

Summary
SNARE (soluble N-ethylmaleimide sensitive factor attachment protein receptor) complex composed of synaptobrevin, syntaxin and SNAP25 forms the essential fusion machinery for neurotransmitter release. Recent studies have reported several mutations in the gene encoding SNAP25 as a causative factor for developmental and epileptic encephalopathies of infancy and childhood with diverse clinical manifestations. However, it remains unclear how SNAP25 mutations give rise to these disorders. Here, we show that while structurally clustered mutations in SNAP25 give rise to related synaptic transmission phenotypes, specific alterations in spontaneous neurotransmitter release are a key factor to account for disease heterogeneity. Importantly, we identified a single mutation that augments spontaneous release without altering evoked release, suggesting that aberrant spontaneous release is sufficient to cause disease in humans.
Keywords: SNAP25, spontaneous release, neurotransmitter, synaptic transmission, exocytosis, synaptotagmin, epilepsy, development, developmental delay, child neurology
eTOC Blurb
Alten et al. show how SNAP25 variants give rise to clinically heterogeneous developmental and epileptic encephalopathies using structural and electrophysiological approaches. Their study identifies aberrant spontaneous neurotransmission as a culprit and suggests that therapies specifically targeting spontaneous release would be beneficial in treatment of these intractable disorders.
Introduction
Developmental and epileptic encephalopathies (DEEs) of infancy and childhood is a group of heterogeneous and treatment-resistant disorders characterized by developmental slowing as a direct result of either the epileptic activity, the underlying cause independent from the epileptic activity, or a combination of both (Berg et al., 2010; Scheffer et al., 2017). Recent work has identified de novo mutations in genes mostly coding for proteins involved in synaptic transmission leading to these disorders, cause of which were previously unknown (Claes et al., 2001; Euro et al., 2014; McTague et al., 2016; Thomas and Berkovic, 2014). Synaptic transmission relies on evoked release, in which depolarization of presynaptic terminal by an invading action potential causes neurotransmitter release. SNARE (Soluble N-ethylmaleimide sensitive factor Attachment Protein Receptor) complex composed of synaptobrevin-2 on synaptic vesicle, and SNAP25 and syntaxin-1 on plasma membrane forms the essential fusion machinery for neurotransmitter release (Sollner et al., 1993a; Sollner et al., 1993b; Weber et al., 1998). With the advent of next-generation sequencing, many de novo mutations in the genes coding for SNAP25 (Deciphering Developmental Disorders, 2015, 2017; Fukuda et al., 2018; Gabriel, 2016; Hamdan et al., 2017; Heyne et al., 2018; Liang, 2018; Rohena et al., 2013; Shen et al., 2014), synaptobrevin-2 (Salpietro et al., 2019; Simmons, 2020), syntaxin-1 (Vardar et al., 2020; Wolking et al., 2019) and synaptotagmin-1 (Syt1) (Baker et al., 2018), the calcium sensor for fast synchronized release, have been identified in patients with DEEs. Among these, SNAP25 comprises the highest number of disease-causing mutations scattered throughout the SNARE complex, making SNAP25 an ideal model to explain how aberrant fusion machinery gives rise to DEEs with diverse clinical manifestations and varying severities. Here, we characterize SNAP25 variant-specific synaptic disturbances by structural, biochemical and electrophysiological approaches.
A total of 10 different heterozygous mutations in the evolutionarily conserved gene encoding SNAP25 has been reported to date (Table 1 and Fig S1). All of these mutations are de novo in origin with the exception of R59P, which was inherited from an asymptomatic mosaic father and shared by two affected siblings (Fukuda et al., 2018). Alternative splicing of exon 5 (consisting of residues R59 and I67) of SNAP25 produces two different isoforms, SNAP25a and SNAP25b, which differ in only 9 of 206 amino acids (Bark and Wilson, 1994). The expression levels of these isoforms vary with the developmental stage. SNAP25a is the predominant isoform during embryonic and early postnatal development, whereas SNAP25b becomes the predominant isoform after early postnatal development through adulthood (Bark et al., 1995). The R59P and I67N mutations are only present in the adult-predominant isoform SNAP25b (Fukuda et al., 2018; Shen et al., 2014). The clinical features vary greatly in terms of seizure type, treatment response to antiepileptic pharmacotherapy, severity of neurodevelopmental delay/intellectual disability and associated cerebellar ataxia and facial dysmorphisms. Of note, the R59P variant is associated with the mildest clinical phenotype (Fukuda et al., 2018), whereas the Q174X and I192N variants were lethal in the first year of life (Deciphering Developmental Disorders, 2015, 2017; Gabriel, 2016). It is unknown whether these disorders are due to a simple loss-of-function or due to SNAP25 variants exerting specific synaptic impairments and interfering with the normal functioning of the wild-type (WT) SNAP25 in a dominant-negative manner.
Table 1. Summary of patients with SNAP25-associated developmental and epileptic encephalopathies.
c. designates a location in the cDNA and p. designates a location in the predicted protein.
| p.Lys40Glu | p.Gly43Arg | p.Val48Phe | p.Leu50Ser | p.Arg59 Pro | p.Ile67Asn | p.Asp166Tyr | p.Gln174* | p.Ile192Asn | p.Ala199Gly | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| c.118A>G | c.127G>C | c.142G>T | c.149T>C | c.176G>C | c.200T>A | c.496G>T | c.520C>T | c.575T>A | c.596C>G | ||
| Sex and age | male / 7y | female / 2y5m | female / 15y | female / 5y10m | female / 11y2m | male / 2y5m | female / 11y | male / 23y | female / 11m | male / 1y | ND / 6y |
| Inheritance | de novo | de novo | de novo | de novo | inherited from mosaic father | de novo | de novo | de novo | de novo | de novo | |
| Seizure type | absence seizures | GTCS and absence seizures | GTCS and focal clonic seizures | infantile spasms and tonic seizures | GTCS, FS and myocloni c seizures | GTCS, FS and focal seizures with impaired awareness | blank stare and unresponsivenes s | GTCS and focal seizures with impaired awareness | GTCS | + but not specified | ND |
| Developmental delay | global | gross motor | global | + but not specified | language | motor clumsines s | global | global | ND | ND | global |
| Intellectual disability | ND | ND | + | + | − | − | + | + | ND | ND | + |
shows gain of a stop codon. Abbreviations: ND not described, GTCS general tonic clonic seizures, FS febrile seizures.
Results
Effects of SNAP25 haploinsufficiency on synaptic transmission
Since loss of one normal allele of SNAP25 is common to all patients, we first investigated the effects of SNAP25 haploinsufficiency on synaptic transmission. The absence of both alleles of SNAP25 is lethal perinatally (Washbourne et al., 2002), and SNAP25+/− (Het) mice have increased susceptibility to kainate-induced seizures even though they do not seize spontaneously (Corradini et al., 2014). Our group previously showed that SNAP25 Het mice showed no significant behavioral impairments except for a marked hypoactivity (Monteggia et al., 2019). To complement our previous behavioral studies, we obtained field recordings from acute hippocampal slices prepared from either WT or Het mice (Fig. 1A). With increasing stimulation intensities applied to Schaffer collaterals, we readily observed a comparable increase in the postsynaptic field potentials recorded from CA1 region of hippocampus in both WT and Het slices (Fig. 1B), suggesting that haploinsufficiency does not change the overall efficiency of evoked synaptic transmission. We observed a small increase in the paired pulse ratio in Het slices, classically associated with a small decrease in release probability (Fig. 1C). In addition, we did not detect alterations in the frequency nor the amplitude of the miniature spontaneous currents recorded from Het neurons (Fig. 1D, E). Given the severity and diversity of the clinical manifestations, we concluded that haploinsufficiency of SNAP25 is unlikely the sole explanation of the disease pathophysiology, but may aggravate the synaptic manifestations of individual SNAP25 variants. This conclusion is further supported by the clinical data from patients with heterozygous deletions of chromosome 20 including SNAP25. Among 7 patients whose phenotypes were reported in the DECIPHER database (Firth et al., 2009) at the time of the preparation of this manuscript, only one patient was reported to have seizures and only four had developmental delays and/or intellectual disability.
Figure 1. Effects of SNAP25 haploinsufficiency on synaptic transmission.
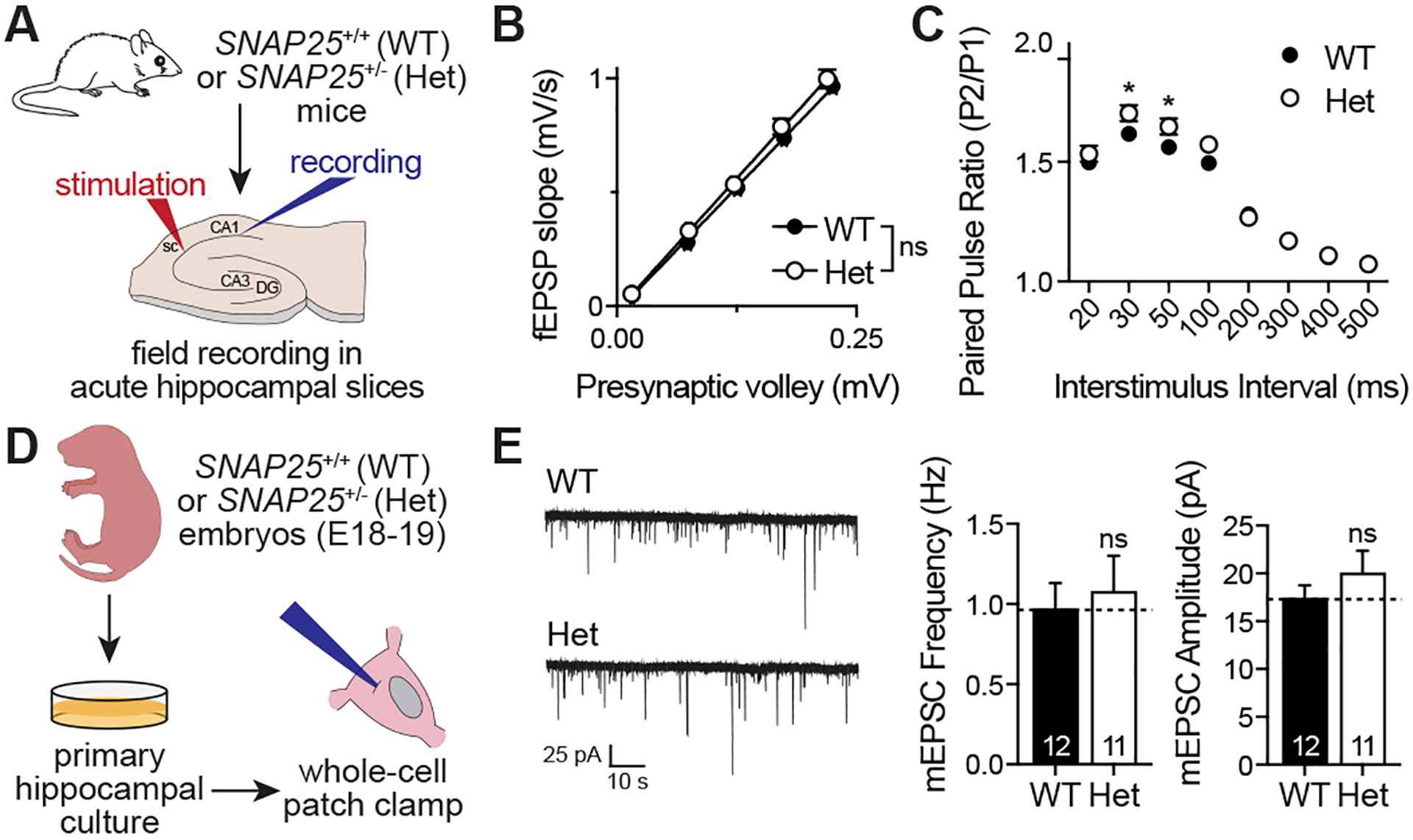
A. Experimental design for acute hippocampal slice field electrophysiology. B. Input-output curves are not statistically different between SNAP25 Het mice and littermate WTs (n= 13 slices from 3 mice/group, slopes of the curves were compared using simple linear regression module of Prism 8, F1,253=2.819, p>0.05). C. Paired pulse ratio was increased in hippocampal slices of SNAP25 Het mice compared to littermate WTs (n= 13 slices from 3 mice/group, compared by multiple t-tests corrected for multiple comparisons using the Holm-Sidak method, p<0.05 for interstimulus intervals of 30 and 50 ms). D. Experimental design for whole cell patch clamp electrophysiology. E. Representative mEPSC traces with quantitative analysis of mEPSC frequencies and amplitudes.
