

Abstract
Diabetic nephropathy (DN) is a major cause of end-stage renal disease, but treatment remains ineffective. C-reactive protein (CRP) is pathogenic in DN, which significantly correlated with dipeptidyl peptidase-4 (DPP4) expression in diabetic patients with unknown reason. Here, using our unique CRPtg-db/db mice, we observed human CRP markedly induced renal DPP4 associated with enhanced kidney injury compared with db/db mice. Interestingly, linagliptin, a US Food and Drug Administration (FDA)-approved specific DPP4 inhibitor, effectively blocked this CRP-driven DN in the CRPtg-db/db mice. Mechanistically, CRP evoked DPP4 in cultured renal tubular epithelial cells, where CD32b/nuclear factor κB (NF-κB) signaling markedly enriched p65 binding on the DPP4 promoter region to increase its transcription. Unexpectedly, we further discovered that CRP triggers dimerization of DPP4 with CD32b at protein level, forming a novel DPP4/CD32b/NF-κB signaling circuit for promoting CRP-mediated DN. More importantly, linagliptin effectively blocked the circuit, thereby inhibiting the CRP/CD32b/NF-κB-driven renal inflammation and fibrosis. Thus, DPP4 may represent a precise druggable target for CRP-driven DN.
Keywords: diabetic nephropathy, dipeptidyl peptidase-4, DPP4, linagliptin, renal inflammation, fibrosis
Graphical Abstract
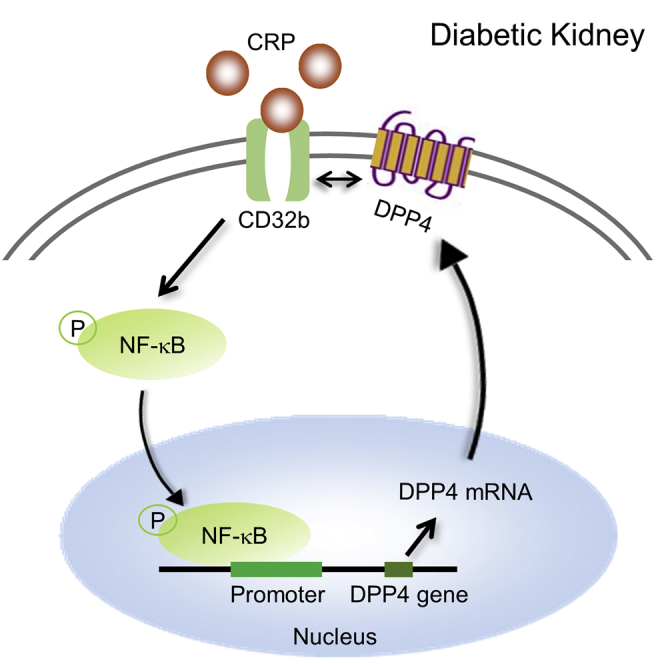
Tang et al. discover a novel DPP4/CD32b/NF-κB circuit and demonstrate its pathogenic role in diabetic nephropathy on type 2 diabetes mice. CRP activates CD32b/NF-κB signaling to increase DPP4 transcription, whose protein dimerizes with CD32b and thereby enhancing the CD32b/NF-κB-mediated nephropathy. DPP4 may represent a precision therapeutic target for CRP-driven diabetic nephropathy.
Introduction
Diabetic nephropathy (DN), the primary microvascular complication of diabetes mellitus (DM), is a leading cause of end-stage renal disease (ESRD), accounting for 30%–47% worldwide. In United States, about 54.4% of patients with DM will eventually enter ESRD.1,2 Until now, effective treatment for DN is still in an urgent need. Increasing evidence shows that type 2 DM (T2DM) is a low-grade inflammatory disease.3 The pathogenesis of DN is complex and is involved in hemodynamics, glycation metabolism, polyol pathway/hexosamine pathway, oxidative stress, low-grade inflammation, and so on.4 Among which, inflammation is well-known to strongly correlate with the development of hyperglycemia and glycated hemoglobin, and is driven by an increased kidney production of chemokines and proinflammatory cytokines in the development of DN. This is particularly important in those with DN.5,6 In patients with T2DM, serum levels of pro-inflammatory cytokines, such as interleukin (IL)-1β, IL-6, and C-reactive protein (CRP), are elevated and serve as predictive markers of T2DM.3,7, 8, 9 Of note, elevated serum levels of CRP are closely associated with an increase in microalbuminuria and renal dysfunction in patients with DN,5,9 suggesting the close link between CRP and the development of diabetic kidney injury.
Among the inflammatory cascade, CRP can induce IL-6 via a nuclear factor κB (NF-κB)-dependent mechanism.10 We also found that human CRP can signal through the CD32-dependent mechanism to activate both NF-κB and transforming growth factor β (TGF-β)/Smad3 pathways, resulting in a significant upregulation of pro-inflammatory cytokines (IL-1β, tumor necrosis factor alpha [TNF-α], monocyte chemoattractant protein-1 [MCP-1]) and pro-fibrotic growth factors (TGF-β1, connective tissue growth factor [CTGF]) in a mouse model of type 1 DN and in vitro, revealing a pathogenic importance for CRP in type 1 diabetic kidney diseases.11 Under diabetic conditions, CRP is induced by high glucose, which in turn synergistically promotes high-glucose-mediated renal inflammation and fibrosis.11 The pathogenic importance for CRP is also found in other disease models, including obstructive nephropathy,12 ischemic kidney injury,13 hypertensive heart disease,14 and atherosclerosis.15
New anti-diabetic agents are sought to diminish diabetic complications, such as glucagon-like peptide-1 (GLP-1) receptor agonists, dipeptidyl peptidase-4 (DPP4) inhibitors, and sodium-glucose cotransporter 2 inhibitors. Several studies, including animal experiments and clinical trials, showed the beneficial effects of DPP4 inhibitors (DPP4is) on DN.16, 17, 18, 19 DPP4 DPP-4 is a member of serine proteases that was first characterized as a T cell surface marker (CD26); its soluble form can be detected in peripheral blood, urine, and other body fluids.20 DPP4 cleaves a wide range of substrates, including growth factors, chemokines, and peptides, in addition to its major role in glucose metabolism.21 DPP4 was highly expressed in kidney under disease conditions found in experimental rodent models, including high-fat-diet-induced diabetes and acute ischemia-reperfusion injury.22,23 It is noted that DPP4i can delay the degradation of incretins in order to rebalance the glycemic control of the patients by prolonging insulin secretion.24,25 Beyond the glucose metabolism, the potential effect of DPP4i on anti-inflammation has been suggested by a number of studies due to the significat reduction of inflammation indicators, such as CRP and IL-6, in the plasma of DPP4i-treated patients.26, 27, 28 However, the pathogenic role and underlying mechanism of DPP4 in diabetic renal injury is still largely unknown.
