

Abstract
Oxidative stress and renal inflammation play a pivotal role in the pathogenesis of hypertension. The redox-sensitive transcription factor, nuclear factor E2-related factor 2 (Nrf2) is the master regulator of phase II antioxidant enzymes that protects against oxidative stress and inflammation. This study aimed to investigate the effect of Nrf2 inhibition on oxidative stress-associated hypertension and renal dopamine 1 receptor (D1R) dysfunction in mice. Male C57BL/6J mice were treated with a pro-oxidant, L-buthionine sulfoximine (BSO) (10 mmol/L in drinking water), and ML385 (10 mg/kg body weight/day, intraperitoneally), a novel Nrf2 inhibitor that blocks Nrf2 regulated downstream target genes expression. Mice treated with BSO exhibited oxidative stress, renal functional impairment, inflammation, and elevated blood pressure. Also, BSO treatment increased the activity of phase II antioxidant enzyme, NAD(P)H: quinone oxidoreductase-1 (NQO-1). BSO and ML385 co-treatment exhibited a robust increase in blood pressure, oxidative stress and intensified the renal function deterioration as indicated by a significant increase in serum creatinine, urinary albumin excretion rate, and albumin to creatinine ratio and decreased glomerular filtration rate (GFR). Also, BSO and ML385 co-treatment downregulated NQO-1 and significantly altered the inflammatory cytokines, IL-1β and IL-10 levels. A D1R agonist SKF38393 failed to promote urinary sodium excretion indicating functional impairment in renal D1R. ML385 per se did not affect mean arterial pressure, GFR, and renal D1R function. Taken together, we concluded that the Nrf2 inhibition aggravated oxidative stress and inflammation by diminishing phase II antioxidant defense that deteriorates renal function and contributes to the development of hypertension in mice.
Keywords: oxidative stress, inflammation, hypertension, Nrf2, ML385
Introduction
Hypertension is a major risk factor for cardiovascular diseases and a leading cause of morbidity and mortality worldwide (1, 2). Hypertension has been the subject of intense research for decades however, the etiology is not yet clear. Several studies have shown oxidative stress and inflammation as a major contributor to the pathogenesis of hypertension (3,4). Increased generation of reactive oxygen species, weakening of antioxidant defense, and accumulation of inflammatory markers are obvious in the kidney and vascular tissues of animals with acquired and genetic hypertension (5, 6).
Nuclear factor erythroid 2-related factor 2 (Nrf2), a basic leucine zipper transcription factor is well recognized for its role in maintaining the intracellular redox homeostasis by regulating the transcription of phase II antioxidant enzymes (7–9). Under basal conditions, Nrf2 is regulated by Kelch-like ECH-associated protein 1, a repressor molecule that facilitates Nrf2 proteasomal degradation. However, in response to stress, the conformation of Kelch-like ECH-associated protein 1/Nrf2 complex changes, resulting in nuclear translocation of Nrf2 protein where it heterodimerizes with small musculoaponeurotic fibrosarcoma proteins. The complex further binds to the antioxidant response element (ARE) sequence to activates the transcription of ARE dependent cytoprotective genes including heme oxygenase-1, NAD(P)H: quinone oxidoreductase-1 (NQO-1), glutathione S-transferase-1, and superoxide dismutase (SOD) that confer protection against oxidative damage and inflammation (10, 11). Recently, activation of the Nrf2-ARE pathway has been documented to be beneficial in many cardiovascular diseases including hypertension (12). Several Nrf2 activators have been tested including sulforaphane (13, 14), resveratrol (15), tert-butylhydroquinone (16), and dimethyl fumarate (17) to study the beneficial role of Nrf2 signaling in hypertension. Despite the significance of Nrf2 in mitigating oxidative stress, little evidence shows impaired Nrf2 function contributes to hypertension (15, 18).
Therefore, in the present study, we hypothesized that the inhibition of Nrf2 transcription factor could aggravate renal dysfunction and the development of hypertension in mice subjected to oxidative stress. To test the hypothesis, we used a specific Nrf2 inhibitor, ML385 that interacts with Nrf2 and inhibits its downstream target gene expression by interfering with the binding of small musculoaponeurotic fibrosarcoma-Nrf2 protein complex to ARE (19, 20). Our results showed that impairment of Nrf2 activity and Nrf2 dependent antioxidant system causes a significant increase in oxidative stress and inflammation that deteriorates renal function and contributes to hypertension in mice.
Materials and methods
Animal treatment
Adult male C57BL/6J mice weighing 30 gm were purchased from The Jackson Laboratory (Bar Harbor, ME) and acclimatized for a week with free access to food and water. Four groups of mice were studied after acclimatization; 1) control, mice kept on tap water; (2) BSO, provided with 10 mmol/L BSO in drinking water; (3) BSO+ML385, mice intraperitoneally implanted with mini-osmotic pumps (Model 1004, Durect Corporation, Cupertino, CA) to dispense ML385 (10 mg/kg bodyweight/day) and supplemented with BSO and (4) ML385, implanted with ML385 filled mini-osmotic pumps. Bodyweight of mice was recorded at the start and completion of the 8-week treatment.
Osmotic pump implantation was performed as detailed by Chang et al. (21). Briefly, anesthesia was induced by 3% isoflurane and maintained by 1.5% isoflurane throughout the surgical procedure using (SomnoSuite, Kent Scientific Corp, Torrington, CT, USA) anesthesia machine. A midline incision was made in the lower abdomen and the drug-filled osmotic pump was inserted, delivery portal first, in the peritoneal cavity.
