

Abstract
Background:
Treatment characteristics, such as cranial radiation therapy (CRT), do not fully explain adiposity risk in childhood acute lymphoblastic leukemia (ALL) survivors. We aimed to characterize genetic variation related to adult body mass index (BMI) among survivors of childhood ALL.
Methods:
We analyzed genetic associations of BMI among 1,458 adult survivors of childhood ALL (median 20 years from diagnosis) using multiple approaches. We conducted a two-stage genome-wide association study in the Childhood Cancer Survivor Study (CCSS) and St Jude Lifetime (SJLIFE) Cohort. BMI is a highly polygenic trait in the general population. Within the known loci we estimated BMI percent variance explained and evaluated additive interactions (X2 test) with CRT in CCSS. We further evaluated the role of DNA methylation in CRT interaction among a subsample of ALL survivors.
Results:
In meta-analysis of CCSS and SJLIFE, we identified two novel loci associated with adult BMI among survivors of childhood ALL (LINC00856 rs575792008 and EMR1 rs62123082; PMeta<5e-8). We estimated that the >700 known loci explain 6.2% of the variation in adult BMI in childhood ALL survivors. Within the known loci we further identified significant main effects for 23 loci and statistical interaction with CRT at 9 loci (P<7.0e-5). At two CRT-interacting loci, DNA methylation patterns may differ by age.
Conclusions:
Adult survivors of childhood ALL have a similar genetic heritability for BMI to that observed in the general population. We provide evidence that treatment with CRT can modify the effect of genetic variants on adult BMI in childhood ALL survivors.
Keywords: childhood acute lymphoblastic leukemia, obesity, body mass index, genetics, survivorship, cranial radiation treatment
Precis:
Adult survivors of childhood acute lymphoblastic leukemia are at increased risk for obesity, particularly those treated with cranial radiation. We provide evidence that body mass index in adult survivors of childhood acute lymphoblastic leukemia is a genetically heritable trait similar to that in the general population and that the effect of genetic loci from the general population may be modified by treatment with cranial radiation.
Children diagnosed with acute lymphoblastic leukemia (ALL) currently have a greater than 90% chance of becoming long-term survivors due to advances in treatment.1 Among survivors of childhood ALL, age at diagnosis, sex, and treatment characteristics modify the risk for becoming obese2 and the greatest obesity risk is observed in females treated with CRT under 4 years of age.3 Treatment with CRT increases body mass index (BMI) by 0.29 and 0.41 kg/m2 per year in male and female ALL survivors, respectively, a significantly greater adipose gain than observed in siblings of survivors.4 Cancer survivors in general face a lifetime excess risk of chronic health conditions as a consequence of their treatment.5 Among these, obesity poses greater risk for metabolic dysfunction, cardiovascular outcomes, and specific subsequent malignancies.6-9
Research has established that demographic and treatment factors alone do not fully predict which survivors are at greatest risk for becoming obese.10 A previous study identified a candidate polymorphism in the leptin gene with sex-specific CRT interaction among overweight and obese ALL survivors.11 BMI is well-characterized in the general adult population as a polygenic trait 12, 13 for which hundreds of independent loci are currently known to explain 6.0% of the variance.13 Taken together, this suggests that in addition to treatment and demographic factors, genetic factors may account for unexplained excess BMI observed in adult survivors of childhood ALL. However, genome-wide investigations to evaluate general population genetic loci or novel genetic loci contributing to adiposity in ALL survivors have not been undertaken.
Here, we use a genome-wide approach in the Childhood Cancer Survivor Study (CCSS) and St. Jude Lifetime (SJLIFE) cohorts to investigate the association of genetic variation in adult BMI in survivors of childhood ALL. We contextualize our findings among childhood ALL survivors to loci identified in the general population, as well as genetic effects that are modified by specific treatment exposures.
Methods
Study populations and phenotype
The CCSS is a multi-center retrospective cohort of 25,664 individuals diagnosed with childhood cancer who survived five or more years after completion of cancer treatment and have been followed prospectively for the late effects of cancer treatment.14 SJLIFE is a clinically assessed cohort of individuals treated for childhood cancer at St. Jude Children’s Research Hospital who survived 10 or more years following primary cancer diagnosis.15 Analyses for this study were restricted to individuals treated for ALL as a primary cancer diagnosis. To investigate genetic variation of adiposity in survivors, we analyzed first adult body mass index (BMI, in kg/m2) for comparability to large-scale studies in individuals without cancer. The study populations and phenotype assessment are described in more detail in the Supplementary Methods.
Genome-wide association analyses
We performed germline genome-wide association study (GWAS) for BMI in survivors of childhood ALL of predominantly European ancestry. The first-stage GWAS was among 1,458 childhood ALL survivors diagnosed 1970-1986 in the CCSS and we sought to replicate variants with P<1e-7 in a second-stage fixed effects meta-analysis with an additional 389 survivors in the CCSS diagnosed 1987-1999 and 398 survivors in SJLIFE (PMeta<5e-8). Linear regression models of BMI were adjusted for traditional and treatment-specific risk factors. GWAS were additionally stratified by treatment with CRT to identify treatment-specific genetic effects. We further validated GWAS findings in 1) nearly 700,000 individuals of European ancestry in the GIANT consortium and UK Biobank 13, and 2) 101 ALL survivors of non-European ancestry in the CCSS diagnosed 1970-1986. Further study-specific information and methodologic details are provided in the Supplementary Methods.