Importantly, mouse CRP is synthesized only in trace amounts and is not an acute-phase reactant, whereas human CRP in mice can activate complements and bind to mouse FcγRs, presumably FcγRIIb (CD32b). Thus, we generated a transgenic human CRP mouse strain (CRPtg mice) that can serve as a convenient and more reliable tool to investigate the biological activities of CRP under different disease conditions.11, 12, 13, 14, 15,29 Interestingly, we found this unique CRPtg-db/db mouse strain shows more severe renal complications than their parental db/db mice.30 In this study, we observed that DPP4 is dramatically triggered in the kidney of CRPtg-db/db mice compared with both db/db and db/m mice, specifically at the early stage of diabetic kidney injury. We demonstrated that CRP is able to trigger DPP4 expression in both murine and human renal cells associated with the induction of renal inflammation and fibrosis markers in vitro. Importantly, treatment with DPP4i effectively ameliorates CRP-driven renal inflammation, fibrosis, and function loss in the CRPtg-db/db mice. Mechanistically, we discovered a novel DPP4/CD32b/NF-κB signaling circuit for CRP-driven T2DN in vivo and in vitro. Thus, DPP4 may represent a novel therapeutic target for CRP-mediated T2DN.
Results
DPP4 Is Increased and Peaked before the Onset of Kidney Injury in CRPtg db/db
The role of DPP4 in CRP-driven kidney injury is still largely unknown. By our human CRP transgenic db/db mice model,31,32 we found that the renal expression level of DPP4 was largely enhanced in glomerulus and tubules of the 20-week-old CRPtg db/db mice compared with the db/db mice, as well as non-diabetic db/m and CRPtg db/m littermates, as shown by immunohistochemistry (IHC) (Figure 1A). The induction of DPP4 in the diabetic kidney of CRPtg db/db mice was highly associated with the human CRP-driven CD32b activation, as shown by IHC, western blot, and real-time polymerase chain reaction (PCR) (Figure 1).
Figure 1.

DPP4 Is Largely Increased in the Diabetic Injured Kidney of CRPtg db/db Mice
(A) IHC of DPP4 and CD32b in the kidney of 20-week-old mice (original magnification ×400). (B–D) Western blot (B) and real-time PCR (C and D) show the induction of renal DPP4 and CD32b in db/db mice is further promoted in the CRPtg db/db mice at 20 weeks old. Each bar represents the mean ± SEM for groups of six mice or three independent experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus db/m mice; #p < 0.05 versus db/db mice.
Then we further investigated the expression pattern of DPP4 in the progression of CRP-driven kidney injury under diabetic condition by a time-course study in vivo. Interestingly, the renal DPP4 level was significantly increased in the CD32b-activated kidney of CRPtg db/db mice since 12 weeks old compared with the db/db and db/m mice, but unexpectedly declined during the development of T2DN shown by western blot (Figure 2A). Interestingly, the renal expression of DPP4 peaked at 12 weeks old and remained high throughout the exacerbation of DN in the CRPtg db/db mice compared with both db/db and db/m mice, as shown by real-time PCR (Figure 2B). Moreover, urine microalbuminuria creatinine ratio (UACR) assay showed that the renal functional loss was significantly promoted in the CRPtg db/db mice since 16 weeks old compared with the db/db mice (Figure 2C), suggesting that renal DPP4 may be important to the onset of CRP-driven T2DN in db/db mice.
Figure 2.

Induction of Renal DPP4 Peaks at the Age of 12 Weeks in CRPtg db/db Mice
(A) Western blot shows the renal expression pattern of DPP4 and CD32b in mice. (B) Real-time PCR examines the renal expression of DPP4 mRNA in mice. (C) ELISA detects the urine microalbuminuria creatinine ratio (UACR) in different groups of mice and indicates the significant promotion of UACR in CRPtg db/db mice compared with db/db mice since age of 20 weeks. Each bar represents the mean ± SEM for groups of six mice. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus db/m mice; #p < 0.05, ##p < 0.01, ###p < 0.001 versus db/db mice.
DPP4 Is Tightly Regulated by CRP/CD32b Signaling in Renal Tubular Epithelial and Mesangial Cells In Vitro
DPP4 was largely increased in both glomerulus and tubules of CRPtg db/db mice associated with the CD32b activation (Figure 1A); thus, we hypothesized that CRP signaling may be capable of triggering DPP4 expression in renal resident cells and tested it on mouse tubular epithelial cells (mTECs) and mesangial cells (mMCs; SV40 MES 13) in vitro. Real-time PCR showed that CRP (10 μg/mL) caused a marked increase of DPP4 mRNA level in both mTECs and mMCs and peaked at 12 h in vitro (Figures 3A and 3B). In addition, we also confirmed this finding on human renal tubular epithelial HK-2 cells. Our result showed that CRP significantly induced CD32b and increased the expression of DPP4 mRNA, resulting in upregulation of inflammatory cytokines (TNF-α, MCP-1) and fibrosis (collagen I, fibronectin) by HK-2 cells (Figures 3C–3H). More importantly, inhibition of DPP4 with specific inhibitor linagliptin (DPP4i) effectively suppressed the expression of renal inflammation and fibrosis markers in the CRP-stimulated HK-2 cells in a dose-dependent manner (Figures 3I and 3J; Figure S1). Thus, we further investigated the functional role and pathogenic mechanism of DPP4 in the CRP-mediated T2DN in vivo.
Figure 3.

CRP Induces DPP4 in Renal Cells Associated with Inflammatory and Fibrotic Response In Vitro
Real-time analysis shows that CRP (10 μg/mL) triggers DPP4 expression in (A) mTEC, (B) mMC, and (C) HK-2 cells; associated with (D) CD32b induction, it induces an inflammatory and fibrotic response in the human HK-2 cell line, including inflammatory markers (E) TNF-α and (F) MCP-1 and fibrotic markers (G) collagen I (Col I) and (H) fibronectin (Fn). DPP4 inhibitor (DPP4i linagliptin) significantly suppresses the CRP-induced expression of (I) TNF-α and (J) collagen I in HK-2 cells, as shown by real-time PCR analysis. Data represent the mean ± SEM for three independent experiments. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus control; #p < 0.05, ##p < 0.01 versus CRP-stimulated HK-2 cells.
DPP4 Inhibition Protects against Renal Injury in CRPtg db/db Mice
To investigate the role of DPP4 in CRP-driven T2DN, we treated the 12-week-old CRPtg db/db mice with DPP4i linagliptin for 12 weeks with effective dosage (3 mg/kg/day orally) supported by the increase in serum GLP-1 level (Figure S2). Our results showed that DPP4i significantly suppressed the progression of diabetic renal injury in the linagliptin-treated 24-week-old CRPtg db/db mice compared with the untreated mice, as suggested by the reduction of histology injury from periodic acid-Schiff (PAS), periodic acid-silver methenamine (PASM), and Masson’s trichrome staining and renal function loss from serum creatinine assay (Figure 4; Figure S3).
