

Abstract
DNAzymes were previously identified by in vitro selection for a variety of chemical reactions, including several biologically relevant peptide modifications. However, finding DNAzymes for peptide lysine acylation is a substantial challenge. By using suitably reactive aryl ester acyl donors as the electrophiles, here we used in vitro selection to identify DNAzymes that acylate amines, including lysine side chains of DNA-anchored peptides. Some of the DNAzymes can transfer a small glutaryl group to an amino group. These results expand the scope of DNAzyme catalysis and suggest the future broader applicability of DNAzymes for sequence-selective lysine acylation of peptide and protein substrates.
Graphical Abstract
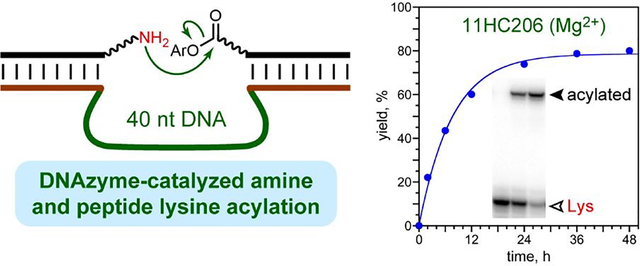
In vitro selection identifies DNAzymes that acylate amino groups, including Lys in tethered peptides, and in some cases by transfer of a small glutaryl group.
INTRODUCTION
DNAzymes, also called DNA enzymes and deoxyribozymes, are specific DNA sequences that catalyze chemical reactions, similar to protein enzymes as catalytic amino acid sequences.1–8 Nature evolved both protein enzymes and RNA enzymes (ribozymes), but to date all DNAzymes have been identified in the laboratory by in vitro selection.9–13 The earliest-reported and most-studied DNAzymes catalyze RNA cleavage by transesterification at phosphorus.1–4,14–18 Since then, DNAzymes for a range of reactions and substrates have been found. Our laboratory is interested in DNAzymes that catalyze reactions relevant to protein post-translational modifications (PTMs),19,20 such as phosphorylation,21–23 dephosphorylation,24 and amide hydrolysis.25,26 One such reaction is lysine (Lys) acylation, where Lys acetylation is critical for histones and in other contexts,27–30 and many longer-chain Lys acylation PTMs31–33 such as malonylation,34,35 succinylation,34,36 and glutarylation37,38 have been discovered yet are poorly understood.39 As an alternative to approaches that include introduction of Lys analogues,40–43 nonsense codon suppression,44–49 bottom-up ligation-based assembly strategies,50–52 or enzymatic methods that typically require creation of a non-native protein by insertion of a specific target sequence,53–58 DNAzymes are promising for top-down introduction of Lys acylation PTMs onto intact native proteins,59–65 but only if DNAzymes can be identified with the fundamental catalytic ability of Lys acylation.
Toward this goal, we previously reported the first DNAzymes that catalyze Lys modification of any kind.66 We used 5′-phosphorimidazolide (5′-Imp) DNA as the electrophile, resulting in the formation of a Lys-phosphoramidite bond (Figure 1A). From that study, a key lesson was the need for a suitably reactive electrophile to react with the amine nucleophile, a consideration that outweighed the value of highly preorganizing the two substrates. Therefore, to achieve Lys acylation in our present work, we carefully considered our options for the acyl donor electrophile. Arguably the most straightforward choice is a thioester, considering that nature often uses thioesters as acyl donors, including for amine acylation (Figure 1B). However, in several new in vitro selection efforts, some of which included modified DNA nucleotides that previously led to amide-hydrolyzing DNAzymes,26 we were unable to identify any amine-acylating DNAzymes using thioesters. We therefore turned to aryl esters (Figure 1C) as acyl donors, among other considerations noting their tunable electrophilicity. Here we report the outcome of these in vitro selection experiments, culminating in new DNAzymes that catalyze Lys acylation of DNA-anchored peptide substrates.
Figure 1.
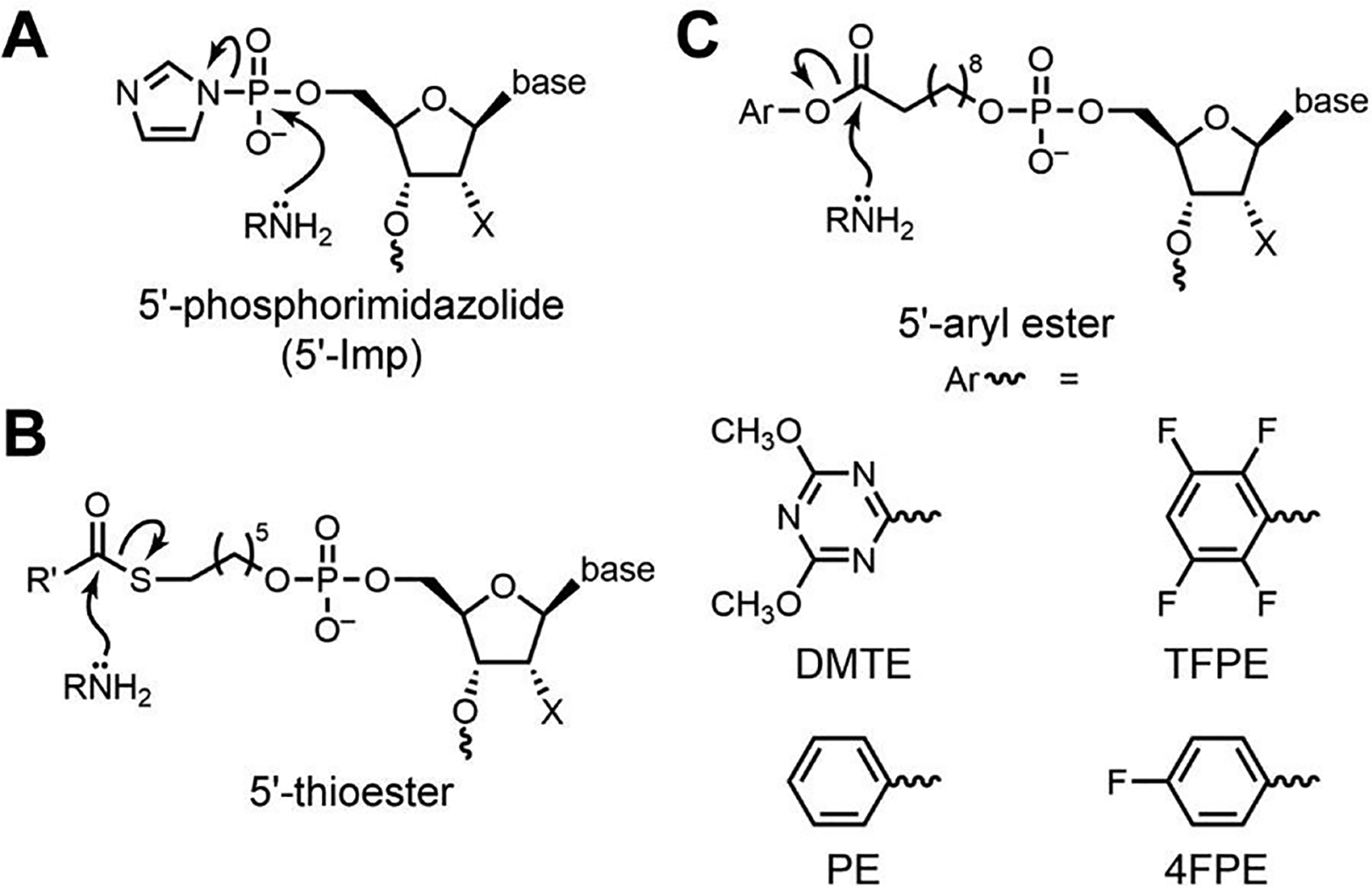
Electrophilic reaction partners for amine nucleophiles in DNAzyme-catalyzed reactions. (A) 5′-Phosphorimidazolide (5′-Imp) DNA, for which we previously found DNAzymes that catalyze Lys-phosphoramidite formation.66 (B) 5′-Thioester DNA, for which here we were unable to identify any amine acylation DNAzymes. (C) 5′-Aryl ester DNA, for which here we describe new DNAzymes that catalyze amine acylation, including with Lys peptides. The DMTE, TFPE, PE, and 4FPE aryl ester substrates were evaluated during this study. DMTE = 4,6-dimethoxy-1,3,5-triazin-2-yl ester; TFPE = 2,3,5,6-tetrafluorophenyl ester; PE = phenyl ester; 4FPE = 4-fluorophenyl ester.
RESULTS AND DISCUSSION
Thioester Acyl Donor Electrophile.
We first performed a set of in vitro selection experiments with an amine nucleophile and a thioester acyl donor electrophile, where both substrates were conjugated to DNA anchor oligonucleotides for standard Watson-Crick binding to the initially random DNAzyme pool (Figure S1). Each experiment used either the four canonical DNA nucleotides, or dT was replaced with one of several modified nucleotides as we reported for DNAzyme-catalyzed amide hydrolysis.26 However, in all cases, after 10 selection rounds no amine acylation activity was observed. We concluded that a thioester is insufficiently reactive as an electrophile to allow the identification of amine-acylating DNAzymes, and a more reactive acyl donor is required.
Highly Reactive Aryl Ester Acyl Donor Electrophiles.
We then performed in vitro selection experiments with acyl donor oligonucleotides activated in situ from their 5′-carboxylic acid (5′-CO2H) precursors using two common amide-forming coupling reagents, 4-(4,6-dimethoxy-1,3,5-triazin-2-yl)-4-methylmorpholinium chloride (DMT-MM)67 or the combination of the water-soluble 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDC) and 2,3,5,6-tetrafluorophenol (TFP).68–71 Before performing in vitro selection, the resulting DMT and TFP esters (here abbreviated DMTE and TFPE; structures in Figure 1C) were assayed for their uncatalyzed background reactivities, using a DNA splint complementary to the DNA-anchored acyl donor and the simple DNA-anchored amine nucleophile (DNA-C3-NH2; Figure 2A,B). Both DMTE and TFPE led to relatively high uncatalyzed background reactivity, with substantial formation of acylation product; e.g., 34% amide formation in 0.5 min (DMTE) and 15% amide formation in 0.5 min (TFPE), each assessed at pH 7.0. Nevertheless, in vitro selection still had the potential to lead to DNAzymes with rate enhancement above this uncatalyzed background reaction. We therefore proceeded to perform in vitro selection experiments using the DMTE and TFPE acyl donors with the DNA-C3-NH2 substrate (Figure 2C).
