

Abstract
Viral infections, such as with cytomegalovirus (CMV), remain a major risk factor for mortality and morbidity of transplant recipients because of their requirement for lifelong immunosuppression (IS). Antiviral drugs often cause toxicity and sometimes fail to control disease. Thus, regeneration of the antiviral immune response by adoptive antiviral T cell therapy is an attractive alternative. Our recent data, however, show only short-term efficacy in some solid organ recipients, possibly because of malfunction in transferred T cells caused by ongoing IS. We developed a vector-free clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9-based good manufacturing practice (GMP)-compliant protocol that efficiently targets and knocks out the gene for the adaptor protein FK506-binding protein 12 (FKBP12), required for the immunosuppressive function of tacrolimus. This was achieved by transient delivery of ribonucleoprotein complexes into CMV-specific T cells by electroporation. We confirmed the tacrolimus resistance of our gene-edited T cell products in vitro and demonstrated performance comparable with non-tacrolimus-treated unmodified T cells. The alternative calcineurin inhibitor cyclosporine A can be administered as a safety switch to shut down tacrolimus-resistant T cell activity in case of adverse effects. Furthermore, we performed safety assessments as a prerequisite for translation to first-in-human applications.
Keywords: adoptive T-cell-therapy, CMV, vector-free gene-editing, CRISPR-Cas9, immunotherapy, drug-resistance, immunosuppression, solid-organ-transplantation
Graphical Abstract
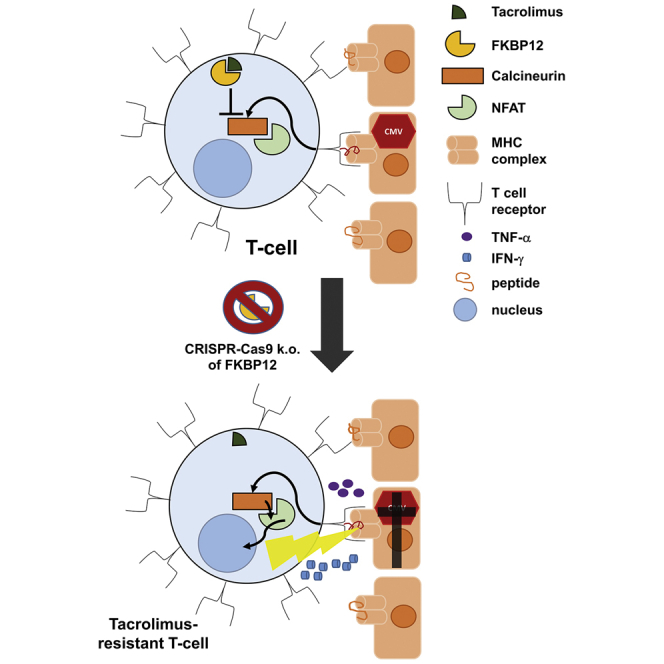
To overcome insufficient long-term efficacy of adoptive T cell therapy for treatment of life-threatening viral complications in immunosuppressed solid organ transplant recipients, an antiviral T cell product resistant to the common immunosuppressant tacrolimus was developed, applying a GMP-compatible vector-free approach based on ribonucleoprotein complexes of Cas9 and FKBP12-specific single guide RNA.
Introduction
Viral infections, e.g. with cytomegalovirus (CMV), Epstein-Barr virus, and BK virus, remain a major morbidity and mortality problem in chronically immunocompromised patients. A representative case is CMV disease, a life-threatening side effect of immunosuppressive medication required for prevention of organ rejection in solid organ transplant (SOT) recipients. CMV disease causes direct and indirect morbidity, including rejection of the transplanted organ1 or chronic allograft nephropathy in kidney transplantation (KTx).2 Classic antiviral medication is used as prophylaxis and preemptive therapy, but careful dosing is crucial to prevent toxicity and resistance.3 Nevertheless, high frequencies of late-onset CMV disease have been observed with increased mortality among CMV-infected KTx recipients compared with uninfected recipients.4 The T cell-mediated anti-CMV response has proven to be a suitable predictive and stratification marker for CMV disease outcome.5,6 Thus, regeneration of the endogenous T cell response by adoptive anti-viral T cell therapy may diminish CMV-associated morbidity and mortality after transplantation.
Antiviral T cell therapy is an established method to reconstitute effective immunity in hematopoietic stem cell transplant (HSCT) recipients.7, 8, 9, 10, 11, 12 We previously confirmed that antiviral T cell products (TCPs) can be successfully generated from immunosuppressed SOT recipients.13,14 Treatment of CMV disease using adoptive T cell transfer to SOT recipients has been reported in a few studies.15, 16, 17 However, despite initial efficient reduction of symptoms and viral load,18,19 the limited persistence and longevity of TCPs are major challenges for successful adoptive cell therapy for CMV disease. Similarly, relapses in Epstein-Barr virus load have been documented in TCP-treated SOT patients.20
One reason for limited long-term efficacy of antiviral T cell therapy may be the ongoing immunosuppression (IS) required to prevent organ rejection in SOT recipients. Conventional IS for KTx (and other SOT patients) is comprised of three classes of immunosuppressants: the proliferation inhibitor mycophenolic acid (MPA), which also inhibits pro-inflammatory cytokine production,21 induces lymphocyte apoptosis and decreases homing of T cells.22 Calcineurin inhibitors (CNIs; e.g. tacrolimus [FK506] and cyclosporine A [CsA]) potently reduce T cell activation, maturation, and cytokine secretion.23 Glucocorticoids blunt cytokine production by T cells24 and provoke lymphocyte apoptosis.25 Preformed allo-antigen-reactive effector T cells are particularly difficult to control in transplant recipients. CNIs are currently the most effective treatment for sustained suppression of these allo-antigen-reactive cells. Therefore, solutions are required to maintain control of allograft-reactive T cells while overcoming suppression of protective antiviral T cell responses. To circumvent the effects of tacrolimus on adoptively transferred virus-specific TCPs, tacrolimus-insensitive forms of calcineurin26,27 have been introduced into cells using retroviruses. Another method to induce tacrolimus insensitivity is use of a retrovirally integrated siRNA expression cassette to knock down the immunophilin fkbp12,28 the adaptor protein needed for the immunosuppressive function of tacrolimus in T cells.29 However, retroviruses integrate into the genome randomly, posing a possible safety risk because of the potential for gene disruption or dysregulation. Furthermore, proteins that may be mutated by insertion are potentially immunogenic. Moreover, RNA interference is often incomplete; thus, partial susceptibility to tacrolimus may be retained. None of these approaches have reached clinical application yet.
Here, we introduce a good manufacturing practice (GMP)-compatible, vector-free gene editing approach targeting the FKBP12 gene to make CMV-specific, tacrolimus-resistant T cells. Efficient gene editing was achieved by transfection with ribonucleoprotein (RNP) complexes consisting of CRISPR-associated protein 9 (Cas9) and respective single guide RNA (sgRNA).30 We implemented a RNP nucleofection step in our existing clinical protocol for generation of antiviral TCPs.19 For safety, our protocol comprises an initial sorting step to solely expand virus-specific T cells, activated upon exposure to the respective viral antigens.14,27,31 Functionally, FKBP12 knockout (KO) antiviral T cells showed superior cytokine production and activation in the presence of tacrolimus compared with unedited antiviral T cells. These properties were inhibited in the presence of an alternative CNI, CsA, demonstrating the specificity of pathway inhibition and providing an optional safety switch to control undesired effects of gene-edited FKBP12 KO T cells in patients. By combining multiple highly refined technologies, we developed a unique approach for efficient and safe generation of clinical-grade, tacrolimus-resistant, virus-specific TCPs suitable for adoptive antiviral T cell therapy.
Results
Integration of FKBP12 KO into a Clinical Protocol for CMV-Specific T Cell Product Generation
We adapted a vector-free protocol for electroporation and RNP-based KO30 of FKBP12 to generate a virus-specific tacrolimus-resistant TCP (FKBP12 KO CMV TCP)19 (Figure 1A). For this, CMV-specific T cells were isolated from peripheral blood mononuclear cells (PBMCs) based on their interferon γ (IFNγ) secretion13 after activation by CMVIE-1/pp-65 overlapping peptide pools (Figure 1B). These cells were expanded for 1 week. FKBP12 KO was achieved by electroporation with RNP complexes of Cas9 and different sgRNAs, selected based on in silico prediction (Figure 1C).32,33 The sgRNAs differed according to the respective target sequence within the gene coding for FKBP12 and the mode of production (in vitro transcribed versus fully synthetic [synt]). The KO efficiencies differed between the tested sgRNAs (Figure 1C). For GMP compatibility reasons, we investigated one synt sgRNA with 2O’-methyl-3′phosphothioate modifications in the first and last 3 nucleotides, which have been reported to enhance editing frequencies in T cells.34 Indeed, the synt version of sgRNA#2 improved the KO efficacy of the sgRNA (Figure 1C). sgRNA#2 was chosen to be produced as a GMP-compliant, fully synt sgRNA because of its superior function in maintaining T cell function in the presence of tacrolimus compared with the other two guide RNAs (Figure 2). We adapted an existing KO protocol for polyclonal T cells30 to our CMV TCPs, employing a different electroporation device suitable for GMP applications, using closed cartridges and electroporation conditions that preserved expansion rates similar to unmodified controls (Figure 1D). The expansion rates were normalized to the unmodified controls, which did not undergo electroporation, to determine whether the electroporation and/or gene editing procedures altered the expansion capability of the cells (Figure 1D). The expansion rates of the electroporated and gene-edited cells were similar to the control, indicating that the electroporation procedure and gene editing did not influence the overall expansion rate of the TCPs (Figure 1D).
Figure 1.

Protocol Outline for a GMP-Compatible Approach to Induce Tacrolimus Resistance in a CMV-Specific TCP Using Vector-Free CRISPR-Cas9 Technology
(A) Individual steps of the production process. (B) Representative purities of CMV-specific IFNγ-producing T cells pre- and post-enrichment from PBMCs. (C) Calculated KO efficiency of FKBP12 in CMV-specific TCPs. (D) Expansion rates of electroporated CMV-specific TCPs normalized to expansion rates of the unmodified control from day 7 to day 21 (following electroporation). (E) Different immunosuppressants used for functional analyses and their affected adaptor proteins or receptors and pathways. Classic triple IS consists of a CNI, prednisolone, and MPA.