A day before sacrifice, urine samples were collected for 24 hours by keeping the mice in standard metabolic cages. All experiments were performed in compliance with Institutional Animal Care and Use Committee and NIH guidelines.
Blood pressure measurement and sample preparation
After 8 weeks of treatment, the mice were used for blood pressure measurement and other biochemical studies. To measure blood pressure, the mice were anesthetized as aforementioned. Under a microscope, a midline neck incision was made and the left carotid artery was carefully separated from the vagus nerve and catheterized with a calibrated pressure catheter (SPR-671 Mikro-Tip®, Millar, Houston, TX) connected to PCU-2000 pressure control unit and blood pressure was recorded using a data acquisition system (PowerLab 35 Series, ADInstruments). After 30 minutes of the stabilization period, the systolic and diastolic blood pressure were recorded. At the end of the blood pressure measurement, kidneys were perfused, harvested, and flash-frozen using liquid nitrogen and stored at −80 °C for further analysis.
Measurement of oxidative stress, inflammation, and antioxidant enzyme activities
Oxidative stress parameters such as renal malondialdehyde, urinary 8-isoprostane levels, and urinary antioxidant capacity, were measured by using commercially available assay kits (Cat# 10009055, Cat# 516351 and Cat# 709001; Cayman Chemicals, Ann Arbor, MI). Inflammatory markers, IL-1β (Cat# DY201, R&D biosystems; Minneapolis, MN, USA) and IL-10 (Cat# DY417, R&D biosystems; Minneapolis, MN, USA) concentrations were measured in renal cortical homogenates by using Duo Set ELISA kit according to the manufacturers’ instructions. The activities of antioxidant enzymes, NQO-1 and SOD were measured by using a commercially available assay kit from Abcam (Cat# ab184867, Cambridge, UK) and Cayman chemicals (Cat# 706002, Ann Arbor, MI) respectively, according to the manufacturers’ instructions.
Real-time quantitative polymerase chain reaction and immunoblotting
Total RNA was extracted from the kidney cortex using an RNAeasy mini kit ( Cat# 74104, Qiagen, Hilden, Germany) and used for cDNA synthesis by using RT2 First Strand Kit (Cat# 330404, Qiagen, Hilden, Germany). The polymerase chain reaction for NQO-1 and β-actin was performed with commercially available TaqMan primers (Integrated DNA Technologies, Coralville, IA). The following sequences of the primers were used: NQO-1 (forward: 5’-CCATGTACTCTCTTCAGG-3’ and reverse:5’-GCCAATGCTGTAAACCAGTTG-3’) and β-actin (forward:5’-GATTACTGCTCTGGCTCCTAG-3’ and reverse: 5’- GACTCATCGTACTCTCCTGCTTG-3’).
Immunoblotting in the renal cortical homogenates was performed using the standard lab method as detailed previously (22). Briefly, tissue homogenates were centrifuged at 12000 rpm for 10 minutes at 4 °C and the supernatant was solubilized in Laemmli buffer, resolved by SDS-PAGE, and transferred to polyvinylidene difluoride membranes at 4°C. The membranes were blocked with 5% nonfat dry milk and incubated with antibodies against Nrf2 (1:500, Cat# 16396-1-AP, Proteintech) and NQO-1 (1:500, Cat# bsm-52830R, Bioss) and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) antibody (1:1000, Cat# 2118, Cell Signaling) followed by corresponding horseradish peroxidase conjugated secondary antibodies.
Renal function markers and SKF38393-induced natriuresis
Serum and urine creatinine concentrations were measured with a picric acid-based method (R & D biosystems Minneapolis, MN, USA). Urine albumin concentration was determined by using an ELISA quantification kit, according to the manufacturer’s protocol (Cat# E99-134, Bethyl Laboratories, Inc).
The effect of SKF38393 on sodium excretion was determined by following a previously detailed protocol (23). Briefly, mice were anesthetized as aforementioned and the right jugular vein was catheterized with PE-10 tubing for infusing saline or SKF38393. A midline abdominal incision was performed, and the urinary bladder was catheterized with PE-50 tubing to collect urine samples. The blood was collected simultaneously through the tail vein. Blood plasma was obtained by centrifuging blood samples at 2000 rpm for 15 minutes at 4 °C. Urine and plasma samples were stored at −80 °C for further analysis. Urine and plasma sodium concentrations were measured by atomic absorption spectroscopy (AAnalyst 400, PerkinElmer, Waltham, MA) to determine urinary sodium excretion (UNaV) and fractional excretion of sodium (FENa).
Statistical analysis
All data are expressed as means ± SE. Data were analyzed by using a commercially available statistical software (GraphPad Prism, version 8.1). Differences between the means were evaluated using unpaired student t-test or one-way ANOVA followed by Tukey’s multiple comparisons test, as appropriate. A two-way ANOVA with Bonferroni’s posttest for repeated measure test was used to compare sodium excretion to respective basal in different groups. A probability level of P< 0.05 was selected as an indicator of statistical significance
Results
Effect of BSO and ML385 treatment on body weight, food and water intake and blood pressure
No significant change in body weight (g, control: 33.3 ± 1.2, BSO: 31.6 ± 2.0, BSO+ML385: 30.5 ± 1.9, ML385: 32.5 ± 1.8), food consumption (g/day, control: 4.31 ± 0.49, BSO: 3.99 ± 0.52, BSO+ML385: 3.90 ± 0.51, ML385: 4.38 ± 0.58), and water intake (ml/day, control: 4.0 ± 0.9, BSO: 3.72 ± 0.56, BSO+ML385: 3.56 ± 0.45, ML385: 3.87 ± 0.53) were observed with treatment in all experimental groups. BSO administration alone caused a significant increase in mean arterial pressure (mmHg; Control-70.78 ± 5.63; BSO-81.30 ± 4.39*, *P<0.05 vs. control). Further, co-treatment of BSO and ML385 robustly increased the mean arterial pressure (mmHg; 95.20 ± 9.57*#, *P<0.05 vs. control; #P<0.05 vs BSO) in comparison to control or BSO administered mice. ML385 per se did not affect the mean arterial pressure (76.61 ± 4.92 mmHg) in the mice (figure 1).