Figure 2.
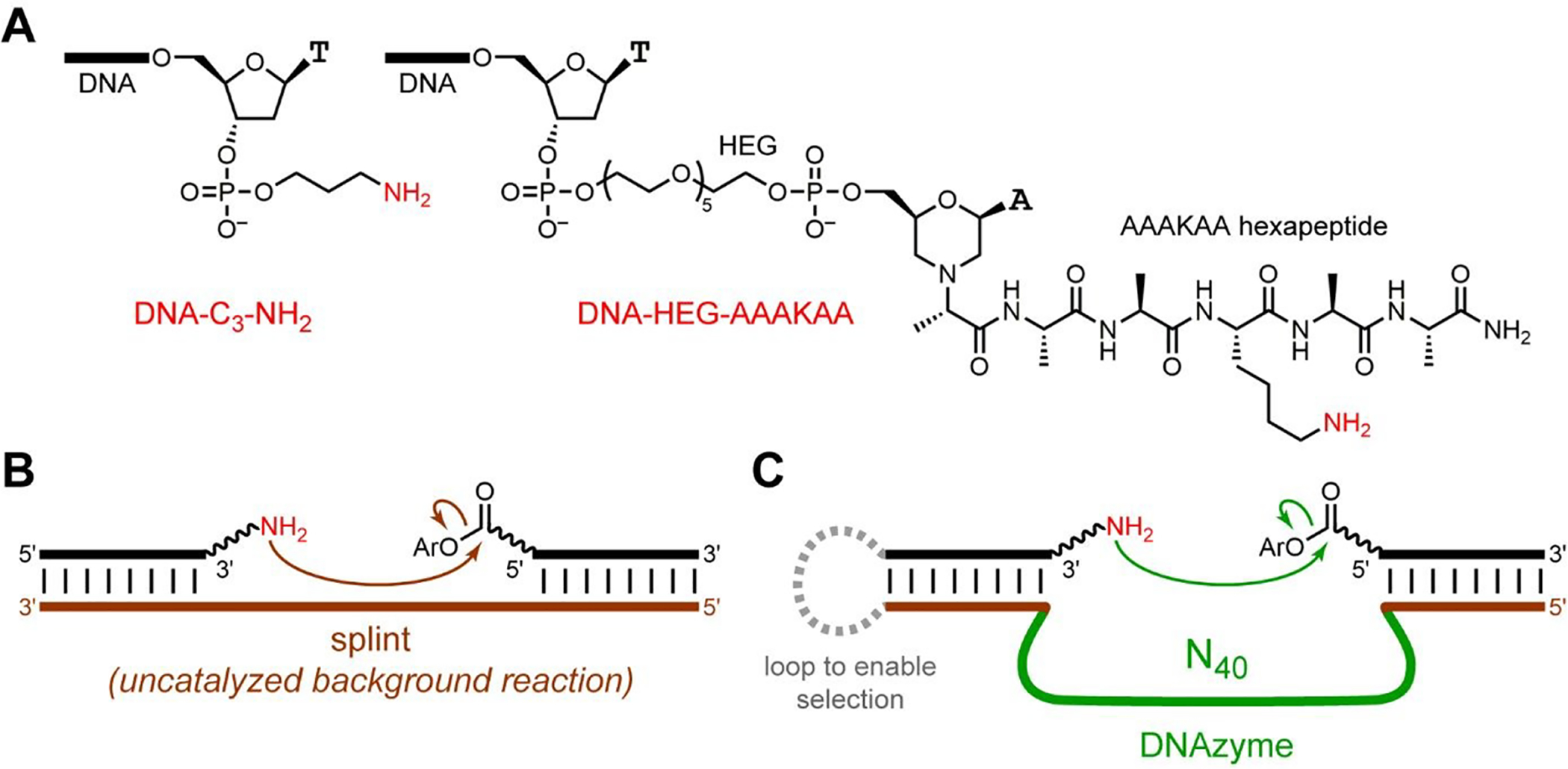
In vitro selection for identifying amine-acylating DNAzymes. (A) Structures of the amine nucleophiles, DNA-C3-NH2 and DNA-HEG-AAAKAA. The latter is formed by reductive amination with the free α-NH2 of the AAAKAA hexapeptide. The HEG-hexapeptide tether is longer than the C3 tether by 35 atoms. (B) Uncatalyzed, splinted background reaction between DNA-anchored amine and aryl ester. (C) Key step of in vitro selection. See Figure S2 and ESI text for details. Any DNAzyme sequences that catalyze amine acylation by the 5′-aryl ester oligonucleotide grow larger by the size of that oligonucleotide, which allows PAGE-shift separation of those DNAzyme sequences. The DNA population is therefore enriched in catalytically active sequences in each round. Iteration for multiple selection rounds is required because some noncatalytic sequences can also survive any particular selection round. The connecting loop on the left side is not included when individual DNAzymes are assayed. The linker joining the aryl ester and the 5′-end of the oligonucleotide is (CH2)9 and a 5′-phosphate.
These in vitro selection experiments used N40 initially random regions (where N40 is a compromise between longer random regions that may be able to form more complex structures, and shorter random regions for which more of sequence space is explored72) along with incubation conditions identical to those used for the background assays. For in vitro selection using the DMTE acyl donor, these incubation conditions included 50 mM DMT-MM, 100 mM MOPS, pH 7.0, 1 mM ZnCl2, and 150 mM NaCl at 37 °C for 2 h. For the TFPE acyl donor, these conditions included 50 mM each EDC and TFP, 100 mM MOPS, pH 7.0, 40 mM MgCl2, 20 mM MnCl2, 1 mM ZnCl2, and 150 mM NaCl at 37 °C for 2 h. For both selection experiments using the DNA-C3-NH2 substrate, the pool yield increased during the selection rounds to 37% (DMTE) or 13% (TFPE) at round 4 (Figure S3A), at which point individual DNA sequences were identified by cloning and sequencing (Figure S4A). For each selection, however, the emergent DNA sequences had no rate enhancement above the uncatalyzed, splinted background reaction under the same incubation conditions. We concluded that each individual DNA sequence likely adopts a combination of secondary and tertiary structure that merely recapitulates a complementary splint. Apparently, rate enhancement beyond the splinting effect cannot be achieved because the DMTE or TFPE electrophile is too reactive.
The same outcome of finding DNA sequences that have no rate enhancement was found for the DMTE and TFPE acyl donors when the amine substrate was instead a hexa(ethylene glycol) [HEG]-tethered AAAKAA hexapeptide that included a single Lys residue (Figures 2A, S3B, and S4B). Therefore, using the considerably less preorganized and presumably less reactive DNA-HEG-AAAKAA substrate did not suppress the too-high background reactivity of the DMTE and TFPE acyl donors.
Intermediate-Reactivity Aryl Ester Acyl Donor Electrophiles and Simple Amine Nucleophile (DNA-C3-NH2).
To this point in our efforts, the observed selection outcomes were divergent. With a thioester acyl donor, no DNAzymes were found due to the insufficiently reactive electrophile. In contrast, with DMTE or TFPE as the acyl donors, specific DNA sequences emerged from the selection process, but they lacked rate enhancement beyond a splint because these electrophiles were too reactive. Therefore, we turned our attention to acyl donors with intermediate reactivity. TFP forms an aryl ester, the parent compound of which is the simple phenyl ester, and the DMT ester formed upon activation of a carboxylic acid by DMT-MM is a multiply substituted aryl ester. We therefore investigated the phenyl ester (PE) and 4-fluorophenyl ester (4FPE) acyl donor substrates (Figure 1C). For these experiments, we decided to synthesize and purify each new 5′-aryl ester oligonucleotide substrate rather than rely upon in situ activation, as we did with the reagents DMT-MM and EDC/TFP because of the high reactivity of the corresponding DMT and TFP esters. The PE and 4FPE oligonucleotide substrates were synthesized from the 5′-CO2H oligonucleotide, EDC, and the appropriate phenol derivative, followed by HPLC purification (Figure S5).
With the PE and 4FPE 5′-aryl ester oligonucleotide substrates in hand, we first evaluated their hydrolytic stabilities and uncatalyzed, splinted background reactivities with the DNA-C3-NH2 substrate under likely incubation conditions for in vitro selection, with quantitative details provided in the ESI (Table S1). Based on the data, we chose two particular incubation conditions for each acyl donor substrate: lower pH of 7.5 (70 mM HEPES, pH 7.5, 40 mM MgCl2, 20 mM MnCl2, 1 mM ZnCl2, and 150 mM NaCl at 37 °C for 16 h) and higher pH of 9.0 (50 mM CHES, pH 9.0, 40 mM MgCl2, and 150 mM NaCl at 37 °C for 16 h). Each of Mg2+, Mn2+, and Zn2+ was included at pH 7.5 on the basis of our many prior successful DNAzyme selection efforts using these metal ions at pH 7.5. However, at pH 9.0 only Mg2+ can be included, because Mn2+ oxidizes and Zn2+ precipitates at this higher pH. With the more reactive 4FPE substrate at the higher pH of 9.0, the acyl donor oligonucleotide was still 74% intact after 16 h, with only 3.5% splinted background yield (Table S1). All other substrate and pH combinations had even higher intact acyl donor and even lower background yield.
