

Abstract
Patients with long-term estrogen-deprived breast cancer (BC), after resistance to tamoxifen or aromatase inhibitors develops, can experience tumor regression when treated with estrogens. Estrogen’s anti-tumor effect is attributed to apoptosis via the estrogen receptor (ER). Estrogen treatment can have unpleasant gynecological and non-gynecological adverse events thus the development of safer estrogenic agents remains a clinical priority. Here, we study synthetic selective estrogen mimics (SEMs) BMI-135 and TTC-352, and the naturally-occurring estrogen estetrol (E4), which are proposed as safer estrogenic agents compared to 17β-estradiol (E2), for the treatment of endocrine-resistant BC. TTC-352 and E4 are being evaluated in BC clinical trials. Cell viability assays, real-time polymerase chain reaction, immunoblotting, ERE DNA pull downs, Mass spectrometry, X-ray crystallography, docking and molecular dynamic simulations, live cell imaging, and annexin V staining were conducted in 11 biologically-different BC models. Results were compared with the potent full agonist E2, less potent full agonist E4, the benchmark partial agonist triphenylethylene bisphenol (BPTPE), and antagonists 4-hydroxytamoxifen and endoxifen. We report ERα’s regulation and coregulators’ binding profiles with SEMs and E4. We describe TTC-352’s pharmacology as a weak full agonist and anti-tumor molecular mechanisms. This study highlights TTC-352’s benzothiophene scaffold that yields an H-bond with Glu353, which allows Asp351-to-helix 12 (H12) interaction; sealing ERα’s ligand binding domain, recruiting E2-enriched coactivators, and triggering rapid ERα-induced unfolded protein response (UPR) and apoptosis, as the basis of its anti-cancer properties. BPTPE’s phenolic OH yields an H-Bond with Thr347, which disrupts Asp351-to-H12 interaction; delaying UPR and apoptosis, and increasing clonal evolution risk.
Keywords: Selective Human Estrogen Receptor Partial Agonists (ShERPAs), estetrol (E4), estrogen receptor (ER), coactivators, unfolded protein response, apoptosis
Introduction
Estrogen therapy can cause tumor regression in breast cancer (BC) patients, who were long-term estrogen-deprived (LTED) with tamoxifen (TAM), which blocks estrogen binding to the BC estrogen receptor (ER), or aromatase inhibitors (AIs), which inhibit estrogen synthesis via the BC aromatase enzyme system (1). Long-term adjuvant TAM therapy (2) became the translational strategy of choice for LTED treatment (3), whereby 5 years of TAM therapy leads to a new phase of endocrine resistance; characterized by TAM-induced BC growth and 17β-estradiol (E2)-induced BC apoptosis (4). Today, AIs are widely used for LTED in treating postmenopausal women with ER-positive BC. The Oxford overview analyses show that at least 50% of BC recurrences occur more than 5 years after diagnosis (5). This prompted investigators to provide a guide (6) to improve the risk-benefit of long-term adjuvant endocrine therapy in concordance with the patient’s individualized risk for early-versus late-distant recurrence.
Estrogen triggers an endoplasmic reticulum (EnR) stress response, the unfolded protein response (UPR), and induce apoptosis in LTED BC models (7). Numerous clinical trials (8-12) demonstrated the benefit of estrogen-induced tumor regression in LTED BC patients. However, estrogen therapy can have unpleasant gynecological and non-gynecological adverse events (AEs). The research and development of safer estrogenic agents for the treatment of drug-resistant or metastatic BC (MBC) remains a clinical priority, especially with BC expected to double by 2030 than it was in 2011 (13), and MBC being associated with significantly higher health care costs (14). The majority of BC will be ER-positive, which has a high risk of recurrence and residual relapse even with clinically low-risk disease (T1N0) (15).
Three Selective Human ER Partial Agonists (ShERPAs; including pilot BMI-135 and clinically-tested TTC-352) (Fig. 1) (16), are proposed as safer estrogen mimics for the treatment of endocrine-resistant BC. In preclinical studies, TTC-352 demonstrated efficacy and tolerability (17). A clinical trial using TTC-352 in hormone receptor-positive MBC patients, who had progressed on at least two lines of endocrine therapy, shows manageable safety and early clinical evidence of activity (11).
Figure 1. Chemical structures of planar estrogens, angular estrogens, SERMS, and ShERPAs.

The box (in green) highlights the benzothiophene scaffold embedded in raloxifene and arzoxifene structures, of which the ShERPAs’ structures were based upon. The continuous box (in yellow) highlights the phenyl ring bearing OH of triphenylethylenes (TPEs): trihydroxytriphenylethylene (3OHTPE) and BPTPE (46), which makes them angular estrogens/partial agonists. The dashed box (in brown) highlights the absence of OH on the phenyl ring of the Z-isomer of dihydroxytriphenylethylene (Z2OHTPE), which makes it an angular estrogen/full agonist like E2 and diethylstilbestrol (DES) (46).
Estetrol (E4) (Fig. 1), a fetal estrogen that activates the nuclear ERα with a vasculoprotective effect, is proposed as a safer estrogen for the treatment of endocrine-resistant BC (12), advanced prostate cancer, and use for hormone replacement therapy as well as oral contraception. The combination of E4 and progestin drospirenone is subject to FDA approval, with the possibility of E4 becoming the first natural estrogen approved in a contraceptive product in the US, and the first new estrogen introduced in the U.S. in 50 years. A clinical trial using E4, to treat advanced BC, shows that the majority of patients experienced favorable subjective effects on wellbeing, and one patient completed the phase 1/IIA with stable disease after 24 weeks of treatment (12).
The ER in LTED BC is at the crossroads of mediating the anti-tumor actions of therapeutics as well as BC growth through ERα-activating mutations (18). Investigating ERα’s regulation and DNA-or-ligand-binding profiles with ShERPAs and E4, enhances our understanding of how these therapeutics influence cancer through ER.
ERα gene ESR1 point mutations in the ligand-binding domain (LBD) lead to constitutive hormone-independent activation of ER, and are identified in approximately 40% of MBC (19). These mutations are especially enriched in BC patients pretreated with AIs (20).
The expression level and stability of ER is modulated by estrogens and anti-estrogens. Two regulatory mechanisms that govern the steady-state of ER messenger RNA (mRNA) and protein levels in BC cells are documented (21). Model I ER regulation reflects the rapid down-regulation of the steady-state of ER mRNA and protein levels upon estrogen exposure, and is exemplified in MCF-7:WS8 BC, ovarian carcinoma (PEO4), and the rat and mouse uterus. Model II ER regulation reflects the up-regulation of the steady-state level of ER mRNA, alongside the maintenance of the ER protein level upon estrogen exposure, and is exemplified in T47D:A18 BC. In MCF-7:WS8 and T47D:A18, the antiestrogen 4-hydroxytamoxifen (4OHT) has little effect on the ER mRNA level, but accumulates the ER protein over time (21). The Selective ER Downregulator ICI 182,780 fulvestrant (ICI), has little effect on the ER mRNA level in MCF-7:WS8, whereas, it causes its reduction in T47D:A18, and ICI dramatically reduces the ER protein level in both (21).
ERα’s transcriptional control of diverse downstream gene expression is dictated by the ability of bound estrogens or anti-estrogens (22) to recruit and assemble primary steroid receptor coactivators (steroid receptor coactivator −3 (SRC-3); also known as nuclear receptor co-activator-3 (NCOA3), and A1B1), followed by secondary coactivators (p300/ E1A binding protein p300 [EP300]); in what is known as minimal receptor-coactivator complex (23). This facilitates chromatin remodeling and transcriptional activation. A model was proposed for the assembly mechanism of the quaternary complex: the two ligand-bound ERα monomers each, independently, recruits one SRC-3 protein through the transactivation domain of ERα, and the two SRC-3s, subsequently, bind to different regions of one p300 protein via multiple contacts (23).
We investigate ShERPAs and E4’s influence on BC through studying their ERα regulation and coactivators’ binding profiles in biologically-different BC models. Furthermore, we present the first X-ray crystallography of TTC-352 with the mutant ER, its pharmacology, and molecular mechanisms of tumor regression in LTED endocrine-resistant BC.
Materials and Methods
Materials
E2, E4, 4OHT, and raloxifene (Ralox) were purchased from Sigma-Aldrich. Endoxifen (Endox) was purchased from Santa Cruz Biotechnology, and ICI from Tocris Bioscience. Triphenylethylene bisphenol (BPTPE) was originally synthesized at the Organic Synthesis Facility, Fox Chase Cancer Center (Philadelphia, PA) (24). The ShERPAs BMI-135 and TTC-352 were a gift from Drs Debra Tonetti and Gregory R. J. Thatcher (University of Chicago, IL). The PERK inhibitor GSK G797800 was purchased from Toronto Research Chemicals. The IRE1α Inhibitor MKC-3946 was purchased from Calbiochem. Thioflavin T (ThT) was purchased from Sigma-Aldrich. For Western blotting, anti-ERα (sc-544), anti-eIF2α (D-3), and anti-β-actin (C-4), were purchased from Santa Cruz Biotechnology. Anti-phospho-eIF2α (Ser51) (#9721), anti-ATF4 (D4B8), anti-CHOP (L63F7), and anti-cleaved PARP (Asp214) (19F4), were purchased from Cell Signaling Technology. Anti-XBP1 (isoforms non-spliced and spliced, ab37152) was purchased from Abcam. For immunoblotting validations for the ERE DNA pull-downs, the antibodies used were: anti-MLL4 (Millipore Sigma, ABE1867), anti-NCOA1 (Santa Cruz Biotechnology, sc-32789), anti-NCOA3 (custom-made in Bert W. O’Malley’s laboratory, BCM (25)), anti-MED6 (Santa Cruz Biotechnology, clone D-2), anti-MED17 (Novus Biologicals, clone 4D4), and anti-ESR1 (Santa Cruz Biotechnology, sc-543). For ChIP’s pull-downs, the antibodies used were: anti-SRC-3 (clone AX15.3, 1 μg/μl; 5 μg per reaction) (Abcam), and normal mouse IgG as IP negative control (2 μg/μl; 5 μg per reaction) (Santa Cruz Biotechnology).