Figure 4.

DPP4 Inhibition Suppresses CRP-Enhanced Kidney Injury in db/db Mice
(A) DPP4 inhibitor treatment suppresses histological renal injury of CRPtg db/db at 24 weeks old, as shown by PAS, PASM, and Masson’s trichrome staining with the (B) quantification analysis of mesangial matrix index and glomerulosclerosis calculated by PASM staining and Masson’s trichrome staining. (C) ELISA results of serum creatinine (Scr) of mice at 24 weeks old, which shows the renoprotective effect of DPP4i treatment, suggested by the significant reduction of Scr in CRPtg db/db mice. Data represent the mean ± SEM for eight mice per group. ∗∗∗p < 0.001 versus db/m mice; #p < 0.05, ##p < 0.01 versus untreated CRPtg db/db mice.
Linagliptin Ameliorates CRP-Driven Renal Inflammation and Fibrosis in db/db Mice
We previously demonstrated that CRP promoted diabetic renal injury with more severe renal inflammation and fibrosis in db/db mice.30 Here, we also detected the enhancement of pro-inflammatory cytokines and chemokine (TNF-α, IL-1β, and MCP-1) and renal fibrotic biomarkers (collagen I and IV) in the diabetic injured kidney of CRPtg db/db mice compared with db/db mice as shown by IHC (Figures 5A and 6A). Interestingly, inhibition of DPP4 significantly reduced the CRP-driven inflammation and fibrosis in the DPP4i-treated CRP-db/db mice compared with their non-treated controls, as shown by the marked reduction of the renal inflammation and fibrosis marker expression, as well as the macrophage infiltration, in the diabetic kidney (Figures 5 and 6). These encouraging data suggested that DPP4 has an important role in the pathogenesis of CRP-driven T2DN.
Figure 5.

DPP4 Inhibition Suppresses Renal Fibrosis in CRPtg db/db Mice
(A) IHC of collagen I (Col I), collagen IV (Col IV), and α-smooth muscle actin (α-SMA) in kidney (original magnification ×400). (B) Real-time PCR analysis of Col I, Col IV, and α-SMA expression in mice at 24 weeks old. (C) Western blot and quantitative analysis of Fn, Col I, Col IV, and α-SMA expression in the kidney of mice at 24 weeks old. Data represent the mean ± SEM for eight mice per group. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus db/m mice; ˆp < 0.05, ˆˆp < 0.01 versus db/db mice; #p < 0.05, ##p < 0.01 versus untreated CRPtg db/db mice.
Figure 6.

DPP4 Inhibition Suppresses Renal Inflammation in CRPtg db/db Mice
(A and B) IHC (A) and real-time PCR (B) result of MCP-1, IL-1β, and TNF-α expression in kidney of mice at 24 weeks old (original magnification ×400). (C) IHC shows the enhanced renal infiltration of F4/80+ macrophages in CRPtg db/db mice is significantly decreased by treatment with DPP4 inhibitor (original magnification ×400). Data represent the mean ± SEM for eight mice per group. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001 versus db/m mice; ˆp < 0.05, ˆˆp < 0.01 versus db/db mice; #p < 0.05, ###p < 0.001 versus untreated CRPtg db/db mice.
A Novel DPP4/CD32b/NF-κB Signaling Circuit in CRP-High Condition
Because our previous study showed that activation of CD32b/NF-κB signaling is important for CRP-driven T2DN,30 we examined its potential role in the transcriptional regulation of DPP4. Interestingly, we observed that CRP markedly induced DPP4 expression in the diabetic injured kidney in vivo (Figure 7A). By evolutionary conserved region (ECR) browser and chromatin immunoprecipitation (ChIP) assay, we identified a direct NF-κB binding site on the promoter region of the DPP4 gene and detected its enhancement in CRP-stimulated HK-2 cells at the genomic level in vitro (Figure 7B). Surprisingly, we further found a strong co-localization of DPP4 and CD32b in the cell membrane of the CRPtg db/db group (Figure 7A), which led us to discover a novel phenomenon where DPP4 can directly dimerize with CD32b at the protein level. We confirmed that CRP was able to induce DPP4:CD32b dimerization in HK-2 cells in vitro by co-immunoprecipitation assay (coIP) assay (Figure 7C). More importantly, inhibition of DPP4 was able to reduce the expression of CD32b at both mRNA and protein levels in a dose-dependent manner as detected by real-time PCR and western blot (Figures 7D and 7E). Interestingly, targeting of either CD32b, NF-κB, or DPP4 also suppressed the other two proteins in the CRP-stimulated HK-2 cells in vitro (Figures 7F and 7G; Figure S3), demonstrating a novel DPP4/NF-κB/CD32b signaling circuit under CRP-rich condition. These results uncovered the existence of a novel DPP4/CD32b/NFκB circuit for promoting CRP-driven kidney injury in vivo and in vitro.
Figure 7.

A Novel DPP4/CD32b/NF-κB Circuit in CRP-Rich Diabetic Condition
(A) Immunofluorescence staining shows the co-localization of DPP4 and CD32b in the diabetic injured kidney of CRPtg db/db mice at 24 weeks old (original magnification ×400). (B) The predicted binding site of NF-κB on the evolutionarily conserved region of DPP4 in human (yellow highlighted at right lower panel) and mouse genomes by ECR browser, where ChIP assay shows that NF-κB physically binds DPP4 promoter in response to CRP (10 μg/mL) in HK-2 cells at 1 h. (C) Co-immunoprecipitation (coIP) assay demonstrates that CRP triggers the binding of DPP4 to CD32b in HK-2 cells at 24 h after stimulation. (D and E) Real-time PCR (D) and western blot (E) analysis detect that DPP4 inhibition suppresses CRP-induced CD32b expression in HK-2 cells. (F and G) Real-time PCR (F) and western blot (G) analysis show the addition of either NF-κB inhibitor BAY11-7085 (NF-κBi, 10 μM), DPP4i, or CD32b-neutralizing antibody (CD32bi, 10 μg/mL) also blocks the activation of CRP-induced DPP4/CD32b/NF-κB signaling circuit in HK-2 cells. Data represent the mean ± SEM for three independent experiments. ∗∗p < 0.01, ∗∗∗p < 0.001 versus control; #p < 0.05, ##p < 0.01 versus CRP-stimulated HK-2 cells.
