoxia-inducible factor 1α (HIF-1α); and the HSP90 pathway. To prove that SCAPs are essential for Snc survival, we targeted the SCAP network nodes with siRNAs in senescent versus nonsenescent human primary adipocyte progenitors and human umbilical vascular endothelial cells (HUVECs) to determine whether the SCAPs impede apoptosis. SCAPs differ between cell types; for example, senescent adipocyte progenitors do not depend on BCL-2 family proteins, whereas senescent HUVECs depend on BCL-xL, PI3K, and the HIF-1α pathways. We next used bioinformatics approaches to identify drugs and natural products that act on critical SCAP nodes. The first senolytic drug reported was D+Q (19). The combination of D+Q is more effective than either agent alone. On the basis of our finding that BCL-xL is a SCAP, 10 months later, we and others reported that navitoclax, a BCL-2 family inhibitor, is senolytic in some but not all Snc types (26). This mechanism-based, hypothesis-driven approach led to the identification of more senolytic drugs, including fisetin and related natural products, other BCL-2 family inhibitors A1331852 and A1155463. A list of senolytics reported to date is provided in Table 1.
4.2. Drug Screening
As an alternative to the bioinformatics approach, drug libraries can be screened for drugs that have the ability to selectively kill a variety of Snc types (Figure 3). For example, libraries have been screened on oncogene-induced senescence using IMR-90 cells (primary human fibroblasts) with suppression of SA-βgal activity and cell proliferation as readouts (69) and for ionizing radiation-induced senescence by enzyme-linked immunosorbent assay (ELISA) to analyze secretion of the SASP factor IL-6 (70). Similarly, screening for kinases that regulate senescence in human telomerase reverse transcriptase immortalized-retinal pigment epithelial 1 (hTERT-RPE1) cells using siRNA also revealed several key kinases important for regulating senescence, including a target for dasatinib (71). We developed a senotherapeutic screening assay using primary DNA-repair-deficient mouse embryonic fibroblasts (MEFs) (Figure 3). The rationale for using these MEFs is that senescence occurs rapidly when the cells are grown at atmospheric oxygen (O2) as a consequence of unrepaired oxidative DNA damage, which is a physiologically relevant driver of senescence in vivo (72). By passage 5 at 21% O2, roughly half of the MEFs are SA-βgal positive, allowing us to test selective killing of Sncs, but not of proliferating cells, in a single well using C12FDG (5-dodecanoylaminofluorescein-di-β-d-galactopyranoside), a fluorescent substrate for SA-βgal used to measure Snc number. The cells are counterstained with Hoechst to quantitate the total number of viable cells. An IN Cell Analyzer 6000, a laser-based line-scanning confocal imager with a large field-of-view sCMOS (scientific complementary metal–oxide–semiconductor) camera detection technology, can be used to quantitate the cells and the C12FDG-positive Sncs.
Figure 3.
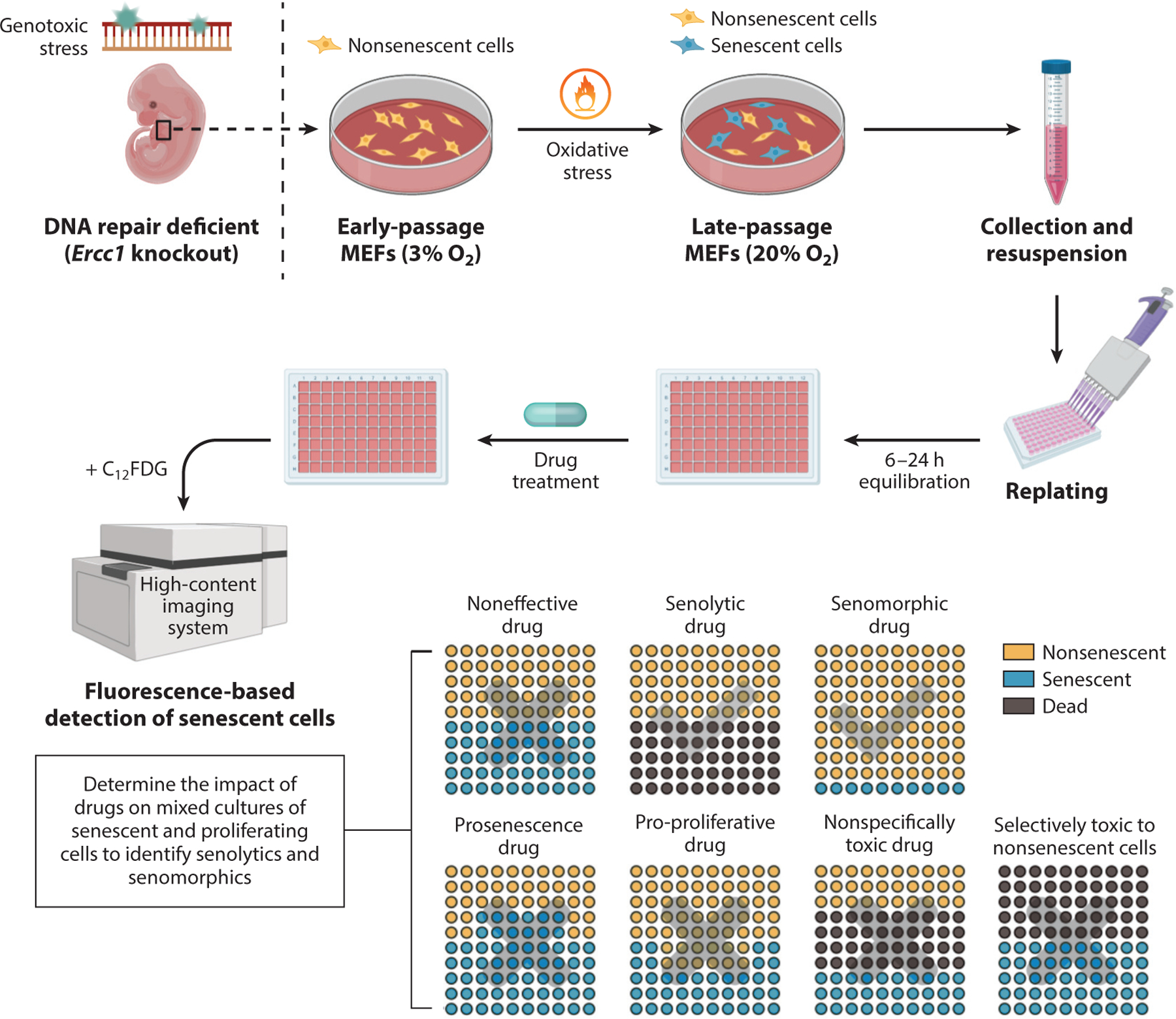
Senescent MEF-based screening assay. MEFs deficient in the DNA repair endonuclease ERCC1-XPF are used to increase physiologically relevant, oxidative stress–induced genotoxic stress and thereby senescence. Greater than 50% of these primary MEFs become SA-βgal positive by passage 5–6 at 20% O2. These cells can then be aliquoted onto a 96- or 384-well plate for drug screening. After drug application, the total number of cells (DAPI-positive nuclei), the number of cells positive for SA-βgal (C12FDG fluorescence), and cell morphology are measured in a high-content fluorescent plate reader. The use of this approach facilitates the identification of senolytic drugs, by which there is a reduction in the number of Sncs preferentially, and senomorphic drugs, by which there is no reduction in the number of cells but a reduction in SA-βgal-positive cells. It is also possible to identify drugs that increase the percent of Sncs on the plate without affecting cell number (prosenescent drugs) and drugs that increase the total number of cells on the plate (pro-proliferative drugs). This approach can address the specificity of the drugs for Sncs over non-Sncs. Subsequent assays are needed to validate the senolytic activity of the hits identified in this screen because some compounds may affect the detection of SA-βgal activity. Figure adapted, with permission, from the original figure by Dr. Rajesh Vyas. Abbreviations: C12FDG, 5-dodecanoylaminofluorescein-di-β-d-galactopyranoside; DAPI, 4′,6-diamidino-2-phenylindole; MEF, mouse embryonic fibroblast; SA-βgal, senescence-associated β-galactosidase; Snc, senescent cell.