Figure 2.

Functional Profiles of CMV-Specific TCPs with Disrupted FKBP12
CMV-specific stimulation of TCPs on day 21 of culture. Immunosuppressants were added where indicated. Tac, tacrolimus; CsA, cyclosporine A; Tac/Pred/MMF, Tac + prednisolone + mycophenolic acid (MPA). ∗p < 0.05. (A) Representative flow cytometry plots showing CD4+ and CD8+ IFNγ and TNF-α producers in unmodified and FKBP12 KO CMV TCPs edited with synt sgRNA#2. Unstimulated and CMV peptide-stimulated TCPs are shown in the presence of the indicated immunosuppressants. (B) Summary of CD4+ IFNγ producers in unmodified and FKBP12-specific sgRNA#1–sgRNA#3 or synt modified sgRNA#2 gene-edited FKBP12 KO CMV TCPs in the presence of the indicated immunosuppressants. (C) Summary of CD8+ IFNγ producers in unmodified and FKBP12-specific sgRNA#1–sgRNA#3 or synt modified sgRNA#2 gene-edited FKBP12 KO CMV-TCPs in the presence of the indicated immunosuppressants. (D) Summary of CD4+ TNF-α and IFNγ double producers in unmodified and FKBP12-specific sgRNA#1–sgRNA#3 or synt modified sgRNA#2 gene-edited FKBP12 KO CMV TCPs in the presence of the indicated immunosuppressants. (E) Summary of CD4+ TNF-α/IFNγ double producers in unmodified and FKBP12-specific sgRNA#1–sgRNA#3 or synt modified sgRNA#2 gene-edited FKBP12 KO CMV TCPs in the presence of the indicated immunosuppressants. (F) Killing capacity of CMV-TCPs: percentage killing of CMV-specific peptide-loaded autologous targets.
For functional analyses, we exposed TCPs to clinical doses of immunosuppressive drugs (Figure 1E). The CNI tacrolimus, which requires FKBP12 as an adaptor protein (Figure 1E), is the main target in this project. To confirm the specificity of the tacrolimus-targeted approach, we used an alternative CNI, CsA, which depends on peptidylprolyl isomerase A (PPIA) as its adaptor protein (Figure 1E). To test the functionality of the modified T cells in the clinical context of SOT recipients receiving triple IS, we used classic triple IS consisting of tacrolimus, prednisolone (requiring the glucocorticoid receptor, which acts as a transcription factor), and MPA (which acts directly on nucleotide synthesis) (Figure 1E). We did not investigate the distinct effects of prednisolone or MPA on TCPs because FKBP12 KO should not interfere with these pathways, and their effects on memory T cells are described elsewhere.22,25,35
Cytokine Production in the Presence of Tacrolimus Is Rescued by FKBP12 KO in CMV-Specific T Cell Products
A major function of effector T cells is production of anti-viral cytokines to instruct other immune cells and induce an antiviral state in infected cells. Producers of multiple cytokines show particularly high antiviral functionality.36 Upon CMV-specific re-stimulation on day 21 of culture, we recorded similar proportions of producers of the antiviral cytokines IFNγ and tumor necrosis factor alpha (TNF-α) among CD4+ and CD8+ T cells in FKBP12 KO CMV and unmodified CMV TCPs (Figures 2A–2E). Addition of immunosuppressive drugs during stimulation significantly decreased IFNγ and TNF-α/IFNγ double producers among CD4+ (Figures 2B and 2D) and CD8+ T cells (Figures 2C and 2E), which was partially rescued by FKBP12 KO (Figures 2B–2E). Although cytokine production improved under all FKBP12 KO conditions in the presence of a clinically relevant dose of tacrolimus, only samples electroporated with sgRNA#2 retained physiological levels of cytokine producers in stimulated TCPs (Figures 2B–2E). We therefore tested sgRNA#2 as a GMP-compliant synt modified sgRNA, with 2O’-methyl-3′phosphothioate modifications in the first and last 3 nucleotides, which have been reported to enhance editing frequencies in T cells.34 Using synt modified sgRNA#2, we achieved higher editing efficacy (Figure 1C) and better recovery of cytokine production, especially in the presence of clinically relevant doses of triple IS, during stimulation (Figures 2A–2E).
FKBP12 KO CMV TCPs Demonstrate a Killing Capacity Comparable with Unmodified CMV TCPs
Because antiviral T cells induce targeted elimination of infected cells by cytotoxicity, we tested the cytotoxic killing capacity of TCPs.37,38 Unlike antiviral cytokines, which were strongly affected by short-term classic immunosuppressive treatment (Figures 2A–2E), T cell-mediated killing of CMV peptide-loaded target cells was unaffected by short-term incubation with immunosuppressive drugs at clinical doses (Figure 2F). Notably, the data demonstrated no differences in the killing capacity of FKBP12 KO CMV TCPs compared with unmodified CMV TCPs, irrespective of the applied sgRNA.
Enhanced Survival of FKBP12 KO CMV-Specific T Cell Products in the Presence of Tacrolimus
Before clinical application, antiviral TCPs must be cryopreserved until all release criteria and safety tests are accomplished. Therefore, we froze all edited and non-edited CMV TCPs. We focused on synt sgRNA#2 in subsequent experiments because it was the most effective (Figure 2) and preferred GMP-compliant material. Synt sgRNA#2-treated FKBP12 KO CMV and unmodified CMV TCPs were thawed and evaluated for viability and functionality post-thawing (Figure 3A). The viability of the T cells did not differ directly after thawing (day 0; Figure 3B); however, we found a slightly bigger proportion of living cells in thawed FKBP12 KO CMV TCPs compared with unmodified CMV TCPs following 24-h incubation with tacrolimus (Figure 3C; p = 0.06, normalized to culture in medium without immunosuppressants). 24-h incubation with other immunosuppressants revealed no differences in viability (Figure 3C).
Figure 3.

FKBP12 KO CMV TCPs after Thawing: Viability, Activation and Exhaustion, and Memory Markers after Culture in Immunosuppressants
Unmodified and FKBP12 KO CMV TCPs (edited with synt sgRNA#2) were frozen (indicated by the ice symbol) and stored in liquid nitrogen until thawing (indicated by the orange wavy lines). Cells were cultured without cytokines in the presence of the indicated immunosuppressant (Tac and CsA) on day 0 and day 2. Addition of drugs (CsA and Tac + Pred + MPA) is indicated by the pipette and a green teardrop. ∗p< 0.05. (A) Representative flow cytometry plot showing L/D and Annexin V staining of single cells after thawing and demonstrating the gating strategy; double-negative staining (bottom left quadrant) indicates the population of viable cells. (B) Viability of single cells in unmodified (green) and FKBP12 KO CMV TCPs (red) directly after thawing (day 0). (C) Viability of single cells in unmodified (green) and FKBP12 KO CMV TCPs (red) cultured in the indicated immunosuppressant 24 h after thawing (day 1). (D) MFI of CD25 on single T cells of unmodified (green) and FKBP12 KO CMV TCPs (red) directly after thawing (day 0). (E) MFI of CD25 on single T cells of unmodified (green) and FKBP12 KO CMV TCPs (red) cultured in the indicated immunosuppressant 24 h after thawing (day 1). (F) MFI of CD25 on single T cells of unmodified (green) and FKBP12 KO CMV TCPs (red) cultured in the indicated immunosuppressant 96 h after thawing (day 4). (G) MFI of PD-1 on single T cells of unmodified (green) and FKBP12 KO CMV TCPs (red) directly after thawing (day 0). (H) MFI of PD-1 on single T cells of unmodified (green) and FKBP12 KO CMV TCPs (red) cultured in the indicated immunosuppressant 24 h after thawing (day 1). (I) MFI of PD-1 on single T cells of unmodified (green) and FKBP12 KO CMV TCPs (red) cultured in the indicated immunosuppressant 96 h after thawing (day 4). (J) MFI of CTLA-4 on single T cells of unmodified (green) and FKBP12 KO CMV TCPs (red) directly after thawing (day 0). (K) MFI of CTLA-4 on single T cells of unmodified (green) and FKBP12 KO CMV TCPs (red) cultured in the indicated immunosuppressant 24 h after thawing (day 1). (L) MFI of CTLA-4 on single T cells of unmodified (green) and FKBP12 KO CMV TCPs (red) cultured in the indicated immunosuppressant 96 h after thawing (day 4).
Exhaustion and Phenotypic Memory Markers of FKBP12 KO and Unmodified CMV-Specific T Cell Products following 4 Days of Consecutive Culture in the Presence of Immunosuppressants
We evaluated the cell surface expression of distinct phenotypic markers for T cell activation (CD25) and exhaustion (programmed death 1 [PD-1] and cytotoxic T lymphocyte-associated protein 4 [CTLA-4] in the presence of clinical doses of immunosuppressants 0, 1, and 4 days post-thawing by flow cytometry. We detected high CD25 expression directly after thawing (Figure 3D), which diminished after 24 h (Figures 3E and 3F). The mean fluorescence intensity (MFI) of CD25 remained significantly higher in TCPs cultured for 24 h with clinical doses of classical triple IS, irrespective of whether FKBP12 was genetically edited (Figure 3D). However, no differences in CD25 expression were recorded following 4 days of culture in the presence of immunosuppressants (added every 48 h) (Figure 3F). Next we assessed exhaustion markers in TCPs. To this end, we determined the expression of PD-1 and CTLA-4 during 4 days of culture post-thawing. For all conditions, PD-1 and CTLA-4 expression increased equally over the observed time course (Figures 3G–3L). Interestingly, CTLA-4 protein levels (MFI) were significantly lower in TCPs treated with triple IS irrespective of FKBP12 editing (Figure 3L).
