

Abstract
Alzheimer’s disease (AD) is the most common neurodegenerative disorder leading to dementia in the elderly, and the mechanisms of AD are not fully defined. MicroRNAs (miRNAs) have been shown to contribute to memory deficits in AD. In this study, we identified that miR-204-3p was downregulated in the hippocampus and plasma of 6-month-old APPswe/PS1dE9 (APP/PS1) mice. miR-204-3p overexpression attenuated memory and synaptic deficits in APP/PS1 mice. The amyloid levels and oxidative stress were decreased in the hippocampus of APP/PS1 mice after miR-204-3p overexpression. Nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 4 (Nox4) was a target of miR-204-3p, and Nox4 inhibition by GLX351322 protected neuronal cells against Aβ1–42-induced neurotoxicity. Furthermore, GLX351322 treatment rescued synaptic and memory deficits, and decreased oxidative stress and amyloid levels in the hippocampus of APP/PS1 mice. These results revealed that miR-204-3p attenuated memory deficits and oxidative stress in APP/PS1 mice by targeting Nox4, and miR-204-3p overexpression and/or Nox4 inhibition might be a potential therapeutic strategy for AD treatment.
Keywords: Alzheimer’s disease, beta amyloid, memory deficits, miR-204-3p, Nox4, oxidative stress, synaptic dysfunction
Graphical Abstract
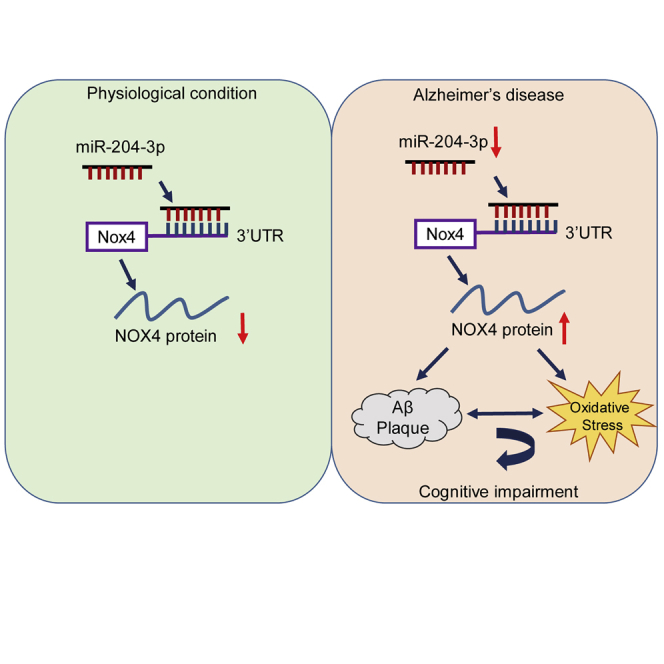
Zhu and colleagues demonstrate that miR-204-3p attenuates memory and synaptic deficits in APPswe/PS1dE9 (APP/PS1) mice via inhibition of NADPH oxidase 4 (Nox4). In addition, Nox4 inhibition by GLX351322 rescues memory deficits in APP/PS1 mice, suggesting that miR-204-3p overexpression and/or Nox4 inhibition is a potential therapeutic strategy for Alzheimer’s disease treatment.