We validated the MEF assay by screening small libraries of compounds. For example, screening a library of drugs that regulate autophagy identified rapamycin and rapamycin derivatives known as rapalogs as senomorphic and the HSP90 inhibitors geldanamycin and tanespimycin (17-AAG) as senolytic. Alvespimycin (17-DMAG) and other HSP90 inhibitors were also found to be senolytic. The senolytic activity of D+Q, fisetin, navitoclax, and other molecules was confirmed in the MEF assay.
4.3. Further Development of Senolytics
What is surprising about the studies to identify senotherapeutic drugs is the number of hits obtained in screens of relatively small libraries. Indeed, many anticancer drugs as well as natural products have some senotherapeutic activity in vitro. The question, then, is which hits to develop further. We intentionally focused our efforts on drugs that (a) are already FDA approved or are natural products with a history of safe use in humans, (b) can be administered orally, (c) cross the blood-brain barrier, and (d) have a short elimination half-life (t½). Initially, dasatinib, quercetin, and fisetin meet these criteria: Dasatinib has been approved for clinical use since 2006. Quercetin and fisetin are natural products with favorable safety profiles. All are effective orally and their elimination t½ is less than 4 h (73, 74) (i.e., they are cleared from the human body in less than 24 h).
The target of senolytics is Sncs, not a single receptor, enzyme, or biochemical pathway. When prosurvival networks instead of single molecules are targeted, specificity for Sncs can be increased, side effect profiles flattened, and off-target effects on non-Sncs reduced. Drugs with single or limited targets, such as Nutlin3a (an MDM2 inhibitor) or navitoclax (a BCL-2 inhibitor), can have off-target apoptotic effects on multiple non-Snc types, such as neutrophils and platelets in the case of navitoclax (75), making them panolytic. Also, navitoclax eliminates only a restricted range of Sncs and is not senolytic against, for example, human adipocyte progenitors, one of the most abundant Snc types in aged humans or in diabetes and obesity (27), and is not senolytic against human astrocytes. Rather than the usual one-target/one-drug/one-disease drug development paradigm, our approaches for developing senolytics have more in common with strategies for developing antibiotic regimens, in which the target is a cell type (not a single molecule), several agents in combination may be more effective than a single compound, and effects are anticipated for numerous disorders rather than for a single disease caused by that pathogen (e.g., skin infections, pneumonias, and urinary tract infections in the case of antibiotics).
4.4. Senolytic Optimization
Although the bioinformatic analysis and Snc screening assay have identified compounds that are senolytic, it is likely that their activity and selectivity for Sncs can be improved. For example, the off-target apoptotic effects of navitoclax on multiple non-Snc types have limited its use systemically. However, the therapeutic window of navitoclax is improved by utilizing an approach that degrades certain BCL-2 family members instead of just inhibiting their function (76). Proteolysistargeting chimera (PROTAC) is an approach that links a compound to an E3 ligase–targeting moiety (Figure 4a). Binding of the compound to its target quickly and efficiently results in polyubiquitination and degradation of the target protein. The use of a navitoclax PROTAC facilitated the rapid and specific degradation of BCL-2, resulting in apoptosis of cancer cells with less off-target lysis.
Figure 4.
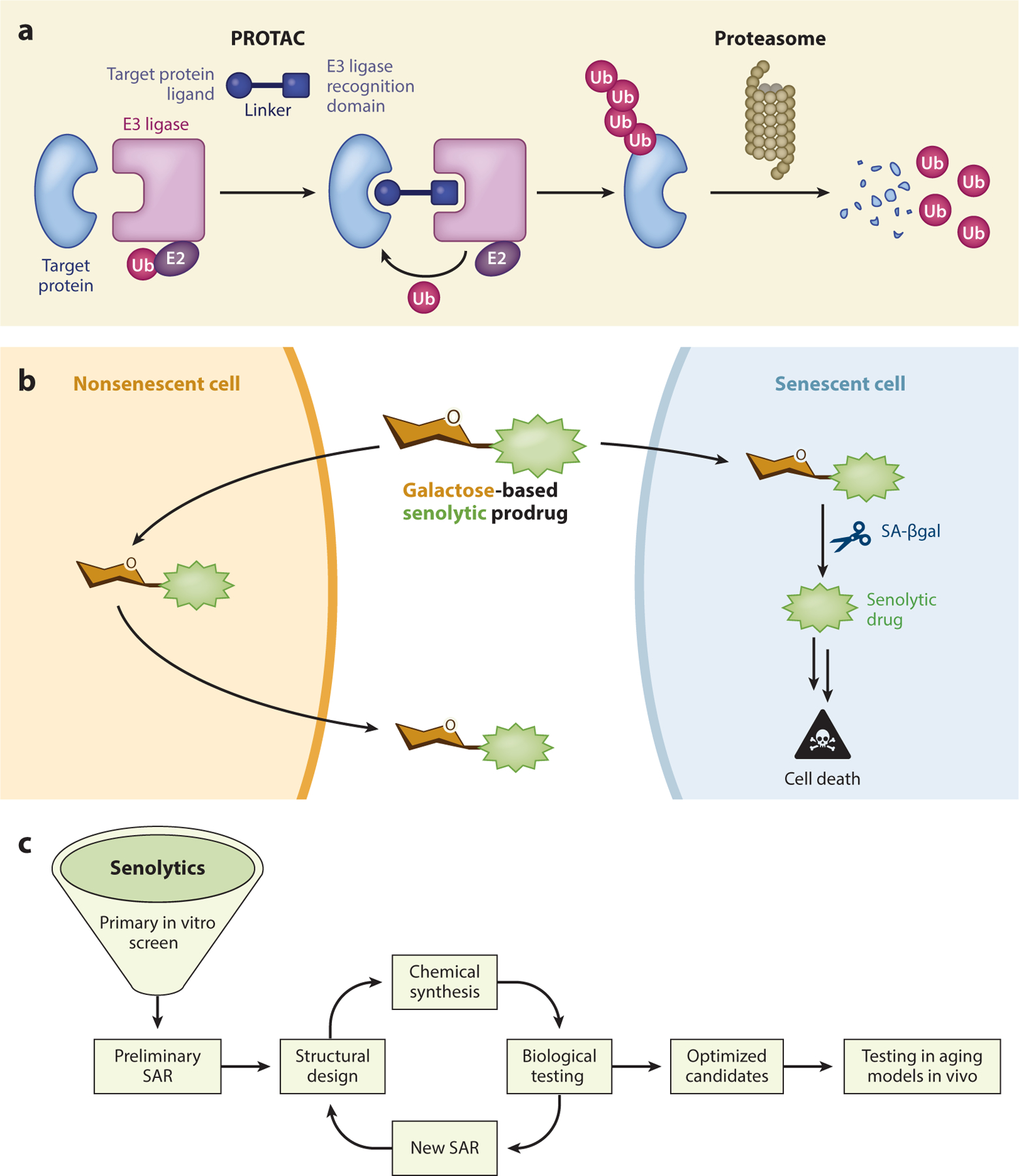
Development of novel approaches to selectively kill Sncs. (a) Selectively targeting specific SCAP factors can be enhanced by PROTAC. Here, a drug that binds a SCAP is linked directly to an E3 ligase–targeting moiety to direct rapid and efficient Ub-dependent proteasomal degradation of the SCAP, rendering the Snc vulnerable to apoptosis. (b) To target a cytotoxic drug specifically to Sncs, one approach involves linking a cytotoxic agent to galactoside, which can be cleaved by lysosomal β-galactosidase, the enzyme selectively increased in Sncs (SA-βgal) to release an active toxin. (c) Optimizing senolytic activity by SAR. To optimize the activity of a senolytic, a series of analogs can be generated for testing in different Snc assays. Additional rounds of SAR can be performed to optimize senolytic activity and to improve the drug-like properties of the senolytic. Figure adapted, with permission, from the original figure by Dr. Lei Zhang and Dr. Carolina Soto Palma. Abbreviations: PROTAC, proteolysis-targeting chimera; SA-βgal, senescence-associated β-galactosidase; SAR, structure-activity relationship; SCAP, senescence-associated antiapoptotic pathway; Snc, senescent cell; Ub, ubiquitin.