BMI variants from the general population
To evaluate whether BMI-associated variants identified in the general population impact BMI among adult survivors of childhood ALL, we identified general population genetic variants from publicly available meta-analysis of individuals of European ancestry in the GIANT consortium and UK Biobank 13 and identified index variants within each known locus using the EasyStrata R package.16 For this study, we defined a genetic locus in a consistent manner with loci reported from general population GWAS of polygenic traits. We defined a genetic locus as a 1 Mb region around an index variant, regardless of linkage structure in the region. The index variant is defined as the top-associated SNP based on p-value within that locus (P<5e-8). Therefore, the SNP with the lowest p-value in each locus is referred to as the general population index variant and the 500kb region flanking that SNP defines the general population known locus. Using this approach, we identified 712 index variants associated with standardized BMI in GIANT and the UK Biobank.
Percent variance of BMI explained
To characterize the genetic heritability of adult BMI in survivors of childhood ALL, we estimated the percent variance of BMI explained by general population index variants in the CCSS Cohort diagnosed 1970-1986 (N=1,458). We used Genome-wide Complex Trait Analysis (GCTA) software 17 to estimate the proportion of variability in adult BMI explained by general population index variants present in the CCSS Cohort diagnosed 1970-1986 genotype data. We then compared our estimated percent variance explained to that reported by Yengo et al 13 among 8,552 older adults in the Health and Retirement Study (HRS).
Evaluation of general population genetic BMI variant-CRT interactions
As there is evidence that general population genetic variants interact with CRT to increase the risk of being overweight or obese in survivors of childhood ALL,11 we aimed to characterize SNP effects that could be modified by CRT treatment. For variants that mapped to the 500kb region flanking each general population index variant, we first assessed the SNP main effect in the overall GWAS among the CCSS Cohort diagnosed 1970-1986. We further performed statistical tests of interaction at all SNPs within the general population loci by incorporating a SNP x CRT interaction term into the previously described model among the overall sample from the CCSS Cohort diagnosed 1970-1986. A 1 df chi-squared test of interaction was used to evaluate genetic effects modified by treatment within loci that are known to be related to BMI in the general population. To assess both the main effects and CRT-modified effects of general population genetic loci, we performed a Bonferroni correction for 712 known loci (P<7.0e-5). We additionally sought to characterize the potential functional link of DNA methylation within loci statistically interacting with CRT, as detailed in the Supplementary Methods.
Results
We identified 1,458 adult survivors enrolled in the CCSS cohort and diagnosed 1970-1986 to characterize the relationship of genetic variation with adult BMI after childhood ALL treatment. To replicate association of individual genetic variants with BMI, we additionally identified 787 adult survivors in SJLIFE (n=398) and the CCSS diagnosed 1987-1999 (n=389). Among the three cohorts, ALL was diagnosed at 5.8-8.0 years, on average (Table 1). Children enrolled in the CCSS and diagnosed with ALL after 1987 were the least likely to receive CRT (28.0%) on newer treatment protocols than in SJLIFE (37.9%) or the CCSS diagnosed 1970-1986 (66.0%). Obesity was a common condition affecting up to one-third of childhood ALL survivors as young adults (Table 1). On average, SJLIFE had the oldest age at first adult follow-up (mean=29.1 years) and also the highest BMI (mean=28.2 kg/m2).
Table 1.
Characteristics of adult survivors of childhood acute lymphoblastic leukemia (ALL) enrolled in the CCSS and SJLIFE and assessed for genome-wide association of adult body mass index (BMI). Continuous traits are presented as mean ± SD and binary traits are presented as n (%).
| CCSS diagnosed 1970-1986 |
SJLIFE | CCSS diagnosed 1987-1999 |
|
|---|---|---|---|
| Characteristic | n=1,458 | n=398 | n=389 |
| Age at ALL diagnosis, years | 5.8 ± 4.4 | 7.0 ± 4.5 | 8.0 ± 5.3 |
| Age at first adult BMI, years | 25.2 ± 4.1 | 29.1 ± 8.4 | 27.5 ± 5.2 |
| First adult BMI, kg/m2 | 26.2 ± 5.8 | 28.2 ± 6.5 | 26.0 ± 5.1 |
| Obese | 305 (20.9%) | 129 (32.4%) | 75 (19.3%) |
| Non-obese | 1,153 (79.1%) | 269 (67.6%) | 314 (80.7%) |
| Female | 775 (53.2%) | 182 (45.7%) | 192 (49.4%) |
| Male | 683 (46.8%) | 216 (54.3%) | 197 (50.6%) |
| No cranial radiation therapy | 496 (34.0%) | 247 (62.1%) | 280 (72.0%) |
| Treated with cranial radiation therapy | 962 (66.0%) | 151 (37.9%) | 109 (28.0%) |
In the first-stage GWAS we identified 23 SNPs in 17 loci associated with adult BMI among childhood ALL survivors enrolled in the CCSS Cohort diagnosed 1970-1986 (P<1e-7; Supplemental Tables 1-3). These SNPs were moved to the second-stage of our GWAS approach, where we identified association of two novel genetic variants in meta-analyses with independent samples of adult survivors of childhood ALL from SJLIFE and the CCSS diagnosed 1987-1999 second-stage cohorts (PMeta<5e-8; Table 2). Among ALL survivors who were treated with CRT in childhood, we observed a statistically significant effect of a low frequency allele at LINC00856 rs575792008 on adult BMI (CCSS MAF=0.006, imputation quality=0.80; PMeta=2.3e-8). Individuals carrying the A allele at rs575792008 had adult BMI measures on average 7.7 kg/m2 greater than GG homozygotes when they received CRT treatment in childhood, whereas we did not observe a statistically significant effect of this allele among ALL survivors who were not treated with CRT. We additionally identified a statistically significant effect of rs62123082, an intronic variant in EMR1, on adult BMI (CCSS MAF=0.011, imputation quality=0.91; PMeta=2.8e-10). Individuals who were not treated with CRT in childhood and carried the G allele on average had an adult BMI that was 7.0 kg/m2 greater than AA homozygous individuals. Three variants in high linkage with this index variant were also assessed in the second stage of our GWAS and provide further support for evidence of BMI-associated genetic variation in this region. Genetic variation within both novel loci were further validated for association with BMI in the general population, as well as minority childhood ALL survivors (SNPs in each region with P-value < 1e-4, Figure 1).