With two acyl donor oligonucleotide substrates (PE and 4FPE) and two incubation conditions (pH 7.5 and 9.0) per substrate, the four selection experiments were each iterated for 7–8 rounds (Figure 3). For each substrate, the pH 7.5 selection gave a substantial and promising increase in pool yield as the rounds progressed; these selections were cloned after round 8 (PE) and 7 (4FPE). Substantial amine acylation activity was observed for many of the resulting individual DNAzymes (Figures 4A and S6; sequences in Figure S4C). The PE and 4FPE selections each gave four DNAzymes, each with ~50% yield in 24 h. The highest rate enhancements, calculated by taking single-turnover kobs for the DNAzyme and dividing by kbkgd for the uncatalyzed, splinted background reaction using the complementary DNA splint, were 1100 and 760 (each ~103) for the 7FN216 and 7FN202 DNAzymes, respectively (Figure 4B), which both use the 4FPE substrate at pH 7.5.73
Figure 3.
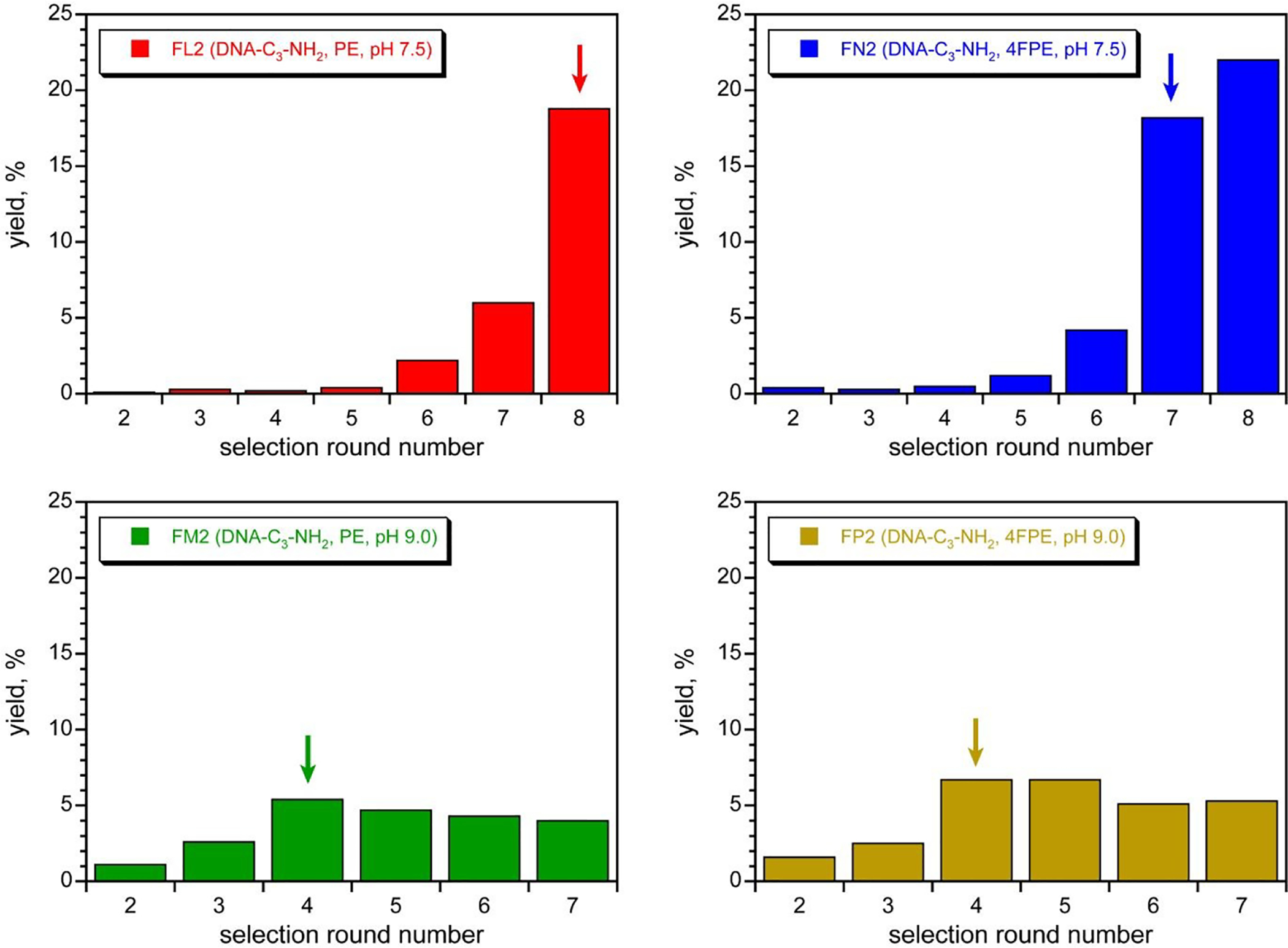
Selection progressions using the DNA-C3-NH2 nucleophile with the PE and 4FPE acyl donors. See text for details of incubation conditions. Arrows mark the cloned rounds. In all cases, the round 1 yield was not quantified because nonradiolabeled pool was used.
Figure 4.
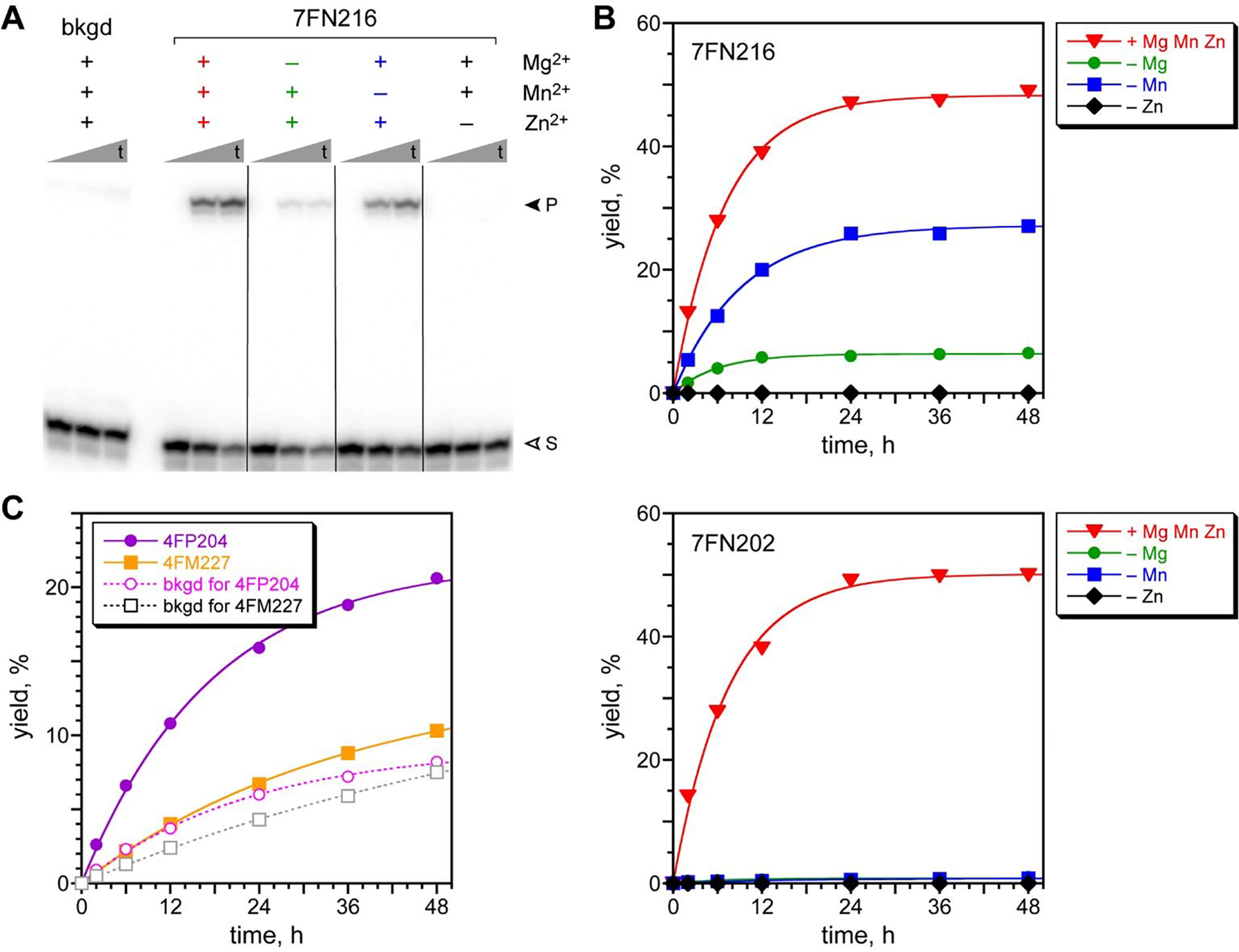
Assays of DNAzymes identified by in vitro selection using the DNA-C3-NH2 substrate. (A) Representative PAGE image for the 7FN216 DNAzyme with its 4FPE acyl donor substrate. Bkgd = complementary DNA splint in place of DNAzyme, to assess the uncatalyzed background reaction. Incubation conditions: 70 mM HEPES, pH 7.5, combinations of 40 mM MgCl2, 20 mM MnCl2, and 1 mM ZnCl2 as indicated, and 150 mM NaCl at 37 °C. The background reaction was with Mg2+/Mn2+/Zn2+. Shown are representative timepoints (t = 0.5 min, 6 h, 48 h; S = substrate, P = product). (B) Kinetic plots for 7FN216 and 7FN202, which have different metal ion dependence. Plots for the other two 7FN2 DNAzymes (4FPE substrate) and all four 8FL2 DNAzymes (PE substrate) are in Figure S6. kobs values (h−1 ± standard deviation, each n = 3, with Mg2+/Mn2+/Zn2+): 7FN216, 0.19 ± 0.06; 7FN202, 0.13 ± 0.02; background (kbkgd), (1.7 ± 0.1) × 10−4. kobs values for the other six DNAzymes are 0.05–0.09 h−1. (C) Kinetic plots for 4FM227 (PE substrate), 4FP204 (4FPE substrate), and a complementary DNA splint as background reaction for each substrate. Incubation conditions: 50 mM CHES, pH 9.0, 40 mM MgCl2, and 150 mM NaCl at 37 °C. kobs values (h−1, n = 1): 4FM227, 0.028; 4FP204, 0.057; background (kbkgd): PE, 0.014; 4FPE, 0.044. Data was similar for the other eleven 4FM2 and 4FP2 DNAzymes (not shown).
