

Abstract
Rationale:
Changing activity of cardiac CaV1.2 channels under basal conditions, during sympathetic activation, and in heart failure is a major determinant of cardiac physiology and pathophysiology. Although cardiac CaV1.2 channels are prominently up-regulated via activation of protein kinase A, essential molecular details remained stubbornly enigmatic.
Objective:
The primary goal of this study was to determine how various factors converging at the CaV1.2 I-II loop interact to regulate channel activity under basal conditions, during β-adrenergic stimulation, and in heart failure.
Methods and Results:
We generated transgenic mice with expression of CaV1.2 α1C subunits with: 1) mutations ablating interaction between α1C and β subunits; 2) flexibility-inducing polyglycine substitutions in the I-II loop (GGG-α1C); or 3) introduction of the alternatively spliced 25-amino acid exon 9* mimicking a splice variant of α1C up-regulated in the hypertrophied heart. Introducing three glycine residues that disrupt a rigid IS6-AID helix markedly reduced basal open probability despite intact binding of CaVβ to α1C I-II loop, and eliminated β-adrenergic agonist stimulation of CaV1.2 current. In contrast, introduction of the exon 9* splice variant in α1C I-II loop, which is increased in ventricles of patients with end-stage heart failure, increased basal open probability but did not attenuate stimulatory response to β-adrenergic agonists when reconstituted heterologously with β2B and Rad or transgenically expressed in cardiomyocytes.
Conclusions:
Ca2+ channel activity is dynamically modulated under basal conditions, during β-adrenergic stimulation, and in heart failure by mechanisms converging at the α1C I-II loop. CaVβ binding to α1C stabilizes an increased channel open probability gating mode by a mechanism that requires an intact rigid linker between the β subunit binding site in the I-II loop and the channel pore. Release of Rad-mediated inhibition of Ca2+ channel activity by β-adrenergic agonists/PKA also requires this rigid linker and β binding to α1C.
Keywords: Calcium, calcium channels, protein kinase A (PKA), adrenergic, cardiac, excitation-contraction coupling, ion channel, physiology
Subject Terms: Basic Science Research, Calcium Cycling/Excitation-Contraction Coupling, Ion Channels/Membrane Transport, Physiology
Graphical Abstract
INTRODUCTION
In cardiomyocytes, Ca2+ influx through L-type CaV1.2 channels commences the process of excitation-contraction coupling via the triggering of Ca2+ release from ryanodine receptors. In the failing heart, dysfunctional regulation of CaV1.2 channels can trigger electrical abnormalities leading to early Ca2+-mediated after-depolarizations, arrhythmias, and sudden death.
Voltage-gated Ca2+ channels are comprised of a pore-forming α1 subunit 1 and a cytosolic β subunit that interacts with the α-interaction domain (AID) in the intracellular linker between domains I and II (I-II loop) of the α1 subunit 2–4 (Figure 1A). When expressed heterologously, binding to the β subunit is obligatory for α1C trafficking to the plasma membrane, and for normalizing channel activation and inactivation gating properties 5–8 via a mechanism that requires a rigid IS6-AID helix linker 9, 10. β-adrenergic agonists, via activation of protein kinase A (PKA), increase Ca2+ influx through CaV1.2 11, 12, an important component of the physiological ‘fight-or-flight’ response that contributes to the increased contractility of the heart during exercise. The overall features of this regulation − PKA-dependent enhanced whole-cell current amplitude, a hyperpolarizing shift in voltage-dependence of channel activation, and increased open probability (Po) − are well-established 11, 12, yet essential molecular details remained stubbornly enigmatic for decades. Recently, we showed that binding of CaVβ subunits to α1C is required for β-adrenergic stimulation of CaV1.2 channels and positive inotropy in the heart 13, and that the Ca2+ channel inhibitor Rad 14, which binds to the β subunit, is the functionally relevant PKA target in the CaV1.2 complex 15.
Figure 1. Electrophysiological properties of pseudo-wild-type and AID-mutant Ca2+ channels.
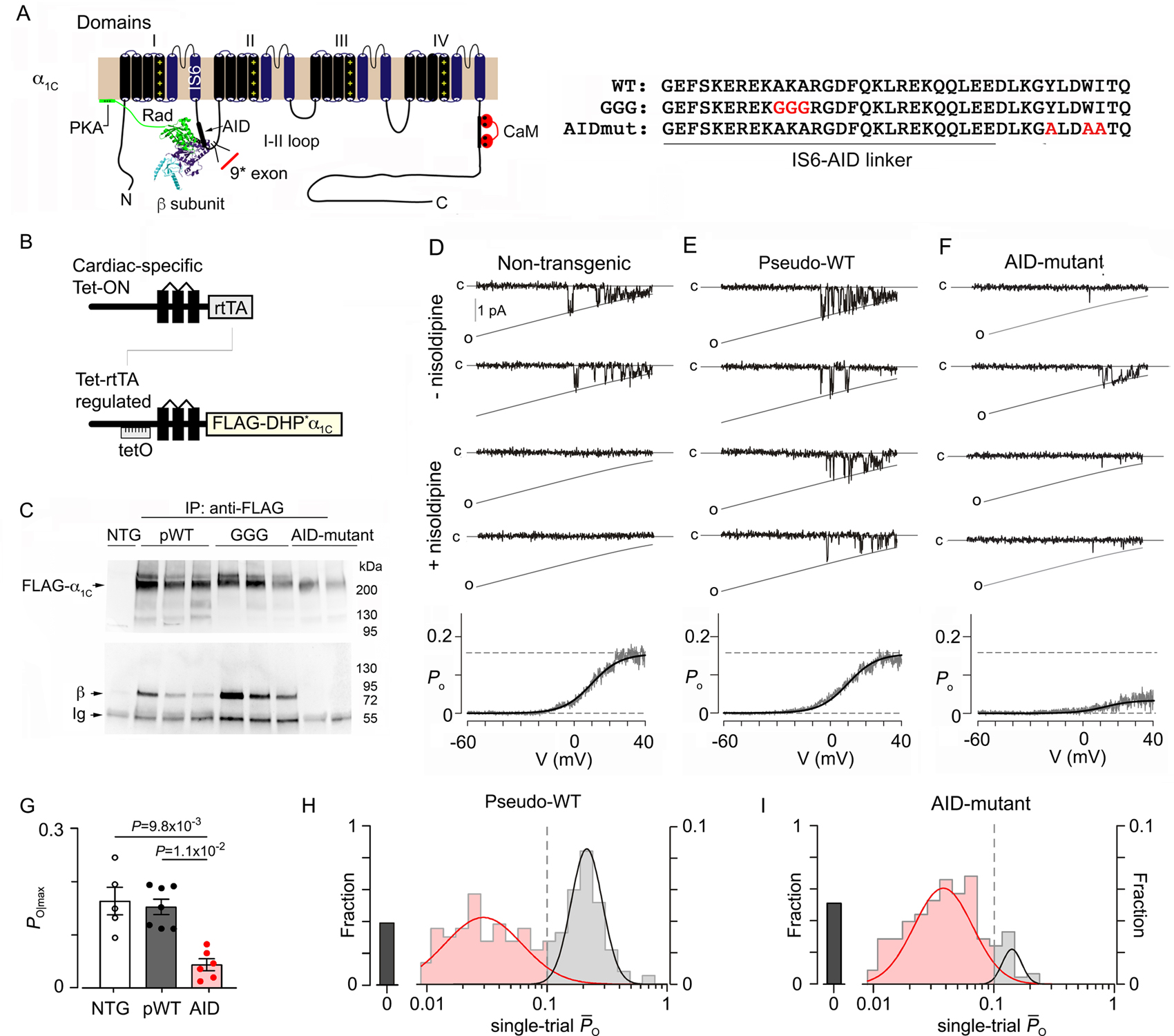
(A) Schematic of rabbit cardiac α1C subunit topology showing β subunit binding to α-interaction domain (AID) motif, and the position of the 9* exon in I-II loop. Rad interaction with both β subunit and the plasma membrane are shown. WT, mutant GGG, and mutant AID motif in the I-II loop of α1C. (B) Diagrams showing the binary transgene system that allow expression of FLAG-DHP-resistant (DHP*) α1C only when both reverse tetracycline-controlled transactivator (rtTA) (top diagram) and doxycycline are present (Tet-ON). The lower diagram shows cDNA for FLAG-DHP-resistant (DHP*) α1C ligated behind seven tandem tetO sequences. (C) Anti-FLAG (upper) and anti-β immunoblots (lower) of anti-FLAG antibody immunoprecipitation of cardiac homogenates of non-transgenic (NTG), pseudo-wild-type α1C, GGG-α1C and AID-mutant α1C mice. Representative of 3 experiments. (D-F) Single channel Ba2+ currents are shown. Channel closures are labeled “c” and openings are downward deflections to the open level (slanted gray curves, labeled “o”) in the absence of nisoldipine (top 2 rows) and presence of nisoldipine (bottom 2 rows). Bottom: Po versus voltage relationship, averaged over multiple patches. N= 5, 7, 6, from left to right. (G) Graph of Po for Ca2+ channels recorded from non-transgenic (NTG), pseudo-wild-type (pWT) α1C and AID-mutant α1C cardiomyocytes. Kruskal-Wallis P= 4 × 10−4; Dunn’s multiple comparison test P-values in panel. (H-I) Histograms show distribution of single-trial average Po obtained from DHP-resistant one-channel patches from pseudo-wild-type α1C and AID-mutant cardiomyocytes.
Beyond these, the I-II loop is also subject to alternative splicing that tunes channel function and interacting proteins in a cell-type specific manner. The inclusion of alternatively-spliced exon 9* is observed at high levels in the smooth muscle and at lower but variable expression in adult heart that increases in animal models of hypertrophy and in the peri-infarct zone after myocardial infarction in mice 16–18. This variant results in the insertion of 25 amino acid residues C-terminal to the AID 18–20 and tunes channel activation. In all, the modulatory landscape supported by the CaV1.2 domain I-II linker appears rich and multifaceted, involving the β subunits, RGK proteins, phosphoregulation by PKA, and alternative splicing, all poised to precisely tune Ca2+ influx into cardiomyocytes.