Table 2.
Novel index variants identified for association with body mass index in meta-analysis (P<5e-8) of adult survivors of childhood acute lymphoblastic leukemia by cranial radiation therapy.
| CCSS Diagnosed 1970-1986 |
SJLIFE | CCSS Diagnosed 1987-1999 |
Meta-Analysis | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Treatment group |
Locus | SNP | Risk allele |
MAF | n | beta | se | p | n | beta | se | p | n | beta | se | p | beta | se | p |
| CRT | LINC00856 | rs575792008 | A | 0.006 | 962 | 7.5 | 1.4 | 9.4E-08 | 151 | 10.8 | 6.8 | 0.12 | 109 | NA | NA | NA | 7.7 | 1.4 | 2.3E-08 |
| no CRT | EMR1 intron | rs62123082 | G | 0.011 | 496 | 7.6 | 1.3 | 5.8E-09 | 247 | −4.7 | 4.3 | 0.28 | 280 | 8.5 | 2.5 | 8.7E-04 | 7.0 | 1.1 | 2.8E-10 |
CRT, cranial radiation therapy; MAF, minor allele frequency; NA, variant not present in cohort; SNP, single nucleotide polymorphism. The units for effect estimates are change in body mass index (kg/m2) per minor allele.
Figure 1.
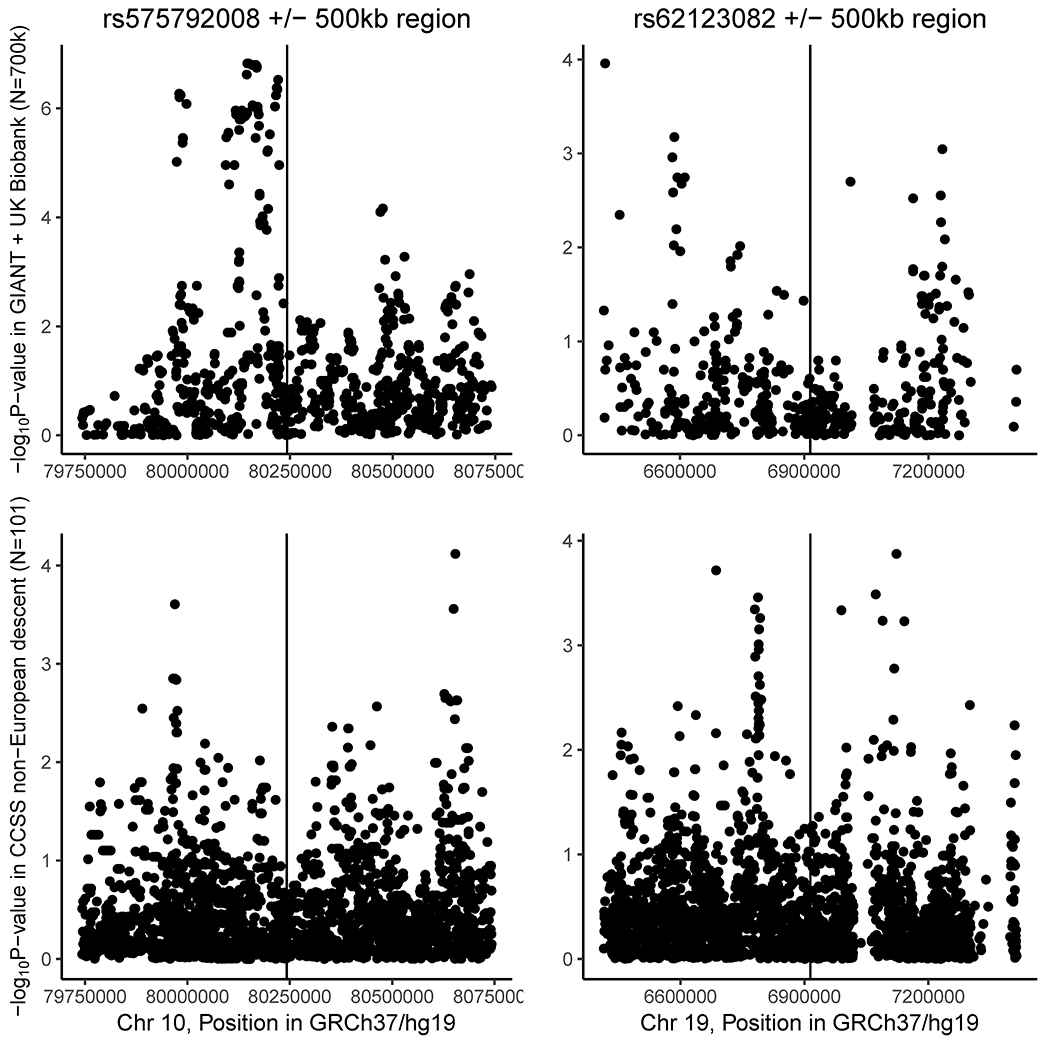
Regional associations for two novel loci associated with body mass index in adult survivors of childhood acute lymphoblastic leukemia among 1) meta-analysis of nearly 700,000 individuals of European ancestry in the GIANT consortium and UK Biobank (upper panels), and 2) 101 individuals of non-European ancestry in the Original CCSS cohort (lower panels).
