

Abstract
Many investigational adoptive immunotherapy regimens utilizing natural killer (NK) cells require the administration of interleukin-2 (IL-2) or IL-15, but these cytokines cause serious dose-dependent toxicities. To reduce or preclude the necessity for IL-2 use, we investigated whether genetic engineering of NK cells to express the erythropoietin (EPO) receptor (EPOR) or thrombopoietin (TPO) receptor (c-MPL) could be used as a method to improve NK cell survival and function. Viral transduction of NK-92 cells to express EPOR or c-MPL receptors conveyed signaling via appropriate pathways, protected cells from apoptosis, augmented cellular proliferation, and increased cell cytotoxic function in response to EPO or TPO ligands in vitro. In the presence of TPO, viral transduction of primary human NK cells to express c-MPL enhanced cellular proliferation and increased degranulation and cytokine production toward target cells in vitro. In contrast, transgenic expression of EPOR did not augment the proliferation of primary NK cells. In immunodeficient mice receiving TPO, in vivo persistence of primary human NK cells genetically modified to express c-MPL was higher compared with control NK cells. These data support the concept that genetic manipulation of NK cells to express hematopoietic growth factor receptors could be used as a strategy to augment NK cell proliferation and antitumor immunity.
Keywords: natural killer cell, cellular immunotherapy, MPL, c-MPL, EPOR, thrombopoietin, TPO, THPO, erythropoietin, EPO
Graphical Abstract
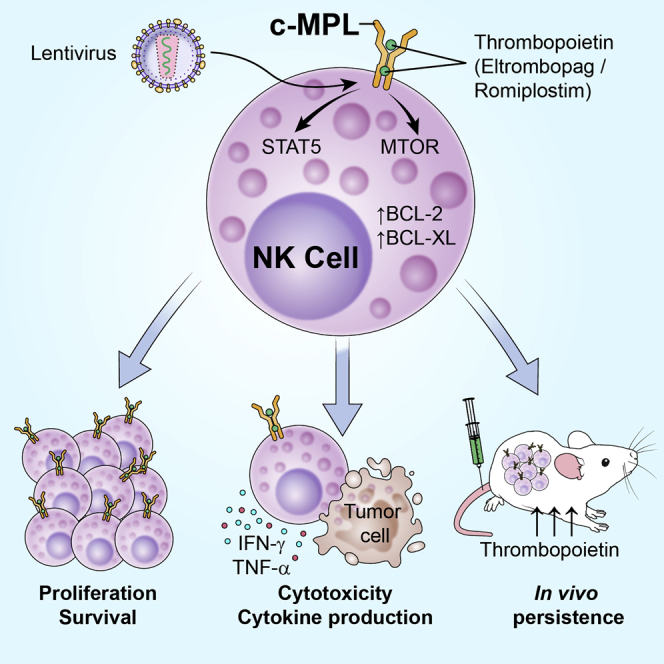
Aiming to reduce IL-2 dependence and toxicity during NK cell immunotherapy, Childs and colleagues investigated the exogenous expression of thrombopoietin and erythropoietin receptors in NK cells and the use of hematopoietic growth factors to specifically stimulate NK cell function and persistence after adoptive transfer.
Introduction
There currently exist a number of different pre-clinical and clinical strategies for utilizing natural killer (NK) cells as an immunotherapeutic to treat cancer.1, 2, 3, 4, 5 NK cells lack the requirement for prior sensitization and can induce tumor cytotoxicity in an antigen-independent manner without causing graft-versus-host disease, making NK cells an attractive cell-based treatment option. Recent advances in genetic engineering techniques now provide the possibility to modify NK cells to enhance their efficacy in treating cancer. In this regard, several pre-clinical reports have shown that the antitumor cytotoxicity of NK cells can be improved by engineering them to target specific tumor antigens.1,4,5 One significant limitation of NK cell-based immunotherapy is their reliance on cytokines, such as interleukin-2 (IL-2), IL-15, or IL-21, which enhance NK cell persistence and expansion in vitro and in vivo. At present, most clinical trials evaluating NK cell therapy in humans utilize the exogenous administration of IL-2 or IL-15 following adoptive cell transfer.3 Although IL-2 promotes activation, proliferation, and cytotoxicity of NK cells, it also has the undesirable effect of inducing the expansion of regulatory T cells (Tregs), which may suppress immune responses. Although IL-15 improves NK cell homeostasis, expansion, and cytolytic capacity without expanding Tregs,6,7 it has cytokine-associated toxicities similar to those observed with IL-2, including capillary leak syndrome, hypotension, fever, and chills.8, 9, 10
Investigators have recently explored a number of approaches to improve NK cell persistence in vivo that minimize toxicities and reduce or avoid the need for exogenous cytokine administration. Genetic modification of NK cell lines to express IL-2 or endoplasmic reticulum-retained IL-2 have been shown to result in autonomous cell proliferation.11,12 Likewise, expressing IL-15 or membrane-bound IL-15 on NK cells can enhance their proliferation and cytotoxicity in the absence of exogenous cytokines.13, 14, 15 Recently, Liu et al.16 demonstrated that NK cells derived from cord blood engineered to express anti-CD19 CAR, IL-15, and a suicide gene had improved anti-tumor activity and long-term persistence. In addition, an IL-15 superagonist complex called ALT-803 exhibited a longer half-life and better immune NK cell activation in vivo compared with wild-type IL-15.17,18
Hematopoietic growth factors, such as recombinant human (rh) erythropoietin (EPO) and thrombopoietin (TPO/THPO) mimetics are US Food and Drug Administration (FDA) approved for use in humans with anemia and thrombocytopenia. In comparison with cytokine-based therapy, these agents tend to be well tolerated by patients.19,20 Binding of EPO to its EPO receptor (EPOR) promotes differentiation, proliferation, and survival of erythroid progenitor cells.21 TPO, a ligand of the c-MPL (MPL) receptor, is essential for megakaryocyte differentiation and expansion, as well as hematopoietic stem cell proliferation and maintenance.22,23 Both the EPO/EPOR and TPO/c-MPL interactions have been shown to transduce signals through three main pathways, JAK-STAT, phosphatidylinositol 3-kinase (PI3K)-AKT, and mitogen-activated protein kinase (MAPK) pathways, having similarities to IL-2 and IL-15 signaling cascades.21,23,24 Recently, Nishimura et al.25 demonstrated that c-MPL-engineered T cell receptor-transgenic T cells showed enhanced proliferation and anti-tumor function in response to TPO. Previous studies showed conflicting results of an EPO effect on the proliferation of EPOR-transduced mouse immortalized T cell lines.26, 27, 28
In this study, we investigated the use of EPO/EPOR and TPO/c-MPL signaling as adjuvant signals to improve NK cell expansion and to minimize the requirement for exogenous IL-2.
Results
Exogenous Expression of EPOR or c-MPL in Human NK Cell Lines Enhances Their Proliferative Response to Low-Dose IL-2 in the Presence of EPO or TPO
To investigate the functional effects of exogenously expressing EPOR or c-MPL in NK cells, we transduced the IL-2-dependent human NK cell line NK-9229, 30, 31, 32 using lentiviral vectors to stably express EPOR or c-MPL, along with an EGFP marker (Figures 1A and 1E). Untransduced parental NK-92 lacked EPOR and c-MPL expression by flow cytometry staining (Figure S1A). Following transduction, cell number expansion in response to different doses of IL-2 was examined. Both EPOR+ NK-92 and c-MPL+ NK-92 cells proliferated in a dose-dependent fashion to varying concentrations of IL-2 (Figures 1B and 1F).
Figure 1.

Exogenous Expression of EPOR or c-MPL in NK Cell Lines Facilitates Their Proliferation in the Presence of EPO or TPO
(A) EPOR and GFP or (E) c-MPL and GFP expression on NK-92 cell lines after fluorescence-activated cell sorting of transduced cells. Fold expansion of (B–D) EPOR+ NK-92 or (F–H) c-MPL+ NK-92 cells in the presence of IL-2, EPO, or TPO supplemented into culture medium as indicated. (B and F) IL-2 at varying concentrations. EPO and TPO were contrasted and combined with (C and G) 1 U/mL IL-2 or (D and H) 5 U/mL of IL-2. Mean and standard deviation (SD) from three to four independent experiments are shown. Significance was analyzed at day 8 using unpaired Student’s t test: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001. Some results are shown in more than one panel to aid comparison. See also Figure S1.