Another approach to improve selectivity is by generating senolytic prodrugs. Here, linking galactose to a cytotoxic compound, in theory, will be preferentially processed in Sncs by SA-βgal, resulting in their selective killing (Figure 4b). The feasibility of this approach has been documented by generating duocarmycin and navitoclax fused to galactose (77). Both of these prodrugs induced selective apoptosis of Sncs that was dependent on the presence of lysosomal β-galactosidase.
Finally, medicinal chemistry can be applied to any of the senolytic compounds to improve their selectivity for a specific type of Snc (Figure 4c). By going through multiple rounds of structure-activity relationship (SAR), senolytic compounds can be made not only more effective at killing Sncs specifically but also more drug-like with improved bioavailability. We have used medicinal chemistry to increase the therapeutic window of several senolytic compounds.
5. MODELS FOR PRECLINICAL TESTING OF SENOLYTICS
Whereas demonstrating senolytic activity in vitro is relatively easy, demonstrating a drug is senolytic in vivo is difficult. Simply measuring a reduction in markers of senescence, such as cell cycle regulators, SASP factors, or SA-βgal activity, in a tissue does not necessarily prove that Sncs are being eliminated. More rigorous approaches, such as the injection of labeled Sncs followed by drug treatment, document that senolytic function in vivo is similar to that in vitro, killing that specific type of transplanted Snc. Eventually, improved methods for colocalizing apoptotic and senescence markers to the same cell in vivo, such as through the use of CyTOF, mass imaging, dual fluorescent reporters, or spatial genomics, will be needed to prove a compound is senolytic. Also, certain senolytics may be cell-type specific or tissue specific, so it will be important to examine multiple tissue and cell types for effects.
One approach to document senolytic activity in vivo is to demonstrate the ability of a drug to confer long-lasting effects after a short period of treatment. For example, we demonstrated the senolytic activity of D+Q by administering one application of D+Q to mice following hind limb irradiation (19). That single administration conferred a therapeutic benefit in terms of treadmill performance that endured for over 1 year. This finding is consistent with a hit-and-run mechanism in which the drug kills disease-causing cells, characteristic of senolytics, rather than inhibiting a molecular target, which requires continuous exposure to the therapeutic.
5.1. Murine Models
Obviously, aged mice can be used for testing the efficacy of senolytics. The advantages of this approach are studying the effects of drugs on physiologically produced Sncs and on the opportunity to initiate therapy late in life, which is viewed as the most translationally relevant use of senolytics. However, the use of aged mice is expensive and slow. Thus, researchers have used alternative approaches to accelerate aging such as whole-body irradiation, treatment with a DNA-damaging agent, or a high-fat diet. Several genetic models of human progeroid syndromes exist. The Ercc1−/Δ mouse has reduced expression of the ERCC1-XPF endonuclease, which is important for multiple types of DNA repair (78). Ercc1−/Δ mice spontaneously develop Sncs in multiple tissues as a result of an accumulation of endogenous DNA damage, mitochondrial dysfunction, and increased abundance of reactive oxygen species (72). Aged wild-type mice and progeroid Ercc1−/Δ mice accumulate Sncs in the same tissues and to the same level (79), making Ercc1−/Δ mice a good model for studying senolysis. The BubR1H/H mice are also progeroid and carry two hypomorphic alleles of the BUB1B gene, which encodes a core component of the mitotic spindle assembly checkpoint (BUBR1) (80). Human aging is associated with decreased expression of BUBR1 in multiple tissues (80). This BubR1H/H progeria model has accelerated and elevated superphysiologic levels of cellular senescence due to chromosome missegregation as well as increasing aneuploidy (8, 80).
5.2. Rat Models
The rat closely recapitulates human physiology and pathophysiology and can be used to study aspects of aging, including longevity, senescence-related pathology, and pharmacologic changes with increasing age. Compared with mouse physiology, rat physiology is easier to monitor and in general more similar to that of humans (81). The larger size of the rat compared with that of mouse provides advantages to a preclinical model in terms of the size of disease lesions and the feasibility of serial blood sampling and surgical procedures. Rats are models highly suited for studying human aging, and they spontaneously develop more age-related diseases common in humans (e.g., diabetes and sarcopenia) than mice do. There are now diverse inbred strains of rats that have unique characteristics of aging.
Rats are more amenable than mice to cognitive behavioral measurements, endocrine (e.g., glucose tolerance) testing, physical function measurements, and cardiovascular assessments. In addition, there are extensive historical data on physiological changes with aging in rats. Cells derived from multiple rat tissues have been used to study cellular senescence. Genetic modification technologies have advanced in the rat, and it is now possible to create rat models with transgenes that enable clearance of p16Ink4a-expressing cells (e.g., an INK-ATTAC rat line).
5.3. Human Tissue Explants
To determine whether senolytics could be effective in humans, researchers have used human tissue explants to prove senolytic efficacy in human tissues. Tissues most amenable to collection include adipose tissue, skin, and peripheral blood mononuclear cells. We demonstrated the effectiveness of this approach using greater omental adipose explants resected during surgery (26). The explant is cut into small pieces and then cultured either with or without a senolytic. The conditioned media can be collected for multiplex protein analysis and the tissue is analyzed for senescence markers by qPCR, SA-βgal staining, and analysis of TAFs. This approach can be expanded to brain and liver sections from cadavers or possibly to skin resected from the elderly.
