

Abstract
Rationale:
Previous translational studies implicate plasma extracellular microRNA-30d (miR-30d) as a biomarker in left ventricular (LV) remodeling and clinical outcome in heart failure (HF) patients, though precise mechanisms remain obscure.
Objective:
To investigate the mechanism of miR-30d-mediated cardioprotection in HF.
Methods and Results:
In rat and mouse models of ischemic HF, we show that miR-30d gain of function (genetic, lentivirus or agomiR-mediated) improves cardiac function, decreases myocardial fibrosis, and attenuates cardiomyocyte (CM) apoptosis. Genetic or locked nucleic acid (LNA)-based knock-down of miR-30d expression potentiates pathological LV remodeling, with increased dysfunction, fibrosis, and CM death. RNA-seq of in vitro miR-30d gain and loss of function, together with bioinformatic prediction and experimental validation in cardiac myocytes and fibroblasts, were used to identify and validate direct targets of miR-30d. miR-30d expression is selectively enriched in CMs, induced by hypoxic stress and is acutely protective, targeting mitogen-associate protein kinase (MAP4K4) to ameliorate apoptosis. Moreover, miR-30d is secreted primarily in extracellular vesicles by CMs and inhibits fibroblast proliferation and activation by directly targeting integrin α5 in the acute phase via paracrine signaling to cardiac fibroblasts. In the chronic phase of ischemic remodeling, lower expression of miR-30d in the heart and plasma EVs is associated with adverse remodeling in rodent models and human subjects, and is linked to whole blood expression of genes implicated in fibrosis and inflammation, consistent with observations in model systems.
Conclusions:
These findings provide the mechanistic underpinning for the cardioprotective association of miR-30d in human HF. More broadly, our findings support an emerging paradigm involving intercellular communication of EV-contained miRNAs to trans regulate distinct signaling pathways across cell types. Functionally validated RNA biomarkers and their signaling networks may warrant further investigation as novel therapeutic targets in HF.
Keywords: Heart failure, miRNA, fibrosis, apoptosis, exosomes, Animal Models of Human Disease, Biomarkers, Cell Signaling/Signal Transduction, Remodeling
Graphical Abstract
INTRODUCTION
Heart failure (HF) is a leading cause of cardiovascular disease morbidity and mortality 1, with more than 26 million individuals affected worldwide 2. Despite advances in prevention and treatment, mortality from advanced HF remains high. As evidenced by novel therapeutics in HF (e.g., SGLT2 inhibition), therapeutic interventions that involve unexplored pathways in myocardial function may offer new avenues of HF management. Drugs favorably impacting outcome in HF with reduced ejection fraction (HFrEF) generally produce reverse left ventricular remodeling 3. In this regard, targeting pathways central to cardiac remodeling is likely to produce the most fruitful therapeutic result.
Cardiac remodeling in response to pathological stress involves inflammation, fibrosis and cell death, ultimately progressing to advanced HF 4. During the last decade, non-coding RNAs have emerged as biomarkers of HF disease progression and prognosis 5, though how non-coding RNAs signal across multiple cell types to impact the heart and cardiac remodeling remains an area of active exploration. One class of non-coding RNAs, miRNAs, regulate gene expression and play an important role in pathological cardiac remodeling, cardiac hypertrophy, fibrosis and dysfunction 5. Studies in other disease processes suggest that miRNAs may be packaged in circulating extracellular vesicles (EVs) and mediate some aspects of cross-talk between cell types6,7. Previous work suggests that higher circulating levels of miR-30d are associated with favorable ventricular remodeling response to advanced device therapies in HF 8 and may be protective against 1 year mortality in HF 9. However, the precise mechanism of how miR-30d impacts cardiac remodeling remains obscure.
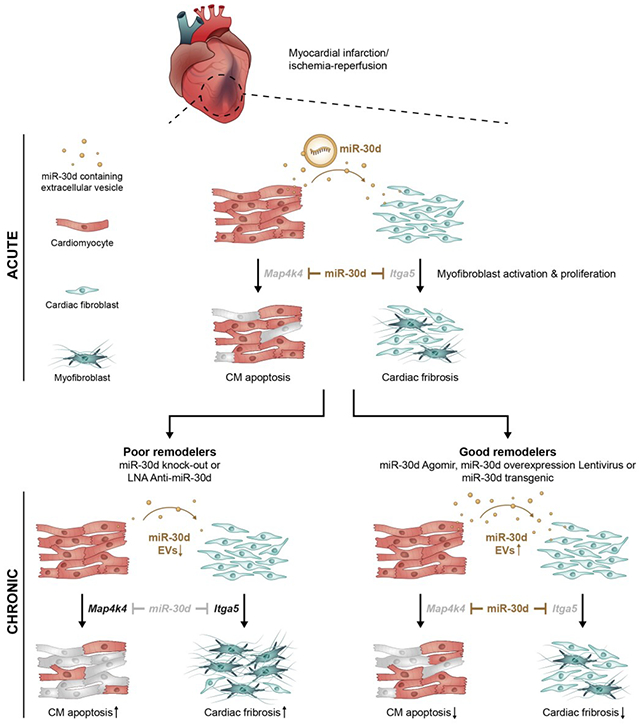
Here, we use multiple models of miR-30d overexpression (OE; genetic, lentivirus and agomiR-mediated) and loss-of-function (locked nucleic acid LNA and genetic) in rodents to delineate the mechanistic underpinnings of specific cellular processes regulated by miR-30d in the context of ischemic HF. We investigate the complex dynamics of miR-30d expression and release in cardiomyocytes, demonstrating an increase acutely after ischemia, with subsequent decrease over time. Increased expression of miR-30d prevents the development of cardiac dysfunction, fibrosis and cardiomyocyte apoptosis in vivo by specifically targeting map4k4 within cardiomyocytes and itga5 in cardiac fibroblasts, while ablation of miR-30d has the opposite effect. Furthermore, we conclusively establish that miR-30d is released by cardiomyocytes primarily in extracellular vesicles (EVs), mediating a novel mode of paracrine signaling between cardiomyocytes and fibroblasts by targeting itga5, a key regulator of fibrosis pathways. Using a novel exosome tracking mouse model, we show that this EV-mediated paracrine signaling between CMs and fibroblasts is activated in vivo in ischemic HF. In chronic ischemic HF, circulating miR-30d-EV levels are reduced in wild-type but not miR-30d overexpressing rodents (where post-MI remodeling is ameliorated). Similarly, in human patients with chronic HF, miR-30d-EV levels are associated with systolic function, with higher levels associated with beneficial remodeling. Co-expression analyses of mRNA in whole blood with plasma circulating miR-30d levels suggest involvement of miR-30d in MAP kinase and fibrosis signaling in humans, providing support to pathways discovered in animal models. Taken together, our results indicate that miR-30d is protective in the acute phase of post-ischemic remodeling, while subsequent decline in cardiac and EV expression of miR-30d in the chronic phase is associated with adverse remodeling in the rodent models. In both the rodent models, and chronic human HF, higher levels of miR-30d are associated with beneficial remodeling. Our study provides the mechanistic underpinnings for clinical observations suggesting its role as a functional biomarker of cardiac remodeling, and a potential therapeutic target in the chronic phase of ventricular remodeling.
METHODS
Detailed and expanded methods are presented in the online Supplemental Material.
Data Availability.
The authors will make our data, study materials and analytic methods available to other researchers. All supporting data are available within the article, supplemental material and datasets provided. If additional data is required, it will be made available by the corresponding author upon reasonable request.
RESULTS
Transgenic rats overexpressing miR-30d are protected from adverse cardiac remodeling as compared to WT in response to myocardial infarction (MI).
Our previous study had suggested a role for miR-30d in ameliorating cardiomyocyte apoptosis in cell culture models 8 as a possible basis for its association with beneficial cardiac remodeling. To definitively determine a critical role for miR-30d in the pathogenesis of cardiac remodeling in vivo, we examined the effects of miR-30d overexpression or loss-of-function in a rodent model of ischemic HF. We generated whole body miR-30d overexpressing SD transgenic rats (Supp. Figure I A–C). Expression of mature miR-30d was increased three-fold in this novel rat transgenic model (Supp. Figure I C). The miR-30d transgenic rats alongside wild-type (WT) SD rats, were subjected to myocardial infarction (MI) or sham surgery, and their cardiac phenotype was determined 3 weeks after induction of MI (Figure 1). Ventricular function, determined by non-invasive echocardiography and quantified by percent fractional shortening, was significantly improved in miR-30d overexpressing rats as compared to WT rats 3 weeks after MI (Figure 1A). We next sought to determine if miR-30d may affect cellular processes such as fibrosis and apoptosis that contribute to cardiac remodeling. Ventricular fibrosis was quantified by staining rat heart cross-sections for Masson’s trichrome, as a direct indicator of collagen deposition. While both WT and miR-30d overexpressing rats showed an increase in interstitial ventricular fibrosis as a result of the MI surgery, rats expressing elevated miR-30d levels presented a significantly lower degree of fibrosis as compared to their WT counterparts (Figure 1B). Additionally, protein levels of α-SMA (alpha- Smooth Muscle Actin), a marker of fibroblast activation and myofibroblast transformation, was significantly augmented in WT rats after MI surgery, while miR-30d overexpression prevented α-SMA upregulation (Figure 1C). Together, these data suggested that miR-30d negatively regulates pro-fibrotic pathways in the myocardium.
Figure 1. Transgenic rats and lentivirus treated mice overexpressing miR-30d are protected from adverse cardiac remodeling as compared to WT in response to myocardial infarction (MI).
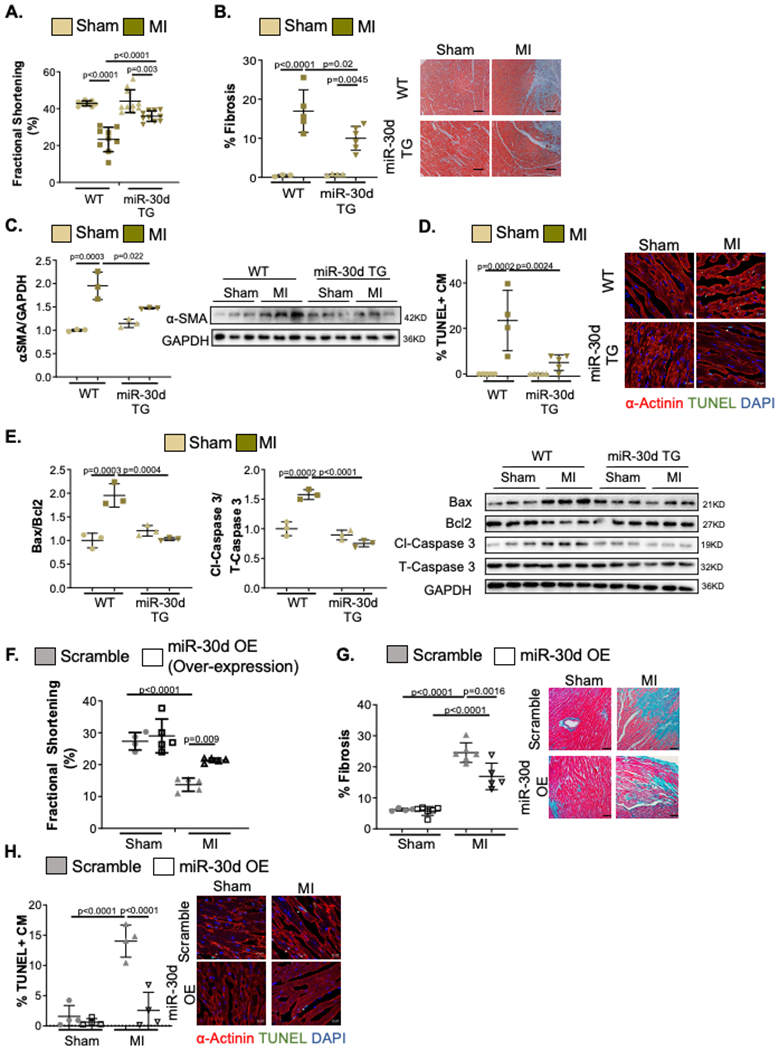
(A-E) WT and miR-30d overexpressing transgenic rats were subjected to Sham or MI surgery and their myocardial remodeling phenotype was evaluated. A. Cardiac function quantified by percent Fractional Shortening determined through echocardiography. WT Sham n=9, WT MI n=9, miR-30d TG Sham n=10, miR-30d TG MI n=10. B. Collagen deposition determined by staining heart cross-sections for Masson’s Trichrome; results are graphed on the left and representative bright field images are shown on the right next by. WT Sham n=3, WT MI n=5, miR-30d TG Sham n=4, miR-30d TG MI n=6. C. Protein levels of α-SMA were determined through western blot; quantification of fold change vs WT Sham is graphed on the left and representative blot is shown on the right. WT Sham n=3, WT MI n=3, miR-30d TG Sham n=3, miR-30d TG MI n=3.D. Cardiac myocyte (CM) apoptosis 24 hours post-surgery is determined by TUNEL and α-actinin co-staining of heart cross-sections, and quantified as % of TUNEL+ cardiac myocytes per field; data is graphed on the left and representative pictures are shown on the right by the graph. White arrows point to representative apoptotic cardiac myocytes. Scale bar 20μM. WT Sham n=5, WT MI n=4, miR-30d TG Sham n=5, miR-30d TG MI n=5. E. Molecular pathways regulating apoptotic pathways were determined through western blotting for Bax, Bcl2, cleaved-Caspase 3 and total Caspase 3; data is graphed on the left as fold change vs WT Sham and representative blots are shown on the right. WT Sham n=3, WT MI n=3, miR-30d TG Sham n=3, miR-30d TG MI n=3. (F-H) C57BL/6 mice treated with 10^9TU/mL miR-30d overexpressing lentivirus or scramble lentivirus, were subjected to Sham or MI surgery and their cardiac remodeling phenotype was analyzed. F. Cardiac function was determined through echocardiography and the percent Fractional Shortening was quantified. Sham scramble lentivirus n=4, Sham miR-30d OE lentivirus n=5, MI scramble lentivirus n=5, MI miR-30d OE lentivirus n=5. G. Myocardial fibrosis was quantified by staining cardiac cross-sections for Masson’s Trichrome; results are represented on the left and representative bright field images are shown on the right next by. Sham scramble lentivirus n=4, Sham miR-30d OE lentivirus n=6, MI scramble lentivirus n=6, MI miR-30d OE lentivirus n=5. H. Cardiac myocyte apoptosis was quantified 24 hours post-surgery by co-staining heart cross-sections for TUNEL and α-actinin; quantification of % of TUNEL+ cardiac myocytes (CM) per field is graphed on the left and representative pictures are shown on the right by the graph. White arrows point to representative TUNEL+ CM. Scale bar 20μM. Sham scramble lentivirus n=4, Sham miR-30d OE lentivirus n=4, MI scramble lentivirus n=4, MI miR-30d OE lentivirus n=4. Data are represented as mean ± SD. Normality was tested using Shapiro-Wilk test. P values calculated with ordinary ANOVA test, corrected using Tukey’s multiple comparison test.