Moreover, we observed that the linagliptin treatment (DPP4i) was capable of blocking the DPP4/CD32b/NF-κB circuit in diabetic kidney under the CRP-rich microenvironment in vivo, as shown by the significant CD32b suppression and NF-κB inactivation in the linagliptin-treated CRPtg db/db mice (Figure 8; Figures S4 and S5). Taken together, we discovered a novel DPP4/CD32b/NF-κB signaling circuit in CRP-mediated T2DN and demonstrated that DPP4 may represent an effective therapeutic target for CRP-driven T2DN (Figure S6).
Figure 8.

Linagliptin Effectively Blocks DPP4/CD32b/NF-κB Signaling Circuit in CRPtg db/db Mice
The 12-week treatment with linagliptin significantly suppresses the CRP-induced DPP4/CD32b/NF-κB signaling circuit in the diabetic kidney of CRPtg db/db mice at 24 weeks old, as shown by (A and B) IHC staining, (C) real-time PCR, and (D) western blot analysis (original magnification ×400). Data represent the mean ± SEM for eight mice per group. ∗∗∗p < 0.001 versus db/m mice; ˆp < 0.05, ˆˆˆp < 0.001 versus db/db mice; #p < 0.05, ###p < 0.001 versus untreated CRPtg db/db mice.
Discussion
DN is a severe complication of T2DM and one of the major causes of ESRD, but effective treatments are still limited because of the complexity of the pathogenic mechanisms. It has been shown that CRP is a risk factor in patients with T2DM and T2DN because elevated serum levels of CRP were closely associated with the increased microalbuminuria and renal dysfunction in T2DM patients.33, 34, 35 DPP4 is highly associated with diabetes because of its physiological function as a primary mechanism for GLP-1 degradation.36 Recently, we also demonstrated the pathogenic role of CRP in renal inflammation and renal fibrosis in experimental diabetic models.30 DPP4 deficiency blocks renal damage in a variety of experimental models,17,37,38 but its potential role in CRP-driven DN is still not investigated. Here, by using our unique human CRP transgenic db/db mice,30 we discovered the importance of a novel DPP4/CD32b/NF-κB signaling circuit in CRP-driven T2DN. More importantly, we further demonstrated that targeting DPP4 was capable of blocking the entire circuit and therefore inhibiting the progression of CRP-mediated T2DN in vitro and in vivo.
DPP4 is an integral membrane glycoprotein and is expressed ubiquitously in most organs and cell types. We found that DPP4 was highly induced in the glomerulus (i.e., podocytes and mesangial cells) and tubules (i.e., brush borders and epithelial cells) in the CRPtg db/db mice, which is consistent with studies on other experimental disease models.31,32 Importantly, we are the first study to discover that DPP4 induction is closely associated with the CRP-driven CD32b activation in mice under diabetic conditions. More surprisingly, the renal DPP4 expression peaked at the age of 12 weeks before the onset of diabetic complications, such as renal function impairment, indicated by the development of microalbuminuria. In vitro study demonstrated that CRP can trigger DPP4 expression on both murine and human renal cells, and is associated with the induction of renal inflammation and fibrosis markers. These results revealed a pathogenic role for DPP4 in the CRP-mediated T2DN.
Indeed, DPP4 exists in soluble and membrane-bound forms, both of which are responsible for proteolytic activity.39 The soluble form is produced from shedding of the membrane DPP4 into the circulation, whereas the membrane-bound form exerts pleiotropic actions and expresses on many cell types, including kidney tubular cells.39 To examine the importance of DPP4 in CRP-driven T2DN, we applied a 12-week-treatment with DPP4i in CRPtg db/db mice. Our results demonstrated that DPP4i treatment effectively suppressed renal function and histology injuries, renal inflammation, and fibrosis in the CRPtg db/db mice compared with the control group.
In this study, we observed that high CRP level not only increased expression of DPP4, but also unexpectedly enhanced the physical binding of membrane DPP4 with CD32b in the diabetic injured kidney in vivo. The CRP-induced DPP4:CD32b dimerization was further confirmed on a human proximal tubule epithelial cell line HK-2 by coIP assay in vitro. More importantly, inhibition of DPP4 significantly suppressed both the activation and expression of CD32b under the CRP-high condition. Our finding revealed that DPP4 acts as a co-stimulatory membrane protein under CRP stimulation, is associated with the activation of CD32b signaling, and may serve as an important promoter of the pathogenesis of DN. Indeed, our data also found that inhibition of DPP4 significantly reduced the kidney-infiltrating B cells in CRPtg db/db mice in vivo and activation of the SDF-1α (CXCL12)/CXCR4 pathway40 in CRP-stimulated HK-2 cells in vitro. The underlying implications would be worth further investigation.
Mechanically, we identified the promoting role of CRP-induced CD32b/NF-κB signaling in the transcriptional regulation of DPP4 expression in the human tubular epithelial cells in vitro, where CRP enhanced the direct binding of the p65 subunit of NF-κB on the promoter region of DPP4 at the genomic level to promote its expression. Surprisingly, we found that the CRP/ CD32b/NF-κB-induced DPP4 dimerized with CD32b at the membrane of renal cells under the CRP-rich condition to maintain the activation and expression of CD32b. Eventually, we found that inhibition of CD32b, NF-κB, and DPP4 individually was also able to alter the expression and activation of the others, demonstrating a novel DPP4/NF-κB/CD32b signaling circuit for CRP-driven T2DN. All of these findings revealed that DPP4 is a crucial regulator for promoting NF-κB activation together with CD32b signaling, and therefore accelerating the development of T2DN via enhancing renal inflammation in the diabetic kidney. More importantly, we demonstrated that targeting DPP4 with an inhibitor is capable of blocking the DPP4/CD32b/NF-κB signaling circuit in the diseased kidney of CRPtg db/db mice, resulting in an improvement of renal dysfunction and renal inflammation and fibrosis. Our results provided evidence and important clinical rationale that targeting of DPP4 may represent a precise therapeutic strategy for T2DN.
Taken together, the present study identified a new pathogenic role of DPP4 in CRP-driven T2DN via controlling a renal DPP4/CD32b/NF-κB signaling circuit. Targeting of DPP4 may represent a novel therapeutic strategy for CRP-driven DN.
Materials and Methods
Animal Model
db/db mice overexpressing human CRP (CRPtg-db/db) were chosen in this study. CRPtg-db/db mice were generated by crossbreeding db/m (C57BL/KSJ background) mice with human CRP transgenic mice (C57BL/6J background) as in our previous study.12,30 Meanwhile, db/m, CRPtg-db, and WT-db/db served as control groups. The genotypes of mice were determined by genotyping as previous described. All mice were fed in Laboratory Animal Services Centre, the Chinese University of Hong Kong and were maintained in a standard animal house with 12 h/12 h light/dark cycle. CRPtg-db/db mice were treated with linagliptin, 3 mg/kg/day for each mouse, from the age of 12 weeks, and all mice were sacrificed at the age of 24 weeks via injection of ketamine/xylene.19 All studies were approved by the Animal Experimentation Ethics Committee, the Chinese University of Hong Kong, and the experimental methods were carried out in accordance with the approved guidelines.