Structural investigation of SNAP25 variants
Synaptic SNAREs (Sollner et al., 1993a; Sollner et al., 1993b; Weber et al., 1998) SNAP25, syntaxin-1, and synaptobrevin-2 zipper into a highly stable complex composed of four parallel intertwined α-helices (each called a SNARE motif) held together by 15 interacting layers of hydrophobic side chains and a central ionic layer (Fig. 2A–B) (Sutton et al., 1998). Zippering of the individual SNAREs starts from the cytoplasmic N-terminus and proceeds towards the C-terminus in a step-wise manner, where it is coupled to fusion driven by the energy liberated from the formation of the complex (Gao et al., 2012; Lai et al., 2017; Ma et al., 2015; Sorensen et al., 2006). SNAP25 contributes two SNARE motifs to the complex and interacts with the Ca2+ sensor synaptotagmin for neurotransmitter release (Zhang et al., 2002; Zhou et al., 2015; Zhou et al., 2017). In particular, Syt1 is the primary Ca2+ sensor for evoked synchronous release with fast kinetics (Bacaj et al., 2013; Brose et al., 1992; Fernandez-Chacon et al., 2001; Geppert et al., 1994; Xu et al., 2007) and its C2B domain interacts with SNAP25 of SNARE complex extensively at the primary interface (Zhou et al., 2015; Zhou et al., 2017). Since the residues mutated in the K40E, V48F and D166Y variants reside in the primary interface, we grouped these variants as SNARE-Syt1 primary interface variants. Interestingly, all of the remaining variants were mutated in either one of the 15 hydrophobic layers or the central ionic layer (with the exception of R59P), emphasizing the importance of the hydrophobic interactions in SNARE-driven membrane fusion. Since the N- and C-terminal “sides” of the SNARE complex have differential functions (Gao et al., 2012; Sorensen et al., 2006; Zorman et al., 2014), we grouped these hydrophobic layer variants according to the side of the SNARE complex they reside in. Thus, G43R and L50S were grouped as N-terminal side hydrophobic layer variants, whereas I67N, I192N and A199G variants were grouped as C-terminal side hydrophobic layer variants. As the gain of a stop codon causes loss of the complete C-terminal side hydrophobic layers of the second SNARE motif of SNAP25 in addition to the central ionic layer, we also included Q174X to the group of C-terminal side hydrophobic layer variants. R59P is the only variant that does not impair a known interaction interface or the hydrophobic interactions of the complex, however, it is expected to disrupt the regularity of the α-helical backbone conformation, as proline cannot donate a hydrogen bond to stabilize an α-helix. Thus, we categorized it as the C-terminal side non-hydrophobic layer variant.
Figure 2. Structural characterization of SNAP25 variants.
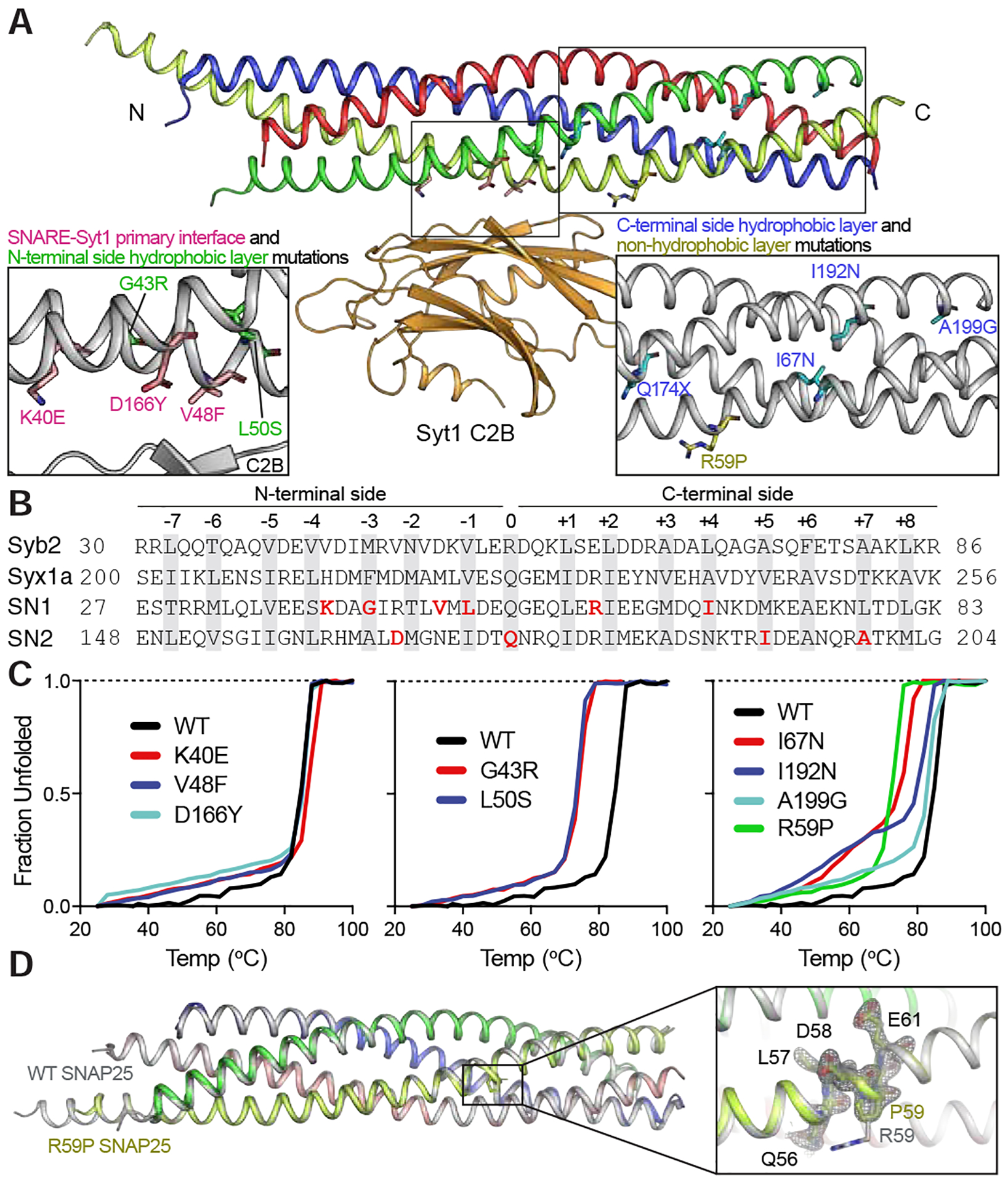
A. Structure of the SNARE complex and its primary interface with C2B domain of Syt1 (synaptobrevin-2 (syb2) blue, syntaxin-1a (syx1a) red, SNAP25 SN1 yellow, SNAP25 SN2 green, Syt1 C2B orange). Mutated residues are marked in insets for better visualization. B. Amino acid sequence map of individual SNARE motifs forming the ternary SNARE complex. 15 hydrophobic layers and the central ionic layer are highlighted in grey, whereas mutated SNAP25 residues are shown in red. C. Thermal melting curves using CD spectroscopy observed at 220 nm. D. Crystal structure of the ternary SNARE complex formed with the R59P SNAP25 variant (in color) superimposed on the complex formed with WT SNAP25 (in grey, PDB ID 1N7S). The omit map (mFo-DFc electron density map) of the residue region 56–61 from SNAP25 countered at 2.5 σ (grey mesh). See also supplementary table 1 and supplementary movies 1 and 2.
Next, we assayed whether the SNAP25 variants are able to form SNARE complexes with the other cognate SNAREs syntaxin-1A and synaptobrevin-2/VAMP2, and if so, how stable the formed complexes are. We used a co-expression system to express all four SNARE motifs of aforementioned SNAREs in Escherichia coli, thus leading to the complex formation in the bacterial host (Zhou et al., 2015). After purification of the formed complexes, with an exception of the complex formed with the Q174X variant which failed to purify, we measured the melting temperature at which the SNARE complex loses alpha-helicity using circular dichroism (CD) spectroscopy, a measure of the stability of the complex (Fig. 2C and Fig. S2). As expected, the SNARE-Syt1 primary interface variants do not change the melting temperature of the complex, consistent with the structure since the side chains of the mutated residues are facing outwards and do not participate in the SNARE complex core. The N-terminal side hydrophobic layer variants, namely G43R and L50S, reduced the melting temperature, followed by the C-terminal side hydrophobic layer variants I67N, I192N and A199G. The R59P variant impaired the SNARE complex stability the most, which is not surprising given that the proline substitution is considered to be an α-helix breaker. However, proline is occasionally found in the helices of both transmembrane (Peralvarez-Marin et al., 2006) and soluble (Karpusas et al., 1990) proteins, where it may cause helix bending and distort ideal helix geometry. If there was a substantial disruption of the structure of the ternary SNARE complex by interruption or distortion of one of the helices, one would expect a severe effect on function. Surprisingly, however, the R59P variant caused the mildest phenotype among all SNAP25 variants. To explain this observation, we determined the crystal structure of the R59P variant containing SNARE complex (Table S1). Consistent with the mild phenotype, the R59P variant retains the α-helical conformation of the SNARE motif of SNAP-25 with the mutant, and consequently, the structure of the mutant SNARE complex is similar to that of wildtype SNARE complex without distortion by the R59P mutation (Fig. 2D, supplementary movies 1 and 2). Considering that the overall geometry of the four-helix bundle is not affected in the R59P variant containing SNARE complex, the force available from C-terminal zippering may not be greatly affected by the mutation.
Effects of SNAP25 variants on evoked release
To elucidate how these SNAP25 variants impact action potential triggered evoked neurotransmitter release, we expressed mutants using a lentiviral expression system on neuronal cultures prepared from SNAP25 knock out (KO) embryos. First, we immunostained variant-expressing KO neurons against both SNAP25 and synaptic marker synapsin-1 to show that all of the variants were expressed and trafficked normally (Fig S3). In addition, to investigate their potential dominant-negative effects, we overexpressed the variants using the same approach but on WT cultures (Fig. 3A). As control, we overexpressed WT SNAP25 on WT cultures and showed that the overexpression of WT SNAP25 is electrophysiologically indistinguishable from WT cultures with endogenous SNAP25 only (Fig S4). Since SNAP25 functions similarly in both excitatory and inhibitory neurotransmission (Bronk et al., 2007), we recorded evoked inhibitory postsynaptic currents (eIPSCs) to prevent recurrent excitation from contaminating the excitatory recordings (Maximov et al., 2007). SNAP25 KO virtually abolished the evoked release, whereas WT SNAP25 was able to rescue the evoked release with fast kinetics as expected (Fig. 3) (Bronk et al., 2007). SNARE-Syt1 primary interface variants were able to rescue the KO phenotype however the eIPSC amplitudes were smaller with slower desynchronized kinetics compared to WT, consistent with the premise that these mutants impair Syt1 interaction (Fig. 3B and Fig. S5). When expressed on WT cultures, all three SNARE-Syt1 primary interface variants exerted a dominant-negative effect on evoked release (Fig. 3C). Previous work showed that the SNARE-Syt1 interaction at the primary interface is essential for emergence of dominant negative effects, as corresponding mutations in Syt1 suppressed a dominant-negative effect on evoked release of the so-called DN mutation of Syt1 C2B (Syt1-DN) in Drosophila (Guan et al., 2017). In response to 10 Hz repetitive stimulation, the K40E variant depressed less, consistent with a decrease in probability of release, whereas the V48F and D166Y variants depressed more, consistent with a small increase in probability of release (Fig. 3C). Among the N-terminal side hydrophobic layer variants, expression of the G43R variant showed a similar but a much milder electrophysiological phenotype compared to the K40E variant in all evoked release parameters assayed (Fig. 3D–E). Interestingly, expression of the L50S variant was able to rescue the KO phenotype of evoked release with fast kinetics, similar to the WT SNAP25, and did not exert any dominant-negative effects when expressed on WT cultures (Fig. 3D–E). Expression of the C-terminal side hydrophobic layer variants showed the most severe impairments in evoked release, consistent with the role of C-terminal side acting as a power stroke to finalize the fusion. The Q174X and I192N variants failed to rescue evoked release at all, the I67N variant was nearly non-functional, and the A199G variant retained some rescue ability (Fig. 3F). When expressed on WT cultures, Q174X exerted the greatest dominant-negative effect on evoked release, whereas the I67N, I192N, and A199G variants showed similar effects (Fig. 3G). R59P, the non-hydrophobic layer C-terminal side variant, was able to rescue evoked release and did not exert a dominant-negative effect, suggesting that force generated from the zippering of the hydrophobic layers of the C-terminal half is essential for the final stages of membrane fusion, while the thermostability of the C-terminal half is less important.
Figure 3. Effects of SNAP25 variants on evoked neurotransmission.