Cell culture
Wild-type (WT) estrogen-dependent BC MCF-7:WS8 (26); mutant p53 estrogen-dependent BC T47D:A18 (27); estrogen-responsive, ER-positive, progesterone receptor (PgR)-positive, and human epidermal growth factor receptor 2 (HER2)-positive luminal B BC BT-474 (28); estrogen-responsive, ER-positive, PgR-positive, and androgen receptor-positive luminal A BC ZR-75-1 (29); the first in vitro cellular model recapitulating acquired-TAM resistance developed in athymic mice in vivo MCF-7:PF (30); anti-hormone-resistant estrogen-independent BC MCF-7:5C (31); anti-hormone-sensitive estrogen-independent BC MCF-7:2A (32); anti-hormone (raloxifene)-resistant, estrogen-independent BC MCF-7:RAL (33); TAM-sensitive, estrogen-independent, ER-positive BC LCC1 (34, 35); TAM-resistant and ICI-sensitive, estrogen-independent, ER-positive BC LCC2 (36); and TAM-and-ICI-cross resistant, ER-positive BC LCC9 (37), were cultured as described previously.
Cell viability and proliferation assays
The biological properties of compounds (E2, BMI-135, TTC-352, BPTPE, 4OHT, endoxifen, and raloxifene) in cells lines, were evaluated by assessing the DNA content of the cells, as a measure of cell viability, using a DNA fluorescence Quantitation kit (Bio-Rad Laboratories) as described previously (38). The DNA fingerprinting pattern of these cell lines is consistent with that reported by ATCC. All cell lines were validated according to their short tandem repeat (STR) profiles at UT MD Anderson Cancer Center Characterized Cell Line Core (CCLC). The STR patterns of all cell lines were consistent with those from the CCLC standard cells (Supplementary Table S1). The calculated half maximal effective concentration (EC50) for all test compounds, used in treating these cell lines, are summarized in Supplementary Table S2.
Quantitative real-time reverse transcription-polymerase chain reaction (RT-PCR)
Total RNA was isolated from MCF-7:WS8 and MCF-7:5C cells using MagMAX-96 Total RNA Isolation Kit (Applied Biosystems), and processed using Kingfisher Duo Prime magnetic particle processor (Thermo Scientific). The cDNA was synthesized using High Capacity cDNA Reverse transcription kit (Applied Bioscience). Quantitative real-time PCR assays were performed using Power SYBR Green PCR Master Mix (Applied Biosystems) and a QuantStudio 6 Flex real-time PCR System (Applied Biosystems). All primers were synthesized by Integrated DNA Technologies Inc. All data were normalized using reference gene 36B4.
Immunoblotting
Cells were treated with different compounds (E2 [1 nM], BMI-135 [1 μM], TTC-352 [1 μM], E4 [1 μM], BPTPE [1 μM], 4OHT [1 μM], endoxifen [1 μM], raloxifene [1 μM], ICI [1 μM], thapsigargin [1 μM], GSK G797800 [10 μM], and MKC-3946 [20 μM]), for different periods, and harvested in cell lysis buffer (Cell Signaling Technology) supplemented with Protease Inhibitor Cocktail Set I and Phosphatase Inhibitor Cocktail Set II (Calbiochem). Immunoblotting was performed as described previously (38). Analysis was validated by densitometry using Image J (National Institutes of Health). Densitometry data is presented in Supplementary Tables S3-4.
ERE DNA pulldowns
MCF-7:WS8 and MCF-7:5C cells were grown in 20-30 15-cm dishes, and nuclear extracts (NEs) were made. The 4x estrogen response element (ERE) DNA pulldown assays were performed; by first immobilizing four copies of the Xenopus Vitellogennin ERE sequences onto Dynabeads M280 streptavidin as described previously (39). One mg of NE from MCF-7:WS8 or MCF-7:5C cells, and 0.5 μg recombinant ERα protein (Invitrogen), were added to 4xERE-beads, with either vehicle controls (ethanol or DMSO), or E2 (100 nM), BMI-135 (1 μM), TTC-352 (1 μM), E4 (1 μM), BPTPE (1 μM), and endoxifen (1 μM), for a 1.5 hour incubation at 4°C. After performing three washes, the final coregulator-ERα-ERE DNA complexes were eluted from the beads in 30 μl 2x SDS-sample buffer for mass spectrometry as described previously (39). The detailed methodology is presented in supplementary materials.
Mass spectrometry (MS)
Label-free liquid chromatography-MS was performed with quantification; by intensity-based absolute quantification (iBAQ) (40), and the ERE/ER coregulator binding reactions were analyzed as described previously (39). Samples were electrophoresed on 10% NuPAGE gels, 4 broad-region bands were excised, and the proteins were in-gel digested with trypsin. For each experiment, the peptides were combined into two pools, and measured on a Thermo Scientific Orbitrap Elite mass spectrometer coupled to an EASY nLC1200 UHPLC system. The raw data were searched in Proteome Discoverer suite (PD2.2) with Mascot 2.5 engine. For peptide quantification, the PD 2.2 Peak Area Detector module was used, and for gene-centric inference and label-free quantitation based on the iBAQ method, the gpGrouper software was used. All raw MS and gpGrouper result files are deposited into the ProteomeXchange Consortium (http://proteomecentral.proteomexchange.org) (41). Compiled results are provided in Supplementary Table S5.
X-ray crystallography
The 6×His-TEV-tagged ER-Y537S LBD mutant was expressed in E.coli BL21 (DE3), and purified as described previously (42). The LBD (5 mg/mL) was incubated with 1 mM TTC-352 and 2.5 mM GRIP peptide at 4°C overnight. The detailed methodology is presented in supplemental materials. Each structure was validated, and deposited in the Protein Data Bank (accession code 7JHD).
Molecular dynamics (MD) simulations
MD simulations were performed for the following systems: hERαLBD in complex with E2, TTC-352, and BPTPE, using the Desmond software (Schrödinger Release 2019-3, Schrödinger, LLC, New York, NY, 2019). The detailed methodology is presented in supplementary materials.
Live cell imaging and analysis to detect cellular stress
MCF-7:5C cells were seeded into three 15 μ-slide 2-well chambered coverslip slides (Ibidi). After 24 hours, cells were treated with vehicle control (DSMO [0.1%]), positive control thapsigargin (1 μM), TTC-352 (1 μM), and 4OHT (1 μM). After 72-hour-treatment, ThT was prepared as described previously (43), and used in co-treating the cells for 1 hour. The Hoechst 33342 dye (Thermo Fisher Scientific), was prepared at a final concertation of 5 μg/mL, and used in staining the cells for 15 minutes. Live cell images were taken at a 38 ms exposure under a 20X/0.7 objective with ZEISS Celldiscoverer 7 (Carl Zeiss AG). Fluorescent images were converted to 12-bit before being quantified by the ZEISS Zen Software Module-Image Analysis. Cells from each image were manually counted to normalize the fluorescent data per cell. Relative intensity per cell =ThT intensity/cell count, was generated for each treatment per image. An average of the relative intensity per cell (using 3 images per treatment) was then calculated to give a final quantification. The excitation and emission settings were: Hoechst 33342 (Ex. 348 nm, Em. 455 nm), and ThT (Ex. 433 nm, Em. 475 nm).
Annexin V binding assays to detect apoptosis
A FITC Annexin V Apoptosis Detection Kit I (BD Pharmingen) was used to quantify apoptosis of cells through flow cytometry. In brief, MCF-7:5C, MCF-7:2A and MCF-7:RAL cells were seeded in 10-cm dishes. After 24 hours, the cells were treated with different compounds (E2 [1 nM], TTC-352 [1 μM], 4OHT [1 μM], raloxifene [1 μM], GSK G797800 [10 μM], and MKC-3946 [20 μM]) for different time periods. Cells were suspended in 1x binding buffer, and 1*105 cells were stained simultaneously with FITC-labeled Annexin V and propidium iodide (PI) for 15 minutes at room temperature. The cells were analyzed using a BD Accuri C6 plus flow cytometer.
Chromatin immunoprecipitation (ChIP) assays
The ChIP assay was performed as described previously (44, 45). The DNA fragments were purified using Qiaquick PCR purification kit (Qiagen). Then, 2 μl of eluted DNA was used for RT-PCR analysis. The primer sequences used are: GREB1 proximal ERE enhancer site amplification: 5’-GTGGCAACTGGGTCATTCTGA-3’ sense, 5’-CGACCCACAGAAATGAAAAGG-3’ anti-sense (Integrated DNA Technologies). The data are expressed as percent input, of starting chromatin material, after subtracting the percent input pull-down of the IP negative control.
Statistical analysis
All reported values are means ± standard deviation. Statistical comparisons were assessed using two-tailed Student t tests. Results were considered statistically significant if the P value was less than 0.05.
Results
Effects of TTC-352 on cell viability in multiple BC models
Cell viability assays were used to test the biological properties of compounds. TTC-352 exhibits a full agonist action, similar to E2, across eight BC cell lines that are estrogen-dependent (MCF-7:WS8, T47D:A18, BT-474, ZR-75-1, and MCF-7:PF), estrogen-independent (MCF-7:5C, MCF-7:2A, and MCF-7:RAL), endocrine-sensitive (MCF-7:2A), endocrine-resistant (MCF-7:PF, MCF-7:5C, and MCF-7:RAL), mutant p53 (T47D:A18), HER2-positive (BT-474), luminal A (ZR-75-1), and luminal B (BT-474).
The concentration 1 μM for TTC-352 achieved either the maximal cellular growth (P-value < 0.05 compared to vehicle control) (Supplementary Fig. S1A-E), or the maximal cellular death (P < 0.05 compared to vehicle control) (Supplementary Fig. S1F-H). TTC-352 was shown to be a less potent full agonist compared to E2 (Supplementary Fig. S1 and Supplementary Table S2). The calculated EC50s are summarized in Supplementary Table S2, and the detailed results are presented in supplementary materials.