Several important mechanistic unknowns persist impeding in-depth pathophysiological understanding. First, although CaVβ – α1 interaction is obligatory for CaV1.2 trafficking in heterologous cells, we found it to be dispensable for trafficking to the dyad, for basal function and for initiating excitation-contraction coupling in adult cardiomyocyte 13, thus raising fundamental questions about the functional role of CaVβ subunits in cardiomyocytes. Second, it is unknown how distal conformational changes involving Rad interaction with the CaVβ subunit and phosphorylation-dependent signaling are ultimately conveyed to the channel pore-domain. Third, how alternative splicing of the domain I-II linker contributes to this overall regulatory scheme including downstream effects of PKA activation remains to be fully-elucidated. Importantly, the pathogenesis of heart failure has been long-suspected to reshape this regulatory framework, although the precise changes remain largely undefined.
To dissect these possibilities, we measured baseline channel gating properties and the strength of adrenergic modulation of CaV1.2 in three transgenic mouse models: (1) Our previously-established AID mutant where CaV1.2 is incapable of binding to the β subunit 13, (2) Triple-glycine substitution (GGG-α1C) of the rigid I-II linker, which connects the pore-domain with the AID. These mutations have been previously shown to disrupt coupling between these two domains 9, 10, 21, and (3) The insertion of 25 amino acid residues C-terminal to the AID corresponding to the introduction of exon 9* variant (9*-α1C) (Figure 1A). Our findings identify the IS6-AID linker as a vital molecular element for transducing the PKA-induced activation of CaV1.2, and as a molecular rheostat for CaV1.2 activity whereby distinct structural changes elicited by β subunits, RGK proteins, and alternative splicing bidirectionally tune Ca2+ influx into the heart in both normal physiology and during heart failure.
METHODS
Data Availability.
All supporting data are available within the article and its online data supplement.
For details on the experimental procedures, see the materials and methods section in the online data supplement.
RESULTS
Generation of inducible, cardiac-specific α1C transgenic mice.
We created several mice lines with inducible, cardiac-specific expression of dihydropyridine (DHP)-resistant, FLAG-epitope-tagged α1C (Figure 1B). We have previously shown that control transgenic FLAG-tagged DHP-resistant α1C subunits, termed pseudo-wild-type α1C have similar properties as native cardiac Ca2+ channels 22. To study properties of CaV1.2 devoid of the β subunit, we used transgenic mice13 expressing rabbit α1C with a disrupted AID via alanine substitutions of 3 conserved residues, Y467, W470 and I471, that are essential for binding of β subunit 21. We further generated transgenic mice (Figure 1B) expressing FLAG-tagged DHP-resistant α1C with a substitution of three glycine residues (GGG) for the AKA motif in the IS6-AID linker 10, designated GGG-α1C (Figure 1A). All three lines, pseudo-WT-, AID- and GGG-α1C were crossed with transgenic mice expressing reverse transcriptional transactivator in the heart (αMHC-rtTA) 23 (Figure 1B), yielding mice with doxycycline-inducible α1C expression. As expected, channels with a disrupted AID motif do not bind β subunits, assessed by anti-FLAG antibody immunoprecipitation of cleared homogenates (Figure 1C). By comparison, GGG-α1C channels exhibit robust β subunit binding similar to pseudo-wild-type α1C (Figure 1C).
Binding of β to α1C enhances channel openings.
Previously, we showed that in cardiomyocytes, CaV1.2 channels without β subunits can be transported to the dyad and can generate currents that mediate normal excitation-contraction coupling 13. To determine whether β binding to α1C alters basal channel gating in cardiomyocytes, we utilized low-noise single-channel recordings of acutely isolated cardiomyocytes, employing Ba2+ as a charge carrier. Stochastic channel openings, which reflect near-steady-state open probability (Po) at each voltage, were elicted by a slow voltage ramp 24, 25. Ca2+ channels in cardiomyocytes from non-transgenic mice were inhibited by 300 nM nisoldipine (Figure 1D, middle). In the transgenic mice, however, there is a mixture of transgenic nisodipine-resistant channels and endogenous nisoldipine-sensitive channels (Figure 1E–F). As dihydropyridines are known to allosterically modify channel gating 26, partial blockade of nisoldipine-resistant channels may be a confounding factor. To obviate this possibility, we obtain ~80–120 stochastic records from each patch and subsequently apply nisoldipine to identify resistant transgenic channels. This process allows us to unambiguously establish baseline function of mutant CaV1.2. Indeed, exemplar traces from DHP-resistant pseudo-wild-type α1C channels confirm channel openings in the presence of nisoldipine. Measurements of steady-state Po as a function of voltage were obtained by averaging many records and by normalizing the unitary current level. Reassuringly, steady-state Po-V relationships of pseudo-wild-type α1C channels are similar to that of non-transgenic channels. The DHP-resistant AID-mutant α1C channels, however, displayed a striking 3.5-fold reduction in maximal Po compared to DHP-sensitive endogenous Ca2+ channels from non-transgenic Ca2+ mice and DHP-resistant channels from pseudo-wild-type α1C mice (Figure 1G). To further elucidate changes in elementary channel gating mechanisms, we scrutinized single-trial average open probabilities () from one-channel patches. A dash-line discriminator with was used to identify low activity versus high-activity traces. Thus analyzed, pseudo-wild-type α1C channels switched between epochs of no openings or blanks (40.6% of traces), low activity (33.0%), and high activity (26.4%) (Figure 1H) consistent with previous studies. By comparison, AID-mutant α1C channels exhibited a distinct pattern (p<0.0001 by χ2 test of independence) with rare sojourns to the high-activity mode (7.1%) and a higher propensity for blank (45.7%) and low activity sweeps (47.2%) (Figure 1I). Thus β subunit binding appears to stabilize the high-activity gating mode. We conclude that the interaction between the β subunit and the α1C subunit essentially modulates CaV1.2 channel activity in cardiomyocytes by enhancing channel openings.
Increased flexibility of IS6 linker reduces basal Ca2+ channel activity in cardiomyocytes.
Having established the importance of β subunits in upregulating CaV1.2 channel activity, we considered whether the rigid I-II linker between the IS6 pore helix and the AID is essential for tuning channel function. Introduction of the three glycine residues in the IS6-AID linker had no effect on the subcellular localization and functional expression of CaV1.2 in cardiomyocytes. Anti-FLAG antibody immunofluorescence studies on fixed cardiomyocytes showed that GGG-α1C CaV1.2 channels demonstrated a striated z-disk pattern consistent with localization to the surface membrane and transverse-tubules (t-tubules) (Figure 2A). Similar to cardiomyocytes expressing pseudo-wild-type α1C or AID-mutant α1C transgenic channels, field-stimulated contraction of cardiomyocytes isolated from GGG-α1C transgenic mice persisted in the presence of 300 nM nisoldipine, which is sufficient to block excitation-contraction coupling induced by endogenous CaV1.2 channels in non-transgenic mice (Figure 2B), indicating that the GGG-α1C CaV1.2 channels are localized correctly and flux sufficient Ca2+ to evoke Ca2+-induced Ca2+ release in cardiomyocytes.
Figure 2. Electrophysiological properties of GGG Ca2+ channels.