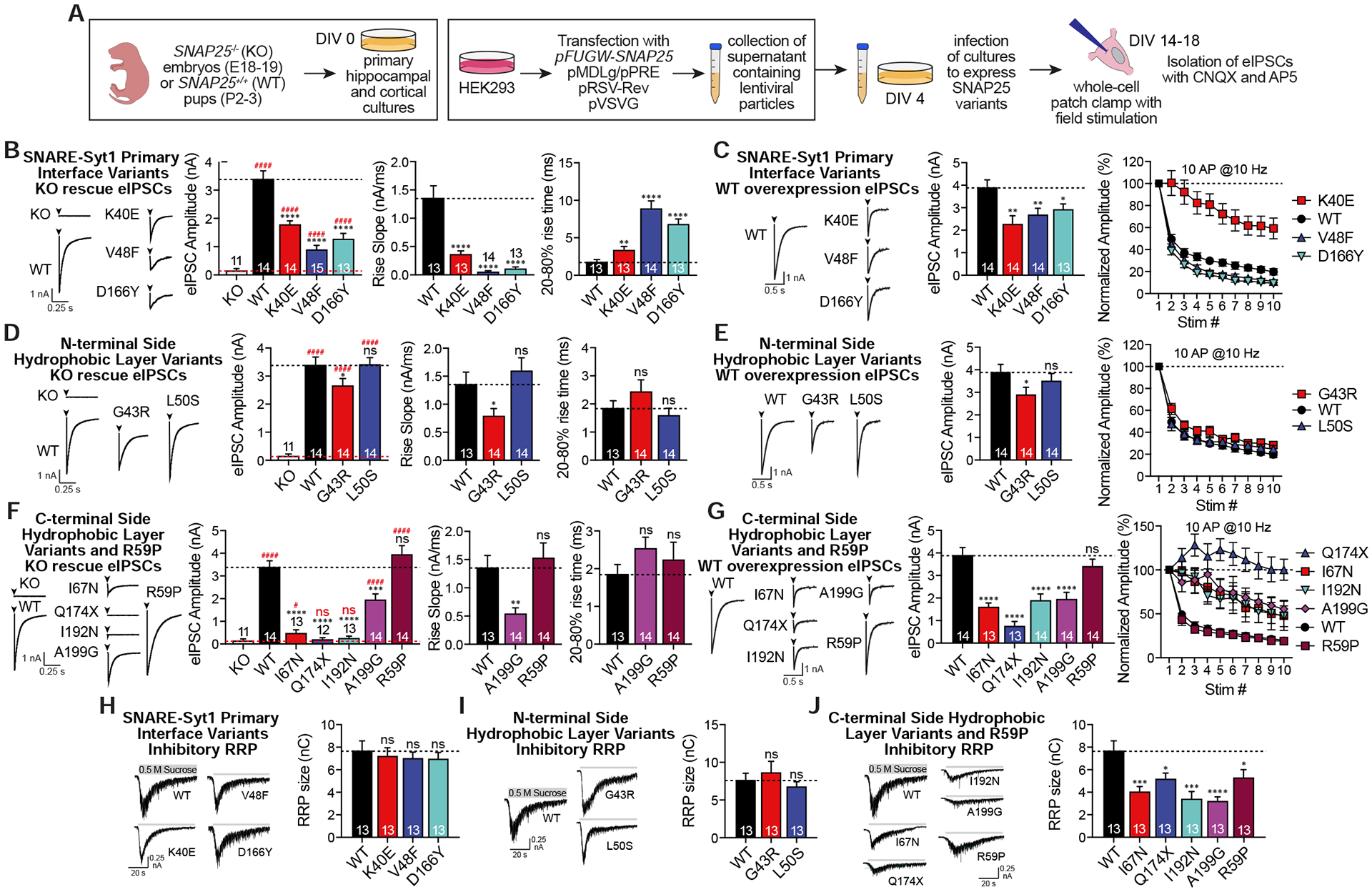
A. Experimental design. B-G. Representative eIPSC traces with the quantitative analysis of the amplitudes, rise slopes and 20–80% rise times of eIPSCs in response to single stimulation and eIPSC responses to repetitive 10 Hz 10 stimulations were presented for each group of SNAP25 variants expressed either on KO background or on WT background. SNAP25 variants were individually compared against either KO (red dashed line, significance denoted with #) or WT (black dashed line, significance denoted with *). Rise slopes and rise times of eIPSCs of the I67N, Q174X and I192N were not analysed since they are either not functional or nearly non-functional. H-J. Effects of SNAP25 variants overexpressed on WT cultures on the size of inhibitory readily releasable pool measured as a response to hypertonic sucrose perfusion.
Another important function of the SNARE complex assembly is to maintain a readily releasable pool (RRP) of vesicles which respond to Ca2+ influx induced by presynaptic depolarization. The aforementioned impairments in evoked release may arise from a decrease in RRP size, since the canonical SNAREs, including SNAP25 (Bronk et al., 2007), are necessary for the maintenance of the RRP size. In contrast, loss of both Syt1 and Syt7 is required to impair RRP size (Bacaj et al., 2015). Consistent with this premise, when overexpressed on WT cultures where Syt7 is abundant (Bacaj et al., 2013), the SNARE-Syt1 primary interface variants did not affect the size of the RRP measured by hyperosmotic stimulation, a standard Ca2+ influx-independent way to assess the RRP size (Fig. 3H) (Rosenmund and Stevens, 1996). Furthermore, N-terminal side hydrophobic layer variants did not impair RRP size (Fig. 3I), while C-terminal side variants, including R59P, significantly reduced it (Fig. 3J), suggesting that overall thermostability of the C-terminal side regardless of the layer mutated is crucial to maintain the RRP.
Effects of SNAP25 variants on spontaneous release
So far, we have shown that almost all of the SNAP25 variants impair either the evoked release or its associated readily releasable pool of vesicles, with the exception of L50S. This finding brings up two intriguing questions: (I) How do global impairments in evoked release give rise to this great heterogeneity in clinical manifestations? and (II) How does the L50S variant give rise to any kind of disease without affecting the action potential-triggered neurotransmitter release? To answer these questions, we focused on a less conventional mode of neurotransmitter release. Neurotransmitter containing synaptic vesicles can fuse with the presynaptic membrane spontaneously in the absence of action potentials in a quantal and regulated manner, hence called the spontaneous release (Kavalali, 2015). Spontaneous release shares certain key components of the fusion machinery with evoked release such as SNAP25 (Bronk et al., 2007). Even though the regulation and functions of spontaneous release are still a matter of ongoing research, it has been proposed to function in synaptic plasticity and homeostasis (Kavalali, 2015).
To examine the effects of SNAP25 variants on spontaneous neurotransmission, we utilized tetrodotoxin to block voltage-gated Na+ channels, preventing action potential firing (Fig. 4A). Similar to its effect on the evoked release, SNAP25 KO almost completely abolished the frequency of spontaneous release events, which was rescued by reintroduction of WT SNAP25 (Fig 4). Even though the identity of the Ca2+ sensor for spontaneous release is still a matter of debate, Syt1 is thought to suppress spontaneous release by clamping a second Ca2+ sensor, which is more sensitive to Ca2+ but has a lower affinity for SNARE complex (Courtney et al., 2018; Xu et al., 2009). All SNARE-Syt1 primary interface variants rescued spontaneous release, and the V48F and D166Y variants caused substantial augmentation of spontaneous release, up to ~40 fold (Fig. 4B), which is far greater than what has been previously reported in case of unclamping of spontaneous release by Syt1 loss-of-function (Bacaj et al., 2013). This trend was also observed when variants were expressed on WT cultures, exerting a dominant-positive effect in a similar fashion for both excitatory and inhibitory spontaneous miniature postsynaptic currents (Fig. 4C and Fig. S6A). We preincubated neurons expressing V48F and D166Y variants with BAPTA-AM, an exogeneous intracellular Ca2+ chelator, to examine whether this augmentation of spontaneous release is driven by an alternate Ca2+ sensor, similar to earlier studies in the case of Syt1 loss-of-function (Xu et al., 2009). However, BAPTA-AM preincubation was ineffective in reducing this robust increase in spontaneous release (Fig S6D), suggesting that the augmentation of spontaneous release by V48F and D166Y variants is due to an intrinsic increased propensity of spontaneous fusion, likely independent of an alternate Ca2+ sensor. The aforementioned DN-Syt1 caused enhanced spontaneous release as a dominant effect which failed to normalize upon disruption of the SNARE-Syt1 interaction at the primary interface through mutations of DN-Syt1 (Guan et al., 2017), further suggesting the V48F and D166Y variants do not simply elicit Syt1 loss-of-function effects to augment spontaneous release.
Figure 4. Effects of SNAP25 variants on spontaneous neurotransmission.
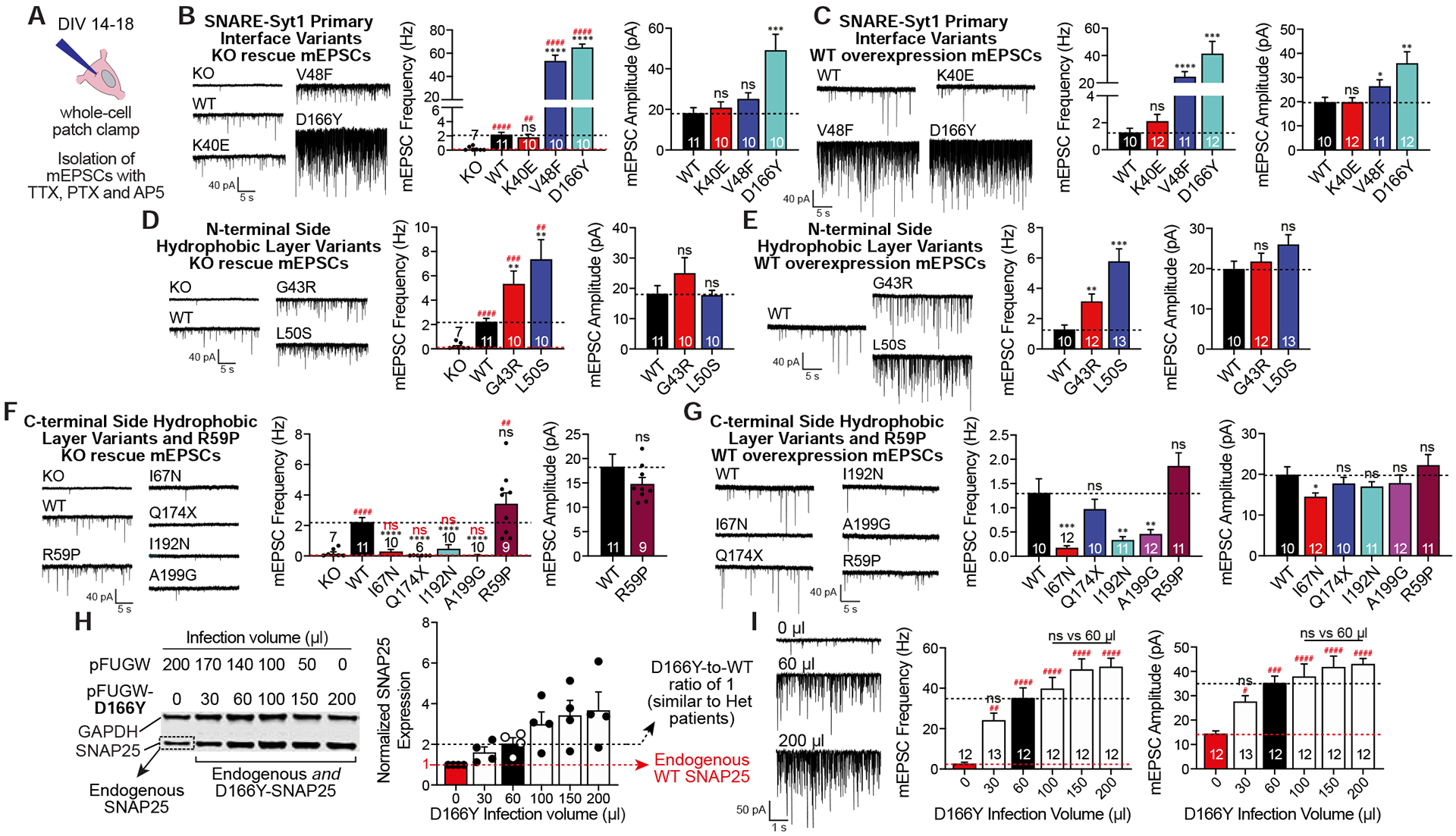
A. Experimental design was similar to that of evoked neurotransmission except for the drugs used to isolate mEPSCs. B-G. Representative mEPSC recordings with the quantitative analysis of the frequency and amplitudes of mEPSCs were presented for each group of SNAP25 variants expressed either on KO background or on WT background. SNAP25 variants were individually compared against either KO (red dashed line, significance denoted with #) or WT (black dashed line, significance denoted with *). Amplitudes of mEPSCs of the KO and the C-terminal side hydrophobic layer variants expressed on the KO cultures were not analysed since they did not have enough number of spontaneous release events. H. Quantification of the SNAP25 protein levels of the D166Y titration experiment. I. Analysis of mEPSC recordings from coverslips increasingly expressing the D166Y variant. mEPSC frequencies (F5, 67=18.71, p<0.0001) and amplitudes (F5, 67=11.17, p<0.0001) were compared using one-way ANOVA with Tukey’s multiple comparisons test. Red dashed line with # for significance denotes comparison against coverslips only expressing WT SNAP25 endogenously, whereas black dashed line denotes comparison against coverslips infected with 60 μl of supernatant (expressing 1:1 D166Y:WT SNAP25 mimicking the heterozygous patients).
Expression of the N-terminal side hydrophobic layer variants, G43R and L50S, rescued spontaneous release with increased frequency and exerted a dominant-positive effect on WT cultures (Fig. 4D–E and Fig. S6B). The L50S variant was associated with greater increase compared to the G43R variant. Interestingly, the augmentation of spontaneous release by these variants was shown to require Ca2+, suggested by their BAPTA-AM responsivity (Fig. S6D), possibly due to involvement of an alternate Ca2+ sensor. Given the close proximity of G43R and L50S to the primary interface and causing instability of the N-terminal side of the SNARE complex (Fig. 2), which houses the primary interface and other important interfaces for Syt1 interaction (Zhou et al., 2015; Zhou et al., 2017), we speculate that these variants may alter the Syt1 interaction with the SNARE complex indirectly, leading to impaired evoked release with slow kinetics in case of G43R, and unclamping of spontaneous release and unchanged RRP size in case of both G43R and L50S. The additional effects on evoked release of G43R variant can be explained either by the mutated residues lying in different hydrophobic layers or the more dramatic difference in the side chains of glycine (small and hydrophobic) vs arginine (large and positively charged) compared to a more subtle difference in case of the L50S variant.
None of the C-terminal side hydrophobic layer variants were able to rescue spontaneous release when expressed on KO cultures (Fig. 4F), consistent with C-terminal half providing a force for membrane fusion irrespective of the mode of release. I67N, I192N and A199G variants showed a dominant-negative effect on spontaneous release, whereas Q174X did not (Fig. 4G). The R59P variant retained the ability to rescue the spontaneous release similar to WT and showed no effect when expressed on WT cultures (Fig. 4F–G).