6. CLINICAL TRIALS
Given that some of the first senolytics identified were either FDA-approved anticancer drugs or natural products, the path to the clinic was relatively straightforward. To date, there have been over one dozen FDA-approved clinical trials with the senolytics D+Q, fisetin, and UBX0101. D+Q and fisetin are being tested for the systemic treatment of multiple age-related conditions and diseases, including AD. UBX0101, an inhibitor of the p53–MDM2 interaction, is being used locally within the joint space for the treatment of osteoarthritis (see the Supplemental Appendix for a list of current clinical trials testing senolytics). The advantage of senolytic drugs is that they only have to be administered intermittently to yield a physiological impact. Thus, in theory, the safety profile of senolytics should be very good, even though they target and kill a subset of cells.
To date, only three trials have reported results. Results of the trial using UBX0101 to treat osteoarthritis have been disappointing to date (https://clinicaltrials.gov/ct2/show/NCT04229225). However, early studies with D+Q appear more promising. D+Q was used in an open-label phase I pilot study to treat subjects with diabetic kidney disease (https://clinicaltrials.gov/ct2/show/NCT02848131). Adipose tissue, skin biopsies, and blood were collected before and after the treatment and analyzed for markers of senescence and the SASP. The abundance of Sncs in adipose tissue and macrophage infiltration were decreased 11 days after the last of 3 daily doses of D+Q (82). A composite score of blood SASP factors was also reduced 11 days after completing the last of the 3 daily doses of D+Q. This result strongly suggests that D+Q decreases the Snc burden in humans. This trial is continuing to ascertain whether senolytics alleviate diabetes and its complications. An open-label, two-center study of intermittent D+Q treatment in patients with idiopathic pulmonary fibrosis (https://clinicaltrials.gov/ct2/show/NCT02874989) also was conducted to demonstrate the feasibility and safety of a senolytic clinical trial (83). Physical function such as gait speed and chair stand time improved in the treated group. Although not significant, there was a trend toward a reduction in certain SASP proteins in the serum of patients in the treated group. Clinical trials testing fisetin for chronic kidney disease (https://clinicaltrials.gov/ct2/show/NCT03325322), skeletal health (https://clinicaltrials.gov/ct2/show/NCT04313634), frailty (https://clinicaltrials.gov/ct2/show/NCT03675724), and osteoarthritis (https://clinicaltrials.gov/ct2/show/NCT04210986) are under way; no results have been reported yet. Finally, two trials testing fisetin for the prevention of disease progression and death in the elderly infected with SARS-CoV-2 (severe acute respiratory syndrome–coronavirus 2) were recently registered with the National Institutes of Health (https://clinicaltrials.gov/ct2/show/NCT04476953 and https://clinicaltrials.gov/ct2/show/NCT04537299) (see the sidebar titled Senolytics in Viral Infection). Taken together, however, these early results suggest that senolytic agents hold promise for treating age-related disease.
7. FUTURE DIRECTIONS
A question that must be addressed to prove the mechanism of action of senolytics and their potential disease applications is whether candidate drugs are truly senolytic for that particular disorder. We have proposed a modified set of Koch’s postulates (84) to address this question. To establish causality, we must determine
whether Sncs are present in animals or humans with the disorder;
whether individuals without Sncs have the disorder;
whether the disorder can be reproduced by inducing local accumulation of Sncs (e.g., by transplantation of Sncs or focal irradiation);
whether eliminating such transplanted or induced Sncs prevents or alleviates the disorder;
whether reducing abundance of naturally occurring Sncs alleviates the disorder;
whether the potential senolytic has few or no effects related to the disorder being tested when administered to individuals with few or no Sncs (e.g., young mice);
whether the potential senolytic alleviates the disorder even if it is given intermittently at intervals longer than the drug’s half-life, because Sncs can take 2–6 weeks to reaccumulate [e.g., monthly administration of D+Q is as effective as daily administration for age-related osteoporosis in mice (16)]; and
whether the candidate senolytic can alleviate multiple age-related or chronic disease-related disorders.
All eight criteria have been met for diabetes, frailty, and age-related osteoporosis in the case of D+Q, and several of the criteria have been met for osteoarthritis and neurodegenerative diseases. This modified set of Koch’s postulates needs to be tested for the new candidate senolytics in Table 1 and for several diseases and conditions believed to be caused by Sncs, particularly before these candidate drugs advance to clinical trials.
Development of drugs that selectively eliminate Sncs is potentially of profound significance. On the basis of our studies of genetically modified progeroid mice, aging mice, chronic disease models, and humans, the prototypical senolytic agents that are now in hand could presage a transformative shift in the management of age-related disorders, but much remains to be done. Senolytics may enhance health span and delay, prevent, or treat multiple chronic diseases, geriatric syndromes, and age-related declines in physical resilience, but this is not a certainty. More information about safety, tolerability, side effects, and target engagement (effectiveness in reducing Snc burden) is needed. Severe, unanticipated side effects could emerge. Unfortunately, there has been premature excitement about senolytics along with efforts to sell them to the public while safety and efficacy measures are still being evaluated. We are concerned about self-medication or physicians prescribing senolytics outside the context of clinical trials. It is imperative to comprehensively study effects and side effects of existing and candidate senolytics. At this juncture, we believe the use of senolytic drugs should be confined to carefully monitored placebo-controlled clinical trials.
8. FUTURE WORK
We must gain a greater understanding of what drives senescence in the human body in order to improve the physiological relevance of in vitro models for studying cellular senescence and developing senotherapeutics. Currently, detection of Sncs involves measuring multiple senescence markers in tissue lysates. It will be necessary to switch to analysis of single cells to fully characterize the heterogeneity of Sncs. This will require new tools and methods to detect Sncs in vivo and to isolate them. These same tools will facilitate clinical trials. Deep phenotyping of Sncs via genomics, proteomics, lipidomics, and metabolomics approaches is needed to better define senescence, the secretory phenotype of Sncs, the dynamic properties of Sncs, and their physiologic impact (good and bad). Senescence is detected in many tissues in vivo with aging, but it is not known which Sncs (lineage and location) drive aging and age-related diseases. Therefore, animal models in which senescence can be induced in a cell-type-and tissue-specific manner are needed in order to identify disease-causing Sncs, which are key targets for senolytics.