To characterize the baseline genetic risk for increased adult adiposity in survivors of childhood ALL, we used multiple approaches to evaluate the known BMI-associated loci from the general population in the context of surviving childhood ALL. We first evaluated the role of these loci on BMI in childhood ALL survivors. Among childhood ALL survivors, 139 SNPs within 23 known BMI loci were associated with adult BMI after correcting for multiple testing (P<7.0e-5; Supplemental Figure 1) and all known general population loci harbored at least one variant that showed nominal association with adult BMI (P<0.05). Next, we estimated the genetic heritability of general population BMI index variants and found these variants explain 6.2% of the variability in adult BMI among survivors of childhood ALL (Figure 2). We further tested whether variants mapped to known general population BMI loci could have effect modification by CRT treatment. We identified 49 SNPs within 9 known loci that showed a statistically significant interaction with CRT on BMI in adult survivors of childhood ALL after correction for multiple testing (P<7e-5; Supplemental Figure 2 and Table 3). Individuals treated with CRT on average had an adult BMI that was 1.8-4.0 kg/m2 different for each minor allele carried when compared to individuals not treated with CRT and those who do not carry a minor allele. We estimated the interaction loci stratified by CRT and identified that common alleles within each of these nine loci had effect estimates in the opposite direction dependent on CRT treatment (Table 3). In an independent sample of individuals never treated for childhood cancer, we identified the 9 index variants showing statistical interaction with CRT also act as meQTLs for DNA methylation at 17 CpGs total (Supplemental Table 4). Of these CpGs, three CpGs mapped to two genomic regions associated with BMI in adult survivors of childhood ALL (Table 4). The rs145623412 CRT interaction index variant was identified to act as an meQTL throughout the lifecourse in an independent sample from the general population never treated with CRT and the associations of rs145623412 with methylation tended to increase from childhood through middle age (meQTL effects: 0.008 to 0.067 percent methylation per risk allele). At the CpGs identified through meQTL analysis, we identified significant associations of methylation with BMI in ALL survivors not treated with CRT (P<0.05). Methylation at these three CpGs was not associated with CRT (P>0.6).
Figure 2.
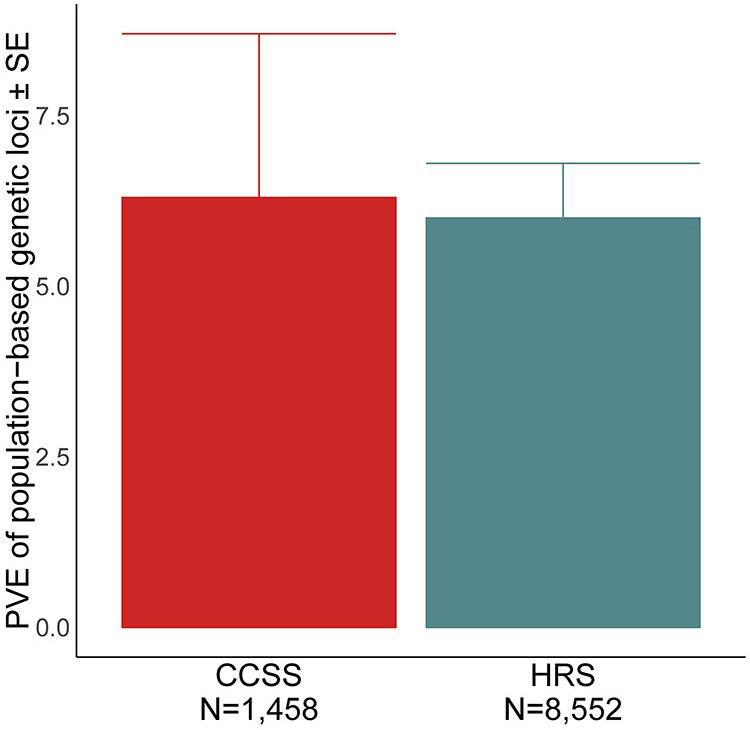
Percent variance (heritability) of adult body mass index explained by general population index variants among survivors of childhood acute lymphoblastic leukemia. Estimates from the Health and Retirement Study (HRS) are among older adults and as reported by Yengo et al.
Table 3.