Introduction
Alzheimer’s disease (AD) is the most common neurodegenerative disorder leading to dementia in the elderly, and the estimated number of AD patients is 5.8 million in the United States in 2019.1 Although the mechanisms of AD are not fully defined, beta amyloid (Aβ) is generally considered to be a trigger of AD.2 Aβ is generated by sequential cleavage of amyloid precursor protein (APP) by β-secretase and γ-secretase in the amyloidogenic pathway. Alternatively, APP is cleaved by α-secretase and γ-secretase in the nonamyloidogenic pathway.3 Persuasive evidence has demonstrated that Aβ-induced mitochondrial dysfunction and oxidative stress play a critical role in the pathogenesis of AD. The products of oxidative damage to lipids, proteins, and nucleic acids are elevated in the brains of AD, and anti-oxidative therapy is shown to be of benefit in AD models.4,5
MicroRNAs (miRNAs) are small non-coding RNAs of approximately 22 nucleotides, which inhibit protein expression mainly by binding to the 3′ untranslated region (UTR) of target mRNAs.6 Increasing evidence demonstrates that miRNAs play an important role in the pathogenesis of AD.7,8 miRNA cluster miR-29a/b-1 is decreased in the brains of sporadic AD and suppresses β-site APP cleaving enzyme 1 (BACE1) activity and Aβ generation.9 miRNA-124 is significantly increased in the hippocampus of AD transgenic mice and AD patients, and inhibition of miR-124 restores synaptic plasticity and memory function by directly regulating tyrosine-protein phosphatase non-receptor type 1 (PTPN1).10 Recently, a meta-analysis has shown that a panel of 10 miRNAs, which are associated with neuron growth factor signaling, Wnt signaling, and cellular senescence, are deregulated in the early stage of AD.11 In this study, we have shown that miR-204-3p was significantly decreased in the hippocampus of 6-month-old APP/PS1 mice.
miR-204 has been shown to contribute to aging and several neurodegenerative diseases.12,13 The level of miR-204 is significantly increased in the brains of Huntington’s disease patients.14 Exosomal miR-204 level in the cerebrospinal fluid (CSF) is decreased in symptomatic genetic frontotemporal dementia patients compared with that in presymptomatic patients.13 miR-204-5p is significantly increased in the hippocampus of 18-month-old C57BL/6J male mice, and it downregulates the surface expression of the N-methyl-d-aspartate receptor (NMDAR) NR1 subunit in hippocampal neurons by inhibiting Ephrin Type-B receptor 2 (EphB2).12 However, most studies of miR-204 focused on exploring the functions of miR-204-5p. The role of miR-204-3p in neurodegenerative diseases, including AD, remains undefined. Here, we found that lentivirus (Lv)-mediated miR-204-3p overexpression in the hippocampus of APP/PS1 mice attenuated synaptic and memory dysfunction, and reduced amyloid load and oxidative stress. In addition, we showed that nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 4 (Nox4) was a target of miR-204-3p, and Nox4 inhibition by GLX351322 protected against memory deficits and oxidative stress in APP/PS1 mice, which indicated that miR-204-3p/Nox4 were potential therapeutic targets for AD treatment.
Results
miR-204-3p Was Decreased in the Hippocampus and Plasma of APP/PS1 Mice
To identify endogenous miRNAs associated with cognitive impairment in AD, we detected the miRNA profiles in the hippocampus of 6-month-old APP/PS1 mice by a mouse miRNA microarray. There were 17 miRNAs upregulated and 47 miRNAs downregulated in the hippocampus of 6-month-old APP/PS1 mice compared with age-matched wild-type (WT) littermates (Figure 1A). Quantitative real-time PCR was performed to verify the result of microarray, and miR-204-3p was significantly decreased in the hippocampus of 6-month-old APP/PS1 mice (Figure S1). In addition, the level of miR-204-3p was significantly decreased in the hippocampus of 6-month-old and 9-month-old APP/PS1 mice (Figure 1B). Furthermore, the level of miR-204-3p was decreased in the plasma of 6-month-old AD mice (Figure 1C), indicating that miR-204-3p might be a potential plasma biomarker of AD.
Figure 1.

miR-204-3p Was Decreased in the Brain and Plasma of APP/PS1 Mice
(A) Heatmap diagram showing different expression patterns of miRNAs in the hippocampus of 6-month-old APP/PS1 and wild-type (WT) mice. n = 3. (B) The levels of miR-204-3p in the hippocampus of 4-, 6-, and 9-month-old APP/PS1 mice were examined by qPCR. n = 4–7. (C) The levels of miR-204-3p in the plasma of 6-month-old WT and APP/PS1 mice were detected by qPCR. n = 5. All data are presented as means ± SEM. ∗p < 0.05, ∗∗p < 0.01. ns, no significance.