Similarly, animal models in which specific Sncs can be removed by cell-type-or tissue-specific expression of a suicide gene from a senescence-associated promoter (e.g., p16Ink4a, p21Cip1) are needed. The burden of Sncs in individuals varies widely and if measurable could be valuable for predicting risk of disease, frailty, or adverse outcomes in infection, surgery, or aggressive chemotherapy. It will be important to develop approaches that can measure the Snc burden of an individual using readily accessible biologic samples. The senotherapeutics identified to date are likely suboptimal with respect to activity, efficacy, and safety. Further, little is known about their specificity for certain cell and tissue types. Therefore, multiple approaches to optimize and identify novel senotherapeutics are needed and include (a) applying medicinal chemistry to known senolytics to improve senolytic/senomorphic activity; (b) screening FDA-approved drug libraries and libraries of natural products with multiple human cell types; (c) using bioinformatics to identify novel SCAPs, which represent discrete molecular targets for novel senolytics, in multiple types of Sncs; (d) developing prodrugs that are activated only in Sncs, for example, via hydrolysis by SA-βgal; (e) testing combinations of senotherapeutics and combinations of senotherapeutics with interventions that target other pillars of aging; and (f) determining the dynamics of Snc appearance in vivo to optimize intermittent dosing with senolytics. These will be key for optimizing the safety of senotherapeutics.
Clinical trials to test senotherapeutics must be particularly thoughtfully designed. This is because aging, frailty, and multimorbidity of old age are not FDA-recognized clinical end points that warrant therapeutic intervention. Diseases with Sncs as part of their pathophysiology are good surrogate primary outcomes to therapeutically target. Secondary and tertiary end points will be critical for learning more about the targets of senolytics (tissues, concurrent diseases), drug interactions, safety in the elderly, and pharmacodynamics.
Supplementary Material
SUMMARY POINTS.
Cellular senescence is a cell fate induced by numerous types of cell stress or damage that leads to irreversible loss of proliferative potential; resistance to cell death; increased metabolic activity; and a senescence-associated secretory phenotype (SASP), which can lead to sterile inflammation, loss of tissue homeostasis and stem cell function, fibrosis, and other adverse effects, although the SASP is also necessary for wound healing.
The accumulation of senescent cells (Sncs) contributes to driving aging and age-related diseases, including hepatic steatosis, diabetes, pulmonary fibrosis, osteoarthritis, osteoporosis, Alzheimer’s disease (AD), cardiovascular disease, chronic kidney disease, intervertebral disc degeneration, and macular degeneration.
Because Sncs are quite heterogeneous, multiple markers of senescence are needed to confirm whether a cell is senescent, especially in vivo. These markers include quantitation of senescence-associated β-galactosidase; elevated expression of p16INK4A, p21CIP1, and SASP factors; an increase in γH2AX foci, telomere-associated foci, senescence-associated distension of satellite DNA, and senescence-associated heterochromatin foci; loss of HMGB1 and Lamin B; and a change in cell morphology. However, not all Sncs have all of these markers.
Transgenic mice expressing suicide genes (INK-ATTAC and p16-3MR) with inducible activity were used to prove that p16-expressing Sncs play a causal role in aging and disease. This finding is consistent with findings resulting from the separate but parallel development of senolytic drugs and their use in aged mice and models of age-related diseases.
FDA-approved anticancer drugs, natural products, and new chemical entities have been identified as senolytics through the use of bioinformatic analysis of Snc profiles and drug screening. The first senolytic drugs reported were the combination of dasatinib and quercetin (D+Q), which were followed by navitoclax and other BCL-2 family member inhibitors, heat shock protein 90 inhibitors, and other natural products.
Senolytic drugs are currently being tested in more than 10 clinical trials for treating conditions in which Sncs play a key role in pathology, including frailty, chronic kidney disease, pulmonary fibrosis, and AD. Initial results suggest that D+Q reduces expression of senescence markers in adipose tissue, skin, and serum.
SENOLYTICS IN VIRAL INFECTION.
The recent COVID-19 (corona virus disease 2019) pandemic has highlighted the fact that the elderly and individuals with preexisting conditions are at a higher risk for adverse outcomes following acute exposure to pathogens, likely driven by cytokine storm. This raises the question whether it is a higher senescent cell (Snc) burden in the aged and those with underlying conditions such as obesity, type 2 diabetes, and chronic kidney disease that renders them more susceptible to pathogen-driven inflammation. We propose that pathogen-associated molecular pattern molecules (PAMPs), which are microbe-derived molecules that activate innate immunity via pattern recognition receptors, amplify the senescence-associated secretory phenotype of preexisting Sncs, resulting in a highly inflammatory, profibrotic secretome. If true, then senolytics could be used to prevent or treat individuals with Sncs to reduce morbidity and mortality following infection with pathogens such as SARS-CoV-2 (severe acute respiratory syndrome coronavirus 2)/COVID-19 or even the flu.
ACKNOWLEDGMENTS
This work was supported by National Institutes of Health grants P01 AG062412 (J.L.K., T.T., S.K., N.K.L., P.D.R., R.J.P., L.J.N.) P01 AG043376 (P.D.R., L.J.N.), R01 AG063543 (L.J.N.), R56 AG059676 (L.J.N.), R56 AG059675 (P.D.R.), U19 AG056278 (P.D.R., L.J.N.), R33 AG062018 (J.L.K., T.T., S.K., N.K.L., P.D.R., R.J.P., L.J.N.), R01 AG068048 (J.F.P., D.J., J.L.K.), and R37 AG 013925 (J.L.K., T.T.); the Robert and Arlene Kogod Center on Aging (J.L.K., T.T., D.J., S.K., N.K.L., J.D.M., J.F.P., R.J.P.); the Connor Group (J.L.K., T.T.); Robert J. and Theresa W. Ryan (J.L.K., T.T., J.D.M.); and the Noaber Foundation (J.L.K., T.T.). The authors would like to thank Drs. Rajesh Vyas, Lei Zhang, and Carolina Soto-Palma for providing figures and Mariah Witt and Makenna Ash for carefully proofreading an earlier draft of this article.
Glossary
- Cellular senescence
a cell fate caused by stress that typically includes irreversible loss of proliferative potential, resistance to apoptosis, increased metabolism, and a proinflammatory secretome
- Snc
senescent cell
- Senescence-associated β-galactosidase (SA-βgal)
a lysosomal enzyme catalyzing the hydrolysis of β-galactosides into monosaccharides detected at a high pH only in Sncs
- Senescence-associated heterochromatin foci (SAHFs)
heterochromatic DNA domains silencing expression of proliferation-promoting genes in Sncs; detected by immunostaining for γH2AX, HMGA1/2, HP1, and H3K9 methylation
- Senescence-associated secretory phenotype (SASP)
factors, including cytokines, chemokines, growth factors, metalloproteinases, certain lipids, and extracellular vesicles, secreted by Sncs
- Telomere-associated foci (TAFs)
DNA damage at telomeres measured by immunodetection of γH2AX and 53BP1 at telomeric DNA
- Senolytic/senostatic
an agent that specifically induces death of Sncs
- Senomorphic
an agent that suppresses phenotypes of senescence, including the SASP
- Dasatinib
a tyrosine kinase inhibitor used to treat leukemias; has senolytic activity, particularly in combination with quercetin
- Quercetin
a plant-derived flavonol found in many fruits, vegetables, leaves, seeds, and grains that has antioxidant and senolytic activity
- Fisetin
a plant-derived flavonol, similar to quercetin, found in fruits and vegetables that has senolytic and senomorphic activity
- Navitoclax (ABT263)
an orally active experimental anticancer drug that blocks the activity of BCL-2, BCL-xL, and BCL-W and is a senolytic
- Senescence-associated distension of satellite DNA (SADS)
the large-scale unraveling of periand centromeric satellite DNA observed in Sncs, detected by fluorescence in situ hybridization and DNA hypomethylation; indicative of derepression
- Senescence cell antiapoptotic pathway (SCAP)
a network or signaling pathway upregulated in Sncs to prevent cell death even though the cell is damaged and exposed to SASP
Footnotes
DISCLOSURE STATEMENT
J.L.K. and T.T. have a financial interest related to this research. Patents on senolytic drugs are held by the Mayo Clinic and the University of Minnesota. This research has been reviewed by the Mayo Clinic Conflict of Interest Review Board and was conducted in compliance with Mayo Clinic Conflict of Interest policies.