Significant statistical interaction with cranial radiation therapy (CRT) of genetic variation within general population known loci in 1,458 adult survivors of childhood acute lymphoblastic leukemia enrolled in CCSS diagnosed 1970-1986 (P<7e-5).
| Locus | Lead SNP | Chr | Position | Risk allele |
MAF | Interaction beta |
P | Significant SNPs in Locus |
CRT treated | Not CRT treated |
||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| beta | P | beta | P | |||||||||
| SRGAP3 intron | rs730901 | 3 | 9,221,399 | A | 0.051 | 3.9 | 6.3E-05 | 2 | 1.8 | 0.0055 | −2.2 | 0.0018 |
| ADCY5 intron | rs34411635 | 3 | 123,052,845 | T | 0.200 | −2.4 | 2.0E-05 | 2 | −0.8 | 0.0210 | 1.5 | 0.0002 |
| BMPR1B intron | rs13113279 | 4 | 95,728,627 | A | 0.182 | −2.5 | 1.6E-05 | 2 | −1.1 | 0.0027 | 1.5 | 0.0004 |
| UNCX-MICALL2 | rs145623412 | 7 | 1,383,830 | A | 0.067 | −4.0 | 9.6E-06 | 17 | −1.5 | 0.0052 | 2.3 | 0.0005 |
| upstream LINGO2 | rs1928654 | 9 | 29,222,438 | C | 0.101 | −2.9 | 6.0E-05 | 8 | −0.9 | 0.0418 | 1.8 | 0.0005 |
| PRDM11 intron | rs11038334 | 11 | 45,174,418 | T | 0.413 | 1.8 | 5.0E-05 | 12 | 0.6 | 0.0260 | −1.2 | 0.0004 |
| CACNG3 intron | rs9922364 | 16 | 24,287,378 | T | 0.470 | 1.9 | 2.7E-05 | 3 | 0.4 | 0.1299 | −1.4 | 1.5E-05 |
| upstream MC4R | rs526653 | 18 | 58,196,139 | G | 0.100 | 3.1 | 3.1E-05 | 2 | 1.0 | 0.0175 | −1.9 | 0.0005 |
| IGLON5 intron | rs8103176 | 19 | 51,820,203 | G | 0.064 | −3.7 | 4.8E-05 | 1 | −0.6 | 0.2972 | 3.0 | 3.2E-06 |
CRT, cranial radiation therapy; MAF, minor allele frequency; SNP, single nucleotide polymorphism. The units for effect estimates are change in body mass index (kg/m2) per minor allele.
Table 4.
Methylation associated with index variants showing significant statistical interaction with cranial radiation therapy (CRT) for effect on body mass index (BMI). CpGs were identified as associated with index variants across the lifecourse in ARIES then associated with BMI in adult survivors of childhood acute lymphoblastic leukemia in the CCSS diagnosed 1970-1986.
| Locus | CRT Interaction Index SNP |
CpG | CpG-SNP Distance |
ARIES meQTL (CpG-SNP) |
CCSS Cohort (CpG-Outcome) |
|||||
|---|---|---|---|---|---|---|---|---|---|---|
| Timepoint | Effect | P | Methylation sample | Outcome | Effect | P | ||||
| SRGAP3 intron | rs730901 | cg24850121 | −43,645 | Middle Age | 0.003 | 6.3E-08 | Overall (n=96) | BMI | 0.0013 | 0.18 |
| CRT treated (n=47) | BMI | −0.0005 | 0.8 | |||||||
| Not CRT treated (n=49) | BMI | 0.0024 | 0.02 | |||||||
| Overall (n=96) | CRT | 0.0057 | 0.6 | |||||||
| UNCX-MICALL2 | rs145623412 | cg18423103 | 68,981 | Pregnancy | 0.058 | 2.1E-15 | Overall (n=96) | BMI | 0.0018 | 0.004 |
| Birth | 0.002 | 5.9E-10 | CRT treated (n=47) | BMI | 0.0014 | 0.16 | ||||
| Childhood | 0.009 | 3.4E-18 | Not CRT treated (n=49) | BMI | 0.0019 | 0.02 | ||||
| Adolescence | 0.046 | 2.5E-19 | Overall (n=96) | CRT | 0.0015 | 0.8 | ||||
| Middle Age | 0.067 | 8.0E-16 | ||||||||
| UNCX-MICALL2 | rs145623412 | cg15701281 | 78,739 | Pregnancy | 0.017 | 1.7E-17 | Overall (n=96) | BMI | −0.0009 | 0.17 |
| Birth | 0.023 | 1.9E-16 | CRT treated (n=47) | BMI | 0.0012 | 0.27 | ||||
| Childhood | 0.008 | 1.3E-11 | Not CRT treated (n=49) | BMI | −0.0021 | 0.006 | ||||
| Adolescence | 0.022 | 4.5E-15 | Overall (n=96) | CRT | 0.0006 | 0.9 | ||||
| Middle Age | 0.019 | 8.2E-12 | ||||||||
BMI, body mass index; CpG, cytosine-guanine dinucleotide; CRT, cranial radiation therapy; meQTL, methylation quantitative trait locus; SNP, single nucleotide polymorphism.
Discussion
Adult BMI in childhood ALL survivors is a genetically heritable trait similar to that estimated in the general population. We now provide evidence that the effect of BMI-associated loci identified in the general population may be further modified by treatment with CRT in survivors of childhood ALL. Within general population adult BMI loci, we evaluated statistical interactions of CRT and observed a BMI-increasing effect of CRT on major, rather than minor, alleles at these loci. Thus, genetic predisposition to excess adiposity may be enhanced when an individual is treated with CRT for childhood ALL.