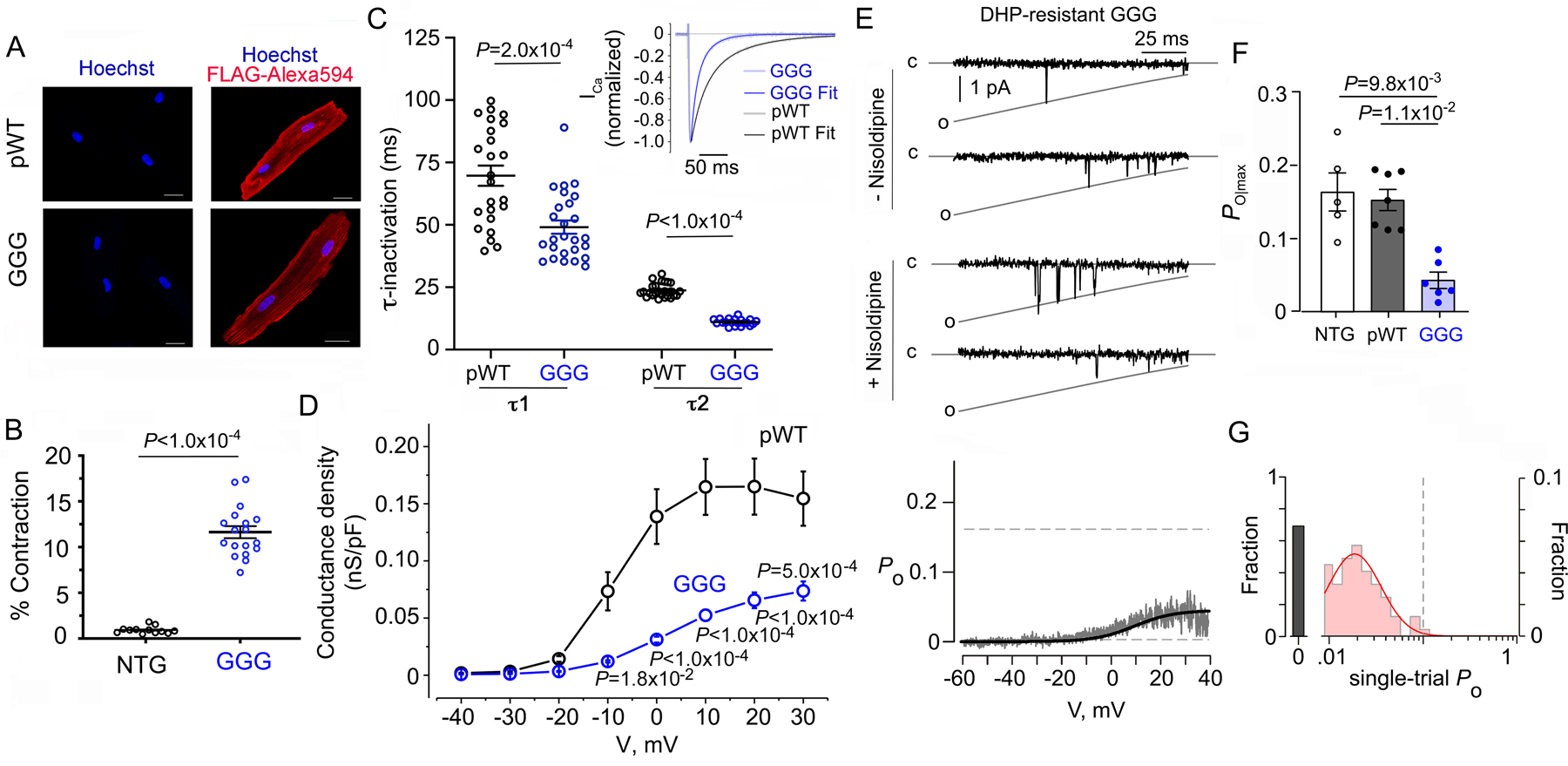
(A) Immunostaining of pseudo-wild-type (pWT) α1C and GGG-α1C cardiomyocytes. Anti-FLAG and Alexa 594-conjugated secondary antibodies, and nuclear labeling with Hoechst stain. Negative control omitted anti-FLAG antibody. Images obtained with confocal microscopy. Scale bar = 20 μm. (B) Percent contraction of sarcomere length in the presence of nisoldipine for cardiomyocytes isolated from NTG and GGG-α1C mice. Cardiomyocytes were field-stimulated at 1-Hz. Unpaired t-test. (C) Graph of τ1 and τ2 inactivation for pseudo-wild-type α1C (n=14 from 4 mice) and GGG-α1C (n=26 from 3 mice) cardiomyocytes. Mann-Whitney test. Inset: Exemplar tracing of normalized Ca2+ current in response to a step depolarization to +10 mV. Fits were obtained using two exponentials by Clampfit. (D) Graph of conductance density-voltage relationship for nisoldipine-resistant Ca2+ channels recorded from pseudo-wild-type α1C (n=22 from 4 mice) and GGG-α1C (n=20 from 3 mice) cardiomyocytes. Mean ± SEM. P < 0.0001 by one-way ANOVA; Sidak’s multiple comparison test P-values in panel. (E) Single-channel Ba2+ currents in absence and presence of nisoldipine. Bottom: Po versus voltage relationship averaged over multiple patches. N=6. (F) Graph of Po for Ca2+ channels recorded from non-transgenic (NTG), pseudo-wild-type (pWT) α1C, and GGG α1C cardiomyocytes. NTG and pWT α1C data are the same as in Figure 1. P= 4 × 10−4 by Kruskal-Wallis. Dunn’s multiple comparison test P-values in panel. (G) Histogram shows distribution of single-trial average Po obtained from DHP-resistant one-channel patches from GGG mutant cardiomyocytes.
When GGG-α1C is co-expressed in Xenopus oocytes with β2B, one of the major β2 isoforms in the heart, channels displayed accelerated voltage-dependent inactivation but slowed Ca2+-dependent inactivation 10. Here, we assessed aggregate inactivation of Ca2+ channels in the heart with Ca2+ as a charge carrier. The inactivation kinetics of nisoldipine-resistant GGG-α1C Ca2+ currents at +10 mV test potential was significantly faster compared to pseudo-wild-type α1C controls (Figure 2C). Therefore, in adult cardiomyocytes CaV1.2 channels comprised of transgenic GGG-α1C have faster overall inactivation kinetics as compared to transgenic pseudo-wild-type CaV1.2 channels likely reflecting accelerated kinetics of voltage-dependent inactivation.
Given that β subunits upregulate CaV1.2 channel Po, we considered whether disruption of the rigid IS6-AID linker might reverse this effect. Consistent with this possibility, the conductance-voltage (G-V) relationships, normalized to cell capacitance, of nisoldipine-resistant transgenic mutant GGG-α1C channels was reduced compared to pseudo-wild-type α1C (Figure 2D). To directly assess changes in Po, we used low-noise single-channel recordings of acutely isolated cardiomyocytes from the GGG-α1C transgenic mice. Exemplar records show DHP-resistant GGG-α1C channels exhibit sparse channel openings (Figure 2E, top), a distinct gating pattern compared to pseudo-wild-type α1C and non-transgenic channels which undergo high-activity flickery openings (Figure 1). Ensemble average Po-V relationship (Figure 2E, bottom) and bar-graph summary of maximal Po (Figure 2F) from individual patches show a striking 3.5-fold reduction in maximal Po compared to both pseudo-wild-type α1C and non-transgenic channels (Figure 2F). Interestingly, this reduced basal activity of GGG-α1C is reminiscent of β-less AID-mutant channels suggesting that disruption of the rigid IS6-AID linker may be akin to the uncoupling of the β subunit from the channel pore 10. To further scrutinize this possibility, we assessed average Po from individual trials for one channel patches of GGG-α1C. Unlike pseudo-wild-type α1C channels, the single-trial distribution of the GGG-α1C channels was restricted to either blank (73.4%) or low activity sweeps (26.6%) with no evidence of high activity traces (Figure 2G). As GGG-α1C are fully capable of β subunit binding (Figure 1), these results suggest that the rigidity of the linker between the pore-domain and I-II loop may be a structural requirement for the high-activity gating configuration. As AID-mutant channels exhibit some propensity for high-activity traces, one attractive possibility is that the IS6-AID linker may switch between rigid and flexible conformations, with β subunit binding to the AID serving to stabilize the rigid helical linker conformation, an outcome also supported by X-ray crystallographic and circular dichroism experiments 2–4.
9*-α1C splice variant increases basal open probability.
Having established the I-II loop as a vital regulator of channel openings, we considered whether alternative splicing in this domain might tune channel gating. The 9* exon, which encodes a 75-nucleotide sequence within the I-II loop (Figure 3A), is expressed at a high level in aortic smooth muscle and has lower expression in non-diseased adult human and rat heart. However, the 9* exon-containing channels are increased in rodent models of hypertrophy and in the perinfarct zone 16–18. This altered pattern of α1C splicing in rodent models raises the possibility of pathological inclusion of exon 9* in human cardiac disease, an outcome yet to be observed clinically. As such, we sought to determine whether the frequency of exon 9* splice variant is changed in humans with end-stage heart failure. Samples from patients undergoing LVAD implantation at Columbia-NY Presbyterian Hospital were acquired in the operating room, and compared to samples obtained from donor hearts without heart failure (Online Tables I–III). Exon 9* transcript expression was increased in humans with heart failure compared to control samples (Figure 3B).
Figure 3. Expression and electrophysiological properties of 9* splice variant-α1C Ca2+ channels in heart.
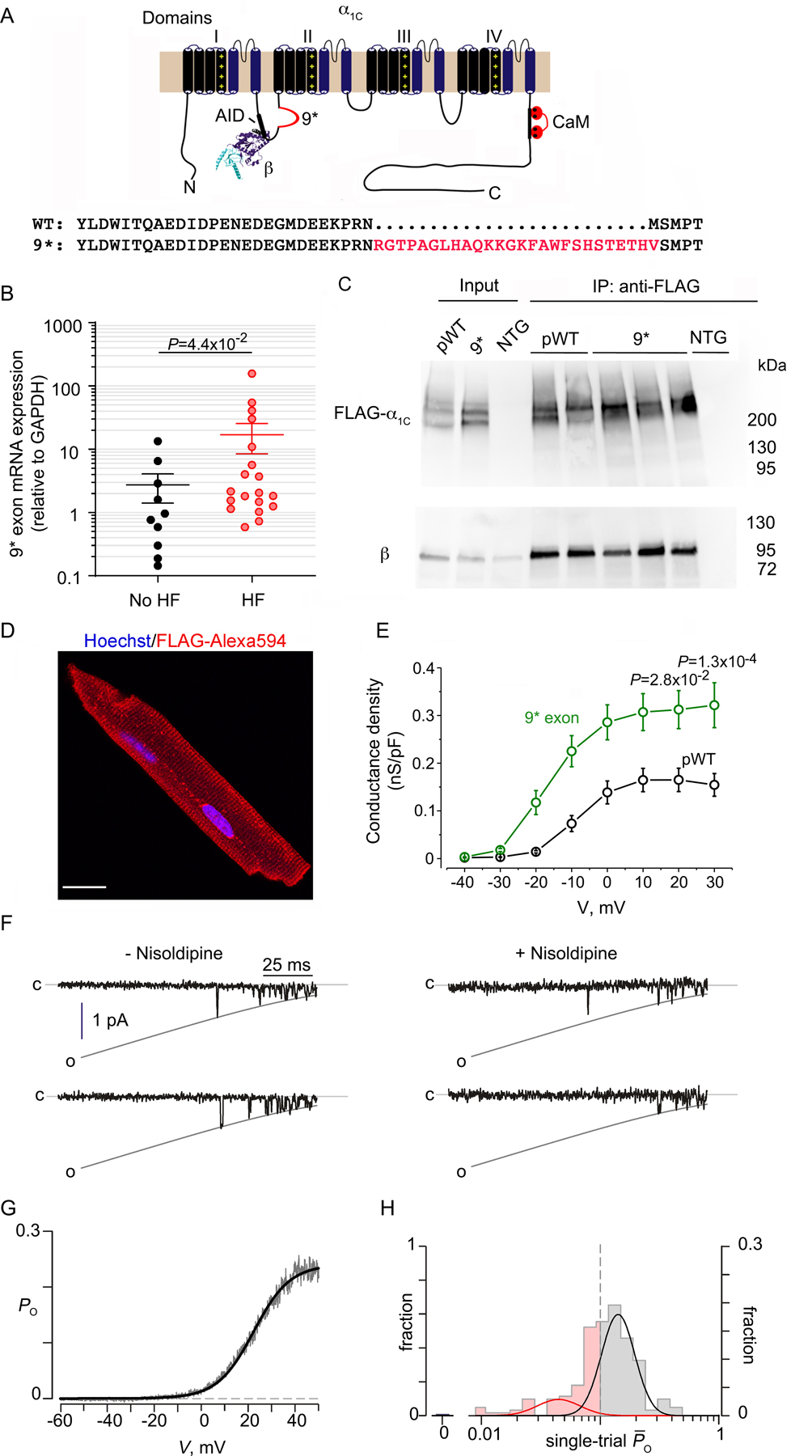
(A) Schematic of rabbit cardiac α1C subunit topology showing β subunit binding to AID motif in I-II loop, and insertion of 9* exon. WT and 9* splice variant sequence in the I-II loop of α1C. (B) Graph of normalized 9* exon mRNA expression from patients with advanced heart failure (HF) undergoing implantation of a left ventricular assist device and control, no-HF patients. Mean ± SEM. N=19 for HF, 10 for no-HF. Mann-Whitney test for P-value in panel. (C) Anti-FLAG (upper) and anti-β immunoblots (lower) of anti-FLAG antibody immunoprecipitation of cardiac homogenates of non-transgenic (NTG), 9*-α1C and pseudo-wild-type (pWT) α1C mice. Representative of 3 experiments. Input is 6% of immunoprecipitation. (D) Immunostaining of 9*-α1C cardiomyocyte. Anti-FLAG and Alexa 594-conjugated secondary antibodies, and nuclear labeling with Hoechst stain. Images obtained with confocal microscopy. Scale bar = 20 μm. (E) Graph of conductance density-voltage relationship for nisoldipine-resistant Ca2+ channels recorded from pseudo-wild-type α1C and 9*-α1C cardiomyocytes. pWT α1C curve is the same as in Figure 2D. Mean ± SEM. N= 20 cells from 3 mice. P < 1.0 ×10−4 by one-way ANOVA; Sidak’s multiple comparison test P-values in panel. (F) Single-channel Ba2+ currents in the absence of nisoldipine (left) and presence of nisoldipine (right). (G) Po versus voltage relationship, averaged over multiple patches. N=10. (H) Histogram shows distribution of single-trial average Po obtained from DHP-resistant one-channel patches from 9*-expressing cardiomyocytes. These channels largely adopt high Po gating mode with a marked reduction in the fraction of blank sweeps.