LITERATURE CITED
- 1.Kennedy BK, Berger SL, Brunet A, Campisi J, Cuervo AM, et al. 2014. Geroscience: linking aging to chronic disease. Cell 159:709–13 [DOI] [PMC free article] [PubMed] [Google Scholar]; Elaborates the geroscience hypothesis of therapeutically targeting fundamental processes of aging.
- 2.López-Otín C, Blasco MA, Partridge L, Serrano M, Kroemer G. 2013. The hallmarks of aging. Cell 153:1194–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.St Sauver JL, Boyd CM, Grossardt BR, Bobo WV, Finney Rutten LJ, et al. 2015. Risk of developing multimorbidity across all ages in an historical cohort study: differences by sex and ethnicity. BMJ Open 5:e006413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Olshansky SJ. 2013. Life expectancy and education: the author replies. Health Aff. 32:822. [DOI] [PubMed] [Google Scholar]
- 5.Tchkonia T, Zhu Y, van Deursen J, Campisi J, Kirkland JL. 2013. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J. Clin. Investig 123:966–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alcorta DA, Xiong Y, Phelps D, Hannon G, Beach D, Barrett JC. 1996. Involvement of the cyclin-dependent kinase inhibitor p16 (INK4a) in replicative senescence of normal human fibroblasts. PNAS 93:13742–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coppe JP, Desprez PY, Krtolica A, Campisi J. 2010. The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol. Mech. Dis 5:99–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, et al. 2011. Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479:232–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, et al. 2016. Naturally occurring p16Ink4a-positive cells shorten healthy lifespan. Nature 530:184–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeon OH, Kim C, Laberge R-M, Demaria M, Rathod S, et al. 2017. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat. Med 23:775–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu M, Palmer AK, Ding H, Weivoda MM, Pirtskhalava T, et al. 2015. Targeting senescent cells enhances adipogenesis and metabolic function in old age. eLife 4:e12997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roos CM, Zhang B, Palmer AK, Ogrodnik MB, Pirtskhalava T, et al. 2016. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 15:973–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, van Deursen JM. 2016. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 354:472–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, et al. 2017. Cellular senescence mediates fibrotic pulmonary disease. Nat. Commun 8:14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ogrodnik M, Miwa S, Tchkonia T, Tiniakos D, Wilson CL, et al. 2017. Cellular senescence drives age-dependent hepatic steatosis. Nat. Commun 8:15691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Farr JN, Xu M, Weivoda MM, Monroe DG, Fraser DG, et al. 2017. Targeting cellular senescence prevents age-related bone loss in mice. Nat. Med 23:1072–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, et al. 2018. Senolytics improve physical function and increase lifespan in old age. Nat. Med 24:1246–56 [DOI] [PMC free article] [PubMed] [Google Scholar]; The senolytic D+Q improves frailty and physical function even when administered late in life.
- 18.Krishnamurthy J, Torrice C, Ramsey MR, Kovalev GI, Al-Regaiey K, et al. 2004. Ink4a/Arf expression is a biomarker of aging. J. Clin. Investig 114:1299–307 [DOI] [PMC free article] [PubMed] [Google Scholar]; First demonstration of the link between Snc accumulation and health span/life span.
- 19.Zhu Y, Tchkonia T, Pirtskhalava T, Gower AC, Ding H, et al. 2015. The Achilles’ heel of senescent cells: from transcriptome to senolytic drugs. Aging Cell 14:644–58 [DOI] [PMC free article] [PubMed] [Google Scholar]; The first report of a senolytic drug (D+Q) and its efficacy in reversing physical decline caused by Sncs.
- 20.Fuhrmann-Stroissnigg H, Ling YY, Zhao J, McGowan SJ, Zhu Y, et al. 2017. Identification of HSP90 inhibitors as a novel class of senolytics. Nat. Commun 8:422. [DOI] [PMC free article] [PubMed] [Google Scholar]; Establishes a screening tool for senolytic drugs that is physiologically relevant and yields HSP90 inhibitors as senolytics.
- 21.Wang Y, Chang J, Liu X, Zhang X, Zhang S, et al. 2016. Discovery of piperlongumine as a potential novel lead for the development of senolytic agents. Aging 8:2915–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu Y, Doornebal EJ, Pirtskhalava T, Giorgadze N, Wentworth M, et al. 2017. New agents that target senescent cells: the flavone, fisetin, and the BCL-XL inhibitors, A1331852 and A1155463. Aging 9:955–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Demaria M, Ohtani N, Youssef Sameh A, Rodier F, Toussaint W, et al. 2014. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev. Cell 31:722–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Laberge R-M, Sun Y, Orjalo AV, Patil CK, Freund A, et al. 2015. MTOR regulates the pro-tumorigenic senescence-associated secretory phenotype by promoting IL1A translation. Nat. Cell Biol 17:1049–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kanigur Sultuybek G, Soydas T, Yenmis G. 2019. NF-κB as the mediator of metformin’s effect on ageing and ageing-related diseases. Clin. Exp. Pharmacol. Physiol 46:413–22 [DOI] [PubMed] [Google Scholar]
- 26.Yousefzadeh MJ, Zhu Y, McGowan SJ, Angelini L, Fuhrmann-Stroissnigg H, et al. 2018. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 36:18–28 [DOI] [PMC free article] [PubMed] [Google Scholar]; Discovers that fisetin extends healthy aging in old mice and triggers senolysis in human adipose tissue.