We identified statistical interaction of CRT with germline genetic variation in cancer survivors. A possible biological mechanism of treatment interaction with inherited genetic variation is through altered transcriptional regulation. The biological mechanisms behind genetic main effects on BMI in the general population is not fully understood, but there is evidence of enrichment for transcriptional regulation through bioinformatic association of genetic variation with gene expression13 as well as involvement of central nervous system pathways that may regulate appetite control12. Alterations to DNA methylation is one potential epigenetic mechanism for radiation exposure to affect gene transcription into survivorship.18 Of the nine loci modified by CRT treatment, data from ENCODE supports variation in these loci belongs to enhancers, promoters, and DNase hypersensitivity sites. Additionally, in our assessment of CRT-interaction index variants in methylation quantitative trait loci estimated among 1,000 mother-child pairs in the ARIES study,19 we found evidence that genetic variation at 7 index variants is associated with altered methylation at 17 CpGs across the lifecourse in individuals not exposed to CRT. At three CpGs, one mapped to the SRGAP3 first intron and two in an intergenic region between UNCX and MICALL2, we identified a significant association of DNA methylation among individuals who survived childhood ALL but were not treated with CRT. Two CpGs were identified as associated with our CRT interaction index variant in UNCX-MICALL2, cg18423103 and cg15701281. The magnitude of association of rs145623412 with both CpGs increases after childhood in the general population. We also estimated methylation at these CpGs is significantly associated with BMI among ALL survivors who were not treated with CRT, but the effects were in the opposite direction (effects=0.0019 and −0.0021). However, we observed non-significant but similar magnitude and direction of effects for each of these CpGs among ALL survivors treated with CRT (effects=0.0014 and 0.0012). Therefore, methylation in these regions appears to change with age and may have differential relationships with BMI based on CRT treatment.
Further mechanistic studies may reveal insights to the connection between enhanced genetic effects on BMI after radiation exposure. Broadly, these regions are also associated with other traits in the general population that can also be late effects of survivorship. SRGAP3 rs730901 is an enhancer across multiple tissues and alters a BRCA1 regulatory motif.20 This variant is associated with serum metabolite levels of phenylacetate,21 which may serve as a functional link between the microbiome, gene expression, and disordered metabolism.22 ADCY5 encodes adenylate cyclase and genetic variation within the gene is consistently associated with multiple traits related to BMI, including birth weight,23 diabetes,24-27 and bone mineral density.28 BMPR1B encodes a bone morphogenetic protein receptor and this signaling pathway contributes to the differentiation of adipocytes from stem cells.29 Genetic variation in PRDM11 is involved in thyroid function30, 31 and blood pressure.32, 33 PRDM11 is further a transcriptional regulator that acts as a tumor suppressor34 and is implicated in leukemias.35 CACNG3 has also been identified as a tumor suppressor gene.36 MC4R is an intronless gene encoding the melanocortin receptor involved in hormonal signaling to stimulate adenylate cyclase. Variants in MC4R can cause autosomal dominant obesity. IGLON5 encodes an immunoglobulin secreted in the brain and syndromic deficiency results in disrupted sleep and ataxia.37
In an overall meta-analysis of three independent sets of ALL survivors (PMeta <5e-8), we identified two genetic loci harboring low frequency variants with relatively large effects that may contribute to some of the excess BMI observed in survivors of childhood ALL. In the general population BMI is a highly polygenic trait, characterized by small effects across hundreds of variants identified in large-scale meta-analyses. The power to identify novel loci in much smaller samples of childhood cancer survivors is limited, and we were only able to detect variants of low frequency that do not have adequate annotation for linkage or function in public databases such as the Genotype-Tissue Expression project. Further, polygenic traits often have inconsistent associations between study populations that are detected in meta-analysis38, as also observed in our data. However, there are multiple sources that corroborate regional adiposity associations for our GWAS findings. First, we validated these regional associations with measures of adult BMI in meta-analysis of GIANT and the UK Biobank, as well as minority survivors of childhood ALL not included in our first-stage GWAS of the CCSS Cohort diagnosed 1970-1986. The loci containing rs575792008 and rs62123082 also have suggestive associations with BMI in additional study populations39, 40 and harbor genome-wide significant associations with other measures of adiposity.40, 41 Therefore, we do not hypothesize these are novel BMI-associated genetic loci unique to cancer survivors, rather adiposity-associated loci that failed to reach genome-wide significance with BMI in our general population reference GWAS from GIANT and the UK Biobank. Both the replication of GWAS findings solely within our meta-analysis of CRT treatment strata and the statistical CRT interaction within general population loci support a background genetic risk for adiposity that is altered by ALL treatment. The modification of candidate gene main effects by radiation treatment is also supported by previous findings in the CCSS for a variant in the leptin gene and overweight or obese survivors of childhood ALL.11
Previous reports from the CCSS have shown that survivors of childhood ALL experience excess adiposity and additional health sequelae as they age into adulthood.2, 3, 8, 42 One of the strongest risk factors for obesity as an adult survivor is having received CRT. Despite contemporary treatment protocols reducing the need for CRT in order to achieve remission, obesity remains a concern among more recent survivors only treated with chemotherapy.43 Additionally, childhood cancer survivors are a unique study population with limited sample sizes. We assessed the genetic determinants of BMI in two large cohorts of childhood cancer survivors, the CCSS and SJLIFE. However, our study has limited power in evaluating both genetic main effects and CRT-interaction effects. As germline DNA samples are collected on additional survivors of childhood cancer, risk prediction models may be developed that incorporate gene-treatment interactions. However, the present study did not have sufficient sample size to validate interaction effects in independent samples due to the reduced need for CRT treatment on contemporary childhood ALL treatment protocols. We also were not powered to examine dose-specific interaction effects, which may further depend on age and sex of the survivor44.