To determine the functional consequence of exon 9* splice inclusion in cardiomyocytes, we created transgenic mice with cardiac-specific expression of DHP-resistant Ca2+ channels containing exon 9*, but with all other mutually exclusive exons typical of cardiac variants. The 9*-α1C channels still bound β subunits (Figure 3C) similar to pseudo-wild-type α1C channels, and trafficked to the surface membrane and t-tubules, as demonstrated by the striated z-disk pattern of immunofluorescence (Figure 3D). Whole-cell electrophysiological analysis of nisoldipine-resistant transgenic mutant 9*-α1C channels revealed a signicant increase in the G-V relationship normalized to cell capacitance in comparison to pseudo-wild-type α1C channels (Figure 3E). Changes in whole cell current may stem from alterations in channel trafficking, unitary conductance, or baseline open probability. To dissect these mechanistic possibilities, we undertook low-noise single-channel recordings to determine whether the increased normalized conductance of 9*-α1C reflected a genuine increase in channel Po. In the presence of nisoldipine, the DHP-resistant 9*-α1C channels exhibited robust channel openings (Figure 3F) with the ensemble average demonstrating a marked increase in maximal Po (0.26 ± 0.03, mean ± s.e.m) (Figure 3G) in comparison to pseudo-wild-type α1C (0.15 ± 0.015, mean ± s.e.m). Furthermore, examination of single-trial distribution revealed a virtual elimination of blank traces (~0% for 9* versus 40.5% for pseudo-wild-type α1C -see Figure 1H), and increased propensity for the high activity gating mode (56.7%) (Figure 3H). Thus, the insertion of the 9* exon into the I-II loop upregulates basal voltage-dependent opening of Ca2+ channels by enhancing channel availability and stabilizing the high Po gating configuration. Interestingly, this functional signature is reminiscent of CaV1.2 behavior in myocytes from failing human hearts, which also show increased availability and Po 27.
β-adrenergic upregulation of CaV1.2 also requires a rigid IS6-AID linker.
Recently, we determined that the mechanism of adrenergic stimulation of CaV1.2 requires constitutive pre-inhibition of CaV1.2 mediated by Rad interaction with the CaV channel β subunit 15. PKA phosphorylation of Rad at conserved sites in its C-terminus alters its interaction with the CaV channel β subunit and relieves constitutive inhibition 15. As increased flexibility of the IS6-AID linker effectively decouples β subunit mediated regulation, we hypothesized that the rigid IS6-AID linker may be also essential for adrenergic upregulation of CaV1.2 currents. Consistent with this possibility, at the single channel level, Rad inhibited CaV1.2 channels have decreased availability and increased propensity for low activity gating mode, akin to GGG-α1C channels 9, 10.
In cardiomyocytes isolated from mice expressing transgenic pseudo-wild-type α1C, forskolin (an adenylyl cyclase activator) induced an increase in the nisoldipine-insensitive maximal conductance (Gmax) (Figure 4A–B, G), and shifted the V50 for activation (Figure 4H), consistent with our prior studies 13, 22, 28, 29. Ca2+ currents through transgenic GGG- α1C channels, however, were not stimulated by forskolin (Fig 4C–D, G–H). By comparison, forskolin increased the Gmax of 9* exon-containing CaV1.2 channels by a mean of 1.4-fold (Fig 4E–F, G), and shifted the V50 for activation (Figure 4H) suggesting that the 9* exon still preserves responsiveness to PKA modulation. For both pseudo-wild-type α1C and 9* CaV1.2 channels, the forskolin-induced enhancement of Ca2+ current was greatest at hyperpolarized potentials and decreased as the test potential approached the reversal potential of Ca2+ (Figure 4I), consistent with prior observations 12. The GGG CaV1.2 channels failed to respond to forskolin at any test potential (Figure 4I).
Figure 4. β-adrenergic regulation of GGG and 9* Ca2+ channels in cardiomyocytes.
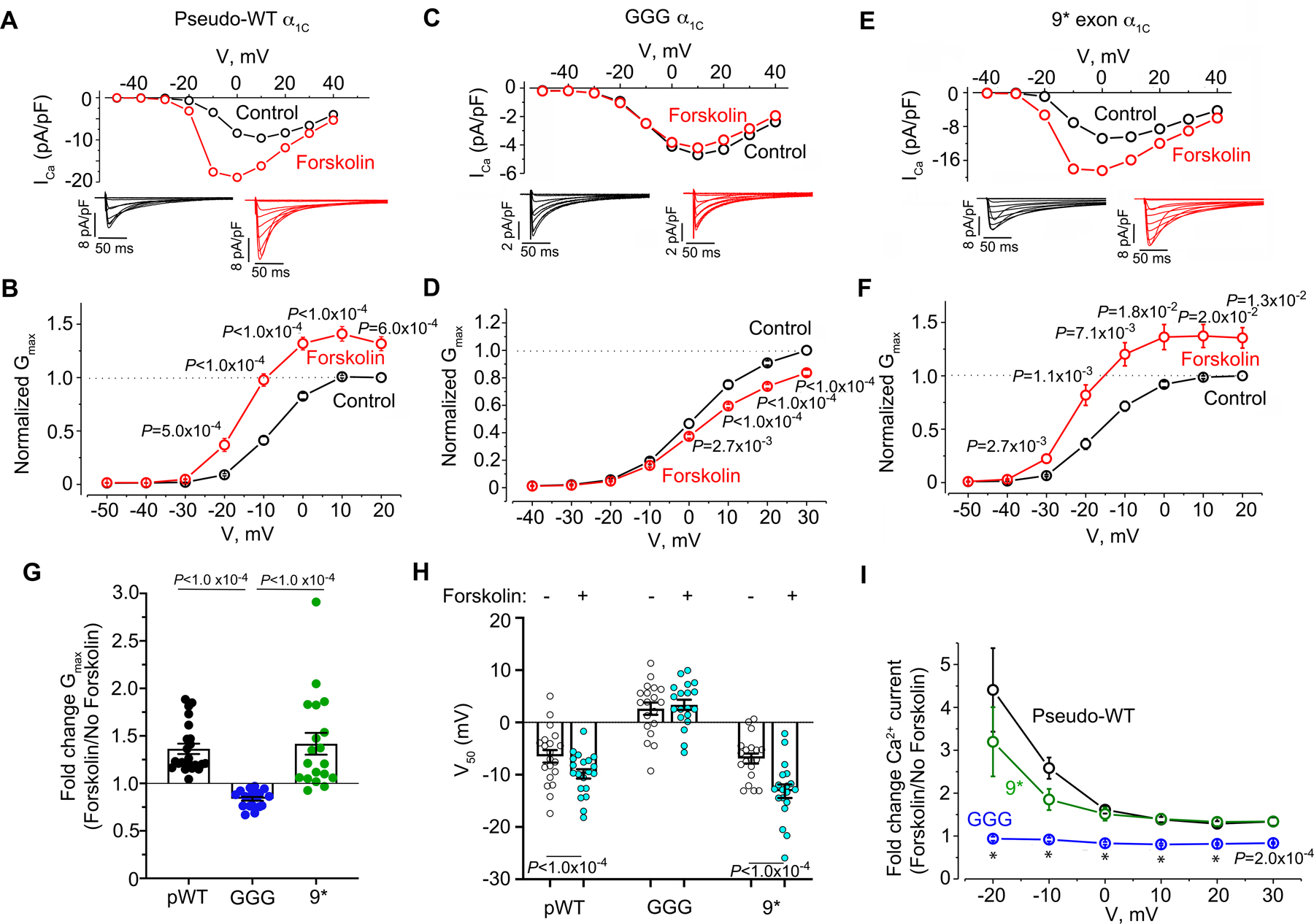
(A, C, E) Current-voltage relationships before and after 10 μM forskolin in the presence of 300 nM nisoldipine. Representative of pseudo-wild-type α1C: n=20, GGG mutant: n= 22 and 9* n=22 cardiomyocytes. Insets: Exemplar whole-cell CaV1.2 currents recorded from freshly dissociated cardiomyocytes of pseudo-wild-type, GGG and 9* α1C transgenic mice. Pulses from −70 mV to +10 mV before (black traces) and 3 minutes after (red traces) 10 μM forskolin in presence of nisoldipine. (B, D, F) Graphs of conductance density-voltage relationship for nisoldipine-resistant Ca2+ channels recorded from pseudo-wild-type α1C, GGG-α1C, and 9*-α1C before (black trace) and after (red trace) forskolin. Mean ± SEM. P<1.0 ×10−4 by repeated measures ANOVA; Sidak’s multiple comparison test P-values are in panels. (G) Fold-change in Gmax. Shown are means ± SEM; P<1.0 ×10−4 by Kruskal-Wallis test; Dunn’s multiple comparison test P-values in panel. (H) Boltzmann function parameter, V50. Shown are means + SEM; Paired two-tailed t-test. (I) Ratio of Ca2+ current after forskolin treatment to Ca2+ current before treatment of cardiomyocytes with forskolin for pseudo-wild-type, 9* and GGG-α1C cardiomyocytes. Mean ± SEM. Pseudo-wild-type n=20, GGG mutant: n= 22 and 9* n=22 cardiomyocytes from at least 3 mice for each group. P<1.0 ×10−4 by Kruskal-Wallis test; Dunn’s multiple comparison test P-values in panel for pseudo-WT vs. GGG. * P<1.0 ×10-4.