In contrast, the pH 9.0 selection for each substrate gave only a modest increase in pool yield as the rounds progressed (Figure 3). Each selection was cloned after round 4 and gave six (PE) or seven (4FPE) DNAzymes, with only 5–20% yields. Each individual DNAzyme had low rate enhancement of at most 2 (representative data in Figure 4C; sequences in Figures S4D), and these DNAzymes were not studied further. An immediate conclusion is that the two pH 7.5 selections, for which the incubation conditions led to lower background yields (0.3–0.6%; Table S1), were more successful at providing DNAzymes than the two pH 9.0 selections, which had higher background yields (2.4–3.5%).
We investigated the metal ion dependence of the eight DNAzymes that use DNA-C3-NH2 and the PE or 4FPE substrates at pH 7.5. By evaluating each DNAzyme with all possible combinations of 40 mM Mg2+, 20 mM Mn2+, and 1 mM Zn2+, which were the concentrations of each ion that were used during the selection process, we found two types of metal ion dependence (Figure 4). All eight DNAzymes worked optimally when all three of Mg2+, Mn2+, and Zn2+ were included. Five of the eight DNAzymes, such as 7FN216, retained substantial activity with only Mg2+ and Zn2+ (omitting Mn2+) and had greatly reduced yield with only Mn2+ and Zn2+ (omitting Mg2+), whereas omitting Zn2+ led to no activity. The other three DNAzymes, such as 7FN202, needed all three of Mg2+, Mn2+, and Zn2+ for catalysis.
For one representative DNAzyme from each of the pH 7.5 selection experiments, the acylation product was isolated by PAGE, and its expected mass was confirmed by MALDI mass spectrometry (see ESI for numerical data).In addition, all eight of the pH 7.5 DNAzymes were assayed using, as a negative control in place of DNA-C3-NH2, an unmodified DNA oligonucleotide lacking the pendant C3-NH2 at its 3′-end. In each case, no product formation was observed (<0.2%; data not shown), consistent with nucleophilic reactivity of the amino group in the DNAzyme-catalyzed acylation reaction.
Intermediate-Reactivity Aryl Ester Acyl Donors and Peptide Lysine Nucleophile (DNA-HEG-AAAKAA).
With success in identifying DNAzymes that acylate the simple amine nucleophile DNA-C3-NH2, especially at the lower pH of 7.5, we shifted our attention to the substrate that presents a peptide Lys nucleophile, DNA-HEG-AAAKAA, where our ultimate goal is DNAzyme-catalyzed acylation of peptide and protein Lys residues. Unsurprisingly based on our previous report with DNA-catalyzed Lys phosphoramidate formation,66 none of the above-described DNAzymes identified using the DNA-C3-NH2 substrate had any detectable activity (<0.2%) when tested with DNA-HEG-AAAKAA (data not shown). Therefore, we performed new selection experiments with DNA-HEG-AAAKAA. We used the same pair of selection conditions as for DNA-C3-NH2, each with the same two PE and 4FPE acyl donor substrates, and the four selections were iterated for 11 rounds (Figure 5). For three of the selections, those with the PE substrate at pH 7.5 and 9.0 as well as that with the 4FPE substrate at pH 9.0, substantial Lys acylation activity was found, and all three selections were cloned after round 11. The fourth selection with the 4FPE substrate at pH 7.5 was not cloned because of poor pool activity (see Figure 5 caption).
Figure 5.
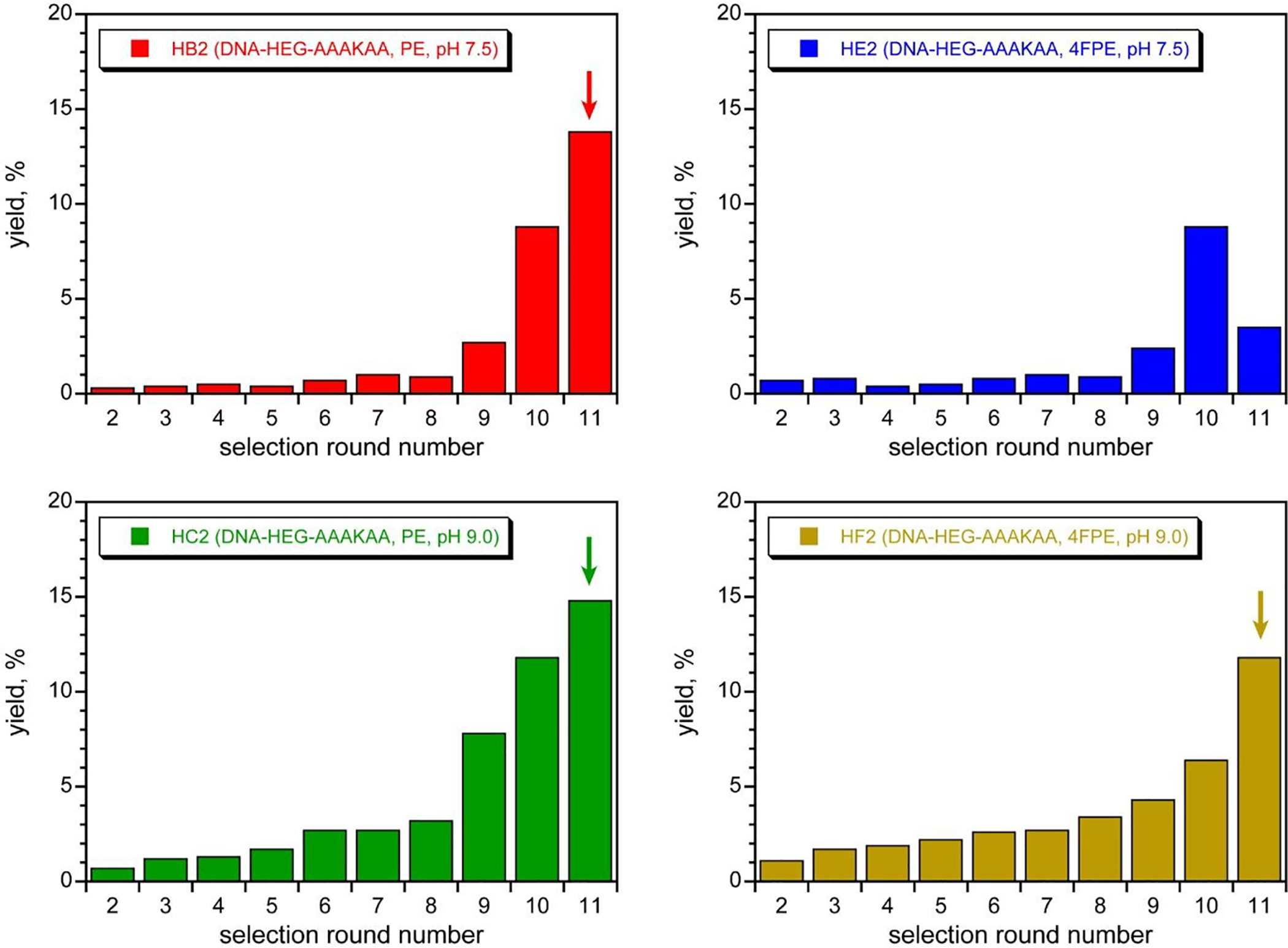
Selection progressions using the DNA-HEG-AAAKAA nucleophile with the PE and 4FPE acyl donors. See text for details of incubation conditions. Arrows mark the cloned rounds. In all cases, the round 1 yield was not quantified because nonradiolabeled pool was used. The HE2 (pH 7.5, 4FPE) selection was not cloned because multiple bands were observed in the product region of the gel, and most of these were assigned to noncatalytic DNA sequences that misfold and therefore migrate aberrantly. Consistent with this decision, each of the 11HB2, 11HC2, and 11HF2 pools was active in trans, i.e., with the DNA pool not ligated to the DNA-HEG-AAAKAA substrate, while in contrast, the 10HE2 pool was inactive in trans.
Lys acylation activity was observed for many of the resulting individual DNAzymes (Figure 6). The selection experiment at pH 7.5 with the PE substrate gave one single DNAzyme sequence, 11HB201 (Figure S4E), with modest 15% yield consistent with that of the uncloned round 11 pool as a whole. In contrast, the two pH 9.0 selections both led to several distinct DNAzymes (Figure S4F), most of which have high yields (>75%). The rate enhancements at pH 9.0 with Mg2+ for 11HC206 (PE substrate) and 11HF210 (4FPE substrate) were 86 and 60, respectively. These values are about an order of magnitude lower than the rate enhancements for the best DNAzymes identified for acylation of the DNA-C3-NH2 substrate, such as 7FN216 and 7FN202. This is due to the different pH values, 9.0 for the DNA-HEG-AAAKAA DNAzymes versus 7.5 for the DNA-C3-NH2 DNAzymes, where kbkgd is substantially greater at the higher pH, and a greater kbkgd leads to a lower rate enhancement. MALDI mass spectrometry was consistent with Lys acylation for the three DNAzymes of Figure 6 (see ESI). Interestingly, in an outcome opposite to that of the above-described selections with the simpler DNA-C3-NH2 substrate, here the selections with DNA-HEG-AAAKAA at the higher pH of 9.0 were more successful in leading to active DNAzymes.
Figure 6.