Strikingly, introduction of soluble EPO to cultures of EPOR+ NK-92 supported the growth of cells independent of IL-2, with a 20-fold expansion occurring by day 8 (Figure 1C). In addition, the combination of EPO and low-dose (LD) IL-2 (1 or 5 U/mL) enhanced the proliferative response of EPOR+ NK-92 cells compared with LD IL-2 alone. For example, on day 8, the addition of EPO plus IL-2 resulted in 60 times greater proliferation of EPOR+ NK-92 cells compared with that observed with 1 U/mL IL-2 alone (25-fold versus 0.41-fold; p < 0.0001) (Figure 1C). This effect was also evident when the IL-2 dose was increased, where the addition of EPO plus 5 U/mL IL-2 resulted in a nine times greater proliferation compared with 5 U/mL IL-2 alone (44-fold versus 4.8-fold; p < 0.0001) (Figure 1D).
In a similar fashion, c-MPL+ NK-92 showed IL-2-independent proliferation in cultures supplemented with TPO alone (7-fold expansion on day 8) (Figure 1G). Furthermore, adding TPO to LD IL-2 significantly enhanced c-MPL+ NK-92 proliferation compared with IL-2 alone or TPO alone (Figures 1G and 1H). For example, the addition of TPO plus 1 U/mL IL-2 increased the c-MPL+ NK-92 expansion to 23-fold compared with only 0.69-fold with 1 U/mL IL-2 alone (day 8; p = 0.01) (Figure 1G). EPO and TPO administration did not alter the expansion of the parental NK92 cell line (Figure S1B). Analogous results were obtained using the KHYG-1 NK cell line transfected to express EPOR or c-MPL receptors (Figures S2 and S3), although in these experiments, EPO alone or TPO alone were insufficient to maintain cell growth of transduced cells (Figure S2). However, EPO or TPO administration synergized with LD IL-2 (1 or 5 U/mL) to enhance the proliferative response of receptor-transduced KHYG-1 cells (Figure S2). Taken altogether, these data show that exogenously expressed EPOR and c-MPL receptors have the capacity to augment the proliferation of NK cell lines in response to stimulation with their cognate ligands.
EPOR- and c-MPL-Expressing NK92 Cells Upregulate the Anti-apoptotic Proteins Bcl-2 and Bcl-xL upon EPO or TPO Stimulation, Enhancing Their Survival
EPOR+ and c-MPL+ NK-92 cells were used for subsequent studies to characterize in more detail the consequences of ligating these exogenous receptors. Simultaneous short-term cell expansion and apoptosis assays examined the mechanisms underlying EPOR- and c-MPL-induced increases in cell numbers (Figures 2A and 2C). By day 3 of culture, NK-92 cells maintained in LD IL-2 alone showed an increasing number of dead or apoptotic cells, determined by flow cytometry (Figures 2B and 2D). In contrast, cultures containing a combination of either EPO or TPO plus LD IL-2 or higher doses of IL-2 (200 U/mL) alone showed significantly lower proportions of dead/apoptotic cells (Figures 2B and 2D). These results mirrored the total number of cells present in these cultures (Figures 2A and 2C).
Figure 2.

EPOR- and c-MPL-Expressing NK-92 Cell Lines Show Lower Levels of Apoptosis and Cell Death When Stimulated with Their Ligands
(A and B) EPOR+ NK-92 and (C and D) c-MPL+ NK-92 cells were cultured in different conditions as indicated. (A and C) Fold expansion of viable cells. (B and D) The percentage of 7-AAD+ and/or Annexin V+ cells detected by flow cytometry at day 2 or 3 of culture. Mean and SD are shown. ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 by unpaired Student’s t test from three or more independent experimental results. See also Figures S5 and S6.
Anti-apoptotic proteins, especially Bcl-xL (BCL2L1), have been shown to be regulated by EPO/EPOR and TPO/c-MPL signaling in erythroid and megakaryocytic cells, respectively.21,33,34 Therefore, transduced NK-92 cells were queried for downstream signaling pathways, including Bcl-2(BCL2) and Bcl-xL. EPOR and c-MPL have been shown to signal via three main pathways, JAK-STAT, PI3K-AKT-mammalian target of rapamycin (mTOR), and MAPK,21,23 which can be assessed using antibodies recognizing pSTAT5, pS6 (pRPS6), and phospho-p44/42 MAPK (pMAPK3/pMAPK1). No alteration of phospho-p44/42 MAPK was detected in EPOR+ and c-MPL+ NK-92 cells when cultures were supplemented with IL-2, EPO, or TPO (Figure S4). However, the addition of IL-2 increased pSTAT5 and pS6 levels in a dose-dependent manner in both EPOR+ and c-MPL+ NK-92 cells, reaching statistical significance when comparing IL-2 at the 200 U/mL concentration compared with no IL-2 cohorts (Figures 3A, 3B, 3E, and 3F). In comparison with the no IL-2 control group, the addition of EPO or TPO appeared to increase pSTAT5 levels in EPOR+ NK-92 or c-MPL+ NK-92 cells in a fashion comparable to LD IL-2 (Figures 3A and 3E). Interestingly, no additive effects on STAT5 phosphorylation were detected when EPO or TPO was combined with LD IL-2 in both EPOR+ and c-MPL+ NK-92 cells (Figures 3A and 3E). There was a trend toward increased quantities of pS6 in TPO-stimulated c-MPL+ NK-92 compared with the group lacking added cytokines (Figure 3F). Moreover, adding TPO to LD IL-2 significantly increased pS6 levels compared with LD IL-2 alone (11.8 versus 7.2; p = 0.04) (Figure 3F). These effects on pS6 were not observed in EPO-treated EPOR+ NK-92 cells (Figure 3B), perhaps reflecting a difference in signaling potential between the two receptors.
Figure 3.

Signaling Pathways in EPOR- and c-MPL-Expressing NK-92 Cells that Increase Levels of Anti-apoptotic Proteins
(A–D) EPOR+ NK-92 and (E–H) c-MPL+ NK-92 cells were cultured without IL-2 overnight, then incubated with different IL-2, EPO, and TPO concentrations for 15 min before analysis of (A and E) pSTAT5 and (B and F) pS6 or for 24 h before (C and G) Bcl-2 and (D and H) Bcl-xL analysis by flow cytometry. The relative mean fluorescent intensities (RMFIs) of the protein expression, normalized to isotype control in each case, are displayed with SD according to the experimental conditions indicated. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001 by paired t test from three to four independent experiments. See also Figure S4.
The Bcl-2 and Bcl-xL proteins were investigated as potential target proteins of EPOR and c-MPL signaling in NK-92 cells. As shown in Figures 3C, 3D, 3G, and 3H, both low and higher doses of IL-2, EPO, and TPO all significantly upregulated these anti-apoptotic proteins in EPOR+ and c-MPL+ NK-92 cells. EPO administration stimulated EPOR+ NK-92 to produce significantly more Bcl-2 and Bcl-xL proteins than no IL-2 control group (Bcl-2 relative mean fluorescent intensity [RMFI]: 81 versus 54, p < 0.0001, and Bcl-xL RMFI: 4.9 versus 3.8, p = 0.003, respectively) (Figures 3C and 3D). Likewise, the addition of TPO activated c-MPL+ NK-92 cells to express significantly higher levels of Bcl-2 and Bcl-xL proteins (Bcl-2 RMFI: 66 versus 50, p = 0.01; Bcl-xL RMFI: 4.5 versus 3.5, p = 0.01) compared with the no IL-2 control group (Figures 3G and 3H).
These results suggest that hematopoietic growth factor receptors exogenously expressed in NK-92 cells can signal to upregulate the anti-apoptotic proteins Bcl-2 and Bcl-xL, which together prolong cell survival. In EPOR+ NK-92 cells, this signaling may proceed through STAT5, whereas c-MPL+ NK-92 cells may utilize both STAT5 and PI3K-AKT-mTOR-S6 pathways. Similar results were obtained in experiments querying other time points of the cell culture (Figures S5 and S6).