In order to investigate the relationship between the variant-to-WT ratio of SNAP25 and observed dominant-negative (or dominant-positive, as some variants augment spontaneous release) synaptic disturbances, we designed a simple titration experiment. The effect of D166Y on mEPSC frequency was the greatest among all parameters we studied so far, allowing enough room to conduct a sensitive titration. We utilized increasing infection volumes of non-tittered lentivirus from the same batch to gradually overexpress the D166Y variant on WT neuronal cultures and measured mEPSC frequency. We achieved the D166Y-to-WT ratio of 1, which mimics the expected expression pattern of heterozygous patients, when cultures were infected with 60 μl of supernatant (Fig 4H). Statistical analysis showed that mEPSC frequency is promptly saturated with increasing infection volumes and the mEPSC frequency obtained with 60 μl of infection volume is no different than frequencies obtained with the other infection volumes (Fig. 4I). This suggests that even a minor presence of mutant SNAP25 is sufficient for the dominant effects to emerge and we were able to mimic the synaptic phenotypes of patients when we overexpressed SNAP25 variants using WT neuronal cultures. For all the remaining variants, we achieved similar overexpression levels to the D166Y variant of the titration experiment (Fig. S4A) with the same amount of lentivirus, suggesting that the stability of variants is comparable with each other. However, the biallelic expression pattern in heterozygous patients may be different than the stoichiometric 1:1 ratio. Our result suggests that, even sub-stoichiometric expression of SNAP25 variants may lead to dominant effects similar to that of the stoichiometric expression.
We also recorded evoked excitatory postsynaptic currents (eEPSC) from WT neurons expressing the SNAP25 variants. To reduce the high background activity and recurrent excitation associated with eEPSC recordings, we lowered Ca2+ concentration as well as stimulus intensity. In this setting, we could show that all variants affect the evoked excitatory neurotransmission similar to their effects on evoked inhibitory neurotransmission, except for V48F and D166Y, where the decreasing trend in response amplitudes did not reach statistical significance (Fig S6E–G).
Effects of SNAP25 variants on synapse density
Even though the brain of SNAP25 KO mouse develops normally (Washbourne et al., 2002), the cultured KO neurons undergo neuronal death within couple weeks. Our group has previously showed that the SNAP25 KO high density cultures have a comparable synapse density with WT neurons on DIV14, but subsequent degeneration causes a 20% reduction in synapse density on DIV21 (Bronk et al., 2007). This finding suggests that SNAP25 is not essential for initial brain development but necessary for neuronal survival in vitro and in vivo (Hoerder-Suabedissen et al., 2019). Therefore, we performed all the functional studies between DIV14 and DIV18, when the synapses are mature, the expression of SNAP25 variants plateaued and synaptic loss due to SNAP25 loss-of-function is negligible. To address whether SNAP25 variants impact synapse numbers under our conditions, we quantified excitatory and inhibitory synapse densities. First, we showed that the expression of SNAP25 variants have already plateaued on DIV14 (Fig 5A). At the same time, we fixed another set of coverslips from the same set of cultures on DIV14 and co-immunostained with antibodies against the excitatory presynaptic protein vesicular Glutamate transport (vGlut) and excitatory postsynaptic density scaffold protein PSD-95 to quantify excitatory synapse density or inhibitory presynaptic marker vesicular GABA transporter (vGAT) and inhibitory postsynaptic marker gephyrin to quantify inhibitory synapse density. When we counted the number of co-localized puncta and normalized to MAP2 (dendritic marker) signal (Fig 5B), we found that expression of SNAP25 variants did not cause significant changes in either excitatory or inhibitory synapse density (Fig 5C–D), suggesting that the electrophysiological alterations are attributable to functional changes but not to changes in synapse density.
Figure 5. Effects on SNAP25 variants on excitatory and inhibitory synapse densities.
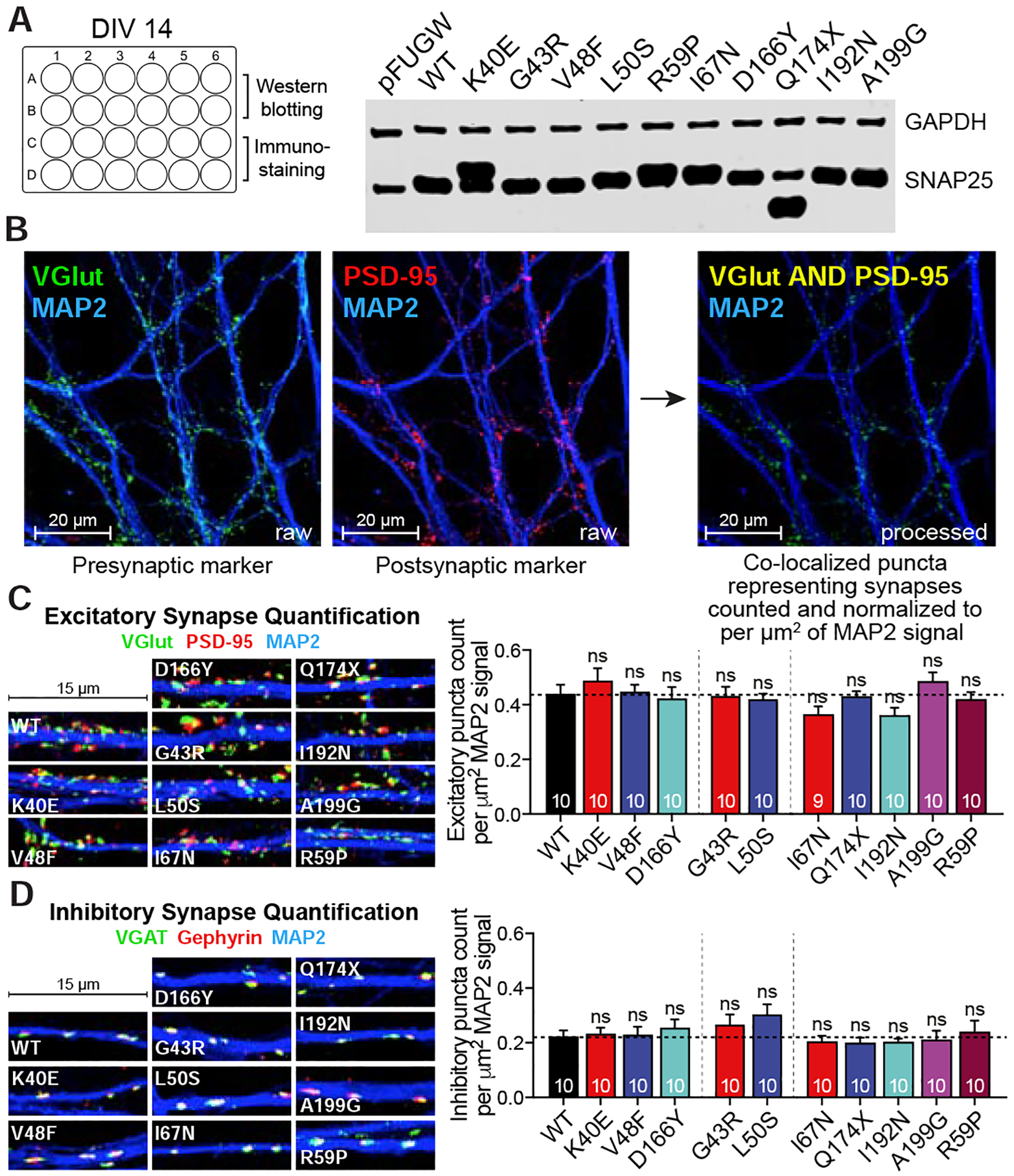
A. Experimental setup with immunoblotting of SNAP25 from samples collected from DIV14 cultures. B. Co-localized puncta representing either inhibitory or excitatory synapses were counted and normalized to MAP2 dendritic signal. C. Quantification of excitatory synapse density. Coverslips expressing SNAP25 variants are co-immunostained with VGlut (excitatory presynaptic marker, green), PSD-95 (excitatory postsynaptic marker, red) and MAP2 (dendritic marker, blue). D. Quantification of inhibitory synapse density. Coverslips expressing SNAP25 variants are co-immunostained with VGAT (inhibitory presynaptic marker, green), gephyrin (inhibitory postsynaptic marker, red) and MAP2 (dendritic marker, blue). Both inhibitory and excitatory puncta are identified using Intellicount software and puncta density per μm2 MAP2 signal of each SNAP25 variant is compared against the density of WT cultures (p>0.05 for all variants vs WT, two-tailed non-paired Student’s t-test).
Effects of variant-specific alterations on network activity
Plotting changes in amplitudes of evoked responses against changes seen in frequencies of spontaneous events reveals a distinct synaptic phenotype for each SNAP25 variant (Fig. 6A–B). Notably, the L50S variant augmented only the spontaneous release without impairing the evoked release whereas the V48F and D166Y variants augmented spontaneous release excessively in addition to relatively modest impairments in evoked release. In the hippocampal cultures, the frequency of mEPSCs is intrinsically higher than the frequency of mIPSCs. All variants augmenting spontaneous release retained this intrinsic ratio of excitatory-to-inhibitory spontaneous release, causing an absolute increase in the excitatory drive (Fig. 6C).
Figure 6. Synaptic phenotypes of SNAP25 variants.
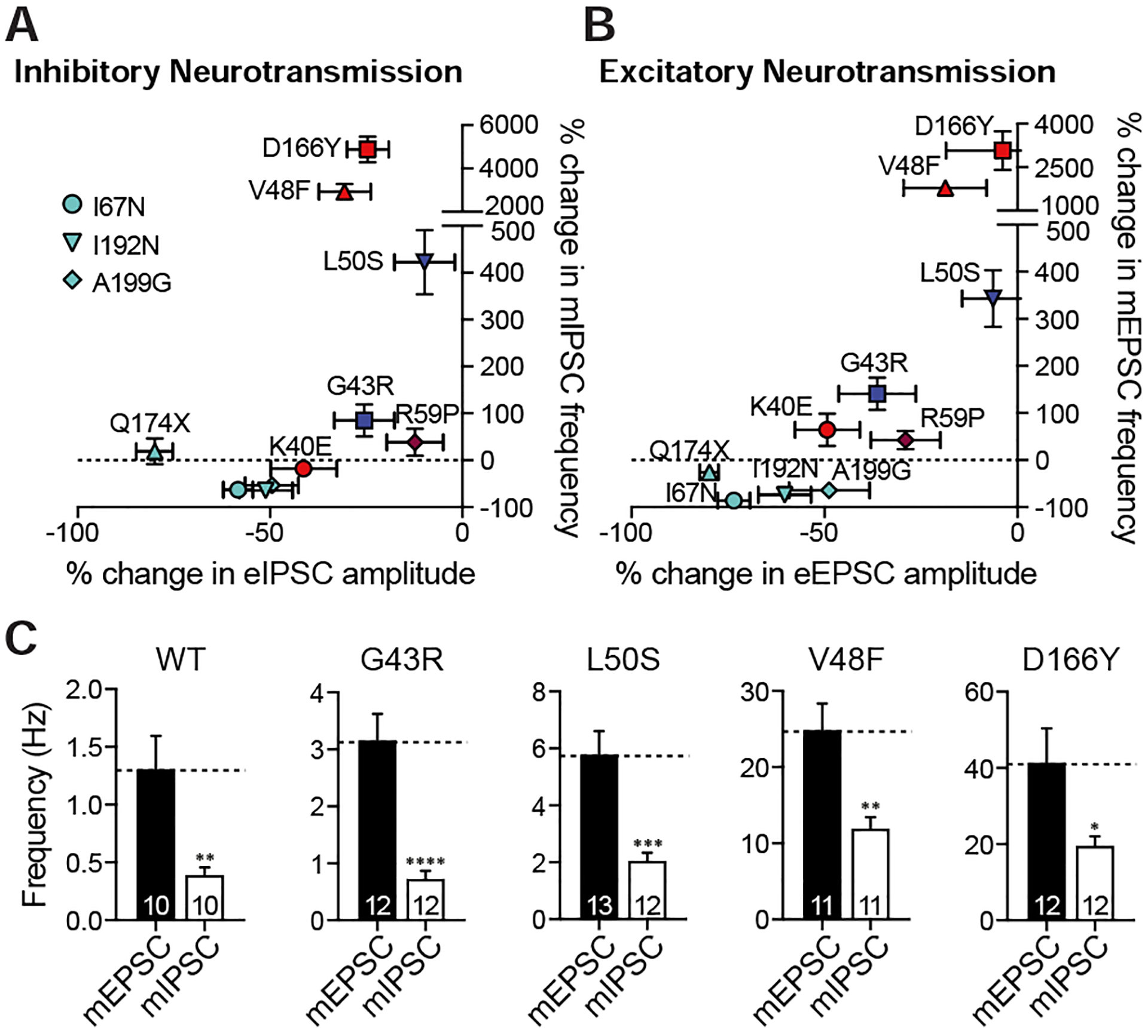
A. Plot of dominant % changes in inhibitory evoked amplitudes against inhibitory miniature frequencies for each SNAP25 variant expressed on WT cultures. B. Plot of dominant % changes in excitatory evoked amplitudes against excitatory miniature frequencies for each SNAP25 variant expressed on WT cultures. C. Excitatory (mEPSC) and inhibitory (mIPSC) miniature event frequencies compared to each other for variants increasing spontaneous release frequencies.
To explore the changes in network activity secondary to the synaptic disturbances we identified, we recorded spontaneous action potential (AP) firings from SNAP25 variant overexpressing WT neurons in whole-cell current clamp mode and analyzed their firing patterns, resting membrane potentials and kinetic characteristics of APs. The K40E variant, by simply impairing evoked release without altering spontaneous release, caused a shift toward a burst-like firing pattern (Fig. 7A) without altering the resting membrane potential. G43R, by impairing evoked release and mildly augmenting spontaneous release, caused a halfway change in the firing pattern between burst-like pattern of the K40E variant and of the WT (Fig. 7A).
Figure 7. Effects of SNAP25 variants on network activity.