In the largest sample to date of genotyped adult survivors of childhood ALL we identified statistical interaction of CRT treatment with genetic variants known to affect the baseline genetic risk for adiposity. We used BMI to assess adiposity of cancer survivors and did not identify novel genetic loci that contribute to an already well-characterized polygenic trait in the general population. Our findings suggest that CRT has a substantial effect on quality of life after surviving cancer at least partially through alterations of genetic effects on BMI, and should inform future studies of heritable metabolic traits in cancer survivors.
Supplementary Material
Acknowledgments
Funding
This work was supported by the Leukemia & Lymphoma Society Translational Research Program (Grant 6314-11: K.Y. Kamdar, principal investigator). This work was also supported in part by the National Institutes of Health (P30 CA125123 for the Dan L. Duncan Comprehensive Cancer Center) and Micaela’s Army Foundation in partnership with St. Baldrick’s Foundation.
CCSS is supported by the National Cancer Institute (U24 CA55727: G.T. Armstrong, principal investigator). SJLIFE is supported by the National Cancer Institute (U01 CA195547: M.M. Hudson and L.L. Robison, principal investigators; Cancer Center Support CORE grant CA21765: C. Roberts, principal investigator) and the American Lebanese Syrian Associated Charities.
Footnotes
Disclosures
The authors have no conflicts of interest.
REFERENCES
- 1.Hunger SP, Mullighan CG. Acute Lymphoblastic Leukemia in Children. New England Journal of Medicine. 2015;373: 1541–1552. [DOI] [PubMed] [Google Scholar]
- 2.Meacham LR, Gurney JG, Mertens AC, et al. Body mass index in long-term adult survivors of childhood cancer: a report of the Childhood Cancer Survivor Study. Cancer. 2005;103: 1730–1739. [DOI] [PubMed] [Google Scholar]
- 3.Oeffinger KC, Mertens AC, Sklar CA, et al. Obesity in adult survivors of childhood acute lymphoblastic leukemia: a report from the Childhood Cancer Survivor Study. Journal of Clinical Oncology. 2003;21: 1359–1365. [DOI] [PubMed] [Google Scholar]
- 4.Garmey EG, Liu Q, Sklar CA, et al. Longitudinal changes in obesity and body mass index among adult survivors of childhood acute lymphoblastic leukemia: a report from the Childhood Cancer Survivor Study. Journal of Clinical Oncology. 2008;26: 4639–4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Robison LL, Hudson MM. Survivors of childhood and adolescent cancer: life-long risks and responsibilities. Nature Reviews: Cancer. 2014;14: 61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mulrooney DA, Yeazel MW, Kawashima T, et al. Cardiac outcomes in a cohort of adult survivors of childhood and adolescent cancer: retrospective analysis of the Childhood Cancer Survivor Study cohort. BMJ. 2009;339: b4606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neville KA, Cohn RJ, Steinbeck KS, Johnston K, Walker JL. Hyperinsulinemia, impaired glucose tolerance, and diabetes mellitus in survivors of childhood cancer: prevalence and risk factors. Journal of Clinical Endocrinology and Metabolism. 2006;91: 4401–4407. [DOI] [PubMed] [Google Scholar]
- 8.Oeffinger KC, Mertens AC, Sklar CA, et al. Chronic health conditions in adult survivors of childhood cancer. New England Journal of Medicine. 2006;355: 1572–1582. [DOI] [PubMed] [Google Scholar]
- 9.Moke DJ, Hamilton AS, Chehab L, Deapen D, Freyer DR. Obesity and Risk for Second Malignant Neoplasms in Childhood Cancer Survivors: A Case-Control Study Utilizing the California Cancer Registry. Cancer Epidemiology, Biomarkers and Prevention. 2019;28: 1612–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wilson CL, Liu W, Yang JJ, et al. Genetic and clinical factors associated with obesity among adult survivors of childhood cancer: A report from the St. Jude Lifetime Cohort. Cancer. 2015;121: 2262–2270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ross JA, Oeffinger KC, Davies SM, et al. Genetic variation in the leptin receptor gene and obesity in survivors of childhood acute lymphoblastic leukemia: a report from the Childhood Cancer Survivor Study. Journal of Clinical Oncology. 2004;22: 3558–3562. [DOI] [PubMed] [Google Scholar]
- 12.Locke AE, Kahali B, Berndt SI, et al. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015;518: 197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yengo L, Sidorenko J, Kemper KE, et al. Meta-analysis of genome-wide association studies for height and body mass index in approximately 700000 individuals of European ancestry. Human Molecular Genetics. 2018;27: 3641–3649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robison LL, Mertens AC, Boice JD, et al. Study design and cohort characteristics of the Childhood Cancer Survivor Study: a multi-institutional collaborative project. Medical and Pediatric Oncology. 2002;38: 229–239. [DOI] [PubMed] [Google Scholar]
- 15.Hudson MM, Ehrhardt MJ, Bhakta N, et al. Approach for Classification and Severity Grading of Long-term and Late-Onset Health Events among Childhood Cancer Survivors in the St. Jude Lifetime Cohort. Cancer Epidemiology, Biomarkers and Prevention. 2017;26: 666–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Winkler TW, Kutalik Z, Gorski M, Lottaz C, Kronenberg F, Heid IM. EasyStrata: evaluation and visualization of stratified genome-wide association meta-analysis data. Bioinformatics. 2015;31: 259–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: a tool for genome-wide complex trait analysis. American Journal of Human Genetics. 2011;88: 76–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rodriguez-Paredes M, Esteller M. Cancer epigenetics reaches mainstream oncology. Nature Medicine. 2011;17: 330–339. [DOI] [PubMed] [Google Scholar]