We used reconstitution studies in HEK293T cells 15 to further gain insights into the mechanisms by which the β subunit and I-II loop modulate adrenergic regulation of CaV1.2. In cells transfected with only α1C + β2B, superfusion of forskolin did not affect the Ba2+ currents (Figure 5A). In contrast, applying forskolin to cells expressing α1C + β2B + Rad increased the current as we have previously described 15. Consistent with our findings in cardiomyocytes, in cells transfected with GGG-α1C forskolin failed to increase Gmax or shift the voltage-dependence of activation in a hyperpolarizing direction (Figure 5C–D, G–H). In contrast, applying forskolin to cells expressing 9*-α1C, β2B, and Rad increased the Gmax by a mean of 1.6-fold, and shifted the V50 for activation (Figure 5E–H).
Figure 5. Rad-mediated β−adrenergic regulation of CaV1.2 requires rigid I-II linker.
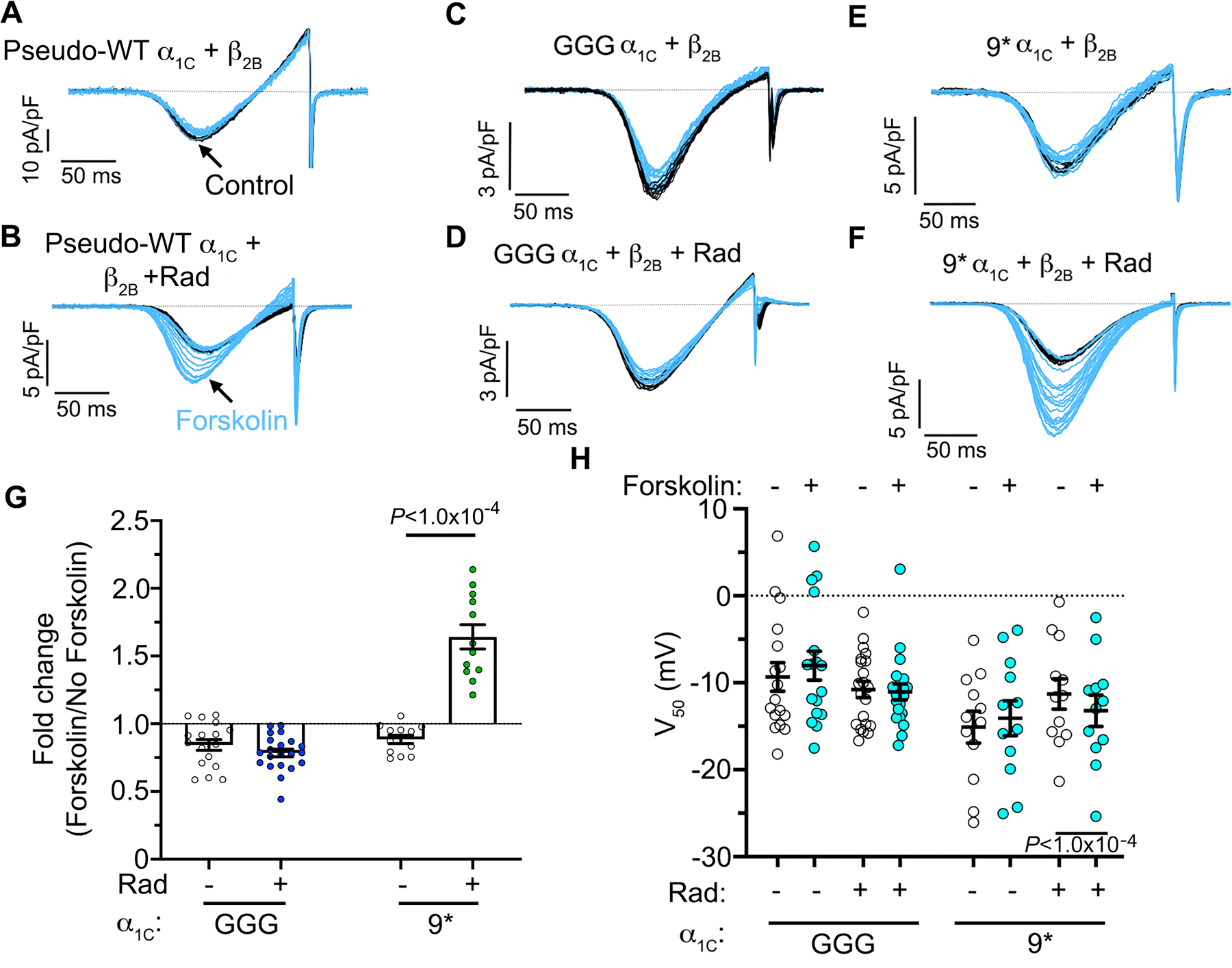
(A–F) Ba2+ current elicited by voltage ramp every 10 s, with black traces obtained before and blue traces obtained after forskolin. The pseudo-WT (pWT) α1C, β2B and Rad were heterologously expressed in HEK293T cells. Representative of top row: 15, 17, 12 cells, left to right; bottom row: 16, 21, 12 cells, left to right. (G) Fold-change in maximum conductance (Gmax) induced by forskolin. Mean ± SEM; Two-tailed unpaired t-test. (H) Boltzmann function parameter V50. Mean ± SEM; Two-tailed paired t-test.
Flow-cytometry Förster resonance energy transfer (FRET) 2-hybrid assay 30 was utilized to determine whether the presence of GGG substitutions or 9* exon altered β subunit binding to the I-II loop, which is required for β-adrenergic regulation of CaV1.2 in heart 13 or the PKA-induced reduction in Rad binding to the β2B subunit. Robust interaction is detected between Cerulean-tagged β2B subunit and Venus-tagged I-II loop (Online Figure I A, E). As would be expected, the mutation of the AID of the I-II loop markedly reduced binding (Online Figure I B), whereas the GGG substitutions or insertion of the 9* exon did not affect binding (Online Figure I C–E). As we previously reported 15, there is a strong interaction between the Cerulean-tagged β2B subunit and Venus-tagged WT Rad (Online Figure II A–B,K). This interaction was markedly weakened, however, by co-expression of PKA catalytic subunit. The basal and PKA-dependent reduction of binding between Rad and β2B was unaffected by co-expression of WT α1C (Online Figure II C–D,K). Similarly, expression of the AID-mutant α1C (Online Figure II E–F), GGG-α1C (Online Figure II G–H) or 9*-α1C (Online Figure II I–J) had no effect on the basal or the PKA-dependent reduction in binding between Rad and β2B (Online Figure II K). These results suggest that the PKA-dependent dissociation of Rad and β2B is neither dependent upon the binding of β to α1C nor is it perturbed by the alterations in the I-II loop induced either by the GGG substitution or insertion of the 9* exon.
DISCUSSION
This work has examined mechanisms of regulation of cardiac CaV1.2 channel gating by three essential factors with important physiological and pathophysiological consequences that converge at the α1C subunit I-II loop— auxiliary CaVβ subunits, sympathetic activation, and alternative splicing. Overall, we find that PKA-modulation of CaV channels in heart and HEK cells is dependent on both Rad phosphorylation and a rigid IS6-AID linker. We discuss our findings on these three inter-related regulatory mechanisms in the context of previously published reports.