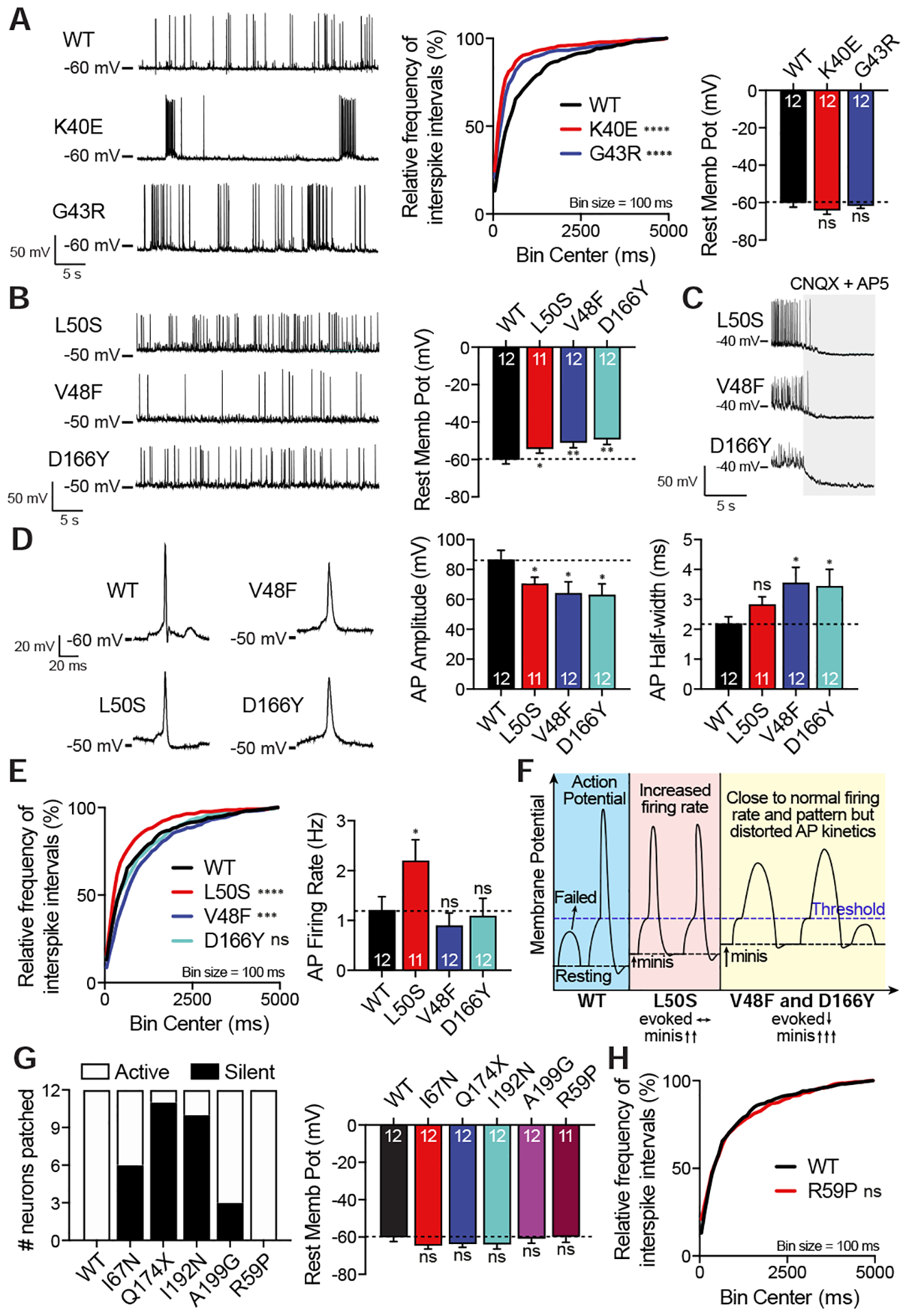
A. Representative traces of spontaneous AP firing patterns of WT SNAP25 and variants K40E and G43R in current-clamp mode along with cumulative histograms of interspike intervals and resting membrane potentials. Cumulative histograms of variants were compared against WT using Kolmogorov-Smirnov test (WT vs K40E Kolmogorov-Smirnov D=0.2762, p<0.0001 and WT vs G43R Kolmogorov-Smirnov D=0.2086, p<0.0001). B. Representative traces of spontaneous AP firings of variants L50S, V48F and D166Y in current-clamp mode along with resting membrane potentials. C. Hyperpolarization of the depolarized resting membrane potential associated with L50S, V48F and D166Y variants when perfused with CNQX (AMPA receptor blocker) and AP5 (NMDA receptor blocker) to block excitatory neurotransmission. D. Representative single action potentials along with the quantitative analysis of their amplitudes and half-width. E. Cumulative histograms of variants were compared against WT using Kolmogorov-Smirnov test (WT vs L50S Kolmogorov-Smirnov D=0.1872, p<0.0001; WT vs V48F Kolmogorov-Smirnov D=0.1337, p=0.0001 and WT vs D166Y Kolmogorov-Smirnov D=0.07846, p>0.05) and spontaneous AP firing rates were analysed. F. Proposed mechanism to explain changes associated with variants L50S, V48F and D166Y, all of which augments spontaneous neurotransmission significantly. G. Neuronal activity of C-terminal side variants were shown along with the resting membrane potentials and H. cumulative histogram of interspike intervals of the R59P variant.
The L50S, V48F and D166Y variants caused a more depolarized resting membrane potential (Fig. 7B), driven by the net increase in excitatory spontaneous neurotransmission (Fig. 7C). Since voltage-gated Na+ channels require a hyperpolarized resting membrane potential to recover from inactivation and respond to the next round of depolarization, this more depolarized resting membrane potential decreased the availability of voltage gated Na+ channels — and presumably voltage-gated K+ channels — leading to wider APs with smaller amplitudes (Fig. 7D). Their effects on membrane potential, AP amplitude and width were in parallel with their effects on spontaneous release, the D166Y variant causing the greatest disturbances followed by the V48F and L50S variants. In the case of the L50S variant, since evoked release is not impaired, subthreshold depolarizations can now trigger APs as the membrane potential is closer to the threshold, which may explain the increased AP firing rate (Fig. 7E–F). The V48F and D166Y variants were expected to have similar firing patterns to the K40E variant only when their effects on evoked release are taken into consideration. However, membrane depolarization due to augmented spontaneous release normalized the firing pattern and rate compared to the K40E variant (Fig. 7E–F). Interestingly, the D166Y variant showed a very similar firing pattern and firing rate relative to WT SNAP25, whereas the V48F variant was not able to normalize the firing pattern completely (Fig. 7E). These observations parallel the severity of clinical manifestations in between patients carrying the V48F and D166 variants. The patient with the V48F variant is globally delayed, nonverbal with severe intellectual disability and seizures being refractory to more than 8 different antiepileptics in up to 3 drug combinations (Rohena et al., 2013). In contrast, the patient with the D166Y variant has milder developmental delays and has caught up with his peers in many of the domains delayed initially, has milder intellectual disability, and seizures in control with a single antiepileptic (Hamdan et al., 2017).
All of the randomly selected neurons expressing the SNARE-Syt1 primary interface and N-terminal side hydrophobic layer variants fired at least one AP during the recording period of 3 mins. However, C-terminal side hydrophobic layer variants caused some of the neurons to be silent, suppressing the network activity (Fig. 7G). The Q174X and I192N variants almost completely silenced the network, which correlates with the fact that the patients carrying these variants died in the first year of life. The I67N variant resulted in half of the recorded neurons to be silent, whereas the A199G variant caused only one fourth of the neurons to be silent. Neither of the variants changed the membrane potential. As expected from the R59P variant, being able to carry out both evoked and spontaneous release similar to WT SNAP25, none of the recorded neurons were silent and they all showed a similar firing rate and pattern with WT (Fig. 7G–H). This finding also correlates with the R59P causing the mildest disease phenotype, probably contributed by haploinsufficiency and decreased RRP size.
Interestingly, wide variety of changes in the network activity driven by the changes in both evoked and spontaneous neurotransmission correlate with the heterogeneity and severity of the clinical manifestations of the patients, suggesting that besides evoked release, dysregulation of spontaneous release is a key factor in the phenotypic diversity among patients with SNAP25-associated encephalopathies.
Discussion
The diagnosis of developmental and epileptic encephalopathies of infancy and childhood is clinically challenging; therefore, their overall incidence is probably underestimated. Whole-exome sequencing and gene panels are offered to affected families to identify a genetic cause of these disorders in the hope of classifying patients into distinct subgroups of DEEs with specific electroclinical features, ultimately allowing application of tailored treatments. De novo mutations of the core fusion machinery components namely SNAP25, synaptobrevin-2 (Salpietro et al., 2019; Simmons, 2020), syntaxin-1 (Vardar et al., 2020; Wolking et al., 2019) and their key interaction partner synaptotagmin-1 (Baker et al., 2018; Bradberry et al., 2020), have been recently identified (Verhage and Sorensen, 2020). In 2018, only 3 out of 24 gene panels from academic and commercial providers for DEEs included SNAP25 and none included synaptobrevin-2 or synaptotagmin-1 (Heyne et al., 2018), showing a need to better recognize mutations of fusion machinery components as a cause of DEEs. We expect that further recognition of genes coding for components of fusion machinery and its interaction partners as a cause of DEEs would uncover more patients in the future.
In this study, we showed that heterozygous de novo mutations in SNAP25 lead to DEEs of infancy and childhood by impairing evoked release and/or modifying spontaneous release in both directions in a dominant manner, giving rise to distinct synaptic phenotypes contributing to the clinical heterogeneity. We found that structurally clustered mutations of SNAP25 give rise to related synaptic phenotypes by either impairing SNARE-synaptotagmin interactions or SNARE complex zippering that drives membrane fusion. Most importantly, we showed that specific dysregulation of the spontaneous release appears to be sufficient to cause disease, and to our knowledge first example implicating aberrant spontaneous neurotransmitter release as a cause of any disease. Identification of the L50S SNAP25 variant serves as a proof of concept that alterations in spontaneous release should not be overlooked in variants of SNARE complex components which alter spontaneous release in addition to impairing evoked release. However, we cannot fully exclude the possibility that L50S SNAP25 variant exerts additional effects on physiological processes other than neurotransmitter release, which may contribute to the disease phenotypes. Furthermore, some variants cause more profound alterations in spontaneous release compared to their effects on evoked release, such as V48F and D166Y. Given that these variants increase mEPSC amplitudes along with comparable eEPSC responses, potential postsynaptic effects of these variants may further augment the depolarizing effect of net increased excitatory spontaneous neurotransmission on postsynaptic membrane.
We show that each variant has a distinct synaptic phenotype leading to distinct network patterns, both of which correlate with the clinical heterogeneity and severity. However, one possibility is that mutations of the alternatively spliced exon of SNAP25, namely R59P and I67N, universally give rise to milder phenotypes secondary to availability of WT SNAP25a along with mutant SNAP25b. Quantification of SNAP25 isoforms in post-mortem adult human brains showed that SNAP25b is expressed at least 2 times higher than SNAP25a depending on the brain region (Prescott and Chamberlain, 2011), suggesting that mutant SNAP25b comprises approximately 70% of total SNAP25. Given that less than 1:1 stoichiometric expression of mutant SNAP25b is sufficient to cause emergence of dominant effects, the differential effects of R59P and I67N on synaptic transmission are the major determinants of the clinical severity, such as I67N causes a more severe phenotype by having a dominant negative effect on both evoked and spontaneous release and silencing the network, while R59P has no dominant negative effects on either forms of release and does not change the network firing characteristics, giving rise to a milder phenotype. These results clearly show that missense mutations of this alternatively spliced exon do not necessarily cause milder phenotypes and availability of WT SNAP25a is not enough to rescue synaptic impairments associated with disease-associated SNAP25b variants.
As DEEs tend to be resistant to the conventional antiepileptic treatments (Berg et al., 2010), understanding the effects of mutations of fusion machinery on synaptic transmission is vital to develop novel targeted treatments. In particular, novel therapeutics that directly target the presynaptic fusion machinery will likely provide significant treatment advance against these disorders (Li and Kavalali, 2017). This is highly crucial since early and aggressive seizure control correlates with greater cognitive outcomes later in life (Berg et al., 2012). We recently identified that aminopyridine class of K+ channel blockers are able to rescue impaired evoked release associated with VAMP2 variants in vitro by prolonging action potentials (Simmons, 2020). Furthermore, a patient with VAMP2 variant treated with off-label aminopyridine treatment showed significant clinical improvement assessed by both parental report and standardized cognitive measures (Simmons, 2020). In addition, this treatment approach is also shown in vitro as a viable option for patients harboring Syt1 variants (Bradberry et al., 2020). However, our results suggest that restoration of impaired evoked release alone in patients harboring variants associated with both aberrant spontaneous and impaired evoked release may not be an adequate approach. Here, a new generation of therapeutic approaches targeting spontaneous release will potentially be beneficial in treating these intractable disorders in addition to their potential benefits for other neurological and neuropsychiatric disorders (Kavalali and Monteggia, 2020). Additionally, as the mutations described here have dominant effects, our study suggests that eliminating the mutant allele instead of a gene addition strategy should be pursued in the future as a gene-based therapeutic approach.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact.
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ege T. Kavalali (ege.kavalali@vanderbilt.edu).
Materials Availability.
This study did not generate new unique reagents.
Data and Code Availability.
All data supporting the findings of this study are available from the lead author Ege T. Kavalali upon reasonable request. The coordinates of the atomic model and corresponding structure factor of the R59P-SNAP25 containing SNARE complex have been deposited in the Protein Data Bank under the accession code 6WVW.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals.