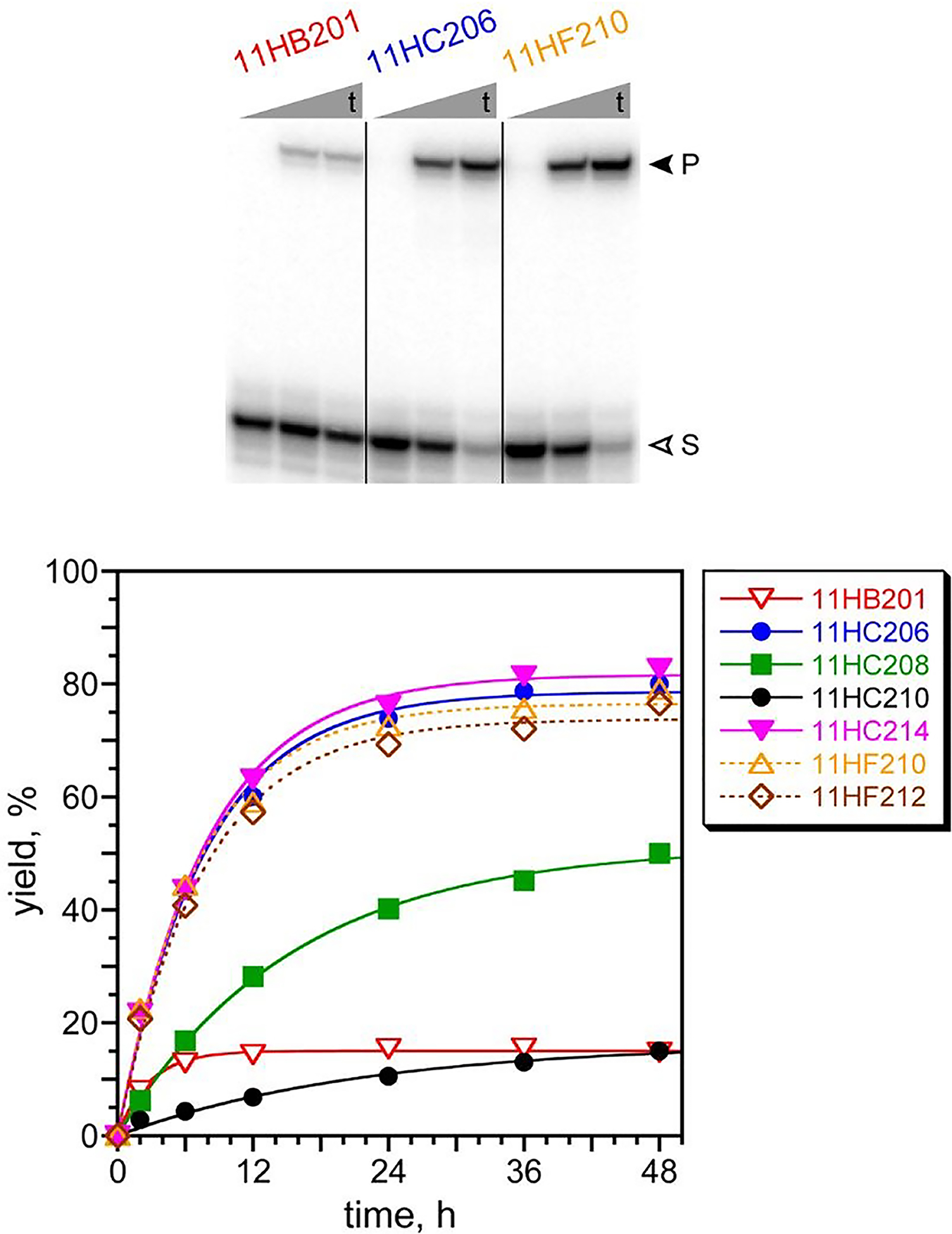
Assays of DNAzymes identified by in vitro selection using the DNA-HEG-AAAKAA substrate with the PE and 4FPE acyl donors. Incubation conditions for 11HB201 (PE substrate): 70 mM HEPES, pH 7.5, 40 mM MgCl2, 20 mM MnCl2, 1 mM ZnCl2, and 150 mM NaCl at 37 °C. Incubation conditions for all 11HC2 (PE substrate) and 11HF2 (4FPE substrate) DNAzymes: 50 mM CHES, pH 9.0, 40 mM MgCl2, and 150 mM NaCl at 37 °C. The PAGE assays for three DNAzymes are shown with representative timepoints (t = 0.5 min, 6 h, 48 h; S = substrate, P = product). Representative kinetic plots are shown for several DNAzymes. kobs values (h−1 ± standard deviation, n = 3): 11HB201, 0.27 ± 0.08; 11HC206, 0.12 ± 0.02; 11HF210, 0.12 ± 0.02. Additional kobs values (h−1 with % yield at 48 h, n = 1): 11HC208, 0.065 (50%); 11HC210, 0.046 (15%); 11HC214, 0.13 (83%); 11HF212, 0.13 (77%). Background assays used the DNA-HEG-AAAKAA substrate and an exactly complementary splint in place of a DNAzyme, with kbkgd values (h−1) and % yield at 48 h as follows: pH 7.5 PE, 0.00013 ± 0.00004 (0.6%; n = 3); pH 9.0 PE, 0.0014 ± 0.0001 (5.8%; n = 3); pH 9.0 4FPE, 0.0020 ± 0.0001 (8.0%; n = 4).
Control experiments were performed using DNA-C3-NH2 in place of DNA-HEG-AAAKAA for all seven of the new DNAzymes. The results with the three DNAzymes of Figure 6 are representative and also consistent with our previous report with DNA-catalyzed Lys phosphoramidate formation.66. In 24 h, the yields with DNA-C3-NH2 were 11HB201, <0.1%; 11HC206, 0.8%; and 11HF210, 1.4%. The corresponding kobs values are calculated to be >6,400-fold, 360-fold, and 210-fold lower, respectively than kobs of the same DNAzymes with DNA-HEG-AAAKAA (Figure 6), which supports the conclusion of selective DNAzyme-catalyzed nucleophilic reactivity of the Lys amino group of the AAAKAA hexapeptide. Because primary amino groups are not indiscriminately acylated by these DNAzymes, productive catalytic interactions are likely between each DNAzyme and its tethered peptide substrate.
Assays with Free Peptide Substrates.
In the longer term, peptide-modifying DNAzymes will have their greatest utility when they can function with free (untethered, not DNA-anchored) peptide substrates. We assayed the DNAzymes that were identified with the DNA-HEG-AAAKAA substrate for their ability to function with 2 mM of free AAAKAA hexapeptide that is not tethered to the DNA anchor oligonucleotide. Unfortunately, in all cases, no activity was observed (<1.5% by PAGE-shift analysis, using 3′−32P-radiolabeled 5′-aryl ester oligonucleotide; the untethered synthetic precursor oligonucleotide that was formerly connected to AAAKAA was included in these experiments). This result is unsurprising, given that the DNA-anchored AAAKAA was presented to the DNAzyme population in every selection round, and an analogous tether requirement by emergent DNAzymes has been encountered in many of our prior selections. In the future, we intend to perform lysine acylation selection experiments in which an azide-modified free peptide is used in the selection step, thereby enforcing a strict pressure for the resulting DNAzymes to function with the free peptide.74
Assays with Acyl Donor for Amine Glutarylation.
By the design of Figure 2C, a successful DNAzyme-catalyzed acylation reaction joins the acyl donor oligonucleotide to the amine acceptor, and the small-molecule phenol derivative of the 5′-aryl ester oligonucleotide is the leaving group. Ideally, amine-acylating DNAzymes will instead be able to use an acyl donor that transfers a small-molecule acyl group rather than a large acyl-oligonucleotide. To explore this possibility, we synthesized a glutaryl donor oligonucleotide (Figure 7A) in which the orientation of the aryl ester functional group was inverted, such that acylation results in glutarylation of the amine nucleophile. This was achieved by first preparing a glutaryl-azide small-molecule compound, which was then used in a CuAAC (copper-catalyzed azide-alkyne cycloaddition) reaction with a 5′-alkyne-modified oligonucleotide, to form the glutaryl donor oligonucleotide.
Figure 7.