Many reconstitution studies in heterologous mammalian cells established the idea that auxiliary CaVβ subunits were necessary for trafficking of CaV1.2 channels to the plasma membrane, and that this depended on high-affinity CaVβ binding to a discrete α1-interaction domain (AID) in the α1C I-II loop 5, 7, 31–37. Beyond trafficking, CaVβ binding also boosted CaV1.2 Po and produced a hyperpolarizing shift in the voltage-dependence of channel activation 36, 38. These gating effects were deduced to require formation of a rigid helix spanning IS6 and AID because they were selectively eliminated by a triple glycine substitution that disrupts the continuous helix 10. In adult cardiomyocytes, cardiac-specific excision of the dominant cardiac CaVβ2 isoform reduced β2 protein levels by 96%, yet resulted in only a 26% reduction in whole-cell CaV1.2 current, providing a first hint that, by contrast to heterologous cells, CaVβ binding to α1C may not be obligatory for forming functional CaV1.2 channels at the cell surface 39. We explicitly confirmed this by showing that transgenic mice expressing a DHP-resistant α1C mutant that does not bind CaVβ, nevertheless, yielded robust nisoldipine-resistant whole-cell Ca2+ currents indicating that the channels made it to the surface sarcolemma 29,13. While CaVβ does not appear necessary for surface trafficking of CaV1.2 in adult cardiomyocytes, it remained unclear whether this also extended to the impact on channel Po. Here, we unambiguously show using single-channel recordings that CaVβ binding to α1C in cardiomyocytes enhances CaV1.2 channel Po by 3.5-fold. The single-channel gating signature of AID-mutant channels was dominated by null (46%) and low-activity (47%) sweeps, with rare sojourns into a high-activity (7%) gating mode. By contrast, CaVβ-bound pseudo-WT channels displayed more high-activity sweeps (26%) and correspondingly lower null (41%) and low-activity (33%) gating modes. GGG-α1C channels displayed only blanks and low-activity gating. These results are consistent with the interpretation that in adult cardiomyocytes CaVβ induces high-Po gating by stabilizing a continuous helix linking IS6 to AID. Absence of CaVβ binding (as occurs with AID-mutation) reduces the propensity for stabilizing a continuous helix and accordingly decreases fractional occupancy of the high-Po gating mode. GGG-α1C completely dispels the continuous helix and, therefore, these channels do not sojourn into the high-Po mode. In the cryo-electron microscopy structure of the homologous CaV1.1 channel, comparison of the two conformations, class Ia and II, revealed signficant shifts between the C-terminal end of IS6 and I-II helix of the α1 subunit, and the β subunit 40. Substitution of the three glycine residues in either one of the two conformations (Figure 6A) likely alters the conformation and increases the flexibility of the I-II loop and the position of the β subunit.
Figure 6. Schematics of role of GGG and 9* in modulating β-adrenergic regulation of CaV1.2.
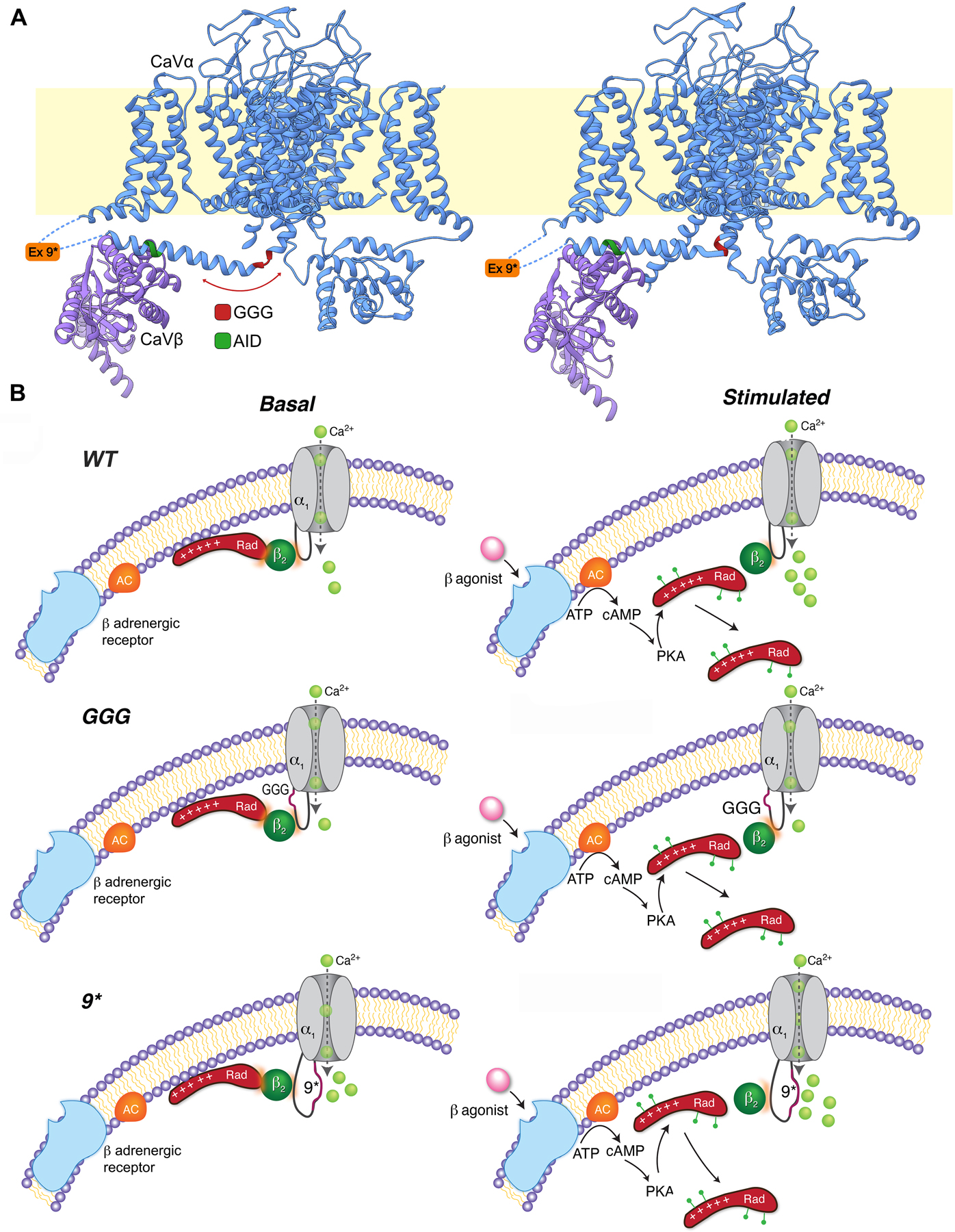
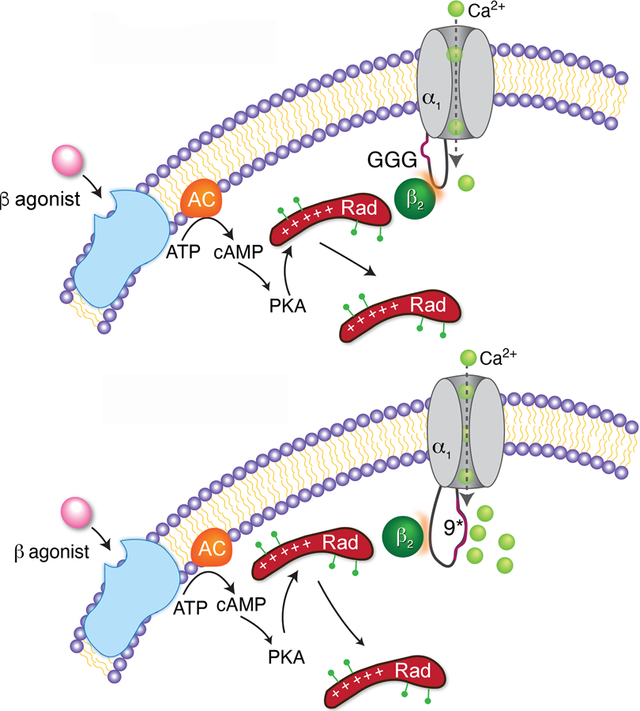
(A) Ribbon representation of the published two CaV1.1 conformations determined by cryo-EM 40 with GGG, AID and exon 9* mutations/insertions in the I-II loop. Left: 5GJV, Right: 5GJW. (B) Proposed models of β−adrenergic regulation of WT, GGG-α1C and 9*-α1C channels. Basal state (left) and after β-adrenergic agonist (stimulated, right). β-agonist-induced activation of adenylyl cyclase (AC) leads to activation of PKA. PKA phosphorylates several residues on Rad, causing dissociationof Rad from the CaV1.2 complex and therefore increased Ca2+ influx. Under basal conditions, the GGG-α1C channels have reduced basal open probability and no response to β-adrenergic stimulation. In contrast to the polyglycine substitution, modifying the I-II loop by introduction of the 9* splice variant increased the basal open probability, yet the stimulatory response to β−adrenergic agonists was preserved.
We recently reported that β-adrenergic regulation of cardiac CaV1.2 channel requires CaVβ binding binding to α1C I-II loop, 13 and PKA phosphorylation of Rad, a small G-protein that inhibits Ca2+ channels via binding to CaVβ subunit 15. Here, we report that GGG-α1C channels expressed in cardiomyocytes, or reconstituted with Rad in heterologous cells, do not display PKA-mediated up-regulation of whole-cell current density, even though they bind CaVβ. This result suggests the rigid IS6-AID helical linker is another essential requirement for transduction of sympathetic regulation of CaV1.2. The exclusively low-Po gating mode of GGG-α1C channels is reminiscent of the behavior of Rad-inhibited channels 15. It is intriguing to speculate that Rad interaction with CaVβ may structurally alter the IS6-AID linker as a mechanism for channel inhibition.
Finally, we examined the impact of the α1C 9*-splice variant that is elevated in the failing heart. Interestingly, the 9*-α1C channels had increased basal Po and were exclusively recorded in high activity gating modes, with no sojourns into mode 0 gating. Given the proximity of the 9* exon to the AID, and the role of the continuous IS6-AID helix in promoting high activity Cav1.2 gating, one explanation is that the 9* exon may increase basal Po by further stabilizing the rigid IS6-AID helical linker. Another explanation is that insertion of the 9* exon may affect the function of the voltage-sensor domain of domain II. Previous single-channel experiments have indicated that CaV1.2 channels in failing hearts have an elevated basal Po which was putatively attributed to enhanced phosphorylation of the channel 41. Our results suggest that the increased CaV1.2 Po observed in heart failure may be due, in part, to the emergence of the alternatively spliced 9*-α1C variant in this condition.