Postnatal day 2–3 Sprague-Dawley rats of either sex were used for the SNAP25 overexpression experiments. Pregnant Sprague-Dawley rats were housed individually until they give birth to a litter. The rats were kept in 12 hours : 12 hours dark:light cycle. The pregnant rats were provided with treats as well as cardboard enrichments. Postnatal day 2–3 littermates were used to prepare primary dissociated neuronal cultures. SNAP25 (Washbourne et al., 2002) KO, Het and WT mice embryos of either sex were used for KO rescue and haploinsufficiency experiments. Briefly, each male heterozygous mouse is paired with two female heterozygous mice and housed together. They were provided with the same treats and the enrichments and kept in the same dark:light cycle as the previously described rats were subjected to. The embryonic day 18 embryos were surgically obtained from the pregnant mice and used to prepare primary dissociated neuronal cultures. All animal procedures were performed in accordance with the guide for the care and use of laboratory animals and were approved by the Institutional Animal Care and Use Committee at Vanderbilt University. Health status of the live animals were periodically checked and confirmed by the veterinary staff of animal facilities of the Vanderbilt University.
Primary dissociated neuronal cultures.
Briefly, either hippocampi or cortical tissue were dissected in ice cold 20% fetal bovine serum (FBS) containing Hanks’ balanced salt solution. Tissues were then washed and treated with 10 mg/ml trypsin and 0.5 mg/ml DNase at 37 °C for 10 mins. The tissues were washed again, dissociated using filtered P1000 tip and centrifuged at 1000 rpm for 10 mins at 4 °C. Pellet containing neurons was resuspended either in plating medium (for postnatal cultures) containing MEM (no phenol red), 5 g/l D-glucose, 0.2 g/l NaHCO3, 0.1 g/l transferrin, 10% FBS, 2 mM L-glutamine and 20 mg/l insulin or in Neurobasal Plus medium (for embryonic cultures) supplemented with GlutaMAX-I and B27 supplement. Neurons were plated onto 12 mm coverslips coated with either 1:50 MEM:Matrigel solution (for postnatal cultures) or poly-D-lysine (for embryonic cultures). Cultures were kept in humidified incubators at 37 °C and gassed with 95% air and 5% CO2. On DIV (day in vitro) 1, the plating medium or the neurobasal plus medium was replaced with 4 μM cytosine arabinoside containing growth medium or neurobasal plus medium. Growth medium is similar to plating medium except for the following: 5% FBS, 0.5 mM L-glutamine, no insulin and supplemented with B27 supplement. On DIV4, the cytosine arabinoside concentration was dropped 2 μM by performing a half media change and lentivirus containing supernatant was added to express SNAP25 variants. Cultures were kept without any disruption until DIV14. All experiments were performed between DIV14–18, when synapses reached maturity and overexpression of the target protein was plateaued (Li et al., 2017). Sample size was not predetermined using statistical methods prior to experimentation. Sample sizes were based on previous studies in the field of molecular & cellular neuroscience.
Cell lines.
Human embryonic kidney-293 (HEK293) cells (ATCC) were used to produce lentiviral particles to infect primary neuronal cultures. HEK293 cultures were kept in humidified incubators at 37 °C and gassed with 95% air and 5% CO2. The cells were split and passaged when they reached 80% confluency. The culture medium consisted of 10% FBS containing Dulbecco’s Modified Eagle Medium supplemented with penicillin and streptavidin.
In vitro studies.
SNAP25a cDNA sequence from rat is used to express SNARE motifs that were used in CD experiments as well as x-ray crystallography. NEB Turbo Competent E. coli (NEB) were used for standard molecular cloning methods, whereas BL21-Gold(DE3) Competent E. coli (Agilent) were used for protein expression.
METHOD DETAILS
Cloning and lentivirus preparation.
SNAP25b was cloned into pFUGW vector containing human ubiquitin promoter and individual mutations were introduced using standard molecular biology methods and verified by sequencing. HEK293 cells were cotransfected by pFUGW and three packaging plasmids (pCMV-VSV-G ((Stewart et al., 2003), pMDLg/pRRE (Dull et al., 1998), pRSV-Rev (Dull et al., 1998)) using FuGENE 6 transfection reagent (Promega). 24 hours after the transfection, HEK cell media containing the transfection cocktail was replaced by either neuronal growth medium or Neurobasal Plus medium. 48 hours after the media change, the media containing lentiviral particles was collected and centrifuged at 2500 rpm for 15 mins at 4 °C to get rid of debris. 200 μl of supernatant was directly added to cultures on DIV4 to infect the neurons.
Electrophysiology and Data Analysis.
Whole-cell patch clamp recordings were performed on pyramidal cells using CV203BU headstage, Axopatch 200B amplifier, Digidata 1320 digitizer and Clampex 8.0 software (Molecular Devices). Recordings were filtered at 1 kHz and sampled at 100 μs. Experiments were conducted at room temperature. For external bath solution, a modified Tyrode’s solution containing the followings was used: (in mM): 150 NaCl, 4 KCl, 1.25 MgCl2, 2 CaCl2, 10 D-glucose, 10 HEPES at pH 7.4. For eEPSCs, the CaCl2 concentration was decreased to 1.25 mM. To isolate mEPSCs, 1 μm TTX, 50 μm PTX, and 50 μm D-AP5 were added. To isolate mIPSCs, 1 μm TTX, 10 μm CNQX, and 50 μm D-AP5 were added. To isolate eIPSCs, 50 μm D-AP5, and 10 μm CNQX were added. To isolate eEPSCs, 50 μm PTX and 50 μm D-AP5 were added. For eIPSC recordings, the field stimulation was provided using a parallel bipolar electrode (FHC) immersed in the external bath solution, delivering 35 mA pulses via a stimulus isolation unit. For eEPSC recordings, the stimulus intensity was decreased to 33 mA. Throughout the evoked experiments where stimulations are delivered via a bipolar electrode, we used a programmable motorized Sutter micromanipulator to make sure that the location of the bipolar electrode is exactly the same in all experiments, making sure that a similar field of coverslip is stimulated each time. For BAPTA-AM experiments, coverslips were incubated either in 100 μM BAPTA-AM in 0.1% DMSO containing external bath solution with 0 mM Ca2+ or in vehicle for 20 mins at room temperature. For voltage clamp experiments, the membrane potential was held at −70 mV and the 3–5 MΩ borosilicate glass patch pipettes were filled with the internal solution contained the following (in mM): 115 Cs-MeSO3, 10 CsCl, 5 NaCl, 10 HEPES, 0.6 EGTA, 20 tetraethylammonium-Cl, 4 Mg-ATP, 0.3 Na3GTP, and 10 QX-314 [N-(2,6-dimethylphenylcarbamoylmethyl)-triethylammonium bromide] at pH 7.35 and 300 mOsm. For current clamp experiments, the 4–6 MΩ borosilicate glass patch pipettes were filled with the internal solution contained the following (in mM): 110 K-Gluconate, 20 KCl, 10 NaCl, 10 HEPES, 4 Mg-ATP, 0.3 Na3GTP, 0.6 EGTA at pH 7.3 and 284 mOsm. For all recordings included for the analysis, the membrane resistance was greater than 100 MΩ, the access resistance was less than 20 MΩ and time constant (τ) was less than 3 ms. Miniature events and spontaneous AP firings were recorded for 3 mins. mPSC frequencies and amplitudes were analyzed and spike analysis was performed by using Mini Analysis software (Synaptosoft). Randomly picked equal number of events (or max number of event if the event number is less) from each recording was included in the cumulative histograms of interevent intervals to make sure that the contribution of each recording is comparable. Evoked IPSCs were analyzed by using Clampfit (Molecular Devices).
Cloning, protein expression and purification.
For SNAP25 variant containing SNARE complexes, the 10x-histidine-tagged and C-terminally truncated rat synaptobrevin-2 (amino-acids 28–89), the rat syntaxin-1A (amino-acids 191–256), the rat SNAP-25_N (amino-acids 7–83), and the rat SNAP-25_C (amino-acids 141–204) fragments were cloned into the Duet expression system (Novagen) as described previously (Zhou et al., 2015; Zhou et al., 2017). As the structure of SNARE complex and its interaction interfaces with synaptotagmin are determined using SNAP25a (Zhou et al., 2015; Zhou et al., 2017) and all the mutated residues are conserved in both of the SNAP25 isoforms, we used SNAP25a for protein experiments. The aforementioned four fragment constructs were co-expressed in Escherichia coli, leading to complex formation in the host. Specifically, E. coli BL21(DE3) were grown overnight at 37 °C using auto-inducing LB medium (Studier, 2005). After harvesting the cells by centrifugation, the pellet was resuspended in lysis buffer (50 mM Tris-HCl, pH 8.0, 300 mM NaCl, 20 mM imidazole, 0.5 mM TCEP) supplemented with lysozyme and DNase I, and subjected to sonication and centrifugation. The cleared lysate was bound to Ni-NTA agarose beads (Qiagen) equilibrated in the lysis buffer. Beads were harvested by centrifugation, poured into a column, and washed with the lysis buffer, urea buffer (50 mM Tris-HCl, pH 8.0, 300 mM NaCl, 60 mM imidazole, 0.5 mM TCEP, 7.5 M urea), and wash buffer (50 mM Tris-HCl, pH 8.0, 300 mM NaCl, 60 mM imidazole, 0.5 mM TCEP). The SNARE complex was then eluted with the lysis buffer supplemented with additional 330 mM imidazole. The fresh eluent of the Ni-NTA affinity purified SNARE complex was supplemented with tobacco etch virus protease and dialyzed against buffer A1 (50 mM Tris-HCl, pH 8.0, 50 mM NaCl, 0.5mM TCEP, 1 mM EDTA) overnight at 4 °C. After removal of uncleaved protein, the His-tag-free complex was subjected to anion exchange chromatography (Buffer A1: 50 mM Tris-HCl, pH 8.0, 50 mM NaCl, 0.5 mM TCEP, 1 mM EDTA; Buffer B1: 50 mM Tris-HCl, pH 8.0, 500 mM NaCl, 0.5 mM TCEP, 1 mM EDTA) using a linear gradient of NaCl starting at 50 mM and ending at 500 mM. The protein eluted at ~280 mM NaCl. The peak fractions were pooled, concentrated and stored at −80 °C for crystallizabon and circular dichroism (CD) experiments.
CD spectroscopy.
Circular dichroism measurements were conducted with circular dichroism spectrometer Model 202–01 (Aviv Biomedical) equipped with a temperature controller. Data were collected with 10 μM samples of WT and mutant SNARE complexes in 10 mM Tris-HCl (pH 8.0), 100 mM NaCl, 0.5 mM EDTA buffer over a wavelength range of 200–260 nm, with 1 nm increments, in a 1 mm path length cell at 25 °C. Temperature melting scans were performed at a wavelength of 220 nm by increasing the temperature from 25 to 100 °C in 3 °C temperature increments, a 2 min temperature equilibration time, and a 3 s averaging time. The fraction of unfolded protein at each temperature was calculated by using the formula (Iobs−If)/(Iu−If), where Iob is the observed mean residue ellipticity, and Iu and If are the mean residue ellipticities of the unfolded and folded states, respectively. Iu and If were estimated by extrapolation of the linear regions of the extremes of the denaturation curves.
Crystallization.
Purified protein sample of the R59P-SNAP25 containing SNARE complex was dialyzed against a buffer solution containing 20 mM HEPES (pH 7.4), 100 mM NaCl and 5 mM DTT. Crystals were grown by the hanging-drop vapour diffusion method at 20 °C by mixing 2 μl protein solution (at a concentration of ~12 mg/ml) with equal volume of reservoir solution containing 100 mM MES (pH 6.0), 100 mM CaCl2, and 20–25% (v/v).
Data collection and structure determination.
Crystals were flash-frozen in a cryo-protecting solution containing the same constituents as the crystallization condition supplemented with 25% (v/v) MPD. Diffraction data were collected at the Stanford Synchrotron Radiation Light Source (SSRL) beam line 9–2 (wavelength 0.97946 Å at 100 K). Diffraction data were indexed, integrated, scaled, and merged using HKL3000 (Minor et al., 2006). The phases for the diffraction data were determined by molecular replacement with Phaser (McCoy et al., 2007) using the rat WT SNARE complex (PDB accession number 1N7S) as a search model. The structure was iteratively rebuilt and refined using the programs Coot (Emsley and Cowtan, 2004) and Phenix (Adams et al., 2002). (Table S1). Ramachandran analysis with MolProbity (Chen et al., 2010) indicated that 100% of the residues are in the favoured regions and none are in disallowed regions. MolProbity was used to evaluate the geometry and quality of the models (Table S1). All structure figures were prepared with PyMol (http://www.pymol.org).
Immunofluorescence.
Coverslips were fixed in 4% PFA/4% sucrose containing PBS solution for 20 mins at room temperature and permeabilized using 0.2% Triton-X containing PBS solution for 30 mins at room temperature. Coverslips were washed and incubated in blocking solution consisting of 1% bovine serum albumin and 2% goat serum in PBS for 2 hours at room temperature. Coverslips were incubated with primary antibodies diluted in the blocking solution: 1:500 Anti-SNAP25 (111–111, Synaptic Systems), 1:500 Anti-synapsin (106–103, Synaptic Systems), 1:1.000 Anti-VGlut (AB5905, EMD Millipore), 1:200 Anti-PSD-95 (7E3–1B8, ThermoFisher), 1:500 Anti-VGAT (131–008, Synaptic Systems), 1:200 Anti-gephyrin (147–021, Synaptic Systems) and anti-MAP2 (188–006, Synaptic Systems) in a humid chamber overnight at 4 °C and with species-appropriate Alexa Fluor secondary antibodies diluted as 1:500 in the blocking solution in a humid chamber for 90 mins at room temperature. Coverslips were mounted on glass slides and imaged via a Zeiss LSM 710 with 63x objective at 1024 × 1024-pixel resolution. The synapse densities were analyzed using Intellicount (Fantuzzo et al., 2017).