We next examined cardiac myocyte (CM) apoptosis by TUNEL staining in α-actinin-immunopositive CMs as an indicator of apoptotic nuclei in rats 24 hours after MI. We found significantly reduced CM apoptosis in miR-30d overexpressing rat hearts as compared to WT rats after MI (Figure 1D); in line with this observation, the ratio of Bax/Bcl2 and cleaved-Caspase 3/ total Caspase 3, protein mediators of apoptotic pathways, were found to be increased in WT rats after MI, in contrast to miR-30d overexpressing rats, in which there was no increase in the ratio of these proteins, suggesting protection from CM apoptosis (Figure 1E). Together, our data in rats overexpressing miR-30d, showed that elevated levels of miR-30d is sufficient to confer protection from developing adverse cardiac remodeling, characterized by cardiac dysfunction, fibrosis and CM apoptosis in this model of ischemic heart disease.
Pharmacological or viral-mediated overexpression of miR-30d in mouse hearts ameliorates adverse cardiac remodeling in ischemic HF.
To complement our studies with genetic overexpression of miR-30d and explore the therapeutic potential of miR-30d mimics in the treatment of ischemic HF, we assessed the role of miR-30d overexpression using pharmacological or viral-gene transfer approaches. A lentivirus mediated cardiac specific miR-30d overexpression approach was next assessed (Supp. Figure I D). Single site myocardial injection of 25μL of viral particles (109TU/mL) during thoracotomy one week before MI surgery resulted in a two-fold increase in miR-30d levels in the heart three weeks after surgery (Supp. Figure I D). Cardiac remodeling 3 weeks after MI was assessed by M-mode echocardiography demonstrated that while the fractional shortening decreased in scramble lentivirus treated animals subjected to MI compared to Sham surgery, lentiviral-mediated miR-30d overexpression (OE) in the heart ameliorated the decline in ventricular contractility after MI as compared to scramble lentivirus-treated mice (Figure 1F). Similar to our findings with genetic overexpression of miR-30d (Figure 1B), cardiac fibrosis following MI was significantly lower in the miR-30d overexpressing mice compared with the scramble lentivirus treated mice (Figure 1G). Correspondingly, while there was upregulation of α-SMA in both miR-30d OE mice as well as the scramble lentiviral control mice after MI (Supp. Figure II A), the increase in α-SMA expression was significantly lower with miR-30d overexpression, as compared to scramble lentivirus treated counterparts. Finally, similar to our results with genetic overexpression of miR-30d, lentiviral-transduced miR-30d overexpression markedly ameliorated CM apoptosis 24 hours following MI (Figure 1H, Supp. Figure II B). TUNEL+ CM quantification showed a significantly reduced number of apoptotic CM in the heart of mice treated with miR-30d overexpressing lentivirus, compared with scramble lentivirus treated mice after MI surgery (Figure 1H). The Bax/Bcl2 and cleaved-Caspase 3/ total Caspase 3 ratios, were accordingly found to be elevated in scramble lentivirus-treated mice after MI, while miR-30d lentivirus treated mice did not demonstrate significant changes in the expression levels of these proteins, confirming miR-30d’s role in preventing CM apoptosis (Supp. Figure II B). As a third method to increase miR-30d expression in the MI model, we injected miR-30d agomiR (RinoBio, 20 nmol/mouse in PBS, via tail vein injection for 3 consecutive days) or a control agomiR to induce whole body overexpression for miR-30d prior to implementation of MI. Sham or MI surgeries were performed one day after the last agomiR dose. Cardiac remodeling as assessed by M-mode echocardiography, histology and biochemical studies showed that miR-30d overexpression, similar to the models described above, abrogated the decline in cardiac contractile function, fibrosis and cardiomyocyte apoptosis (Supp. Figures II C–F). Taken together, our studies demonstrated that miR-30d overexpression, both in a whole body as well as cardiac-restricted manner, achieved using genetic, viral gene transfer and pharmacological agomiR approaches in rodent models, protected against adverse remodeling after MI, ameliorating the decline in cardiac function, and the degree of fibrosis and apoptosis compared to the appropriate controls. These findings suggest a mechanistic basis for our previous clinical observation of association between elevated circulating miR-30d levels and ventricular remodeling in human heart failure patients8.
LNA mediated or genetic miR-30d loss of function results in exaggerated adverse cardiac remodeling in the ischemic HF model.
Heart failure patients with lower levels of circulating miR-30d have worsened cardiac remodeling and increased mortality8,9. To determine the cardiac consequences of reducing miR-30d levels on cardiac remodeling, mice were treated with either an anti-miR-30d Locked Nucleic Acid (LNA, Qiagen) or scramble LNA and subjected to Ischemia – Reperfusion (IR) surgery (Supp. Figure III A). Treatment with anti-miR-30d LNA markedly attenuated expression of mature miR-30d at the time of surgery when compared to scramble LNA (Supp. Figure III A). We chose a milder form of stress (IR surgery compared to MI) to better distinguish the hypothesized deleterious effects of miR-30d silencing on cardiac remodeling. Quantification of cardiac function through M-mode echocardiography showed a worsened cardiac function (as assessed by fractional shortening) in LNA anti-miR-30d treated mice as compared to scramble LNA treated mice in response to IR (Figure 2A). As described above, pro-fibrotic pathways were studied by quantifying collagen deposition in the cardiac interstitium (Figure 2B) and expression of α-SMA (Figure 2C). While both LNA anti-miR-30d and scramble LNA treated mice had a significant increase in interstitial fibrosis in the heart after IR surgery as compared to Sham operated mice, reduced expression of miR-30d leaded to a greater degree of fibrosis as compared to LNA scramble treated mice (Figure 2B). In line with this observation, α-SMA protein expression in the heart was greater in LNA anti-miR-30d treated mice as compared to the LNA scramble group after IR (Figure 2C). Finally, the number of TUNEL+ CM nuclei number was markedly greater in LNA anti-miR-30d mice group after IR, while LNA scramble treated mice had a lower degree of CM apoptosis after IR surgery (Figure 2D). Following the pattern observed with the TUNEL+ CM, the Bax/Bcl2 and the cleaved Caspase 3/total Caspase 3 ratios were increased in LNA anti-miR-30d treated mice as compared to LNA scramble mice group after IR (Figure 2E). As LNA-anti-miRs may have off-target effects, to further confirm the detrimental effect of miR-30d’s loss of function in the cardiac remodeling phenotype, a genetic approach was utilized in SD rats to generate whole body miR-30d knock out (KO) rats (Supp. Figure III B–D). Similar to the phenotype observed in LNA anti-miR-30d treated mice, cardiac function was found to be decreased in miR-30d KO rats as compared to WT rats after MI (Figure 2F). In accordance with the phenotype described above, collagen deposition (Figure 2G) and CM apoptosis (Figure 2H, Supp. Figure IV) were aggravated in miR-30d KO rats after MI to a higher extent than WT rats. Together, miR-30d loss of function data indicate that silencing of miR-30d leads to a higher degree of adverse cardiac remodeling after ischemic insult, defined by severe cardiac dysfunction, increased cardiac fibrosis and exacerbated CM apoptosis.
Figure 2. LNA mediated or genetic miR-30d loss of function results in exaggerated adverse cardiac remodeling after cardiac ischemia.
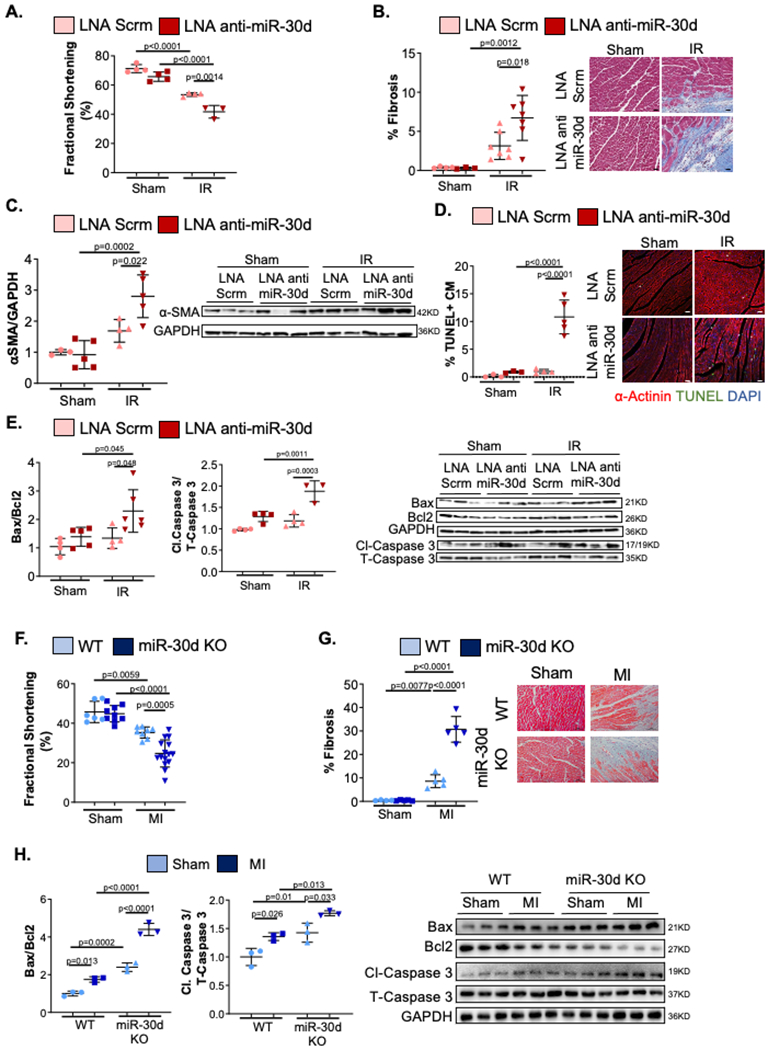
A-E. C57BL/6 mice treated with either Scramble or LNA anti-miR-30d were subjected to Sham or IR surgery and their myocardial remodeling phenotype was evaluated. (A) Cardiac function quantified by percent Fractional Shortening determined through echocardiography. Sham LNA Scrm n= 4, Sham LNA anti-miR30d n= 4, IR LNA Scrm n= 4, IR LNA anti-miR-30d n= 3. (B) Collagen deposition determined by staining heart cross-sections for Masson’s Trichrome; results are graphed on the left and representative bright field images are shown on the right. Scale bar 25μm. Sham LNA Scrm n= 4, Sham LNA anti-miR30d n= 3, IR LNA Scrm n= 7, IR LNA anti-miR-30d n= 7. (C) Protein levels of α-SMA were determined through western blot; quantification of fold change vs Sham LNA Scramble is graphed on the left and representative blot is shown on the right. Sham LNA Scrm n= 3, Sham LNA anti-miR30d n= 5, IR LNA Scrm n= 4, IR LNA anti-miR-30d n= 5. (D) Cardiac myocyte (CM) apoptosis determined by TUNEL and α-actinin co-staining of heart cross-sections, is quantified as % of TUNEL+ CM per field; data is graphed on the left and representative pictures are shown on the right by the graph. White arrows point to representative apoptotic cardiac myocytes. Scale bar 25μm. Sham LNA Scrm n= 3, Sham LNA anti-miR30d n= 3, IR LNA Scrm n= 4, IR LNA anti-miR-30d n= 5. (E) Molecular pathways regulating apoptotic pathways were determined through western blotting for Bax, Bcl2(Sham LNA Scrm n= 4, Sham LNA anti-miR30d n= 5, IR LNA Scrm n= 4, IR LNA anti-miR-30d n= 6), cleaved-Caspase 3 and total Caspase 3(Sham LNA Scrm n= 4, Sham LNA anti-miR30d n= 4, IR LNA Scrm n= 4, IR LNA anti-miR-30d n= 3); data is graphed on the left as fold change vs Sham LNA Scramble and representative blots are shown on the right. F-H. Transgenic miR-30d KO SD rats and WT counterparts were subjected to Sham or MI surgery and their cardiac remodeling phenotype was evaluated. (F) % Fractional Shortening was determined through echocardiography. WT Sham n=6, miR-30d KO Sham n=9, WT MI n=8, miR-30d KO MI n=15. (G) Collagen deposition determined by staining heart cross-sections for Masson’s Trichrome; results are graphed on the left and representative bright field images are shown on the right next by. WT Sham n=4, miR-30d KO Sham n=5, WT MI n=5, miR-30d KO MI n=5. (H) Molecular pathways regulating apoptotic pathways were determined in miR-30d KO and WT rats through western blotting for Bax, Bcl2, cleaved Caspase 3 and total Caspase 3; data is graphed on the left as fold change vs WT Sham and representative blots are shown on the right. WT Sham n=3, miR-30d KO Sham n=3, WT MI n=3, miR-30d KO MI n=3. Data are represented as mean ± SD. Normality was tested using Shapiro-Wilk test. P values calculated with ordinary ANOVA test, corrected using Tukey’s multiple comparison test.
As miR-30d overexpression is protective against cardiomyocyte cell death, we determined the effect of miR-30d levels on acute infarct size at 24 post-surgery by triphenyltetrazolium (TTC) staining. Interestingly, overexpression of miR-30d resulted in a lower infarct size/ area at risk at 24 hours (Supp. Figure V A–B), while the opposite was observed in miR-30d loss of function (Supp. Figure V C–D). Collectively, these data strongly support the role of miR-30d in preventing pathological ventricular remodeling, at least in part by acutely protecting against cardiomyocyte cell death and restricting the initial infarct size in the acute phase. Continued subsequent expression of miR-30d in the heart protects against fibrosis and cardiomyocyte cell death in the chronic remodeling phase.
miR-30d targets specific molecules within the MAPK and integrin signaling pathways.