While powerful tools for electrophysiological assessment of α1C mutants in vivo, certain limitations of our transgenic inducible over-expression models prevent examination of whether and how these α1C mutant contribute to both basal and sympathetic regulation of cardiac contractility, and arrhythmogenesis in vivo. Chief among these is that same strategy that allows us to isolate the DHP-resistant transgenic channels for electrophysiological assessment in vivo—application of nisoldipine—blocks endogenous CaV1.2 channels in both heart and vasculature, thus leading to hypotension and confounding the experiments. Furthermore, modest over-expression of WT α1C can initiate hypertrophy and heart failure 42, 43. A direct assessment of whether exon 9* is detrimental could theoretically be achieved with an inducible 9*-α1C knock-in or knock-out strategy, yet due to the genomic structure of exons 9 and 10 in α1C, creating inducible 9*-α1C knock-in or knock-out mice lines would be quite challenging, likely requiring the insertion of a large minigene.
Notwithstanding these limitations, these results provide key insight into the mechanism underlying the β-adrenergic stimulation of Ca2+ current and contractility in the heart. β subunit binding to the α1C subunit promotes transitions to a high Po state. Upon β-adrenergic stimulation, PKA phosphorylation of Rad releases the Rad-induced inhibition of the Ca2+ channels (Figure 6B). The Rad-inhibited channels are the heart’s functional reserve of Ca2+ channels, likely having minimal effects on excitation-contraction coupling at rest, but primed to respond to β-adrenergic agonists upon release of Rad-induced inhibition. Thus, therapeutic release of Rad-mediated inhibition of Ca2+ channels could be inotropic.
Supplementary Material
Major Resources Table
| Animals (in vivo studies) | ||||
|---|---|---|---|---|
| Species | Vendor or Source | Background Strain | Sex | Persistent ID / URL |
| NA | NA | NA | NA | NA |
| Genetically Modified Animals | |||||
|---|---|---|---|---|---|
| Species | Vendor or Source | Background Strain | Other Information | Persistent ID / URL | |
| Pseudo-WT-α1C | Mouse | Transgenic Mouse Facility Columbia University | B6CBAF2 | Generated at Columbia University | NA |
| GGG-α1C | Mouse | Transgenic Mouse Facility Columbia University | B6CBAF2 | Generated at Columbia University | NA |
| AID-α1C | Mouse | Transgenic Mouse Facility Columbia University | B6CBAF2 | Generated at Columbia University | NA |
| 9*-α1C | Mouse | Transgenic Mouse Facility Columbia University | B6CBAF2 | Generated at Columbia University | NA |
| FVB/N-Tg(Myh6-rtTA)8585Jam/Mmmh | Mouse | MMRC | FVB | NA | RRID: MMRRC_ 010478-MU |
| Antibodies | |||||
|---|---|---|---|---|---|
| Target antigen | Vendor or Source | Catalog # | Working concentration | Lot # (preferred but not required) | Persistent ID / URL |
| FLAG (rabbit) | Sigma | F7425 | 1:200 (ICC) | https://www.sigmaaldrich.com/catalog/product/sigma/f7425 | |
| Monoclonal Anti-FLAG M2-Peroxidase (HRP) antibody | Sigma | A8592 | 1:1000 (WB) | https://www.sigmaaldrich.com/catalog/product/sigma/a8592?lang=en®ion=US | |
| Beta common (custom) | Yen-Zym | 1:1000 | epitope: mouse residues 120–138:DSYTSRPS-DSDVSLEEDRE | ||
| AlexaFluor 594-goat anti rabbit | Thermo | A11037 | 1:200 | 2079421 | https://www.thermofisher.com/antibody/product/Goat-anti-Rabbit-IgG-H-L-Highly-Cross-Adsorbed-Secondary-Antibody-Polyclonal/A-11037 |
| DNA/cDNA Clones | |||||
|---|---|---|---|---|---|
| Clone Name | Sequence | Source / Repository | Persistent ID / URL | ||
| mouse Rrad mRNA | NCBI/GenBank | XM_006531206 | |||
| human Xacnb2 mRNA | NCBI/GenBank | NM_201590.3 | |||
| rabbit cardiac Cacna1c mRNA | NCBI/GenBank | X15539 | |||
| rabbit smooth muscle Cacna1c mRNA | NCBI/GenBank | X55763 | |||
| Cultured Cells | |||
|---|---|---|---|
| Name | Vendor or Source | Sex (F, M, or unknown) | Persistent ID / URL |
| HEK293T cells | ATCC | CRL-3216 | |
| HEK293 cells | ATCC | CRL-1573 | |
| Data & Code Availability | ||
|---|---|---|
| Description | Source / Repository | Persistent ID / URL |
NOVELTY AND SIGNIFICANCE.
What Is Known?
Changing activity of cardiac CaV1.2 channels under basal conditions, during sympathetic activation, and in heart failure is a major determinant of cardiac physiology and pathophysiology.
Activation of β-adrenergic receptors results in a multi-fold upregulation of CaV1.2 currents, which is dependent upon PKA phosphorylation of Rad.
It is unknown how distal conformational changes involving Rad interaction with the CaVβ subunit and phosphorylation-dependent signaling are ultimately conveyed to the channel pore-domain.
How alternative splicing of the I-II linker of the pore-forming a1C subunit contributes to this regulatory scheme remains to be fully-elucidated.
What New Information Does This Article Contribute?
Introducing flexibility into the rigid IS6-AID helix markedly reduced basal open probability despite intact binding of CaVβ to α1C I-II loop, and eliminated β-adrenergic agonist stimulation of CaV1.2 current.
The α1C 9*-splice variant, which is elevated in the failing human heart, causes an increased basal open probability but did not attenuate stimulatory response to β-adrenergic agonists.
PKA-modulation of CaV channels in heart and HEK cells is dependent on both Rad phosphorylation and a rigid IS6-AID linker
β-adrenergic stimulation of CaV1.2 current is vital for sympathetic nervous system regulation of cardiac contractility. Recently, we identified Rad, not CaV1.2, as the functionally-relevant target of PKA. At baseline, Rad inhibits CaV1.2 by binding to CaV β subunit. Upon β-adrenergic activation, PKA phosphorylation of Rad releases this interaction and inhibition. How is the signal from Rad and the β subunit transmitted to the channel’s gate? To answer this question, we created transgenic mice featuring the introduction of three glycine residues that disrupt the rigid helix between pore and the α-interaction-domain. Ca2+ channels from these mice displayed markedly reduced basal open probability and were insensitive to β-adrenergic agonist stimulation. In contrast, introduction in the α1C I-II loop of the exon 9* splice variant, which is increased in ventricles of patients with end-stage heart failure, increased basal open probability but did not attenuate stimulatory response to β-adrenergic agonists in cardiomyocytes. We speculate that the increased CaV1.2 open probability observed in heart failure may be due, in part, to the emergence of the alternatively spliced 9*-α1C variant. Taken together, adrenergic stimulation of CaV1.2 requires an intact rigid linker between the binding site of β subunit in the I-II loop and the channel pore.
ACKNOWLEDGEMENTS
Images were collected (and analysed) in the Confocal and Specialized Microscopy Shared Resource of the Herbert Irving Comprehensive Cancer Center at Columbia University, supported by NIH grant P30 CA013696 (National Cancer Institute). Research reported in this publication was performed in the CCTI Flow Cytometry Core, supported in part by the Office of the Director, National Institutes of Health under awards S10RR027050. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
SOURCES OF FUNDING
This work was supported by NIH R01 HL121253, R01 HL126735, R01 HL146149. Arianne Papa was supported by T32HL120826 and NSF 1644869. Jared Kushner was supported by T32 HL007343 and the NY Academy of Medicine Glorney-Raisbeck Fellowship. Jessica Hennessey was supported by T32 HL007854.