Protein quantification.
To quantify protein levels, western blotting was carried out. Briefly, protein samples were prepared from coverslips using Laemmli Buffer containing protease and phosphatase inhibitor cocktails (Roche) and beta-mercaptoethanol. Samples were sonicated and boiled for 5 min at 95 °C to dissociate SNARE complexes and loaded on SDS-PAGE gels and transferred to nitrocellulose membranes. Membranes were incubated with primary antibodies at 4 °C overnight in following dilutions: 1:2000 Anti-SNAP25 mouse (111–111, Synaptic Systems), 1:10000 Anti-GAPDH rabbit (14C10, Cell Signaling), 1:2000 Anti-syntaxin-1 mouse (110–011, Synaptic Systems) and 1:2000 Anti-synaptobrevin-2 mouse (104–211, Synaptic Systems). After incubation with fluorescent secondary anti-rabbit and anti-mouse antibodies (IRDye Secondary Antibodies, Licor), membranes were imaged using an Odyssey CLx imaging system (Licor). Band intensities were analysed using ImageJ and normalized to loading controls.
Hippocampal slice preparation and field recording.
The mice were anesthetized with isoflurane before decapitation. The brains were removed and immersed in ice-cold dissection buffer containing the following (in mM): 2.6 KCl, 1.25 NaH2PO4, 26 NaHCO3, 0.5 CaCl2, 5 MgCl2, 212 sucrose and 10 D-glucose for 2–3 min. The hippocampi were dissected out and cut with a vibratome into 400 μm thick transverse sections in ice-cold dissection buffer continuously aerated with 95% O2 and 5% CO2. Sections were recovered in oxygenated ACSF containing the following (in mM): 124 NaCl, 5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 2 CaCl2, 2 MgCl2 and 10 D-glucose at pH 7.4 (continuously equilibrated with 95% O2 and 5% CO2) for 2–3 hours at 30 °C. Hippocampal slices were transferred to the recording chamber and perfused with ACSF at a rate of 2–3 ml/min at 30 °C. Field EPSPs (fEPSPs) were evoked by inserting a concentric bipolar stimulating electrode (FHC) to Schaffer collateral/commissural afferents. 1–2 MΩ Extracellular recording electrodes filled with ACSF were inserted into the CA1 area proximally below the molecular layer. Baseline responses were collected every 30 s using an input stimulus intensity that induced 30–40% of the maximum response. Paired-pulse facilitation (PPF) was elicited by giving paired-pulse stimulations at decreasing interstimulus intervals (ISIs) of 500, 400, 200, 100, 50, 30 and 20 ms and analyzed by dividing the fEPSP slope of pulse 2 (P2) by that of pulse 1 (P1).
QUANTIFICATION AND STATISTICAL ANALYSIS
The data was presented as mean ± standard error of mean, unless stated otherwise in the figure legends. The sample size for each experiment was stated either in the figure or in the legend and was not predetermined using statistical methods prior to experimentation. For patch-clamp electrophysiology experiments, the sample size corresponds to number of cells patched. Sample size was not predetermined using statistical methods prior to experimentation. Sample sizes were based on previous studies in the field of molecular & cellular neuroscience. To ensure reproducibility of experimental findings, each set of experiments was performed at least twice (and at least in two different sets of neuronal cultures if appropriate) to confirm the results. Prism 8 (Graphpad) was used to run statistical tests. A test of normality was not conducted and throughout the study, two-tailed non-paired t-test was used to compare effects of SNAP25 variants against either WT SNAP25 or SNAP25 KO. Field recordings were analyzed using multiple t-tests corrected with Holm-Sidak correction for multiple comparisons were used. Kolmogorov–Smirnov test was used to compare cumulative histograms of interevent intervals. Outliers were identified with Robust regression and Outlier removal (ROUT) method. p value less than 0.05 was considered as statistically significant. Significance levels were stated as follows: *p<0.05, **p<0.01, ***p<0.001 and ****p<0.0001. ns denotes non-significance.
Supplementary Material
Supplementary movie 1. 360o rotation of 2Fo-Fc map of R59P-SNAP25 containing SNARE complex. Synaptobrevin-2 is shown as blue, syntaxin-1a is shown as red, SNAP25 SN1 is shown as yellow and SNAP25 SN2 is shown as green.
Supplementary movie 2. 360o rotation of omit map (2.5 σ) of R59P-SNAP25 containing SNARE complex aligned with WT SNARE complex, focused on the mutated residue. Synaptobrevin-2 is shown as blue, syntaxin-1a is shown as red, SNAP25 SN1 is shown as yellow and SNAP25 SN2 is shown as green.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-SNAP25 (mouse monoclonal) | Synaptic Systems | Catalog # 111 111 RRID:AB_887792 |
| Anti-synapsin (rabbit polyclonal) | Synaptic Systems | Catalog # 106 103 RRID:AB_11042000 |
| Anti-syntaxin1 (mouse monoclonal) | Synaptic Systems | Catalog # 110 011 RRID:AB_887844 |
| Anti-synaptobrevin2 (mouse monoclonal) | Synaptic Systems | Catalog # 104 211 RRID:AB_887811 |
| Anti-GAPDH (rabbit monoclonal) | Cell signaling | Catalog # 14C10 RRID:AB_561053 |
| Anti-MAP2 (chicken polyclonal) | Synaptic Systems | Catalog # 188 006 RRID:AB_2619881 |
| Anti-VGlut (guinea pig polyclonal) | EMD Millipore | Catalog # AB5905 RRID:AB_2301751 |
| Anti-PSD95 (mouse monoclonal) | ThermoFisher | Catalog # 7E3 1B8 RRID:AB_2092361 |
| Anti-VGAT (rabbit monoclonal) | Synaptic Systems | Catalog # 131 008 RRID:AB_2800534 |
| Anti-gephyrin (mouse monoclonal) | Synaptic Systems | Catalog # 147 021 RRID:AB_2232546 |
| Bacterial and Virus Strains | ||
| NEB Turbo Competent E. coli | NEB | Catalog # C2984H |
| BL21-Gold(DE3) Competent E. coli | Agilent | Catalog # 230132 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| 6-Cyano-7-nitroquinoxaline-2,3-dione disodium salt hydrate (CNQX) | Sigma-Aldrich | Catalog # C239 |
| D(−)-2-Amino-5-phosphonopentanoic acid (AP-5) | Sigma-Aldrich | Catalog # A8054 |
| Picrotoxin (PTX) | Sigma-Aldrich | Catalog # P1675 |
| BAPTA-AM | Abcam | Catalog # ab120503 |
| Lysozyme | Sigma-Aldrich | Catalog # L6876 |
| DNase I | Sigma-Aldrich | Catalog # D5025 |
| TEV Protease | NEB | Catalog # P8112S |
| Neurobasal Plus Medium | GIBCO | Catalog # A3582901 |
| Tetrodotoxin (TTX) | Enzo Life Sciences | Catalog # BML-NA120–0001 |
| Transferrin | Calbiochem | Catalog # 616420 |
| Cytosine Arabinoside (Ara-C) | Sigma | Catalog # C6645 |
| B-27 supplement | GIBCO | Catalog # 17504–010 |
| Deposited Data | ||
| R59P-SNAP25 containing SNARE complex | This paper | PDB code 6WVW |
| Experimental Models: Cell Lines | ||
| Human embryonic kidney-293 (HEK293) cells | ATCC | Catalog # CRL-1573 RRID: CVCL_0045 |
| Experimental Models: Organisms/Strains | ||
| Sprague-Dawley rat pups (P2–P3) | Charles River | Strain code: 400 |
| Homozygous and heterozygous SNAP25 KO mice and WT littermates | (Washbourne et al., 2002) | N/A |
| Recombinant DNA | ||
| Plasmid: pRSV-REV (lentiviral packaging) | (Dull et al., 1998) | Addgene # 12253 |
| Plasmid: pCMV-VSV-G (lentiviral packaging) | (Stewart et al., 2003) | Addgene # 8454 |
| Plasmid: pMDLg/pRRE (lentiviral packaging) | (Dull et al., 1998) | Addgene # 12251 |
| Plasmid: pFUGW-SNAP25b (WT and variants) | This paper | N/A |
| Plasmid: Duet expression system containing 10x-histidine tagged synaptobrevin-2 (amino acids 28–89), syntaxin-1a (amino acids 191–256), SNAP25_N (amino acids 7–83) and SNAP25_C (amino acids 141–204) (WT and variants) | Modified from (Zhou et al., 2015) | N/A |
| Software and Algorithms | ||
| MiniAnalysis | Synaptosoft | http://www.synaptosoft.com/MiniAnalysis |
| Clampfit | Molecular Devices | http://www.moleculardevices.com |
| Axopatch | Molecular Devices | http://www.moleculardevices.com |
| Prism | GraphPad | http://www.graphpad.com |
| HKL3000 | (Minor et al., 2006) | N/A |
| Coot | (Emsley and Cowtan, 2004) | N/A |
| Phenix | (Adams et al., 2002) | N/A |
| MolProbity | (Chen et al., 2010) | N/A |
| PyMol | http://www.pymol.org | |
| Phaser | (McCoy et al., 2007) | N/A |
| Intellicount | (Fantuzzo et al., 2017) | |
Highlights.
SNAP25 variants cause clinically variable developmental-epileptic encephalopathies
Structurally clustered mutations give rise to similar synaptic phenotypes
Haploinsufficiency and impaired evoked release cannot fully explain the pathology
Aberrant spontaneous release phenotypes are associated with disease heterogeneity
Acknowledgements
We thank Ms. Elizabeth Dellureficio for her encouragement with the initiation of this study. This research was supported by National Institutes of Health grants R01MH66198 and R01AG055577 (ETK), K99MH113764 and R00MH113764 (Q.Z.), R01MH081060 and R01MH070727 (LMM) and R37MH63105 (ATB). This study makes use of data generated by the DECIPHER community. A full list of centres who contributed to the generation of the data is available from https://decipher.sanger.ac.uk and via email from decipher@sanger.ac.uk. Funding for the project was provided by Wellcome Trust. Crystal diffraction screening and data collection were performed at synchrotron facilities provided by the Stanford Synchrotron Radiation Lightsource (SSRL), SLAC National Accelerator Laboratory supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences under Contract No. DEAC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by the National Institutes of Health, National Institute of General Medical Sciences (including P41GM103393). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of NIGMS or NIH. We also thank the staff Clyde Smith at SSRL beamline 9-2 for help with diffraction data collection.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Adams PD, Grosse-Kunstleve RW, Hung LW, Ioerger TR, McCoy AJ, Moriarty NW, Read RJ, Sacchettini JC, Sauter NK, and Terwilliger TC (2002). PHENIX: building new software for automated crystallographic structure determination. Acta Crystallogr D Biol Crystallogr 58, 1948–1954. [DOI] [PubMed] [Google Scholar]
- Bacaj T, Wu D, Burre J, Malenka RC, Liu X, and Sudhof TC (2015). Synaptotagmin-1 and −7 Are Redundantly Essential for Maintaining the Capacity of the Readily-Releasable Pool of Synaptic Vesicles. PLoS Biol 13, e1002267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacaj T, Wu D, Yang X, Morishita W, Zhou P, Xu W, Malenka RC, and Sudhof TC (2013). Synaptotagmin-1 and synaptotagmin-7 trigger synchronous and asynchronous phases of neurotransmitter release. Neuron 80, 947–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker K, Gordon SL, Melland H, Bumbak F, Scott DJ, Jiang TJ, Owen D, Turner BJ, Boyd SG, Rossi M, et al. (2018). SYT1-associated neurodevelopmental disorder: a case series. Brain 141, 2576–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bark IC, Hahn KM, Ryabinin AE, and Wilson MC (1995). Differential expression of SNAP-25 protein isoforms during divergent vesicle fusion events of neural development. Proc Natl Acad Sci U S A 92, 1510–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bark IC, and Wilson MC (1994). Human cDNA clones encoding two different isoforms of the nerve terminal protein SNAP-25. Gene 139, 291–292. [DOI] [PubMed] [Google Scholar]
- Berg AT, Berkovic SF, Brodie MJ, Buchhalter J, Cross JH, van Emde Boas W, Engel J, French J, Glauser TA, Mathern GW, et al. (2010). Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia 51, 676–685. [DOI] [PubMed] [Google Scholar]
- Berg AT, Zelko FA, Levy SR, and Testa FM (2012). Age at onset of epilepsy, pharmacoresistance, and cognitive outcomes: a prospective cohort study. Neurology 79, 1384–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradberry MM, Courtney NA, Dominguez MJ, Lofquist SM, Knox AT, Sutton RB, and Chapman ER (2020). Molecular Basis for Synaptotagmin-1-Associated Neurodevelopmental Disorder. Neuron 107, 52–64 e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronk P, Deak F, Wilson MC, Liu X, Sudhof TC, and Kavalali ET (2007). Differential effects of SNAP-25 deletion on Ca2+ -dependent and Ca2+ -independent neurotransmission. J Neurophysiol 98, 794–806. [DOI] [PubMed] [Google Scholar]
- Brose N, Petrenko AG, Sudhof TC, and Jahn R (1992). Synaptotagmin: a calcium sensor on the synaptic vesicle surface. Science 256, 1021–1025. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, and Richardson DC (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, and De Jonghe P (2001). De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet 68, 1327–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradini I, Donzelli A, Antonucci F, Welzl H, Loos M, Martucci R, De Astis S, Pattini L, Inverardi F, Wolfer D, et al. (2014). Epileptiform activity and cognitive deficits in SNAP-25(+/−) mice are normalized by antiepileptic drugs. Cereb Cortex 24, 364–376. [DOI] [PubMed] [Google Scholar]