Functional Assessment of CMV-Specific TCPs Exposed to Tacrolimus
Next we assessed the functional capacity of TCPs for virus-specific effector functions after the freezing and thawing process and 5-day culture in immunosuppressants, similar to the environment faced upon injection into an immunosuppressed patient. To this end, TCPs were stimulated with CMV-specific peptide pools and assessed for intracellular accumulation of cytokines as well as the presence of activation markers.
Granzyme B (GZB) mediates apoptosis of virus-infected target cells in conjunction with perforin. Therefore, we measured intracellular accumulation of GZB in TCPs upon CMV-specific re-stimulation and found a stimulation-dependent increase in GZB in CD4+ T cells (Figure 4A). Notably, the majority of CD8+ T cells presented with pre-formed GZB even before re-stimulation (Figure 4C). Neither gene editing nor culture with immunosuppressants at clinical doses affected the proportion of CMV-stimulated GZB+ CD4+ (Figure 4B) or GZB+ CD8+ T cells (Figure 4D) in CMV TCPs. Next we measured IFNγ and TNFα production in response to CMV-specific stimulation after thawing of TCPs and culture in immunosuppressants (Figures 4E–4J). CD4+ and CD8+ T cells produced IFNγ in response to CMV stimulus after culture with tacrolimus, although IFNγ producers among CD4+ and CD8+ T cells were significantly more frequent in FKBP12 KO CMV TCPs compared with unmodified CMV TCPs after culture with tacrolimus (Figures 4F and 4H). Remarkably, CD4+ and CD8+ IFNγ/TNFα double producers were also significantly more abundant among CD4+ and CD8+ T cells in FKBP12 KO compared with unmodified CMV TCPs, in which they were almost absent (Figures 4I and 4J). CD8+ IFNγ/TNFα double producers were significantly more frequent among CD8+ T cells from FKBP12 KO CMV TCPs cultured in tacrolimus compared with CsA, which was not evident with unmodified CMV TCPs (Figure 4J). The tendency of enhanced cytokine producers among cells cultured in tacrolimus versus cell cultured in CsA was also observed in CD4+ IFNγ/TNFα double producers and CD4+ and CD8+ IFNγ producers from FKBP12 KO CMV but not unmodified TCPs (Figures 4F, 4H, and 4I). In contrast, in unmodified TCPs, IFNγ producers were more frequent among CsA-treated compared with the tacrolimus-treated cultures (Figures 4F and 4H). CD4+ and CD8+ IFNγ producers (Figures 4F and 4H) and CD8+ IFNγ/TNFα double producers (Figure 4J) were more frequent in cultures of FKBP12 KO CMV TCPs cultured with triple IS compared with their unmodified counterpart.
Figure 4.

FKBP12 KO CMV TCPs after Thawing: Superior Response to CMV-Specific Stimulation in the Presence of Tac Compared with Unmodified CMV TCPs
Unmodified and FKBP12 KO CMV TCPs (edited with synt sgRNA#2) were cultured in the indicated immunosuppressants (Tac, CsA, or Tac + Pred + MPA) for 5 days after thawing and re-stimulated with CMV on day 5 after thawing. ∗p < 0.05. (A) Representative flow cytometry plot illustrating intracellular GZB and IFNγ staining in unstimulated (blue) and CMV peptide-stimulated CD4+ T cells (red) on day 5 after thawing. The plot illustrates gating for GZB+ CD4+ T cells. (B) Summary of proportions of GZB+ T cells within the CD4+ T cell population (gated as illustrated in A) of unmodified (green) and FKBP12 KO CMV TCPs (red) following CMV-specific re-stimulation after 5 days of culture in the indicated immunosuppressants. (C) Representative flow cytometry plot illustrating intracellular GZB and IFNγ staining in unstimulated (blue) and CMV peptide-stimulated CD8+ T cells (red) on day 5 after thawing. The gate illustrates gating for GZB+ CD8+ T cells. (D) Summary of proportions of GZB+ T cells within the CD8+ T cell population (gated as illustrated in C) of unmodified (green) and FKBP12 KO CMV TCPs (red) following CMV-specific re-stimulation after 5 days of culture in the indicated immunosuppressants. (E) Representative flow cytometry plot illustrating intracellular TNFα and IFNγ staining in unstimulated (blue) and CMV peptide-stimulated CD4+ T cells (red) on day 5 after thawing. The gates illustrate gating for IFNγ+ (sum of the two right quadrants) and TNFα+ IFNγ+ CD4+ T cells (top right quadrant). (F) Summary of proportions of IFNγ+ T cells within the CD4+ T cell population (gated as illustrated in E) of unmodified (green) and FKBP12 KO CMV TCPs (red) following CMV-specific stimulation after 5 days of culture in the indicated immunosuppressants. The background (see E, blue population) was subtracted to determine exclusively CMV-specific IFNγ producers. (G) Representative flow cytometry plot illustrating intracellular TNFα and IFNγ staining in unstimulated (blue) and CMV peptide-stimulated CD8+ T cells (red) on day 5 after thawing. The gates illustrate gating for IFNγ+ (sum of the two right quadrants) and TNFα+IFNγ + CD8+ T cells (top right quadrant). (H) Summary of proportions of IFNγ+ T cells within the CD8+ T cell population (gated as illustrated in G) of unmodified (green) and FKBP12 KO CMV TCPs (red) following CMV-specific stimulation after 5 days of culture in the indicated immunosuppressants. The background (see G, blue population) was subtracted to determine exclusively CMV-specific IFNγ producers. (I) Summary of proportions of TNFα+IFNγ+ T cells within the CD4+ T cell population (gated as illustrated in E) of unmodified (green) and FKBP12 KO CMV TCPs (red) following CMV-specific stimulation after 5 days of culture in the indicated immunosuppressants. The background (see E, blue population) was subtracted to determine exclusively CMV-specific TNFα IFNγ double producers. (J) Summary of proportions of TNFα+IFNγ+ T cells within the CD8+ T cell population (gated as illustrated in G) of unmodified (green) and FKBP12 KO CMV TCPs (red) following CMV-specific stimulation after 5 days of culture in the indicated immunosuppressants. The background (see G, blue population) was subtracted to determine exclusively CMV-specific TNFα+IFNγ+ double producers. (K) Representative flow cytometry plot illustrating intracellular IL-2 and IFNγ staining in unstimulated (blue) and CMV peptide-stimulated CD4+ T cells (red) on day 5 after thawing. The gates illustrate gating for IL-2+ (sum of the two right quadrants). (L) Summary of proportions of IL-2+ T cells within the CD4+ T cell population (gated as illustrated in K) of unmodified (green) and FKBP12 KO CMV TCPs (red) following CMV-specific stimulation after 5 days of culture in the indicated immunosuppressants. The background (see K, blue population) was subtracted to determine exclusively CMV-specific IFNγ producers. (M) Representative flow cytometry plot illustrating intracellular IL-2 and IFNγ staining in unstimulated (blue) and CMV peptide-stimulated CD8+ T cells (red) on day 5 after thawing. The gates illustrate gating for IL-2+ (sum of the two right quadrants). (N) Summary of proportions of IL-2+ T cells within the CD8+ T cell population (gated as illustrated in M) of unmodified (green) and FKBP12 KO CMV TCPs (red) following CMV-specific stimulation after 5 days of culture in the indicated immunosuppressants. The background (see M, blue population) was subtracted to determine exclusively CMV-specific IFNγ producers. (O) Representative flow cytometry plot illustrating intracellular CD154 and CD137 staining in unstimulated (blue) and CMV peptide-stimulated CD4+ T cells (red) on day 5 after thawing. The gates illustrate gating for CD137+ (sum of the two right quadrants) and CD154+ CD4+ T cells (sum of the two top quadrants). (P) Summary of proportions of CD154+ T cells within the CD4+ T cell population (gated as illustrated in O) of unmodified (green) and FKBP12 KO CMV TCPs (red) following CMV-specific stimulation after 5 days of culture in the indicated immunosuppressants. The background (see O, blue population) was subtracted to determine exclusively CMV-specific CD154+ CD4+ T cells. (Q) Representative flow cytometry plot illustrating intracellular CD154 and CD137 staining in unstimulated (blue) and CMV peptide-stimulated CD8+ T cells (red) on day 5 after thawing. The gates illustrate gating for CD137+ (sum of the two right quadrants) and CD154+ CD8+ T cells (sum of the two top quadrants). (R) Summary of proportions of CD154+ T cells within the CD8+ T cell population (gated as illustrated in Q) of unmodified (green) and FKBP12 KO CMV TCPs (red) following CMV-specific stimulation after 5 days of culture in the indicated immunosuppressants. The background (see Q, blue population) was subtracted to determine exclusively CMV-specific CD154+ CD8+ T cells. (S) Summary of proportions of CD137+ T cells within the CD4+ T cell population (gated as illustrated in O) of unmodified (green) and FKBP12 KO CMV TCPs (red) following CMV-specific stimulation after 5 days of culture in the indicated immunosuppressants. The background (see O, blue population) was subtracted to determine exclusively CMV-specific CD137+ CD4+ T cells. (T) Summary of proportions of CD137+ T cells within the CD8+ T cell population (gated as illustrated in Q) of unmodified (green) and FKBP12 KO CMV TCPs (red) following CMV-specific stimulation after 5 days of culture in the indicated immunosuppressants. The background (see Q, blue population) was subtracted to determine exclusively CMV-specific CD137+ CD4+ T cells.
Interleukin-2 (IL-2) is mainly produced by memory T cells with a low differentiation state and high proliferative potential, which contribute to establishment of long-term memory.39 A fraction of CD4+ (Figure 4K) and CD8+ T cells (Figure 4M) in thawed TCPs produced IL-2 upon CMV-specific stimulation. CD4+ IL-2 producers were more frequent in FKBP12 KO CMV TCPs cultured in tacrolimus compared with the corresponding unedited CMV TCPs (Figure 4L). This observation was significant for CD8+ IL-2 producers (Figure 4N). CD8+ IL-2-producers from FKBP12 KO CMV TCPs cultured in tacrolimus were significantly more frequent compared with CD8+ IL-2 producers from FKBP12 KO CMV TCPs cultured in CsA or with triple IS (Figure 4N).