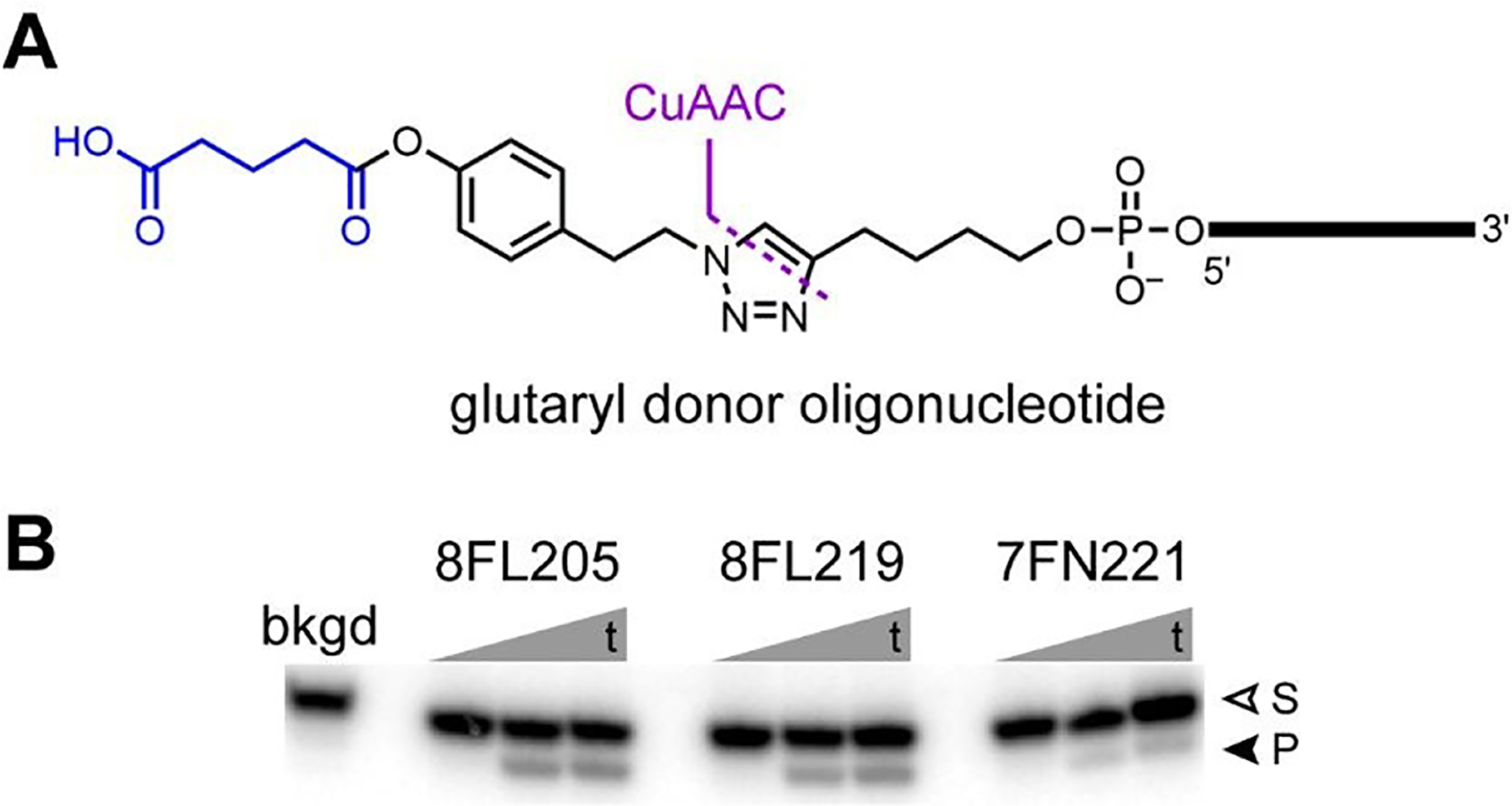
DNAzyme assays with the glutaryl donor. (A) Glutaryl donor oligonucleotide structure, as prepared by CuAAC between a synthesized glutaryl-azide small-molecule compound and a 5′-alkyne-modified oligonucleotide. The glutaryl fragment (blue) is transferred during an amine acylation reaction. (B) Assay results using the glutaryl donor with three DNAzymes identified by in vitro selection for acylation of DNA-C3-NH2 with the PE (8FL205, 8FL219) or 4FPE (7FN221) acyl donor substrate. Bkgd = complementary DNA splint in place of DNAzyme, to assess the uncatalyzed background glutarylation reaction. Incubation conditions: 70 mM HEPES, pH 7.5, 40 mM MgCl2, 20 mM MnCl2, 1 mM ZnCl2, and 150 mM NaCl at 37 °C. Shown are representative timepoints (t = 0.5 min, 16 h, 48 h; S = substrate, P = product). The respective yields at 48 h were 19%, 14%, and 5.4%, versus <0.8% for splinted background (no product band detectable). For each DNAzyme, no product was detectable with unmodified DNA in place of DNA-C3-NH2.
The glutaryl donor oligonucleotide is a p-alkyl-substituted aryl ester of glutaric acid, where the p-alkyl group is inherently electron-donating. Therefore, we expect the glutaryl donor oligonucleotide to be somewhat less reactive than the PE substrate and perhaps substantially less reactive than the fluoro-substituted esters of Figure 1C, including 4FPE. Also, the spatial presentations of the acyl donors are different (e.g., the linkers to the oligonucleotide are not the same), which may suppress the ability of any DNAzymes identified by in vitro selection with the Figure 1C acyl donors to function with the glutaryl donor oligonucleotide. Nevertheless, assaying the eight pH 7.5 DNAzymes of Figure 4 with the glutaryl donor oligonucleotide revealed that three of these DNAzymes catalyze substantial glutarylation of the DNA-C3-NH2 substrate (Figure 7B; see ESI for mass spectrometry product confirmation). The glutarylation yield was as high as 19% for 8FL205, as compared to the splinted background yield of <0.8%. In contrast, none of the seven DNAzymes of Figure 6 catalyzed observable glutarylation of DNA-HEG-AAAKAA above the splinted background (data not shown). These findings establish the feasibility of DNAzyme-catalyzed amine glutarylation, and in ongoing work, we are performing new selection experiments aimed at directly identifying DNAzymes that catalyze Lys glutarylation.
Structural and Mechanistic Considerations.
Using mfold,75 we systematically predicted the secondary structures of all 28 of the new DNAzymes reported in this study. Each DNAzyme is predicted to have 1–7 plausible secondary structures, each with typically modest folding free energy in the range of −4 to −1 kcal/mol, although with three examples of folding free energies in the −9 to −5 kcal/mol range (Table S2 and Figure S7). In many cases, the various mfold-predicted secondary structures for a single DNAzyme are incompatible with one another. We have not endeavored to synthesize and study the large number of DNAzyme mutants (including covariations) that would be required to assess experimentally the validity of the predicted secondary structures. Importantly, doing so would not provide much if any useful, actionable information for our future studies of amine acylation DNAzymes. In parallel, the three-dimensional structures of these new DNAzymes are unknown, in the context that only two DNAzyme structures of any kind have been reported.76,77 Comprehensive analysis of the secondary and tertiary structures and mechanisms of amine acylation DNAzymes will require efforts beyond the scope of the present study.
CONCLUSIONS
In this study we established experimentally that DNAzymes can catalyze amine acylation, including acylation of a Lys residue in a short DNA-anchored peptide. Key to this success was identifying suitably substituted aryl esters (PE and 4FPE) as the electrophilic acyl donors, along with appropriate incubation conditions that balance electrophile stability and reactivity. Thioesters were too unreactive to support DNAzyme catalysis, whereas the more electrophilic DMTE and TFPE were too reactive. The observation that different pH values (and therefore different degrees of uncatalyzed background reactivity) supported emergence of the best DNAzymes for the two different substrates, DNA-C3-NH2 and DNA-HEG-AAAKAA, suggests that pH is an important experimental variable to explore in our future studies. The observation that several DNAzymes can function for amine glutarylation, even though these DNAzymes were not directly identified by selection for amine acylation using the glutaryl donor substrate, bodes well for the longer-term prospects of DNAzymes for amine and Lys acylation with biologically relevant small acyl groups. Finally, we anticipate that new selection experiments involving azide-modified peptides that are not anchored to a DNA oligonucleotide should enable identification of DNAzymes that function with free peptides, as we found for a tyrosine modification reaction.74 Such DNAzymes may also be able to accept larger protein substrates for modification of surface-exposed side chains.
For several other DNAzyme-catalyzed activities, we previously found that performing in vitro selection using a peptide substrate with mixed amino acid composition led to DNAzymes that require those specific peptide sequences in their substrates.22,74,78 By analogy, we anticipate that for DNAzyme-catalyzed peptide Lys acylation, future selection experiments using mixed-composition Lys-containing peptides will provide sequence-selective Lys-acylating DNAzymes, including those that function with free peptide substrates when an appropriate selection pressure is imposed.74 Expanding the substrate tolerance of such DNAzymes from peptides to proteins is a further challenge, but worth undertaking considering the difficulty inherent to achieving nonenzymatic site-selective Lys modification of native proteins.59–65 We are currently pursuing such experiments.
EXPERIMENTAL SECTION
DNA oligonucleotides were obtained from Integrated DNA Technologies (Coralville, IA) or prepared by solid-phase synthesis on an ABI 394 instrument using reagents from Glen Research, including the 5′-CO2H modifier (5′-carboxy-modifier C10, 10–1935). All oligonucleotides and conjugates except for 5′-CO2H, 5′-aryl ester, and glutaryl donor oligonucleotides were purified by 7 M urea denaturing 20% or 8% PAGE with running buffer 1× TBE (89 mM each Tris and boric acid and 2 mM EDTA, pH 8.3), extracted from the polyacrylamide with TEN buffer (10 mM Tris, pH 8.0, 1 mM EDTA, 300 mM NaCl), and precipitated with ethanol. 5′-Aryl ester (PE and 4FPE) oligonucleotides were prepared from the HPLC-purified 5′-CO2H oligonucleotide by treatment with EDC and phenol or 4-fluorophenol followed by HPLC purification; see ESI for details. The glutaryl donor oligonucleotide was prepared by CuAAC using a 5′-alkyne oligonucleotide, as described in the ESI. The AAAKAA hexapeptide was prepared by solid-phase synthesis using Fmoc Rink amide MBHA resin as described.24 The peptide was coupled to the DNA anchor oligonucleotide by reductive amination with a periodate-oxidized 3′-terminal rA nucleotide as described.24 After the DNA-anchored hexapeptide was precipitated with ethanol, the Lys(Tfa) protecting group was removed by incubation in 30% aqueous NH4OH at room temperature for 1 h, dried by SpeedVac, and purified by 20% PAGE. Procedures for selection, cloning, and initial analysis of individual clones are in the ESI.