Figure 1: Effect of L-buthionine sulfoximine (BSO) and ML385 treatment on blood pressure in mice.
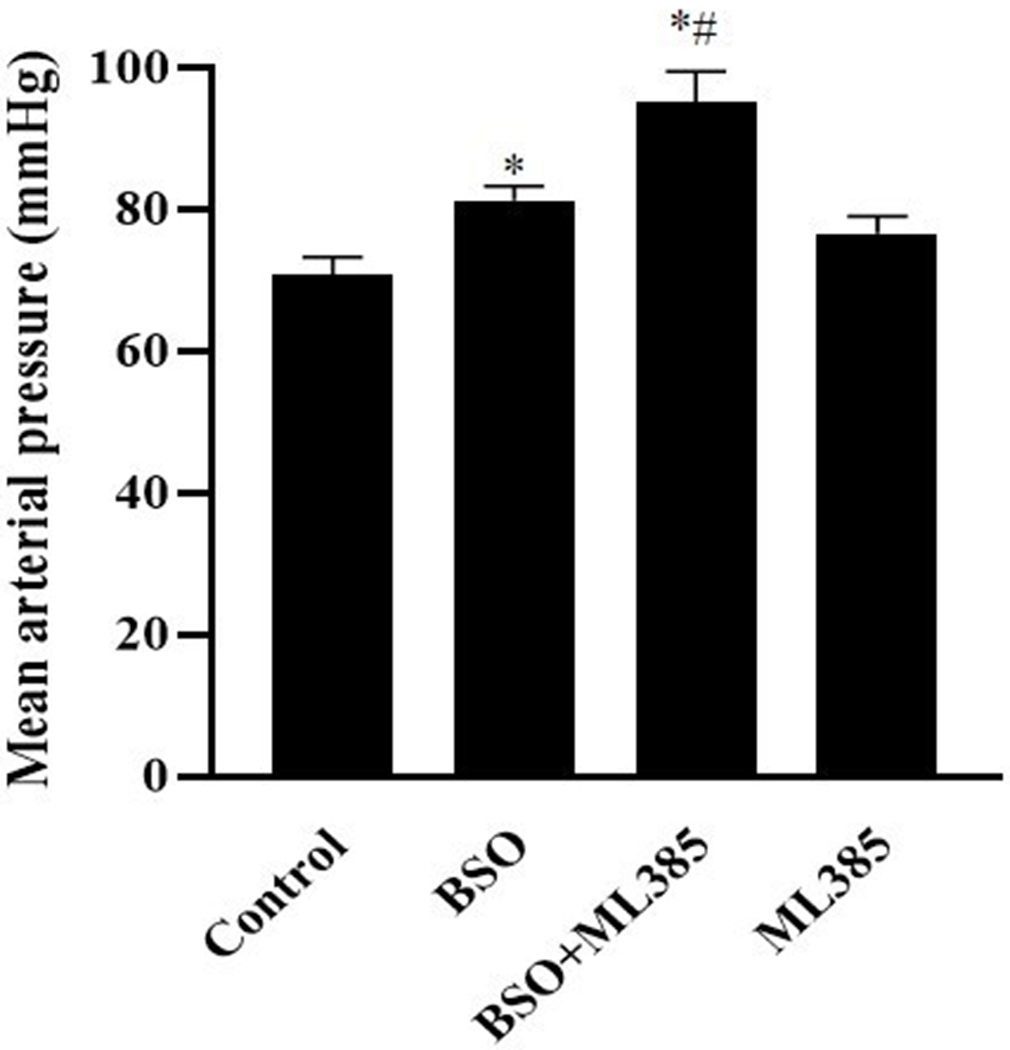
Blood pressure (mmHg) was recorded via intra-arterial catheterization method in control, mice kept on tap water; BSO, mice provided with 10 mmol/L BSO; ML385, mice provided with 10 mg/kg body weight/day of ML385; BSO+ML385, mice provided with both BSO and ML385.
Data are expressed as means ± SE of n=6 mice/group. * P<0.05 vs. control and #P<0.05 vs. BSO.
Effect of BSO and ML385 treatment on oxidative stress and renal function markers
Mice treated with BSO exhibited renal oxidative stress as manifested by increased renal malondialdehyde and urinary 8-isoprostane levels and reduced urinary total antioxidant capacity in comparison to control mice (table 1). ML385 treatment intensified the severity of BSO-induced oxidative stress as demonstrated by the significant increase in renal malondialdehyde and decreased total antioxidant capacity in BSO+ML385 group mice. BSO administration also resulted in impaired renal function as evidenced by increased serum creatinine, urine albumin excretion rate, urine albumin to creatinine ratio, and decreased GFR compared to control mice (table 1). Concomitant treatment of BSO and ML385 increased serum creatinine and albuminuria in comparison to BSO-treated mice indicating aggravation of BSO elicited functional impairments in the kidney. The BSO-induced decrease in GFR was also worsened in the BSO+ML385 combination group (table 1).