Histology and IHC
Kidneys sections were fixed in 4% paraformaldehyde and stained with the PAS, PASM, and Masson’s trichrome staining method. IHC was carried out by using a microwave-based antigen retrieval technique.30,41 The primary antibodies used in this study included DPP4 (sc-52642; Santa Cruz) and CD32b (sc-166711; Santa Cruz). Other antibodies were used in the current study as previously described.41,42 The nuclei were counterstained with hematoxylin. The positive cells were counted under the microscope power field in 10 random areas of kidney tissues with the expected cells per millimeter, and the percentage of positive staining areas was quantified using the Image-Pro Plus software (Media Cybernetics, Bethesda, MD, USA) in 10 consecutive fields.
Western Blot Analysis
Protein from renal cortex and HK-2 cells was extracted using the radioimmunoprecipitation assay (RIPA) lysis buffer. Western blot analysis was performed as described previously.41,43 Antibodies used in the current study include DPP4 (sc-52642; Santa Cruz) and CD32b (MAB14601; R&D), and other antibodies involved, such as fibronectin, collagen I, collagen IV, α-smooth muscle actin, phospho-NF-kB/p65, NF-kB/p65, phospho-Smad3, Smad3, and β-actin, were described previously. Then IRDye800-conjugated secondary antibodies (Rockland Immunochemicals, Gilbertsville, PA, USA) were used as secondary antibodies. Signals were detected using the Li-Cor/Odyssey infrared image system (LI-COR Biosciences, Lincoln, NE, USA), followed by quantitative analysis using the ImageJ program.44
RNA Extraction and Quantitative Real-Time PCR
Total RNA was extracted from the renal cortical tissues and cultured HK-2 cells. Real-time PCR was carried out with a machine (Option 2; Bio-Rad, Hercules, CA, USA) by using IQ SYBR green supermix reagent (Bio-Rad).45, 46, 47 The housekeeping gene β-actin was used as internal control. Primer sequences were: mouse DPP4, forward 5′ TTGTGGATAGCAAGCGAGTTG 3′, reverse 5′ CACAGCTATTCCGCACTTGAA 3′; human DPP4, forward 5′ GCACGGCAACACATTGAA 3′, reverse 5′ TGAGGTTCTGAAGGCCTAAATC 3′; human CD32b, forward 5′ ACTCATCCAAGCCTGTGACC 3′, reverse 5′ ATTGTGTTCTCAGCCCCAAC 3′; human SDF-1α, forward 5′ ACTCCAAACTGTGCCCTTCA 3′, reverse 5′ CCACTTTAGCTTCGGGTCAAT 3′; human CXCR4, forward 5′ CCCTCCTGCTGACTATTCCC 3′, reverse 5′ TAAGGCCAACCATGATGTGC 3′. The ratio of specific mRNA to β-actin mRNA was calculated using the 2−ΔCt method and is expressed as the mean ± SEM.
Cell Culture
The normal adult human TEC line (HK-2 cells) and mTECs were cultured in DMEM/F12 (GIBCO, CA, USA), supplemented with 10% FBS (GIBCO, CA, USA) and 1% antibiotic/antimycotic solution (Life Technologies, USA). SV40-transformed mMCs (MES13) (ATCC, Manassas, VA, USA) were maintained in a 3:1 mixture of DMEM and Ham’s F-12 medium containing 5% FBS and 1% antibiotic/antimycotic solution (Life Technologies, Grand Island, NY, USA).42,48 We use anti-CD32b neutralizing antibody (5 μg/ml; R&D) to block CD32b, DPP4i (50 μg/mL; R&D) to downregulate DPP4 expression, and BAY11-7082 (10 μM) to inhibit the phosphorylation of NF-kB, respectively, for 2 h before addition of CRP (10 μg/mL) for 24 h. All of the experiments were repeated independently at least three times.
Renal Function Measurement
Twenty-four-hour urinary samples were collected in metabolic cages every 4 weeks from the age of 12 weeks. Urinary microalbumin was measured by competitive ELISA according to the manufacturer’s instructions (Exocell, PA, USA). Levels of blood and serum creatinine were determined accordingly with the enzymatic method (Stanbio Laboratories, TX, USA). Urinary albumin excretion was expressed as total urinary albumin/creatinine (μg/mg) as in a previous study.48
Immunofluorescence Staining
Immunofluorescence staining was performed with 5-μm periodate-lysine-paraformaldehyde (PLP)-fixed frozen sections. Primary antibodies used for immunofluorescence staining included DPP4 (sc-52642; Santa Cruz) and CD32b (sc-28842; Santa Cruz), followed by RHO-conjugated anti-mouse and fluorescein isothiocyanate (FITC)-conjugated anti-rabbit secondary antibodies, respectively.49 All slides were mounted with DAPI-containing mounting medium and then analyzed with a fluorescence microscope (Leica Microsystems, Wetzlar, Germany).
Immunoprecipitation
HK-2 cells were extracted in ice-cold RIPA lysis buffer containing protease inhibitor cocktail. Primary antibodies, including DPP4 (sc-52642; Santa Cruz) and CD32b (sc-28842; Santa Cruz), were added into the equal lysis buffer supernatant. After incubating for 4 h at 4°C, the immune complex is captured, or precipitated, on a beaded support to which an antibody-binding protein is immobilized (such as protein A or G). Finally, the immune complexes were washed with lysis buffer three times and then boiled in Laemmli SDS sample buffer for 5 min, followed by western blotting as described above.50
ChIP Analysis
ChIP was performed with Simple Chip Enzymatic Chromatin IP Kit (magnetic beads) (#9003; Cell Signaling, Danvers, MA, USA) as previously described.42,51 The NF-κB binding site on DPP4 genomic sequence was identified by the ECR browser (https://ecrbrowser.dcode.org/) as in our previous study.52 Immunoprecipitation was performed with the antibody against NF-κB (#8242; Cell Signaling) or a normal IgG as negative control. Precipitated DNA fragments were detected by PCR using a specific primer of the promoter region of DPP4: forward 5′-AACCCAGGACTCCGTCTCTT-3′, reverse 5′-AGCACCTGGGAAAAAGTCAA-3′.
Statistical Analysis
All of the data are expressed as mean ± SEM. Statistical analyses were performed with one-way analysis of variance (ANOVA), followed by Newman-Keuls multiple comparison from GraphPad Prism 5.0 (GraphPad Software, San Diego, CA, USA). In addition, a repeated ANOVA was used for albumin excretion analysis.