To identify the molecular pathways specifically targeted by miR-30d which contribute to the cardiac remodeling phenotype described above (Figures 1–2), we performed RNA sequencing on neonatal rat ventricular myocytes (NRVM) transfected with either miR-30d overexpressing adenovirus (overexpression) or LNA anti-miR-30d (loss-of-function). On average, about 81% of the reads mapped uniquely to the rat genome (Rnor 6.0); the full mapping statistics of the RNA sequencing is presented in Supp. Table I. The majority of the reads and the genes detected (greater than 90%, see Supp. Figure VI A–B, Supp. Tables II–III) were assigned to the protein coding regions of the genome. A principal components analysis plot (Supp. Figure VI C) based on the overall gene expression profiles shows a clear distinction between the miR-30d adenovirus, the control and LNA anti-miR-30d NRVM samples. Differential expression analysis was performed to discover genes that were dysregulated with miR-30d: a) Over-expression: miR-30d overexpressing adenovirus versus control and b) Loss of function: LNA Anti-miR-30d versus controls. For miR-30d overexpression, there were 3334 genes differentially expressed (949 upregulated and 2385 downregulated at a two-fold change and padj<0.05); for LNA Anti-miR-30d versus control, there were 1004 genes downregulated and 1633 genes upregulated (the full list of differentially expressed genes is presented in Supp. Table IV). Of the genes that were differentially regulated in both the overexpression and loss-of-function experiments, most of the genes (512 out of 650 genes) were dysregulated in opposite directions with respect to the controls (Supp. Figure VI D). Gene set enrichment analysis with KEGG shows that there are 69 pathways dysregulated (51 pathways upregulated and 18 downregulated) in miR-30d overexpression and 109 pathways dysregulated (33 upregulated and 76 downregulated) in miR-30d loss-of-function (the full list of pathways is presented in Supp. Table V). When restricted to pathways relevant to cardiac remodeling, the Focal Adhesion pathway (relevant to fibrosis) and mitogen associated kinase (MAPK) signaling pathways were of particular interest and significantly dysregulated in opposite directions between the miR-30d overexpression and loss of function conditions. Genes in the KEGG pathways that were discordantly regulated (i.e. either significantly downregulated/unchanged in the miR-30d overexpression condition and changed in the opposite direction with the LNA anti-miR-30d) and also contain the seed sequence for miR-30d were chosen for more detailed interrogation as possible direct targets of miR-30d in the heart. These included itga5, itga6, itgaV, itbl1 and itgb3 from the KEGG Focal Adhesion pathway and mitogen associated protein kinase 4 (map4k4) from the KEGG MAPK Signaling pathway. Our previous studies had implicated mitogen associated protein kinase 4 (map4k4) as a gene regulated by miR-30d, which conferred protection against cardiomyocyte apoptosis in cell culture models8. We therefore sought to validate these possible targets as direct targets and key mediators of miR-30d effects on cardiac remodeling.
Map4k4 is a direct target of miR-30d and mediator of its protective effects on cardiomyocytes.
Luciferase assays were performed by cloning 500bp fragments of the 3’UTR region of the map4k4 encoding gene, which contained the putative miR-30d target complementary to its seed sequence. In addition, the same 500bp fragment of the 3’UTR region of the map4k4 encoding gene was mutated to remove the seed sequence and cloned into vectors, to serve as a control for the luciferase assay. Co-transfection of WT or mutant map4k4 3’UTR containing vectors into HEK293 cells, showed that the luciferase expression was decreased in WT map4k4 3’UTR bearing vectors but not the mutant map4k4 3’UTR containing vectors when co-transfected with miR-30d mimic (Figure 3A). In NRVMs, transfection of miR-30d mimic resulted in a decreased expression of map4k4, while conversely, transfection with LNA anti-miR-30d led to its increased expression (Figure 3B). Given the well described role of MAP4K4 in cellular apoptosis 10, and the phenotypic effect in CM death observed in our different models of miR-30d overexpression and loss of function described above (Figures 1–2), we evaluated the specific role of miR-30d in NRVM apoptosis in a cellular model of ischemia/reperfusion. 24h after NRVM transfection with miR-30d mimic, LNA anti-miR-30d or their respective negative controls, cells were incubated in a hypoxic chamber in glucose/serum free media for 8h, followed by culturing in normal aerobic and complete medium conditions for 12h. TUNEL and α-actinin co-staining showed a decreased number of TUNEL+ NRVMs in miR-30d mimic transfected cells. In contrast, LNA-anti-miR-30d treated cells demonstrated enhanced myocyte apoptosis under hypoxic and glucose/serum free conditions (Figure 3C). These phenotypical features coincided with a decreased Bax/Bcl2 and cleaved Caspase 3/ total Caspase 3 ratio in miR-30d mimic transfected cells (Figure 3D), while LNA-anti-miR-30d transfection resulted in an increased expression of these apoptotic pathways (Figure 3E). Reassuringly, while NRVM transfection with anti-map4k4 siRNA lead to a decrease in apoptotic NRVMs and transfection with LNA Anti-miR-30d resulted in a significant increase in TUNEL+ cells in response to nutrient deprivation and hypoxia, co-transfection of anti-map4k4 siRNA and LNA Anti-miR-30d rescued the apoptotic phenotype in response to stress (Figure 3F). These data demonstrate that miR-30d mediates its effects on cardiomyocyte cell death by targeting map4k4 in our in vitro model of cellular hypoxia/reperfusion.
Figure 3. Map4k4 is a direct target of miR-30d and mediator of its protective effects on cardiomyocytes.
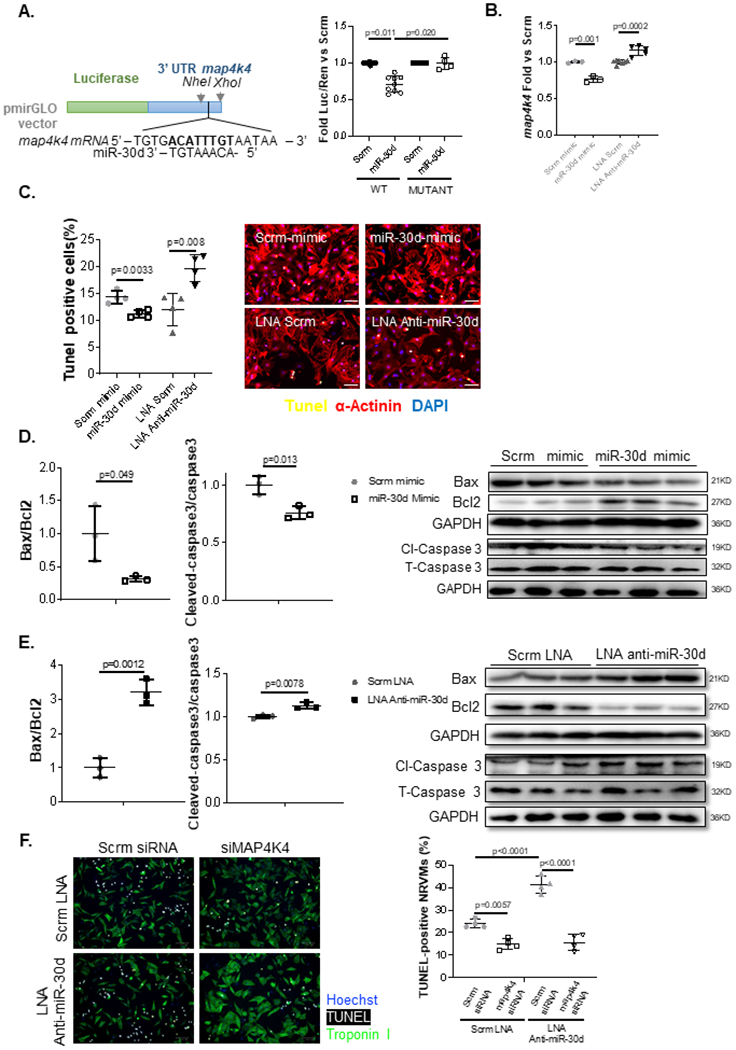
A. 500bp of map4k4’s 3’UTR fragments containing WT or mutated seed sequences were cloned into pmirGLO vectors, and transfected into HEK293 cells in the presence of miR-30d mimic or scramble. Luciferase expression quantification are shown for map4k4. WT Scrm n=9, WT miR-30d n= 9, Mutant Scrm n=4, Mutant miR-30d n=4. P values calculated with non-parametric ANOVA test, corrected using Dunn’s multiple comparison test. B. NRVMs were transfected with either miR-30d mimic or LNA anti-miR-30d and map4k4 mRNA expression was determined 48h later through qPCR; data is represented as fold change vs respective scramble condition. Scrm mimic n=3, miR-30d mimic n=3, LNA Scrm n=7, LNA Anti-miR-30d n=5. C. Role of miR-30d in apoptotic pathways in NRVMs was first determined by transfecting miR-30d mimic or LNA anti-miR30d into NRVMs cultured under anaerobic and serum/glucose free conditions, and co-staining them for TUNEL and α-actinin; results are graphed on the left and representative pictures are shown on the right. Scale bar= 50μm. Scrm mimic n=4, miR-30d mimic n=4, LNA Scrm n=4, LNA Anti-miR-30d n=4. D-E. Role of miR-30d in the Bax/Bcl2 and Caspase-3 apoptotic pathways were determined by transfecting NRVMs with miR-30d mimic (D) or LNA anti-miR-30d (E), and determining the protein levels of these molecules by western blot; quantifications are shown on the left, and representative blots on the right. Scrm mimic n=3, miR-30d mimic n=3, LNA Scrm n=3, LNA Anti-miR-30d n=3. Normality of data (D-G) determined with Shapiro-Wilks test. P values (D-G) calculated with unpaired parametric t-test. F. Quantification of apoptotic NRVMs in response to co-transfection of siRNA anti-map4k4 and LNA Anti-miR-30d under nutrient deprivation and hypoxia. NRVMs transfected with siRNA anti-map4k4 and/or LNA Anti-miR-30d, or their respective negative controls, cultured under glucose and serum deprivation were exposed to hypoxia for 8h, followed by 12h of normoxia in complete media. TUNEL+ Troponin+ NRVMs were quantified through immunofluorescence. Representative images are shown on the left and quantification on the right. Scrm LNA + Scrm siRNA n=4, Scrm LNA + map4k4 siRNA n=4, LNA Anti-miR-30d + Scrm siRNA n=4, LNA Anti-miR-30d + map4k4 siRNA n=4. P values calculated with ordinary ANOVA test, corrected using Tukey’s multiple comparison test. Data are represented as mean ± SD.
To further confirm that map4k4 is a target of miR-30d in vivo, its cardiac mRNA and protein expressions were measured in the different miR-30d overexpression and loss of function conditions in our rodent models of ischemic HF. In mice, lentiviral-mediated overexpression of miR-30d in the heart (Figure 4A–B) as well as systemic administration of the agomiR (Supp. Figure VII) markedly abrogated the increase in MAP4K4 expression at the protein level in response to MI (compared with control lentivirus or agomiR). On the contrary, miR-30d loss of function induced by LNA anti-miR-30d treatment lead to an increased mRNA and protein expression of MAP4K4 as compared to LNA scramble treated mice after IR surgery (Figure 4A–B). Finally, genetic overexpression of miR-30d in rats ameliorated the increase in MAP4K4 protein levels after MI surgery as compared with WT control littermates (Figure 4C). Together with our in vitro and biochemical data, these data confirm that map4k4 is a direct miR-30d target in vivo and may be a key mediator of miR-30d effects on cardiomyocyte cell death and subsequent cardiac remodeling after cardiac ischemia.
Figure 4. miR-30d regulates map4k4 expression levels in vivo.
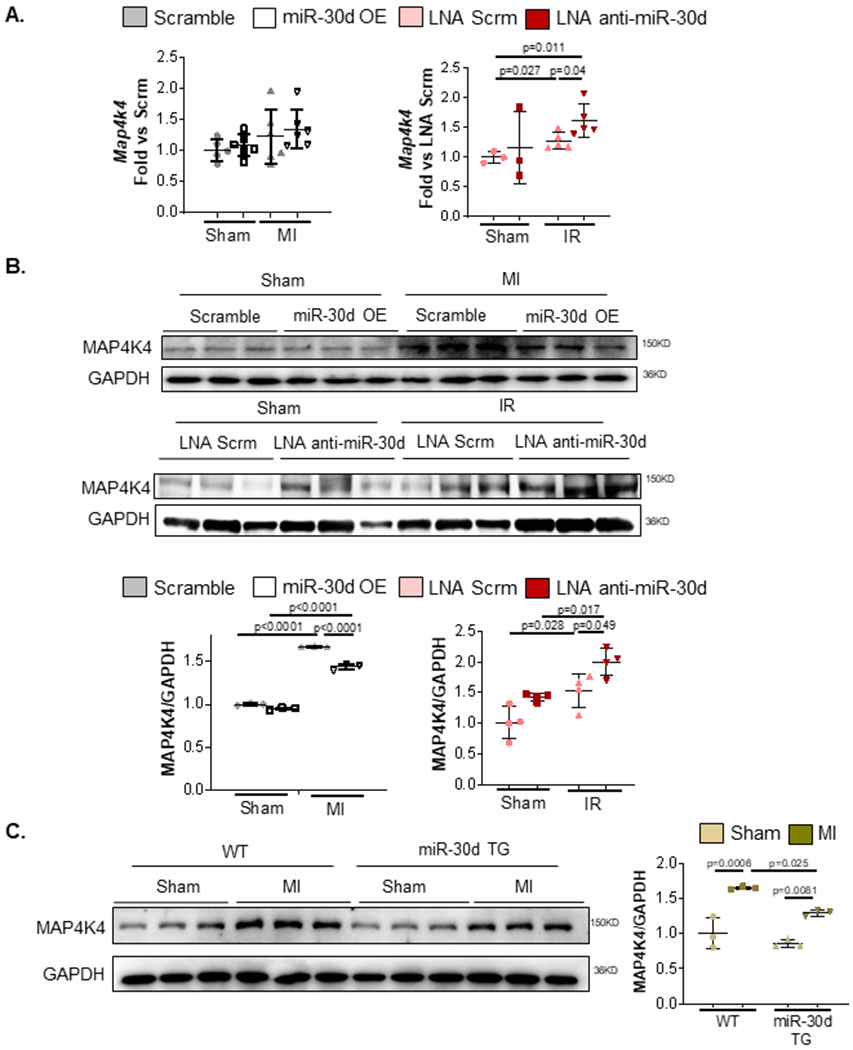
Map4k4 expression levels were determined in miR-30d overexpression and loss of function conditions in C57BL/6 mice undergoing Sham, MI or IR surgery. A. mRNA levels of map4k4 were quantified through qPCR in the heart tissue of mice with lentivirus induced miR-30d OE or LNA mediated miR-30d loss of function. Data is represented as fold versus scramble lentivirus or LNA scramble conditions, respectively. Sham Scrm Lentivirus n=5, Sham miR-30d OE Lentivirus n=6, MI Scrm Lentivirus n=6, MI miR-30d OE Lentivirus n=6 .Sham LNA Scrm n=3, Sham LNA Anti-miR-30d n=3, IR LNA Scrm n=5, IR LNA Anti-miR-30d n=5. Normality of data determined with Shapiro-Wilks test. P values calculated with unpaired parametric t-test. B. Protein levels of MAP4K4 were quantified via western blot in the cardiac tissue of either lentivirus induced miR-30d overexpression, or LNA anti-miR-30d mediated loss of function. Representative blots are shown on the top; quantification as fold versus the respective control condition is graphed on the bottom. Sham Scrm Lentivirus n=3, Sham miR-30d OE Lentivirus n=3, MI Scrm Lentivirus n=3, MI miR-30d OE Lentivirus n=3. Sham LNA Scrm n=4, Sham LNA Anti-miR-30d n=4, IR LNA Scrm n=4, IR LNA Anti-miR-30d n=4. C. Protein levels of MAP4K4 were quantified via western blot in the cardiac tissue of either WT or miR-30d overexpression transgenic rats. Representative blots are shown on the left; quantification as fold versus WT Sham is graphed on the right. WT Sham n=3, WT MI n=3, miR-30d TG Sham n=3, miR-30d TG MI n=3. Normality of data (B-C) determined with Shapiro-Wilks test. P values (B-C) calculated with ordinary ANOVA test, corrected using Tukey’s multiple comparison test. Data are represented as mean ± SD.
