

Abstract
Bladder cancer patients with lymph node (LN) metastasis have an extremely poor prognosis and no effective treatment. The alternative splicing of precursor (pre-)mRNA participates in the progression of various tumors. However, the precise mechanisms of splicing factors and cancer-related variants in LN metastasis of bladder cancer remain largely unknown. The present study identified a splicing factor, non-POU domain-containing octamer-binding protein (NONO), that was significantly downregulated in bladder cancer tissues and correlated with LN metastasis status, tumor stage, and prognosis. Functionally, NONO markedly inhibited bladder cancer cell migration and invasion in vitro and LN metastasis in vivo. Mechanistically, NONO regulated the exon skipping of SETMAR by binding to its motif, mainly through the RRM2 domain. NONO directly interacted with splicing factor proline/glutamine rich (SFPQ) to regulate the splicing of SETMAR, and it induced metastasis suppression of bladder cancer cells. SETMAR-L overexpression significantly reversed the metastasis of NONO-knockdown bladder cancer cells, both in vitro and in vivo. The further analysis revealed that NONO-mediated SETMAR-L can induce H3K27me3 at the promotor of metastatic oncogenes and inhibit their transcription, ultimately resulting in metastasis suppression. Therefore, the present findings uncover the molecular mechanism of lymphatic metastasis in bladder cancer, which may provide novel clinical markers and therapeutic strategies for LN-metastatic bladder cancer.
Keywords: NONO, bladder cancer, lymphatic metastasis, alternative splicing, SETMAR
Graphical Abstract
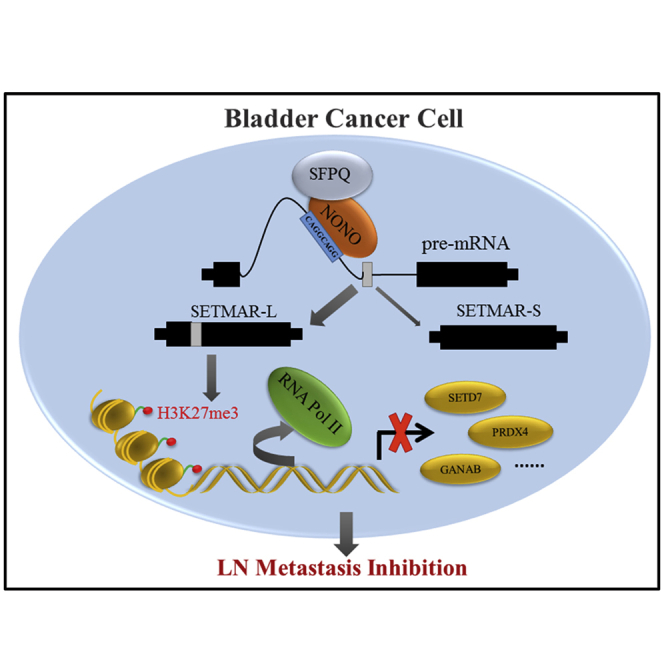
Xie et al. identify that NONO is a lymphatic metastasis-related splicing factor and a prognostic marker in bladder cancer. NONO inhibits invasion in vitro and lymph node metastasis in vivo via regulating splicing of SETMAR. These findings may provide novel clinical markers and therapeutic strategies for bladder cancer.
Introduction
Bladder cancer (BCa) represents the most common genitourinary malignancy worldwide and causes the most deaths in patients with urinary tract disease, which is approximately 165,100 deaths annually.1,2 Non-muscle-invasive BCa (NMIBC) and muscle-invasive BCa (MIBC) are two different types of BCa, and these are determined by their clinically heterogeneous condition. Unlike the recurrence of NMIBC, MIBC is inclined to progress that could spread from the bladder to the pelvic lymph nodes (LNs) and, subsequently, to other organs.3 The probability of death from MIBC with LN metastasis dramatically increases when compared to that from MIBC without LN metastasis, and the death rate increases from 18.6% to 77.6% within 5 years even after receiving radical cystectomy.4 LN metastasis is a critical prognostic factor in BCa, which has been confirmed through a series of independent studies.4, 5, 6 Nonetheless, there are limited treatment options for BCa patients with LN metastasis.7 Therefore, studies on the molecular mechanisms underlying LN metastasis and the identification of novel markers or targets are needed for the diagnosis and therapy of BCa.
Alternative splicing (AS) is a widespread procedure related to structural transcript variation and proteome complexity, which could have entirely divergent functions. Splicing events are commonly disrupted in cancer and contribute to cancer progression.8,9 AS is executed by trans-acting splicing factors, and the dysregulation of splicing factors plays a significant role in the malignant transformation of cancer through modulating the oncogenic variants.10 Splicing factors and cancer-related variants have been described as oncogenic or suppressive drivers in multiple malignancies.11, 12, 13 Furthermore, emergent evidence has shown that cancer-specific splicing factors and variants can provide novel strategies for cancer treatment.14,15
Non-POU domain-containing octamer-binding protein (NONO) is an RNA-binding protein that belongs to the Drosophila behavior human splicing (DBHS) protein family. NONO contains two N-terminal RNA recognition motifs (RRMs), a NonA/paraspeckle domain (NOPS), and a C-terminal coiled coil.16 It is a multifunctional nuclear protein that regulates gene expression in numerous ways, including RNA splicing and stabilization, and transcriptional regulation.16 Previous studies have revealed that NONO can interact with pre-mRNA splicing factors, heterogeneous nuclear ribonucleoproteins (hnRNPs), and transcription factors, and it was associated with cancer progression, such as prostate cancer17 and melanoma.18 However, the differential expression and functional involvement of NONO in the development of BCa have not been clarified.
The present study revealed that NONO was significantly downregulated in LN-metastatic BCa and was a prognostic marker. The inhibition of NONO promoted the lymphatic metastasis of BCa cells, both in vitro and in vivo. Mechanistically, NONO regulated the AS of SETMAR by interacting with splicing factor proline/glutamine rich (SFPQ), ultimately leading to the upregulation of H3K27me3 and the transcriptional inhibition of metastasis-related oncogenes. These findings uncover the new mechanism of NONO-mediated AS in LN metastasis, and offer a novel therapeutic strategy for BCa patients.
Results
NONO Correlates with LN Metastasis and Is a Prognostic Marker in BCa
In order to explore the general expression of NONO in different types of human cancers, we analyzed the Ramaswamy multi-cancer cohort data obtained from the Oncomine database. Interestingly, it was found that the expression of NONO in BCa was the lowest among all cancers, and it was significantly decreased when compared with more than 70% of the other tumors (Figure 1A). Alternatively, NONO expression in bladder normal tissues (BNTs) was higher than most of the other human normal tissues and BCa tissues (Figures 1B and S1A), and its special expression indicates that NONO might exert different functions in BCa. In order to determine whether NONO is involved in the carcinogenesis of BCa, immunohistochemistry (IHC) was initially performed to investigate its expression alteration in BCa tissues (Figure 1C). Consistently, it was found that NONO was significantly downregulated in BCa tissues, when compared to normal adjacent tissues (NATs) (Figure 1D). In addition, the expression of NONO was remarkably lower in LN-metastatic BCa tissues than in LN-negative tissues (Figure 1D). Further analysis revealed that NONO expression was also negatively associated with advanced stage (Figure 1E). The analysis of the Als bladder, Sanchez-Carbayo bladder, and Dyrskjot bladder cohort from the Oncomine database and the Hoglund cohort obtained from the R2 genomics platform also revealed that NONO was notably downregulated in LN-positive and MIBC tissues (Figures 1F–1H and S1B). Importantly, low NONO expression was significantly correlated with poor overall survival (OS) and disease-free survival (DFS) in BCa patients from cohort 1 (Figures 1I and 1J). Similarly, the Kaplan-Meier analysis of The Cancer Genome Atlas (TCGA) database also revealed that patients with lower expression levels of NONO had shorter OS (Figure 1K). In addition, the Kaplan-Meier analysis of the Hoglund cohort also revealed that patients with low NONO expression had shorter OS, but the difference was not statistically significant (Figure S1C). Furthermore, the univariate analysis indicated that NONO expression was significantly associated with OS and DFS in cohort 1 (Tables S3 and S4). Collectively, these data show that the expression of NONO is negatively correlated with LN metastasis and predicts the prognosis outcome of BCa.
Figure 1.