Next we assessed expression of the activation markers CD154 (CD40L) and CD137 (41BB) on CMV-stimulated TCPs. CD154 increases activation of antigen presenting cells and is important for antibody production by B cells.40 CD154 is also induced upon antigen recognition by the T cell receptor (TCR)41 and has been reported to be expressed on T cells protective against CMV.42 We found CMV-specific CD154 upregulation on CD4+ (Figure 4O) but also on a subset of CD8+ T cells (Figure 4Q). CD154-expressing CD4+ (Figure 4P) and CD8+ T cells (Figure 4R) were more frequent among FKBP12 KO CMV TCPs cultured in tacrolimus compared with their unmodified counterpart (Figures 4P and 4R). After culture with triple IS, CD154-expressing CD4+ T cells increased in FKBP12 KO compared with unmodified CMV TCPs upon CMV-specific stimulation (Figure 4P). After culture in CsA and with triple IS, CMV-specific CD154 expressing CD8+ T cells in FKBP12 KO CMV TCPs were significantly reduced compared with tacrolimus-treated cultures of FKBP12 KO CMV TCPs (Figure 4R). CD137 (41BB) is another antigen-dependent activation marker on T cells (Figures 4O and 4Q). Except for a significant reduction in CMV-specific CD137-expressing CD4+ T cells from FKBP12 KO CMV TCPs upon culture in CsA compared with culture without immunosuppressants (Figure 4S), we did not detect differences in CMV-specific CD137-expressing T cells among CMV TCPs, after culture in immunosuppressants, or upon FKBP12 KO (Figures 4O, 4Q, 4S, and 4T).
Taken together, these results show that, after culture in tacrolimus, FKBP12 KO CMV TCPs mediate superior effector function and activation compared with unedited CMV TCPs and indicate that FKBP12 KO CMV TCPs acquired resistance to tacrolimus.
FKBP12 KO CMV TCPs Have a Similar TCRβ Repertoire as CMV TCPs after Expansion
We performed TCRβ sequencing to compare the clonality and total number of clones included in expanded FKBP12 KO versus CMV TCPs. There was no similarity between the TCRβ repertoires of different donors, defined by the Morisita index (Figure 5A). However, we detected high similarity between FKBP12 KO and CMV TCPs generated from the same donor (Figure 5A). We detected comparable clonality (Figure 5B), percentage of total sequence reads covered by the top 10 clones (Figure 5C), and total numbers of clones represented (Figure 5D) in FKBP12 KO versus CMV TCPs. Hence, we conclude that gene editing did not drastically skew the TCRβ repertoire or induce excessive clonal expansion.
Figure 5.

FKBP12 KO CMV TCPs and Unedited Products Have a Similar TCRβ Repertoire
TCRβ sequencing was performed to compare the clonality and number of clones included in the expanded FKBP12 KO versus CMV TCPs. (A) Similarity matrix based on the Morisita index for FKBP12 KO CMV TCPs and CMV TCPs. n = 2 (day 1 and day 2). (B) Clonality of FKBP12 KO CMV TCPs and CMV TCPs. n = 2. Clonality of 1, monoclonal; the lower the clonality, the more diverse the clonal composition of the product. (C) Proportion of total productive sequence reads covered by the top 10 most represented T cell clones in the product. n = 2. (D) Total number of clones. n = 2. (E) Total productive frequencies divided by the number of clones.
Cas9 Protein Is Undetectable in Edited TCPs after 5 Days of Culture
Studies of human T cell reactivity toward the Cas9 protein43,44 may raise the concern that Cas9-edited cells are potentially immunogenic. Therefore, we stained edited T cells for Cas9 protein after electroporation. We detected Cas9 protein expression in low proportions of T cells, which progressively decreased over time. Cas9 protein was undetectable by day 5 post-electroporation (Figure 6).
Figure 6.

Cas9 Protein Is Undetectable in FKBP12 KO CMV TCPs
Shown are proportions of edited T cells stained for Cas9 protein at different time points after electroporation.
Differentiation States of the Different TCPs at Different Stages of Production
The majority of CD8+ T cells have a terminally differentiated phenotype after isolation of CMV-reactive T cells (CCR7−CD45RA+) and, thus, may give rise to the concern that these TCPs have limited longevity (Figure 1B; Figure S1). In fact, after expansion and thawing, this population is almost undetectable in the majority of TCPs, allowing the assumption that less differentiated memory T cells expanded and represent the mass of the expanded TCPs (Figure S1B). Knockout of FKBP12 did not have a major effect on the T cell memory subset composition of the TCPs, and there was no clear trend toward a particular subset preference (Figure S1B).
Discussion
We seek to optimize antiviral T cell therapy for use in a setting requiring IS; for example, SOT. Thus, we developed a GMP-compliant approach for targeted gene editing of virus-specific T cells to achieve tacrolimus resistance. Our approach contains a viral antigen-specific T cell isolation step14,27,31 that is probably superior, in terms of safety, to other published protocols that start with a mixed PBMC population.26,28 This safety step is crucial to exclude an allograft-reactive, IS-resistant T cell contamination that could cause damage to the transplanted organ. We generated a functional FKBP12 KO CMV TCP with superior cytokine production and activation in the presence of tacrolimus and triple IS (used in SOT as standard IS) compared with unedited CMV TCPs. The retained sensitivity of the gene-edited FKBP12 KO CMV TCPs to CsA is an important safety switch. CsA is a clinically approved drug that could be used to limit any toxicity associated with the tacrolimus-resistant FKBP12 KO CMV TCP. Our protocol utilizes a cartridge-based, GMP-compatible electroporation system and adapted electroporation conditions. Although gene KO using multiple sgRNAs is more efficient,30 we performed FKBP12 KO using a single sgRNA to minimize potential off-target effects and to ensure safety and GMP compatibility. Moreover, we applied a GMP-compatible synt sgRNA with 2O’-methyl-3′phosphothioate bonds.34 The synt sgRNA enhanced KO efficiency and functionality of the product in the presence of tacrolimus and triple IS. Recently, similar processes have been reported for production of glucocorticoid-resistant antiviral T cells.45,46 All materials used for the described process are available at GMP grade or have sufficient documentation and quality to be accepted by regulatory authorities for GMP production. This qualifies the whole process as a GMP-compliant, ready-to-translate process.
We detected no effect of classic immunosuppressive drugs on cytotoxic killing capacity, as reported previously.47,48 Cytotoxicity in the “VITAL” assay used for determination of killing capacity is mainly based on perforin and granzyme-mediated killing.37 Transcription of perforin in T cells is reported to be induced by IL-2 and IL-15; thus, is not exclusively dependent on calcineurin.49,50 Substantial release of granzymes has also been reported in the presence of tacrolimus,51 which is in line with our findings of intracellularly captured GZB and increased GZB producers among CD4+ T cells upon viral stimulus in the presence of immunosuppressants.
Addition of CNIs decreased IFNγ producers among CD4+ and CD8+ T cells, but IFNγ production was not completely abolished. This has also been observed by others for CD4+ memory T cells at higher doses of IS than used here.23 Indeed, it has been reported that IFNγ can be induced by a calcineurin-independent pathway in T cells.52,53 This explains continued IFNγ production even in the presence of CNIs. Additionally, some downstream signaling events of the TCR are unaffected by inhibition of calcineurin and may lead to IFNγ induction.54 However, tacrolimus significantly reduced the proportion of IFNγ producers. Cytokine production in the presence of tacrolimus was rescued after FKBP12 KO, even in the presence of triple IS, although MPA and corticosteroids have been reported to further downregulate TCR-mediated IFNγ production.21,24 In contrast, TNFα producers were much more sensitive to inhibition by calcineurin inhibitors. Nonetheless, FKBP12 KO CMV TCPs recovered their ability of TNFα production in the presence of tacrolimus, confirming reports identifying calcineurin as the crucial inducer of TNFα.55 As reported previously,56 we found IL-2 production by T cells to be highly sensitive to immunosuppressants. Our data revealed significant recovery of IL-2 producers in FKBP12 KO CMV TCPs in the presence of tacrolimus. Notably, IL-2 is primarily produced by less differentiated long-lived memory T cells with high proliferative potential, which are a very attractive cell subset for antiviral T cell therapy approaches.39 Polyfunctional IFNγ+ TNFα+ IL-2+ CD4+ and CD8+ anti-viral T cells have been found to be protective against CMV disease after SOT.57 Our strategy for FKBP12 KO retained IFNγ/TNFα double producers and IL-2-producers despite IS; hence, FKBP12 KO CMV TCP is an attractive adoptive T cell transfer approach for induction of protective immunity in vivo.57 The activation marker CD154, which is associated with polyfunctionality58 and protection from CMV after SOT,42 was also highly sensitive to immunosuppressants. CMV-specific CD154 upregulation in the presence of tacrolimus could be restored by FKBP12 KO, underpinning the high antiviral potential of FKBP12 KO CMV TCPs in the presence of tacrolimus. CD154 is also expressed on a small subset of virus-specific CD8+ T cells (here, <10%), which may have helper-like functions.41 We did not observe any effect of CNIs on CMV stimulus-induced upregulation of the activation marker CD137 (4-1BB) on the examined T cells.
Glucocorticoid treatment has been reported to transiently increase the proportions of CD25+CD4+ T cells.59 Indeed, following 24 h of incubation with triple IS, including prednisolone, we recorded transiently elevated expression of CD25 that progressively declined by day 4 of culture with triple IS. MPA has been reported to downregulate CD25 expression on T cells.60 This possibly counteracted the glucocorticoid-mediated upregulation, resulting in the decline observed by day 4 of culture with triple IS. We could not confirm data implying glucocorticoid- or MPA-mediated upregulation of PD-1 on T cells.21,61 MPA and dexamethasone have been suggested to upregulate expression of CTLA-4 on CD4+ T cells.21,62 However, we observed significant attenuation of CTLA-4 after 4 days of culture with triple IS. CTLA-4 expression requires antigen stimulation.63 We presume that single initial antigenic T cell stimulation on day 0 was insufficient to sustain CTLA-4 expression.