Itga5 is a direct miR-30d target and may mediate its effects on fibrosis following myocardial infarction.
Considering the cardiac fibrosis phenotype observed in miR-30d overexpression and loss of function rodent models (in opposite directions) in the models of ischemic heart failure (Figure 1–2), we focused on several components of the integrin pathway that were differentially expressed in the RNA sequencing analysis (Figure 3A–B, Supp. Tables I–V). Integrins constitute an essential family of extracellular matrix receptors in the heart, where they contribute to cell proliferation and myocardial fibrosis development, among other functions 11,12. Luciferase assays were performed by cloning 3’UTR of different integrin alpha or beta genes derived from the bioinformatically predicted targets from the RNA sequencing data (Figure 3A–B, Supp. Tables I–V) in to pmirGLO vectors. These were transfected into HEK 293 cells together with scramble or miR-30d mimic to determine functional effects miR-30d in the presence of its putative seed sequence. From the different integrin chains tested (Supp. Figure VIII), the Luciferase expression of the vector containing the WT form of integrin α5’s (itga5) 3’UTR was the most significantly decreased by miR-30d (Figure 5A); to a lower extent, Luciferase expression was also found decreased when vectors cloned with itgaV’s 3’UTR were co-transfected with miR-30d (Supp. Figure VIII C). Mutation of the miR-30d seed sequence in itga5’s 3’UTR abrogated the miR-30d mediated decrease in Luciferase activity (Figure 5A). To understand the role of miR-30d in the regulation of itga5 expression in cardiac fibroblasts stimulated by TGFβ1, adult mouse cardiac fibroblasts were transfected with miR-30d mimic in the presence or absence of TGFβ1. As expected, itga5 expression was increased as a result of TGFβ1 treatment; however, transfection with miR-30d significantly reduced upregulation of itga5 under pro-fibrotic conditions (Figure 5B).
Figure 5. Itga5 is a direct miR-30d target and may mediate its effects on fibrosis following myocardial infarction.
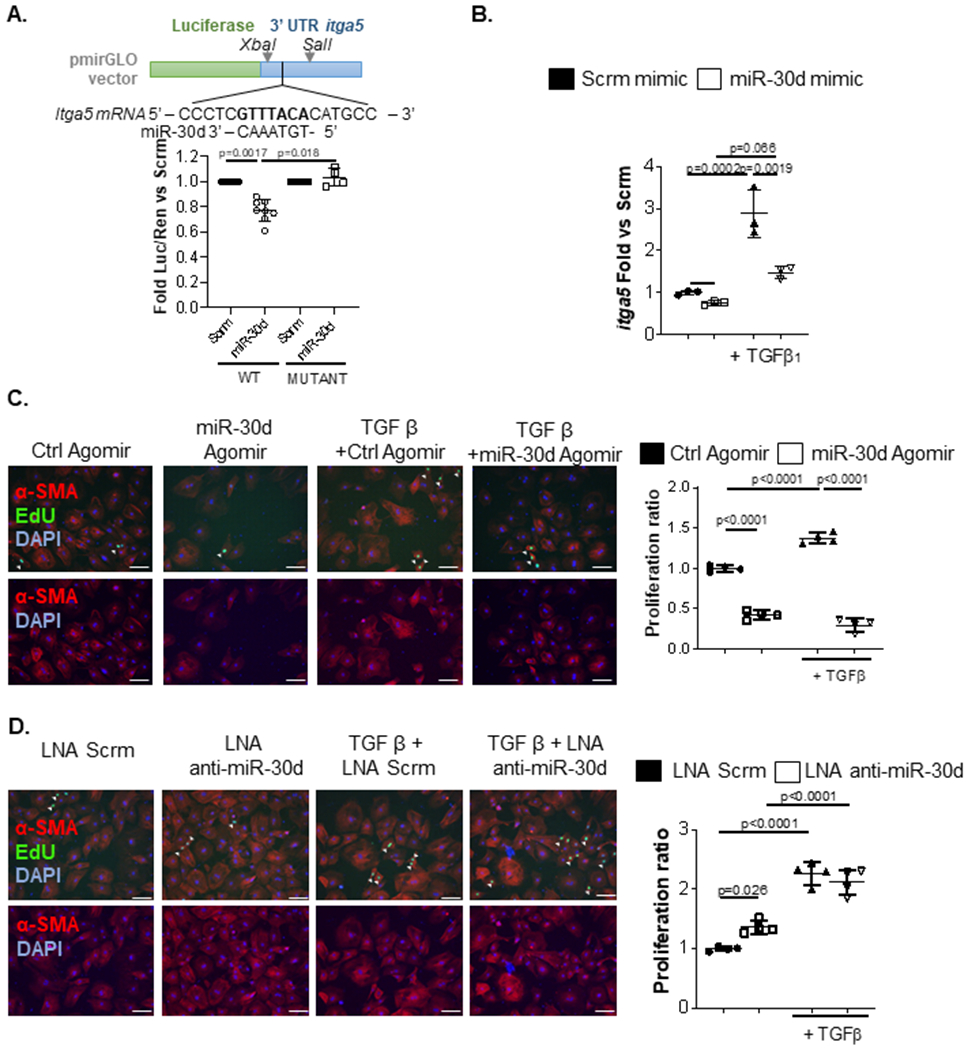
A. 500bp of itga5’s 3’UTR fragments containing WT or mutated seed sequences were cloned into pmirGLO vectors and transfected into HEK293 cells in the presence of miR-30d mimic or scramble. Luciferase expression quantification are shown for itga5. WT Scrm n=9, WT miR-30d n= 9, Mutant Scrm n=4, Mutant miR-30d n=4. P values calculated with non-parametric ANOVA test, corrected using Dunn’s multiple comparison test. B. Adult mouse cardiac fibroblasts were transfected with 50pmol miR-30d mimic and treated with 10ng/mL TGFβ, and mRNA expression of itga5 was determined through qRT-PCR. Fold change vs scramble is represented. Scrm mimic n=3, miR-30d mimic n=3, Scrm mimic + TGFβ = 3, miR-30d mimic + TGFβ n=3. C-D. Role of miR-30d in Neonatal Rat Ventricular Fibroblast (NRVF) proliferation was determined by transfecting either (C) miR-30d agomiR (Ctrl Agomir n=4, miR-30d Agomir n=4, Ctrl Agomir + TGFβ n=4, miR-30d Agomir + TGFβ n=4) or (D) miR-30d inhibitor (LNA Scrm n=4, LNA Anti-miR-30d n=4, LNA Scrm + TGFβ n=4, LNA Anti-miR-30d + TGFβ n=4) and treated with TGFβ. Representative pictures of EdU uptake by NRVF in the different conditions are shown on the left, and proliferation quantification is graphed on the right. Scale bar= 100μm. Normality of data (B-D) determined with Shapiro-Wilks test. P values (B-D) calculated with ordinary ANOVA test, corrected using Tukey’s multiple comparison test. Data are represented as mean ± SD.
Next, the function of miR-30d in regulating cardiac fibroblast proliferation was investigated. Neonatal rat ventricular fibroblasts were transfected with miR-30d agomiR (Figure 5C) or miR-30d inhibitor (Figure 5D), and their proliferation was determined by quantifying proliferation through EdU uptake as a result of TGFβ stimulation. miR-30d agomiR blocked fibroblast proliferation with and without TGFβ treatment as compared to control agomiR treated fibroblasts, preventing TGFβ mediated expansion in cell proliferation (Figure 5C). Transfection with miR-30d inhibitor increased fibroblast proliferation only at baseline without TGFβ, but not further after TGFβ stimulation (Figure 5D). In totality, these data indicate that miR-30d plays a role in cardiac fibroblast proliferation by directly targeting itga5.
miR-30d regulates key pro-fibrotic pathways in the myocardium in response to ischemic heart failure.
In order to characterize the in vivo role of miR-30d in itga5 expression and myocardial fibrosis, mRNA levels of itga5 were first determined in the murine overexpression and loss of function conditions after subjecting them to ischemic heart failure or sham control surgeries. While the mRNA expression of itga5 did not decrease significantly in the miR-30d-agomiR treated mice (Figure 6A) or in the hearts subjected to miR-30d overexpressing lentivirus following MI surgery (Supp. Figure IX A), protein levels of integrin α5 were significantly lower in agomiR treated mice compared to scramble control treated mice after MI surgery (Figure 6B). These findings were also recapitulated in the transgenic rats overexpressing miR-30d (Figure 6C), or in the mice with the viral-mediated miR-30d overexpression (Supp. Figure IX B). On the contrary, LNA mediated miR-30d downregulation resulted in increased itga5 mRNA expression (Figure 6A) as well as increased protein expression of integrin α5 after IR (Figure 6D). Given that integrins can enhance signaling pathways from certain growth factors, such as TGFβ1 13, and consistent with the negative regulatory effects of miR-30d on fibroblast proliferation in vitro, (Figure 5C–D), we found that the TGFβ1 mediated canonical signaling pathways were also regulated by miR-30d in our in vivo models. Thus, overexpression of miR-30d in mice mediated by either agomiR (Figure 6B), lentivirus (Supp. Figure IX B), or in transgenic rats (Figure 6C) showed a decrease in TGFβ1 expression in the myocardium after MI, and therefore a reduced phosphorylation of Smad2/3. On the contrary, LNA mediated miR-30d loss of function resulted in increased TGFβ1 protein levels and subsequently elevated phosphorylation of downstream mediators Smad2/3 (Figure 6D). Altogether, these data indicate that miR-30d plays a key role in modulating the fibrotic response in ischemic heart failure.
Figure 6. miR-30d regulates key pro-fibrotic pathways in the myocardium in response to ischemic heart failure.
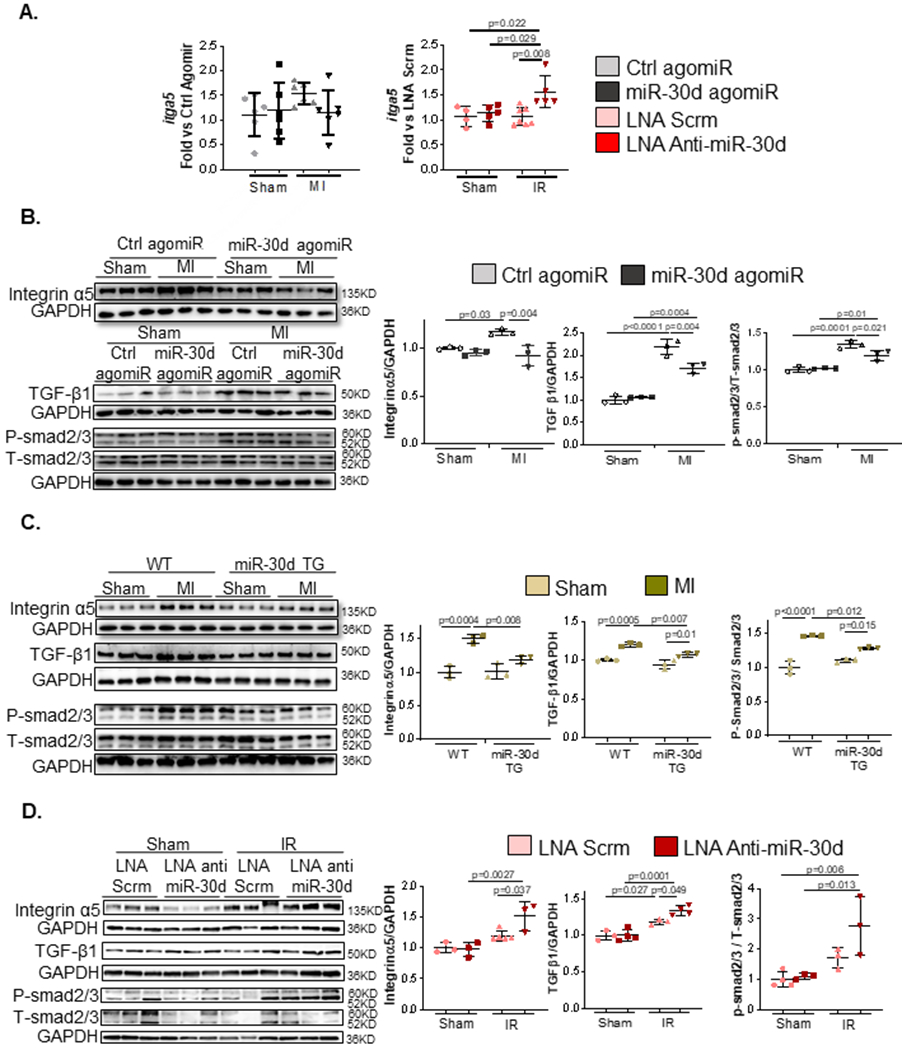
A. mRNA levels of itga5 were quantified through qPCR in the heart tissue of mice with miR-30d agomiR (Sham Ctrl agomiR n=6, Sham miR-30d agomiR n=6, MI Ctrl agomiR n=6, MI miR-30d agomiR n=6) or treated with LNA anti-miR-30d (Sham LNA Scrm n=4, Sham LNA Anti-miR-30d n= 5, IR LNA Scrm n=7, IR LNA Anti-miR-30d n=5). Data is represented as fold versus scramble lentivirus or LNA scramble conditions, respectively.B-C. Protein levels of integrin α5, as well as key proteins in the TGFβ pro-fibrotic pathways were determined by western blot in (B) agomiR-mediated miR-30d overexpression (Sham Ctrl agomiR n=3, Sham miR-30d agomiR n=3, MI Ctrl agomiR n=3, MI miR-30d agomiR n=3) and (C) transgenic rats overexpressing miR-30d (Sham WT n= 3, MI WT n=3, Sham miR-30d TG n=3, MI miR-30d TG n=3); representative blots are shown on the left and quantification of each marker is represented on the right. D. Protein levels of integrin α5, as well as key proteins in the TGFβ pro-fibrotic pathways were determined by western blot in LNA anti-miR-30d treated mice; representative blots are shown on the left and quantification of each marker is represented on the right. Sham LNA Scrm n=3, Sham LNA Anti-miR-30d n= 3–4, IR LNA Scrm n=3–5, IR LNA Anti-miR-30d n=3–4. Normality of data (A-D) determined with Shapiro-Wilks test. P values (A-D) calculated with ordinary ANOVA test, corrected using Tukey’s multiple comparison test. Data are represented as mean ± SD.
miR-30d is expressed in cardiomyocytes in response to acute ischemic stress and released in EVs to mediate intercellular signaling to fibroblasts.