Overexpression of Exogenous miR-204-3p Protects against Memory and Synaptic Impairment in APP/PS1 Mice
To explore whether miR-204-3p contributed to the pathogenesis of AD, we overexpressed exogenous miR-204-3p in the hippocampus of 6-month-old APP/PS1 mice using a GFP-tagged Lv (Lv-miR-204). The level of miR-204-3p was significantly elevated in the hippocampus (Figure 2A) and plasma (Figure 2B) after Lv-miR-204 treatment. To investigate the effect of miR-204-3p overexpression on memory function of APP/PS1 mice, we performed behavior tests, including open field, novel-object recognition (NOR), fear condition (FC), and Morris water maze (MWM) tests, 30 days after the Lv injection. miR-204-3p overexpression did not affect the traveling distance or time spent in the corner and center area of APP/PS1 mice (Figures S2A–S2C), suggesting that miR-204-3p did not affect the motor performance and exploration behavior of APP/PS1 mice in the open field tests. Lv-miR-204-injected APP/PS1 mice showed a significant increase of the discrimination index compared with Lv-con-injected mice during the testing phase in the NOR tests (Figure 2C). In addition, the freezing times of APP/PS1 mice were significantly decreased in both the contextual and the cued tests, which were increased after Lv-miR-204 treatment, indicating miR-204-3p overexpression could rescue fear memory deficits in APP/PS1 mice (Figures 2D and 2E). Spatial memory was determined by the MWM tests. During the training trial, the escape latency was increased in APP/PS1 mice compared with that in WT mice, whereas it was significantly reduced in Lv-miR-204-injected APP/PS1 mice (Figure 2F). During the probe trial, there was no significant difference in the total travel distance and swimming speed of mice in each group (Figures S2D and S2E). However, the crossing platform times (Figure 2G) and time in target quadrant (Figure 2H) of Lv-miR-204-injected APP/PS1 were significantly increased, and latency to target quadrant was decreased (Figure 2I) compared with those in Lv-con-injected mice. These results indicated that miR-204-3p overexpression attenuated memory deficits in APP/PS1 mice.
Figure 2.

Overexpression of Exogenous miR-204-3p Protected against Synaptic and Memory Impairment in APP/PS1 Mice
(A) The levels of miR-204-3p in the hippocampus after Lv-miR-204 injection were determined by qPCR. n = 6–7. (B) The levels of miR-204-3p in the plasma after Lv-miR-204 injection were determined by qPCR. n = 8. (C) In NOR tests, the percentage of time to explore new objects was recorded. (D and E) In FC tests, the percentage of freezing time in contextual (D) and cued (E) tests was recorded. n = 17–18. (F) In the acquisition trial, the escape latency was analyzed in the MWM tests. (G–I) The number of target platform crossings (G), time in the target quadrant (H), and latency to the target quadrant (I) were recorded in the probe trial. n = 17–18. (J) The input-output curve (I-O curve) of hippocampal CA1 in Lv-WT, Lv-con, and Lv-miR-204 mice. n = 6–8 slices from three mice in each group. (K and L) LTP induced by high-frequency stimulation was evaluated in hippocampal CA1. n = 6–8 slices from three mice in each group. (M–O) The levels of synaptic proteins in the hippocampus were determined by western blotting, and quantification of the intensities normalized to β-actin was shown. n = 3. (P and Q) Representative images of dendrite morphology and quantitative analysis of dendritic spine density. n = 6 slices from three mice in each group. Scale bars: 10 μm. (R) The percentage of mushroom spines per 10 μm dendrite length in each group. n = 10 slices from three mice in each group. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. ns, no significance.