Gene editing may raise concerns about transformation of the edited cells, which could lead to uncontrolled proliferation and, eventually, tumor formation. An increase in clonality of TCPs could be an indicator of transformation because certain transformed clones gain a growth advantage and are able to outgrow other clones. Thus, we performed TCR sequencing to exclude that there were any transformative events in TCPs. We found that the FKBP12 KO CMV TCPs did not have higher clonality than their non-edited counterparts, implying that the cells were not transformed. Furthermore, the percentage covered by the top 10 most frequent clones was also not increased, confirming that none of the clones acquired proliferative advantages as a result of gene editing.
Use of microbial compounds for genetic engineering and introduction of genetically modified cells into humans may elicit immune reactions.64 We demonstrated that FKBP12 KO CMV-specific T cells degrade the Cas9 protein within 5 days post-electroporation. Thus, recombinant bacterial Cas9, which has been reported to elicit adaptive immune responses,43,44 is unlikely to cause immune reactions in patients treated with the TCP described here. In general, use of autologous cells bears a low risk of immunogenicity.19 Nonetheless, potential mutations inserted during repair of double strand breaks may result in immunogenicity of the edited cells and cause T cell activation. The major concern when applying CRISPR-Cas9 technology for genetic engineering remains off-target gene editing posing the risk of uncontrollable mutagenesis.65 In-depth characterization applying next-generation sequencing technology to investigate potential off-target editing events66 predicted by the “CRISPR Off-target Sites with Mismatches, Insertions, and Deletions” (COSMID) tool in silico32 and using in vitro assays67,68 is currently ongoing to ensure a complete risk-safety calculation for clinical application.69
The initially sorted CMV-specific CD8+ T cells comprise a high percentage of terminally differentiated CD45RA-expressing effector memory T cells (TEMRA). This could raise concerns about the efficacy, longevity, and induction of functional memory by the resulting TCP. However, TEMRA are strongly associated with CMV infection, which induces them at high levels.70 These cells have been shown not to expand in polyclonally activated chimeric antigen-receptor (CAR)-TCPs when cultured alone or in bulk cultures of different T cell memory subsets.71 This is in line with our findings for expanded and thawed TCPs, which likely result from proliferation of less differentiated memory T cell subsets. Particularly in donors 4 and 5, we observed an accumulation of less differentiated memory T cells after thawing. This may be due to selective survival of earlier memory T cell subsets, which are known to show higher expression of anti-apoptotic molecules compared with their later differentiated counterparts and have also been reported to have high proliferative capacity.72, 73, 74 Another possibility would be de-differentiation from further differentiated memory T cells induced by the freezing and thawing cycle, which we would need to investigate in further experiments. Especially stem-cell memory T cells (TSCM) have been reported to re-acquire their naive-like phenotype during culture72, which could explain the emergence of naive-like cells after thawing, although they were previously underrepresented.
In summary, we developed a GMP-compatible protocol for manufacture of tacrolimus-resistant CMV-specific TCPs that are functionally superior to unmodified antiviral TCPs in the presence of tacrolimus and triple IS. Furthermore, they contain features of highly potent protective early memory T cells. The sensitivity to CsA represents an inherent safety switch that is exploitable in case the product should elicit adverse effects. Additionally, compared with previous approaches to generate CNI-resistant TCPs,26, 27, 28 our product is vector free and utilizes targeted mutagenesis. Therefore, it could be considered a safer methodology for TCP generation. This manufacturing process could be applied to generate effector TCPs directed against other viruses or tumors using the respective peptide pools for initial sorting of T cells with the desired specificity. This preclinical study paves the way for a first-in-human application of our tacrolimus-resistant anti-viral TCPs in the future.
Materials and Methods
Blood Sampling, Isolation, and Culture of Virus-Specific T Cells
The study was approved by the Charité-Universitätsmedizin Berlin Ethics Committee, and peripheral blood was obtained from healthy donors, who had given their written informed consent. PBMCs were isolated using Biocoll (Biochrom) gradient centrifugation. Virus-specific T cells were isolated following 6-h stimulation with overlapping CMV-specific phosphoprotein (pp) 65 and immediate-early (IE)-1 peptide pools (JPT Peptide Technologies, 0.5 μg/mL each) using the IFNγ Secretion Assay—Cell Enrichment and Detection Kit according to the manufacturer’s instructions (Miltenyi Biotec) and cultured in complete medium (very low endotoxin RPMI 1640 medium supplemented with penicillin (100 IU/mL) and streptomycin (all from Biochrom) and 10% fetal calf serum (FCS; PAA Laboratories), supplemented with 10 ng/mL recombinant human (rh) IL-7 and rhIL-15 (CellGenix) on 96- or 24-well plates, respectively, in humidified incubators at 37°C and 5% CO2 as described previously.13,14 Cells were split 1:1 upon reaching 100% confluency. After 21 days, TCPs were frozen in FCS with 10% dimethyl sulfoxide (Sigma-Aldrich) at – 80°C and transferred to liquid nitrogen after 1 day. TCPs were thawed in a water bath at 37°C and washed twice with complete medium. One half was analyzed immediately, and the other half was seeded into 24-well plates and cultured in complete medium with or without immunosuppressive drugs at clinically relevant doses (6 ng/mL tacrolimus [Prograf, Astellas], 120 ng/mL CsA [Sandimmun, Novartis], triple IS = 6 ng/mL tacrolimus + 0.57 μg/mL prednisolone [Urbason Solubile, Sanofi] and 2.7 μg/mL MPA [active substance of mycophenolate mofetil, Sigma-Aldrich]) until further analysis.
Knockout Procedure
FKBP12-specific sgRNAs (Table 1) were in vitro transcribed from DNA templates as described previously,75 using the Hi-Scribe T7 High Yield RNA Synthesis Kit (New England Biosciences). Where indicated, synt modified sgRNA (Synthego) with 2O’-methyl-3′phosphothioate modifications between the first and last 3 nucleotides was used.
Table 1.
Target Sequences of FKBP12-Specific sgRNAs
| FKBP12-Specific sgRNA | Target Sequence |
|---|---|
| sgRNA#1 | 5′-GGACTACACCGGTGAGTCGG-3′ |
| sgRNA#2 | 5′-GGGCGCACCTTCCCCAAGCG-3′ |
| sgRNA#3 | 5′-GGGTGAGTAGTGGCGCGCGG-3′ |
2.5 million antiviral T cells were electroporated on day 7 after isolation using the Amaxa P3 Primary Cell 4D-Nucleofector X Kit L and the Amaxa-Nucleofector-4D (Lonza, program CO-115) to transfer RNP complexes of 30 μg of recombinant Alt-R Streptococcus pyogenes Cas9 protein V3 (Integrated DNA Technologies) precomplexed with 30 μg of in-vitro-transcribed or 15 synt sgRNA (Synthego). The same numbers of unmodified antiviral T cells and antiviral T cells electroporated without additives were expanded as controls.
Phenotypic and Functional Assays Assessed by Flow Cytometry
For assessment of CMV-specific cytokine production and activation, LCLs were generated as described previously76 and used as antigen-presenting cells at a 1:10 ratio for a 6-h CMV-specific stimulation with 0.5 μg/mL CMV-specific peptide pools (IE-1 and pp-65 each) in the presence or absence of immunosuppressants at the clinical doses indicated above. Unstimulated controls included LCLs without CMV peptides. Intracellular cytokine production was captured by addition of 2 μg/mL of Brefeldin A (Sigma-Aldrich) after 1 h of stimulation, and cells were stained using antibodies (all from BioLegend unless stated otherwise) and the FoxP3/Transcription Factor Staining Buffer Set (eBioscience). Staining was performed using fluorophore-conjugated human anti-CD3 (OKT3), anti-CD4 (SK3), anti-CD8 (RPA-T8), anti-IFNγ (4S.B3, eBioscience), anti-TNF-α (MAb11), anti-IL-2 (MQ1-17H12), anti-CD137 (4B4-1), anti-CD154 (24-31), and anti-GZB (GB11, BD Pharmingen) antibodies. LIVE/DEAD Fixable Blue Dead Cell Stain (L/D; Invitrogen) was used to identify living cells and for determination of viability in combination with fluorescently labeled Annexin V (BioLegend).
A VITAL assay was performed to assess the killing capacity of TCPs.14,37,38 Briefly, cells from TCPs were incubated at distinct ratios with CMV peptide-loaded autologous LCLs stained with 10 μM carboxyfluorescein diacetate succinimidyl ester (CFSE-DA; Sigma-Aldrich) for 4 min and unloaded allogenic LCLs stained with 5 μM CellTrace Far Red (Invitrogen) for 10 min. T cell-free LCL mixtures served as internal controls to calculate the CMV-specific killing capacity.37 After 14 h of incubation, co-cultures were stained with L/D.
All flow cytometry samples were analyzed using an LSR-II-Fortessa flow cytometer (Becton Dickinson) and FlowJo-10 software (Tree Star).
Efficiency Analysis
Analysis of on-target editing was performed from isolated DNA (Zymo Research) of day 21 cell samples. The FKBP12 locus was amplified using KAPA HiFi HotStart ReadyMix (Roche) and the following primer pairs: 5′-TCTGACGGGTCAGATAACACCTAG-3′ (F) and 5′-TCTTCCGGAGGCCTGGGTTT-3′ (R) for sgRNA#1 and sgRNA#2 and 5′-ACAGCTGTATCCGGAGGCCT-3′ (F’) and 5′-TCACAGCCGCCGATTCAGAC-3′ (R’) for sgRNA#3 (Eurofins Scientific) with the following touchdown PCR program in an automated thermocycler: (1) 95°C, 3 min; (2) 98°C, 30 s; (3) 72°C–64°C for sgRNA#1 and sgRNA#22 and 68°C–64°C for sgRNA#3, 15 s (−0.5°C for each cycle starting at the highest until the lowest temperature was reached; 20 cycles, 64°C); (4) 72°C, 15 s; (5) repeat of step 2 with a decreasing annealing temperature (as specified); (6) 72°C, 1 min; and (7) 4°C. PCR products were purified using a DNA purification and enrichment kit (Zymo Research) prior to Sanger sequencing with primer F/F’ by LGC Genomics. Editing frequencies were calculated using the inference of CRISPR edits (ICE) algorithm (Synthego).66
TCRβ Sequencing
TCRβ sequencing was performed from genomic DNA by Adaptive Biotechnologies and analyzed using ImmunoSEQ ANALYZER 3.0 (Adaptive Biotechnologies). The data are available at the immuneACCESS platform (http://clients.adaptivebiotech.com/; https://doi.org/10.21417/LA2020MT).