Our previous work had shown that miR-30d is expressed at highest levels in cultured cardiomyocytes compared with other cardiac cell types and can be packaged into extracellular vesicles (EVs) by cardiomyocytes subjected to pathological stress 8,14. We therefore hypothesized that miR-30d contained in EVs may be a novel form of paracrine signaling between cardiomyocytes and fibroblasts in the setting of acute ischemic stress. In NRVMs exposed to hypoxia we noted an increase in the relative copy numbers of both pre-miR-30d and mature miR-30d-5p molecules in the cellular compartment as quantified through ddPCR (droplet digital PCR) (Figure 7A). We isolated EVs from NRVM conditioned media, isolated using ExoRNAse (Qiagen) or Size Exclusion Chromatography and characterized them as suggested by the MISEV (Minimal Information for Studies of Extracellular Vesicles) guidelines 15 (Supp. Figure X A). miR-30d-5p was clearly detected in EVs from NRVMs, even after RNAse treatment and was increased following exposure of the cells to hypoxia (Figure 7A, Supp. Figure X A). To delineate the distribution of miR-30d among different RNA sub-carriers in NRVM media, EV fractions, Argonaute (Ago)-bound RNAs and lipoproteins were isolated and miR-30d expression levels were quantified in each of them through ddPCR. EV fractions 7-10 were highly enriched in miR-30d expression (Supp. Figure X B), while fractions 3-6 and 11-13 contained considerably lower levels of miR-30d. miR-30d levels in Ago and lipoprotein fractions (VLDL and HDL) were insignificant.
Figure 7. miR-30d is expressed in cardiomyocytes in response to acute ischemic stress and released in EVs to mediate intercellular signaling to fibroblasts.
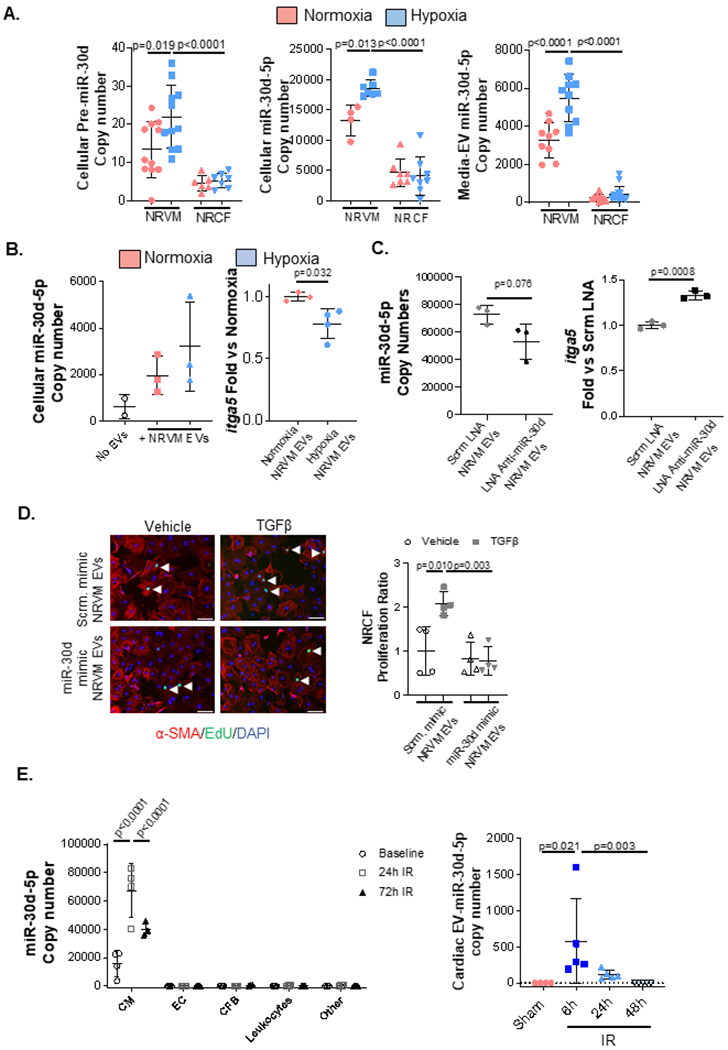
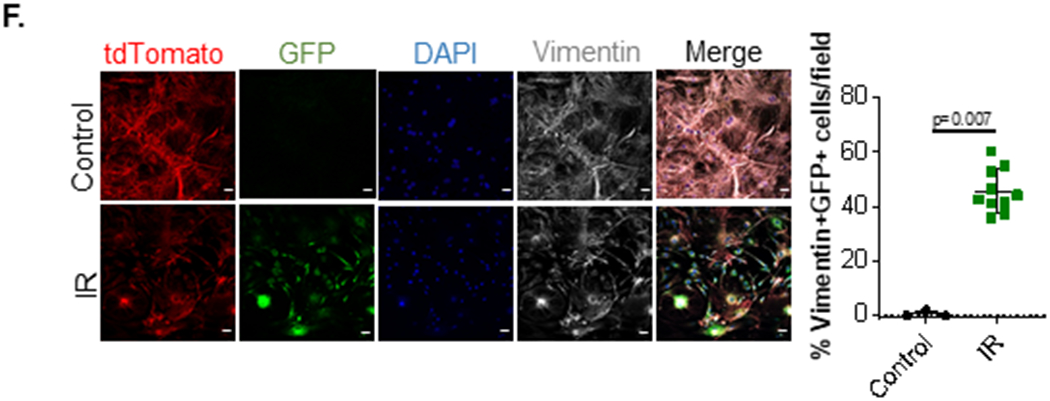
A. 24h post-isolation, NRVMs and NRCFs were exposed to hypoxia (0.2% O2) or normoxia in serum-free media for 24h and cellular pre-miR-30d(NRVM normoxia n=11, NRVM hypoxia n=11, NRCF normoxia n=6, NRCF hypoxia n=7), miR-30d-5p(NRVM normoxia n=4, NRVM hypoxia n=7, NRCF normoxia n=7, NRCF hypoxia n=8), and media EV-miR-30d-5p (NRVM normoxia n=9, NRVM hypoxia n=10, NRCF normoxia n=12, NRCF hypoxia n=14) expression were quantified through droplet digital PCR (ddPCR), normalized to RNA input. Expression of either pre-miR-30d or mature miR-30d-5p in the cellular compartment and mature miR-30d-5p in the EV component is represented as copy numbers per ng RNA input. Normality of data determined with Shapiro-Wilks test. P values calculated with ordinary ANOVA test, corrected using Tukey’s multiple comparison test. B. On the left, miR-30d-5p expression in NRCFs treated with EVs extracted from media of NRVMs exposed to normoxia or hypoxia(n=3), was quantified through ddPCR, and normalized to RNA input. On the right, itga5 expression was determined through qRT-PCR in cardiac fibroblasts treated with EVs extracted from media of NRVMs exposed to normoxia or hypoxia. n=3. Normality of data determined with Shapiro-Wilks test. P value calculated with unpaired parametric t-test. C. NRVMs were transfected with 150pmol scramble or Anti-miR-30d-5p LNA for 48h, after which their conditioned media EVs were collected and used for NRCF treatment for 24h. ddPCR quantification of miR-30d-5p in NRCF treated with EVs derived from LNA-treated NRVMs is graphed on the left, and qRT-PCR for itga5 in this NRCFs is quantified on the right as fold change vs scramble LNA transfected NRVM-EV treated NRCF. n=3. Normality of data determined with Shapiro-Wilks test. P value calculated with unpaired parametric t-test. D. NRCFs were treated with EV fractions 7-10 derived from NRVMs transfected with 150pmol miR-30d mimic or scramble mimic for 48h. After additional 24h treatment of NRCF with 10ng/mL TGFβ, cell proliferation was quantified through EdU staining of NRCF treated with NRVM-derived EVs and TGFβ (or vehicle): representative immunofluorescence pictures are shown on the left (white arrows pointing to proliferating fibroblasts) and proliferation quantification on the right. n=4. Scale bar= 100μm. Normality of data determined with Shapiro-Wilks test. P value calculated with unpaired parametric t-test. E left. At baseline and 24h and 72h post-IR, cardiac resident cells (CM, EC, CFB and leukocytes) were isolated and their miR-30d-5p expression was quantified through ddPCR based on the same input RNA amount per time point and cell type. Data is represented as copy numbers of miR-30d-5p. CM baseline n=4, CM 24h n=4, CM 72h n=3; EC baseline n=4, EC 24h n=4, EC 72h n=4; CFB baseline n=4, CFB 24h n=4, CFB 72h n=3; Leukocytes baseline n=4, Leukocytes 24h n=4, Leukocytes 72h n=4; “other” baseline n=4, “other” 24h n=4, “other” 72h n=4 . Normality of data determined with Shapiro-Wilks test. P values calculated with ordinary ANOVA test, corrected using Tukey’s multiple comparison test. E right. Quantification of miR-30d-5p through ddPCR in myocardial EVs 6h, 24h and 48h after IR or Sham surgery, normalized to U6 and to RNA input. Sham n= 4, IR (6h, 24h, 48h) n= 5. P values calculated with non-parametric ANOVA test, corrected using Dunn’s multiple comparison test. F. Cardiac fibroblasts from αMHC-MerCreMer-RosamTmG mice undergoing IR surgery or control mice were isolated, cultured and stained for Vimentin. Cardiac fibroblasts that were the target of cardiomyocyte-derived EVs had cre-mediated recombination and mGFP expression. Representative pictures of GFP+ Vimentin+ cardiac fibroblasts in vitro are shown on the left (scale bar 25μm), and quantification on the right. Control n=3, IR=10. P value calculated with unpaired non-parametric Mann Whitney t-test. Data are represented as mean ± SD.
To directly quantitate miR-30d within EVs using an alternative method to ddPCR, we utilized molecular beacons (MBs), which have been recently used to directly image intercellular and as well as EV-contained miRNAs 16–18. When bound to their miRNA target, a fluorescent probe is unquenched, leading to ‘unmasking’ of the fluorescent signal. Our custom designed MB coupled to a cell-penetrating peptide (see Methods section) was validated in cardiomyocyte-derived EVs (see details in Methods and Supp. Methods Figure I). EVs in conditioned medium from NRVMs subjected to hypoxia or normoxia were imaged using nano flow cytometry 19 after incubation with the miR-30d-MB coupled to a cell-penetrating peptide. In concordance with our ddPCR results based on two different methods of EV isolation (Figure 7A, Supp. Figure X A and X B), there was an increase in EV-miR-30d levels in conditioned media from NRVMs in response to hypoxia (Supp. Figure X C).
In contrast to NRVMs, NRCFs showed a significantly lower expression of cellular pre-miR-30d, mature miR-30d-5p and EV packaged miR-30d-5p (Figure 7A) at baseline, with no appreciable change with hypoxic stress, suggesting that cardiomyocytes may be the local source for miR-30d action in cardiac fibroblasts. To prove this hypothesis, cardiac fibroblast were treated in vitro with EVs derived from NRVMs exposed to normoxia or hypoxia, (which contain far greater levels of miR-30d levels than fibroblasts), resulting in increased miR-30d levels in fibroblasts (Figure 7B), and subsequent reduction in itga5 expression. On the contrary, NRFC treatment with EVs derived from NRVMs transfected with either scramble or anti-miR-30d-5p LNA (Supp. Figure X D), resulted in decreased transfer of the miRNA in NRCFs, and therefore an increased expression of its target itga5 (Figure 7C). To definitively demonstrate the biological activity of EV-contained miR-30d derived from NRVMs as a paracrine mediator of anti-fibrotic effects, culture media was collected from NRVMs 48 hours after transfection with miR-30d mimic or scramble mimic. EV fractions 7-10 (demonstrated to be most enriched in miR-30d, see Supp. Figure X B) were isolated by SEC. Cardiac fibroblasts were incubated with EVs derived from the miR-30d mimic or scramble mimic treated NRVMs in the presence or absence of the pro-fibrotic factors AngII or TGFβ. While TGFβ and AngII induced a significant increase in the proliferation of cardiac fibroblasts cultured with EVs from the NRVMs transfected with the scramble mimic, EVs from the miR-30d overexpressing NRVMS markedly abrogated the increase in proliferation induced by either TGFβ or AngII (Figure 7D, Supp. Figure X E). Complementary to this data, treatment with miR-30d containing EVs reduced cardiac fibroblast expression of itga5 (direct target of miR-30d, Figure 5A), fibronectin, periostin, CTGF and αSMA (Supp. Figure X F). These experiments suggest that regulation of key fibrotic pathways in cardiac fibroblasts is mediated by transfer of miR-30d derived from cardiomyocyte-EVs.