Expression of c-MPL in NK-92 Cells Augments Their Killing of Raji Tumor Cells in the Presence of TPO
In order to test the functional consequences of expressing EPO or TPO receptors, we expanded transgenic EPOR+ and c-MPL+ NK-92 cells in the presence of EPO or TPO, and cytotoxicity assays were performed in vitro with three different tumor cell lines (K562 and 721.221, representing prototypic NK cell targets, and Raji Burkitt’s lymphoma cell line, previously used as targets for NK-92).29 Compared with LD IL-2 alone, there was a trend toward higher Raji killing by EPOR+ NK-92 cells stimulated with EPO and LD IL-2, while killing between LD IL-2 and EPO-stimulated EPOR+ NK-92 cells against 721.221 or K562 tumor cell lines was similar (Figure 4A). Remarkably, there was a striking increase in Raji tumor killing by c-MPL+ NK-92 cells exposed to TPO plus LD IL-2 compared with IL-2 alone at both lower and higher doses; the mean percentages of dead Raji cells in TPO + LD IL-2 cohorts were 84% (1:1) and 85% (10:1) compared with LD IL-2 alone (51% [1:1], p = 0.02, and 62% [10:1], p = 0.03) and the higher dose of IL-2 (200 U/mL) alone (58% [1:1], p = 0.01, and 67% [10:1], p = 0.04), respectively (Figure 4B). In contrast, no significant killing augmentation against 721.221 or K562 tumor targets was observed with TPO administration (Figure 4B).
Figure 4.

c-MPL-Activated NK-92 Cells Show Significantly Greater Cytotoxic Activity In Vitro versus Raji Tumor Cells
EPOR+ NK-92 and c-MPL+ NK-92 were cultured in different media conditions as indicated for 24–40 h before performing cytotoxicity assays. The mean (and SD) percentages of non-viable Raji, K562, or 721.221 target cells after 4-h incubation with (A) EPOR+ NK-92 or (B) c-MPL+ NK-92 effector cells are presented for each effector-to-target (E:T) ratio. ∗p < 0.05 by unpaired Student’s t test of three to five independent experiments; none of the comparisons in (A) were p < 0.05.
Transgenic Expression of c-MPL in Primary NK Cells Promotes Proliferation in the Presence of TPO
Data from the above experiments indicate proliferative, survival, and functional benefits of overexpressing EPOR or c-MPL in NK-92 cells in the presence of EPO or TPO and LD IL-2. Therefore, we extended our studies to primary NK cells collected from the peripheral blood of healthy donors. Using the same lentiviral constructs as used to transduce NK cell lines, we successfully transduced 4%–37% of primary NK cells (activated before transduction with IL-2 plus an Epstein-Barr virus [EBV]-transformed lymphoblastic cell line) to express either EPOR/GFP or c-MPL/GFP (Figure 5A). Similarly activated but untransduced (Mock) NK cells were used as controls and expanded in an IL-2 concentration-dependent fashion (Figure 5B). During a 6-day competitive expansion culture, GFP+ c-MPL-transduced NK cells supplemented with TPO plus LD IL-2 showed an increase in cell number compared with GFP– untransduced NK cells in the same culture, which is consistent with the c-MPL+ NK cells having a TPO-induced proliferative advantage (Figure S7B). In the presence of 10 U/mL LD IL-2, the addition of TPO increased the fold change of GFP+ c-MPL+ NK cells from 7.2-fold to 24-fold (p = 0.03) (Figure 5E). With a slightly higher concentration of IL-2 (25 U/mL), the impact of TPO was further magnified, increasing the GFP+ fold change from 14-fold to 34-fold (p = 0.047) (Figure 5F). Remarkably, the proliferation level with TPO + 25 U/mL IL-2 (34-fold) was comparable to that observed with much higher doses of IL-2 (500 U/mL) (36-fold) (p = 0.85). In contrast, EPOR+ NK cells did not show an increase in the mean GFP+ fold expansion in the presence of EPO plus LD IL-2 compared with LD IL-2 alone (Figures 5C and 5D; Figure S7A), perhaps because there was a lower level of EPOR expression on the surface of transduced primary NK cells (Figure 5A) or different EPOR signaling capacity in primary NK cells compared with the NK-92 cell line. Nonetheless, c-MPL ligation in primary NK cells clearly enhanced their ability to proliferate.
Figure 5.

Effects on Proliferation of Primary Human NK Cells Genetically Modified to Express EPOR or c-MPL
Human NK cells were stimulated with irradiated LCL feeder cells and IL-2 for 4–5 days and then underwent lentiviral transduction. Three days later, transduced cells were (A) examined for relative transgene expression by flow cytometry (data from one donor) and (B–F) cultured with varying concentrations of the indicated cytokines. (B) Activated but untransduced (Mock) NK cell expansion in cell number in response to IL-2. The mean fold expansion of GFP+ (virally transduced) NK cells expressing (C and D) EPOR and (E and F) c-MPL is shown with SD. Statistical significance with samples cultured in varying media was measured with unpaired Student’s t test using data of three experiments from three healthy donors on day 6 of culture (equivalent to day 14 from the initial NK cell isolation [D+14]). ∗p < 0.05. See also Figure S7.
c-MPL-Transduced Primary NK Cells Show Increased Anti-tumor Function upon TPO Stimulation
To investigate the effects on NK cell function, we transduced primary NK cells with a c-MPL/GFP encoding lentiviral vector, cultured them for 4 days in different cytokine combinations, then co-cultured with K562 target cells. CD107a degranulation and cytokine production were investigated in GFP+-transduced cells and untransduced controls. There was a significantly higher amount of degranulation in GFP+ c-MPL+ cells cultured in media with TPO and 25 U/mL IL-2 (Figure 6A) compared with 25 U/mL IL-2 alone. Strikingly, the level of cellular degranulation observed in these cells with TPO and 25 U/mL IL-2 was comparable to that observed with a much higher dose of IL-2 (500 U/mL); degranulation in GFP+ c-MPL+ NK cells averaged 48% with 25 U/mL IL-2 alone, 83% with the addition of TPO, and 86% with the higher 500 U/mL IL-2 dose (Figure 6A). Similarly, the percent of GFP+ c-MPL+ transduced NK cells that produced interferon gamma (IFN-γ) or tumor necrosis factor alpha (TNF-α) when co-cultured with K562 cells was considerably augmented by the addition of TPO to 25 U/mL IL-2 compared with 25 U/mL IL-2 alone (Figure 6B). For this comparison, p = 0.04 for IFN-γ production and p = 0.01 for TNF-α production, using data normalized to cytokine production of untransduced control cells in 500 U/mL IL-2 to mitigate inter-donor variation (data not shown). Administration of TPO had no measured effects on degranulation or cytokine production of untransduced control cells (Figures 6A and 6B). These results clearly indicate that c-MPL receptors that are exogenously expressed in primary human NK cells can augment commonly measured anti-tumor functions in response to ligand stimulation.
Figure 6.

Transduced c-MPL Receptors Augment the Function of Primary Human NK Cells
Human NK cells, transduced with c-MPL/GFP lentiviral vectors, were subdivided into cultures containing IL-2 and/or TPO at the indicated concentrations. (A and B) After 4 days, cellular function was assessed by mixing at a 1:1 ratio with K562 target cells and assessing (A) NK cell degranulation (by surface anti-CD107a staining) or (B) intracellular IFN-γ and TNF-α. Mean and SD for gated GFP+ transduced cells or independent untransduced cultures from the same donors are shown. (C) The cell surface phenotype of the NK cells was also assessed after 4 days of culture with growth factors as indicated. (D) To control for inter-donor variation, percentages of lymph node homing receptor CD62L+ cells were normalized to values obtained in untransduced NK cells cultured with 500 U/mL IL-2 from each donor. Mean and SD are shown. (A, B, and D) Results of four independent experiments from four blood donors. ∗p < 0.05 by paired t test.
The overall NK cell phenotype of the c-MPL-transduced primary NK cells was also investigated using a number of antibodies recognizing NK cell receptors and molecules important for NK cell function. With the exception of CD62L (L-selectin), no large phenotypic differences were observed in gated GFP+ (MPL-expressing) NK cells when TPO was added to media containing 25 U/mL IL-2 (Figure 6C). The overall percentage of NK cells expressing the lymphoid-tissue homing receptor CD62L (L-selectin) was higher in GFP+ c-MPL+ transduced cells when TPO was added to media containing 25 U/mL IL-2 compared with IL-2 alone at either a 25 or 500 U/mL concentration (Figures 6C and 6D). Although the overall percentage of CD62L positivity varied considerably between donors, when normalized to each donor, supplementing media with TPO significantly increased CD62L expression in GFP+ c-MPL+ transduced cells, but not untransduced cells (Figure 6D).