TTC-352 induces the transcriptional activity of ER similar to E2 in WT BC MCF-7:WS8 and apoptotic-type BC MCF-7:5C
qRT-PCR was used to assess the transcriptional activity of ERα on estrogen-responsive genes (TFF1 and GREB1) with TTC-352. After 24-hour-treatment in MCF-7:WS8, TTC-352 significantly (P < 0.05) increased the levels of TFF1 and GREB1 mRNAs compared to vehicle controls (Fig. 2A-B). On the other hand, BPTPE induced a partial increase in the levels of TFF1 and GREB1 mRNAs, significantly (P < 0.05) less than that of E2 and TTC-352 (Fig. 2A-B). The minimal concentration that produced a complete increase in the levels of TFF1 and GREB1 was at 10−6 M for TTC-352 (P < 0.05 compared to vehicle control).
Figure 2. Transcriptional activity of well-characterized estrogen-responsive genes TFF1 and GREB1 in WT MCF-7:WS8 and LTED endocrine-resistant MCF-7:5C with test compounds.

A, mRNA expression of TFF1 (or pS2) in MCF-7:WS8 cells after 24-hour-treatment with 1 nM E2, and 1 μM for other test compounds. B, mRNA expression of GREB1 in MCF-7:WS8 cells after 24-hour-treatment with 1 nM E2, and 1 μM for other test compounds. C, mRNA expression of TFF1 in MCF-7:5C cells after 24-hour-treatment with 1 nM E2, and 1 μM for other test compounds. D, mRNA expression of GREB1 in MCF-7:5C cells after 24-hour-treatment with 1 nM E2, and 1 μM for other test compounds.
After 24-hour-treatment in MCF-7:5C, TTC-352 significantly (P < 0.05) increased the levels of TFF1 and GREB1 mRNAs compared to vehicle controls (Fig. 2C-D). On the other hand, BPTPE induced a partial increase in the levels of TFF1 and GREB1 mRNAs, significantly (P < 0.05) less than that of E2 and TTC-352 (Fig. 2C-D). The minimal concentration that produced a complete increase in the levels of TFF1 and GREB1 was at 10−6 M for TTC-352 (P < 0.05 compared to vehicle control).
Overall, the induction of the levels of TFF1 and GREB1 mRNAs by TTC-352 in MCF-7:WS8 and MCF-7:5C is similar to that by full agonist E2, only at a higher concentration.
Effects of TTC-352, BMI-135, and E4 on ERα regulation in multiple BC models
Western blotting and densitometry were used to assess the regulation of ERα protein levels with compounds. In MCF-7:WS8, TTC-352 was able to down-regulate the protein levels of ERα after 72-hour-treatment, compared to vehicle control, and similar to E2 and E4 (Model I) (Fig. 3A and Supplementary Table. S3). Whereas, BMI-135 seems to have a different effect by slightly downregulating ERα’s protein levels by 72 hours, compared to vehicle control (Supplementary Fig. S2A and Supplementary Table. S4). This down-regulation is less than that with BPTPE, nonetheless, BMI-135 does not accumulate the receptor compared to 4OHT and endoxifen (Supplementary Fig. S2A and Supplementary Table. S4). This regulation trend with TTC-352, BMI-135, and E4 in MCF-7:WS8 is replicated in MCF-7 ATCC (Supplementary Fig. S2B and Supplementary Table. S3).
Figure 3. ERα protein levels in multiple BC cell lines after 24, 48, and 72-hour-treatments with test compounds.

A, ERα protein levels in MCF-7:WS8. B, ERα protein levels in T47D:A18. C, ERα protein levels in BT-474. D, ERα protein levels in ZR-75-1. E, ERα protein levels in MCF-7:5C. F, ERα protein levels in MCF-7:2A. G, ERα protein levels in MCF-7:RAL. H, ERα protein levels in LCC1. I, ERα protein levels in LCC2. J, ERα protein levels in LCC9. Densitometry data are presented in Supplementary Table. S3.
In T47D:A18, TTC-352 and BMI-135 maintain the protein levels of ERα (Model II), compared to vehicle control, and similar to E2 and E4 (Fig. 3B, Supplementary Fig. S2C and Supplementary Tables. S3-S4). BPTPE, 4OHT, and endoxifen accumulate the receptor by 72 hours (Supplementary Fig. S2C and Supplementary Table. S4).
In BT474, TTC-352 down-regulates the protein levels of ERα by 72 hours, compared to vehicle control, and similar to E2 and E4 (Model I) (Fig. 3C, Supplementary Fig. S2E, and Supplementary Tables. S3-S4). Whereas, BMI-135 seems to have a similar trend except that the protein levels by 72 hours are similar to vehicle control (Supplementary Fig. S2E and Supplementary Table. S4). The protein levels are up-regulated by 72 hours with BPTPE, and more so with endoxifen and 4OHT (Supplementary Fig. S2E and Supplementary Table. S4).
In ZR-75-1, TTC-352 slightly down-regulates the protein levels of ERα after 72-hour-treatment, compared to vehicle control, and similar to E2 and E4 (Model I) (Fig. 3D, Supplementary Fig. S2D, and Supplementary Tables. S3-S4). Whereas, BMI-135 seems to have a different trend whereby by 72 hours the protein levels become similar to vehicle control (Supplementary Fig. S2D and Supplementary Table. S4). The protein levels are maintained by 72 hours with BPTPE, 4OHT, and endoxifen, compared to vehicle control (Supplementary Fig. S2D and Supplementary Table. S4).
In MCF-7:5C, MCF-7:2A, and MCF-7:RAL, TTC-352, BMI-135, and E4 down-regulate the protein levels of ERα by 72 hours, compared to vehicle control, and similar to E2 (Model I) (Fig. 3E-G, and Supplementary Fig. S3D-F). In MCF-7:2A, ER66 and ER77 proteins levels with BMI-135, TTC-352, and E4 are similarly regulated over time (Model I) (Fig. 3F, and Supplementary Fig. S3E) (32). In these cell lines, the protein levels are slightly down-regulated with BPTPE, and maintained or accumulated with endoxifen, 4OHT, and raloxifene (in MCF-7:RAL) (Supplementary Fig. S3D-F).
In LCC1, LCC2 and LCC9, TTC-352, BMI-135, and E4 down-regulate the protein levels of ERα by 72 hours, compared to vehicle control, and similar to E2 (Model I) (Fig. 3H-J, Supplementary Fig. S3A-C, and Supplementary Table. S3). In these cell lines, the protein levels are down-regulated with BPTPE, and maintained or accumulated with endoxifen and 4OHT (Supplementary Fig. S3A-C, and Supplementary Table. S3).
In eleven BC cell lines, TTC-352 regulated the protein levels of ERα in a similar manner to E2 (Fig. 3 and Supplementary Table. S3), and different from that with BPTPE (Supplementary Figs. S2-3, and Supplementary Table. S4), and ICI significantly down-regulated the protein levels of ERα (Fig. 3, and Supplementary Figs. S2-S3).
Effects of TTC-352, BMI-135, and E4 on coregulator recruitment to DNA-bound ER in WT BC MCF-7:WS8 and apoptotic-type BC MCF-7:5C
Cell-free ERE DNA pulldown (39) and liquid chromatography-MS assays were used to assess the composition of coregulators recruited to ER bound to ERE DNA with compounds, using E2 and endoxifen as positive and negative controls, respectively, for coactivator binding.
In MCF-7:WS8, E2 recruited major coactivators such as: NCOA1-3, the Mediator complex (MED; see subunits in Fig. 4), and Lysine Methyltransferase 2D (KMT2D or MLL4) (Fig. 4A), which is consistent with prior proteomic publications (39). Endoxifen did not recruit these coactivators (Fig. 4A). BPTPE did not recruit many of the E2-enriched coactivators, and only a subset of endoxifen-enriched coregulators (Fig. 4A). Estetrol recruited NCOAs and KMT2D in a similar fashion to E2, but failed to recruit many MED subunits at the level promoted by E2 (Fig. 4A). TTC-352 and BMI-135 are different from BPTPE in terms of coactivator recruitment; chiefly recruiting NCOA1-2 and MED subunits, with NCOA3 not readily-enriched compared to E2 and E4 (Fig. 4A).
Figure 4. Proteomics of major ER coregulators in MCF-7:WS8 and MCF-7:5C BC with test ligands, and immunoblots of KMT2D, NCOA1 and 3, and MED17 to validate MS data.

A, Proteomics of major coregulators differentially- recruited to DNA-bound recombinant ER in MCF-7:WS8 and MCF-7:5C cells, treated with E2 (100 nM), E4 (1 μM), TTC-352 (1 μM), BMI-135 (1 μM), BPTPE (1 μM), and endoxifen (1 μM). Ethanol or DMSO served as the vehicle control. ERE DNA pulldown cell-free reactions were performed, and MS data are depicted as a heatmap for coregulator enrichment (light to dark red color) or repulsion (light to dark blue color). The values represent quantification with label free iBAQ method, normalized to ESR1 amount, and are shown (as estimated % relative to ESR1). Official gene symbols are shown (on the leftmost column). NCOA1-3, KMT2D, and MEDs were defined previously as E2-enriched coactivators (25, 39). B, Immunoblotting to validate MS data of KMT2D, NCOA1 and 3, and MED17, recruited in MCF-7:WS8 and MCF-7:5C NE to DNA-bound ER (ESR1), when treated with E2 (100 nM), E4 (1 μM), TTC-352 (1 μM), and BMI-135 (1 μM). Protein size standards are shown (on the left-side as kDa), and 3% input is shown (on the left-side; representing 3% of WS8 or 5C NE that was added to each 4xERE DNA bead).
In MCF-7:5C, E2 recruited NCOA2-3, KMT2D, and the MED subunits, with slightly different distribution of affinities, while endoxifen repelled them, as expected for this Selective ER Modulator (SERM), and BPTPE did not have much of a coactivator binding (Fig. 4A). Estetrol recruited similar levels of NCOA3 and KMT2D, and many of the same MED subunits (Fig. 4A). TTC-352 and BMI-135 recruited NCOA3 and KMT2D at much lower levels than E2 and E4 (Fig. 4A). Interestingly, TTC-352 recruited more MED subunits than E2, while BMI-135 displayed shared MED subunit recruitment with E4 (Fig. 4A).
The recruitment of KMT2D, NCOA1, NCOA3, and MED17, in MCF-7:WS8 and MCF-7:5C, was further validated the by immunoblotting (Fig. 4B, and Supplementary Fig. S9).