To investigate the in vivo source of miR-30d in the heart acutely after ischemia, cardiac resident cells (CM, endothelial cells, cardiac fibroblasts and leukocytes) were isolated from mice following IR surgery (Supp. Figure XI) and their miR-30d expression levels were quantified at different time points after IR. As suggested by the in vitro observations, CMs were the predominant source of mature miR-30d both at baseline and after ischemia as compared to the rest of cardiac resident cells. In parallel with our cell culture data, miR-30d levels peaked acutely after ischemic stress at 24 hours, with a subsequent decrease by 72 hours (Figure 7E, left). Notably expression was negligible in fibroblasts early, although an increase in mature miR-30d, but not pre-miR-30d was noted at 72 hours, most consistent with intercellular transfer rather than de novo synthesis in fibroblasts (Figure 7E left, Supp. Figure XI C).
In order to complement our in vitro EV findings and our cellular miR-30d data above, miR-30d levels were determined in cardiac tissue EVs of mice 6h, 24h and 48h after IR surgery (Figure 7E, right). In line with the MISEV (Minimal Information for Studies of Extracellular Vesicles) guidelines for extracellular vesicle research15, EV size profiling by NanoSight (Supp. Figure XII A) demonstrated that the majority of cardiac EVs were in the 100-300nm size range, although some larger species were also seen, which could correspond to apoptotic bodies. EV markers (Alix, CD9, CD63) were detected through DotBlot confirming presence of the exosomal fraction in the cardiac secretome component isolated (Supp. Figure XII B), although this does not exclude the presence of certain contaminants of other membranes as shown by the weak positivity of Calnexin. This time course after ischemic stress showed a significant increase in miR-30d containing cardiac EVs acutely 6h after IR surgery, which was maintained for 24 post-ischemia, and was finally decreased to baseline levels after 48h post-IR (Figure 7E, right), suggesting that miR-30d is released into the cardiac parenchyma in EVs following acute ischemic stress 20, mirroring the dynamics followed by the cellular expression levels of miR-30d (Figure 7E, left).
As suggested above, CM derived EVs can be up taken by cardiac fibroblasts21. To visualize this process in vivo, we leveraged a model that was previously characterized to study EV-mediated intercellular communication 22,23 (Supp. Figure XIII). In this model, EVs released by Cre-expressing tissues contain functional Cre mRNA or protein, which when transferred to EV-recipient cells in Cre-reporter mice, mediated Cre-dependent recombination. In the background of a Cre-dependent reporter (such as the Rosa26-lacZ mouse 23) EV-targeted cells were identified by expression of the reporter. Having recently validated the use of this mouse model to study intercellular communication 24, we demonstrated the validity of this model in αMHC-MerCreMer - RosamTmG mice (Supp. Figure XIII A). In this model, CMs express Cre (following tamoxifen injection) and therefore undergo Cre-dependent recombination to express membrane-targeted GFP (mGFP, Supp. Figure XIII B). Plasma from these mice contains CM derived GFP+ EVs (Supp. Figure XIII C), which in addition to EV markers Alix and CD9 also express cardiac Troponin T due to their CM origin, as well as Cre recombinase protein and GFP (Supp. Figure XIII D). In this murine model for tracking EV-mediated signaling, Cre recombinase packaged in EVs is transferred to cellular targets of EVs 23, and the subsequent Cre-mediated recombination marks target cells with mGFP expression. In this case, a significant number of Vimentin+ cardiac fibroblast cultured ex vivo from αMHC-MerCreMer - RosamTmG mice undergoing IR surgery (but not from sham controls) were GFP+ following 4 weeks of IR surgery (Figure 7F), suggesting that transfer of CM-derived EVs to fibroblasts occurred in vivo following the stress of IR. These results were further validated in heart cross-sections of αMHC-MerCreMer - RosamTmG mice, where an increase in GFP+Vimentin+ cardiac fibroblasts was demonstrated after IR (Supp. Figure XIIIE). Together, our in vitro and in vivo data demonstrate that EV-mediated communication between cardiomyocytes and fibroblasts in the setting of acute ischemic stress results in miR-30d transfer and signaling.
miR-30d in cardiomyocytes and cardiomyocyte-derived-EVs are decreased in chronic ischemic HF, correlating with adverse cardiac remodeling in HF patients.
Our data suggest that following the acute increase in miR-30d after ischemic stress, miR-30d levels decrease in the heart and plasma in wild-type mice. In order to quantify miR-30d expression in plasma EVs in chronic ischemic HF and determine its potential use as a circulating biomarker of cardiac remodeling, miR-30d levels were quantified in plasma EVs of mice and humans with HF.
We again leveraged the αMHC-MerCreMer - RosamTmG mice, where mGFP is expressed on cardiomyocyte membranes (after treatment with tamoxifen), and hence on any EVs derived from CMs. CM-derived GFP+ EVs of larger and smaller sizes (Supp. Figure XIII C–D) that contain miR-30d were detected in plasma in these mice at baseline (Figure 8A, Supp. Figure XIV A). 4 weeks after IR, levels of miR-30d EVs from CMs were decreased (Figure 8A, Supp. Figure XIV A) suggesting local parenchymal uptake of these EVs, or a lack of sustained chronic increase in miR-30d EVs in this murine ischemic HF model of adverse ventricular remodeling. In contrast, in the miR-30d overexpressing rats (Supp. Figure I C), where the increased cardiac expression of miR-30d is correlated with increased miR-30d in plasma EVs, post-ischemic LV remodeling is ameliorated.
Figure 8. miR-30d in cardiomyocytes and cardiomyocyte-derived-EVs are decreased in chronic ischemic HF, correlating with adverse cardiac remodeling in HF patients (A-C).
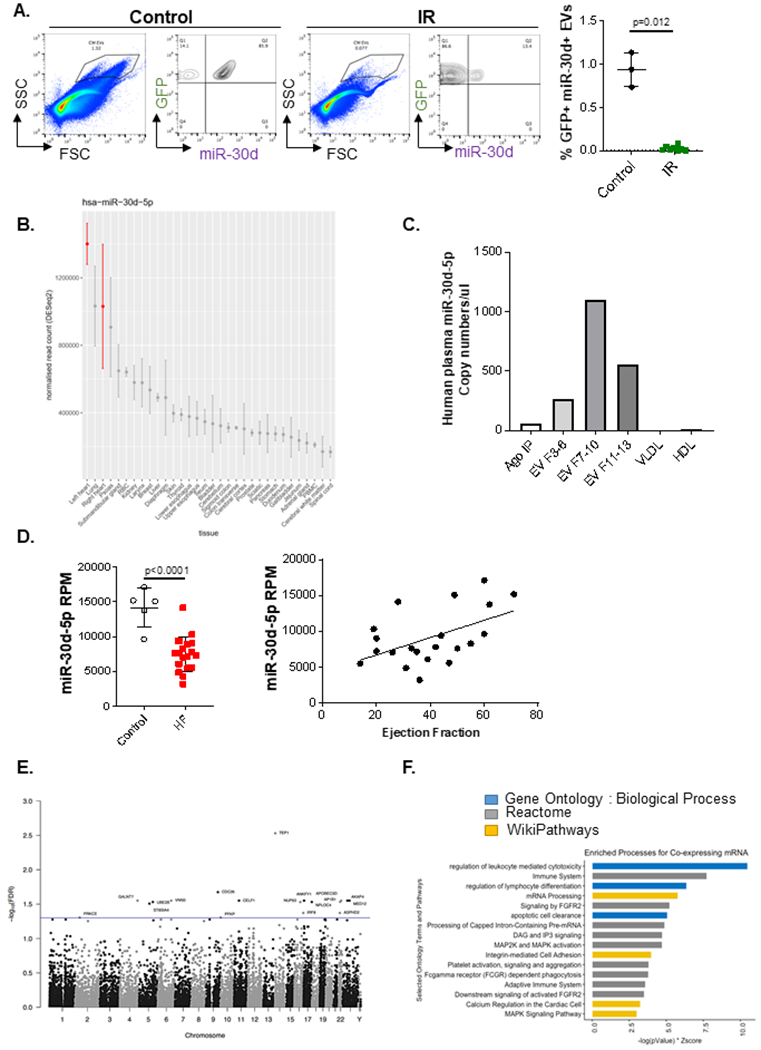
A. Presence of miR-30d in CM derived ‘larger-sized’ circulating EVs in plasma (diluted 1/100) of αMHC-MerCreMer - RosamTmG mice subjected to IR is determined through nano flow cytometry, using a specific molecular beacon conjugated to Alexa 647. Representative FACS plots are shown on the left indicating miR-30d expression in GFP+ EVs. Quantification of GFP+ miR-30d+ EVs is represented on the right, Control n=3, IR n= 8. P value calculated with unpaired non-parametric Mann Whitney t-test. Data are represented as mean ± SD. B. Tissue origin of miR-30d based on the Small RNA Atlas analysis. RNA from a variety of tissues were isolated and their small RNA content was sequenced. Normalized read counts are represented from all the tissues investigated. C. miR-30d expression levels associated to different carriers (EV fractions 3-6/ 7-10/ 11-13, Ago-bound RNA and lipoproteins VLDL and HDL) in human pooled plasma quantified by ddPCR. D. On the left, RNA from plasma EVs of patients with HF or controls was sequenced through Illumina HiSeq platform. Reads per million (RPM) are represented for each patient population. N= 5 control, 18 HF. Normality of data determined with Shapiro-Wilks test. P value calculated with unpaired parametric t-test. Data are represented as mean ± SD. On the right, Correlation between miR-30d-5p RPM in plasma EVs of HF patients and controls with their respective ejection fraction (EF) was determined, resulting in r= 0.5085, r2= 0.2586 and two-tailed P value of 0.0186. n=21. Data are represented as mean ± SD. (E-F) In the populational level, co-expression analysis of whole blood mRNA and plasma circulating miRNA in humans suggests a role for miR-30d in integrin and MAPK pathways, among other immune pathways. E. Manhattan plot of all measured genes by chromosome position. Y-axis is −log10(FDR) with a threshold illustrated at FDR < 0.05 (blue line) highlighting 18 genes (labeled). F. Using a relaxed threshold of FDR < 0.10, functional enrichment analysis was performed on 72 genes against Gene Ontology: Biological Process (GO:BP, blue), Reactome (gray) and WikiPathways (gold) using Enrichr. Bar length is a combined score of −log(P-Value) and Z-Score.
To determine if our new findings demonstrating the critical importance of miR-30d contained in cardiomyocyte-derived EVs in the regulation of post-ischemic ventricular remodeling was relevant to human HF, we assessed the distribution of miR-30d across tissues, and plasma subcarrier compartments in human subjects. miR-30d read counts were quantified across human tissues (Small RNA Atlas) (Figure 8B) and in Argonaute (Ago2), EV fractions collected by SEC and lipoproteins (VLDL and HDL) in human plasma (Figure 8C Supp. Figure XIV B, C). As indicated in the Small RNA Atlas tissue origin distribution, the highest expression of miR-30d is found in the left ventricle (Figure 8B). When assessing the plasma distribution of miR-30d levels across different carriers, the majority of miR-30d was found in different EV fractions (mainly in fractions 7-10), with a smaller amount detected in the Ago2 pulldowns, while none was found in lipoproteins (Figure 8C, Supp. Figure XIV B). This observation is consistent with the distribution of miR-30d in NRVM media (Supp. Figure X B). The presence of miR-30d in EVs in plasma was further validated from previously available data we had generated as part of a study assessing biases of RNA isolation platforms25. Using specific pull-downs for EV markers (CD63, CD81 and CD9) followed by RNASeq: as shown in Supp. Figure XIV C, miR-30d was found present in all CD63, CD81 and CD9 pull-down compartments.
Having confirmed that the majority of circulating miR-30d is present in EVs and is likely derived from the heart (which has the greatest expression), we assessed any association of EV-miR-30d with ventricular remodeling in chronic HF patients. RNA sequencing of plasma EVs from patients with chronic HF (Supp. Table VI) showed that the average miR-30d reads were decreased in the chronic HF patients (average LVEF 33%) as compared to controls (Figure 8D, left). However, within this group, the miR-30d-5p level in plasma EVs (quantified with reads per million reads) was strongly correlated with ventricular function indicating that low expression of miR-30d in EVs is associated with adverse ventricular remodeling in chronic HF, while higher levels are associated with beneficial remodeling (Figure 8D, right). Together, our observations indicate that CM derived EVs containing miR-30d can be taken up by cardiac fibroblasts, therefore playing a role in the local signaling networks that mediate fibroblast proliferation and myocardial fibrosis during cardiac remodeling. Our mechanistic cell culture and animal studies are complemented by the data in human HF patients that demonstrate an association between plasma EV-miR-30d levels and cardiac remodeling.
Co-expression analysis of whole blood mRNA and plasma circulating miRNA in humans suggests a role for miR-30d in integrin and MAPK pathways, among other immune pathways.
Plasma miR-30d levels have been studied as an accessible biomarker of LV remodeling in multiple contexts8,9 , with higher levels associated with clinical protection. The mechanistic relations between miR-30d and gene expression in human subjects (that may support its pathogenic role), however, have not been widely studied. In a co-expression analysis of whole blood gene expression (mRNAs) and plasma circulating miR-30d-5p in the Framingham Heart Study (FHS), we found 72 genes whose expression in whole blood was associated with plasma miR-30d-5p level (at a false-discovery rate of 10%; Supp. Table VII). In pathway analyses of these 72 co-expressed genes (Figure 8E–F), we found that immune, apoptosis, FGFR2, MAPK, and integrin-mediated cell adhesion signaling were among the top pathways from Reactome and WikiPathways, consistent with our findings in rodent models.
In conclusion, this work shows dynamic regulation of miR-30d in ischemic HF. MiR-30d in CMs, and CM-derived EVs increases acutely with ischemic stress, providing protection against CM apoptosis and myofibroblast activation through EV-mediated paracrine signaling. In chronic ischemic HF, miR-30d expression in CMs and EVs decreases, contemporaneous with progression of adverse cardiac remodeling in mice, rats and humans (see Graphical Abstract). In chronic human HF patients, EV-miR-30d levels show a range of values with lower levels associated with adverse remodeling, positioning miR-30d as an optimal biomarker to predict pathological ventricular remodeling in HF.