- 19.Gaunt TR, Shihab HA, Hemani G, et al. Systematic identification of genetic influences on methylation across the human life course. Genome Biology. 2016;17: 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kheradpour P, Kellis M. Systematic discovery and characterization of regulatory motifs in ENCODE TF binding experiments. Nucleic Acids Research. 2014;42: 2976–2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Suhre K, Shin SY, Petersen AK, et al. Human metabolic individuality in biomedical and pharmaceutical research. Nature. 2011;477: 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoyles L, Fernandez-Real JM, Federici M, et al. Molecular phenomics and metagenomics of hepatic steatosis in non-diabetic obese women. Nature Medicine. 2018;24: 1070–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Warrington NM, Beaumont RN, Horikoshi M, et al. Maternal and fetal genetic effects on birth weight and their relevance to cardio-metabolic risk factors. Nature Genetics. 2019;51: 804–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dupuis J, Langenberg C, Prokopenko I, et al. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nature Genetics. 2010;42: 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Morris AP, Voight BF, Teslovich TM, et al. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nature Genetics. 2012;44: 981–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao W, Rasheed A, Tikkanen E, et al. Identification of new susceptibility loci for type 2 diabetes and shared etiological pathways with coronary heart disease. Nature Genetics. 2017;49: 1450–1457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Manning AK, Hivert MF, Scott RA, et al. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nature Genetics. 2012;44: 659–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Morris JA, Kemp JP, Youlten SE, et al. An atlas of genetic influences on osteoporosis in humans and mice. Nature Genetics. 2019;51: 258–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang H, Song TJ, Li X, et al. BMP signaling pathway is required for commitment of C3H10T1/2 pluripotent stem cells to the adipocyte lineage. Proceedings of the National Academy of Sciences of the United States of America. 2009;106: 12670–12675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Teumer A, Chaker L, Groeneweg S, et al. Genome-wide analyses identify a role for SLC17A4 and AADAT in thyroid hormone regulation. Nat Commun. 2018;9: 4455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Porcu E, Medici M, Pistis G, et al. A meta-analysis of thyroid-related traits reveals novel loci and gender-specific differences in the regulation of thyroid function. Plos Genetics. 2013;9: e1003266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoffmann TJ, Ehret GB, Nandakumar P, et al. Genome-wide association analyses using electronic health records identify new loci influencing blood pressure variation. Nature Genetics. 2017;49: 54–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Giri A, Hellwege JN, Keaton JM, et al. Trans-ethnic association study of blood pressure determinants in over 750,000 individuals. Nature Genetics. 2019;51: 51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fog CK, Asmar F, Come C, et al. Loss of PRDM11 promotes MYC-driven lymphomagenesis. Blood. 2015;125: 1272–1281. [DOI] [PubMed] [Google Scholar]
- 35.Chen C, Bartenhagen C, Gombert M, et al. Next-generation-sequencing-based risk stratification and identification of new genes involved in structural and sequence variations in near haploid lymphoblastic leukemia. Genes, Chromosomes and Cancer. 2013;52: 564–579. [DOI] [PubMed] [Google Scholar]
- 36.Kumar RD, Searleman AC, Swamidass SJ, Griffith OL, Bose R. Statistically identifying tumor suppressors and oncogenes from pan-cancer genome-sequencing data. Bioinformatics. 2015;31: 3561–3568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wenninger S Expanding the Clinical Spectrum of IgLON5-Syndrome. J Neuromuscul Dis. 2017;4: 337–339. [DOI] [PubMed] [Google Scholar]
- 38.Panagiotou OA, Willer CJ, Hirschhorn JN, Ioannidis JP. The power of meta-analysis in genome-wide association studies. Annu Rev Genomics Hum Genet. 2013;14: 441–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Anderson D, Cordell HJ, Fakiola M, et al. First genome-wide association study in an Australian aboriginal population provides insights into genetic risk factors for body mass index and type 2 diabetes. PloS One. 2015;10: e0119333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kichaev G, Bhatia G, Loh PR, et al. Leveraging Polygenic Functional Enrichment to Improve GWAS Power. American Journal of Human Genetics. 2019;104: 65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shungin D, Winkler TW, Croteau-Chonka DC, et al. New genetic loci link adipose and insulin biology to body fat distribution. Nature. 2015;518: 187–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Meacham LR, Sklar CA, Li S, et al. Diabetes mellitus in long-term survivors of childhood cancer. Increased risk associated with radiation therapy: a report for the childhood cancer survivor study. Archives of Internal Medicine. 2009;169: 1381–1388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Foster KL, Kern KD, Chambers TM, et al. Weight trends in a multiethnic cohort of pediatric acute lymphoblastic leukemia survivors: A longitudinal analysis. PloS One. 2019;14: e0217932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Armstrong GT, Stovall M, Robison LL. Long-term effects of radiation exposure among adult survivors of childhood cancer: results from the childhood cancer survivor study. Radiation Research. 2010;174: 840–850. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.