Statistics
The p values were determined by tests for normal distribution (Shapiro-Wilk and Kolmogorov-Smirnov tests), followed by one-way ANOVA (normally distributed datasets) or Friedman test (not normally distributed datasets) and paired t tests (normally distributed datasets) or Wilcoxon matched-pairs signed-rank tests (not normally distributed datasets) as post-tests.
Author Contributions
L.A., H.-D.V., P.R., and M.S.-H. conceptualized and designed the study. D.L.W. and L.F.W. performed pre-experiments with polyclonal T cells and identified sgRNAs in silico. L.A. performed the experiments with support from D.L.W., T.V., D.J.W., G.Z., and U.R. L.A. acquired, analyzed, and interpreted data. L.A. and M.S.-H. composed the figures and manuscript. D.L.W., D.J.W., H.-D.V., and P.R. critically revised and all authors approved the final version of the manuscript.
Conflicts of Interest
L.A., M.S.-H., D.L.W., H.D.V., and P.R. have a patent pending (“Immunosuppressant-resistant T-cells for adoptive immunotherapy”).
Acknowledgments
We thank all voluntary blood donors for their donations. We thank Dr. Nicola Brindle for language editing of the manuscript. We thank Rebecca Noster for performing PCRs and Sanger sequencing preparation for KO efficiency measurements.
The study was generously supported in parts by the Deutsche Forschungsgemeinschaft (DFG-SFB-TR36-project A3 to H.-D.V., P.R., and M.S.H.), the German Federal Ministry of Education and Research (BIH Center for Regenerative Therapies, 10178 Berlin, to L.A., T.V., D.J.W., P.R., H.-D.V., and M.S.-H.), a kick-box grant for young scientists and a research grant from the Einstein Center for Regenerative Therapies (to L.A., D.L.W., U.R., G.Z., and M.S.-H.), and a Berlin Institute of Health (BIH) Translation Mission Fund and a Crossfield project fund from the BIH Research Focus Regenerative Medicine (to L.A., D.J.W., P.R., H.-D.V., and M.S.-H.). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.09.011.
Supplemental Information
References
- 1.Chen J.H., Mao Y.Y., He Q., Wu J.Y., Lv R. The impact of pretransplant cytomegalovirus infection on acute renal allograft rejection. Transplant. Proc. 2005;37:4203–4207. doi: 10.1016/j.transproceed.2005.11.036. [DOI] [PubMed] [Google Scholar]
- 2.Tong C.Y., Bakran A., Peiris J.S., Muir P., Herrington C.S. The association of viral infection and chronic allograft nephropathy with graft dysfunction after renal transplantation. Transplantation. 2002;74:576–578. doi: 10.1097/00007890-200208270-00026. [DOI] [PubMed] [Google Scholar]
- 3.Grossi P.A., Costa A.N., Fehily D., Blumberg E.A., Kuehnert M.J., Fishman J.A., Ison M.G., Lattes R., Kotton C.N., Lilleri D. Infections and organ transplantation: new challenges for prevention and treatment--a colloquium. Transplantation. 2012;93(5, Suppl):S4–S39. doi: 10.1097/TP.0b013e3182481347. [DOI] [PubMed] [Google Scholar]
- 4.Harvala H., Stewart C., Muller K., Burns S., Marson L., MacGilchrist A., Johannessen I. High risk of cytomegalovirus infection following solid organ transplantation despite prophylactic therapy. J. Med. Virol. 2013;85:893–898. doi: 10.1002/jmv.23539. [DOI] [PubMed] [Google Scholar]
- 5.Kumar D., Chernenko S., Moussa G., Cobos I., Manuel O., Preiksaitis J., Venkataraman S., Humar A. Cell-mediated immunity to predict cytomegalovirus disease in high-risk solid organ transplant recipients. Am. J. Transplant. 2009;9:1214–1222. doi: 10.1111/j.1600-6143.2009.02618.x. [DOI] [PubMed] [Google Scholar]
- 6.Schachtner T., Stein M., Reinke P. CMV-Specific T Cell Monitoring Offers Superior Risk Stratification of CMV-Seronegative Kidney Transplant Recipients of a CMV-Seropositive Donor. Transplantation. 2017;101:e315–e325. doi: 10.1097/TP.0000000000001825. [DOI] [PubMed] [Google Scholar]
- 7.Hill G.R., Tey S.K., Beagley L., Crough T., Morton J.A., Clouston A.D., Whiting P., Khanna R. Successful immunotherapy of HCMV disease using virus-specific T cells expanded from an allogeneic stem cell transplant recipient. Am. J. Transplant. 2010;10:173–179. doi: 10.1111/j.1600-6143.2009.02872.x. [DOI] [PubMed] [Google Scholar]
- 8.Clancy L.E., Blyth E., Simms R.M., Micklethwaite K.P., Ma C.K., Burgess J.S., Antonenas V., Shaw P.J., Gottlieb D.J. Cytomegalovirus-specific cytotoxic T lymphocytes can be efficiently expanded from granulocyte colony-stimulating factor-mobilized hemopoietic progenitor cell products ex vivo and safely transferred to stem cell transplantation recipients to facilitate immune reconstitution. Biol. Blood Marrow Transplant. 2013;19:725–734. doi: 10.1016/j.bbmt.2013.01.021. [DOI] [PubMed] [Google Scholar]
- 9.Stemberger C., Graef P., Odendahl M., Albrecht J., Dössinger G., Anderl F., Buchholz V.R., Gasteiger G., Schiemann M., Grigoleit G.U. Lowest numbers of primary CD8(+) T cells can reconstitute protective immunity upon adoptive immunotherapy. Blood. 2014;124:628–637. doi: 10.1182/blood-2013-12-547349. [DOI] [PubMed] [Google Scholar]
- 10.Stuehler C., Stüssi G., Halter J., Nowakowska J., Schibli A., Battegay M., Dirks J., Passweg J., Heim D., Rovo A. Combination therapy for multidrug-resistant cytomegalovirus disease. Transpl. Infect. Dis. 2015;17:751–755. doi: 10.1111/tid.12435. [DOI] [PubMed] [Google Scholar]
- 11.Neuenhahn M., Albrecht J., Odendahl M., Schlott F., Dössinger G., Schiemann M., Lakshmipathi S., Martin K., Bunjes D., Harsdorf S. Transfer of minimally manipulated CMV-specific T cells from stem cell or third-party donors to treat CMV infection after allo-HSCT. Leukemia. 2017;31:2161–2171. doi: 10.1038/leu.2017.16. [DOI] [PubMed] [Google Scholar]
- 12.Riddell S.R., Watanabe K.S., Goodrich J.M., Li C.R., Agha M.E., Greenberg P.D. Restoration of viral immunity in immunodeficient humans by the adoptive transfer of T cell clones. Science. 1992;257:238–241. doi: 10.1126/science.1352912. [DOI] [PubMed] [Google Scholar]
- 13.Brestrich G., Zwinger S., Roemhild A., Noutsias M., Rohde M., Keeren K., Sawitzki B., Volk H.D., Reinke P., Hammer M.H. Generation of HCMV-specific T-cell lines from seropositive solid-organ-transplant recipients for adoptive T-cell therapy. J. Immunother. 2009;32:932–940. doi: 10.1097/CJI.0b013e3181b88fda. [DOI] [PubMed] [Google Scholar]
- 14.Amini L., Vollmer T., Wendering D.J., Jurisch A., Landwehr-Kenzel S., Otto N.M., Jürchott K., Volk H.D., Reinke P., Schmueck-Henneresse M. Comprehensive Characterization of a Next-Generation Antiviral T-Cell Product and Feasibility for Application in Immunosuppressed Transplant Patients. Front. Immunol. 2019;10:1148. doi: 10.3389/fimmu.2019.01148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Macesic N., Langsford D., Nicholls K., Hughes P., Gottlieb D.J., Clancy L., Blyth E., Micklethwaite K., Withers B., Majumdar S. Adoptive T cell immunotherapy for treatment of ganciclovir-resistant cytomegalovirus disease in a renal transplant recipient. Am. J. Transplant. 2015;15:827–832. doi: 10.1111/ajt.13023. [DOI] [PubMed] [Google Scholar]
- 16.Holmes-Liew C.L., Holmes M., Beagley L., Hopkins P., Chambers D., Smith C., Khanna R. Adoptive T-cell immunotherapy for ganciclovir-resistant CMV disease after lung transplantation. Clin. Transl. Immunology. 2015;4:e35. doi: 10.1038/cti.2015.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Smith C., Beagley L., Rehan S., Neller M.A., Crooks P., Solomon M. Autologous adoptive T-cell therapy for recurrent or drug-resistant cytomegalovirus complications in solid organ transplant patients: A single-arm open-label phase I clinical trial. Clin. Infect. Dis. 2018;68:632–640. doi: 10.1093/cid/ciy549. [DOI] [PubMed] [Google Scholar]
- 18.Einsele H., Roosnek E., Rufer N., Sinzger C., Riegler S., Löffler J., Grigoleit U., Moris A., Rammensee H.G., Kanz L. Infusion of cytomegalovirus (CMV)-specific T cells for the treatment of CMV infection not responding to antiviral chemotherapy. Blood. 2002;99:3916–3922. doi: 10.1182/blood.v99.11.3916. [DOI] [PubMed] [Google Scholar]
- 19.Brestrich G., Zwinger S., Fischer A., Schmück M., Röhmhild A., Hammer M.H., Kurtz A., Uharek L., Knosalla C., Lehmkuhl H. Adoptive T-cell therapy of a lung transplanted patient with severe CMV disease and resistance to antiviral therapy. Am. J. Transplant. 2009;9:1679–1684. doi: 10.1111/j.1600-6143.2009.02672.x. [DOI] [PubMed] [Google Scholar]