Table 1:
Effect of L-buthionine sulfoximine (BSO) and ML385 treatment on oxidative stress and renal function markers
| Parameters | Control | BSO | BSO+ML385 | ML385 |
|---|---|---|---|---|
| Renal malondialdehyde (nmol/mg protein) | 0.35 ± 0.04 | 0.64 ± 0.04* | 0.93± 0.05* | 0.44 ± 0.06 |
| Urinary 8-isoprostane (ng/ml) | 1.04 ± 0.14 | 2.17 ± 0.60* | 1.89 ± 0.26* | 1.03 ± 0.08 |
| Total antioxidant capacity (mmol/L) | 6.08 ± 0.52 | 4.12 ± 0.31 | 2.98 ± 0.26*# | 5.81 ± 0.68 |
| Serum creatinine (mg/dl) | 1.35 ± 0.50 | 2.41 ± 0.55* | 2.65 ± 0.43*# | 1.72 ± 0.39 |
| Urinary albumin excretion rate (µg/day) | 10.77 ± 2.84 | 13.34 ± 2.19* | 18.32 ± 3.89*# | 9.94 ± 1.82 |
| Albumin/Creatinine (mg/g) | 4.35 ± 0.58 | 5.77 ± 0.54 | 8.99 ± 0.89*# | 4.38 ± 0.64 |
| GFR (ml/minute) | 0.23 ± 0.04 | 0.16 ± 0.07 | 0.11 ± 0.03*# | 0.21 ± 0.05 |
Control, mice kept on tap water; BSO, mice provided with 10 mmol/L BSO; ML385, mice provided with 10 mg/kg body weight/day of ML385; BSO+ML385; mice provided with both BSO and ML385. The glomerular filtration rate (GFR) (ml/minute) was calculated from creatinine clearance.
Data are expressed as means ± SE of n=6 mice/group.
P<0.05 vs. control
P <0.05 vs. BSO.
Effect of BSO and ML385 treatment on renal inflammation
As illustrated in figure 2a and 2b, BSO treatment alone did not show any significant alterations on pro-inflammatory and anti-inflammatory cytokines IL-1 β and IL-10, respectively. Co-treatment of BSO and ML385 resulted in the onset of renal inflammation as indicated by significant elevation of IL-1β and a decline in IL-10 levels in cortical homogenates (figure 2a and 2b). ML385 per se did not cause any change in the IL-1β and IL-10 levels (figure 2a and 2b).
Figure 2: Effect of L-buthionine sulfoximine (BSO) and ML385 treatment on inflammatory cytokines in renal cortical homogenates.
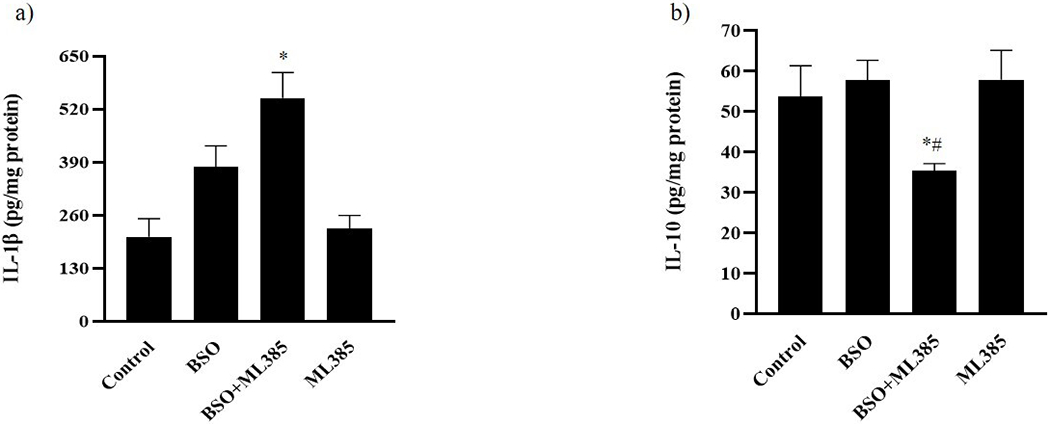
(a) IL-1β and (b) IL-10 levels were measured in pg/mg protein in control, mice kept on tap water; BSO), mice provided with 10 mmol/L BSO; ML385, mice provided with 10 mg/kg body weight/day of ML385; BSO+ML385, mice provided with both BSO and ML385.
Data are expressed as means ± SE of n=6 mice/group. *P<0.05 vs. control and #P<0.05 vs. BSO.
Effect of BSO and ML385 treatment on Nrf2 protein expression and its downstream antioxidant enzymes
As shown in figure 3a, BSO alone did not affect renal Nrf2 protein expression, however, BSO+ML385 co-treatment increased renal protein expression of Nrf2 compared to control mice. The phase II antioxidant enzyme, NQO-1 mRNA, and protein expression increased significantly after BSO treatment. However, ML385 treatment to BSO-administered mice significantly reduced BSO-induced increase in NQO-1 mRNA and protein in these mice (figure 3b and 3c). Also, the activities of NQO-1 and SOD antioxidant enzymes significantly decreased after BSO and ML385 co-treatment in comparison to BSO-treated mice (figure 3d and 3e).
Figure 3: Effect of L-buthionine sulfoximine (BSO) and ML385 treatment on Nrf2 and phase II antioxidant enzymes in renal cortical homogenates.