NONO Correlates with LN Metastasis and Is a Prognostic Marker in BCa
(A) NONO expression was analyzed in acute myeloid leukemia (AML) (n = 10), B cell acute lymphoblastic leukemia (BCALL) (n = 10), bladder cancer (BLCA) (n = 11), breast cancer (BRCA) (n = 12), colorectal adenocarcinoma (COAD) (n = 12), diffuse large B cell lymphoma (DLBC) (n = 11), endometrial adenocarcinoma (ENAD) (n = 10), follicular lymphoma (FOLY) (n = 11), glioblastoma (GBM) (n = 10), lung adenocarcinoma (LUAD) (n = 12), medulloblastoma (MBM) (n = 10), melanoma (MEM) (n = 10), ovarian cancer (OVCA) (n = 12), pancreatic adenocarcinoma (PAAD) (n = 11), pleural mesothelioma (PLMA) (n = 11), prostate cancer (PRCA) (n = 14), renal cell carcinoma (RCCA) (n = 11), and T cell acute lymphoblastic leukemia (TBALL) (n = 10). The red line represents the mean value of the NONO expression in BCa. (B) NONO expression was analyzed between BCa tissues (n = 11) and bladder normal tissues (BNTs, n = 7). (C) Representative IHC images of the NONO expression in the paraffin-embedded NAT and tumor sections of BCa, with or without LN metastasis. Scale bars: black, 200 μm; red, 50 μm. (D) IHC staining of cohort 1 shows the NONO expression among NAT (n = 35), LN-negative (n = 84), and LN-positive (n = 29) tumor tissues. (E) NONO expression was analyzed between NMIBC (n = 54) and MIBC (n = 59) tissues in cohort 1. (F–H) NONO mRNA expression was analyzed between LN-negative (n = 4) versus LN-positive (n = 13) tissues in the Als bladder cohort (F), NMIBC (n = 28) versus MIBC (n = 81) in the Sanchez-Carbayo cohort (G), and NMIBC (n = 28) versus MIBC (n = 13) in the Dyrskjot bladder cohort (H). (I and J) Kaplan-Meier curves for the OS (I) and DFS (J) of BCa patients with high versus low expression of NONO in cohort 1. Patients were divided into the NONO-low (n = 55) and NONO-high (n = 58) groups. (K) Kaplan-Meier curves for OS of BCa patients with high (n = 293) versus low (n = 111) expression of NONO in the TCGA cohort. Statistical significance was assessed using two-tailed t tests or one-way analysis of variance (ANOVA). ∗p < 0.05, ∗∗p < 0.01.
NONO Suppresses the Migration and Invasion of BCa Cells In Vitro
Prompted by the above findings, we investigated whether NONO exerted metastasis-associated functions in BCa. Initially, we transiently silenced the expression of NONO with two independent small interfering RNAs (siRNAs), and the efficient knockdown of NONO was confirmed by qPCR and western blot, both in the T24 and UM-UC-3 cell lines (Figures S2A and S2B). Compared with the control group, the migratory speed of T24 and UM-UC-3 cells was remarkably upregulated following NONO knockdown, as gauged by the wound healing assays (Figures S2C and S2D). In addition, NONO knockdown significantly promoted the migration and invasion abilities of BCa cells (Figures S2E and S2F). Meanwhile, the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay was conducted to determine whether NONO is involved in the proliferation of BCa. The results revealed that NONO silencing did not affect the growth of BCa cells (Figure S2G).
In order to further confirm whether NONO is critical for metastasis roles in BCa, we constructed the stably NONO-knockdown cell lines by lentiviral transfection. Then, we infected these stably NONO-knockdown cell lines with the short hairpin RNA (shRNA)-resistant synonymous mutant of NONO, in order to restore NONO expression, as determined by western blot (Figure 2A). The 3D epithelial Matrigel culture models that mimicked the invasion process of cancer cells revealed that NONO knockdown could accelerate BCa cell invasion, while NONO restoration inhibited the invasive capability of BCa cells (Figure 2D). Consistently, the stable NONO knockdown by shRNA enhanced the migration, invasion, and motility abilities of BCa cells, while NONO restoration could notably reverse these above effects (Figures 2B, 2C, and 2E). Taken together, these results demonstrate that NONO inhibits the migration and invasion of BCa cells in vitro.
Figure 2.

NONO Inhibits the Migration and Invasion of BCa Cells In Vitro and In Vitro
(A) Western blot analysis of NONO expression levels in the scramble, NONO-sh, and NONO-sh+NONO re-expression groups. (B and C) Representative images and quantification of Transwell migration (B) and invasion (C) in T24 and UM-UC-3 cells. Cells were treated as indicated. (D) Representative images of the 3D culture of UM-UC-3 cells embedded in Matrigel for 4 days. Cells were treated as indicated. Scale bar, 50 μm. (E) Representative images and quantification of wound-healing assays using T24 and UM-UC-3 cells. Cells were treated as indicated. (F and G) Representative bioluminescence images (F) and histogram analysis (G) of the popliteal metastatic LNs from nude mice treated as indicated (n = 8 per group). The red arrow show the footpad tumor and metastatic popliteal LN. (H) Kaplan-Meier survival analysis of mice that were inoculated with the UM-UC-3 treatment as indicated. (I) Representative image of the popliteal LN metastasis model. (J and K) Representative images of the dissected popliteal LNs (J) and histogram analysis (K) of the LN volume. (L) Representative images of the H&E staining confirming the LN status. Scale bars: black, 500 μm; red, 100 μm. (M) Percentage of LN status in all groups (n = 8). Statistical significance was assessed using a two-tailed t test or one-way ANOVA. Error bars represent the standard deviations of three independent experiments. ∗p < 0.05, ∗∗p < 0.01.
NONO Knockdown Promotes the Lymphatic Metastasis of BCa Cells In Vivo
In order to further determine the function of NONO in the LN metastasis of BCa, an in vivo nude mouse popliteal LN metastasis model was used according to the previous study19. The UM-UC-3/luc BCa cell line with the indicated lentiviral transfection was inoculated into the footpads of nude mice. It was found that NONO knockdown clearly promoted BCa cell LN metastasis, while NONO restoration notably reversed the ability of these BCa cells to metastasize to LNs, as determined by luminescence intensity (Figures 2F and 2G). In addition, the survival times were much shorter in mice that bared the NONO-knockdown tumors, when compared to the corresponding control group, while these markedly increased in the NONO restoration group (Figure 2H). The primary footpad tumors and popliteal LNs were dissected and collected (Figure 2I). The volumes of LNs were notably larger in the NONO-silenced tumor group than in the controls, while the NONO restoration group reversed this effect (Figures 2J and 2K). The dissected popliteal LNs were all validated by H&E staining, which further verifies that the NONO knockdown facilitates LN metastasis (the ratio of metastatic LNs increased from 37.5% to 100%, Figures 3L and 3M), while NONO re-expression inhibited the metastasis (the ratio decreased from 100% to 25.0%, Figures 2L and 2M). Collectively, these results demonstrate that NONO suppresses the LN metastasis of BCa cells in vivo.
Figure 3.

The Validation of NONO-Mediated Splicing Events in BCa Cells
(A) Quantification of AS events after UM-UC-3 cells were treated with two independent siRNAs that targeted NONO. AS events are classified into five categories: skipped exon (SE), alternative 5′ splice site (A5SS), alternative 3′ splice site (A3SS), mutually exclusive exon (MXE), and retained intron (RI). (B and C) Scatterplots show that the NONO knockdown affected the SE events (B, NONO-si1; C, NONO-si2). (D) Validation of candidate genes by qRT-PCR in T24 and UM-UC-3 cells. (E and F) Representative images (E) and quantification (F) of the SETMAR-L/SETMAR-S ratio in T24 and UM-UC-3 cells by RT-PCR. (G) Representative images of RNA-FISH assay. Scale bar, 20 μm. (H and I) Representative images (I) and quantification (H) of RT-PCR analysis of SETMAR from the RIP assay of T24 and UM-UC-3 cells using the anti-NONO antibody. RNA enrichment was determined relative to the non-targeting IgG control. U1 was used as a non-specific control. (J) RNA pull-down assay was performed using potential binding sequences in the intron segment of SETAMR pre-mRNA and the mutation of the potential binding sequence. NONO was detected by western blot. GAPDH was detected as a non-specific control. (K) Schematic diagram of NONO domains and constructions of three NONO mutants: ΔRRM1 (deleting RRM1), ΔRRM2 (deleting RRM2), and ΔRRM1&ΔRRM2 (deleting RRM1 and RRM2). All mutants were FLAG tagged. (L) Western blot of exogenous NONO and its mutants using the anti-FLAG antibody. (M and N) Representative images (M) and quantification (N) of RT-PCR analysis of SETMAR-L/SETMAR-S PSI in T24 cells with NONO knockdown, and re-overexpression of NONO deletion mutants. Statistical significance was assessed using a two-tailed t test or one-way ANOVA. Error bars represent the standard deviations of three independent experiments. ∗p < 0.05, ∗∗p < 0.01.