Primary Human NK Cells Genetically Modified to Express c-MPL Preferentially Persist upon Adoptive Transfer into Immunodeficient Mice Receiving TPO
We next examined the capacity for exogenous c-MPL receptor expression to support adoptively transferred NK cells in vivo. Primary human NK cells transduced with lentiviral constructs encoding GFP and either c-MPL or truncated surface-expressed human CD34 (as a control) were mixed at a 1:1 ratio, then were injected intravenously into non-obese diabetic (NOD) severe combined immunodeficiency (SCID) Il2rg–/– (NSG) immunodeficient mice. Following NK cell infusion, mice received daily intraperitoneal injections of TPO (50 μg/kg) or human IL-2 (10,000 IU/mouse; a dose previously reported to represent “LD IL-2” treatment in vivo).35 After 4 days, GFP+ c-MPL+ cells represented a significantly higher proportion of human cells detected in all the organs of mice that had received TPO (Figure 7A). In contrast, mice that had been administered IL-2 showed roughly equal proportions of GFP+ c-MPL+ and GFP+ CD34+ cells (Figure 7A). GFP+ c-MPL+ cells constituted approximately 10% of the mixed cells that were administered to mice. However, in mice that received TPO, the normalized average percentage of GFP+ c-MPL+ cells rose among the recovered human cells to 15% in the spleen, 15% in blood, 20% in the liver, and 20% in the lung (all p < 0.02 versus injected cells by paired t test without data normalization). These results suggest that c-MPL-expressing NK cells achieved a selective persistence advantage in vivo when TPO was administered, in comparison with both control CD34-transduced cells and bystander untransduced NK cells. Parallel cultures of the same mixed cells supplemented in vitro with only TPO showed an analogous increase in the GFP+ c-MPL+ population to 31% (Figure 7A). For a less comparative measure of the numbers of in vivo persisting NK cells, populations were quantitated as a percentage of the total live cells (mouse or human) isolated from each organ (Figure 7B). This analysis similarly revealed that TPO-treated mice had significantly more GFP+ c-MPL+ cells compared with GFP+ CD34+ control cells (Figure 7B); in TPO-treated mice, GFP+ CD34+ cells would not be expected to receive any exogenous cytokine support. Importantly, GFP+ c-MPL+ cells in TPO-treated mice approached percentages observed in IL-2-treated mice (Figure 7B). These data suggest that c-MPL ligation on transduced NK cells preferentially supported their persistence in vivo in a fashion comparable with IL-2, although perhaps somewhat less effective under these experimental conditions.
Figure 7.

c-MPL-Expressing Human NK Cells Persist Preferentially in Immunodeficient Mice Administered TPO
NK cells from human PBMCs were stimulated and then transduced with matched lentiviral vectors encoding GFP/c-MPL or GFP/CD34 as a control. Cells averaged approximately 20% GFP+. Both populations were then mixed at a 1:1 ratio immediately before injection intravenously into NSG mice (such that GFP+ c-MPL+ cells and GFP+ CD34+ cells each constituted ∼10% of the mixed cells). Mice received daily injections of human TPO (50 μg/kg) or human IL-2 (10,000 IU/mouse) for 4 days, after which the presence of transduced human cells was quantified in various organs via flow cytometry. c-MPL-transduced NK cells were identified as GFP+ c-MPL+, while CD34-transduced NK cells were gated as GFP+ CD34+. The frequency of transduced cells is presented (A) as a percentage of MHC-I+ human cells isolated from organs or (B) as percent of total live mononuclear cells recovered from each organ. The legend applies to both (A) and (B). Mean and SD from nine mice per group are shown with NK cells derived from three human blood donors. Data were normalized to account for unequal transduction or subset mixing before injection using the following equation: , where the last term represents the mean of all c-MPL+ and CD34+ groups from all donors. Groups were compared by paired t test as indicated. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. The significance of indicated comparisons was unaffected by data normalization (with the exception of bone marrow samples).
TPO-Mimetics Eltrombopag and Romiplostim Support c-MPL-Expressing Human NK Cells In Vitro
Eltrombopag (a small molecule) and romiplostim (a biologic) are FDA-approved drugs that stimulate the c-MPL receptor. After transduction with c-MPL/GFP encoding lentiviral vector, the response of c-MPL-expressing primary human NK cells to these molecules was examined over a 4-day culture. Strikingly, the percentage of GFP was significantly augmented in cultures containing eltrombopag and romiplostim, compared with cells grown in medium alone or supplemented with IL-2 (Figures S8A and S8C), indicative of preferential proliferation and/or survival. Numbers of GFP+ (c-MPL+) cells, measured relative to parallel cultures with 500 U/mL IL-2, were also significantly higher than medium alone when eltrombopag or romiplostim was added (Figures S8B and S8D). The maximum effect of eltrombopag on cell number appeared to be at approximately 0.25–0.75 μg/mL depending on the blood donor. Romiplostim had marked effects even at approximately 50 pg/mL but continued to augment numbers to 2 μg/mL (Figure S8D). These results raise the possibility of replacing or reducing the dose of costly IL-2 used in protocols to expand ex vivo c-MPL-expressing NK cell numbers. Furthermore, they point toward a potential clinical treatment strategy wherein c-MPL-expressing NK cells may be supported in their persistence and function by pharmaceutical ligands after adoptive transfer, with reduced or absent requirement for hazardous IL-2 administration.
Primary T Cells Transduced to Express c-MPL Have Augmented Cell Numbers in the Presence of Ligand
The effects of transgenic expression of EPOR and c-MPL were also evaluated in primary human T cells. Efficient transduction of EPOR/GFP or c-MPL/GFP in activated T cells from healthy donors was observed, ranging from 40% to 60% positive (Figure S9A). Activated but untransduced (Mock) T cells were used as controls and were observed to proliferate in vitro in an IL-2 concentration-dependent fashion (Figure S9B). A 6-day competitive culture was used to examine the fold expansion of GFP+ (transduced) cells. EPOR+ T cells in the presence of EPO plus LD IL-2 appeared to show a slightly higher mean GFP+ fold expansion compared with LD IL-2 alone (day 6; 5.6-fold versus 2.8-fold, p = 0.05) (Figure S9C); however, this was not different from GFP– untransduced NK cells in the same culture (Figure S10). c-MPL transduced T cells in the presence of TPO alone appeared to show acquisition of an IL-2-independent expansion capacity; the GFP+ fold expansion on day 6 was 6.0-fold in the presence of TPO alone versus 3.0-fold in the presence of LD IL-2 (5 U/mL) alone (p = 0.006). In addition, the proliferation of c-MPL+ activated T cells increased on day 6 to 11.4-fold when exposed to TPO plus LD IL-2 (p = 0.004 compared with LD IL-2 alone [Figure S9D] and p = 0.02 compared with GFP– untransduced cells [Figure S10)]. Remarkably, the level of T cell proliferation with TPO + LD IL-2 was comparable to that achieved with much higher doses of IL-2 (300 U/mL). Taken altogether, transgenic expression of c-MPL appeared to have quantitatively greater functional activity when expressed in primary T cells compared with EPOR.
Discussion
Our results support the concept that genetic manipulation of NK cells to exogenously express hematopoietic growth factor receptors can provide adjuvant signals to improve their proliferative capacity, survival, and tumor cytotoxicity in vitro, while simultaneously reducing their requirement for IL-2. In the presence of relatively LDs of IL-2, the cell yields of both EPOR- and c-MPL-expressing NK-92 NK cells were improved when cells were cultured in the presence of EPO or TPO, respectively (Figure 1). Similar effects were also observed in the KHYG-1 cell line, albeit these effects were less pronounced, perhaps because of its higher baseline proliferative rate and greater dependence on higher concentrations of IL-2. Importantly, we also observed enhanced in vitro tumor cytotoxicity and an augmentation in proliferation in primary human NK cells transduced to express the c-MPL receptor (Figures 5 and 6). Remarkably, proliferation was augmented in c-MPL+ transduced NK cells cultured in TPO plus 25 U/mL IL-2 to a level comparable to that observed in NK cell populations cultured in 20-fold higher doses of IL-2 (500 U/mL). Although EPO administration enhanced the proliferative capacity of EPOR transduced NK-92 cells, it did not augment the proliferation of EPOR transduced primary NK cells, possibly because of limitations in our experiments where surface expression of the EPOR on primary NK cells appeared to not be induced to the higher levels we achieved with the c-MPL receptor.