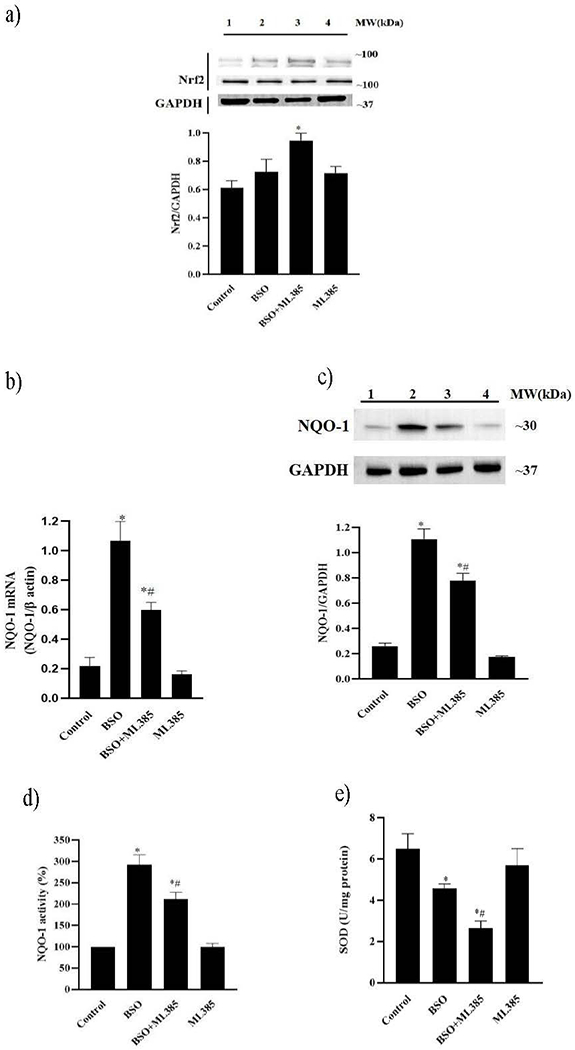
(a) two representative nuclear factor E2-related factor 2 (Nrf2) protein blots and a glyceraldehyde 3-phosphate dehydrogenase (GAPDH) blot. lane 1, control, mice kept on tap water; lane 2, BSO, mice provided with 10 mmol/L BSO; lane 3, BSO+ML385, mice provided with both BSO and ML385.; lane 4, ML385, mice provided with 10 mg/kg body weight/day of ML385; Molecular weight (MW); (b) NAD(P)H: quinone oxidoreductase-1 (NQO-1) mRNA levels, (c) bar representing NQO-1 protein content; top: a representative NQO-1 and GAPDH blot: lane 1, control; lane 2, BSO; lane 3, BSO+ML385; lane 4, ML385 and enzymatic activity of (d) NQO-1 and (e) superoxide dismutase (SOD) in mice renal cortical homogenates.
Data are expressed as means ± SE of n=6 mice/group. *P<0.05 vs. control and #P<0.05 vs. BSO.
Effect of BSO and ML385 treatment on SKF3839-induced natriuresis
The effect of BSO or ML385 alone or in combination was observed on D1R agonist SKF38393-induced sodium excretion. Infusion of SKF38393 caused a significant increase in UNaV and FENa in control and ML385-treated mice (figure 4a and 4b). However, SKF38393 failed to increase UNaV and FENa in BSO alone and BSO+ML385 co-treated mice (figure 4a and 4b).
Figure 4: Effect of L-buthionine sulfoximine (BSO) and ML385 treatment on SKF38393-induced natriuresis.
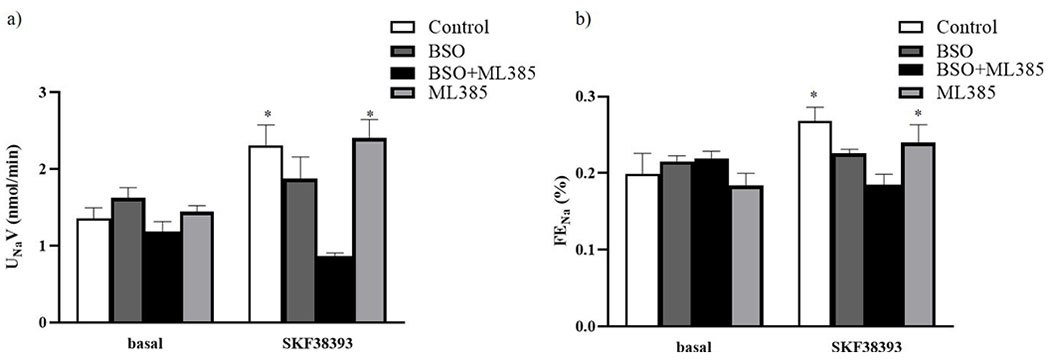
SKF38393-induced a) urinary sodium excretion (UNaV) and (b) fractional excretion of sodium (FENa) in control, mice kept on tap water; BSO, mice provided with 10 mmol/L BSO; ML385, mice provided with 10 mg/kg body weight/day of ML385; BSO+ML385, mice provided with both BSO and ML385.
Data are expressed as means ± SE of n= 6 mice/group. *P<0.05 vs. respective basal.