NONO Mediates the AS of SETMAR in BCa Cells
In order to identify the NONO-regulated AS events in BCa, next-generation RNA sequencing (RNA-seq) was conducted on the UM-UC-3 cell line under the treatment of the NONO siRNAs and control siRNA. More than thousands of NONO-mediated AS events were identified in both independent siRNAs, when compared with the controls, which could be classified into five AS categories, as shown in Figure 3A. The skipped exon (SE) constituted more than half of the total AS events, indicating that NONO mainly modulated SEs. The subsequent analysis indicated the dual role of NONO as a splicing activator or repressor, since this caused a similar percentage of exon inclusion and exclusion splicing events among SEs (Figures 3B and 3C). Among all of the NONO-affected AS events, 102 genes were found to be significantly changed (Figure S3A). 62 genes with a junction that read less than 20 were excluded due to their low expression. In addition, 18 genes that were false positive were also excluded. Finally, 32 genes were identified (Supplementary Table), and we selected the significantly changed and cancer-related genes to initially verify via qPCR. As shown in the Figures 3D and S3B, the splicing of SETMAR was one of the most significantly changed genes. A previous study indicated that SETMAR was a histone methylase with a broad effect on gene expression,20 and that it might exert important functions in cancer. Hence, SETMAR was chosen for further investigation. NONO knockdown significantly promoted the exon2 exclusion of SETMAR (SETMAR-S) and decreased the exon2 inclusion of SETMAR (SETMAR-L), as detected by RT-PCR and qPCR (Figures 3E, 3F, and S3C). Consistently, NONO restoration could significantly rescue the exon2 inclusion of SETMAR (Figures S3D and S3E). In order to observe the splicing more directly, we detected the expression of SETMAR-L and SETMAR-S via RNA fluorescence in situ hybridization (RNA-FISH). It was found that NONO silencing significantly changed the expression of the SETMAR isoform (Figure 3G), which was consistent with the results of RT-PCR.
In order to further determine whether NONO regulates exon skipping by directly binding to the pre-mRNA of SETMAR, RNA immunoprecipitation (RIP) assays initially were conducted. Compared to the negative control, NONO could notably enrich the intron fragments that flanked the exon2 of SETMAR (Figures 3H and 3I). Furthermore, RNA pull-down assays revealed that oligonucleotide 2 (Oligo 2) derived from the SETMAR intron sequence could bind to NONO, but not the other oligonucleotides (Figure 3J and S3F). Intriguingly, Oligo 2 contains the CAGGCAGG sequence, and this is consistent with a previous study that reported that CAGGCAGG is the RNA-binding motif of NONO and is mainly distributed in introns, as analyzed through the RIP-sequencing results.21 In addition, the mutation of this sequence in Oligo 2 could notably weaken the binding between Oligo 2 and NONO, further verifying their direct interaction (Figures 3J and S3F).
Given the critical part of RRMs in splicing, we further determined their roles in NONO. Since NONO contains two N-terminal RRMs that mainly function in splicing, we constructed several deletion mutations of NONO by deleting the sequence of RRM1, RRM2, or both RRM1 and RRM2 (Figure 3K). Then, these mutants were transfected into T24 and UM-UC-3 cells with the NONO shRNA (Figures 3L and S3G). These results show that the RRM1 deletion rescued the SETMAR exon2 inclusion, which is similar to the NONO full-length overexpression, while the RRM2 deletion or RRM1 and RRM2 combined deletion mutants could not reverse the exon2 inclusion of SETMAR (Figures 3M, 3N, S3H, and S3I), indicating that RRM2 is the predominant domain for splicing. Taken together, these data demonstrate that NONO modulates SETMAR exon skipping by binding to its motif in the intron, mainly depending on the RRM2 domain.
NONO Interacts with SFPQ to Regulate SETMAR Splicing in BCa
In order to determine whether NONO interacts with other splicing factors to form the spliceosome complex, we initially identified the potential interacting proteins via GeneCards (Figure 4A). Then, we performed Co-Immunoprecipitation (Co-IP) assays, followed by silver staining. As shown in Figure 4B, it was identified that there is an apparent differential band, and this was further verified as SFPQ by mass spectrometry (Figure 4C; Table S5), which is consistent with the prediction of GeneCards. The interaction between NONO and SFPQ was confirmed by western blot, following Co-IP (Figure 4D). Interestingly, a positive correlation was found between the NONO and SFPQ expression in BCa from TCGA cohort (Figure 4E). Furthermore, immunofluorescence detection displayed their co-localization in the nucleus of BCa tissues (Figure 4F). In order to further determine whether SFPQ is involved in SETMAR splicing, SFPQ knockdown was conducted in BCa cells. Interestingly, SFPQ silencing significantly suppressed the inclusion of SETMAR exon2, which is similar to the effect induced by NONO knockdown (Figures 4G and 4H), indicating that SFPQ and NONO might regulate the splicing of SETMAR together through constituting spliceosome complex. In addition, the SFPQ silencing could also significantly improve the migration and invasion abilities of BCa cells as well (Figures 4I, S4A, and S4B). The analysis of the Als bladder, Sanchez-Carbayo bladder, and Dyrskjot bladder cohorts obtained from Oncomine revealed that the SFPQ expression was significantly downregulated in LN-positive and MIBC tissues (Figures 4J–4L). Meanwhile, note that the underexpression of SFPQ was correlated with shorter OS in TCGA cohort (Figure 4M), which is similar to the outcome of NONO. Collectively, these results indicate that NONO directly interacts with SFPQ to regulate the LN metastasis of BCa.
Figure 4.

NONO Interacts with SFPQ to Regulate SETMAR Splicing in BCa
(A) Predicted interacting proteins of NONO through GeneCards. (B) Co-immunoprecipitation using anti-NONO or control IgG antibody, followed by sliver staining. The black arrows show the position of SFPQ (above) and NONO (below). (C) Mass spectrometry (MS) identification of NONO-interacting proteins. (D) Co-immunoprecipitation analysis shows the interaction between endogenous NONO and SFPQ. (E) Pearson correlations between the expression of NONO and SFPQ in TCGA cohort. (F) Representative immunofluorescence images of NONO and SFPQ localization in BCa patient tissues. Scale bars, 100 μm. (G and H) Representative images (G) and quantification (H) of the SETMAR-L/SETMAR-S ratio after SFPQ knockdown in T24 and UM-UC-3 cells by RT-PCR. (I) Histogram analysis of migrated or invaded cells after SFPQ knockdown. (J–L) SFPQ expression analysis between LN-negative (n = 4) and LN-positive (n = 13) tissues in the Als bladder cohort (J), LN-negative (n = 38) and LN-positive (n = 12) tissues in the Stransky bladder cohort (K), and NMIBC (n = 30) and MIBC (n = 10) tissues in the Dyrskjot bladder cohort (L). (M) Kaplan-Meier curves for the OS of BCa patients with the high versus low expression of SFPQ in TCGA cohort. Patients were divided into SFPQ-low (n = 198) and SFPQ-high (n = 206) groups. Statistical significance was assessed using a two-tailed t test or one-way ANOVA. The error bars represent the standard deviations of three independent experiments. ∗p < 0.05, ∗∗p < 0.01.