Author Contributions
P.M.-K.T., Y.-Y.Z., and H.-Y.L. designed the experiments. P.M.-K.T., Y.-Y.Z., J.S.-C.H., and J.Y.-F.C. carried out the experiments. X.-R.H. supervised the animal models. P.M.-K.T. and Y.-Y.Z. wrote the manuscript. P.M.-K.T. and J.Y.-F.C. revised the manuscript. H.-Y.L. supervised the whole study.
Declaration of Interests
The authors declare no competing interests.
Acknowledgments
This work is supported by grants from the Health and Medical Research Fund (0314048, 05161326, 06173986, and 14152321), Lui Che Woo Institute of Innovative Medicine (CARE program), Research Grants Council of Hong Kong (RGC; 14106518, 14111019, 14111720, 14121816, 14163317, 14117418, 14104019, C7018-16G, R4012-18), Faculty Innovation Award and Direct Grant for Research of The Chinese University of Hong Kong CUHK (4620528 and 4054510), Lui Che Woo Institute of Innovative Medicine (CARE program), and The Guangdong-Hong Kong-Macao-Joint Labs Program from Guangdong Science and Technology (2019B121205005).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.08.017.
Contributor Information
Patrick Ming-Kuen Tang, Email: patrick.tang@cuhk.edu.hk.
Hui-Yao Lan, Email: hylan@cuhk.edu.hk.
Supplemental Information
References
- 1.Lin Y.C., Chang Y.H., Yang S.Y., Wu K.D., Chu T.S. Update of pathophysiology and management of diabetic kidney disease. J. Formos. Med. Assoc. 2018;117:662–675. doi: 10.1016/j.jfma.2018.02.007. [DOI] [PubMed] [Google Scholar]
- 2.de Boer I.H., Rue T.C., Cleary P.A., Lachin J.M., Molitch M.E., Steffes M.W., Sun W., Zinman B., Brunzell J.D., White N.H., Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications Study Research Group Long-term renal outcomes of patients with type 1 diabetes mellitus and microalbuminuria: an analysis of the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications cohort. Arch. Intern. Med. 2011;171:412–420. doi: 10.1001/archinternmed.2011.16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Donath M.Y., Shoelson S.E. Type 2 diabetes as an inflammatory disease. Nat. Rev. Immunol. 2011;11:98–107. doi: 10.1038/nri2925. [DOI] [PubMed] [Google Scholar]
- 4.Wada J., Makino H. Inflammation and the pathogenesis of diabetic nephropathy. Clin. Sci. (Lond.) 2013;124:139–152. doi: 10.1042/CS20120198. [DOI] [PubMed] [Google Scholar]
- 5.Overgaard A.J., McGuire J.N., Hovind P., Parving H.H., Rossing P., Pociot F. Serum amyloid A and C-reactive protein levels may predict microalbuminuria and macroalbuminuria in newly diagnosed type 1 diabetic patients. J. Diabetes Complications. 2013;27:59–63. doi: 10.1016/j.jdiacomp.2012.06.016. [DOI] [PubMed] [Google Scholar]
- 6.Zambrano-Galvan G., Rodríguez-Morán M., Simental-Mendía L.E., Lazalde B., Reyes-Romero M.A., Guerrero-Romero F. C-reactive protein is directly associated with urinary albumin-to-creatinine ratio. Arch. Med. Res. 2011;42:451–456. doi: 10.1016/j.arcmed.2011.09.009. [DOI] [PubMed] [Google Scholar]
- 7.Pradhan A.D., Manson J.E., Rifai N., Buring J.E., Ridker P.M. C-reactive protein, interleukin 6, and risk of developing type 2 diabetes mellitus. JAMA. 2001;286:327–334. doi: 10.1001/jama.286.3.327. [DOI] [PubMed] [Google Scholar]
- 8.Wang X., Bao W., Liu J., Ouyang Y.Y., Wang D., Rong S., Xiao X., Shan Z.L., Zhang Y., Yao P., Liu L.G. Inflammatory markers and risk of type 2 diabetes: a systematic review and meta-analysis. Diabetes Care. 2013;36:166–175. doi: 10.2337/dc12-0702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen F.Q., Wang J., Liu X.B., Ma X.Y., Zhang X.B., Huang T., Ma D.W., Wang Q.Y. Levels of inflammatory cytokines in type 2 diabetes patients with different urinary albumin excretion rates and their correlation with clinical variables. J. Diabetes Res. 2013;2013:138969. doi: 10.1155/2013/138969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang H.R., Chen D.L., Zhao M., Shu S.W., Xiong S.X., Gan X.D., Chao S.P., Cao J.L. C-reactive protein induces interleukin-6 and thrombospondin-1 protein and mRNA expression through activation of nuclear factor-κB in HK-2 cells. Kidney Blood Press. Res. 2012;35:211–219. doi: 10.1159/000332402. [DOI] [PubMed] [Google Scholar]
- 11.Liu F., Chen H.Y., Huang X.R., Chung A.C., Zhou L., Fu P., Szalai A.J., Lan H.Y. C-reactive protein promotes diabetic kidney disease in a mouse model of type 1 diabetes. Diabetologia. 2011;54:2713–2723. doi: 10.1007/s00125-011-2237-y. [DOI] [PubMed] [Google Scholar]
- 12.Li Z.I., Chung A.C., Zhou L., Huang X.R., Liu F., Fu P., Fan J.M., Szalai A.J., Lan H.Y. C-reactive protein promotes acute renal inflammation and fibrosis in unilateral ureteral obstructive nephropathy in mice. Lab. Invest. 2011;91:837–851. doi: 10.1038/labinvest.2011.42. [DOI] [PubMed] [Google Scholar]
- 13.Tang Y., Huang X.R., Lv J., Chung A.C., Zhang Y., Chen J.Z., Szalai A.J., Xu A., Lan H.Y. C-reactive protein promotes acute kidney injury by impairing G1/S-dependent tubular epithelium cell regeneration. Clin. Sci. (Lond.) 2014;126:645–659. doi: 10.1042/CS20130471. [DOI] [PubMed] [Google Scholar]
- 14.Zhang R., Zhang Y.Y., Huang X.R., Wu Y., Chung A.C., Wu E.X., Szalai A.J., Wong B.C., Lau C.P., Lan H.Y. C-reactive protein promotes cardiac fibrosis and inflammation in angiotensin II-induced hypertensive cardiac disease. Hypertension. 2010;55:953–960. doi: 10.1161/HYPERTENSIONAHA.109.140608. [DOI] [PubMed] [Google Scholar]
- 15.Paul A., Ko K.W., Li L., Yechoor V., McCrory M.A., Szalai A.J., Chan L. C-reactive protein accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Circulation. 2004;109:647–655. doi: 10.1161/01.CIR.0000114526.50618.24. [DOI] [PubMed] [Google Scholar]