The general single-turnover assay procedure for each DNAzyme using a 5′-aryl ester oligonucleotide substrate was as follows. The DNA-anchored amine substrate was 5′−32P-radiolabeled using γ−32P-ATP and T4 polynucleotide kinase. A 14 μL sample containing 0.5 pmol of 5′−32P radiolabeled amine substrate, 10 pmol of DNAzyme, and 20 pmol of 5′-aryl ester substrate was annealed in 5 mM HEPES, pH 7.5 or 5 mM CHES, pH 9.0, 15 mM NaCl, and 0.1 mM EDTA by heating at 95 °C for 3 min and cooling on ice 5 min. The DNAzyme-catalyzed reaction was initiated by bringing the sample to 20 μL total volume containing 70 mM HEPES, pH 7.5, 1 mM ZnCl2, 20 mM MnCl2, 40 mM MgCl2 and 150 mM NaCl or 50 mM CHES, pH 9.0, 40 mM MgCl2, and 150 mM NaCl. For the DMTE and TFPE substrates, the 5′-CO2H substrate was used, and either DMT-MM or EDC/TFP were included for activation of the 5′-CO2H group. The sample was incubated at 37 °C. At each time points, a 2 μL aliquot was quenched with 5 μL of stop solution (80% formamide, 1× TBE [89 mM each Tris and boric acid and 2 mM EDTA, pH 8.3], 50 mM EDTA, 0.025% bromophenol blue, 0.025% xylene cyanol). Before PAGE for most assays, to each quenched sample was added 80 pmol of a “decoy oligonucleotide”, which was a 60-mer complementary to the DNAzyme’s initially random region (40 nt) along with 10 nt of binding arm on either side. This decoy oligonucleotide was added to displace the DNAzyme from the substrate and product. In these cases when the decoy was omitted, gel bands were noticeably smeared, which inhibited proper quantification. Quenched samples were separated by 20% PAGE and quantified using a Phosphorimager. Values of kobs were obtained by fitting the yield versus time data directly to first-order kinetics; i.e., yield = Y•(1 − e−kt), where k = kobs and Y is the final yield. Each kobs value is reported with error calculated as the standard deviation from the indicated number of independent determinations. When kobs was sufficiently low such that an exponential fit was not meaningful (e.g., for the background reactions), the initial points were fit to a straight line, and kobs was taken as the slope of the line.
Supplementary Material
ACKNOWLEDGMENT
The early part of this research was partially supported by NIH grant GM065966 to S.K.S.
REFERENCES
- (1).Schlosser K; Li Y Biologically inspired synthetic enzymes made from DNA. Chem. Biol 2009, 16, 311–322. [DOI] [PubMed] [Google Scholar]
- (2).Liu J; Cao Z; Lu Y Functional nucleic acid sensors. Chem. Rev 2009, 109, 1948–1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Liu M; Chang D; Li Y Discovery and Biosensing Applications of Diverse RNA-Cleaving DNAzymes. Acc. Chem. Res 2017, 50, 2273–2283. [DOI] [PubMed] [Google Scholar]
- (4).Cepeda-Plaza M; Peracchi A Insights into DNA catalysis from structural and functional studies of the 8–17 DNAzyme. Org. Biomol. Chem 2020, 18, 1697–1709. [DOI] [PubMed] [Google Scholar]
- (5).Hollenstein M DNA Catalysis: The Chemical Repertoire of DNAzymes. Molecules 2015, 20, 20777–20804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Morrison D; Rothenbroker M; Li Y DNAzymes: Selected for Applications. Small Methods 2018, 2, 1700319. [Google Scholar]
- (7).Hollenstein M Nucleic acid enzymes based on functionalized nucleosides. Curr. Opin. Chem. Biol 2019, 52, 93–101. [DOI] [PubMed] [Google Scholar]
- (8).Ma L; Liu J Catalytic Nucleic Acids: Biochemistry, Chemical Biology, Biosensors, and Nanotechnology. iScience 2020, 23, 100815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Tuerk C; Gold L Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [DOI] [PubMed] [Google Scholar]
- (10).Ellington AD; Szostak JW In vitro selection of RNA molecules that bind specific ligands. Nature 1990, 346, 818–822. [DOI] [PubMed] [Google Scholar]
- (11).Robertson DL; Joyce GF Selection in vitro of an RNA enzyme that specifically cleaves single-stranded DNA. Nature 1990, 344, 467–468. [DOI] [PubMed] [Google Scholar]
- (12).Joyce GF Directed Evolution of Nucleic Acid Enzymes. Annu. Rev. Biochem 2004, 73, 791–836. [DOI] [PubMed] [Google Scholar]
- (13).Joyce GF Forty Years of In Vitro Evolution. Angew. Chem. Int. Ed 2007, 46, 6420–6436. [DOI] [PubMed] [Google Scholar]
- (14).Breaker RR; Joyce GF A DNA enzyme that cleaves RNA. Chem. Biol 1994, 1, 223–229. [DOI] [PubMed] [Google Scholar]
- (15).Faulhammer D; Famulok M The Ca2+ Ion as a Cofactor for a Novel RNA-Cleaving Deoxyribozyme. Angew. Chem. Int. Ed. Engl 1996, 35, 2837–2841. [Google Scholar]
- (16).Santoro SW; Joyce GF A general purpose RNA-cleaving DNA enzyme. Proc. Natl. Acad. Sci. USA 1997, 94, 4262–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Li J; Zheng W; Kwon AH; Lu Y In vitro selection and characterization of a highly efficient Zn(II)-dependent RNA-cleaving deoxyribozyme. Nucleic Acids Res 2000, 28, 481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Silverman SK In vitro selection, characterization, and application of deoxyribozymes that cleave RNA. Nucleic Acids Res 2005, 33, 6151–6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Silverman SK Pursuing DNA Catalysts for Protein Modification. Acc. Chem. Res 2015, 48, 1369–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Silverman SK Catalytic DNA: Scope, Applications, and Biochemistry of Deoxyribozymes. Trends Biochem. Sci 2016, 41, 595–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Walsh SM; Sachdeva A; Silverman SK DNA Catalysts with Tyrosine Kinase Activity. J. Am. Chem. Soc 2013, 135, 14928–14931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Walsh SM; Konecki SN; Silverman SK Identification of Sequence-Selective Tyrosine Kinase Deoxyribozymes. J. Mol. Evol 2015, 81, 218–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Li Y; Breaker RR Phosphorylating DNA with DNA. Proc. Natl. Acad. Sci. USA 1999, 96, 2746–2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Chandrasekar J; Silverman SK Catalytic DNA with Phosphatase Activity. Proc. Natl. Acad. Sci. USA 2013, 110, 5315–5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Brandsen BM; Hesser AR; Castner MA; Chandra M; Silverman SK DNA-Catalyzed Hydrolysis of Esters and Aromatic Amides. J. Am. Chem. Soc 2013, 135, 16014–16017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Zhou C; Avins JL; Klauser PC; Brandsen BM; Lee Y; Silverman SK DNA-Catalyzed Amide Hydrolysis. J. Am. Chem. Soc 2016, 138, 2106–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Choudhary C; Weinert BT; Nishida Y; Verdin E; Mann M The growing landscape of lysine acetylation links metabolism and cell signalling. Nat. Rev. Mol. Cell Biol 2014, 15, 536–550. [DOI] [PubMed] [Google Scholar]
- (28).Verdin E; Ott M 50 years of protein acetylation: from gene regulation to epigenetics, metabolism and beyond. Nat. Rev. Mol. Cell Biol 2015, 16, 258–264. [DOI] [PubMed] [Google Scholar]
- (29).Ali I; Conrad RJ; Verdin E; Ott M Lysine Acetylation Goes Global: From Epigenetics to Metabolism and Therapeutics. Chem. Rev 2018, 118, 1216–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Narita T; Weinert BT; Choudhary C Functions and mechanisms of non-histone protein acetylation. Nat. Rev. Mol. Cell Biol 2019, 20, 156–174. [DOI] [PubMed] [Google Scholar]
- (31).Olsen CA Expansion of the lysine acylation landscape. Angew. Chem. Int. Ed 2012, 51, 3755–3756. [DOI] [PubMed] [Google Scholar]
- (32).Hirschey MD; Zhao Y Metabolic Regulation by Lysine Malonylation, Succinylation, and Glutarylation. Mol. Cell. Proteomics 2015, 14, 2308–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Dutta A; Abmayr SM; Workman JL Diverse Activities of Histone Acylations Connect Metabolism to Chromatin Function. Mol. Cell 2016, 63, 547–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Du J; Zhou Y; Su X; Yu JJ; Khan S; Jiang H; Kim J; Woo J; Kim JH; Choi BH; He B; Chen W; Zhang S; Cerione RA; Auwerx J; Hao Q; Lin H Sirt5 is a NAD-dependent protein lysine demalonylase and desuccinylase. Science 2011, 334, 806–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Peng C; Lu Z; Xie Z; Cheng Z; Chen Y; Tan M; Luo H; Zhang Y; He W; Yang K; Zwaans BM; Tishkoff D; Ho L; Lombard D; He TC; Dai J; Verdin E; Ye Y; Zhao Y The first identification of lysine malonylation substrates and its regulatory enzyme. Mol. Cell. Proteomics 2011, 10, M111.012658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Zhang Z; Tan M; Xie Z; Dai L; Chen Y; Zhao Y Identification of lysine succinylation as a new post-translational modification. Nat. Chem. Biol 2011, 7, 58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Tan M; Peng C; Anderson KA; Chhoy P; Xie Z; Dai L; Park J; Chen Y; Huang H; Zhang Y; Ro J; Wagner GR; Green MF; Madsen AS; Schmiesing J; Peterson BS; Xu G; Ilkayeva OR; Muehlbauer MJ; Braulke T; Muhlhausen C; Backos DS; Olsen CA; McGuire PJ; Pletcher SD; Lombard DB; Hirschey MD; Zhao Y Lysine glutarylation is a protein posttranslational modification regulated by SIRT5. Cell Metab 2014, 19, 605–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Bao X; Liu Z; Zhang W; Gladysz K; Fung YME; Tian G; Xiong Y; Wong JWH; Yuen KWY; Li XD Glutarylation of Histone H4 Lysine 91 Regulates Chromatin Dynamics. Mol. Cell 2019, 76, 660–675 e669. [DOI] [PubMed] [Google Scholar]