SETMAR-L/SETMAR-S Is Correlated with the LN Metastasis of BCa
In order to investigate whether the SETMAR variants participated in the LN metastasis of BCa, we initially detected the expression of SETMAR-L and SETMAR-S through RT-PCR. As presented in Figure 5A, SETMAR-L was apparently decreased in BCa tissues, but increased in bladder normal tissues, while an opposite phenomenon was observed in SETMAR-S. Significantly, the SETMAR-L/SETMAR-S ratio was downregulated in BCa tissues, when compared with NATs (Figure 5B). A positive correlation was observed between the NONO expression and the SETMAR-L/SETMAR-S ratio (Figure 5C), in accordance with the above results that the SETAMR variant switch was under the modulation of NONO. Furthermore, it was also found that the SETMAR-L/SETMAR-S ratio was markedly decreased in LN-positive BCa tissues when compared with LN-negative tissues, and in MIBC tissues when compared with NMIBC tissues (Figures 5D and 5E), indicating that SETMAR variants might be involved in the metastasis of BCa. Therefore, we further determined the functions of SETMAR-L in BCa cells. Two different siRNAs targeting SETMAR exon2 were designed, and their isoform-specific knockdown effects on SETMAR-L were confirmed by both RT-PCR and qPCR, without affecting SETMAR-S (Figures 5F and 5G). It was found that SETMAR-L knockdown considerably promoted the motility, migration, and invasion capacities of BCa cells in vitro (Figures 5H–5J), while this did not affect the proliferation ability of BCa cells (Figure S5A). Taken together, these data demonstrate that SETMAR variant expression is associated with the LN metastasis of BCa, and SETMAR-L plays a suppressive role in the metastasis of BCa cells.
Figure 5.

SETMAR-L And SETMAR-S Were Associated with LN Metastasis of BCa
(A) Representative images of SETMAR-L and SETMAR-S by RT-PCR in BCa tissues. (B) SETMAR-L/SETMAR-S expression analysis between NAT and BCa tissues in cohort 2. (C) Pearson correlations between mRNA expression of NONO and SETMAR-L/SETMAR-S in cohort 2. (D and E) SETMAR-L/SETMAR-S expression analysis in LN-positive versus LN-negative (D) and NMIBC versus MIBC (E) tissues in cohort 2. (F and G) Representative images (F) and quantification (G) of RT-PCR analysis of SETMAR isoform expression following SETMAR-L knockdown. (H) Representative images and quantification of wound-healing assays using T24 and UM-UC-3 cells after SETMAR-L knockdown. (I and J) Representative images and quantification of Transwell migration (I) and invasion (J) in T24 and UM-UC-3 cells after SETMAR-L knockdown. Statistical significance was assessed using a two-tailed t test or one-way ANOVA. Error bars represent the standard deviations of three independent experiments. ∗p < 0.05, ∗∗p < 0.01.
The Restoration of SETMAR-L Reverses the Pro-metastasis Effects of NONO Knockdown
In order to further determine whether NONO regulates the LN metastasis of BCa by inducing the SETMAR isoform switch, we infected NONO-knockdown cell lines with the control lentivirus or SETMAR-L-overexpressing lentivirus (Figures 6A and 6B). Interestingly, it was found that the restoration of SETMAR-L could efficiently reverse the migratory and invasive abilities of BCa cells induced by NONO knockdown (Figures 6C–6E). Furthermore, the effects of the overexpression of SETMAR-L on NONO knockdown-induced LN metastasis were also determined in vivo. It was found that SETMAR-L overexpression could reduce the NONO-shRNA (sh-)transduced tumor burden, which led to the prolonged survival times of tumor-bearing mice (Figure 6F). The SETMAR-L overexpression significantly decreased the capacity of UM-UC-3 cells to metastasize from the footpad to the LNs, as determined by the luminescence and the volume of metastatic LNs (Figures 6G–6J), suggesting that the restoration of SETMAR-L can inhibit the LN metastasis caused by NONO knockdown. Meanwhile, it was found that SETMAR-L overexpression only partially suppressed the LN-metastatic capability of control BCa cells (the ratio of metastatic LNs decreased from 37.50% to 25.00%), but this markedly inhibited the LN-metastatic capability of NONO knockdown cells (the ratio decreased from 87.50% to 37.50%, Figure 6K). Taken together, these data provide evidence that NONO regulates the LN metastasis of BCa in a SETMAR-L-dependent manner.
Figure 6.

Restoration of SETMAR-L Reverses the Pro-metastasis Effects of the NONO Knockdown
(A) Relative expression of SETMAR-L was detected by qPCR after T24 and UM-UC-3 cells were treated as indicated. (B) NONO expression was analyzed by western blot after T24 and UM-UC-3 cells were treated as indicated. (C–E) Representative images of migrated (D) and invaded (E) cells, and the quantification (C) after T24 and UM-UC-3 cells were treated as indicated. (F) Kaplan-Meier survival analysis of mice inoculated with UM-UC-3 cell treatment as indicated. (G and H) Representative images of dissected popliteal LNs (G) and histogram analysis of the LN volume (H). (I) Representative images of the bioluminescence and H&E staining, confirming the LN status. The red arrows show the footpad tumor and metastatic popliteal LN. Scale bars: black, 500 μm; red, 100 μm. (J) Histogram analysis of bioluminescence from the treated nude mice as indicated (n = 8 per group). (K) Percentage of LN status in all groups. Statistical significance was assessed using a one-way ANOVA, followed by a Dunnett’s test. Error bars represent the standard deviations of three independent experiments. ∗p < 0.05, ∗∗p < 0.01.
NONO Regulates Gene Expression via SETMAR-L-Induced H3K27me3
Apart from the splicing regulation, we also analyzed the global gene expression change under the influence of the NONO knockdown. A total of 926 genes exhibiting significant expression changes (fold change >2.0) were identified through the transcriptomic sequence analysis (Figure 7A), and KEGG pathway analysis revealed that the important cancer-related pathways were significantly enriched by NONO knockdown (Figure 7B). Several important oncogenes were selected and validated by qPCR and western blot. We found that the expression of SETD7, PRDX4, and GANAB, which were previously identified as metastatic oncogenes, significantly increased at both the mRNA and protein levels following NONO silencing (Figures 7C and 7D). Consistently, the protein expression levels of SETD7, PRDX4, and GANAB also markedly increased in the NONO-silenced xenograft footpad tumor (Figure S6A and S6B). Importantly, the elevated expression levels of SETD7, PRDX4, and GANAB were significantly correlated with the LN metastasis and tumor stage, as well as the OS and DFS of BCa in TCGA cohort (Figures S7A–S7D). Intriguingly, the expression levels of SETD7, PRDX4, and GANAB were similarly upregulated after SETMAR-L knockdown (Figures 7E and 7F), indicating that NONO might mediate the gene expression in a SETMAR-L-dependent manner. Considering the histone methylation function of SETMAR, we detected several histone methylation statuses that were mainly involved in transcriptional repression. Interestingly, the H3K27me3 expression remarkably decreased following SETMAR-L knockdown, while the modified status of other histones remained unchanged (Figure 7G). It was also found that NONO knockdown decreased the H3K27me3 expression, and that this effect could be rescued by SETMAR-L overexpression (Figure 7H). In order to confirm that the SETMAR-L knockdown activated the target gene expression by H3K27me3, chromatin immunoprecipitation (ChIP) assay and qPCR analysis were performed. SETMAR-L knockdown resulted in a decrease in the location of H3K27me3, but an increase in the location of RNA polymerase II, at the promoter regions of SETD7, PRDX4, and GANAB (Figure 7I). Conversely, SETMAR-L overexpression significantly increased H3K27me3 and decreased RNA polymerase II at the SETD7, PRDX4, and GANAB promoters (Figure 7J), suggesting the transcriptional inhibition of these genes. Taken together, these data suggest that NONO regulates the gene expression through SETMAR-L-mediated H3K27 trimethylation.
Figure 7.

NONO Modulates the Gene Expression through SETMAR-L-Induced H3K27me3
(A) Heatmap representing the unsupervised hierarchical clustering of mRNA expression levels in UM-UC-3 cells transfected with control siRNA or NONO siRNAs. Red and green indicate high and low expression, respectively. (B) KEGG pathway analysis of expression-changed genes. (C and D) Expression of SETD7, PRDX4, and GANAB was detected after NONO knockdown by qPCR (C) and western blot (D). (E and F) Expression of SETD7, PRDX4, and GANAB was detected after SETMAR-L knockdown by qPCR (E) and western blot (F). (G) Histone methylation statuses were analyzed by western blot after SETMAR-L knockdown. (H) H3K27me3 expression was detected by western blot after cells were treated as indicated. (I and J) CHIP-qPCR of RNA polymerase II and H3K27me3 at the promotor region of SETD7, PRDX4, and GANAB following SETMAR-L knockdown (I) or overexpression (J). Two siRNAs in equal proportions were mixed when performing RNAi. (K) Schematic model of the mechanism underlying the role of NONO in BCa LN metastasis. Statistical significance was assessed using a two-tailed t test or one-way ANOVA. Error bars represent the standard deviations of three independent experiments. ∗p < 0.05, ∗∗p < 0.01.