Comparative analysis of the X-ray structures of hERαLBD in complex with ligands E2, TTC-352, and BPTPE
The experimental X-ray structure (Fig. 5G-I) of hERαLBD in complex with TTC-352 shows the ligand binding to the agonist conformation of ERα (i.e., helix 12 (H12) is docked over the active site, in a grove between helices H5 and H11, leaning on H3, and closing the ligand inside the hydrophobic binding pocket) (Fig. 5A). The superposition with hERαLBD in complex with E2 indicates minor differences between these two structures, with an average root-mean-square-deviation (RMSD) of 0.55 Å; calculated based on Cα atoms. However, a difference has been noticed in the positioning of H12; with it being slightly displaced in the TTC-352:ERα structure by an average RMSD of 0.85Å, compared to the E2:ERα structure (Fig. 5A). The binding mode of TTC-352 to the active site shares similar features with that of E2. The benzothiophene moiety of TTC-352 overlaps well with the A and B rings of E2, being involved in π-π stacking interactions with Phe404, and forming the H-bond network between the hydroxyl group and the side-chains of residues Glu353, Arg394, and a crystallization water molecule. The phenoxy ring occupies the same region of the binding pocket as the D ring of E2, but it is buried slightly deeper in the active site, and oriented parallel with the imidazole ring of His524, favoring the formation of the H-bond with the hydroxyl group, like E2 (Fig. 5B).
Figure 5. Representation of ERα-LBD with E2, TTC-352, and BPTPE.

Comparison of the WT structures of E2 (yellow), TTC-352 (blue), and BPTPE (green) bound to hERα. The superposition of the X-ray structures of hERαLBD:E2 with hERαLBD:TTC-352 (A), and of hERαLBD:E2 with hERαLBD:BPTPE (D). The closer views of the active sites show the binding modes of E2 (yellow), in comparison with TTC-352 (blue) (B) and BPTPE (green) (E), together with the H-bond contacts between H3 and H12 for E2, TTC-352 (C) and BPTPE (F). The superposition of the mutant Tyr537Ser X-ray structures of hERαLBD in complex with E2 (orange) and TTC-352 (teal) (G), together with the binding site alignment of the ligands (H), and a close view of the H-bond interactions between Asp351 and Ser537 (I). Amino acid residues involved in direct interactions (i.e., H-bonds and hydrophobic contacts) are labeled and shown in sticks, together with the amino acids directly involved in H-bonds with H12. In the overall structures, H12 is labeled together with the helices that form the ligand binding site (i.e., H3, H6, H8, H11,) and part of the coactivator site (i.e., H5, H9, H10). The H-bonds are displayed (dashed lines) in yellow, blue, and green in the structures of E2, TTC-352, and BPTPE, respectively.
The analysis of the X-ray structure of hERαLBD in complex with BPTPE, reveals good overlapping with the E2:ERα structure with an average RMSD of 1.28 Å (Fig. 5D). Regarding the ligand binding mode, BPTPE’s alignment in the active site is comparable with E2 and TTC-352 (Fig. 5E), preserving the same interactions with the exception of few noteworthy changes. First, the absence of the H-bond to His524, the residue’s sidechain is flipped out of the binding site, and the space freed is partially-occupied by the phenyl ring of BPTPE. This observation could explain the displacement of BPTPE by 0.7Å towards H11, compared with TTC-352. Second, the presence of an H-bond between the phenoxy ring of BPTPE and Thr347 (Fig. 5E-F). This contact leads to an alternative orientation of Thr347, with the methyl group pointing towards Tyr537, which results in a different conformation, and the displacement of Tyr537 relative to its position, compared with TTC-352:ERα or E2:ERα (Fig. 5C and 5F ). This repositioning at the base of H12, together with the reorientation of Leu540 due to the large phenoxy ring of BPTPE, alters the H-bond pattern in the vicinity. As a result, the H-bonds between Asp351 (H3) and the backbone of Leu539 and Leu540 (H12), which normally stabilize the orientation of H12 in the agonist conformation with E2:ERα (Fig. 5C), are absent with BPTPE (Fig. 5F), but are present with TTC-352 (Fig. 5C).
MD simulations were performed to investigate the dynamics of hERαLBD and of ligand binding, specifically, the interactions responsible for binding; to highlight the differences that could discriminate between the ligands, and explain their observed biological behaviors in tumor cells. The influence of the ligands on H12 conformation was also investigated, by monitoring the key interactions that are known to stabilize H12 in the agonist conformation (Fig. 5C and 5F).
Evaluation of the trajectory stability from MD simulations
165 nanoseconds (ns) MD simulations were performed for all systems. The RMSDs of the protein backbone atoms, relative to their position in the first frame, were monitored to evaluate the equilibration and stability of the simulations. The RMSD evolution for all simulations, together with the stability of H12 and amino acids of the binding sites, are displayed (Supplementary Fig. S4A-C), and detailed in supplementary materials. Briefly, the data indicates that all systems reached equilibrium, and that the trajectories were stable (Supplementary Fig. S4A). Similar trends were observed for the systems of TTC-352 and E2, whereas, BPTPE showed more conformational changes in the segment corresponding to H12 (Supplementary Fig. S4B). No significant conformational changes were detected in the active site of all structures (Supplementary Fig. S4C).
To gain insights into the local flexibility of the receptor chain, root mean square fluctuation (RMSF) was monitored along the trajectories of all complexes (Supplementary Fig. S4D-F), with detailed results presented in supplementary materials. In summary, the data suggests that the large fluorophenyl moiety of TTC-352 induces more flexibility in the loop between H11 and H12, than the phenoxy ring of BPTPE. However, this is not translating into larger flexibility of H12, mainly because of the stabilizing effect of the H-bonds between Asp351, and the backbone of Leu539 and Leu540 with TTC-352, but not with BPTPE (Fig.5C and 5F). Moreover, in the system of BPTPE, increasingly large fluctuations have been detected at the terminal residues of H12. This indicates more mobility in this region of the helix, which destabilizes and hinders the proper closing of H12 over the binding pocket, leading to the inability of BPTPE complex to reach the full-agonist conformation of the receptor.
Analysis of E2, TTC-352, and BPTPE’s binding modes from MD simulations
The interaction maps of the ligands with key residues of the binding site, together with the occurrences for specific contacts, are displayed (Fig. 6), and the detailed results are presented in supplementary materials. The MD simulations confirmed that the H-bonds to Glu353 and His524 are highly-stable for TTC-352 and E2 (Fig. 6A-B). As reported previously (46), the H-bond to Thr347 is the most stable interaction for BPTPE; being maintained during the entire simulation, which indicates a strong bond, while the H-bond to Glu353 shows a significantly-decreased frequency, which indicates a weaker bond (Fig. 6C).
Figure 6. 2D ligand-ERα interaction maps highlighting key interactions between the ligands and ERα’s amino acids during the simulations.

These maps were generated from the recorded trajectories and the occurrences of the key contributing interactions between E2 (A), TTC-352 (B), BPTPE (C), and the amino acids in the binding site of ERα, as shown.
Analysis of the binding free energy decomposition for E2, TTC-352, and BPTPE
To gain a deeper understanding of the ligand-receptor interactions, and highlight the subtle differences that could discriminate between these ligands, we performed ligand binding energy calculations using the MM-GBSA method for the simulated systems. The contribution of each residue to the binding process was analyzed; by decomposing the binding energy into ligand-residue pairs, with the results displayed (Supplementary Fig. S5), and detailed in supplementary materials.
The data shows that the H-bonding to Glu353 is the driving force of binding for TTC-352, similar to E2. This interaction is crucial for agonist binding. The H-bond between BPTPE and Glu353 is weaker, and the binding of BPTPE is governed by the H-bond to Thr347 (Fig. 6C). This leads to instability in this region of H3, which could have an impact on the conformation of Asp351 that is found in close proximity, and could explain why the H-bonds to Leu539 and Leu540 are missing for BPTPE:ERα. In addition, the strong interaction with Thr347 stabilizes the ligand in the active site, but perturbs the local environment, and disrupts the H-bond between Tyr537 and Asn348. Finally, the stability of H12, and proper closing specific for the agonist conformation, are affected in the structure of BPTPE:ERα.
TTC-352 induces ThT fluorescence as a marker of UPR
Thioflavin T was used to detect and quantify the EnR stress or UPR in living cells, as it interacts directly with the accumulated misfolded protein amyloid during the UPR (43). The “blue” Hoechst 33342 fluorescent dye was used as a nuclear counterstaining dye in MCF-7:5C living cells (Fig. 7, channel A), the “green” ThT fluorescent dye was used as a UPR-indicative dye (channel B), and a co-localization of ThT and Hoechst 33342 dyes is shown (channel C).
Figure 7. Detection of EnR stress in live MCF-7:5C cells using ThT dye.

A, Hoechst 33342 dye single panel (blue). B, ThT dye single panel (green). C, A co-localization panel of ThT and Hoechst 33342 dyes (blue and green). Treatments included vehicle control (DMSO [0.1%]), positive control thapsigargin (1 μM), and TTC-352 (1 μM). After 72-hour-treatment, cells were co-treated with ThT (5 μM) for 1 hour. The previous culture media was then swiped with Hoechst 33342 dye (5 μg/ml) in warm PBS for 15 minutes. Live cell imaging was done by the ZEISS Celldiscoverer 7 microscope. Scale bar = 50 μM.
TTC-352 induced ThT fluorescence by 72 hours compared to vehicle control, and somewhat similar to the induction seen with positive control thapsigargin (i.e., triggers EnR stress by disrupting EnR Ca2 + homeostasis) (Fig. 7B). The ThT relative intensity/cell for thapsigargin was 3.320555, and 2.025762 for TTC-352, compared to 0.4725 for vehicle control.
TTC-352 triggers apoptosis in multiple estrogen-independent and endocrine-resistant BC models
Flow cytometry was used to assess if the type of stress-induced cell death was actually apoptosis, when treated with 1 μM TTC-352. In MCF-7:5C, TTC-352 induces apoptosis (annexin staining 22.9% versus vehicle control 6.9%) (Fig. 8A), similar to the time course of 1 nM E2 (annexin staining 23.4% versus control 6.7%) (Supplementary Fig. S7A), which is in 3 days.