- 27.Zhu Y, Tchkonia T, Fuhrmann-Stroissnigg H, Dai HM, Ling YY, et al. 2015. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 15(3):428–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chang J, Wang Y, Shao L, Laberge R-M, Demaria M, et al. 2016. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med 22:78–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Baar MP, Brandt RM, Putavet DA, Klein JD, Derks KW, et al. 2017. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell 169:132–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, et al. 2019. Cellular senescence: defining a path forward. Cell 179:813–27 [DOI] [PubMed] [Google Scholar]
- 31.Hernandez-Segura A, de Jong TV, Melov S, Guryev V, Campisi J, Demaria M. 2017. Unmasking transcriptional heterogeneity in senescent cells. Curr. Biol 27:2652–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muñoz-Espín D, Cañamero M, Maraver A, Gómez-López G, Contreras J, et al. 2013. Programmed cell senescence during mammalian embryonic development. Cell 155:1104–18 [DOI] [PubMed] [Google Scholar]
- 33.Hall BM, Balan V, Gleiberman AS, Strom E, Krasnov P, et al. 2016. Aging of mice is associated with p16(Ink4a)- and β-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging 8:1294–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ogrodnik M, Zhu Y, Langhi LGP, Tchkonia T, Krüger P, et al. 2019. Obesity-induced cellular senescence drives anxiety and impairs neurogenesis. Cell Metab. 29:1061–77.e8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anderson R, Lagnado A, Maggiorani D, Walaszczyk A, Dookun E, et al. 2019. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 38(5):e100492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ma S, Sun S, Geng L, Song M, Wang W, et al. 2020. Caloric restriction reprograms the single-cell transcriptional landscape of Rattus norvegicus aging. Cell 180:984–1001 [DOI] [PubMed] [Google Scholar]
- 37.Kirkland JL, Cartwright M, Tchkonia T, Lenburg M, Schlauch K, et al. 2007. Aging, fat depot origin, and preadipocyte expression profiles: setting the stage for fat tissue dysfunction. Int. J. Obes 31(Suppl. 1):S18 [Google Scholar]
- 38.Olefsky JM, Glass CK. 2010. Macrophages, inflammation, and insulin resistance. Annu. Rev. Physiol 72:219–46 [DOI] [PubMed] [Google Scholar]
- 39.Xu M, Tchkonia T, Ding H, Ogrodnik M, Lubbers ER, et al. 2015. JAK inhibition alleviates the cellular senescence-associated secretory phenotype and frailty in old age. PNAS 112:E6301–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zaragosi LE, Wdziekonski B, Villageois P, Keophiphath M, Maumus M, et al. 2010. Activin a plays a critical role in proliferation and differentiation of human adipose progenitors. Diabetes 59:2513–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Palmer AK, Gustafson B, Kirkland JL, Smith U. 2019. Cellular senescence: at the nexus between ageing and diabetes. Diabetologia 62:1835–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Palmer AK, Xu M, Zhu Y, Pirtskhalava T, Weivoda MM, et al. 2019. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 18(3):e12950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wright NC, Looker AC, Saag KG, Curtis JR, Delzell ES, et al. 2014. The recent prevalence of osteoporosis and low bone mass in the United States based on bone mineral density at the femoral neck or lumbar spine. J. Bone Miner. Res 29:2520–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Burge R, Dawson-Hughes B, Solomon DH, Wong JB, King A, Tosteson A. 2007. Incidence and economic burden of osteoporosis-related fractures in the United States, 2005–2025. J. Bone Miner. Res 22:465–75 [DOI] [PubMed] [Google Scholar]
- 45.Farr JN, Fraser DG, Wang H, Jaehn K, Ogrodnik MB, et al. 2016. Identification of senescent cells in the bone microenvironment. J. Bone Miner. Res 31:1920–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Baumgartner RN, Koehler KM, Gallagher D, Romero L, Heymsfield SB, et al. 1998. Epidemiology of sarcopenia among the elderly in New Mexico. Am. J. Epidemiol 147:755–63 [DOI] [PubMed] [Google Scholar]
- 47.Aversa Z, Zhang X, Fielding RA, Lanza I, LeBrasseur NK. 2019. The clinical impact and biological mechanisms of skeletal muscle aging. Bone 127:26–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Krell RW, Kaul DR, Martin AR, Englesbe MJ, Sonnenday CJ, et al. 2013. Association between sarcopenia and the risk of serious infection among adults undergoing liver transplantation. Liver Transpl. 19:1396–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lieffers JR, Bathe OF, Fassbender K, Winget M, Baracos VE. 2012. Sarcopenia is associated with postop-erative infection and delayed recovery from colorectal cancer resection surgery. Br. J. Cancer 107:931–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Prado CM, Baracos VE, McCargar LJ, Reiman T, Mourtzakis M, et al. 2009. Sarcopenia as a determinant of chemotherapy toxicity and time to tumor progression in metastatic breast cancer patients receiving capecitabine treatment. Clin. Cancer Res 15:2920–26 [DOI] [PubMed] [Google Scholar]
- 51.Psutka SP, Carrasco A, Schmit GD, Moynagh MR, Boorjian SA, et al. 2014. Sarcopenia in patients with bladder cancer undergoing radical cystectomy: impact on cancer-specific and all-cause mortality. Cancer 120:2910–18 [DOI] [PubMed] [Google Scholar]
- 52.Justice JN, Gregory H, Tchkonia T, LeBrasseur NK, Kirkland JL, et al. 2017. Cellular senescence biomarker p16INK4a+ cell burden in thigh adipose is associated with poor physical function in older women. J. Gerontol. A Biol. Sci. Med. Sci 73:939–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sousa-Victor P, Perdiguero E, Muñoz-Cánoves P. 2014. Geroconversion of aged muscle stem cells under regenerative pressure. Cell Cycle 13:3183–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bentzinger CF, Wang YX, Dumont NA, Rudnicki MA. 2013. Cellular dynamics in the muscle satellite cell niche. EMBO Rep. 14:1062–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hebert LE, Weuve J, Scherr PA, Evans DA. 2013. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology 80:1778–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kelley AS, McGarry K, Gorges R, Skinner JS. 2015. The burden of health care costs for patients with dementia in the last 5 years of life. Ann. Intern. Med 163:729–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Musi N, Valentine JM, Sickora KR, Baeuerle E, Thompson CS, et al. 2018. Tau protein aggregation is associated with cellular senescence in the brain. Aging Cell 17(6):e12840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, Baker DJ. 2018. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 562:578–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhang P, Kishimoto Y, Grammatikakis I, Gottimukkala K, Cutler RG, et al. 2019. Senolytic therapy alleviates Aβ-associated oligodendrocyte progenitor cell senescence and cognitive deficits in an Alzheimer’s disease model. Nat. Neurosci 22:719–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Julien C, Tremblay C, Phivilay A, Berthiaume L, Émond V, et al. 2010. High-fat diet aggravates amyloidbeta and tau pathologies in the 3xTg-AD mouse model. Neurobiol. Aging 31:1516–31 [DOI] [PubMed] [Google Scholar]
- 61.McEwen JE, Zimniak P, Mehta JL, Shmookler Reis RJ. 2005. Molecular pathology of aging and its implications for senescent coronary atherosclerosis. Curr. Opin. Cardiol 20:399–406 [DOI] [PubMed] [Google Scholar]
- 62.Burton DG, Matsubara H, Ikeda K. 2010. Pathophysiology of vascular calcification: pivotal role of cellular senescence in vascular smooth muscle cells. Exp. Gerontol 45:819–24 [DOI] [PubMed] [Google Scholar]