Using transgenic NK-92 cells, we identified signaling through EPOR and c-MPL that upregulated the anti-apoptotic proteins Bcl-2 and Bcl-xL, which provided transduced NK-92 cells with a survival advantage compared with their untransduced counterparts (Figures 2 and 3). In hematopoietic precursor cells, EPOR and TPO are known to transduce signals through three main pathways: JAK-STAT, PI3K-AKT, and MAPK. In transduced NK-92 cells, engagement of either EPOR or c-MPL with their cognate ligands appeared to induce STAT5 phosphorylation. In contrast, binding of TPO to c-MPL mediated S6 phosphorylation (often used as an indicator of activity of mTOR complex 1 [mTORC1]), whereas EPOR ligation did not. Differences in the pathways triggered by c-MPL versus EPOR may account for the different functional effects observed in transduced NK cell populations in this study (i.e., enhanced cytotoxic activity was observed with c-MPL+ NK92 cells, and greater proliferation was seen with c-MPL-transduced primary NK cells; Figures 4 and 5, respectively). mTORC1 is one of the major regulators of metabolism in NK cells.36 Overnight stimulation of human NK cells with IL-2 leads to increased pS6 and enhanced rates of glycolysis and oxidative phosphorylation; increased glycolysis could be inhibited by rapamycin (inhibitor of mTORC1).37 Strategies that impair NK cell metabolism inhibit cytotoxic activity, IFN-γ production, and proliferation.36,37 Our observation that c-MPL ligation increased pS6 in NK-92 cells (Figure 3F) implies signaling through this pathway may augment NK cell metabolic activity, which potentially could promote NK cell proliferation and resistance to apoptosis.
Stimulating c-MPL and IL-2 receptors together on transduced NK-92 cells augmented the anti-tumor function against Raji cells, which remarkably was superior to IL-2 alone, even when cells were cultured in high doses of this cytokine (200 U/mL). The mechanism of this additional anti-tumor activity provided by c-MPL signaling to NK-92 cells is unknown, although a recent study showed improvement of several measures of immune synapse formation in c-MPL-expressing T cells.25 c-MPL-transduced human NK cells co-incubated with K562 showed striking functional increases in degranulation and cytokine production in response to TPO administration (Figure 6). These data suggest that c-MPL ligation in NK cells with transgenic c-MPL expression could facilitate NK cell activation and tumor killing.
An unexpected finding was that primary NK cells engineered to express c-MPL and treated with TPO showed higher percentages of the lymphoid tissue homing receptor CD62L (L-selectin) (Figure 6). IL-2 activation and ex vivo expansion have previously been shown to downregulate CD62L. Following lymphocyte activation, CD62L downregulation can be mediated by increased proteolytic cleavage from the cell surface, through the action of metalloprotease ADAM17, or changes in RNA transcription and RNA stability.38,39 It would be interesting to dissect the relative mechanisms at play and the degree to which c-MPL-ligation can counteract IL-2-mediated effects (as also noted for IL-21 in NK T cells).40 It would also be intriguing to determine whether NK cells stimulated via transgenic c-MPL share features with CD56dim CD62L+ NK cells from human peripheral blood, reported to be a polyfunctional NK cell subset or developmental intermediate, capable of cytokine-triggered proliferation and IFN-γ production (like CD56bright NK cells), along with higher activating receptor-mediated cytotoxicity and cytokine production (like CD56dim CD62L– NK cells).41 Although the implications of c-MPL signaling increasing CD62L expression on activated and expanded NK cells are unknown, they will be studied in future experiments because they could directly impact NK cell homing to target tissues or amplify CXCR4 signaling, which could alter NK cell homing to the bone marrow.42,43
In both primary T cells and primary NK cells, we observed that transduction and signaling through c-MPL resulted in superior cellular proliferation compared with signaling through the EPOR (Figure 5; Figure S9). Our findings in T cells are consistent with similar findings by Nishimura et al.25 where cellular proliferation was augmented and IL-2 interdependent growth was achieved in T cells that expressed c-MPL exposed to TPO. These observations are also consistent with previous studies in mouse T cell lines (CTLL-2 and CTL-D) expressing EPOR that did not show a proliferation advantage upon stimulation with EPO alone.27,28 Another potential pitfall of using an EPOR-based approach includes a prior report showing very high doses of EPO (1,000–2,000 U/mL) are immunosuppressive in vitro to T lymphocytes expressing EPOR activated through anti-CD3/anti-CD28 stimulation44 (although this effect was not observed with the lower doses of EPO that we used in our experiments). One potential avenue to enhance the effectiveness of EPOR signaling in T or NK cells could be the use of a chimeric EPOR, where an EPOR extracellular region is linked to intracellular signaling domains from receptors that are more effective in inducing T/NK cell proliferation, such as the IL-2 receptor.28
This study represents an initial attempt to explore the use of alternative receptors that are transgenically expressed in NK cells to improve their ability to proliferate in vitro and to bolster anti-tumor effects following adoptive infusion. Our results in immunodeficient mice indicate that c-MPL-transduced primary NK cells have superior in vivo survival in response to systemic TPO administration compared with control populations. The numbers of c-MPL+ NK cells recovered in TPO-treated mice approached those detected in mice administered IL-2 (Figure 7). Data from our study could potentially be translated to a clinical trial setting, where the infusion of genetically modified c-MPL-expressing NK cells is followed by the off-label administration of a TPO-mimetic drug (e.g., eltrombopag, romiplostim) combined with LD IL-2. Eltrombopag and romiplostim have been FDA approved for more than 10 years, with a history of being safely tolerated, and would likely contribute less to therapeutic costs than IL-2. In our studies, using concentrations that are close to those obtained pharmacologically in humans,45, 46, 47 both romiplostim and eltrombopag triggered augmented numbers of c-MPL-expressing human NK cells, although high concentrations of eltrombopag had diminishing effects (Figure S8) perhaps because of drug toxicity.25 Future studies and animal models will be utilized to inform optimal concentrations of TPO-mimetic agents and IL-2 to facilitate anti-tumor activity of c-MPL+ NK cells. Such regimens might avoid the substantial side effects caused by intermediate or high-dose IL-2 while inducing a selective cellular proliferative effect that is restricted to the genetically modified NK cells, avoiding the induction of proliferation of Tregs, which inhibit the antitumor effects of NK and T cells. Expression of c-MPL could be combined with other genetic modifications, such as chimeric antigen receptors or enhancers of anti-tumor activity.
In summary, we demonstrate that hematopoietic growth factor receptors, in particular, c-MPL, when transgenically expressed in NK cells are able to function and signal in response to their cognate ligands, augmenting NK cell proliferation, survival, anti-tumor capacity in vitro, and persistence in vivo. These data provide a strategy for future clinical trials aimed at incorporating methods to augment the anti-tumor immunity and proliferation of adoptively infused NK cells while avoiding Treg proliferation and the considerable side effects associated with the use of exogenous IL-2 administration.
Materials and Methods
Cell Culture and Cell Lines
NK-92, Raji, 721.221, and K562 cell lines were purchased from ATCC (Manassas, VA, USA). The KHYG-1 cell line was purchased from Leibniz Institute DSMZ (Braunschweig, Germany). NK-92 cells were cultured in RPMI medium supplemented with 12.5% heat-inactivated fetal bovine serum (FBS), 12.5% horse serum, and 2 mM l-glutamine (Life Technologies, Thermo Fisher Scientific, Waltham, MA, USA). The KHYG-1 cells were cultured in RPMI medium containing 10% FBS, penicillin/streptomycin (100 μg/mL), and 2 mM l-glutamine. Both NK-92 and KHYG-1 were supplemented with 200 U/mL rhIL-2 (Roche, Basel, Switzerland). Raji, K562, and 721.221 were also cultured in RPMI medium with 10% FBS, while 293T cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) with 10% FBS and 2 mM l-glutamine.
Lentiviral Production and Transduction
Lentivirus gene expression vectors encoding EGFP-P2A linker-human EPOR or EGFP-P2A-human c-MPL were cloned according to our design by VectorBuilder (Chicago, IL, USA). Plasmid DNA for the above and pCMV-dR8.2 dvpr and pCMV-VSV-G packaging plasmids was purified using ZymoPUREII-EndoZero plasmid maxiprep kit (Zymo Research, Irvine, CA, USA). Lentiviral particles were generated with HEK293T cells and Lipofectamine 3000 reagent (Thermo Fisher Scientific) following manufacturers’ instructions. Viral supernatant was harvested at 24 and 48 h, pooled, and concentrated with lenti-X concentrator (Takara Bio, Kusatsu, Japan) or by centrifugation at 18,600 rpm for 2 h at 4°C. The target cells were transduced with lentivirus at a multiplicity of infection (MOI) of 20 using a retronectin (Takara Bio)-coated plate protocol. NK-92 and KHYG-1 showed >50% transduction. After 5 days post-transduction, NK-92 or KHYG-1 cells were stained and sorted with fluorescence-activated cell sorting (FACS) using a BD FACSAria II to purify GFP+EPOR+ and GFP+c-MPL+ NK cell populations.