Figure 8. Flow cytometry and Western blotting in MCF-7:5C cells treated with TTC-352 and its combination with a PERK inhibitor and an IRE1α inhibitor.

A, MCF-7:5C cells were treated with vehicle control (DMSO [0.1%]), GSK G797800 (10 μM), TTC-352 (1 μM), and TTC-352 plus GSK G797800, for 3 days, and then stained with annexin V–FITC and propidium iodide, and analysed by flow cytometry. Viable cells (left lower quadrant) are annexin V–FITC− and PI−; early apoptotic cells (right lower quadrant) are annexin V–FITC+ and PI−; dead cells (left upper quadrant) are PI+ and late apoptotic cells (right upper quadrant) are annexin V–FITC+ and PI+. An increased, late apoptotic effect is observed in the right upper quadrant. B, p-eIF2α, total eIF2α, ATF4, CHOP, and cleaved PARP protein levels, in MCF-7:5C, after 72-hour treatments with vehicle control DMSO [0.1%], UPR +ve control thapsigargin [1 μM], TTC-352 [1 μM], and TTC-352 [1 μM] plus GSK 797800 [10 μM]. C, MCF-7:5C cells were treated with vehicle control (DMSO [0.1%]), MKC-3946 (20 μM), TTC-352 (1 μM), and TTC-352 plus MKC-3946, and analysed by flow cytometry. D, XBP1s, and XBP1 protein levels, in MCF-7:5C, after 72-hour treatments with vehicle control DMSO [0.1%], TTC-352 [1 μM], and TTC-352 [1 μM] plus MKC-3946 [20 μM]. A and C, ***P < 0.001.
In MCF-7:2A, TTC-352 induces apoptosis (annexin staining 21.2% versus control 2.7%), similar to the time course of E2 (annexin staining 20.4% versus control 2.7%) (Supplementary Fig. S7B), which is in 9 days.
In MCF-7:RAL, TTC-352 induces apoptosis (annexin staining 8.4% versus control 1.5%), similar to the time course of E2 (annexin staining 9.1% versus control 1.5%) (Supplementary Fig. S7C), which is in 14 days.
Inhibition of PERK UPR pathway blocks apoptosis in MCF-7:5C with TTC-352 treatment.
Blocking the UPR transducer PERK with 10 μM GSK G797800 in combination with 1 μM TTC-352 by 72 hours, inhibited apoptosis (annexin staining 6.9% versus control 6.9%) (Fig. 8A), compared to TTC-352 alone treatment (Fig. 8A) (annexin staining 22.9% versus vehicle control 6.9%), and compared to 10 μM GSK G797800 alone treatment (annexin staining 8.3% versus control 6.9%) (Fig. 8A).
PERK downstream targets p-eIF2α, ATF4, and CHOP as well as apoptosis target cleaved PARP, are up-regulated after 72-hour treatment with TTC-352, whereas, the addition of GSK 797800 inhibits this UPR/apoptosis effect (Fig. 8B).
Inhibition of IRE1α:XBP1s UPR pathway enhances apoptosis in MCF-7:5C with TTC-352 treatment.
Inhibiting the UPR transducer IRE1α, by inhibiting basal XBP1 splicing, with 20 μM MKC-3946 in combination with 1 μM TTC-352 by 72 hours, enhanced apoptosis (annexin staining 35.5% versus vehicle control 1.4%) (Fig. 8C), compared to TTC-352 alone treatment (annexin staining 27.9% versus vehicle control 1.4%) (Fig. 8C), and compared to 20 μM MKC-3946 alone treatment (annexin staining 8.8% versus vehicle control 1.4%) (Fig. 8C).
IRE1α downstream target XBP1s (or spliced XBP1) is up-regulated after 72-hour treatment with TTC-352, whereas, the addition of MKC-3946 inhibits this splicing effect (Fig. 8D).
Transcriptional-translational, UPR, and apoptotic effects of TTC-352 are mediated via ERα.
TTC-352 was shown to function via ERα. The combination of TTC-352 and 4OHT blocked SRC-3 recruitment compared to TTC-352 alone treatment (Fig. 9A); inhibited ERE activation compared to TTC-352 alone treatment (Fig. 9B); blocked the anti-proliferative effects of TTC-352 alone treatment (Supplementary Fig. S8A-C); inhibited the UPR PERK pathway activation (Fig. 9C-D); and prevented apoptosis (Fig. 9C, and E-G)
Figure 9. Mediation of the transcriptional-translational, UPR, and apoptotic effects of TTC-352 via ERα.
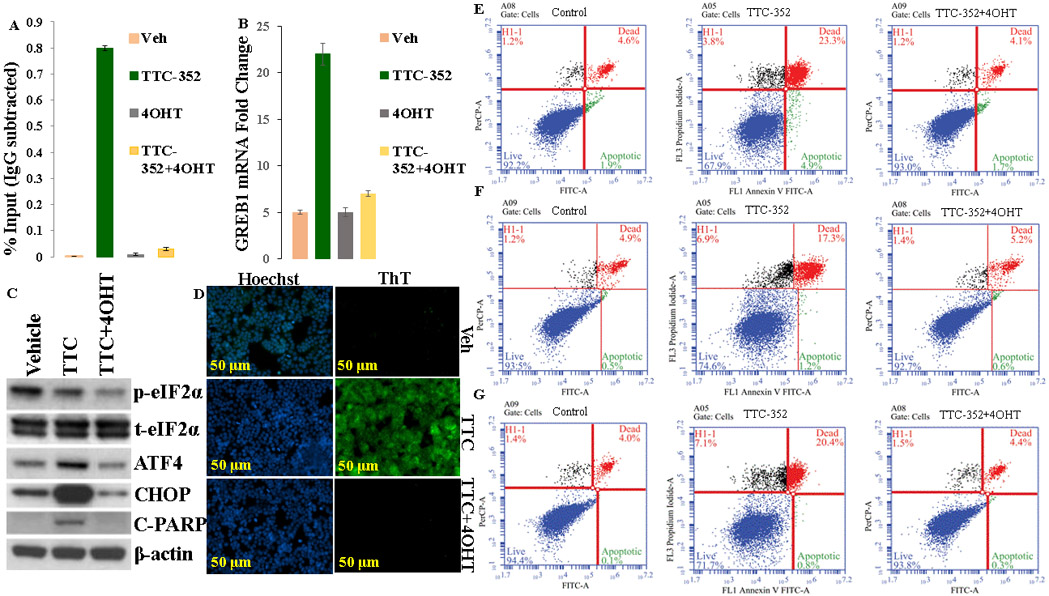
A, Recruitment of SRC-3, in MCF-7:5C cells, after 45-minute-treatment with indicated ligands (1 μM). Recruitment of SRC-3 was calculated as percentage of the total input after subtracting the IgG recruitment. B, mRNA expression of GREB1, in MCF-7:5C cells, after 24-hour-treatment with ligands (1 μM). C, p-eIF2α, total eIF2α, ATF4, CHOP, and cleaved PARP protein levels, in MCF-7:5C, after 72-hour treatments with vehicle control DMSO [0.1%], TTC-352 [1 μM], and TTC-352 [1 μM] plus 4OHT [1 μM]. D, Detection of UPR, in live MCF-7:5C cells, using ThT dye, after 72-hour treatments with vehicle control DMSO [0.1%], TTC-352 [1 μM], and TTC-352 [1 μM] plus 4OHT [1 μM]. Scale bar = 50 μM. E, MCF-7:5C cells were treated with vehicle control (DMSO [0.1%]), TTC-352 [1 μM], and TTC-352 [1 μM] plus 4OHT [1 μM] for 3 days, and analysed by flow cytometry. F, MCF-7:2A cells were treated with vehicle control (DMSO [0.1%]), TTC-352 [1 μM], and TTC-352 [1 μM] plus 4OHT [1 μM] for 9 days, and analysed by flow cytometry. G, MCF-7:RAL cells were treated with vehicle control (DMSO [0.1%]), TTC-352 [1 μM], and TTC-352 [1 μM] plus 4OHT [1 μM] for 14 days, and analysed by flow cytometry. E-G, ***P < 0.001.
Discussion
TTC-352 is a member of a new class of estrogen mimics (Fig. 1) (16), which is currently being evaluated in endocrine-resistant MBC clinical trials (11). This work reports: (i) the X-ray crystallography of TTC-352:mutant ER with clinical implications given that many MBC patients harbor ER mutations (Fig. 5G-I) (19, 20, 47); (ii) the molecular mechanisms of TTC-352’s BC regression in LTED patients (Figs.7-10); and (iii) the key interactions at the molecular and atomic levels of the benchmark partial agonist BPTPE:WT ER, involving Asp351 and H12 (Figs.5-6), which explains the delayed ERα-induced UPR and apoptosis (48) compared to TTC-352 (Fig. 8).
Figure 10. Schematic representation of the study’s conclusions highlighting major ER coactivator-binding differences in MCF-7:5C between E2, E4, ShERPAs, and BPTPE, and with ShERPA TTC-352 between WT MCF-7:WS8 and LTED endocrine-resistant MCF-7:5C BC.

A, E2, E4, ShERPA BMI-135, ShERPA TTC-352, and BPTPE major ER coactivators’ recruitment, and anti-tumor molecular mechanism in MCF-7:5C. E2:ERα and E4:ERα complexes mainly recruit NCOA3, KMT2D, and many of the same MEDs, and induce a high threshold of stress; through the synthesis of unfolded and/ or misfolded proteins, leading to rapid apoptosis. TTC-352 (referred to as TT in the illustration) recruited more MED subunits than E2, but less NCOA3 and KMT2D than E2 and E4, with a similar threshold of stress and timing of apoptosis to E2 and E4. BMI-135 (referred to as BM in the illustration) recruited less NCOA3 and KMT2D than E2 and E4, and shared MED subunit recruitment with E4, which generated a lower threshold of stress and delayed apoptosis (48) compared to E2, E4, and TTC-352. BPTPE (referred to as BP in the illustration) did not have much of a coactivator-recruitment, which generates a very low threshold of stress and a much more delayed course of apoptosis (48) compared to E2, E4, TTC-352, and BMI-135. This differential ligand:ERα:coactivator-reucitment-and-induced EnR stress, sets the therapeutics apart, in terms of the timing of activating the UPR, followed by inducing apoptosis. The box (in gray) highlights the observed recruitment patterns: thick arrow (in red) represents relatively more recruitment, thin arrow (in burgundy) is relatively less recruitment, thin arrow (in burgundy with green border) is shared subunit recruitment, and thin arrow (in blue) is no recruitment. B, TTC-352’s paradoxical effect in WT growth-inducing BC MCF-7:WS8 versus LTED apoptosis-inducing BC MCF-7:5C. NCOA3, KMT2D, and many MEDs (especially MED12-16 and MED23) are recruited to ER, in MCF-7:5C treated with TTC-352 (thick arrow in maroon), compared to E2, but these same coactivators are reduced upon the treatment of MCF-7:WS8 with TTC-352 (thin arrow in rose). This differential ligand:ERα:coactivator-reucitment-and-induced EnR stress, phenotypically sets the two BC models apart.