- 63.Rudolph D, Yeh W-C, Wakeham A, Rudolph B, Nallainathan D, et al. 2000. Severe liver degeneration and lack of NF-κB activation in NEMO/IKKγ-deficient mice. Genes Dev. 14:854–62 [PMC free article] [PubMed] [Google Scholar]
- 64.Jurk D, Wilson C, Passos JF, Oakley F, Correia-Melo C, et al. 2014. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat. Commun 2:4172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wilson CL, Jurk D, Fullard N, Banks P, Page A, et al. 2015. NFκB1 is a suppressor of neutrophil-driven hepatocellular carcinoma. Nat. Commun 6:6818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sekoguchi S, Nakajima T, Moriguchi M, Jo M, Nishikawa T, et al. 2007. Role of cell-cycle turnover and oxidative stress in telomere shortening and cellular senescence in patients with chronic hepatitis C. J. Gastroenterol. Hepatol 22:182–90 [DOI] [PubMed] [Google Scholar]
- 67.Aravinthan A, Scarpini C, Tachtatzis P, Verma S, Penrhyn-Lowe S, et al. 2013. Hepatocyte senescence predicts progression in non-alcohol-related fatty liver disease. J. Hepatol 58:549–56 [DOI] [PubMed] [Google Scholar]
- 68.Wood MJ, Gadd VL, Powell LW, Ramm GA, Clouston AD. 2014. Ductular reaction in hereditary hemochromatosis: the link between hepatocyte senescence and fibrosis progression. Hepatology 59:848–57 [DOI] [PubMed] [Google Scholar]
- 69.Bitler BG, Fink LS, Wei Z, Peterson JR, Zhang R. 2013. A high-content screening assay for small-molecule modulators of oncogene-induced senescence. J. Biomol. Screen 18:1054–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Laberge R-M, Zhou L, Sarantos MR, Rodier F, Freund A, et al. 2012. Glucocorticoids suppress selected components of the senescence-associated secretory phenotype. Aging Cell 11:569–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lahtela J, Corson LB, Hemmes A, Brauer MJ, Koopal S, et al. 2013. A high-content cellular senescence screen identifies candidate tumor suppressors, including EPHA3. Cell Cycle 12:625–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Robinson AR, Yousefzadeh MJ, Rozgaja TA, Wang J, Li X, et al. 2018. Spontaneous DNA damage to the nuclear genome promotes senescence, redox imbalance and aging. Redox Biol. 17:259–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Christopher LJ, Cui D, Li W, Barros A Jr., Arora VK, et al. 2008. Biotransformation of [14C]dasatinib: in vitro studies in rat, monkey, and human and disposition after administration to rats and monkeys. Drug Metab. Dispos 36:1341–56 [DOI] [PubMed] [Google Scholar]
- 74.Graefe EU, Wittig J, Mueller S, Riethling AK, Uehleke B, et al. 2001. Pharmacokinetics and bioavailability of quercetin glycosides in humans. J. Clin. Pharmacol 41:492–99 [DOI] [PubMed] [Google Scholar]
- 75.Wilson WH, O’Connor OA, Czuczman MS, LaCasce AS, Gerecitano JF, et al. 2010. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 11:1149–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Khan S, Zhang X, Lv D, Zhang Q, He Y, et al. 2019. A selective BCL-XL PROTAC degrader achieves safe and potent antitumor activity. Nat. Med 25:1938–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guerrero A, Guiho R, Herranz N, Uren A, Withers DJ, et al. 2020. Galactose-modified duocarmycin prodrugs as senolytics. Aging Cell 19(4):e13133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Gregg SQ, Robinson AR, Niedernhofer LJ. 2011. Physiological consequences of defects in ERCC1-XPF DNA repair endonuclease. DNA Repair 10:781–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Yousefzadeh MJ, Zhao J, Bukata C, Wade EA, McGowan SJ, et al. 2020. Tissue specificity of senescent cell accumulation during physiologic and accelerated aging of mice. Aging Cell 19(3):e13094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S, et al. 2004. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nat. Genet 36:744–49 [DOI] [PubMed] [Google Scholar]
- 81.Carter CS, Richardson A, Huffman DM, Austad S. 2020. Bring back the rat! J. Gerontol. A Biol. Sci. Med. Sci 75:405–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hickson LJ, Langhi Prata LGP, Bobart SA, Evans TK, Giorgadze N, et al. 2019. Senolytics decrease senescent cells in humans: preliminary report from a clinical trial of dasatinib plus quercetin in individuals with diabetic kidney disease. EBioMedicine 47:446–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Justice JN, Nambiar AM, Tchkonia T, LeBrasseur NK, Pascual R, et al. 2019. Senolytics in idiopathic pulmonary fibrosis: results from a first-in-human, open-label, pilot study. EBioMedicine 40:554–63 [DOI] [PMC free article] [PubMed] [Google Scholar]; One of the first clinic trials to use senolytics.
- 84.Wissler Gerdes EO, Zhu Y, Tchkonia T, Kirkland JL. 2020. Discovery, development and future application of senolytics: theories and predictions. FEBS J. 287(12):2418–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li W, He Y, Zhang R, Zheng G, Zhou D. 2019. The curcumin analog EF24 is a novel senolytic agent. Aging 11:771–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Liu X, Wang Y, Zhang X, Gao Z, Zhang S, et al. 2018. Senolytic activity of piperlongumine analogues: synthesis and biological evaluation. Bioorg. Med. Chem 26:3925–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Xu X, Liu Q, Zhang C, Ren S, Xu L, et al. 2019. Inhibition of DYRK1A-EGFR axis by p53-MDM2 cascade mediates the induction of cellular senescence. Cell Death Dis. 10:282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vilgelm AE, Pawlikowski JS, Liu Y, Hawkins OE, Davis TA, et al. 2015. Mdm2 and aurora kinase A inhibitors synergize to block melanoma growth by driving apoptosis and immune clearance of tumor cells. Cancer Res. 75:181–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Triana-Martínez F, Picallos-Rabina P, Da Silva-Álvarez S, Pietrocola F, Llanos S, et al. 2019. Identification and characterization of cardiac glycosides as senolytic compounds. Nat. Commun 10:4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Guerrero A, Herranz N, Sun B, Wagner V, Gallage S, et al. 2019. Cardiac glycosides are broad-spectrum senolytics. Nat. Metab 1:1074–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Feng M, Kim J, Field K, Reid C, Chatzistamou I, Shim M. 2019. Aspirin ameliorates the long-term adverse effects of doxorubicin through suppression of cellular senescence. FASEB Bioadv. 1:579–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fuhrmann-Stroissnigg H, Santiago FE, Grassi D, Ling Y, Niedernhofer LJ, Robbins PD. 2019. SA-β-galactosidase-based screening assay for the identification of senotherapeutic drugs. J. Vis. Exp 2019:148. [DOI] [PubMed] [Google Scholar]
- 93.Weiland T, Lampe J, Essmann F, Venturelli S, Berger A, et al. 2014. Enhanced killing of therapy-induced senescent tumor cells by oncolytic measles vaccine viruses. Int. J. Cancer 134:235–43 [DOI] [PubMed] [Google Scholar]
- 94.Chen Z, Hu K, Feng L, Su R, Lai N, et al. 2018. Senescent cells re-engineered to express soluble programmed death receptor-1 for inhibiting programmed death receptor-1/programmed death ligand-1 as a vaccination approach against breast cancer. Cancer Sci. 109:1753–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Muñoz-Espín D, Rovira M, Galiana I, Giménez C, Lozano-Torres B, et al. 2018. A versatile drug delivery system targeting senescent cells. EMBO Mol. Med 10:e9355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nakagami H 2020. Cellular senescence and senescence-associated T cells as a potential therapeutic target. Geriatr. Gerontol. Int 20:97–100 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.