Discussion
Oxidative stress defined as an imbalance between the generation of oxidants and antioxidant is a key player in the progression of hypertension (24, 25). Although the exact mechanism underlying the generation of reactive oxygen species and perturbation of antioxidant defense remains unclear, an impairment of Nrf2 signaling that regulates a plethora of cytoprotective genes including those encoding antioxidant and phase-II detoxifying enzymes plays an important role (26). Considering the previous reports on the involvement of oxidative stress in the development of hypertension and Nrf2 activation in mitigating oxidative stress (3, 27), we investigated the role of Nrf2 inhibition on oxidative stress-induced renal functional impairment and blood pressure in mice. We used a specific Nrf2 inhibitor ML385, that interferes with Nrf2 binding to ARE sequence and thus, hinders with the normal functioning of the Nrf2 signaling pathway (19, 28). In this study, we showed that under oxidative stress conditions, inhibition of Nrf2 transcription factor led to the suppression of Nrf2 regulated phase II antioxidant enzymes resulting in aggravated renal oxidative stress and inflammation, attenuated kidney function, and elevated blood pressure in mice.
Administration of a pro-oxidant compound, BSO caused an increase in mean arterial pressure in comparison to control group mice. ML385 alone did not change mean arterial pressure however, concomitant treatment of BSO and ML385 resulted in a robust increase in mean arterial pressure. In agreement with earlier findings, BSO administration significantly increased renal malondialdehyde and urinary 8-isoprostane levels accompanied by a decrease in total antioxidant capacity, suggesting the induction of oxidative stress (29). A marked elevation in serum creatinine, urinary albumin, and albumin to creatinine ratio together with decreased GFR was also observed in BSO administered mice. Co-treatment of BSO and ML385 significantly augmented BSO-induced oxidative stress parameters. BSO-induced deterioration of renal function was further intensified as revealed by elevated serum creatinine and albuminuria after Nrf2 inhibition in BSO+ML385 co-treated mice. These results indicate that the impaired Nrf2 function amplified oxidative stress and altered renal function which could be a contributing factor in the development of hypertension.
Oxidative stress is known to modulate numerous factors that contribute to hypertension such as inflammation, imbalance of salt and water homeostasis, and disturbances in the renin-angiotensin-aldosterone system (15, 30, 31). Several studies reported oxidative stress and inflammation as synergistic partners in the pathogenesis of hypertension (4). Therefore, we studied the effect of BSO and ML385 alone and their combination on renal inflammation. The results demonstrated that BSO alone did not cause any significant alterations in renal IL-1β and IL-10 levels. In contrast, Nrf2 inhibition by ML385 treatment in BSO administered mice resulted in a significant increase in IL-1β and a decrease in IL-10 levels in these mice suggesting the role of Nrf2 inhibition in renal inflammation which could contribute to the rise of blood pressure.
Nrf2 via activation of phase II antioxidant genes protects the cells against oxidative stress (32, 33). Herein, we measured the expression of Nrf2 and its downstream target genes in ML385 and BSO+ML385-treated mice. We found that oxidative stress-induced by BSO resulted in an insignificant increase in the Nrf2 protein but a significant rise in phase II antioxidant enzyme NQO-1 mRNA, protein expression, and its activity in the renal cortex. The SOD activity decreased significantly after BSO treatment pointing a maladaptive response to oxidative stress in these mice. ML385 per se did not affect the expression of Nrf2 and phase II antioxidant enzyme. However, BSO+ML385 co-treatment showed a significant increase in Nrf2 expression compared to control mice but a significant reduction in NQO-1 mRNA/protein expression and the enzymatic activities of NQO-1 and SOD in comparison to BSO-treated mice. Decreased expression/activities of phase II antioxidant enzymes after ML385 treatment in BSO-treated mice suggests the suppression of antioxidant defense in these mice. Taken together, these results illustrate the effectiveness of ML385 in inhibiting Nrf2 activity and point to the role of impaired Nrf2 function and phase II antioxidant defense in the pathogenesis of oxidative stress and inflammation in mice kidney.
Increased generation of reactive oxygen species and secretion of inflammatory cytokines from immune and renal epithelial cells has been implicated in disruption of renal sodium handling leading to alterations in pressure natriuresis and sodium and water balance that could lead to blood pressure elevation (4, 29, 34). Studies reported that the reactive oxygen species attenuates renal proximal tubular D1R function, which disrupts sodium regulation and leads to hypertension (35–37). In our study, we tried to investigate the role of Nrf2 inhibition in D1R function as it relates to oxidative stress. Concordant with our previous findings, an impairment in SKF38393 mediated natriuretic effect was observed after BSO administration reflecting the loss of D1R function to regulate sodium excretion (14, 29). The effect of ML385-mediated inhibition of Nrf2 signaling on urinary sodium excretion and FENa was also studied. The concomitant treatment of BSO and ML385 resulted in a prominent decline in urinary sodium and FENa in response to SKF38393 infusion compared to BSO alone treatment. The failure of SKF38393 to promote sodium excretion in BSO+ML385-treated mice might be due to loss of renal D1R function as a result of aggravated oxidative stress caused by impaired Nrf2 and phase II antioxidant enzymes. Therefore, we suggest that inhibition of Nrf2 function could lead to oxidative stress-mediated loss of renal D1R function that could be a significant risk factor for oxidative stress-associated hypertension.
The present study has translational potential as the data provide evidence in support of Nrf2 transcription factor as a therapeutic target in treating hypertension. The use of drugs/chemicals that directly activate Nrf2 and phase II antioxidant enzymes could be of great benefit in treating hypertensive patients by rescuing against oxidative stress-induced renal function alterations and renal inflammation. However, the interpretation of inflammation data warrants caution as we did not measure IL-6 which is known to play an important role in renal proximal tubular inflammation especially following the BSO administration (38).