- 16.Abd El Motteleb D.M., Elshazly S.M. Renoprotective effect of sitagliptin against hypertensive nephropathy induced by chronic administration of L-NAME in rats: role of GLP-1 and GLP-1 receptor. Eur. J. Pharmacol. 2013;720:158–165. doi: 10.1016/j.ejphar.2013.10.033. [DOI] [PubMed] [Google Scholar]
- 17.Alter M.L., Ott I.M., von Websky K., Tsuprykov O., Sharkovska Y., Krause-Relle K., Raila J., Henze A., Klein T., Hocher B. DPP-4 inhibition on top of angiotensin receptor blockade offers a new therapeutic approach for diabetic nephropathy. Kidney Blood Press. Res. 2012;36:119–130. doi: 10.1159/000341487. [DOI] [PubMed] [Google Scholar]
- 18.Kanasaki K., Shi S., Kanasaki M., He J., Nagai T., Nakamura Y., Ishigaki Y., Kitada M., Srivastava S.P., Koya D. Linagliptin-mediated DPP-4 inhibition ameliorates kidney fibrosis in streptozotocin-induced diabetic mice by inhibiting endothelial-to-mesenchymal transition in a therapeutic regimen. Diabetes. 2014;63:2120–2131. doi: 10.2337/db13-1029. [DOI] [PubMed] [Google Scholar]
- 19.Sharkovska Y., Reichetzeder C., Alter M., Tsuprykov O., Bachmann S., Secher T., Klein T., Hocher B. Blood pressure and glucose independent renoprotective effects of dipeptidyl peptidase-4 inhibition in a mouse model of type-2 diabetic nephropathy. J. Hypertens. 2014;32:2211–2223. doi: 10.1097/HJH.0000000000000328. discussion 2223. [DOI] [PubMed] [Google Scholar]
- 20.Gupta S., Sen U. More than just an enzyme: Dipeptidyl peptidase-4 (DPP-4) and its association with diabetic kidney remodelling. Pharmacol. Res. 2019;147:104391. doi: 10.1016/j.phrs.2019.104391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Higashijima Y., Tanaka T., Yamaguchi J., Tanaka S., Nangaku M. Anti-inflammatory role of DPP-4 inhibitors in a nondiabetic model of glomerular injury. Am. J. Physiol. Renal Physiol. 2015;308:F878–F887. doi: 10.1152/ajprenal.00590.2014. [DOI] [PubMed] [Google Scholar]
- 22.Yang J., Campitelli J., Hu G., Lin Y., Luo J., Xue C. Increase in DPP-IV in the intestine, liver and kidney of the rat treated with high fat diet and streptozotocin. Life Sci. 2007;81:272–279. doi: 10.1016/j.lfs.2007.04.040. [DOI] [PubMed] [Google Scholar]
- 23.Chen Y.T., Wallace C.G., Yang C.C., Chen C.H., Chen K.H., Sung P.H., Chen Y.L., Chai H.T., Chung S.Y., Chua S. DPP-4 enzyme deficiency protects kidney from acute ischemia-reperfusion injury: role for remote intermittent bowel ischemia-reperfusion preconditioning. Oncotarget. 2017;8:54821–54837. doi: 10.18632/oncotarget.18962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lan H.Y. Transforming growth factor-β/Smad signalling in diabetic nephropathy. Clin. Exp. Pharmacol. Physiol. 2012;39:731–738. doi: 10.1111/j.1440-1681.2011.05663.x. [DOI] [PubMed] [Google Scholar]
- 25.Pérez-Morales R.E., Del Pino M.D., Valdivielso J.M., Ortiz A., Mora-Fernández C., Navarro-González J.F. Inflammation in Diabetic Kidney Disease. Nephron. 2019;143:12–16. doi: 10.1159/000493278. [DOI] [PubMed] [Google Scholar]
- 26.Blagosklonny M.V. TOR-centric view on insulin resistance and diabetic complications: perspective for endocrinologists and gerontologists. Cell Death Dis. 2013;4:e964. doi: 10.1038/cddis.2013.506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lloberas N., Cruzado J.M., Franquesa M., Herrero-Fresneda I., Torras J., Alperovich G., Rama I., Vidal A., Grinyó J.M. Mammalian target of rapamycin pathway blockade slows progression of diabetic kidney disease in rats. J. Am. Soc. Nephrol. 2006;17:1395–1404. doi: 10.1681/ASN.2005050549. [DOI] [PubMed] [Google Scholar]
- 28.Liu X., Men P., Wang B., Cai G., Zhao Z. Effect of dipeptidyl-peptidase-4 inhibitors on C-reactive protein in patients with type 2 diabetes: a systematic review and meta-analysis. Lipids Health Dis. 2019;18:144. doi: 10.1186/s12944-019-1086-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lai W., Tang Y., Huang X.R., Ming-Kuen Tang P., Xu A., Szalai A.J., Lou T.Q., Lan H.Y. C-reactive protein promotes acute kidney injury via Smad3-dependent inhibition of CDK2/cyclin E. Kidney Int. 2016;90:610–626. doi: 10.1016/j.kint.2016.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.You Y.K., Huang X.R., Chen H.Y., Lyu X.F., Liu H.F., Lan H.Y. C-Reactive Protein Promotes Diabetic Kidney Disease in db/db Mice via the CD32b-Smad3-mTOR signaling Pathway. Sci. Rep. 2016;6:26740. doi: 10.1038/srep26740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tiruppathi C., Miyamoto Y., Ganapathy V., Roesel R.A., Whitford G.M., Leibach F.H. Hydrolysis and transport of proline-containing peptides in renal brush-border membrane vesicles from dipeptidyl peptidase IV-positive and dipeptidyl peptidase IV-negative rat strains. J. Biol. Chem. 1990;265:1476–1483. [PubMed] [Google Scholar]
- 32.Klemann C., Wagner L., Stephan M., von Hörsten S. Cut to the chase: a review of CD26/dipeptidyl peptidase-4's (DPP4) entanglement in the immune system. Clin. Exp. Immunol. 2016;185:1–21. doi: 10.1111/cei.12781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu Q., Jiang C.Y., Chen B.X., Zhao W., Meng D. The association between high-sensitivity C-reactive protein concentration and diabetic nephropathy: a meta-analysis. Eur. Rev. Med. Pharmacol. Sci. 2015;19:4558–4568. [PubMed] [Google Scholar]
- 34.Hayashino Y., Mashitani T., Tsujii S., Ishii H., Diabetes Distress and Care Registry at Tenri Study Group Serum high-sensitivity C-reactive protein levels are associated with high risk of development, not progression, of diabetic nephropathy among Japanese type 2 diabetic patients: a prospective cohort study (Diabetes Distress and Care Registry at Tenri [DDCRT7]) Diabetes Care. 2014;37:2947–2952. doi: 10.2337/dc14-1357. [DOI] [PubMed] [Google Scholar]