Primary NK Cell Activation and Transduction
Peripheral blood NK cells and T cells from healthy volunteers were isolated from de-identified buffy coat samples from Department of Transfusion Medicine, NIH Clinical Center. NIH Office of Human Subjects Research Protections determined that this material is exempt from the requirements of Institutional Review Board (IRB) review. NK cells were separated by immune-density negative selection and density gradient centrifugation using RosetteSep human NK cell enrichment cocktail (STEMCELL Technologies, Vancouver, BC, Canada) and lymphocyte separation medium (MP Biomedicals, Irvine, CA, USA). The primary NK cells were activated with irradiated EBV-transformed lymphoblastic cell line (SMI-LCL)48 and maintained in X-Vivo20 medium (Lonza, Basel, Switzerland) with 10% heat-inactivated human serum, 2 mM l-glutamine, and 500 U/mL IL-2. After 4–5 days of NK cell activation, NK cells were transduced with the previously described lentiviral particles (MOI = 20) in combination with 1.5 μM BX795 (InvivoGen, San Diego, CA, USA). On day 2–3 post-transduction, cells were evaluated for their expression of CD3, GFP, CD56, EPOR, and c-MPL, and cultured with varied growth factors as indicated. The transduction efficiency (measured as GFP+ cells) of primary NK cells ranged from 4% to 37%. Both GFP+ and GFP− NK cells were utilized in the competitive cell expansion assay. Greater than 95% of cells resulting from this protocol were CD56+ CD3–.
In Vitro Cell Expansion Cultures
The NK-92 (5 × 104 cells/mL) and KHYG-1 (1 × 104 cells/mL) cells were cultured in 1 mL of complete medium in a 24-well plate. The NK cell lines were supplemented with different IL-2 doses, 5 U/mL rhEPO (R&D Systems, Minneapolis, MN, USA), and/or 50 ng/mL rhTPO (R&D Systems) depending on experiments. Trypan blue-negative cells were counted every other day. On day 4, the medium was changed and replaced with fresh IL-2/EPO/TPO as their initial conditions. The short-term cell expansion assay was performed in similar fashion, except cells were initially cultured without IL-2 overnight before adding different cytokines and enumerating the cell number. For competitive primary NK cell expansion assay, all transduced cells (5-10 × 105/mL) containing a mixture of GFP+ (transduced) and GFP− (untransduced) cells were plated. Fresh medium with appropriate supplements was replaced every other day. Viable cells were counted, and percentage of GFP+ cells was acquired on days 4 and 6. The fold change of GFP+ cells was calculated by multiplying the counted cell number times the fraction GFP+. Cultures were supplemented as indicated with eltrombopag (Selleck Chemicals, Houston, TX, USA) solubilized in DMSO or romiplostim (Nplate; Amgen, Thousand Oaks, CA, USA) reconstituted as recommended.
Flow Cytometry, Reagents, and Pathway Analysis
Fluorochrome-conjugated monoclonal antibodies used were as follows: CD56-phycoerythrin (PE)-Cy7 (NCAM16.2), CD3-PE (UCHT1), c-MPL-PE (1.6.1), STAT5(pY694)-Alexa Fluor 647 (47/stat5(pY694)), NKp46-allophycocyanin (APC) (9E2), NKp44-PE (p44-8), FAS-PE (DX2), and CD62L-V450 (DREG-56) were from BD Biosciences (San Jose, CA, USA); EPOR-PE (FAB307P) was from R&D Systems; Bcl-2-PE (100), TRAIL-APC (RIK-2), CXCR4-BV421 (12G5), and KIR3DL1-BV421 (DX9) were from BioLegend (San Diego, CA, USA); Bcl-xL-PE (ab208747) was from Abcam (Cambridge, UK); P-p44/42 MAPK-Alexa Fluor 647 (E10) and p-S6 ribosomal protein-PE (D57.2.2E) were from Cell Signaling Technology (Danvers, MA, USA); NKG2A-PE (Z199), KIR2DL1/S1-PE (EB6B), and KIR2DL2/3-PE/Cy5.5 (GL183) were from Beckman Coulter (Brea, CA, USA). Staining was performed in phosphate-buffered saline (PBS) with 2% FBS and 2 mM EDTA and analyzed on a BD LSRFortessa machine. If intracellular staining was required, BD Cytofix/Cytoperm (BD Biosciences) was applied for cell fixation and permeabilization after surface staining. The apoptosis assay was performed using PE Annexin V apoptosis detection kit (BD Biosciences). The data were analyzed on FlowJo software.
For measurement of cell signaling and anti-apoptotic proteins, cells were cultured overnight without IL-2 and then incubated in appropriate medium supplemented with different IL-2/EPO/TPO conditions for 15 min for phospho-protein detection and 24 h for anti-apoptotic protein studies. Expression of target proteins was evaluated afterward by flow cytometry as described above.
Flow Cytometry-Based Killing Assay
The effector cells were prepared by culturing EPOR+ NK-92 and c-MPL+ NK-92 cells in different IL-2/EPO/TPO concentrations for 24–40 h before the experiment. The target cells were labeled with 1 μM CellTrace Violet in some experiments (Thermo Fisher Scientific). The effector and target cells were incubated together at specific effector-to-target (E:T) ratios in complete medium without cytokines for 4 h. 7-aminoactinomycin D (7-AAD) and Sphero AccuCount Fluorescent particles (Spherotech, Lake Forest, IL, USA) were added before analysis by flow cytometry. The absolute number of viable 7-AAD-negative target cells was calculated according to bead numbers added.49 The percentage of dead cells was calculated relative to target cell only control wells.
Degranulation Assay and Cytokine Production
Transduced human NK cells or control untransduced cells were cultured with IL-2 and/or TPO for 4 days, then mixed at a 1:1 ratio with K562 target in medium lacking added cytokines. To examine degranulation, we washed cells after 2 h and stained them with anti-CD107a (H4A3) (BioLegend). To assay cytokine production, we added GogiPlug and GolgiStop (BD Biosciences) at the manufacturer’s recommended concentrations within the first hour of co-culture with K562. After 4 h, cells were washed and stained for cell surface markers, then stained intracellularly with monoclonal antibodies recognizing TNF-α (Mab 11) and IFN-γ (B27) (BD Biosciences).
Persistence of Transduced Human NK Cells in Immunodeficient Mice
Experiments with animals were approved by the National Heart Lung and Blood Institute (NHLBI) Animal Care and Use Committee. NK cells from human PBMCs were stimulated for 4 days with LCL feeder cells and IL-2, then transduced with matched lentiviral vectors encoding GFP/MPL or GFP/CD34. Cells were maintained in IL-2-containing medium for 3–4 additional days; then both populations were mixed at a 1:1 ratio and injected intravenously into NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice (5–32 weeks of age; from Jackson Laboratory, Bar Harbor, ME, USA) (2.2 × 107 total NK cells/mouse). Mice received daily intraperitoneal injections of human TPO (50 μg/kg/mouse) or human IL-2 (10,000 IU/mouse) for 4 days. After euthanasia, blood was drawn by cardiac puncture; bone marrow was harvested by flushing the tibia; and spleen, liver, and lung fragments were processed by grinding over 70-μM nylon mesh. Blood, bone marrow, lung, and spleen samples received 1–2 rounds of 5-min incubations with ACK lysis buffer (Quality Biological, Gaithersburg, MD, USA). Liver samples were purified by centrifugation over 35% Percoll (Sigma) solution in RPMI. The presence of transduced human cells was quantified in various organs via flow cytometry using antibodies to human major histocompatibility complex (MHC) class I (W6/32) (BioLegend), c-MPL (1.6.1), CD34 (581) (BD Biosciences), GFP fluorescence, and Live/Dead Aqua viability dye (Thermo Fisher Scientific).
Statistical Analysis
Data were analyzed and presented using GraphPad Prism software. The mean, standard deviation (SD), and p values were displayed with statistical tests as indicated.
Author Contributions
C.C. designed and conducted the experiments and wrote the paper. D.S.J.A. initiated the hypothesis, designed the experiments, conducted some experiments, and wrote the paper. M.C. conducted some experiments and gave advice. R.N.R. conducted some experiments and gave advice. R.W.C. initiated the hypothesis, designed the experiments, gave advice, and wrote the paper.