Earlier pharmacological studies classified ER binding ligands into agonists, partial agonists, and antagonists (49), and complimented the subsequent X-ray crystallography studies of the agonist and antagonist ER complexes of the LBD (22, 50). Earlier biological studies described E2-induced apoptosis (51). Current study concludes that TTC-352 is a less potent full estrogen agonist in numerous biologically-different BC models (Figs.2-6 and 9, Supplementary Fig. S1, and Supplementary Table. S2), with a rapid apoptotic effect (through the UPR) in estrogen-independent and endocrine-resistant BC models (Figs.7-10, and Supplementary Fig. S7B-C).
This research area is of particular importance given that BC is projected to double by 2030 than it was in 2011 (13). The majority will be ER-positive with a high risk of recurrence, even with clinically low-risk disease (T1N0) (15). Moreover, treated metastases often harbor private ‘driver’ mutations, compared to untreated metastases (52). In the case of ER-positive HER2-negative BC, metastases treated with endocrine therapy, acquire somatic single-nucleotide variants (47). This highlights the need to evaluate and develop new rapidly-acting BC therapeutics such as estrogens. The recent long-term follow-up results of the Women’s Health Initiative Trials (53, 54) reaffirm the clinical potential of novel and safe estrogenic therapy in significantly reducing BC incidence and mortality in LTED patients.
MD simulations and MM-GBSA calculations for WT ERα in complex with TTC-352, E2, and BPTPE are valuable methodologies to discover key ligand-receptor interactions, which aids the pharmacological classification of TTC-352. Most importantly, they identify key structural components of estrogenic therapeutics that ensure the appropriate closure of the ERα LBD by H12 in LTED BC. This closure is a prerequisite for the activation of ERE-mediated UPR and apoptosis, which is the basis of their anti-tumor properties.
The H-bond of TTC-352’s benzothiophene scaffold to Glu353, followed by the H-bond to His524, are the most stable contacts contributing to the binding mechanism of TTC-352, which are also the two binding features specific for the estrogenic activity of E2 (E2’s A ring to Glu353) (Fig. 6A-B). Such strong H-bond between TTC-352 and Glu353, induces stability in the LBD and, consequently, to H3. This supports the formation of the H-bonds between the side-chain of Asp351 (H3) to the backbone of Leu539 and Leu540 (H12), which stabilizes H12 in the full-agonist conformation (Fig. 5C). Such LBD-stabilizing network of H-bonds is preserved with TTC-352, but more so with E2, which explains the altered potency of TTC-352:ERα compared to E2:ERα. By contrast, BPTPE’s binding mechanism is governed by the H-bond of BPTPE’s angular phenolic OH to Thr347, followed by the hydrophobic contacts, and a significantly weaker H-bond to Glu353 (Fig. 5E and Fig. 6C). Although the H-bond between BPTPE’s angular phenolic group and Thr347 stabilizes its binding, it disturbs the H-bond network within H3 and the stabilizing H-bonds to H12 (i.e., Asp351 to Leu 539 and Leu540). As a result, H12 is prevented from adopting the proper orientation specific for the ER full-agonist conformation (Fig. 5F). This is consistent with BPTPE’s reduced recruitment of coactivators (Fig. 4A), and our previous reports (48) of its delayed activation of ERα-induced UPR and apoptosis as well as its functional modulation from a partial agonist (3OHTPE) to a full agonist (Z2OHTPE) by the removal of the para-phenol substitution (46).
Both, Asp351 and H12, play a critical role in modulating the estrogenic and anti-estrogenic intrinsic efficacy of the ligand-ER complex. The natural mutation Asp351Tyr was discovered; overexpressed in TAM-stimulated MCF-7 tumors grown in athymic mice (55). The molecular pharmacology of the WT ER (56) and Asp351Tyr ER (57) was established by stable transfection into ER-negative MDA-MB-231 BC. Unexpectedly, Asp351Try ER converted the raloxifene:WT Asp351 ER complex from anti-estrogenic to estrogenic (58). Subsequent X-ray crystallography of the raloxifene:ER LBD (22), demonstrated the critical role of the anti-estrogenic side chain containing a piperidine ring N to shield and neutralize Asp351, which prevented the closure of H12, and the subsequent ERE activation. Subsequent interrogation of the structural modulation of raloxifene and its interactions with Asp351, demonstrated how Asp351 modulates the estrogenic and anti-estrogenic efficacy of the ligand-ER complex (59).
ESR1 somatic mutations, Y537S and D538G, stabilize ERα in the agonist state, and are linked to acquired resistance to endocrine therapies (60). Mutations Tyr537Ser and Asp538Gly were most prevalent in BC metastases (47), especially AI-resistant BC patients. These mutations improve the closure of H12 over ERα’s LBD, through interacting with Asp351, and recruiting coactivators in the absence of estrogen, which increases the estrogen-like properties of the complex (47).
ERE DNA pull downs and MS are valuable methodologies to determine if TTC-352 has an ERα:coactivators’ binding profile of a full or partial agonist, and better understand why the TTC-352:ERα:coactivators’ complex in the LTED endocrine-resistant MCF-7:5C is phenotypically apoptosis-promoting, whereas, such complex in WT MCF-7 is phenotypically growth-promoting.
In MCF-7:WS8 BC, TTC-352 and BMI-135 are different from BPTPE in terms of NCOA1-2 (or SRC1-2) and MED subunit recruitments, with NCOA3 (or SRC-3) not being readily-enriched with either ShERPAs compared to E2 and E4 (Fig. 4A). BPTPE did not recruit many of the E2-enriched coactivators, and only a subset of endoxifen-enriched coregulators (Fig. 4A). In MCF-7:5C BC, TTC-352 and BMI-135 recruited NCOA3 and KMT2D at much lower levels than E2 and E4, with TTC-352 recruiting more MED subunits than E2, while BMI-135 displaying shared MED subunit recruitment with E4 (Fig. 4A). BPTPE did not have much of coactivator binding (Fig. 4A).
TTC-352’s differential recruitment of MED subunit types alongside their differential enrichment levels, could explain its ability to cause a higher threshold of stress of ER-mediated unfolded proteins followed by earlier apoptosis, compared to that with BMI-135 (Fig. 10A). In MCF-7:5C, NCOA3, KMT2D, and many MEDs (especially MED12-16 and MED23) are recruited to ERα with TTC-352, compared to E2, but these coactivators are reduced upon the treatment of MCF-7:WS8 with TTC-352 (Supplementary Table. S5). TTC-352’s higher recruitment of major ER coactivators in MCF-7:5C, compared to MCF-7:WS8, can explain its ability to cause a high threshold of stress of ER-mediated unfolded proteins followed by apoptosis; making it phenotypically apoptosis-promoting, versus the growth-promoting MCF-7:WS8 (Fig. 10B). The altered recruitment patterns of major ER coactivators for transcriptional activation, with TTC-352, BMI-135, and E4, in comparison to the levels promoted by E2 or BPTPE, can better explain the observed differences in their potency and ERα-mediated UPR.
Our MD simulations data complements the MS data whereby the dynamics of H12 orchestrate ER’s coactivator-mediated transcriptional activity. In the agonist complex structure, H12 forms one side of a hydrophobic coactivator binding pocket, which allows the recruitment of an LXXLL motif present in many transcriptional cofactors (61). This is, especially, true with SRC1-3 that possess three LXXLL motifs, two of which bridge across the ER dimer (at least for an extended polypeptide containing all three motifs), which accounts for the 100-fold higher affinity relative to the single LXXLL-containing peptide (62). The overexpression of SRC-3 is observed in over 50% of BCs, and leads to constitutive ER-mediated transcriptional activity in the agonist confirmation, conferring endocrine-resistance in preclinical models and in patients treated with TAM (63). E2, E4, TTC-352, and BMI-135 in complex with ERα, yield an agonist confirmation of the ligand-ER complex (Figs. 5-6) (48), and, subsequently, recruit more coactivators (Fig. 4A), opposite to BPTPE. H12 acts as a molecular switch with the contribution of those H-Bond networks (Fig. 6), and such coactivators (Fig. 4A).
We demonstrate that the structure-function model of the synthetic estrogen mimic TTC-352 is a less potent full estrogen agonist compared to E2, allowing H12 to seal the LBD, which recruits many E2-enriched coactivators, and induces rapid ERα-mediated UPR and apoptosis. This contradicts the model of the benchmark partial agonist BPTPE, not allowing H12 to seal the LBD properly, which does not recruit many E2-enriched coactivators, and induces delayed ERα-mediated UPR and apoptosis. These data suggest that BC patients would potentially benefit more from full agonists like TTC-352 rather than partial agonists, because of BPTPE’s delayed UPR-apoptotic effect. A partial agonist with delayed apoptosis might create a higher probability of tumor clonal evolution and acquired-resistance (64).
Supplementary Material
Acknowledgments
We thank Dr. Robert Clarke for providing the LCC BC cell line series. BA holds a dual position: the Dallas/Ft. Worth Living Legend Fellow of Cancer Research at the Department of Breast Medical Oncology, the University of Texas MD Anderson Cancer Center (TX, USA) and a split-site PhD trainee under Model C “Applicants of Very High Quality” at the Faculty of Biological Sciences, the University of Leeds (West Yorkshire, UK).