- 35.Fu C.C., Wu D.A., Wang J.H., Yang W.C., Tseng C.H. Association of C-reactive protein and hyperuricemia with diabetic nephropathy in Chinese type 2 diabetic patients. Acta Diabetol. 2009;46:127–134. doi: 10.1007/s00592-008-0069-0. [DOI] [PubMed] [Google Scholar]
- 36.Deacon C.F., Johnsen A.H., Holst J.J. Degradation of glucagon-like peptide-1 by human plasma in vitro yields an N-terminally truncated peptide that is a major endogenous metabolite in vivo. J. Clin. Endocrinol. Metab. 1995;80:952–957. doi: 10.1210/jcem.80.3.7883856. [DOI] [PubMed] [Google Scholar]
- 37.Matsui T., Nakashima S., Nishino Y., Ojima A., Nakamura N., Arima K., Fukami K., Okuda S., Yamagishi S. Dipeptidyl peptidase-4 deficiency protects against experimental diabetic nephropathy partly by blocking the advanced glycation end products-receptor axis. Lab. Invest. 2015;95:525–533. doi: 10.1038/labinvest.2015.35. [DOI] [PubMed] [Google Scholar]
- 38.Nakashima S., Matsui T., Takeuchi M., Yamagishi S.I. Linagliptin blocks renal damage in type 1 diabetic rats by suppressing advanced glycation end products-receptor axis. Horm. Metab. Res. 2014;46:717–721. doi: 10.1055/s-0034-1371892. [DOI] [PubMed] [Google Scholar]
- 39.Hasan A.A., Hocher B. Role of soluble and membrane-bound dipeptidyl peptidase-4 in diabetic nephropathy. J. Mol. Endocrinol. 2017;59:R1–R10. doi: 10.1530/JME-17-0005. [DOI] [PubMed] [Google Scholar]
- 40.Takashima S., Fujita H., Fujishima H., Shimizu T., Sato T., Morii T., Tsukiyama K., Narita T., Takahashi T., Drucker D.J. Stromal cell-derived factor-1 is upregulated by dipeptidyl peptidase-4 inhibition and has protective roles in progressive diabetic nephropathy. Kidney Int. 2016;90:783–796. doi: 10.1016/j.kint.2016.06.012. [DOI] [PubMed] [Google Scholar]
- 41.Sun S.F., Tang P.M.K., Feng M., Xiao J., Huang X.R., Li P., Ma R.C.W., Lan H.Y. Novel lncRNA Erbb4-IR Promotes Diabetic Kidney Injury in db/db Mice by Targeting miR-29b. Diabetes. 2018;67:731–744. doi: 10.2337/db17-0816. [DOI] [PubMed] [Google Scholar]
- 42.Zhou Q., Huang X.R., Yu J., Yu X., Lan H.Y. Long Noncoding RNA Arid2-IR Is a Novel Therapeutic Target for Renal Inflammation. Mol. Ther. 2015;23:1034–1043. doi: 10.1038/mt.2015.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tang P.M., Zhou S., Li C.J., Liao J., Xiao J., Wang Q.M., Lian G.Y., Li J., Huang X.R., To K.F. The proto-oncogene tyrosine protein kinase Src is essential for macrophage-myofibroblast transition during renal scarring. Kidney Int. 2018;93:173–187. doi: 10.1016/j.kint.2017.07.026. [DOI] [PubMed] [Google Scholar]
- 44.Tang P.M.-K., Zhang Y., Xiao J., Tang P.C.-T., Chung J.Y.-F., Li J., Xue V.W., Huang X.-R., Chong C.C.-N., Ng C.-F. Neural transcription factor Pou4f1 promotes renal fibrosis via macrophage–myofibroblast transition. Proc. Natl Acad. Sci. U S A. 2020;117:20741–20752. doi: 10.1073/pnas.1917663117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tang P.M., Zhou S., Meng X.M., Wang Q.M., Li C.J., Lian G.Y., Huang X.R., Tang Y.J., Guan X.Y., Yan B.P. Smad3 promotes cancer progression by inhibiting E4BP4-mediated NK cell development. Nat. Commun. 2017;8:14677. doi: 10.1038/ncomms14677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tang P.M., Tang P.C., Chung J.Y., Hung J.S.C., Wang Q.M., Lian G.Y., Sheng J., Huang X.R., To K.F., Lan H.Y. A Novel Feeder-free System for Mass Production of Murine Natural Killer Cells In Vitro. J. Vis. Exp. 2018;131 doi: 10.3791/56785. 56785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li C., Xue V.W., Wang Q.M., Lian G.Y., Huang X.R., Lee T.L., To K.F., Tang P.M., Lan H.Y. The Mincle/Syk/NF-κB Signaling Circuit Is Essential for Maintaining the Protumoral Activities of Tumor-Associated Macrophages. Cancer Immunol. Res. 2020;8:1004–1017. doi: 10.1158/2326-6066.CIR-19-0782. [DOI] [PubMed] [Google Scholar]
- 48.You Y.-K., Huang X.-R., Chen H.-Y., Lyu X.-F., Liu H.-F., Lan H.Y. C-Reactive Protein Promotes Diabetic Kidney Disease in db/db Mice via the CD32b-Smad3-mTOR signaling Pathway. Sci. Rep. 2016;6 doi: 10.1038/srep26740. 26740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang S., Meng X.M., Ng Y.Y., Ma F.Y., Zhou S., Zhang Y., Yang C., Huang X.R., Xiao J., Wang Y.Y. TGF-β/Smad3 signalling regulates the transition of bone marrow-derived macrophages into myofibroblasts during tissue fibrosis. Oncotarget. 2016;7:8809–8822. doi: 10.18632/oncotarget.6604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang Y., Huang J., Liu X., Niu Y., Zhao L., Yu Y., Zhou L., Lu L., Yu C. β-Arrestin-biased AT1R stimulation promotes extracellular matrix synthesis in renal fibrosis. Am. J. Physiol. Renal Physiol. 2017;313:F1–F8. doi: 10.1152/ajprenal.00588.2016. [DOI] [PubMed] [Google Scholar]
- 51.Lv L.L., Tang P.M., Li C.J., You Y.K., Li J., Huang X.R., Ni J., Feng M., Liu B.C., Lan H.Y. The pattern recognition receptor, Mincle, is essential for maintaining the M1 macrophage phenotype in acute renal inflammation. Kidney Int. 2017;91:587–602. doi: 10.1016/j.kint.2016.10.020. [DOI] [PubMed] [Google Scholar]
- 52.Zhou Q., Chung A.C., Huang X.R., Dong Y., Yu X., Lan H.Y. Identification of novel long noncoding RNAs associated with TGF-β/Smad3-mediated renal inflammation and fibrosis by RNA sequencing. Am. J. Pathol. 2014;184:409–417. doi: 10.1016/j.ajpath.2013.10.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.