Discussion
The prognosis for BCa patients with LN metastasis is not optimistic, since merely a slight improvement has been reached through present treatment modalities.16 Thus, the elucidation of the molecular mechanism underlying LN metastasis may provide clinical prevention and therapeutic strategies for BCa patients. The present study identified the LN-metastatic-associated splicing factor NONO, which remarkably suppressed the metastasis capacities of BCa cells, both in vitro and in vivo. Mechanically, NONO directly interacted with SFPQ to regulate the AS of SETMAR, which was mainly through binding to its motif, depending on the RRM2 domain. Furthermore, NONO-induced SETMAR-L could markedly suppress the metastasis of BCa cells by inducing H3K27me3 at the promotor of the target genes, and suppress their transcription (Figure 7K). These novel findings uncover the splicing factor-mediated mechanism in LN metastasis of BCa, which might provide novel diagnosis and therapy options in the clinic.
DBHS family proteins engage in almost every procedure of gene regulation, including RNA processing, transport, transcriptional regulation and DNA repair.16 There are three members of this family: NONO, SFPQ, and paraspeckle protein component 1 (PSPC1). DBHS family members exert functions by mediating protein-protein and protein-nucleic acid interactions through post-translational modifications and the availability of interaction partners.16 In cancer, DBHS proteins can function as tumor oncogenes or suppressors in different contexts. For instance, the loss of NONO is correlated with hormonal phenotype and tumor size in breast cancer,22,23 which is consistent with the present results. Similarly, the dysregulation of the SFPQ/PTBPQ complex can induce growth and metastasis in colorectal cancer.24 Meanwhile, NONO overexpression was also found to be associated with tumor progression and involved in regulating the proliferation, migration, and chemoresistance abilities of cancer cells.17,18,25 However, the clinical and biological functions of NONO in the progression of BCa remains unknown. The present study revealed that NONO was remarkably stepwise decreased from adjacent normal tissues, to LN-negative BCa, and to LN-positive BCa. In addition, the loss of NONO and SFPQ correlated with the worse survival prognosis of BCa patients, respectively. Furthermore, NONO significantly suppressed the metastasis behavior of BCa cells, both in vitro and in vivo. Considering the different functions and clinical implications that NONO may display among different cancers, further studies are necessary.
NONO has been previously identified as a critical splicing factor.11 Through the AS of phosphodiesterase mRNA, NONO regulates cyclic AMP-dependent glucocorticoid production.26 Benegiamo et al.21 reported that NONO binds to the pre-mRNAs of various genes involved in glucose and fat metabolism in the liver, affecting their rhythmic expression. However, it remains unknown whether and how NONO modulates the AS in BCa. The present study revealed that NONO regulated the AS of SETMAR by binding to the motif in the intron, which is in agreement with a previous finding that NONO primarily binds to introns.21 In addition, the RNA pull-down assay further provided evidence that CAGGCAGG might represent the binding motif of NONO, as initially identified by a previous study via RIP sequencing (RIP-seq) analysis.21 In addition, it was also verified that RRM2 is the predominant domain that regulated the SETMAR isoform switch. Meanwhile, it was identified that NONO directly interacted with SFPQ in BCa cells, probably forming the spliceosome to regulate the splicing of important isoforms. Consistently, two previous studies indicated that NONO and SFPQ can act as heterodimers, in order to promote the DNA double-strand break repair and ensure telomere integrity.27,28 On the basis of these present findings, we speculated that NONO might function as a molecular scaffold on the target RNA sequence, and assemble spliceosome machinery proteins such as SFPQ in order to exert the AS regulation.
SETMAR, which is also known as Metnase, is a fusion protein between SET domain protein methylase and Hsmar1 transposase.20 It has been reported that the SET domain is responsible for the methylation of histone H3 lysine 36, while the transposase domain is critical for both the binding with DNA and the DNA cleavage activity.29 The mRNA expression levels of several SETMAR transcript variants in hematologic patients were determined in a previous study, showing that SETMAR-L decreased in acute myeloid leukemia patients.30 Previous studies have revealed that SETMAR exerts critical functions in several cellular processes, including non-homologous end joining (NHEJ), the integration of transfected plasmids and lentiviruses, the restart of stalled replication forks, and chromosomal decatenation.29,31,32 However, the role and mechanism of SETMAR variants in BCa remain unclear. Importantly, the present study was the first to show that NONO regulates the AS of SETMAR, and that the SETMAR-L/SETMAR-S ratio is significantly downregulated in the LN-positive metastasis BCa and MIBC, when compared to the corresponding groups. These present results also show that the SETMAR-L knockdown notably enhanced the metastasis abilities of BCa cells. Intriguingly, a previous study revealed that the NONO knockout resulted in the significant reduction of H3K27me3,33 but the underlying mechanism was unclear. The present study provided a novel discovery, in which the NONO-mediated SETMAR-L could increase the expression of H3K27me3, probably via the methylation function of the SET domain, ultimately leading to the transcriptional repression of multiple oncogenes, such as PRDX4, GANAB, and SETD7, and this was previously reported to be associated with the metastasis of cancer.34, 35, 36, 37 Hence, we found a novel mechanism in which NONO inhibited the LN metastasis of BCa via SETMAR-L-mediated H3K27me3 on the target genes.
Given the critical part the AS plays in cancer progression, multiple studies have focused on the development of therapeutic strategies to target the abnormal splicing incidents in cancer. For instance, the small molecule E7107 could impair the aberrant splicing without affecting other constitutive splicing events.38 Another orally available small molecule, H3B-8800, could potently and preferentially kill spliceosome mutant epithelial and hematologic cancer cells by directly interacting with the SF3b complex, and this molecule is presently in a phase 1 clinical study (ClinicalTrials.gov: NCT02841540).39 In addition, oligonucleotide-based therapies that target aberrant splicing events were also promising, and clinical trials for patients with Duchenne muscular dystrophy have already been established (ClinicalTrials.gov: NCT00844597).40, 41, 42 Thus, the investigation of spicing factors and cancer-related variants could shed light on the novel therapeutic strategies for BCa.
In summary, the present study provides a novel discovery that NONO downregulation is clinically and functionally correlated to the LN metastasis of human BCa through the SETMAR-L-mediated regulation of various oncogene expression levels. Understanding the LN metastasis mechanism in an AS manner would enable the development of potential diagnostic approaches and therapeutic strategies for BCa patients with LN metastasis.
Materials and Methods
Human Tissue Samples
A total of 113 formalin-fixed, paraffin-embedded BCa specimens and 35 NAT samples, termed cohort 1, and a total of 75 snap-frozen fresh BCa tissues and 25 NATs, termed cohort 2, were obtained with a written informed consent obtained from patients who underwent surgery at Sun Yat-sen Memorial Hospital of Sun Yat-sen University (Guangzhou, China) between January 2004 and February 2017. Two pathologists pathologically confirmed each sample by H&E staining. Ethical consent was approved by the Committees for Ethical Review of Research Involving Human Subjects of Sun Yat-sen University. The patient demographics and clinical characteristics are provided in Tables S1 and S2.
IHC Analysis
IHC was performed according to a previously described method.43 Briefly, anti-NONO (ab70335, 1:1000), anti-SETD7 (ab189347, 1:100), anti-PRDX4 (ab184167, 1:200), and anti-GANAB (ab96757, 1:200) antibodies were used to detect the corresponding gene expression in BCa tissues and the xenograft footpad tumor. The expression of NONO in primary carcinomas and xenograft tumor specimens obtained from nude mice was blindly quantified by two pathologists, according to a staining scoring system. Briefly, the percentage of tumor cells with positive staining was designated as follows: 0 (no positive), 1 (positive ≤ 10%), 2 (10% < positive ≤ 30%), 3 (30% < positive ≤ 70%), and 4 (positive > 70%). The staining intensity was graded as follows: 1 (no staining), 2 (weak staining, light yellow), 3 (moderate staining, brown), and 4 (strong staining, brown red). The staining index (SI) was calculated by multiplying the proportion of positive tumor cells and the staining intensity score with the possible scores of 0, 1, 2, 3, 4, 6, 8, 9, 12, and 16. The median value, which was SI = 8, was chosen as the cutoff value. Thus, samples with an SI of ≥8 had a high expression, and samples with an SI of <8 had a low expression. The IHC analysis was independently performed by two pathologists who were blinded to the tissue information, in order to avoid evaluation biases. Cases with discrepancies were jointly reevaluated until a consensus was reached. The images were visualized using a Nikon Eclipse Ti microscope system (Nikon, Japan), and processed using Nikon software.