DISCUSSION
Considerable advances have been made towards understanding the complex biological pathways regulating adverse cardiac remodeling in heart failure (HF). Recently, substantial recognition has been given to non-coding RNAs, in particular to plasma miRNAs, as possible functional biomarkers that may serve as key post-transcriptional regulators of gene expression networks in various clinical scenarios, including HF 5,26. miRNAs play an important role as markers of disease progression 27,28 and have recently been implicated as couriers of intercellular communication, including paracrine cross-talk between cardiac resident cells 29–32. Within the context of both ischemic and non-ischemic HF, miRNAs have been comprehensively investigated as regulators of the classical features of adverse cardiac remodeling 33, with dysregulated miRNA expression patterns demonstrated in hypertrophied and fibrotic myocardial tissues, pointing to an association between specific miRNA levels and development of pathological cardiac remodeling 5,34,35. Given recent studies that point to an important role for extracellular RNAs (exRNAs) in mediating disease pathogenesis, we hypothesized that exRNAs that serve as biomarkers of cardiac remodeling may in fact serve in a functional role to mediate key processes in remodeling. Characterization of these functional biomarkers may not only elucidate novel signaling mechanisms, but may also lead to their identification as novel therapeutic targets that may alter the trajectory of pathological heart remodeling 5,36.
Previously, we described that in HF patients, lower circulating miR-30d was associated with adverse clinical responses, while higher levels were beneficial89. While preliminary work from these studies raised the possibility that miR-30d may be cardioprotective in cell culture models, no prior work has conclusively demonstrated a functional role for miR-30d in the cardiac remodeling process in vivo. The current work sought to delineate the ‘functional’ role of miR-30d in regulating ventricular remodeling, fibrosis and cellular apoptosis and to identify the unique molecular targets of miR-30d that mediate the protective effects of elevated miR-30d levels noted in our previous clinical studies. Furthermore, using innovative technologies, as well as an exosome-tracking mouse model, we show that cardiomyocytes are the major cell type that express miR-30d and secrete them packaged in EVs. To achieve these goals, we utilized genetic, pharmacologic and lentiviral transfer mediated approaches to achieve overexpression (OE) or loss of function (LOF) experimental conditions, both in mice and in rats, having generated to our knowledge the first transgenic rats overexpressing or deficient in a specific miRNA. A major strength of our study is the presence of these multiple complementary models that validate our phenotypic and mechanistic findings, limiting off-target effects seen in studies that use a single approach. Our studies confirmed that elevated miR-30d levels confer protection against pathologic ventricular remodeling in the context of ischemic HF by preventing myocardial dysfunction, cardiac fibrosis and cardiomyocyte apoptosis. On the contrary, these phenotypical features were found pathologically aggravated in the miR-30d loss of function rodent models. Together, these data confirm that miR-30d is associated with beneficial cardiac remodeling and plays a functional role in disease pathogenesis.
An unbiased approach to discovering miR-30d targets in the heart by RNA sequencing of miR-30d overexpression and loss of function in cardiomyocytes led to the identification of putative targets by bioinformatic predictions that were validated using specific Luciferase assays utilizing the 3’-UTR of these targets. Map4k4 and itga5 were corroborated as molecules directly regulated by miR-30d both in our cell culture as well as in all the rodent models of miR-30d OE or LOF. Importantly, our results conclusively provided in vivo validation for previous studies that had demonstrated an association between miR-30d levels and map4k4 expression in cardiomyocyte cultures8 , as well as in the context of insulin resistance 37. Additionally, the pro-apoptotic role of map4k4 has been described by others in relations to a variety of pathological conditions 10,38. In the present work, map4k4 is further characterized as a direct target of miR-30d specifically in CM, and silencing MAP4K4 can mitigate some of the deleterious effects of miR-30d ablation during ischemic stress, which could explain the reduced CM apoptosis phenotype observed in miR-30d OE conditions both in vitro under stress conditions as well as in vivo in the context of ischemic HF. Interestingly, a recent study independently confirmed MAP4K4 as necessary for cardiomyocyte apoptosis in the setting of oxidative stress and myocardial infarction 39, and a small molecule therapy targeting MAP4K4 was found to decrease infarct size in the murine IR model. This aligns with our finding that miR-30d is acutely increased in cardiomyocytes subjected to hypoxia as a protective response. Therefore, the smaller infarct sizes with miR-30d OE (and larger infarct sizes with LOF), may be one key factor that may explain the beneficial effects of miR-30d on cardiac remodeling.
While the beneficial effects on fibrosis seen in the OE models may in part be secondary to less CM cell death, our studies suggest that there is a novel form of epigenetic paracrine signaling between CMs and fibroblasts that may further contribute to this phenotype. Interestingly miR-30d expression is highest in cardiomyocytes at baseline and increases further with ischemic stress. Cardiomyocyte secretion of miR-30d packaged in EVs also increases with stress, and these EVs serve as a novel signaling entity that can transfer their cargo miR-30d to fibroblasts. Our experiments demonstrate the integrin itga5 as a novel target for miR-30d in fibroblasts, serving to regulate fibroblast proliferation and activation, particularly early after ischemic stress. Integrins have been described as cell surface receptors with a critical role in cell adhesion, proliferation, migration, molecular signaling and mechanotransduction 12. Furthermore, integrins can interact with extracellular matrix (ECM) proteins, as well as enhance signals from pro-fibrotic molecules as TGFβ1 13, both mechanisms that have long been characterized as key players of pro-fibrotic phenotypes, including myocardial fibrosis.
The presence of miR-30d in CM-derived EVs, demonstrated using a variety of complementary technologies, including nanoflow acquisition of plasma from αMHC-MerCreMer - RosamTmG reporter mice, support the existence of a paracrine miR-30d trafficking between CMs and other cell types, including cardiac fibroblasts, enabled by EV release and uptake. Prior studies had demonstrated EV-mediated signaling between cardiomyocytes and cardiac fibroblasts in cell culture models or indirectly conferred in murine models 29,31. In this study, we leverage a novel murine model of EV-target mapping to demonstrate the definitive uptake of CM-derived EVs by fibroblasts in the context of ischemic insult. This crosstalk may explain the miR-30d mediated itga5 expression regulation in cardiac fibroblasts, which results in a decreased activation of TGFβ1 pro-fibrotic pathway, diminished fibroblast proliferation and therefore ameliorated collagen deposition and myocardial fibrosis.
The dynamics of miR-30d expression and release in EVs is complex. Our studies suggest an increase in miR-30d expression both in CMs and their secreted EVs in the acute phase following ischemic insult, serving to protect against cardiomyocyte apoptosis and myofibroblast activation. In the chronic ischemic remodeling stage, there is a decrease in cardiomyocyte and EV-contained miR-30d coinciding with adverse remodeling and increased fibrosis (see Graphical Abstract). Forced sustained expression of miR-30d protects against adverse remodeling, while further suppression of miR-30d worsens ventricular remodeling. In addition to the rodent animal models that provide the mechanistic underpinnings for the cardioprotection afforded by miR-30d, we translate our findings back to human subjects, where our data show that miR-30d expression is highest in ventricular tissue in a new small RNA tissue atlas, and is predominantly present in EVs in the plasma. The findings in our animal models also mirror clinical observations. While EV-miR-30d levels decrease in our cohort of patients with chronic HF with adverse remodeling, the relationship between higher EV-miR-30d expression and beneficial remodeling is preserved, consistent with our finding across multiple studies. EV-miR-30d levels is also linked to whole blood expression of genes implicated in fibrosis and inflammation, providing human context to our animal studies. Future studies will be needed to define the timing of miR-30d-based therapeutic approaches to target fibrosis and remodeling.
The protective phenotype observed in the context of ischemic HF conferred by elevated levels of miR-30d, is mirrored in other models of disease. In the oncology field, miR-30 has been well described as an attenuator of cancerous cell metastasis through targeting itga5, among other molecules 40. In fibrotic pathologies, other than myocardial fibrosis, miR-30d over-expression in the presence of pulmonary fibrosis is shown to block TGFβ1-induced activation of pro-fibrotic pathways 41. In addition, data from the FHS in this study suggest a relationship between circulating plasma miR-30d levels and expression of genes in cellular sources (whole blood) that have been implicated in MAPK activation, integrin-mediated signaling, and apoptosis, providing correlative evidence from human studies to support our findings. The previous evidence, together with the data shown in the current manuscript suggest that the strong evidence for miR-30d as a predictive or prognostic biomarker for cardiac remodeling in human studies may be based on its role as a functional mediator of processes implicated in cardiac remodeling. More generally, this study provides a paradigm for investigating non-coding RNAs as functional biomarkers that may not only serve as direct therapeutic targets but may also uncover pathways that are therapeutically tractable to improve ventricular remodeling.
Limitations.
Our study establishes a paradigm for investigation of ‘functional’ extracellular RNA biomarkers, including as mediators of intercellular communication important for disease pathogenesis. While we were able to provide compelling evidence of the presence and regulation of miR-30d in cardiomyocyte-derived EVs, and demonstrate their transfer to, and biological effect in cardiac fibroblasts in vitro, our in vivo studies are suggestive of this novel signaling mechanism, but do not definitively prove the critical importance of EV-mediated communication in the pathogenesis of cardiac fibrosis. Currently available tools that inhibit EV release significantly affect other cellular processes (such as intracellular vesicle trafficking or membrane biology) and would confound the results if administered in vivo. Furthermore, none of these compounds can selectively block cardiomyocyte EV release; hence definitive proof of the relative importance of this mode of paracrine signaling is not feasible until the development of new tools. Finally, miR-30d may also be associated to a lesser degree with other types of carriers (e.g. bound to Ago 2 or in other types of EVs such as exomeres); a detailed investigation of the functional role of these entities was beyond the scope of this study.
An alternative approach with therapeutic implications would be to transfuse wild-type mice with CM-derived EVs from miR-30d overexpressing mice. However, technical limitations on isolation of CM-derived EVs (e.g. GFP-positive EVs from the αMHC-MerCreMer - RosamTmG mice) by nanoflow sorting present a significant hurdle to this approach. Future studies with transfusion of engineered EVs loaded with miR-30d may be instructive.
While our study provides a compelling rationale for therapeutic approaches to target miR-30d by overexpression approaches, future studies will need to determine the feasibility and timing of these therapies. The lentiviral gene transfer approach to miR-30d OE suggests that delivery of the therapy may confer protection from MI when miR-30d can be directly targeted to the heart; however, there may be a window in the trajectory of cardiac remodeling when increasing miR-30d levels may be effective. As with other HF therapies, it is possible that increasing miR-30d levels in end-stage HF may not be sufficient to improve remodeling. Future studies with administration of miR-30d mimics or analogs at the time of MI or during the remodeling phase may clarify these issues. However, as with all RNA therapies, delivery to cardiac tissue with systemic injections remains a challenge and a current area of investigation, but beyond the scope of this study. In this regard, continued development and approval of RNA therapies for human diseases serves as a source of encouragement.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
The heart undergoes a remodeling process after an initial injury that includes cellular processes like cell death and fibrosis.
RNAs, including microRNAs (miRs), can be secreted by cells in response to stress and can serve as potential biomarkers and signaling molecules in disease pathogenesis.
Previous studies have shown that lower levels of circulating miR-30d are associated with adverse remodeling and worse outcome in patients with heart failure (HF).
What New Information Does This Article Contribute?
Overexpression of miR-30d in the heart protects against cardiomyocyte apoptosis, fibrosis and improves remodeling, while silencing increases apoptosis, fibrosis and worsens remodeling in a rodent model of ischemic HF.
miR-30d targets mitogen-associated protein kinase 4 (MAP4K4) in cardiomyocytes ameliorating cell death, and EV-contained miR-30d secreted by cardiomyocytes mediates silencing of integrin α5 (itga5) in fibroblasts, abrogating fibrosis.
MiR-30d expression in cardiomyocytes and secreted EVs is acutely increased with hypoxic stress, but subsequently decreases in chronic ischemic HF, coincident with adverse remodeling in rodent models and ischemic HF.
Extracellular RNAs have potential as biomarkers and novel mediators of intercellular signaling. Plasma microRNA-30d level is associated with ventricular remodeling and clinical outcome in HF patients, but its functional role is unknown. Here we demonstrate a functional role for miR-30d in the pathogenesis of ventricular remodeling in ischemic HF. Overexpression of miR-30d is associated with beneficial remodeling, decreased cardiomyocyte apoptosis and fibrosis in rodent models of ischemic HF, while the converse is noted with silencing of miR-30d. MiR-30d expression in cardiomyocytes and its secretion in EVs increases with acute hypoxic stress. MiR-30d targets MAP4K4 within cardiomyocytes to ameliorate apoptosis, while secreted miR-30d mediates paracrine signaling in fibroblasts, targeting ITGA5 and counteracting myofibroblast activation. In chronic ischemic HF, miR-30d in cardiomyocytes and EVs decrease, coincident with adverse remodeling. EV-miR-30d levels are associated with cardiac function in chronic human HF and EV-miR-30d expression is associated with MAP4K4 and integrin pathways. These studies support the paradigm that circulating RNA biomarkers play a role in disease pathogenesis and specifically, provide the mechanistic underpinning for the association between EV-miR-30d and cardiac remodeling. Our study has broad implication for EV-RNAs as prognostic biomarkers or as tools to define molecular pathways for therapeutic targeting.
Acknowledgments
SOURCES OF FUNDING
AMS is supported by an American Heart Association postdoctoral fellowship (18POST34030167). SD is supported by grants from NIH (R01 HL122547, R01HL102368, R35HL150807) as well as from NCATS (UH3 TR000901). RS is supported from grants from the NIH (K23-HL127099). Additionally, this work was supported by the grants from National Natural Science Foundation of China (81722008 to J.J.X., 81470515 and 81670362 to J.H.X., 91639101 and 81570362 to J.J.X.), Innovation Program of Shanghai Municipal Education Commission (2017-01-07-00-09-E00042 to J.J.X.), the grant from Science and Technology Commission of Shanghai Municipality (17010500100, 18410722200 to J.J.X.).
This work was part of the Extracellular RNA Communication Consortium.
DISCLOSURES
Dr. Shah has received funds from Amgen (scientific advisory board), Myokardia (consulting), and Best Doctors (consulting). Dr. Das and Dr. Shah are co-inventors on a patent for ex-RNAs signatures of cardiac remodeling. Dr. Das is a founder of Switch Therapeutics, which did not play any role in this study and has consulted for Amgen.