- 20.Savoldo B., Goss J.A., Hammer M.M., Zhang L., Lopez T., Gee A.P., Lin Y.F., Quiros-Tejeira R.E., Reinke P., Schubert S. Treatment of solid organ transplant recipients with autologous Epstein Barr virus-specific cytotoxic T lymphocytes (CTLs) Blood. 2006;108:2942–2949. doi: 10.1182/blood-2006-05-021782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.He X., Smeets R.L., Koenen H.J., Vink P.M., Wagenaars J., Boots A.M., Joosten I. Mycophenolic acid-mediated suppression of human CD4+ T cells: more than mere guanine nucleotide deprivation. Am. J. Transplant. 2011;11:439–449. doi: 10.1111/j.1600-6143.2010.03413.x. [DOI] [PubMed] [Google Scholar]
- 22.Allison A.C., Eugui E.M. Mycophenolate mofetil and its mechanisms of action. Immunopharmacology. 2000;47:85–118. doi: 10.1016/s0162-3109(00)00188-0. [DOI] [PubMed] [Google Scholar]
- 23.Tsuda K., Yamanaka K., Kitagawa H., Akeda T., Naka M., Niwa K., Nakanishi T., Kakeda M., Gabazza E.C., Mizutani H. Calcineurin inhibitors suppress cytokine production from memory T cells and differentiation of naïve T cells into cytokine-producing mature T cells. PLoS ONE. 2012;7:e31465. doi: 10.1371/journal.pone.0031465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brinkmann V., Kristofic C. Regulation by corticosteroids of Th1 and Th2 cytokine production in human CD4+ effector T cells generated from CD45RO- and CD45RO+ subsets. J. Immunol. 1995;155:3322–3328. [PubMed] [Google Scholar]
- 25.Herold M.J., McPherson K.G., Reichardt H.M. Glucocorticoids in T cell apoptosis and function. Cell. Mol. Life Sci. 2006;63:60–72. doi: 10.1007/s00018-005-5390-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brewin J., Mancao C., Straathof K., Karlsson H., Samarasinghe S., Amrolia P.J., Pule M. Generation of EBV-specific cytotoxic T cells that are resistant to calcineurin inhibitors for the treatment of posttransplantation lymphoproliferative disease. Blood. 2009;114:4792–4803. doi: 10.1182/blood-2009-07-228387. [DOI] [PubMed] [Google Scholar]
- 27.Ricciardelli I., Brewin J., Lugthart G., Albon S.J., Pule M., Amrolia P.J. Rapid generation of EBV-specific cytotoxic T lymphocytes resistant to calcineurin inhibitors for adoptive immunotherapy. Am. J. Transplant. 2013;13:3244–3252. doi: 10.1111/ajt.12475. [DOI] [PubMed] [Google Scholar]
- 28.De Angelis B., Dotti G., Quintarelli C., Huye L.E., Zhang L., Zhang M., Pane F., Heslop H.E., Brenner M.K., Rooney C.M., Savoldo B. Generation of Epstein-Barr virus-specific cytotoxic T lymphocytes resistant to the immunosuppressive drug tacrolimus (FK506) Blood. 2009;114:4784–4791. doi: 10.1182/blood-2009-07-230482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Xu X., Su B., Barndt R.J., Chen H., Xin H., Yan G., Chen L., Cheng D., Heitman J., Zhuang Y. FKBP12 is the only FK506 binding protein mediating T-cell inhibition by the immunosuppressant FK506. Transplantation. 2002;73:1835–1838. doi: 10.1097/00007890-200206150-00023. [DOI] [PubMed] [Google Scholar]
- 30.Gundry M.C., Brunetti L., Lin A., Mayle A.E., Kitano A., Wagner D., Hsu J.I., Hoegenauer K.A., Rooney C.M., Goodell M.A., Nakada D. Highly Efficient Genome Editing of Murine and Human Hematopoietic Progenitor Cells by CRISPR/Cas9. Cell Rep. 2016;17:1453–1461. doi: 10.1016/j.celrep.2016.09.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schmueck M., Fischer A.M., Hammoud B., Brestrich G., Fuehrer H., Luu S.H., Mueller K., Babel N., Volk H.D., Reinke P. Preferential expansion of human virus-specific multifunctional central memory T cells by partial targeting of the IL-2 receptor signaling pathway: the key role of CD4+ T cells. J. Immunol. 2012;188:5189–5198. doi: 10.4049/jimmunol.1103763. [DOI] [PubMed] [Google Scholar]
- 32.Cradick T.J., Qiu P., Lee C.M., Fine E.J., Bao G. COSMID: A Web-based Tool for Identifying and Validating CRISPR/Cas Off-target Sites. Mol. Ther. Nucleic Acids. 2014;3:e214. doi: 10.1038/mtna.2014.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Casper J., Zweig A.S., Villarreal C., Tyner C., Speir M.L., Rosenbloom K.R., Raney B.J., Lee C.M., Lee B.T., Karolchik D. The UCSC Genome Browser database: 2018 update. Nucleic Acids Res. 2018;46(D1):D762–D769. doi: 10.1093/nar/gkx1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hendel A., Bak R.O., Clark J.T., Kennedy A.B., Ryan D.E., Roy S., Steinfeld I., Lunstad B.D., Kaiser R.J., Wilkens A.B. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 2015;33:985–989. doi: 10.1038/nbt.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Weist B.J., Wehler P., El Ahmad L., Schmueck-Henneresse M., Millward J.M., Nienen M., Neumann A.U., Reinke P., Babel N. A revised strategy for monitoring BKV-specific cellular immunity in kidney transplant patients. Kidney Int. 2015;88:1293–1303. doi: 10.1038/ki.2015.215. [DOI] [PubMed] [Google Scholar]
- 36.Kannanganat S., Ibegbu C., Chennareddi L., Robinson H.L., Amara R.R. Multiple-cytokine-producing antiviral CD4 T cells are functionally superior to single-cytokine-producing cells. J. Virol. 2007;81:8468–8476. doi: 10.1128/JVI.00228-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hammoud B., Schmueck M., Fischer A.M., Fuehrer H., Park S.J., Akyuez L., Schefold J.C., Raftery M.J., Schönrich G., Kaufmann A.M. HCMV-specific T-cell therapy: do not forget supply of help. J. Immunother. 2013;36:93–101. doi: 10.1097/CJI.0b013e31827b87cc. [DOI] [PubMed] [Google Scholar]
- 38.Hermans I.F., Silk J.D., Yang J., Palmowski M.J., Gileadi U., McCarthy C., Salio M., Ronchese F., Cerundolo V. The VITAL assay: a versatile fluorometric technique for assessing CTL- and NKT-mediated cytotoxicity against multiple targets in vitro and in vivo. J. Immunol. Methods. 2004;285:25–40. doi: 10.1016/j.jim.2003.10.017. [DOI] [PubMed] [Google Scholar]
- 39.Sallusto F., Geginat J., Lanzavecchia A. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu. Rev. Immunol. 2004;22:745–763. doi: 10.1146/annurev.immunol.22.012703.104702. [DOI] [PubMed] [Google Scholar]
- 40.Arce S., Nawar H.F., Muehlinghaus G., Russell M.W., Connell T.D. In vitro induction of immunoglobulin A (IgA)- and IgM-secreting plasma blasts by cholera toxin depends on T-cell help and is mediated by CD154 up-regulation and inhibition of gamma interferon synthesis. Infect. Immun. 2007;75:1413–1423. doi: 10.1128/IAI.01367-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frentsch M., Stark R., Matzmohr N., Meier S., Durlanik S., Schulz A.R., Stervbo U., Jürchott K., Gebhardt F., Heine G. CD40L expression permits CD8+ T cells to execute immunologic helper functions. Blood. 2013;122:405–412. doi: 10.1182/blood-2013-02-483586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ashokkumar C., Green M., Soltys K., Michaels M., Mazariegos G., Reyes-Mugica M., Higgs B.W., Spishock B., Zaccagnini M., Sethi P. CD154-expressing CMV-specific T cells associate with freedom from DNAemia and may be protective in seronegative recipients after liver or intestine transplantation. Pediatr. Transplant. 2020;24:e13601. doi: 10.1111/petr.13601. [DOI] [PubMed] [Google Scholar]
- 43.Wagner D.L., Amini L., Wendering D.J., Burkhardt L.M., Akyüz L., Reinke P., Volk H.D., Schmueck-Henneresse M. High prevalence of Streptococcus pyogenes Cas9-reactive T cells within the adult human population. Nat. Med. 2018;25:242–248. doi: 10.1038/s41591-018-0204-6. [DOI] [PubMed] [Google Scholar]
- 44.Charlesworth C.T., Deshpande P.S., Dever D.P., Camarena J., Lemgart V.T., Cromer M.K., Vakulskas C.A., Collingwood M.A., Zhang L., Bode N.M. Identification of preexisting adaptive immunity to Cas9 proteins in humans. Nat. Med. 2019;25:249–254. doi: 10.1038/s41591-018-0326-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kaeuferle T., Deisenberger L., Jablonowski L., Stief T.A., Blaeschke F., Willier S., Feuchtinger T. CRISPR-Cas9-Mediated Glucocorticoid Resistance in Virus-Specific T Cells for Adoptive T Cell Therapy Posttransplantation. Mol. Ther. 2020;28:1965–1973. doi: 10.1016/j.ymthe.2020.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Basar R., Daher M., Uprety N., Gokdemir E., Alsuliman A., Ensley E., Ozcan G., Mendt M., Hernandez Sanabria M., Kerbauy L.N. Large-scale GMP-compliant CRISPR-Cas9-mediated deletion of the glucocorticoid receptor in multivirus-specific T cells. Blood Adv. 2020;4:3357–3367. doi: 10.1182/bloodadvances.2020001977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Strauss G., Osen W., Debatin K.M. Induction of apoptosis and modulation of activation and effector function in T cells by immunosuppressive drugs. Clin. Exp. Immunol. 2002;128:255–266. doi: 10.1046/j.1365-2249.2002.01777.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Savoldo B., Goss J., Liu Z., Huls M.H., Doster S., Gee A.P., Brenner M.K., Heslop H.E., Rooney C.M. Generation of autologous Epstein-Barr virus-specific cytotoxic T cells for adoptive immunotherapy in solid organ transplant recipients. Transplantation. 2001;72:1078–1086. doi: 10.1097/00007890-200109270-00017. [DOI] [PubMed] [Google Scholar]