To summarize, the present investigation demonstrates a direct role of Nrf2 inhibition on renal function impairment and in the development of hypertension. BSO administration induced oxidative stress and renal dysfunction in mice which contributes to a rise in blood pressure. BSO-induced oxidative stress per se activates Nrf2 and phase II antioxidant enzymes. However, under oxidative stress conditions, Nrf2 transcription factor inhibition by ML385 resulted in exacerbation of oxidative stress and inflammation that further deteriorates renal function, sodium excretion and aggravates hypertension in mice via diminution of the phase II antioxidant defense mechanism.
Acknowledgments
This work was supported by National Institutes of Health (National Heart, Lung and Blood Institute) Grant HL-139808 to M.F.L.
Footnotes
Disclosure of interest
The authors declare no conflict of interest.
References
- 1.Wu CY, Hu HY, Chou YJ, Huang N, Chou YC, Li CP. High Blood Pressure and All-Cause and Cardiovascular Disease Mortalities in Community-Dwelling Older Adults. Medicine (Baltimore). 2015;94(47):e2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.James PA, Oparil S, Carter BL, Cushman WC, Dennison-Himmelfarb C, Handler J, et al. 2014 evidence-based guideline for the management of high blood pressure in adults: report from the panel members appointed to the Eighth Joint National Committee (JNC 8). JAMA. 2014;311(5):507–520. [DOI] [PubMed] [Google Scholar]
- 3.Briones AM, Touyz RM. Oxidative stress and hypertension: current concepts. Curr Hypertens Rep. 2010;12(2):135–142. [DOI] [PubMed] [Google Scholar]
- 4.Crowley SD. The cooperative roles of inflammation and oxidative stress in the pathogenesis of hypertension. Antioxid Redox Signal. 2014;20(1):102–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Steven S, Frenis K, Oelze M, Kalinovic S, Kuntic M, Bayo Jimenez MT, et al. Vascular Inflammation and Oxidative Stress: Major Triggers for Cardiovascular Disease. Oxid Med Cell Longev. 2019;2019:7092151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dinh QN, Drummond GR, Sobey CG, Chrissobolis S. Roles of inflammation, oxidative stress, and vascular dysfunction in hypertension. Biomed Res Int. 2014;2014:406960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kaspar JW, Niture SK, Jaiswal AK. Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic Biol Med. 2009;47(9):1304–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Luo Z, Aslam S, Welch WJ, Wilcox CS. Activation of nuclear factor erythroid 2-related factor 2 coordinates dimethylarginine dimethylaminohydrolase/PPAR-gamma/endothelial nitric oxide synthase pathways that enhance nitric oxide generation in human glomerular endothelial cells. Hypertension. 2015;65(4):896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang Z, Han N, Zhao K, Li Y, Chi Y, Wang B. Protective effects of pyrroloquinoline quinine against oxidative stress-induced cellular senescence and inflammation in human renal tubular epithelial cells via Keap1/Nrf2 signaling pathway. Int Immunopharmacol. 2019;72:445–453. [DOI] [PubMed] [Google Scholar]
- 10.Ma JQ, Ding J, Xiao ZH, Liu CM. Puerarin ameliorates carbon tetrachloride-induced oxidative DNA damage and inflammation in mouse kidney through ERK/Nrf2/ARE pathway. Food Chem Toxicol. 2014;71:264–271. [DOI] [PubMed] [Google Scholar]
- 11.Tu W, Wang H, Li S, Liu Q, Sha H. The Anti-Inflammatory and Anti-Oxidant Mechanisms of the Keap1/Nrf2/ARE Signaling Pathway in Chronic Diseases. Aging Dis. 2019;10(3):637–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Barancik M, Gresova L, Bartekova M, Dovinova I. Nrf2 as a key player of redox regulation in cardiovascular diseases. Physiol Res. 2016;65 Suppl 1:S1–S10. [DOI] [PubMed] [Google Scholar]
- 13.Senanayake GV, Banigesh A, Wu L, Lee P, Juurlink BH. The dietary phase 2 protein inducer sulforaphane can normalize the kidney epigenome and improve blood pressure in hypertensive rats. Am J Hypertens. 2012;25(2):229–235. [DOI] [PubMed] [Google Scholar]
- 14.Banday AA, Lokhandwala MF. Transcription factor Nrf2 protects renal dopamine D1 receptor function during oxidative stress. Hypertension. 2013;62(3):512–517. [DOI] [PubMed] [Google Scholar]
- 15.Javkhedkar AA, Quiroz Y, Rodriguez-Iturbe B, Vaziri ND, Lokhandwala MF, Banday AA. Resveratrol restored Nrf2 function, reduced renal inflammation, and mitigated hypertension in spontaneously hypertensive rats. Am J Physiol Regul Integr Comp Physiol. 2015;308(10):R840–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bai J, Yu XJ, Liu KL, Wang FF, Jing GX, Li HB, et al. Central administration of tert-butylhydroquinone attenuates hypertension via regulating Nrf2 signaling in the hypothalamic paraventricular nucleus of hypertensive rats. Toxicol Appl Pharmacol. 2017;333:100–109. [DOI] [PubMed] [Google Scholar]