Conflicts of Interest
The authors declare a provisional patent application to be filed.
Acknowledgments
We thank Keyvan Keyvanfar (NHLBI Clinical Flow Cytometry Facility) for his invaluable assistance with flow cytometry sorting. We are grateful to John Tisdale, Naoya Uchida, and Joseph Clara (NHLBI) for thoughtful insights. We thank Kanon Yamanaka for performing experiments that extended our understanding but are not shown. This research was supported by the Intramural Research Program of the NIH, NHLBI, Cellular and Molecular Therapeutics Branch and Hematology Branch, and the Commissioned Corps of the US Public Health Service. C.C. was supported by the Research Unit in Translational Hematology, Chulalongkorn University, Bangkok, Thailand.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.09.023.
Supplemental Information
References
- 1.Fang F., Xiao W., Tian Z. NK cell-based immunotherapy for cancer. Semin. Immunol. 2017;31:37–54. doi: 10.1016/j.smim.2017.07.009. [DOI] [PubMed] [Google Scholar]
- 2.Shimasaki N., Coustan-Smith E., Kamiya T., Campana D. Expanded and armed natural killer cells for cancer treatment. Cytotherapy. 2016;18:1422–1434. doi: 10.1016/j.jcyt.2016.06.013. [DOI] [PubMed] [Google Scholar]
- 3.Childs R.W., Carlsten M. Therapeutic approaches to enhance natural killer cell cytotoxicity against cancer: the force awakens. Nat. Rev. Drug Discov. 2015;14:487–498. doi: 10.1038/nrd4506. [DOI] [PubMed] [Google Scholar]
- 4.Rezvani K., Rouce R., Liu E., Shpall E. Engineering Natural Killer Cells for Cancer Immunotherapy. Mol. Ther. 2017;25:1769–1781. doi: 10.1016/j.ymthe.2017.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carlsten M., Childs R.W. Genetic Manipulation of NK Cells for Cancer Immunotherapy: Techniques and Clinical Implications. Front. Immunol. 2015;6:266. doi: 10.3389/fimmu.2015.00266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rautela J., Huntington N.D. IL-15 signaling in NK cell cancer immunotherapy. Curr. Opin. Immunol. 2017;44:1–6. doi: 10.1016/j.coi.2016.10.004. [DOI] [PubMed] [Google Scholar]
- 7.Marçais A., Cherfils-Vicini J., Viant C., Degouve S., Viel S., Fenis A., Rabilloud J., Mayol K., Tavares A., Bienvenu J. The metabolic checkpoint kinase mTOR is essential for IL-15 signaling during the development and activation of NK cells. Nat. Immunol. 2014;15:749–757. doi: 10.1038/ni.2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwartzentruber D.J. Guidelines for the safe administration of high-dose interleukin-2. J. Immunother. 2001;24:287–293. doi: 10.1097/00002371-200107000-00004. [DOI] [PubMed] [Google Scholar]
- 9.Conlon K.C., Lugli E., Welles H.C., Rosenberg S.A., Fojo A.T., Morris J.C., Fleisher T.A., Dubois S.P., Perera L.P., Stewart D.M. Redistribution, hyperproliferation, activation of natural killer cells and CD8 T cells, and cytokine production during first-in-human clinical trial of recombinant human interleukin-15 in patients with cancer. J. Clin. Oncol. 2015;33:74–82. doi: 10.1200/JCO.2014.57.3329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Anderson P.M., Sorenson M.A. Effects of route and formulation on clinical pharmacokinetics of interleukin-2. Clin. Pharmacokinet. 1994;27:19–31. doi: 10.2165/00003088-199427010-00003. [DOI] [PubMed] [Google Scholar]
- 11.Nagashima S., Mailliard R., Kashii Y., Reichert T.E., Herberman R.B., Robbins P., Whiteside T.L. Stable transduction of the interleukin-2 gene into human natural killer cell lines and their phenotypic and functional characterization in vitro and in vivo. Blood. 1998;91:3850–3861. [PubMed] [Google Scholar]
- 12.Konstantinidis K.V., Alici E., Aints A., Christensson B., Ljunggren H.G., Dilber M.S. Targeting IL-2 to the endoplasmic reticulum confines autocrine growth stimulation to NK-92 cells. Exp. Hematol. 2005;33:159–164. doi: 10.1016/j.exphem.2004.11.003. [DOI] [PubMed] [Google Scholar]
- 13.Zhang J., Sun R., Wei H., Zhang J., Tian Z. Characterization of interleukin-15 gene-modified human natural killer cells: implications for adoptive cellular immunotherapy. Haematologica. 2004;89:338–347. [PubMed] [Google Scholar]
- 14.Sahm C., Schönfeld K., Wels W.S. Expression of IL-15 in NK cells results in rapid enrichment and selective cytotoxicity of gene-modified effectors that carry a tumor-specific antigen receptor. Cancer Immunol. Immunother. 2012;61:1451–1461. doi: 10.1007/s00262-012-1212-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Imamura M., Shook D., Kamiya T., Shimasaki N., Chai S.M., Coustan-Smith E., Imai C., Campana D. Autonomous growth and increased cytotoxicity of natural killer cells expressing membrane-bound interleukin-15. Blood. 2014;124:1081–1088. doi: 10.1182/blood-2014-02-556837. [DOI] [PubMed] [Google Scholar]
- 16.Liu E., Tong Y., Dotti G., Shaim H., Savoldo B., Mukherjee M., Orange J., Wan X., Lu X., Reynolds A. Cord blood NK cells engineered to express IL-15 and a CD19-targeted CAR show long-term persistence and potent antitumor activity. Leukemia. 2018;32:520–531. doi: 10.1038/leu.2017.226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xu W., Jones M., Liu B., Zhu X., Johnson C.B., Edwards A.C., Kong L., Jeng E.K., Han K., Marcus W.D. Efficacy and mechanism-of-action of a novel superagonist interleukin-15: interleukin-15 receptor αSu/Fc fusion complex in syngeneic murine models of multiple myeloma. Cancer Res. 2013;73:3075–3086. doi: 10.1158/0008-5472.CAN-12-2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Romee R., Cooley S., Berrien-Elliott M.M., Westervelt P., Verneris M.R., Wagner J.E., Weisdorf D.J., Blazar B.R., Ustun C., DeFor T.E. First-in-human phase 1 clinical study of the IL-15 superagonist complex ALT-803 to treat relapse after transplantation. Blood. 2018;131:2515–2527. doi: 10.1182/blood-2017-12-823757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sheikh S., Littlewood T.J. Erythropoiesis-stimulating agents for anemic patients with cancer. Expert Rev. Hematol. 2010;3:697–704. doi: 10.1586/ehm.10.64. [DOI] [PubMed] [Google Scholar]
- 20.Rodeghiero F., Carli G. Beyond immune thrombocytopenia: the evolving role of thrombopoietin receptor agonists. Ann. Hematol. 2017;96:1421–1434. doi: 10.1007/s00277-017-2953-6. [DOI] [PubMed] [Google Scholar]
- 21.Jelkmann W., Bohlius J., Hallek M., Sytkowski A.J. The erythropoietin receptor in normal and cancer tissues. Crit. Rev. Oncol. Hematol. 2008;67:39–61. doi: 10.1016/j.critrevonc.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 22.Kaushansky K., Lok S., Holly R.D., Broudy V.C., Lin N., Bailey M.C., Forstrom J.W., Buddle M.M., Oort P.J., Hagen F.S. Promotion of megakaryocyte progenitor expansion and differentiation by the c-Mpl ligand thrombopoietin. Nature. 1994;369:568–571. doi: 10.1038/369568a0. [DOI] [PubMed] [Google Scholar]
- 23.Hitchcock I.S., Kaushansky K. Thrombopoietin from beginning to end. Br. J. Haematol. 2014;165:259–268. doi: 10.1111/bjh.12772. [DOI] [PubMed] [Google Scholar]
- 24.Sim G.C., Radvanyi L. The IL-2 cytokine family in cancer immunotherapy. Cytokine Growth Factor Rev. 2014;25:377–390. doi: 10.1016/j.cytogfr.2014.07.018. [DOI] [PubMed] [Google Scholar]