Nonstandard Abbreviations and Acronyms:
- AID
α-interaction domain
- αMHC
α-myosin heavy chain
- CaM
calmodulin
- DHP
Dihydropyridine
- FRET
Förster resonance energy transfer
- G-V
conductance-voltage
- Gmax
maximal conductance
- HF
heart failure
- Po
open probability
- PKA
protein kinase A
- pWT
pseudo-wild-type
- RGK
Rad, Gem, Kir proteins
- rtTA
reverse transcriptional transactivator
Footnotes
DISCLOSURES
None
REFERENCES
- 1.Catterall WA. Structure and regulation of voltage-gated ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–555 [DOI] [PubMed] [Google Scholar]
- 2.Chen YH, Li MH, Zhang Y, He LL, Yamada Y, Fitzmaurice A, Shen Y, Zhang H, Tong L, Yang J. Structural basis of the alpha1-beta subunit interaction of voltage-gated ca2+ channels. Nature. 2004;429:675–680 [DOI] [PubMed] [Google Scholar]
- 3.Opatowsky Y, Chen CC, Campbell KP, Hirsch JA. Structural analysis of the voltage-dependent calcium channel beta subunit functional core and its complex with the alpha 1 interaction domain. Neuron. 2004;42:387–399 [DOI] [PubMed] [Google Scholar]
- 4.Van Petegem F, Clark KA, Chatelain FC, Minor DL Jr. Structure of a complex between a voltage-gated calcium channel beta-subunit and an alpha-subunit domain. Nature. 2004;429:671–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perez-Reyes E, Castellano A, Kim HS, Bertrand P, Baggstrom E, Lacerda AE, Wei XY, Birnbaumer L. Cloning and expression of a cardiac/brain beta subunit of the l-type calcium channel. J Biol Chem. 1992;267:1792–1797 [PubMed] [Google Scholar]
- 6.Singer D, Biel M, Lotan I, Flockerzi V, Hofmann F, Dascal N. The roles of the subunits in the function of the calcium channel. Science. 1991;253:1553–1557 [DOI] [PubMed] [Google Scholar]
- 7.Buraei Z, Yang J. The beta subunit of voltage-gated ca2+ channels. Physiol Rev. 2010;90:1461–1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Takahashi SX, Miriyala J, Colecraft HM. Membrane-associated guanylate kinase-like properties of beta-subunits required for modulation of voltage-dependent ca2+ channels. Proc Natl Acad Sci U S A. 2004;101:7193–7198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arias JM, Murbartian J, Vitko I, Lee JH, Perez-Reyes E. Transfer of beta subunit regulation from high to low voltage-gated ca2+ channels. FEBS Lett. 2005;579:3907–3912 [DOI] [PubMed] [Google Scholar]
- 10.Findeisen F, Minor DL Jr. Disruption of the is6-aid linker affects voltage-gated calcium channel inactivation and facilitation. J Gen Physiol. 2009;133:327–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reuter H, Scholz H. The regulation of the calcium conductance of cardiac muscle by adrenaline. J Physiol. 1977;264:49–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bean BP, Nowycky MC, Tsien RW. Beta-adrenergic modulation of calcium channels in frog ventricular heart cells. Nature. 1984;307:371–375 [DOI] [PubMed] [Google Scholar]
- 13.Yang L, Katchman A, Kushner J, Kushnir A, Zakharov SI, Chen BX, Shuja Z, Subramanyam P, Liu G, Papa A, Roybal D, Pitt GS, Colecraft HM, Marx SO. Cardiac cav1.2 channels require beta subunits for beta-adrenergic-mediated modulation but not trafficking. J Clin Invest. 2019;129:647–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Finlin BS, Crump SM, Satin J, Andres DA. Regulation of voltage-gated calcium channel activity by the rem and rad gtpases. Proc Natl Acad Sci U S A. 2003;100:14469–14474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu G, Papa A, Katchman AN, Zakharov SI, Roybal D, Hennessey JA, Kushner J, Yang L, Chen BX, Kushnir A, Dangas K, Gygi SP, Pitt GS, Colecraft HM, Ben-Johny M, Kalocsay M, Marx SO. Mechanism of adrenergic cav1.2 stimulation revealed by proximity proteomics. Nature. 2020;577:695–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fan J, Fan W, Lei J, Zhou Y, Xu H, Kapoor I, Zhu G, Wang J. Galectin-1 attenuates cardiomyocyte hypertrophy through splice-variant specific modulation of cav1.2 calcium channel. Biochim Biophys Acta Mol Basis Dis. 2019;1865:218–229 [DOI] [PubMed] [Google Scholar]
- 17.Tang ZZ, Liao P, Li G, Jiang FL, Yu D, Hong X, Yong TF, Tan G, Lu S, Wang J, Soong TW. Differential splicing patterns of l-type calcium channel cav1.2 subunit in hearts of spontaneously hypertensive rats and wistar kyoto rats. Biochim Biophys Acta. 2008;1783:118–130 [DOI] [PubMed] [Google Scholar]
- 18.Liao P, Li G, Yu DJ, Yong TF, Wang JJ, Wang J, Soong TW. Molecular alteration of ca(v)1.2 calcium channel in chronic myocardial infarction. Pflugers Arch. 2009;458:701–711 [DOI] [PubMed] [Google Scholar]
- 19.Liao P, Yu D, Lu S, Tang Z, Liang MC, Zeng S, Lin W, Soong TW. Smooth muscle-selective alternatively spliced exon generates functional variation in cav1.2 calcium channels. J Biol Chem. 2004;279:50329–50335 [DOI] [PubMed] [Google Scholar]
- 20.Liao P, Zhang HY, Soong TW. Alternative splicing of voltage-gated calcium channels: From molecular biology to disease. Pflugers Arch. 2009;458:481–487 [DOI] [PubMed] [Google Scholar]
- 21.Van Petegem F, Duderstadt KE, Clark KA, Wang M, Minor DL Jr. Alanine-scanning mutagenesis defines a conserved energetic hotspot in the cavalpha1 aid-cavbeta interaction site that is critical for channel modulation. Structure. 2008;16:280–294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang L, Katchman A, Samad T, Morrow JP, Weinberg RL, Marx SO. Beta-adrenergic regulation of the l-type ca2+ channel does not require phosphorylation of alpha1c ser1700. Circ Res. 2013;113:871–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Valencik ML, McDonald JA. Codon optimization markedly improves doxycycline regulated gene expression in the mouse heart. Transgenic Res. 2001;10:269–275 [DOI] [PubMed] [Google Scholar]
- 24.Adams PJ, Ben-Johny M, Dick IE, Inoue T, Yue DT. Apocalmodulin itself promotes ion channel opening and ca(2+) regulation. Cell. 2014;159:608–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Banerjee R, Yoder JB, Yue DT, Amzel LM, Tomaselli GF, Gabelli SB, Ben-Johny M. Bilobal architecture is a requirement for calmodulin signaling to cav1.3 channels. Proc Natl Acad Sci U S A. 2018;115:E3026–E3035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hess P, Lansman JB, Tsien RW. Different modes of ca channel gating behaviour favoured by dihydropyridine ca agonists and antagonists. Nature. 1984;311:538–544 [DOI] [PubMed] [Google Scholar]
- 27.Schroder F, Handrock R, Beuckelmann DJ, Hirt S, Hullin R, Priebe L, Schwinger RH, Weil J, Herzig S. Increased availability and open probability of single l-type calcium channels from failing compared with nonfailing human ventricle. Circulation. 1998;98:969–976 [DOI] [PubMed] [Google Scholar]
- 28.Yang L, Katchman A, Weinberg RL, Abrams J, Samad T, Wan E, Pitt GS, Marx SO. The pdz motif of the alpha1c subunit is not required for surface trafficking and adrenergic modulation of cav1.2 channel in the heart. J Biol Chem. 2015;290:2166–2174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Katchman A, Yang L, Zakharov SI, Kushner J, Abrams J, Chen BX, Liu G, Pitt GS, Colecraft HM, Marx SO. Proteolytic cleavage and pka phosphorylation of alpha1c subunit are not required for adrenergic regulation of cav1.2 in the heart. Proc Natl Acad Sci U S A. 2017;114:9194–9199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee SR, Sang L, Yue DT. Uncovering aberrant mutant pka function with flow cytometric fret. Cell Rep. 2016;14:3019–3029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Castellano A, Wei X, Birnbaumer L, Perez-Reyes E. Cloning and expression of a neuronal calcium channel beta subunit. J Biol Chem. 1993;268:12359–12366 [PubMed] [Google Scholar]
- 32.Lacerda AE, Kim HS, Ruth P, Perez-Reyes E, Flockerzi V, Hofmann F, Birnbaumer L, Brown AM. Normalization of current kinetics by interaction between the alpha 1 and beta subunits of the skeletal muscle dihydropyridine-sensitive ca2+ channel. Nature. 1991;352:527–530 [DOI] [PubMed] [Google Scholar]
- 33.Bichet D, Cornet V, Geib S, Carlier E, Volsen S, Hoshi T, Mori Y, De Waard M. The i-ii loop of the ca2+ channel alpha1 subunit contains an endoplasmic reticulum retention signal antagonized by the beta subunit. Neuron. 2000;25:177–190 [DOI] [PubMed] [Google Scholar]
- 34.Chien AJ, Zhao X, Shirokov RE, Puri TS, Chang CF, Sun D, Rios E, Hosey MM. Roles of a membrane-localized beta subunit in the formation and targeting of functional l-type ca2+ channels. J Biol Chem. 1995;270:30036–30044 [DOI] [PubMed] [Google Scholar]
- 35.Brice NL, Berrow NS, Campbell V, Page KM, Brickley K, Tedder I, Dolphin AC. Importance of the different beta subunits in the membrane expression of the alpha1a and alpha2 calcium channel subunits: Studies using a depolarization-sensitive alpha1a antibody. Eur J Neurosci. 1997;9:749–759 [DOI] [PubMed] [Google Scholar]
- 36.Dolphin AC. Beta subunits of voltage-gated calcium channels. J Bioenerg Biomembr. 2003;35:599–620 [DOI] [PubMed] [Google Scholar]
- 37.Arikkath J, Campbell KP. Auxiliary subunits: Essential components of the voltage-gated calcium channel complex. Curr Opin Neurobiol. 2003;13:298–307 [DOI] [PubMed] [Google Scholar]
- 38.Miriyala J, Nguyen T, Yue DT, Colecraft HM. Role of cavbeta subunits, and lack of functional reserve, in protein kinase a modulation of cardiac cav1.2 channels. Circ Res. 2008;102:e54–64 [DOI] [PubMed] [Google Scholar]
- 39.Meissner M, Weissgerber P, Londono JE, Prenen J, Link S, Ruppenthal S, Molkentin JD, Lipp P, Nilius B, Freichel M, Flockerzi V. Moderate calcium channel dysfunction in adult mice with inducible cardiomyocyte-specific excision of the cacnb2 gene. J Biol Chem. 2011;286:15875–15882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wu J, Yan Z, Li Z, Qian X, Lu S, Dong M, Zhou Q, Yan N. Structure of the voltage-gated calcium channel ca(v)1.1 at 3.6 a resolution. Nature. 2016;537:191–196 [DOI] [PubMed] [Google Scholar]
- 41.Chen X, Piacentino V 3rd, Furukawa S, Goldman B, Margulies KB, Houser SR. L-type ca2+ channel density and regulation are altered in failing human ventricular myocytes and recover after support with mechanical assist devices. Circ Res. 2002;91:517–524 [DOI] [PubMed] [Google Scholar]
- 42.Muth JN, Bodi I, Lewis W, Varadi G, Schwartz A. A ca(2+)-dependent transgenic model of cardiac hypertrophy: A role for protein kinase calpha. Circulation. 2001;103:140–147 [DOI] [PubMed] [Google Scholar]
- 43.Wang S, Ziman B, Bodi I, Rubio M, Zhou YY, D’Souza K, Bishopric NH, Schwartz A, Lakatta EG. Dilated cardiomyopathy with increased sr ca2+ loading preceded by a hypercontractile state and diastolic failure in the alpha(1c)tg mouse. PLoS One. 2009;4:e4133. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All supporting data are available within the article and its online data supplement.
For details on the experimental procedures, see the materials and methods section in the online data supplement.