- 17.Hsu CN, Lin YJ, Yu HR, Lin IC, Sheen JM, Huang LT, et al. Protection of Male Rat Offspring against Hypertension Programmed by Prenatal Dexamethasone Administration and Postnatal High-Fat Diet with the Nrf2 Activator Dimethyl Fumarate during Pregnancy. Int J Mol Sci. 2019;20(16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu KLH, Wu CW, Chao YM, Hung CY, Chan JYH. Impaired Nrf2 regulation of mitochondrial biogenesis in rostral ventrolateral medulla on hypertension induced by systemic inflammation. Free Radic Biol Med. 2016;97:58–74. [DOI] [PubMed] [Google Scholar]
- 19.Singh A, Venkannagari S, Oh KH, Zhang YQ, Rohde JM, Liu L, et al. Small Molecule Inhibitor of NRF2 Selectively Intervenes Therapeutic Resistance in KEAP1-Deficient NSCLC Tumors. ACS Chem Biol. 2016;11(11):3214–3225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu X, Zhu Q, Zhang M, Yin T, Xu R, Xiao W, et al. Isoliquiritigenin Ameliorates Acute Pancreatitis in Mice via Inhibition of Oxidative Stress and Modulation of the Nrf2/HO-1 Pathway. Oxid Med Cell Longev. 2018;2018:7161592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang SC, Ren S, Rau CD, Wang JJ. Isoproterenol-Induced Heart Failure Mouse Model Using Osmotic Pump Implantation. Methods Mol Biol. 2018;1816:207–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Banday AA, Fazili FR, Marwaha A, Lokhandwala MF. Mitogen-activated protein kinase upregulation reduces renal D1 receptor affinity and G-protein coupling in obese rats. Kidney Int. 2007;71(5):397–406. [DOI] [PubMed] [Google Scholar]
- 23.Marwaha A, Banday AA, Lokhandwala MF. Reduced renal dopamine D1 receptor function in streptozotocin-induced diabetic rats. Am J Physiol Renal Physiol. 2004;286(3):F451–457. [DOI] [PubMed] [Google Scholar]
- 24.Baradaran A, Nasri H, Rafieian-Kopaei M. Oxidative stress and hypertension: Possibility of hypertension therapy with antioxidants. J Res Med Sci. 2014;19(4):358–367. [PMC free article] [PubMed] [Google Scholar]
- 25.Rodrigo R, Gonzalez J, Paoletto F. The role of oxidative stress in the pathophysiology of hypertension. Hypertens Res. 2011;34(4):431–440. [DOI] [PubMed] [Google Scholar]
- 26.Lopes RA, Neves KB, Tostes RC, Montezano AC, Touyz RM. Downregulation of Nuclear Factor Erythroid 2-Related Factor and Associated Antioxidant Genes Contributes to Redox-Sensitive Vascular Dysfunction in Hypertension. Hypertension. 2015;66(6):1240–1250. [DOI] [PubMed] [Google Scholar]
- 27.Sun W, Yan C, Frost B, Wang X, Hou C, Zeng M, et al. Pomegranate extract decreases oxidative stress and alleviates mitochondrial impairment by activating AMPK-Nrf2 in hypothalamic paraventricular nucleus of spontaneously hypertensive rats. Sci Rep. 2016;6:34246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xiao D, Yuan D, Tan B, Wang J, Liu Y, Tan B. The Role of Nrf2 Signaling Pathway in Eucommia ulmoides Flavones Regulating Oxidative Stress in the Intestine of Piglets. Oxid Med Cell Longev. 2019;2019:9719618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Banday AA, Lokhandwala MF. Transcriptional regulation of renal dopamine D1 receptor function during oxidative stress. Hypertension. 2015;65(5):1064–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Korsager Larsen M, Matchkov VV. Hypertension and physical exercise: The role of oxidative stress. Medicina (Kaunas). 2016;52(1):19–27. [DOI] [PubMed] [Google Scholar]
- 31.Tanase DM, Gosav EM, Radu S, Ouatu A, Rezus C, Ciocoiu M, et al. Arterial Hypertension and Interleukins: Potential Therapeutic Target or Future Diagnostic Marker? Int J Hypertens. 2019;2019:3159283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yang JH, Shin BY, Han JY, Kim MG, Wi JE, Kim YW, et al. Isorhamnetin protects against oxidative stress by activating Nrf2 and inducing the expression of its target genes. Toxicol Appl Pharmacol. 2014;274(2):293–301. [DOI] [PubMed] [Google Scholar]
- 33.Lee JM, Johnson JA. An important role of Nrf2-ARE pathway in the cellular defense mechanism. J Biochem Mol Biol. 2004;37(2):139–143. [DOI] [PubMed] [Google Scholar]
- 34.Norlander AE, Madhur MS. Inflammatory cytokines regulate renal sodium transporters: how, where, and why? Am J Physiol Renal Physiol. 2017;313(2):F141–F4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zeng C, Felder RA, Jose PA. A new approach for treatment of hypertension: modifying D1 dopamine receptor function. Cardiovasc Hematol Agents Med Chem. 2006;4(4):369–377. [DOI] [PubMed] [Google Scholar]
- 36.Banday AA, Marwaha A, Tallam LS, Lokhandwala MF. Tempol reduces oxidative stress, improves insulin sensitivity, decreases renal dopamine D1 receptor hyperphosphorylation, and restores D1 receptor-G-protein coupling and function in obese Zucker rats. Diabetes. 2005;54(7):2219–2226. [DOI] [PubMed] [Google Scholar]
- 37.Cuevas S, Villar VA, Jose PA, Armando I. Renal dopamine receptors, oxidative stress, and hypertension. Int J Mol Sci. 2013;14(9):17553–17572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Banday AA, Lokhandwala MF. Renal dopamine oxidation and inflammation in high salt fed rats. J. Am. Heart Assoc. 2020;9(1): e014977. [DOI] [PMC free article] [PubMed] [Google Scholar]