- Courtney NA, Briguglio JS, Bradberry MM, Greer C, and Chapman ER (2018). Excitatory and Inhibitory Neurons Utilize Different Ca(2+) Sensors and Sources to Regulate Spontaneous Release. Neuron 98, 977–991 e975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deciphering Developmental Disorders, S. (2015). Large-scale discovery of novel genetic causes of developmental disorders. Nature 519, 223–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deciphering Developmental Disorders, S. (2017). Prevalence and architecture of de novo mutations in developmental disorders. Nature 542, 433–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dull T, Zufferey R, Kelly M, Mandel RJ, Nguyen M, Trono D, and Naldini L (1998). A third-generation lentivirus vector with a conditional packaging system. J Virol 72, 8463–8471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Euro, E.-R.E.S.C., Epilepsy Phenome/Genome, P., and Epi, K.C. (2014). De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am J Hum Genet 95, 360–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fantuzzo JA, Mirabella VR, Hamod AH, Hart RP, Zahn JD, and Pang ZP (2017). Intellicount: High-Throughput Quantification of Fluorescent Synaptic Protein Puncta by Machine Learning. eNeuro 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Chacon R, Konigstorfer A, Gerber SH, Garcia J, Matos MF, Stevens CF, Brose N, Rizo J, Rosenmund C, and Sudhof TC (2001). Synaptotagmin I functions as a calcium regulator of release probability. Nature 410, 41–49. [DOI] [PubMed] [Google Scholar]
- Firth HV, Richards SM, Bevan AP, Clayton S, Corpas M, Rajan D, Van Vooren S, Moreau Y, Pettett RM, and Carter NP (2009). DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am J Hum Genet 84, 524–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda H, Imagawa E, Hamanaka K, Fujita A, Mitsuhashi S, Miyatake S, Mizuguchi T, Takata A, Miyake N, Kramer U, et al. (2018). A novel missense SNAP25b mutation in two affected siblings from an Israeli family showing seizures and cerebellar ataxia. J Hum Genet 63, 673–676. [DOI] [PubMed] [Google Scholar]
- Gabriel HF,S; Stoebe P; Hoertnagel K; Biskup S; Schell-Apacik C (2016). A de novo mutation in the SNAP25 gene in a patient with epileptic encephalopathy, hypotonia and contractures identified by trio-based exome sequencing In European Human Genetics Conference (Barcelona, Spain: ). [Google Scholar]
- Gao Y, Zorman S, Gundersen G, Xi Z, Ma L, Sirinakis G, Rothman JE, and Zhang Y (2012). Single reconstituted neuronal SNARE complexes zipper in three distinct stages. Science 337, 1340–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, and Sudhof TC (1994). Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell 79, 717–727. [DOI] [PubMed] [Google Scholar]
- Guan Z, Bykhovskaia M, Jorquera RA, Sutton RB, Akbergenova Y, and Littleton JT (2017). A synaptotagmin suppressor screen indicates SNARE binding controls the timing and Ca(2+) cooperativity of vesicle fusion. Elife 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamdan FF, Myers CT, Cossette P, Lemay P, Spiegelman D, Laporte AD, Nassif C, Diallo O, Monlong J, Cadieux-Dion M, et al. (2017). High Rate of Recurrent De Novo Mutations in Developmental and Epileptic Encephalopathies. Am J Hum Genet 101, 664–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyne HO, Singh T, Stamberger H, Abou Jamra R, Caglayan H, Craiu D, De Jonghe P, Guerrini R, Helbig KL, Koeleman BPC, et al. (2018). De novo variants in neurodevelopmental disorders with epilepsy. Nat Genet 50, 1048–1053. [DOI] [PubMed] [Google Scholar]
- Hoerder-Suabedissen A, Korrell KV, Hayashi S, Jeans A, Ramirez DMO, Grant E, Christian HC, Kavalali ET, Wilson MC, and Molnar Z (2019). Cell-Specific Loss of SNAP25 from Cortical Projection Neurons Allows Normal Development but Causes Subsequent Neurodegeneration. Cereb Cortex 29, 2148–2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpusas M, Branchaud B, and Remington SJ (1990). Proposed mechanism for the condensation reaction of citrate synthase: 1.9-A structure of the ternary complex with oxaloacetate and carboxymethyl coenzyme A. Biochemistry 29, 2213–2219. [PubMed] [Google Scholar]
- Kavalali ET (2015). The mechanisms and functions of spontaneous neurotransmitter release. Nat Rev Neurosci 16, 5–16. [DOI] [PubMed] [Google Scholar]
- Kavalali ET, and Monteggia LM (2020). Targeting Homeostatic Synaptic Plasticity for Treatment of Mood Disorders. Neuron 106, 715–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai Y, Choi UB, Leitz J, Rhee HJ, Lee C, Altas B, Zhao M, Pfuetzner RA, Wang AL, Brose N, et al. (2017). Molecular Mechanisms of Synaptic Vesicle Priming by Munc13 and Munc18. Neuron 95, 591–607 e510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YC, Chanaday NL, Xu W, and Kavalali ET (2017). Synaptotagmin-1- and Synaptotagmin-7-Dependent Fusion Mechanisms Target Synaptic Vesicles to Kinetically Distinct Endocytic Pathways. Neuron 93, 616–631 e613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YC, and Kavalali ET (2017). Synaptic Vesicle-Recycling Machinery Components as Potential Therapeutic Targets. Pharmacol Rev 69, 141–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang JSW,JS; Lin LJ; Yang MT; Hung KL; Lu JF (2018). Genetic Diagnosis in Children with Epilepsy and Developmental Delay/Mental Retardation Using Targeted Gene Panel Analysis. Neuropsychiatry (London) 8, 1577–1585. [Google Scholar]
- Ma L, Rebane AA, Yang G, Xi Z, Kang Y, Gao Y, and Zhang Y (2015). Munc18–1-regulated stage-wise SNARE assembly underlying synaptic exocytosis. Elife 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Pang ZP, Tervo DG, and Sudhof TC (2007). Monitoring synaptic transmission in primary neuronal cultures using local extracellular stimulation. J Neurosci Methods 161, 75–87. [DOI] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007). Phaser crystallographic software. J Appl Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McTague A, Howell KB, Cross JH, Kurian MA, and Scheffer IE (2016). The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol 15, 304–316. [DOI] [PubMed] [Google Scholar]
- Minor W, Cymborowski M, Otwinowski Z, and Chruszcz M (2006). HKL-3000: the integration of data reduction and structure solution--from diffraction images to an initial model in minutes. Acta Crystallogr D Biol Crystallogr 62, 859–866. [DOI] [PubMed] [Google Scholar]
- Monteggia LM, Lin PY, Adachi M, and Kavalali ET (2019). Behavioral Analysis of SNAP-25 and Synaptobrevin-2 Haploinsufficiency in Mice. Neuroscience 420, 129–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peralvarez-Marin A, Bourdelande JL, Querol E, and Padros E (2006). The role of proline residues in the dynamics of transmembrane helices: the case of bacteriorhodopsin. Mol Membr Biol 23, 127–135. [DOI] [PubMed] [Google Scholar]
- Prescott GR, and Chamberlain LH (2011). Regional and developmental brain expression patterns of SNAP25 splice variants. BMC Neurosci 12, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohena L, Neidich J, Truitt Cho M, Gonzalez KD, Tang S, Devinsky O, and Chung WK (2013). Mutation in SNAP25 as a novel genetic cause of epilepsy and intellectual disability. Rare Dis 1, e26314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, and Stevens CF (1996). Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron 16, 1197–1207. [DOI] [PubMed] [Google Scholar]
- Salpietro V, Malintan NT, Llano-Rivas I, Spaeth CG, Efthymiou S, Striano P, Vandrovcova J, Cutrupi MC, Chimenz R, David E, et al. (2019). Mutations in the Neuronal Vesicular SNARE VAMP2 Affect Synaptic Membrane Fusion and Impair Human Neurodevelopment. Am J Hum Genet 104, 721–730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, Hirsch E, Jain S, Mathern GW, Moshe SL, et al. (2017). ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 58, 512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen XM, Selcen D, Brengman J, and Engel AG (2014). Mutant SNAP25B causes myasthenia, cortical hyperexcitability, ataxia, and intellectual disability. Neurology 83, 2247–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons RLL,H; Alten B; Santos MS; Jiang R; Paul B; Lalani SJ; Cortesi A; Parks K; Khandelwal N; Smith-Packard B,; Phoong MA; Chez M; Fisher H; Scheuerle AE; Shinawi M; Hussain SA; Kavalali ET; Sherr EH; Voglmaier SM (2020). Overcoming Presynaptic Effects of VAMP2 Mutations with 4-Aminopyridine Treatment. Human Mutation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollner T, Bennett MK, Whiteheart SW, Scheller RH, and Rothman JE (1993a). A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell 75, 409–418. [DOI] [PubMed] [Google Scholar]
- Sollner T, Whiteheart SW, Brunner M, Erdjument-Bromage H, Geromanos S, Tempst P, and Rothman JE (1993b). SNAP receptors implicated in vesicle targeting and fusion. Nature 362, 318–324. [DOI] [PubMed] [Google Scholar]
- Sorensen JB, Wiederhold K, Muller EM, Milosevic I, Nagy G, de Groot BL, Grubmuller H, and Fasshauer D (2006). Sequential N- to C-terminal SNARE complex assembly drives priming and fusion of secretory vesicles. EMBO J 25, 955–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PA, et al. (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studier FW (2005). Protein production by auto-induction in high density shaking cultures. Protein Expr Purif 41, 207–234. [DOI] [PubMed] [Google Scholar]
- Sutton RB, Fasshauer D, Jahn R, and Brunger AT (1998). Crystal structure of a SNARE complex involved in synaptic exocytosis at 2.4 A resolution. Nature 395, 347–353. [DOI] [PubMed] [Google Scholar]
- Thomas RH, and Berkovic SF (2014). The hidden genetics of epilepsy-a clinically important new paradigm. Nat Rev Neurol 10, 283–292. [DOI] [PubMed] [Google Scholar]
- Vardar G, Gerth F, Schmitt XJ, Rautenstrauch P, Trimbuch T, Schubert J, Lerche H, Rosenmund C, and Freund C (2020). Epilepsy-causing STX1B mutations translate altered protein functions into distinct phenotypes in mouse neurons. Brain 143, 2119–2138. [DOI] [PubMed] [Google Scholar]
- Verhage M, and Sorensen JB (2020). SNAREopathies: Diversity in Mechanisms and Symptoms. Neuron 107, 22–37. [DOI] [PubMed] [Google Scholar]
- Washbourne P, Thompson PM, Carta M, Costa ET, Mathews JR, Lopez-Bendito G, Molnar Z, Becher MW, Valenzuela CF, Partridge LD, and Wilson MC (2002). Genetic ablation of the t-SNARE SNAP-25 distinguishes mechanisms of neuroexocytosis. Nat Neurosci 5, 19–26. [DOI] [PubMed] [Google Scholar]
- Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Sollner TH, and Rothman JE (1998). SNAREpins: minimal machinery for membrane fusion. Cell 92, 759–772. [DOI] [PubMed] [Google Scholar]
- Wolking S, May P, Mei D, Moller RS, Balestrini S, Helbig KL, Altuzarra CD, Chatron N, Kaiwar C, Stohr K, et al. (2019). Clinical spectrum of STX1B-related epileptic disorders. Neurology 92, e1238–e1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Mashimo T, and Sudhof TC (2007). Synaptotagmin-1, −2, and −9: Ca(2+) sensors for fast release that specify distinct presynaptic properties in subsets of neurons. Neuron 54, 567–581. [DOI] [PubMed] [Google Scholar]
- Xu J, Pang ZP, Shin OH, and Sudhof TC (2009). Synaptotagmin-1 functions as a Ca2+ sensor for spontaneous release. Nat Neurosci 12, 759–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Kim-Miller MJ, Fukuda M, Kowalchyk JA, and Martin TF (2002). Ca2+-dependent synaptotagmin binding to SNAP-25 is essential for Ca2+-triggered exocytosis. Neuron 34, 599–611. [DOI] [PubMed] [Google Scholar]
- Zhou Q, Lai Y, Bacaj T, Zhao M, Lyubimov AY, Uervirojnangkoorn M, Zeldin OB, Brewster AS, Sauter NK, Cohen AE, et al. (2015). Architecture of the synaptotagmin-SNARE machinery for neuronal exocytosis. Nature 525, 62–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Zhou P, Wang AL, Wu D, Zhao M, Sudhof TC, and Brunger AT (2017). The primed SNARE-complexin-synaptotagmin complex for neuronal exocytosis. Nature 548, 420–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorman S, Rebane AA, Ma L, Yang G, Molski MA, Coleman J, Pincet F, Rothman JE, and Zhang Y (2014). Common intermediates and kinetics, but different energetics, in the assembly of SNARE proteins. Elife 3, e03348. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary movie 1. 360o rotation of 2Fo-Fc map of R59P-SNAP25 containing SNARE complex. Synaptobrevin-2 is shown as blue, syntaxin-1a is shown as red, SNAP25 SN1 is shown as yellow and SNAP25 SN2 is shown as green.
Supplementary movie 2. 360o rotation of omit map (2.5 σ) of R59P-SNAP25 containing SNARE complex aligned with WT SNARE complex, focused on the mutated residue. Synaptobrevin-2 is shown as blue, syntaxin-1a is shown as red, SNAP25 SN1 is shown as yellow and SNAP25 SN2 is shown as green.
Data Availability Statement
All data supporting the findings of this study are available from the lead author Ege T. Kavalali upon reasonable request. The coordinates of the atomic model and corresponding structure factor of the R59P-SNAP25 containing SNARE complex have been deposited in the Protein Data Bank under the accession code 6WVW.