- 25.Nishimura C.D., Brenner D.A., Mukherjee M., Hirsch R.A., Ott L., Wu M.F., Liu H., Dakhova O., Orange J.S., Brenner M.K. c-MPL provides tumor-targeted T-cell receptor-transgenic T cells with costimulation and cytokine signals. Blood. 2017;130:2739–2749. doi: 10.1182/blood-2017-02-769463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Showers M.O., Moreau J.F., Linnekin D., Druker B., D’Andrea A.D. Activation of the erythropoietin receptor by the Friend spleen focus-forming virus gp55 glycoprotein induces constitutive protein tyrosine phosphorylation. Blood. 1992;80:3070–3078. [PubMed] [Google Scholar]
- 27.Yamamura Y., Kageyama Y., Matuzaki T., Noda M., Ikawa Y. Distinct downstream signaling mechanism between erythropoietin receptor and interleukin-2 receptor. EMBO J. 1992;11:4909–4915. doi: 10.1002/j.1460-2075.1992.tb05597.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Minamoto S., Treisman J., Hankins W.D., Sugamura K., Rosenberg S.A. Acquired erythropoietin responsiveness of interleukin-2-dependent T lymphocytes retrovirally transduced with genes encoding chimeric erythropoietin/interleukin-2 receptors. Blood. 1995;86:2281–2287. [PubMed] [Google Scholar]
- 29.Gunesch J.T., Angelo L.S., Mahapatra S., Deering R.P., Kowalko J.E., Sleiman P., Tobias J.W., Monaco-Shawver L., Orange J.S., Mace E.M. Genome-wide analyses and functional profiling of human NK cell lines. Mol. Immunol. 2019;115:64–75. doi: 10.1016/j.molimm.2018.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tang X., Yang L., Li Z., Nalin A.P., Dai H., Xu T., Yin J., You F., Zhu M., Shen W. First-in-man clinical trial of CAR NK-92 cells: safety test of CD33-CAR NK-92 cells in patients with relapsed and refractory acute myeloid leukemia. Am. J. Cancer Res. 2018;8:1083–1089. [PMC free article] [PubMed] [Google Scholar]
- 31.Arai S., Meagher R., Swearingen M., Myint H., Rich E., Martinson J., Klingemann H. Infusion of the allogeneic cell line NK-92 in patients with advanced renal cell cancer or melanoma: a phase I trial. Cytotherapy. 2008;10:625–632. doi: 10.1080/14653240802301872. [DOI] [PubMed] [Google Scholar]
- 32.Tonn T., Schwabe D., Klingemann H.G., Becker S., Esser R., Koehl U., Suttorp M., Seifried E., Ottmann O.G., Bug G. Treatment of patients with advanced cancer with the natural killer cell line NK-92. Cytotherapy. 2013;15:1563–1570. doi: 10.1016/j.jcyt.2013.06.017. [DOI] [PubMed] [Google Scholar]
- 33.Kapur R., Zhang L. A novel mechanism of cooperation between c-Kit and erythropoietin receptor. Stem cell factor induces the expression of Stat5 and erythropoietin receptor, resulting in efficient proliferation and survival by erythropoietin. J. Biol. Chem. 2001;276:1099–1106. doi: 10.1074/jbc.M007442200. [DOI] [PubMed] [Google Scholar]
- 34.Kirito K., Watanabe T., Sawada K., Endo H., Ozawa K., Komatsu N. Thrombopoietin regulates Bcl-xL gene expression through Stat5 and phosphatidylinositol 3-kinase activation pathways. J. Biol. Chem. 2002;277:8329–8337. doi: 10.1074/jbc.M109824200. [DOI] [PubMed] [Google Scholar]
- 35.Granzin M., Stojanovic A., Miller M., Childs R., Huppert V., Cerwenka A. Highly efficient IL-21 and feeder cell-driven ex vivo expansion of human NK cells with therapeutic activity in a xenograft mouse model of melanoma. OncoImmunology. 2016;5:e1219007. doi: 10.1080/2162402X.2016.1219007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.O’Brien K.L., Finlay D.K. Immunometabolism and natural killer cell responses. Nat. Rev. Immunol. 2019;19:282–290. doi: 10.1038/s41577-019-0139-2. [DOI] [PubMed] [Google Scholar]
- 37.Keating S.E., Zaiatz-Bittencourt V., Loftus R.M., Keane C., Brennan K., Finlay D.K., Gardiner C.M. Metabolic Reprogramming Supports IFN-γ Production by CD56bright NK Cells. J. Immunol. 2016;196:2552–2560. doi: 10.4049/jimmunol.1501783. [DOI] [PubMed] [Google Scholar]
- 38.Chao C.C., Jensen R., Dailey M.O. Mechanisms of L-selectin regulation by activated T cells. J. Immunol. 1997;159:1686–1694. [PubMed] [Google Scholar]
- 39.Peschon J.J., Slack J.L., Reddy P., Stocking K.L., Sunnarborg S.W., Lee D.C., Russell W.E., Castner B.J., Johnson R.S., Fitzner J.N. An essential role for ectodomain shedding in mammalian development. Science. 1998;282:1281–1284. doi: 10.1126/science.282.5392.1281. [DOI] [PubMed] [Google Scholar]
- 40.Ngai H., Tian G., Courtney A.N., Ravari S.B., Guo L., Liu B., Jin J., Shen E.T., Di Pierro E.J., Metelitsa L.S. IL-21 Selectively Protects CD62L+ NKT Cells and Enhances Their Effector Functions for Adoptive Immunotherapy. J. Immunol. 2018;201:2141–2153. doi: 10.4049/jimmunol.1800429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Juelke K., Killig M., Luetke-Eversloh M., Parente E., Gruen J., Morandi B., Ferlazzo G., Thiel A., Schmitt-Knosalla I., Romagnani C. CD62L expression identifies a unique subset of polyfunctional CD56dim NK cells. Blood. 2010;116:1299–1307. doi: 10.1182/blood-2009-11-253286. [DOI] [PubMed] [Google Scholar]
- 42.Ding Z., Issekutz T.B., Downey G.P., Waddell T.K. L-selectin stimulation enhances functional expression of surface CXCR4 in lymphocytes: implications for cellular activation during adhesion and migration. Blood. 2003;101:4245–4252. doi: 10.1182/blood-2002-06-1782. [DOI] [PubMed] [Google Scholar]
- 43.Levy E., Reger R., Segerberg F., Lambert M., Leijonhufvud C., Baumer Y., Carlsten M., Childs R. Enhanced Bone Marrow Homing of Natural Killer Cells Following mRNA Transfection With Gain-of-Function Variant CXCR4R334X. Front. Immunol. 2019;10:1262. doi: 10.3389/fimmu.2019.01262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cravedi P., Manrique J., Hanlon K.E., Reid-Adam J., Brody J., Prathuangsuk P., Mehrotra A., Heeger P.S. Immunosuppressive effects of erythropoietin on human alloreactive T cells. J. Am. Soc. Nephrol. 2014;25:2003–2015. doi: 10.1681/ASN.2013090945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jenkins J.M., Williams D., Deng Y., Uhl J., Kitchen V., Collins D., Erickson-Miller C.L. Phase 1 clinical study of eltrombopag, an oral, nonpeptide thrombopoietin receptor agonist. Blood. 2007;109:4739–4741. doi: 10.1182/blood-2006-11-057968. [DOI] [PubMed] [Google Scholar]
- 46.Wang B., Nichol J.L., Sullivan J.T. Pharmacodynamics and pharmacokinetics of AMG 531, a novel thrombopoietin receptor ligand. Clin. Pharmacol. Ther. 2004;76:628–638. doi: 10.1016/j.clpt.2004.08.010. [DOI] [PubMed] [Google Scholar]
- 47.Erickson-Miller C.L., Delorme E., Tian S.S., Hopson C.B., Landis A.J., Valoret E.I., Sellers T.S., Rosen J., Miller S.G., Luengo J.I. Preclinical activity of eltrombopag (SB-497115), an oral, nonpeptide thrombopoietin receptor agonist. Stem Cells. 2009;27:424–430. doi: 10.1634/stemcells.2008-0366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Berg M., Lundqvist A., McCoy P., Jr., Samsel L., Fan Y., Tawab A., Childs R. Clinical-grade ex vivo-expanded human natural killer cells up-regulate activating receptors and death receptor ligands and have enhanced cytolytic activity against tumor cells. Cytotherapy. 2009;11:341–355. doi: 10.1080/14653240902807034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cooper M.L., Choi J., Staser K., Ritchey J.K., Devenport J.M., Eckardt K., Rettig M.P., Wang B., Eissenberg L.G., Ghobadi A. An “off-the-shelf” fratricide-resistant CAR-T for the treatment of T cell hematologic malignancies. Leukemia. 2018;32:1970–1983. doi: 10.1038/s41375-018-0065-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.