- (39).Wang ZA; Cole PA The Chemical Biology of Reversible Lysine Post-translational Modifications. Cell Chem. Biol 2020, 27, 953–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Wright TH; Bower BJ; Chalker JM; Bernardes GJ; Wiewiora R; Ng W-L; Raj R; Faulkner S; Vallée MR; Phanumartwiwath A; Coleman OD; Thézénas M-L; Khan M; Galan SRG; Lercher L; Schombs MW; Gerstberger S; Palm-Espling ME; Baldwin AJ; Kessler BM; Claridge TDW; Mohammed S; Davis BG Posttranslational mutagenesis: A chemical strategy for exploring protein side-chain diversity. Science 2016, 354. [DOI] [PubMed] [Google Scholar]
- (41).Yang A; Ha S; Ahn J; Kim R; Kim S; Lee Y; Kim J; Söll D; Lee H-Y; Park H-S A chemical biology route to site-specific authentic protein modifications. Science 2016, 354, 623–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Bhat S; Hwang Y; Gibson MD; Morgan MT; Taverna SD; Zhao Y; Wolberger C; Poirier MG; Cole PA Hydrazide Mimics for Protein Lysine Acylation To Assess Nucleosome Dynamics and Deubiquitinase Action. J. Am. Chem. Soc 2018, 140, 9478–9485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Jing Y; Liu Z; Tian G; Bao X; Ishibashi T; Li XD Site-Specific Installation of Succinyl Lysine Analog into Histones Reveals the Effect of H2BK34 Succinylation on Nucleosome Dynamics. Cell Chem. Biol 2018, 25, 166–174. [DOI] [PubMed] [Google Scholar]
- (44).Neumann H; Peak-Chew SY; Chin JW Genetically encoding Ne-acetyllysine in recombinant proteins. Nat. Chem. Biol 2008, 4, 232–234. [DOI] [PubMed] [Google Scholar]
- (45).Chalker JM; Bernardes GJL; Davis BGA “tag-and-modify” approach to site-selective protein modification. Acc. Chem. Res 2011, 44, 730–741. [DOI] [PubMed] [Google Scholar]
- (46).Lee YJ; Wu B; Raymond JE; Zeng Y; Fang X; Wooley KL; Liu WR A genetically encoded acrylamide functionality. ACS Chem. Biol 2013, 8, 1664–1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).de Boor S; Knyphausen P; Kuhlmann N; Wroblowski S; Brenig J; Scislowski L; Baldus L; Nolte H; Krüger M; Lammers M Small GTP-binding protein Ran is regulated by posttranslational lysine acetylation. Proc. Natl. Acad. Sci. USA 2015, 112, E3679–3688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Wang ZA; Kurra Y; Wang X; Zeng Y; Lee Y-J; Sharma V; Lin H; Dai SY; Liu WR A Versatile Approach for Site-Specific Lysine Acylation in Proteins. Angew. Chem. Int. Ed 2017, 56, 1643–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Fu C; Chen Q; Zheng F; Yang L; Li H; Zhao Q; Wang X; Wang L; Wang Q Genetically Encoding a Lipidated Amino Acid for Extension of Protein Half-Life in vivo. Angew. Chem. Int. Ed 2019, 58, 1392–1396. [DOI] [PubMed] [Google Scholar]
- (50).Siman P; Karthikeyan SV; Nikolov M; Fischle W; Brik A Convergent chemical synthesis of histone H2B protein for the site-specific ubiquitination at Lys34. Angew. Chem. Int. Ed 2013, 52, 8059–8063. [DOI] [PubMed] [Google Scholar]
- (51).Yu RR; Mahto SK; Justus K; Alexander MM; Howard CJ; Ottesen JJ Hybrid phase ligation for efficient synthesis of histone proteins. Org. Biomol. Chem 2016, 14, 2603–2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Liu H; Liu H; Li X Use of Serine/Threonine Ligation for the Total Chemical Synthesis of HMGA1a Protein with Site-Specific Lysine Acetylations. ChemPlusChem 2019, 84, 779–785. [DOI] [PubMed] [Google Scholar]
- (53).Bellucci JJ; Bhattacharyya J; Chilkoti A A noncanonical function of sortase enables site-specific conjugation of small molecules to lysine residues in proteins. Angew. Chem. Int. Ed 2015, 54, 441–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).McConnell SA; Amer BR; Muroski J; Fu J; Chang C; Ogorzalek Loo RR; Loo JA; Osipiuk J; Ton-That H; Clubb RT Protein Labeling via a Specific Lysine-Isopeptide Bond Using the Pilin Polymerizing Sortase from Corynebacterium diphtheriae. J. Am. Chem. Soc 2018, 140, 8420–8423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Zhang Y; Park K-Y; Suazo KF; Distefano MD Recent progress in enzymatic protein labelling techniques and their applications. Chem. Soc. Rev 2018, 47, 9106–9136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Marculescu C; Lakshminarayanan A; Gault J; Knight JC; Folkes LK; Spink T; Robinson CV; Vallis K; Davis BG; Cornelissen B Probing the limits of Q-tag bioconjugation of antibodies. Chem. Commun 2019, 55, 11342–11345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Ebenig A; Juettner NE; Deweid L; Avrutina O; Fuchsbauer H-L; Kolmar H Efficient Site-Specific Antibody-Drug Conjugation by Engineering a Nature-Derived Recognition Tag for Microbial Transglutaminase. ChemBioChem 2019, 20, 2411–2419. [DOI] [PubMed] [Google Scholar]
- (58).Hofmann R; Akimoto G; Wucherpfennig TG; Zeymer C; Bode JW Lysine acylation using conjugating enzymes for site-specific modification and ubiquitination of recombinant proteins. Nat Chem 2020, 10.1038/s41557-41020-40528-y. [DOI] [PubMed] [Google Scholar]
- (59).Takaoka Y; Nishikawa Y; Hashimoto Y; Sasaki K; Hamachi I Ligand-directed dibromophenyl benzoate chemistry for rapid and selective acylation of intracellular natural proteins. Chem. Sci 2015, 6, 3217–3224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Chilamari M; Kalra N; Shukla S; Rai V Single-site labeling of lysine in proteins through a metal-free multicomponent approach. Chem. Commun 2018, 54, 7302–7305. [DOI] [PubMed] [Google Scholar]
- (61).Adusumalli SR; Rawale DG; Singh U; Tripathi P; Paul R; Kalra N; Mishra RK; Shukla S; Rai V Single-Site Labeling of Native Proteins Enabled by a Chemoselective and Site-Selective Chemical Technology. J. Am. Chem. Soc 2018, 140, 15114–15123. [DOI] [PubMed] [Google Scholar]
- (62).Forte N; Benni I; Karu K; Chudasama V; Baker JR Cysteine-to-lysine transfer antibody fragment conjugation. Chem. Sci 2019, 10, 10919–10924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Mortensen MR; Skovsgaard MB; Gothelf KV Considerations on probe design for affinity guided protein conjugation. ChemBioChem 2019, 20, 2711–2728. [DOI] [PubMed] [Google Scholar]
- (64).Yamada K; Ito Y Recent Chemical Approaches for Site-Specific Conjugation of Native Antibodies: Technologies toward Next-Generation Antibody-Drug Conjugates. ChemBioChem 2019, 20, 2729–2737. [DOI] [PubMed] [Google Scholar]
- (65).Nielsen T; Märcher A; Drobňáková Z; Hučko M; Štengl M; Balšánek V; Wiberg C; Nielsen PF; Nielsen TE; Gothelf KV; Cló E Disulphide-mediated site-directed modification of proteins. Org. Biomol. Chem 2020, 18, 4717–4722. [DOI] [PubMed] [Google Scholar]
- (66).Brandsen BM; Velez TE; Sachdeva A; Ibrahim NA; Silverman SK DNA-Catalyzed Lysine Side Chain Modification. Angew. Chem. Int. Ed 2014, 53, 9045–9050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (67).Kunishima M; Kawachi C; Hioki K; Terao K; Tani S Formation of carboxamides by direct condensation of carboxylic acids and amines in alcohols using a new alcohol- and water-soluble condensing agent: DMT-MM. Tetrahedron 2001, 57, 1551–1558. [Google Scholar]
- (68).Hui KY; Holleran EM; Kovacs J Protected amino acid tetrafluorophenyl esters for peptide synthesis. Int. J. Pept. Protein Res 1988, 31, 205–211. [Google Scholar]
- (69).Lockett MR; Phillips MF; Jarecki JL; Peelen D; Smith LM A tetrafluorophenyl activated ester self-assembled monolayer for the immobilization of amine-modified oligonucleotides. Langmuir 2008, 24, 69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Pham GH; Ou W; Bursulaya B; DiDonato M; Herath A; Jin Y; Hao X; Loren J; Spraggon G; Brock A; Uno T; Geierstanger BH; Cellitti SE Tuning a Protein-Labeling Reaction to Achieve Highly Site Selective Lysine Conjugation. ChemBioChem 2018, 19, 799–804. [DOI] [PubMed] [Google Scholar]
- (71).Haskali MB; Farnsworth AL; Roselt PD; Hutton CA 4-Nitrophenyl activated esters are superior synthons for indirect radiofluorination of biomolecules. RSC Med. Chem 2020, 11, 919–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Silverman SK Catalytic DNA (deoxyribozymes) for synthetic applications—current abilities and future prospects. Chem. Commun 2008, 3467–3485. [DOI] [PubMed] [Google Scholar]
- (73). Each DNAzyme has an alphanumeric designation that includes the round number from which it was cloned (here, 7), the systematically assigned selection designation (here, FN2), and the individual clone number (here, 02 and 16).
- (74).Chu C; Wong O; Silverman SK A Generalizable DNA-Catalyzed Approach to Peptide-Nucleic Acid Conjugation. ChemBioChem 2014, 15, 1905–1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Zuker M Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res 2003, 31, 3406–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Ponce-Salvatierra A; Wawrzyniak-Turek K; Steuerwald U; Höbartner C; Pena V Crystal structure of a DNA catalyst. Nature 2016, 529, 231–234. [DOI] [PubMed] [Google Scholar]
- (77).Liu H; Yu X; Chen Y; Zhang J; Wu B; Zheng L; Haruehanroengra P; Wang R; Li S; Lin J; Li J; Sheng J; Huang Z; Ma J; Gan J Crystal structure of an RNA-cleaving DNAzyme. Nat. Commun 2017, 8, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Wang P; Silverman SK DNA-Catalyzed Introduction of Azide at Tyrosine for Peptide Modification. Angew. Chem. Int. Ed 2016, 55, 10052–10056. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.