Oncomine and TCGA Data Mining
The clinical profiles of patients in the Als bladder, Sanchez-Carbayo bladder, and Dyrskjot bladder cohorts were available in the Oncomine database. The clinical profiles of patients in the Hoglund cohort were available at the R2 genomics platform. The clinical profiles of patients in TCGA cohort were available at https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga. The Kaplan-Meier survival analysis of NONO and SFPQ in 402 cases in TCGA cohort was obtained from the Kaplan-Meier plotter (http://kmplot.com/analysis/).
Cell Culture
Human BCa cell lines T24 and UM-UC-3 were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). T24 cells were cultured in RPMI 1640 (Gibco, Shanghai, China), and UM-UC-3 cells were cultured in DMEM (Gibco, Shanghai, China). All media was supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (HyClone, Thermo Scientific, USA). Cells were cultured in an incubator with 5% CO2 at 37°C. All cell lines used in the present study tested negative for mycoplasma contamination.
RNAi
The siRNA oligonucleotides targeting NONO, SFPQ, SETMAR-L, and negative control siRNA were purchased from GenePharma (Shanghai, China) and are listed in Table S6. The siRNA transfections were performed according to the manufacturer’s instructions and as previously described.44
Lentivirus Transduction
In order to establish the stable overexpression and knockdown of cell lines, the full-length NONO or shRNA sequences that specifically targeted NONO were cloned into the pCDH-CMV-MCS-EF1-Puro or pLKO.1-Puro vectors, respectively. Bidirectional sequencing was performed to verify the correct sequences. The sequences of all shRNAs are listed in Table S6. Lentivirus production and introduction of infection were performed as previously described.45
RNA Isolation, qRT-PCR, and Western Blot
Total RNA was extracted from cells using TRIzol reagent (Invitrogen), according to the manufacturer’s instructions, and was used as a template for reverse transcription using the PrimerScript RT-PCR kit (TaKaRa Biotechnology, Dalian, China). qRT-PCR was conducted using a standard SYBR Green PCR kit (Roche) and the protocol with a LightCycler real-time instrument (Roche). The relative gene expression was calculated using the 2−ΔΔCt method (Ct, cycle threshold). All specific primers are listed in Table S7. The western blot analysis was performed, as previously described.19 Primary antibodies specific to NONO (ab70335, 1:1,000), SFPQ (ab11825, 1:1,000), SETD7 (ab189347, 1:1,000), PRDX4 (ab184167, 1:1,000), GANAB (ab96757, 1:1,000), H3K27me3 (ab192985, 1:1,000), H3K27me2 (ab6147, 1:1,000), H3K9me2 (ab32521, 1:1,000), H3K9me3 (ab176916, 1:1,000), H4K20me (ab78517, 1:1,000), and GAPDH (ab8245, 1:1,000) were used. The protein bands were visualized using enhanced chemiluminescence.
In Vitro Cell Metastasis Assays and In Vivo Popliteal LN Metastasis
A wound healing assay, Transwell assay, and MTT assay were performed to detect the cell migration and invasion abilities. The details are described in a previous study.19 All animal experimental procedures were approved by the Institutional Animal Care and Use Committee of Sun Yat-sen University. The male BALB/c nude mice (4–5 weeks old) were purchased from the Experimental Animal Center of Sun Yat-sen University and housed in specific pathogen-free (SPF) barrier facilities. Eight mice were included for each group. The footpads of mice were inoculated with 50 μL of PBS suspensions of BCa cells that were transduced with different plasmid. The lymphatic metastasis was monitored and imaged using a bioluminescence imaging system (PerkinElmer, IVIS Spectrum in vivo imaging system) at 4 weeks after tumor cell injection. The primary tumors and popliteal LNs were enucleated and embedded in paraffin. The LN volumes were calculated using the following formula: LN volume (mm3) = (length [mm]) × (width [mm])2 × 0.52. The images were captured using the Nikon Eclipse Ti microscope system (Nikon, Japan), and processed with Nikon software.
RNA-Seq Analysis
Cells were transfected with si-NONO (siRNA-1 and siRNA-2) or control siRNA for 48 h. Then, total RNA was extracted from cells using TRIzol (Invitrogen) and subjected to RNA-Seq, according to the manufacturer’s instruction. The sequencing library was built and sequenced by Annoroad gene technology (Annoroad, Beijing, China). rMATS was used to analyze the different AS events between the control and si-NONO groups. The percent spliced in (PSI) index indicates the efficiency of splicing a specific exon into the transcript population of a gene. In addition, DEGseq was used for the differential gene expression analysis between the control and si-NONO samples. All primary data in the RNA-seq analysis were uploaded to Gene Expression Omnibus (GEO: GSE147323).
RNA-FISH
RNA-FISH was performed as previously described.45 Briefly, T24 and UM-UC-3 cells were fixed with 4% paraformaldehyde and treated with 0.5% Triton X-100, followed by pre-hybridization. Then, these cells were hybridized with the probe (5 mM) overnight. The Cy3-labeled SETMAR-L and FAM-labeled SETMAR-S probes were synthesized by GenePharma (Shanghai, China). The cells were visualized using a confocal microscope (Zeiss, Munich, Germany). The sequences of the probes are listed in Table S8.
Co-IP
The Co-IP was performed using a Pierce crosslink magnetic Co-IP kit (Thermo Scientific), according to the manufacturer’s instructions and our previous study.44 Briefly, the nuclear extracts of cells were incubated with the anti-NONO, anti-SFPQ antibody, or immunoglobulin G (IgG), and were rotated for 3 h at room temperature. Then, the mixture was treated with A+G magnet beads and rotated for 2 h. The precipitated beads were washed three times with IP buffer containing a protease inhibitor cocktail. The protein lysates were detected by western blot.
RIP and RNA Pull-Down
RIP was performed using the EZ-Magna RIP kit (Millipore), according to the manufacturer’s instructions and as previously described.46 Then, 107 cells were lysed with lysis buffer with one freeze-thaw cycle. The cell extracts were co-immunoprecipitated with anti-NONO, and the retrieved RNA was subjected to real-time qPCR analysis.
The RNA pull-down assay was performed using a magnetic RNA-protein pull-down kit (Thermo Scientific), according to the manufacturer’s instructions and as previously described.45 The samples were separated using electrophoresis, and the NONO-specific bands were identified by western blot. The oligonucleotides for the RNA pull-down are listed in Table S9.
ChIP
ChIP was conducted according to the manufacturer’s instructions and as previously reported.47 The stable transfected cells were treated with 1% formaldehyde for 10 min and lysed with SDS lysis buffer, followed by ultrasonication and incubation with the appropriate antibody (anti-IgG, anti-H3K27me3, or RNA polymerase II). After washing with low salt, high salt, and the LiCl buffer, the elution buffer was used to harvest the chromatin fragments. Finally, de-crosslinking was performed, and the enrichment was examined using qPCR. The primers for ChIP-qPCR are listed in Table S7.
Statistical Analysis
The quantitative data are presented as the mean ± SD of three independent experiments. The differences between two groups were analyzed using unpaired/paired Student’s t tests (two-tailed tests) and one-way ANOVA, followed by a Dunnett’s multiple comparisons test, which was performed when more than two groups were compared. A Pearson’s chi-square test was used to analyze the clinical variables. Spearman’s correlation analysis was performed to determine the correlation between two variables. Cumulative survival time was calculated using the Kaplan-Meier method, and analyzed using the log-rank test. The best point cutoff value was used to define the gene expression level (low versus high) for all survival analyses in the present study. A multivariate Cox proportional hazards model was used to estimate the adjusted hazard ratios (HRs) and 95% confidence intervals, and identify independent prognostic factors. All statistical analyses in the present study were performed using the SPSS 19.0 software. A p value of <0.05 was considered statistically significant.