Financial support: This work was supported by the National Institutes of Health MD Anderson Cancer Center Support Grant (P. W. Pisters CA016672); the George and Barbara Bush Foundation for Innovative Cancer Research (V.C. Jordan); the Cancer Prevention Research Institute of Texas (CPRIT) for the STARs and STARs Plus Awards (V.C. Jordan); the Dallas/Ft. Worth Living Legend Fellowship of Cancer Research (B. Abderrahman); the Coriolan Dragulescu Institute of Chemistry of the Romanian Academy (R.F. Curpan Project no. 1.1/2020); the Adrienne Helis Malvin Medical Research Foundation through direct engagement with the continuous active conduct of medical research in conjunction with BCM (C. E. Foulds and Y. Chen); the Baylor College of Medicine Mass Spectrometry Proteomics Core supported by the Dan L. Duncan Comprehensive Cancer Center grant (A. Malovannaya NIH P30 CA125123) and CPRIT Proteomics and Metabolomics Core Facility Award (A. Malovannaya RP170005); and the Argonne National Laboratory, Structural Biology Center (SBC), at the Advanced Photon Source operated by UChicago Argonne, LLC, for the U.S. Department of Energy, and supported by the Office of Biological and Environmental Research (G.L. Greene DE-AC02-06CH11357).
Abbreviation list:
- 4OHT
4-hydroxytamoxifen
- AEs
Adverse events
- AIs
Aromatase inhibitors
- BC
Breast cancer
- BPTPE
Triphenylethylene bisphenol
- E2
17β-estradiol
- E4
Estetrol
- EC50
Effective concentration
- Endox
Endoxifen
- EnR
Endoplasmic reticulum
- EP300
E1A binding protein p300
- ER
Estrogen receptor
- EREs
Estrogen responsive elements
- HER2
Human epidermal growth factor receptor 2
- iBAQ
Intensity-based absolute quantification
- ICI
ICI 182,780 fulvestrant
- IRE1α
Inositol-requiring enzyme 1
- KMT2D
Lysine Methyltransferase 2D
- LBD
Ligand-binding domain
- LTED
Long-term estrogen deprivation
- MBC
Metastatic breast cancer
- MED
Mediator complex
- mRNA
ER messenger RNA
- NCOA3
Nuclear receptor co-activator-3
- NE
Nuclear extract
- PERK
Protein kinase regulated by RNA-like endoplasmic reticulum kinase
- PgR
Progesterone receptor
- Ralox
Raloxifene
- RMSD
Root-mean-square deviations
- RMSF
Root-mean-square fluctuation
- SEM
Selective estrogen mimic
- SERMs
Selective Estrogen Receptor Modulators
- ShERPAs
Selective Human Estrogen Receptor Partial Agonists
- SRC-3
Steroid receptor coactivator −3
- TAM
Tamoxifen
- ThT
Thioflavin T
- UPR
Unfolded protein response
Footnotes
Disclosure of potential conflicts of interest: C. E. Foulds discloses an equity position in Coactigon, Inc. D. A. Tonetti and G. R. J. Thatcher disclose equity positions in TTC Oncology, LLC.
References:
- 1.Jordan VC, Brodie AM. Development and evolution of therapies targeted to the estrogen receptor for the treatment and prevention of breast cancer. Steroids. 2007;72(1):7–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jordan V,C, Dix CJ, Allen KE. The effectiveness of long-term treatment in a laboratory model for adjuvant hormone therapy of breast cancer In: Salmon SE, Jones SE, editors. Adjuvant Therpy of Cancer II. New York Greene and Stratton; 1979. p. 19–26. [Google Scholar]
- 3.Early Breast Cancer Trialists' Collaborative Group. Tamoxifen for early breast cancer: an overview of the randomised trials. Early Breast Cancer Trialists' Collaborative Group. Lancet. 1998;351(9114):1451–67. [PubMed] [Google Scholar]
- 4.Yao K, Lee ES, Bentrem DJ, England G, Schafer JI, O'Regan RM, et al. Antitumor action of physiological estradiol on tamoxifen-stimulated breast tumors grown in athymic mice. Clin Cancer Res. 2000;6(5):2028–36. [PubMed] [Google Scholar]
- 5.Early Breast Cancer Trialists' Collaborative G, Davies C, Godwin J, Gray R, Clarke M, Cutter D, et al. Relevance of breast cancer hormone receptors and other factors to the efficacy of adjuvant tamoxifen: patient-level meta-analysis of randomised trials. Lancet. 2011;378(9793):771–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sgroi DC, Sestak I, Cuzick J, Zhang Y, Schnabel CA, Schroeder B, et al. Prediction of late distant recurrence in patients with oestrogen-receptor-positive breast cancer: a prospective comparison of the breast-cancer index (BCI) assay, 21-gene recurrence score, and IHC4 in the TransATAC study population. Lancet Oncol. 2013;14(11):1067–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ariazi EA, Cunliffe HE, Lewis-Wambi JS, Slifker MJ, Willis AL, Ramos P, et al. Estrogen induces apoptosis in estrogen deprivation-resistant breast cancer through stress responses as identified by global gene expression across time. Proc Natl Acad Sci U S A. 2011;108(47):18879–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lonning PE, Taylor PD, Anker G, Iddon J, Wie L, Jorgensen LM, et al. High-dose estrogen treatment in postmenopausal breast cancer patients heavily exposed to endocrine therapy. Breast Cancer Res Treat. 2001;67(2):111–6. [DOI] [PubMed] [Google Scholar]
- 9.Ellis MJ, Gao F, Dehdashti F, Jeffe DB, Marcom PK, Carey LA, et al. Lower-dose vs high-dose oral estradiol therapy of hormone receptor-positive, aromatase inhibitor-resistant advanced breast cancer: a phase 2 randomized study. JAMA. 2009;302(7):774–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iwase H, Yamamoto Y, Yamamoto-Ibusuki M, Murakami KI, Okumura Y, Tomita S, et al. Ethinylestradiol is beneficial for postmenopausal patients with heavily pre-treated metastatic breast cancer after prior aromatase inhibitor treatment: a prospective study. Br J Cancer. 2013;109(6):1537–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dudek AZ, Liu LC, Fischer JH, Wiley EL, Sachdev JC, Bleeker J, et al. Phase 1 study of TTC-352 in patients with metastatic breast cancer progressing on endocrine and CDK4/6 inhibitor therapy. Breast Cancer Res Treat. 2020. [DOI] [PubMed] [Google Scholar]
- 12.Schmidt M, Hönig A, Zimmerman Y, Verhoeven C, Almstedt K, Battista M, Lenhard HG, Krijgh J, Bennink HC. Abstract P5-11-15: Estetrol for treatment of advanced ER+/HER2- breast cancer. Cancer research. 2020;80((4 Suppl) ). [Google Scholar]
- 13.Rosenberg PS, Barker KA, Anderson WF. Estrogen Receptor Status and the Future Burden of Invasive and In Situ Breast Cancers in the United States. Journal of the National Cancer Institute. 2015;107(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Reyes C, Engel-Nitz NM, DaCosta Byfield S, Ravelo A, Ogale S, Bancroft T, et al. Cost of Disease Progression in Patients with Metastatic Breast, Lung, and Colorectal Cancer. Oncologist. 2019;24(9):1209–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schroeder B, Zhang Y, Stal O, Fornander T, Brufsky A, Sgroi DC, et al. Risk stratification with Breast Cancer Index for late distant recurrence in patients with clinically low-risk (T1N0) estrogen receptor-positive breast cancer. Npj Breast Cancer. 2017;3:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xiong R, Patel HK, Gutgesell LM, Zhao J, Delgado-Rivera L, Pham TND, et al. Selective Human Estrogen Receptor Partial Agonists (ShERPAs) for Tamoxifen-Resistant Breast Cancer. J Med Chem. 2016;59(1):219–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tonetti DA, Xiong R, Zhao J, Gutgesell L, Dudek AZ, Thatcher GR. (Abstract e14090) Efficacy, maximal tolerated dose, and toxicokinetics of TTC-352 in rats and dogs, a partial ER agonist for metastatic ER+ breast cancer. J Clin Oncol. 2017. [Google Scholar]
- 18.Jordan VC, Curpan R, Maximov PY. Estrogen Receptor Mutations Found in Breast Cancer Metastases Integrated With the Molecular Pharmacology of Selective ER Modulators. Jnci-J Natl Cancer I. 2015;107(6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pejerrey SM, Dustin D, Kim JA, Gu G, Rechoum Y, Fuqua SAW. The Impact of ESR1 Mutations on the Treatment of Metastatic Breast Cancer. Horm Cancer. 2018;9(4):215–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Niu J, Andres G, Kramer K, Kundranda MN, Alvarez RH, Klimant E, et al. Incidence and clinical significance of ESR1 mutations in heavily pretreated metastatic breast cancer patients. Onco Targets Ther. 2015;8:3323–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pink JJ, Jordan VC. Models of estrogen receptor regulation by estrogens and antiestrogens in breast cancer cell lines. Cancer research. 1996;56(10):2321–30. [PubMed] [Google Scholar]
- 22.Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, Engstrom O, et al. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389(6652):753–8. [DOI] [PubMed] [Google Scholar]
- 23.Yi P, Wang Z, Feng Q, Pintilie GD, Foulds CE, Lanz RB, et al. Structure of a biologically active estrogen receptor-coactivator complex on DNA. Mol Cell. 2015;57(6):1047–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maximov PY, Myers CB, Curpan RF, Lewis-Wambi JS, Jordan VC. Structure-function relationships of estrogenic triphenylethylenes related to endoxifen and 4-hydroxytamoxifen. J Med Chem. 2010;53(8):3273–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gates LA, Gu G, Chen Y, Rohira AD, Lei JT, Hamilton RA, et al. Proteomic profiling identifies key coactivators utilized by mutant ERalpha proteins as potential new therapeutic targets. Oncogene. 2018;37(33):4581–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jiang SY, Wolf DM, Yingling JM, Chang C, Jordan VC. An estrogen receptor positive MCF-7 clone that is resistant to antiestrogens and estradiol. Mol Cell Endocrinol. 1992;90(1):77–86. [DOI] [PubMed] [Google Scholar]