Nonstandard Abbreviations and Acronyms:
- AAR
area-at-risk
- Ago2
Argonaute-2
- AngII
angiotensin II
- αMHC
alpha- myosin heavy chain
- Apo
apolipoprotein
- αSMA
alpha smooth muscle actin
- BSA
bovine serum albumin
- CD
cluster of differentiation
- cDNA
complementary DNA
- CM
cardiac myocyte
- CTGF
connective tissue growth factor
- Ctrl
control
- ddPCR
Droplet digital PCR
- DMEM
Dulbecco’s Modified Eagle Medium
- EC
endothelial cells
- EdU
5-Ethynyl-2’-deoxyuridine
- ES
enrichment score
- EV
extracellular vesicles
- ExRNA
extracellular RNA
- FBS
fetal bovine serum
- FC
fold change
- FDR
false discovery rate
- FFPE
Formalin-fixed-paraffin-embedded
- FGFR2
fibroblast growth factor receptor 2
- FHS
Framingham Heart Study
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- GFP
green fluorescent protein
- GSEA
Gene Set Enrichment Analysis
- HDL
high-density lipoprotein
- HEK293
human embryonic kidney 293 cells
- HF
heart failure
- IGV
Integrative Genome Viewer
- ip
intra-peritoneal
- IP
immuno-precipitation
- IR
ischemia-reperfusion
- ITGA5
integrin alpha-5
- iv
intravenous
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- KO
knock out
- LAD
left anterior descending coronary artery
- LNA
locked nucleic acid
- LV
left ventricle
- LOF
loss of function
- MAP4K4
mitogen-associate protein kinase kinase kinase kinase 4
- MB
molecular beacon
- MerCreMer
tamoxifen inducible Cre recombinase
- MI
myocardial infarction
- miR
microRNA
- NRVM
neonatal rat ventricular myocytes
- OE
overexpression
- OGD/R
Oxygen-Glucose Deprivation/Reperfusion
- ON
over night
- PBS
Phosphate-buffered saline
- PCR
polymerase chain reaction
- PECAM1
platelet endotelial cell adhesion molecule 1
- PFA
paraformaldehyde
- PMSF
phenylmethylsulfonyl fluoride
- SEM
standard error of the mean
- RNA
ribonucleic acid
- RNA-seq
RNA sequencing
- Rno
Rattus Norvegicus
- RT
reverse transcription
- RT
room temperature
- Scrm
scramble
- SD
Sprague Dawley
- SDS-PAGE
sodium dodecyl sulphate-polyacrylamide gel electrophoresis
- SEC
size exclusion chromatography
- TGFβ1
transforming growth factor beta 1
- TG
transgenic
- TPMs
transcripts per million reads
- TTC
triphenyltetrazolium chloride
- TUNEL
deoxyribonucleotidyl transferase (TdT)-mediated dUTP nick end labeling
- UTR
untranslated region
- VLDL
very low density lipoprotein
- WT
wild-type
REFERENCES
- 1.Benjamin EJ, Virani SS, Callaway CW, et al. Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation. 2018;137:e67–e492. [DOI] [PubMed] [Google Scholar]
- 2.Ambrosy AP, Fonarow GC, Butler J, et al. The Global Health and Economic Burden of Hospitalizations for Heart Failure: Lessons Learned From Hospitalized Heart Failure Registries. J Am Coll Cardiol. 2014;63:1123–1133. [DOI] [PubMed] [Google Scholar]
- 3.Januzzi JL, Prescott MF, Butler J, et al. Association of Change in N-Terminal Pro-B-Type Natriuretic Peptide Following Initiation of Sacubitril-Valsartan Treatment With Cardiac Structure and Function in Patients With Heart Failure With Reduced Ejection Fraction. JAMA. 2019;1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Da Costa Martins PA, De Windt LJ. MicroRNAs in control of cardiac hypertrophy. Cardiovasc Res. 2012;93:563–572. [DOI] [PubMed] [Google Scholar]
- 5.Melman YF, Shah R, Das S. MicroRNAs in heart failure: is the picture becoming less miRky? Circ Heart Fail. 2014;7:203–214. [DOI] [PubMed] [Google Scholar]
- 6.Khan M, Nickoloff E, Abramova T, et al. Embryonic stem cell-derived exosomes promote endogenous repair mechanisms and enhance cardiac function following myocardial infarction. Circ Res. 2015;117:52–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Thomou T, Mori MA, Dreyfuss JM, et al. Adipose-derived circulating miRNAs regulate gene expression in other tissues. Nature. 2017;542:450–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Melman YF, Shah R, Danielson K, et al. Circulating MicroRNA-30d Is Associated With Response to Cardiac Resynchronization Therapy in Heart Failure and Regulates Cardiomyocyte Apoptosis: A Translational Pilot Study. Circulation. 2015;131:2202–2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xiao J, Gao R, Bei Y, et al. Circulating miR-30d Predicts Survival in Patients with Acute Heart Failure. Cell Physiol Biochem Int J Exp Cell Physiol Biochem Pharmacol. 2017;41:865–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu Y-F, Qu G-Q, Lu Y-M, Kong W-M, Liu Y, Chen W-X, Liao X-H. Silencing of MAP4K4 by short hairpin RNA suppresses proliferation, induces G1 cell cycle arrest and induces apoptosis in gastric cancer cells. Mol Med Rep. 2016;13:41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ross RS, Borg TK. Integrins and the myocardium. Circ Res. 2001;88:1112–1119. [DOI] [PubMed] [Google Scholar]
- 12.Chen C, Li R, Ross RS, Manso AM. Integrins and integrin-related proteins in cardiac fibrosis. J Mol Cell Cardiol. 2016;93:162–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Frangogiannis NG. Matricellular proteins in cardiac adaptation and disease. Physiol Rev. 2012;92:635–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Danielson KM, Shah R, Yeri A, et al. Plasma Circulating Extracellular RNAs in Left Ventricular Remodeling Post-Myocardial Infarction. EBioMedicine. 2018;32:172–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Théry C, Witwer KW, Aikawa E, et al. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J Extracell Vesicles. 2018;7:1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baker MB, Bao G, Searles CD. In vitro quantification of specific microRNA using molecular beacons. Nucleic Acids Res. 2012;40:e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ko HY, Lee J, Joo JY, Lee YS, Heo H, Ko JJ, Kim S. A color-tunable molecular beacon to sense miRNA-9 expression during neurogenesis. Sci Rep. 2014;4:4626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee JH, Kim JA, Kwon MH, Kang JY, Rhee WJ. In situ single step detection of exosome microRNA using molecular beacon. Biomaterials. 2015;54:116–125. [DOI] [PubMed] [Google Scholar]
- 19.Camacho V, Toxavidis V, Tigges JC. Characterization of Extracellular Vesicles by Flow Cytometry. Methods Mol Biol Clifton NJ. 2017;1660:175–190. [DOI] [PubMed] [Google Scholar]
- 20.Loyer X, Zlatanova I, Devue C, et al. Intra-Cardiac Release of Extracellular Vesicles Shapes Inflammation Following Myocardial Infarction. Circ Res. 2018;123:100–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bollini S, Smits AM, Balbi C, Lazzarini E, Ameri P. Triggering Endogenous Cardiac Repair and Regeneration via Extracellular Vesicle-Mediated Communication. Front Physiol. 2018;9:1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Herring BP, Hoggatt AM, Burlak C, Offermanns S. Previously differentiated medial vascular smooth muscle cells contribute to neointima formation following vascular injury. Vasc Cell. 2014;6:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ridder K, Keller S, Dams M, et al. Extracellular vesicle-mediated transfer of genetic information between the hematopoietic system and the brain in response to inflammation. PLoS Biol. 2014;12:e1001874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kur I-M, Prouvot P-H, Fu T, et al. Neuronal activity triggers uptake of hematopoietic extracellular vesicles in vivo. PLOS Biol. 2020;18:e3000643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Srinivasan S, Yeri A, Cheah PS, et al. Small RNA Sequencing across Diverse Biofluids Identifies Optimal Methods for exRNA Isolation. Cell. 2019;177:446–462.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shah R, Ziegler O, Yeri A, et al. MicroRNAs Associated With Reverse Left Ventricular Remodeling in Humans Identify Pathways of Heart Failure Progression. Circ Heart Fail. 2018;11:e004278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.D’Alessandra Y, Devanna P, Limana F, et al. Circulating microRNAs are new and sensitive biomarkers of myocardial infarction. Eur Heart J. 2010;31:2765–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Widera C, Gupta SK, Lorenzen JM, Bang C, Bauersachs J, Bethmann K, Kempf T, Wollert KC, Thum T. Diagnostic and prognostic impact of six circulating microRNAs in acute coronary syndrome. J Mol Cell Cardiol. 2011;51:872–875. [DOI] [PubMed] [Google Scholar]
- 29.Bang C, Batkai S, Dangwal S, et al. Cardiac fibroblast-derived microRNA passenger strand-enriched exosomes mediate cardiomyocyte hypertrophy. J Clin Invest. 2014;124:2136–2146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen X, Liang H, Zhang J, Zen K, Zhang C- Y. Secreted microRNAs: a new form of intercellular communication. Trends Cell Biol. 2012;22:125–132. [DOI] [PubMed] [Google Scholar]
- 31.Pironti G, Strachan RT, Abraham D, Mon-Wei Yu S, Chen M, Chen W, Hanada K, Mao L, Watson LJ, Rockman HA. Circulating Exosomes Induced by Cardiac Pressure Overload Contain Functional Angiotensin II Type 1 Receptors. Circulation. 2015;131:2120–2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Viereck J, Bang C, Foinquinos A, Thum T. Regulatory RNAs and paracrine networks in the heart. Cardiovasc Res. 2014;102:290–301. [DOI] [PubMed] [Google Scholar]
- 33.Li Y, Liang Y, Zhu Y, Zhang Y, Bei Y. Noncoding RNAs in Cardiac Hypertrophy. J Cardiovasc Transl Res. 2018; [DOI] [PubMed] [Google Scholar]
- 34.Thum T, Gross C, Fiedler J, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–984. [DOI] [PubMed] [Google Scholar]
- 35.Ucar A, Gupta SK, Fiedler J, et al. The miRNA-212/132 family regulates both cardiac hypertrophy and cardiomyocyte autophagy. Nat Commun. 2012;3:1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Rooij E, Olson EN. MicroRNA therapeutics for cardiovascular disease: opportunities and obstacles. Nat Rev Drug Discov. 2012;11:860–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao X, Mohan R, Özcan S, Tang X. MicroRNA-30d induces insulin transcription factor MafA and insulin production by targeting mitogen-activated protein 4 kinase 4 (MAP4K4) in pancreatic β-cells. J Biol Chem. 2012;287:31155–31164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chen S, Xuan J, Wan L, Lin H, Couch L, Mei N, Dobrovolsky VN, Guo L. Sertraline, an antidepressant, induces apoptosis in hepatic cells through the mitogen-activated protein kinase pathway. Toxicol Sci Off J Soc Toxicol. 2014;137:404–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fiedler LR, Chapman K, Xie M, et al. MAP4K4 Inhibition Promotes Survival of Human Stem Cell-Derived Cardiomyocytes and Reduces Infarct Size In Vivo. Cell Stem Cell. 2019;24:579–591.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Croset M, Pantano F, Kan CWS, et al. miRNA-30 Family Members Inhibit Breast Cancer Invasion, Osteomimicry, and Bone Destruction by Directly Targeting Multiple Bone Metastasis-Associated Genes. Cancer Res. 2018;78:5259–5273. [DOI] [PubMed] [Google Scholar]
- 41.Zhao S, Xiao X, Sun S, Li D, Wang W, Fu Y, Fan F. MicroRNA-30d/JAG1 axis modulates pulmonary fibrosis through Notch signaling pathway. Pathol Res Pract. 2018;214:1315–1323. [DOI] [PubMed] [Google Scholar]
- 42.Matsui T, Li L, Wu JC, Cook SA, Nagoshi T, Picard MH, Liao R, Rosenzweig A. Phenotypic spectrum caused by transgenic overexpression of activated Akt in the heart. J Biol Chem. 2002;277:22896–22901. [DOI] [PubMed] [Google Scholar]
- 43.Yeri A, Courtright A, Danielson K, et al. Evaluation of commercially available small RNASeq library preparation kits using low input RNA. BMC Genomics. 2018;19:331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Andrews S FastQC: A quality control tool for high throughput sequence data. Http://Www.Bioinformatics.Babraham.Ac.Uk/Projects/Fastqc/. 2010;http://www.bioinformatics.babraham.ac.uk/projects/.
- 45.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinforma Oxf Engl. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29:24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Patro R, Duggal G, Kingsford C. Salmon: Accurate, Versatile and Ultrafast Quantification from RNA-seq Data using Lightweight-Alignment. bioRxiv. 2015;021592. [Google Scholar]
- 48.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mootha VK, Lindgren CM, Eriksson KF, et al. PGC-1α-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat Genet. 2003;34:267–273. [DOI] [PubMed] [Google Scholar]
- 50.Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: A knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci. 2005;102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rozowsky J, Kitchen RR, Park JJ, Galeev TR, Diao J, Warrell J, Thistlethwaite W, Subramanian SL, Milosavljevic A, Gerstein M. exceRpt: A Comprehensive Analytic Platform for Extracellular RNA Profiling. Cell Syst. 2019;8:352–357.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Joehanes R, Ying S, Huan T, et al. Gene expression signatures of coronary heart disease. Arterioscler Thromb Vasc Biol. 2013;33:1418–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Freedman JE, Gerstein M, Mick E, et al. Diverse human extracellular RNAs are widely detected in human plasma. Nat Commun. 2016;7:11106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Almasy L, Blangero J. Multipoint quantitative-trait linkage analysis in general pedigrees. Am J Hum Genet. 1998;62:1198–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Joehanes R, Zhang X, Huan T, et al. Integrated genome-wide analysis of expression quantitative trait loci aids interpretation of genomic association studies. Genome Biol. 2017;18:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kuleshov MV, Jones MR, Rouillard AD,et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016;44:W90–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fabregat A, Jupe S, Matthews L, et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018;46:D649–D655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Slenter DN, Kutmon M, Hanspers K, et al. WikiPathways: a multifaceted pathway database bridging metabolomics to other omics research. Nucleic Acids Res. 2018;46:D661–D667. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors will make our data, study materials and analytic methods available to other researchers. All supporting data are available within the article, supplemental material and datasets provided. If additional data is required, it will be made available by the corresponding author upon reasonable request.