Author Contributions
R.X. and X.C. conceived and designed the study. L.C. and M.H. performed the data analysis. Q.Z. and J.Z. performed cell culture. Y.C. and S.P. performed the animal experiments. Z.C. contributed to IHC staining. W.D. performed bioinformatics analysis. R.X., X.C., J.H., and T.L. wrote and reviewed the manuscript. All authors read and approved the final version of the manuscript. Authorship order among the co-first authors was assigned based on their relative contributions.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This study was supported by the National Key Research and Development Program of China (Grant no. 2018YFA0902803), the National Natural Science Foundation of China (Grant nos. 81825016, 81961128027, 81702523, 81772719, 81772728, 81972383), the Key Areas Research and Development Program of Guangdong (Grant no. 2018B010109006), the Guangdong Basic and Applied Basic Research Foundation (Grant nos. 2020A1515010888, 2019A1515010188), the Science and Technology Planning Project of Guangdong Province (Grant no. 2017B020227007), the Guangdong Special Support Program (2017TX04R246), the Fundamental Research Funds for the Central Universities (for X.C., 18ykpy18), the Project Supported by Guangdong Province Higher Vocational Colleges & Schools Pearl River Scholar Funded Scheme (for T.L.), and by the Yat-Sen Scholarship for Young Scientists (for X.C.).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.08.018.
Contributor Information
Xu Chen, Email: chenx457@mail.sysu.edu.cn.
Jian Huang, Email: huangj8@mail.sysu.edu.cn.
Tianxin Lin, Email: lintx@mail.sysu.edu.cn.
Supplemental Information
References
- 1.Torre L.A., Bray F., Siegel R.L., Ferlay J., Lortet-Tieulent J., Jemal A. Global cancer statistics, 2012. CA Cancer J. Clin. 2015;65:87–108. doi: 10.3322/caac.21262. [DOI] [PubMed] [Google Scholar]
- 2.Van Batavia J., Yamany T., Molotkov A., Dan H., Mansukhani M., Batourina E., Schneider K., Oyon D., Dunlop M., Wu X.-R. Bladder cancers arise from distinct urothelial sub-populations. Nat. Cell Biol. 2014;16:982–991. doi: 10.1038/ncb3038. [DOI] [PubMed] [Google Scholar]
- 3.Youssef R.F., Raj G.V. Lymphadenectomy in management of invasive bladder cancer. Int. J. Surg. Oncol. 2011;2011:758189. doi: 10.1155/2011/758189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hautmann R.E., de Petriconi R.C., Pfeiffer C., Volkmer B.G. Radical cystectomy for urothelial carcinoma of the bladder without neoadjuvant or adjuvant therapy: long-term results in 1100 patients. Eur. Urol. 2012;61:1039–1047. doi: 10.1016/j.eururo.2012.02.028. [DOI] [PubMed] [Google Scholar]
- 5.Zargar-Shoshtari K., Zargar H., Lotan Y., Shah J.B., van Rhijn B.W., Daneshmand S., Spiess P.E., Black P.C. A multi-institutional analysis of outcomes of patients with clinically node positive urothelial bladder cancer treated with induction chemotherapy and radical cystectomy. J. Urol. 2016;195:53–59. doi: 10.1016/j.juro.2015.07.085. [DOI] [PubMed] [Google Scholar]
- 6.Raza S.J., Al-Daghmin A., Zhuo S., Mehboob Z., Wang K., Wilding G., Kauffman E., Guru K.A. Oncologic outcomes following robot-assisted radical cystectomy with minimum 5-year follow-up: the Roswell Park cancer institute experience. Eur. Urol. 2014;66:920–928. doi: 10.1016/j.eururo.2014.03.015. [DOI] [PubMed] [Google Scholar]
- 7.May M., Herrmann E., Bolenz C., Tiemann A., Brookman-May S., Fritsche H.M., Burger M., Buchner A., Gratzke C., Wülfing C. Lymph node density affects cancer-specific survival in patients with lymph node-positive urothelial bladder cancer following radical cystectomy. Eur. Urol. 2011;59:712–718. doi: 10.1016/j.eururo.2011.01.030. [DOI] [PubMed] [Google Scholar]
- 8.Scotti M.M., Swanson M.S. RNA mis-splicing in disease. Nat. Rev. Genet. 2016;17:19–32. doi: 10.1038/nrg.2015.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu X., Si W., Liu X., He L., Ren J., Yang Z., Yang J., Li W., Liu S., Pei F. JMJD6 promotes melanoma carcinogenesis through regulation of the alternative splicing of PAK1, a key MAPK signaling component. Mol. Cancer. 2017;16:175. doi: 10.1186/s12943-017-0744-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Venables J.P., Klinck R., Koh C., Gervais-Bird J., Bramard A., Inkel L., Durand M., Couture S., Froehlich U., Lapointe E. Cancer-associated regulation of alternative splicing. Nat. Struct. Mol. Biol. 2009;16:670–676. doi: 10.1038/nsmb.1608. [DOI] [PubMed] [Google Scholar]
- 11.Sveen A., Kilpinen S., Ruusulehto A., Lothe R.A., Skotheim R.I. Aberrant RNA splicing in cancer; expression changes and driver mutations of splicing factor genes. Oncogene. 2016;35:2413–2427. doi: 10.1038/onc.2015.318. [DOI] [PubMed] [Google Scholar]
- 12.Zhou X., Wang R., Li X., Yu L., Hua D., Sun C., Shi C., Luo W., Rao C., Jiang Z. Splicing factor SRSF1 promotes gliomagenesis via oncogenic splice-switching of MYO1B. J. Clin. Invest. 2019;129:676–693. doi: 10.1172/JCI120279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Choi S., Wang D., Chen X., Tang L.H., Verma A., Chen Z., Kim B.J., Selesner L., Robzyk K., Zhang G. Function and clinical relevance of RHAMM isoforms in pancreatic tumor progression. Mol. Cancer. 2019;18:92. doi: 10.1186/s12943-019-1018-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Venables J.P. Unbalanced alternative splicing and its significance in cancer. BioEssays. 2006;28:378–386. doi: 10.1002/bies.20390. [DOI] [PubMed] [Google Scholar]
- 15.Slansky J.E., Spellman P.T. Alternative splicing in tumors—a path to immunogenicity? N. Engl. J. Med. 2019;380:877–880. doi: 10.1056/NEJMcibr1814237. [DOI] [PubMed] [Google Scholar]
- 16.Knott G.J., Bond C.S., Fox A.H. The DBHS proteins SFPQ, NONO and PSPC1: a multipurpose molecular scaffold. Nucleic Acids Res. 2016;44:3989–4004. doi: 10.1093/nar/gkw271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ishiguro H., Uemura H., Fujinami K., Ikeda N., Ohta S., Kubota Y. 55 kDa nuclear matrix protein (nmt55) mRNA is expressed in human prostate cancer tissue and is associated with the androgen receptor. Int. J. Cancer. 2003;105:26–32. doi: 10.1002/ijc.11021. [DOI] [PubMed] [Google Scholar]
- 18.Schiffner S., Zimara N., Schmid R., Bosserhoff A.K. p54nrb is a new regulator of progression of malignant melanoma. Carcinogenesis. 2011;32:1176–1182. doi: 10.1093/carcin/bgr103. [DOI] [PubMed] [Google Scholar]
- 19.Xie R., Chen X., Chen Z., Huang M., Dong W., Gu P., Zhang J., Zhou Q., Dong W., Han J. Polypyrimidine tract binding protein 1 promotes lymphatic metastasis and proliferation of bladder cancer via alternative splicing of MEIS2 and PKM. Cancer Lett. 2019;449:31–44. doi: 10.1016/j.canlet.2019.01.041. [DOI] [PubMed] [Google Scholar]
- 20.Tellier M., Chalmers R. Human SETMAR is a DNA sequence-specific histone-methylase with a broad effect on the transcriptome. Nucleic Acids Res. 2019;47:122–133. doi: 10.1093/nar/gky937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Benegiamo G., Mure L.S., Erikson G., Le H.D., Moriggi E., Brown S.A., Panda S. The RNA-binding protein NONO coordinates hepatic adaptation to feeding. Cell Metab. 2018;27:404–418.e7. doi: 10.1016/j.cmet.2017.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Traish A.M., Huang Y.H., Ashba J., Pronovost M., Pavao M., McAneny D.B., Moreland R.B. Loss of expression of a 55 kDa nuclear protein (nmt55) in estrogen receptor-negative human breast cancer. Diagn. Mol. Pathol. 1997;6:209–221. doi: 10.1097/00019606-199708000-00005. [DOI] [PubMed] [Google Scholar]