- 27.Murphy CS, Pink JJ, Jordan VC. Characterization of a receptor-negative, hormone-nonresponsive clone derived from a T47D human breast cancer cell line kept under estrogen-free conditions. Cancer research. 1990;50(22):7285–92. [PubMed] [Google Scholar]
- 28.Lasfargues EY, Coutinho WG, Redfield ES. Isolation of two human tumor epithelial cell lines from solid breast carcinomas. Journal of the National Cancer Institute. 1978;61(4):967–78. [PubMed] [Google Scholar]
- 29.Engel LW, Young NA, Tralka TS, Lippman ME, O'Brien SJ, Joyce MJ. Establishment and characterization of three new continuous cell lines derived from human breast carcinomas. Cancer research. 1978;38(10):3352–64. [PubMed] [Google Scholar]
- 30.Fan P, Agboke FA, Cunliffe HE, Ramos P, Jordan VC. A molecular model for the mechanism of acquired tamoxifen resistance in breast cancer. Eur J Cancer. 2014;50(16):2866–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lewis JS, Osipo C, Meeke K, Jordan VC. Estrogen-induced apoptosis in a breast cancer model resistant to long-term estrogen withdrawal. J Steroid Biochem Mol Biol. 2005;94(1-3):131–41. [DOI] [PubMed] [Google Scholar]
- 32.Pink JJ, Jiang SY, Fritsch M, Jordan VC. An Estrogen-Independent Mcf-7 Breast-Cancer Cell-Line Which Contains a Novel 80-Kilodalton Estrogen Receptor-Related Protein. Cancer research. 1995;55(12):2583–90. [PubMed] [Google Scholar]
- 33.Liu H, Lee ES, Gajdos C, Pearce ST, Chen B, Osipo C, et al. Apoptotic action of 17beta-estradiol in raloxifene-resistant MCF-7 cells in vitro and in vivo. Journal of the National Cancer Institute. 2003;95(21):1586–97. [DOI] [PubMed] [Google Scholar]
- 34.Clarke R, Brunner N, Katzenellenbogen BS, Thompson EW, Norman MJ, Koppi C, et al. Progression of human breast cancer cells from hormone-dependent to hormone-independent growth both in vitro and in vivo. Proc Natl Acad Sci U S A. 1989;86(10):3649–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brunner N, Boulay V, Fojo A, Freter CE, Lippman ME, Clarke R. Acquisition of hormone-independent growth in MCF-7 cells is accompanied by increased expression of estrogen-regulated genes but without detectable DNA amplifications. Cancer research. 1993;53(2):283–90. [PubMed] [Google Scholar]
- 36.Brunner N, Frandsen TL, Holsthansen C, Bei M, Thompson EW, Wakeling AE, et al. Mcf7/Lcc2 - a 4-Hydroxytamoxifen Resistant Human Breast-Cancer Variant That Retains Sensitivity to the Steroidal Antiestrogen Ici-182,780. Cancer research. 1993;53(14):3229–32. [PubMed] [Google Scholar]
- 37.Brunner N, Boysen B, Jirus S, Skaar TC, HolstHansen C, Lippman J, et al. MCF7/LCC9: An antiestrogen-resistant MCF-7 variant in which acquired resistance to the steroidal antiestrogen ICI 182,780 confers an early cross-resistance to the nonsteroidal antiestrogen tamoxifen. Cancer research. 1997;57(16):3486–93. [PubMed] [Google Scholar]
- 38.Fan P, Griffith OL, Agboke FA, Anur P, Zou X, McDaniel RE, et al. c-Src modulates estrogen-induced stress and apoptosis in estrogen-deprived breast cancer cells. Cancer research. 2013;73(14):4510–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Foulds CE, Feng Q, Ding C, Bailey S, Hunsaker TL, Malovannaya A, et al. Proteomic analysis of coregulators bound to ERalpha on DNA and nucleosomes reveals coregulator dynamics. Mol Cell. 2013;51(2):185–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Malovannaya A, Lanz RB, Jung SY, Bulynko Y, Le NT, Chan DW, et al. Analysis of the human endogenous coregulator complexome. Cell. 2011;145(5):787–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saltzman AB, Leng M, Bhatt B, Singh P, Chan DW, Dobrolecki L, et al. gpGrouper: A Peptide Grouping Algorithm for Gene-Centric Inference and Quantitation of Bottom-Up Proteomics Data. Molecular & Cellular Proteomics. 2018;17(11):2270–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Speltz TE, Fanning SW, Mayne CG, Fowler C, Tajkhorshid E, Greene GL, et al. Stapled Peptides with gamma-Methylated Hydrocarbon Chains for the Estrogen Receptor/Coactivator Interaction. Angew Chem Int Ed Engl. 2016;55(13):4252–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Beriault DR, Werstuck GH. Detection and quantification of endoplasmic reticulum stress in living cells using the fluorescent compound, Thioflavin T. Bba-Mol Cell Res. 2013;1833(10):2293–301. [DOI] [PubMed] [Google Scholar]
- 44.Sengupta S, Sharma CG, Jordan VC. Estrogen regulation of X-box binding protein-1 and its role in estrogen induced growth of breast and endometrial cancer cells. Horm Mol Biol Clin Investig. 2010;2(2):235–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Obiorah I, Sengupta S, Curpan R, Jordan VC. Defining the Conformation of the Estrogen Receptor Complex That Controls Estrogen-Induced Apoptosis in Breast Cancer. Molecular pharmacology. 2014;85(5):789–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maximov PY, Abderrahman B, Hawsawi YM, Chen Y, Foulds CE, Jain A, et al. The Structure-Function Relationship of Angular Estrogens and Estrogen Receptor Alpha to Initiate Estrogen-Induced Apoptosis in Breast Cancer Cells. Molecular pharmacology. 2020;98(1):24–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Toy W, Shen Y, Won H, Green B, Sakr RA, Will M, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nature Genetics. 2013;45(12):1439–U189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Abderrahman B, Maximov PY, Curpan RF, Hanspal JS, Fan P, Xiong R, et al. Pharmacology and Molecular Mechanisms of Clinically-Relevant Estrogen Estetrol and Estrogen Mimic BMI-135 for the Treatment of Endocrine-Resistant Breast Cancer Molecular pharmacology. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jordan VC, Lieberman ME. Estrogen-stimulated prolactin synthesis in vitro. Classification of agonist, partial agonist, and antagonist actions based on structure. Molecular pharmacology. 1984;26(2):279–85. [PubMed] [Google Scholar]
- 50.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, Agard DA, et al. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95(7):927–37. [DOI] [PubMed] [Google Scholar]
- 51.Jordan VC. The new biology of estrogen-induced apoptosis applied to treat and prevent breast cancer. Endocrine-related cancer. 2015;22(1):R1–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hu Z, Li Z, Ma Z, Curtis C. Multi-cancer analysis of clonality and the timing of systemic spread in paired primary tumors and metastases. Nat Genet. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Chlebowski RT, Anderson GL, Aragaki AK, Manson JE, Stefanick ML, Pan K, et al. Association of Menopausal Hormone Therapy With Breast Cancer Incidence and Mortality During Long-term Follow-up of the Women's Health Initiative Randomized Clinical Trials. JAMA. 2020;324(4):369–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jordan VC. Molecular Mechanism for Breast Cancer Incidence in the Women's Health Initiative. Cancer Prev Res (Phila). 2020. [DOI] [PubMed] [Google Scholar]
- 55.Wolf DM, Jordan VC. The estrogen receptor from a tamoxifen stimulated MCF-7 tumor variant contains a point mutation in the ligand binding domain. Breast Cancer Res Treat. 1994;31(1):129–38. [DOI] [PubMed] [Google Scholar]
- 56.Jiang SY, Jordan VC. Growth regulation of estrogen receptor-negative breast cancer cells transfected with complementary DNAs for estrogen receptor. Journal of the National Cancer Institute. 1992;84(8):580–91. [DOI] [PubMed] [Google Scholar]
- 57.Catherino WH, Wolf DM, Jordan VC. A Naturally-Occurring Estrogen-Receptor Mutation Results in Increased Estrogenicity of a Tamoxifen Analog. Molecular Endocrinology. 1995;9(8):1053–63. [DOI] [PubMed] [Google Scholar]
- 58.Levenson AS, Catherino WH, Jordan VC. Estrogenic activity is increased for an antiestrogen by a natural mutation of the estrogen receptor. J Steroid Biochem. 1997;60(5-6):261–8. [DOI] [PubMed] [Google Scholar]
- 59.Liu H, Park WC, Bentrem DJ, McKian KP, Reyes Ade L, Loweth JA, et al. Structure-function relationships of the raloxifene-estrogen receptor-alpha complex for regulating transforming growth factor-alpha expression in breast cancer cells. J Biol Chem. 2002;277(11):9189–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fanning SW, Mayne CG, Dharmarajan V, Carlson KE, Martin TA, Novick SJ, et al. Estrogen receptor alpha somatic mutations Y537S and D538G confer breast cancer endocrine resistance by stabilizing the activating function-2 binding conformation. Elife. 2016;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shiau AK, Barstad D, Radek JT, Meyers MJ, Nettles KW, Katzenellenbogen BS, et al. Structural characterization of a subtype-selective ligand reveals a novel mode of estrogen receptor antagonism. Nat Struct Biol. 2002;9(5):359–64. [DOI] [PubMed] [Google Scholar]
- 62.Nolte RT, Wisely GB, Westin S, Cobb JE, Lambert MH, Kurokawa R, et al. Ligand binding and co-activator assembly of the peroxisome proliferator-activated receptor-gamma. Nature. 1998;395(6698):137–43. [DOI] [PubMed] [Google Scholar]
- 63.Osborne CK, Bardou V, Hopp TA, Chamness GC, Hilsenbeck SG, Fuqua SAW, et al. Role of the estrogen receptor coactivator AIB1 (SRC-3) and HER-2/neu in tamoxifen resistance in breast cancer. Journal of the National Cancer Institute. 2003;95(5):353–61. [DOI] [PubMed] [Google Scholar]
- 64.Rodriguez-Brenes IA, Wodarz D. Preventing clonal evolutionary processes in cancer: Insights from mathematical models. Proc Natl Acad Sci U S A. 2015;112(29):8843–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.