- 49.Zhang J., Scordi I., Smyth M.J., Lichtenheld M.G. Interleukin 2 receptor signaling regulates the perforin gene through signal transducer and activator of transcription (Stat)5 activation of two enhancers. J. Exp. Med. 1999;190:1297–1308. doi: 10.1084/jem.190.9.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Alves N.L., Hooibrink B., Arosa F.A., van Lier R.A. IL-15 induces antigen-independent expansion and differentiation of human naive CD8+ T cells in vitro. Blood. 2003;102:2541–2546. doi: 10.1182/blood-2003-01-0183. [DOI] [PubMed] [Google Scholar]
- 51.Kataoka T., Nagai K. Involvement of FK506-sensitive and insensitive granule exocytosis pathways in perforin-dependent target cell lysis mediated by a CD8+ CTL clone. Immunol. Lett. 2000;72:49–52. doi: 10.1016/s0165-2478(00)00160-7. [DOI] [PubMed] [Google Scholar]
- 52.Yang J., Murphy T.L., Ouyang W., Murphy K.M. Induction of interferon-γ production in Th1 CD4+ T cells: evidence for two distinct pathways for promoter activation. Eur. J. Immunol. 1999;29:548–555. doi: 10.1002/(SICI)1521-4141(199902)29:02<548::AID-IMMU548>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 53.Balasubramani A., Shibata Y., Crawford G.E., Baldwin A.S., Hatton R.D., Weaver C.T. Modular utilization of distal cis-regulatory elements controls Ifng gene expression in T cells activated by distinct stimuli. Immunity. 2010;33:35–47. doi: 10.1016/j.immuni.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Dutta D., Barr V.A., Akpan I., Mittelstadt P.R., Singha L.I., Samelson L.E., Ashwell J.D. Recruitment of calcineurin to the TCR positively regulates T cell activation. Nat. Immunol. 2017;18:196–204. doi: 10.1038/ni.3640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Goldfeld A.E., Tsai E., Kincaid R., Belshaw P.J., Schrieber S.L., Strominger J.L., Rao A. Calcineurin mediates human tumor necrosis factor alpha gene induction in stimulated T and B cells. J. Exp. Med. 1994;180:763–768. doi: 10.1084/jem.180.2.763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.O’Keefe S.J., Tamura J., Kincaid R.L., Tocci M.J., O’Neill E.A. FK-506- and CsA-sensitive activation of the interleukin-2 promoter by calcineurin. Nature. 1992;357:692–694. doi: 10.1038/357692a0. [DOI] [PubMed] [Google Scholar]
- 57.Snyder L.D., Chan C., Kwon D., Yi J.S., Martissa J.A., Copeland C.A., Osborne R.J., Sparks S.D., Palmer S.M., Weinhold K.J. Polyfunctional T-Cell Signatures to Predict Protection from Cytomegalovirus after Lung Transplantation. Am. J. Respir. Crit. Care Med. 2016;193:78–85. doi: 10.1164/rccm.201504-0733OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Cytomegalovirus. Am. J. Transplant. 2004;4(Suppl 10):51–58. doi: 10.1111/j.1600-6135.2004.00727.x. [DOI] [PubMed] [Google Scholar]
- 59.Braitch M., Harikrishnan S., Robins R.A., Nichols C., Fahey A.J., Showe L., Constantinescu C.S. Glucocorticoids increase CD4CD25 cell percentage and Foxp3 expression in patients with multiple sclerosis. Acta Neurol. Scand. 2009;119:239–245. doi: 10.1111/j.1600-0404.2008.01090.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Prémaud A., Rousseau A., Johnson G., Canivet C., Gandia P., Muscari F., Peron J.M., Rostaing L., Marquet P., Kamar N. Inhibition of T-cell activation and proliferation by mycophenolic acid in patients awaiting liver transplantation: PK/PD relationships. Pharmacol. Res. 2011;63:432–438. doi: 10.1016/j.phrs.2011.01.005. [DOI] [PubMed] [Google Scholar]
- 61.Maeda N., Maruhashi T., Sugiura D., Shimizu K., Okazaki I.M., Okazaki T. Glucocorticoids potentiate the inhibitory capacity of programmed cell death 1 by up-regulating its expression on T cells. J. Biol. Chem. 2019;294:19896–19906. doi: 10.1074/jbc.RA119.010379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xia M., Gasser J., Feige U. Dexamethasone enhances CTLA-4 expression during T cell activation. Cell. Mol. Life Sci. 1999;55:1649–1656. doi: 10.1007/s000180050403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gibson H.M., Hedgcock C.J., Aufiero B.M., Wilson A.J., Hafner M.S., Tsokos G.C., Wong H.K. Induction of the CTLA-4 gene in human lymphocytes is dependent on NFAT binding the proximal promoter. J. Immunol. 2007;179:3831–3840. doi: 10.4049/jimmunol.179.6.3831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Riddell S.R., Elliott M., Lewinsohn D.A., Gilbert M.J., Wilson L., Manley S.A., Lupton S.D., Overell R.W., Reynolds T.C., Corey L., Greenberg P.D. T-cell mediated rejection of gene-modified HIV-specific cytotoxic T lymphocytes in HIV-infected patients. Nat. Med. 1996;2:216–223. doi: 10.1038/nm0296-216. [DOI] [PubMed] [Google Scholar]
- 65.Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hsiau T., Conant D., Rossi N., Maures T., Waite K., Yang J., Joshi S., Kelso R., Holden K., Enzmann B.L., Stoner R. Inference of CRISPR Edits from Sanger Trace Data. bioRxiv. 2019 doi: 10.1101/251082. [DOI] [PubMed] [Google Scholar]
- 67.Turchiano G., Andrieux G., Blattner G., Pennucci V., Klermund J., Monaco G., Poddar S., Mussolino C., Cornu T.I., Boerries M., Cathomen T. Quantitative Evaluation of Chromosomal Rearrangements in Primary Gene-Edited Human Stem Cells by Preclinical CAST-Seq. Cell Stem Cell. 2020 doi: 10.2139/ssrn.3565007. Published online April 9, 2020. [DOI] [PubMed] [Google Scholar]
- 68.Cameron P., Fuller C.K., Donohoue P.D., Jones B.N., Thompson M.S., Carter M.M., Gradia S., Vidal B., Garner E., Slorach E.M. Mapping the genomic landscape of CRISPR-Cas9 cleavage. Nat. Methods. 2017;14:600–606. doi: 10.1038/nmeth.4284. [DOI] [PubMed] [Google Scholar]
- 69.Cathomen T., Schüle S., Schüßler-Lenz M., Abou-El-Enein M. The Human Genome Editing Race: Loosening Regulatory Standards for Commercial Advantage? Trends Biotechnol. 2019;37:120–123. doi: 10.1016/j.tibtech.2018.06.005. [DOI] [PubMed] [Google Scholar]
- 70.Di Benedetto S., Derhovanessian E., Steinhagen-Thiessen E., Goldeck D., Müller L., Pawelec G. Impact of age, sex and CMV-infection on peripheral T cell phenotypes: results from the Berlin BASE-II Study. Biogerontology. 2015;16:631–643. doi: 10.1007/s10522-015-9563-2. [DOI] [PubMed] [Google Scholar]
- 71.Schmueck-Henneresse M., Omer B., Shum T., Tashiro H., Mamonkin M., Lapteva N., Sharma S., Rollins L., Dotti G., Reinke P. Comprehensive Approach for Identifying the T Cell Subset Origin of CD3 and CD28 Antibody-Activated Chimeric Antigen Receptor-Modified T Cells. J. Immunol. 2017;199:348–362. doi: 10.4049/jimmunol.1601494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schmueck-Henneresse M., Sharaf R., Vogt K., Weist B.J., Landwehr-Kenzel S., Fuehrer H., Jurisch A., Babel N., Rooney C.M., Reinke P., Volk H.D. Peripheral blood-derived virus-specific memory stem T cells mature to functional effector memory subsets with self-renewal potency. J. Immunol. 2015;194:5559–5567. doi: 10.4049/jimmunol.1402090. [DOI] [PubMed] [Google Scholar]
- 73.Gattinoni L., Lugli E., Ji Y., Pos Z., Paulos C.M., Quigley M.F., Almeida J.R., Gostick E., Yu Z., Carpenito C. A human memory T cell subset with stem cell-like properties. Nat. Med. 2011;17:1290–1297. doi: 10.1038/nm.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sallusto F., Lenig D., Förster R., Lipp M., Lanzavecchia A. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature. 1999;401:708–712. doi: 10.1038/44385. [DOI] [PubMed] [Google Scholar]
- 75.Li D., Qiu Z., Shao Y., Chen Y., Guan Y., Liu M., Li Y., Gao N., Wang L., Lu X. Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat. Biotechnol. 2013;31:681–683. doi: 10.1038/nbt.2661. [DOI] [PubMed] [Google Scholar]
- 76.Moosmann A., Khan N., Cobbold M., Zentz C., Delecluse H.J., Hollweck G., Hislop A.D., Blake N.W., Croom-Carter D., Wollenberg B. B cells immortalized by a mini-Epstein-Barr virus encoding a foreign antigen efficiently reactivate specific cytotoxic T cells. Blood. 2002;100:1755–1764. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.