- 23.Pavao M., Huang Y.H., Hafer L.J., Moreland R.B., Traish A.M. Immunodetection of nmt55/p54nrb isoforms in human breast cancer. BMC Cancer. 2001;1:15. doi: 10.1186/1471-2407-1-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ji Q., Zhang L., Liu X., Zhou L., Wang W., Han Z., Sui H., Tang Y., Wang Y., Liu N. Long non-coding RNA MALAT1 promotes tumour growth and metastasis in colorectal cancer through binding to SFPQ and releasing oncogene PTBP2 from SFPQ/PTBP2 complex. Br. J. Cancer. 2014;111:736–748. doi: 10.1038/bjc.2014.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tsofack S.P., Garand C., Sereduk C., Chow D., Aziz M., Guay D., Yin H.H., Lebel M. NONO and RALY proteins are required for YB-1 oxaliplatin induced resistance in colon adenocarcinoma cell lines. Mol. Cancer. 2011;10:145. doi: 10.1186/1476-4598-10-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu J.Y., Sewer M.B. p54nrb/NONO regulates cyclic AMP-dependent glucocorticoid production by modulating phosphodiesterase mRNA splicing and degradation. Mol. Cell. Biol. 2015;35:1223–1237. doi: 10.1128/MCB.00993-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jaafar L., Li Z., Li S., Dynan W.S. SFPQ•NONO and XLF function separately and together to promote DNA double-strand break repair via canonical nonhomologous end joining. Nucleic Acids Res. 2017;45:1848–1859. doi: 10.1093/nar/gkw1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petti E., Buemi V., Zappone A., Schillaci O., Broccia P.V., Dinami R., Matteoni S., Benetti R., Schoeftner S. SFPQ and NONO suppress RNA:DNA-hybrid-related telomere instability. Nat. Commun. 2019;10:1001. doi: 10.1038/s41467-019-08863-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee S.H., Oshige M., Durant S.T., Rasila K.K., Williamson E.A., Ramsey H., Kwan L., Nickoloff J.A., Hromas R. The SET domain protein Metnase mediates foreign DNA integration and links integration to nonhomologous end-joining repair. Proc. Natl. Acad. Sci. USA. 2005;102:18075–18080. doi: 10.1073/pnas.0503676102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jeyaratnam D.C., Baduin B.S., Hansen M.C., Hansen M., Jørgensen J.M., Aggerholm A., Ommen H.B., Hokland P., Nyvold C.G. Delineation of known and new transcript variants of the SETMAR (Metnase) gene and the expression profile in hematologic neoplasms. Exp. Hematol. 2014;42:448–456.e4. doi: 10.1016/j.exphem.2014.02.005. [DOI] [PubMed] [Google Scholar]
- 31.Williamson E.A., Farrington J., Martinez L., Ness S., O’Rourke J., Lee S.H., Nickoloff J., Hromas R. Expression levels of the human DNA repair protein Metnase influence lentiviral genomic integration. Biochimie. 2008;90:1422–1426. doi: 10.1016/j.biochi.2008.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wray J., Williamson E.A., Sheema S., Lee S.H., Libby E., Willman C.L., Nickoloff J.A., Hromas R. Metnase mediates chromosome decatenation in acute leukemia cells. Blood. 2009;114:1852–1858. doi: 10.1182/blood-2008-08-175760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ma C., Karwacki-Neisius V., Tang H., Li W., Shi Z., Hu H., Xu W., Wang Z., Kong L., Lv R. Nono, a bivalent domain factor, regulates Erk signaling and mouse embryonic stem cell pluripotency. Cell Rep. 2016;17:997–1007. doi: 10.1016/j.celrep.2016.09.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Boussadia Z., Lamberti J., Mattei F., Pizzi E., Puglisi R., Zanetti C., Pasquini L., Fratini F., Fantozzi L., Felicetti F. Acidic microenvironment plays a key role in human melanoma progression through a sustained exosome mediated transfer of clinically relevant metastatic molecules. J. Exp. Clin. Cancer Res. 2018;37:245. doi: 10.1186/s13046-018-0915-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang K.P., Yu J.S., Chien K.Y., Lee C.W., Liang Y., Liao C.T., Yen T.C., Lee L.Y., Huang L.L., Liu S.C. Identification of PRDX4 and P4HA2 as metastasis-associated proteins in oral cavity squamous cell carcinoma by comparative tissue proteomics of microdissected specimens using iTRAQ technology. J. Proteome Res. 2011;10:4935–4947. doi: 10.1021/pr200311p. [DOI] [PubMed] [Google Scholar]
- 36.Guo T., Wen X.Z., Li Z.Y., Han H.B., Zhang C.G., Bai Y.H., Xing X.F., Cheng X.J., Du H., Hu Y. ISL1 predicts poor outcomes for patients with gastric cancer and drives tumor progression through binding to the ZEB1 promoter together with SETD7. Cell Death Dis. 2019;10:33. doi: 10.1038/s41419-018-1278-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rafiei S., Tiedemann K., Tabariès S., Siegel P.M., Komarova S.V. Peroxiredoxin 4: a novel secreted mediator of cancer induced osteoclastogenesis. Cancer Lett. 2015;361:262–270. doi: 10.1016/j.canlet.2015.03.012. [DOI] [PubMed] [Google Scholar]
- 38.Lee S.C., Dvinge H., Kim E., Cho H., Micol J.B., Chung Y.R., Durham B.H., Yoshimi A., Kim Y.J., Thomas M. Modulation of splicing catalysis for therapeutic targeting of leukemia with mutations in genes encoding spliceosomal proteins. Nat. Med. 2016;22:672–678. doi: 10.1038/nm.4097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seiler M., Yoshimi A., Darman R., Chan B., Keaney G., Thomas M., Agrawal A.A., Caleb B., Csibi A., Sean E. H3B-8800, an orally available small-molecule splicing modulator, induces lethality in spliceosome-mutant cancers. Nat. Med. 2018;24:497–504. doi: 10.1038/nm.4493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cirak S., Arechavala-Gomeza V., Guglieri M., Feng L., Torelli S., Anthony K., Abbs S., Garralda M.E., Bourke J., Wells D.J. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet. 2011;378:595–605. doi: 10.1016/S0140-6736(11)60756-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goyenvalle A., Babbs A., van Ommen G.J., Garcia L., Davies K.E. Enhanced exon-skipping induced by U7 snRNA carrying a splicing silencer sequence: Promising tool for DMD therapy. Mol. Ther. 2009;17:1234–1240. doi: 10.1038/mt.2009.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kole R. Splicing: hear and now! Mol. Ther. 2013;21:920–921. doi: 10.1038/mt.2013.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen X., Gu P., Xie R., Han J., Liu H., Wang B., Xie W., Xie W., Zhong G., Chen C. Heterogeneous nuclear ribonucleoprotein K is associated with poor prognosis and regulates proliferation and apoptosis in bladder cancer. J. Cell. Mol. Med. 2017;21:1266–1279. doi: 10.1111/jcmm.12999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen X., Xie R., Gu P., Huang M., Han J., Dong W., Xie W., Wang B., Wang He W., Zhong G. Long noncoding RNA LBCS inhibits self-renewal and chemoresistance of bladder cancer stem cells through epigenetic silencing of SOX2. Clin. Cancer Res. 2019;25:1389–1403. doi: 10.1158/1078-0432.CCR-18-1656. [DOI] [PubMed] [Google Scholar]
- 45.Chen Z., Chen X., Xie R., Huang M., Dong W., Han J., Zhang J, Zhou Q, Li H, Huang J and, Lin T DANCR promotes metastasis and proliferation in bladder cancer cells by enhancing IL-11-STAT3 signaling and CCND1 expression. Mol. Ther. 2019;27:326–341. doi: 10.1016/j.ymthe.2018.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gu P., Chen X., Xie R., Xie W., Huang L., Dong W., Han J., Liu X., Shen J., Huang J., Lin T. A novel AR translational regulator lncRNA LBCS inhibits castration resistance of prostate cancer. Mol. Cancer. 2019;18:109. doi: 10.1186/s12943-019-1037-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gu P., Chen X., Xie R., Han J., Xie W., Wang B., Dong W., Chen C., Yang M., Jiang J lncRNA HOXD-AS1 regulates proliferation and chemo-resistance of castration-resistant prostate cancer via recruiting WDR5. Mol. Ther. 2017;25:1959–1973. doi: 10.1016/j.ymthe.2017.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.