Because synaptic plasticity is fundamental to learning and memory functions,15 we explored the effects of miR-204-3p overexpression on basal synaptic transmission and synaptic plasticity of hippocampal neurons. The slope of hippocampus slices from Lv-miR-204-treated mice was moderately increased compared with that in Lv-con-treated mice, although this trend did not reach statistical significance (Figure 2J). In addition, Lv-miR-204-treated mice showed higher long-term potentiation (LTP) magnitudes compared with Lv-con-treated mice (Figures 2K and 2L). These data demonstrated that miR-204-3p overexpression improved synaptic transmission in APP/PS1 mice. In addition, miR-204-3p overexpression led to a significant increase of the NMDAR subunit 2A (NMDAR2A), α-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA) receptor subunit GluA1, GluA2, and postsynaptic density protein 95 (PSD-95), whereas the levels of NMDAR1, NMDAR2B, calcium/calmodulin-dependent protein kinase II (CaMK II), and synaptophysin were not significantly changed (Figures 2M–2O). Furthermore, the dendritic spine density in the CA1 region of APP/PS1 mice was significantly compromised, and miR-204-3p overexpression could promote dendrite branching and increase the percentage of mushroom spines (Figures 2P–2R).
miR-204-3p Overexpression Decreased Amyloid Levels and Oxidative Stress in the Hippocampus of APP/PS1 Mice
To investigate whether miR-204-3p overexpression affected Aβ depositions, we examined the amount of Aβ plaques in the hippocampus of APP/PS1 mice after injection of Lv-miR-204 by immunofluorescence. The amounts of amyloid plaques were significantly decreased in the hippocampus of Lv-miR-204-treated APP/PS1 mice (Figures 3A and 3B). In addition, miR-204-3p overexpression significantly reduced the levels of Tris-buffered saline (TBS) soluble, Triton X-100/TBS (TBS-T) soluble, and formic acid (FA) soluble Aβ1–40 and Aβ1–42 in the hippocampus of APP/PS1 mice (Figures 3C and 3D). To determine whether miR-204-3p modulated the generation of Aβ, we detected the protein levels of APP and secretases. The level of a disintegrin and metalloproteinase domain 10 (ADAM10) was increased and presenilin 1 (PSEN1) was decreased, while the levels of APP, ADAM17, and BACE1 were not changed in the hippocampus of Lv-miR-204 treated APP/PS1 mice (Figures 3E and 3F), which might be associated with the Aβ reduction.
Figure 3.

Overexpression of miR-204-3p Decreased the Amyloid Levels and Oxidative Stress in the Hippocampus of APP/PS1 Mice
(A) Representative images of Aβ staining in the hippocampus of Lv-miR-204-treated APP/PS1 mice and (B) quantification of the area percentage of Aβ plaque load. n = 10 slices from three mice in each group. Scale bars: 250 μm. (C and D) The levels of TBS-soluble, TBS-T-soluble, and FA-soluble Aβ1–40 (C) and Aβ1–42 (D) were determined by ELISA in the hippocampus of APP/PS1 mice. n = 5. (E and F) The levels of APP and secretases in the hippocampus were examined by western blotting (E), and quantification of the intensities normalized to GAPDH as a loading control was shown (F). n = 3. (G) The level of ROS in the hippocampus of Lv-miR-204-treated APP/PS1 mice was detected by fluorescence. Scale bars: 250 μm. (H) The level of H2O2 in the hippocampus of Lv-miR-204 treated APP/PS1 mice was examined by spectrophotometry. n = 6. (I–K) The levels of 4-HNE (I), 3-NT (J), and 8-OHdG (K) in the hippocampus of Lv-miR-204-treated APP/PS1 mice were measured by ELISA. n = 4–5. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. ns, no significance.
Emerging evidence has shown that reactive oxygen species (ROS) overproduction plays a critical role in the onset and progression of AD.5 To explore whether miR-204-3p regulated the level of oxidative stress in the hippocampus of AD, we tested the level of ROS production by fluorescence in the hippocampus of APP/PS1 mice and found that the level of ROS was reduced after Lv-miR-204 treatment (Figure 3G). The level of H2O2 was also significantly decreased in the hippocampus of Lv-miR-204-treated APP/PS1 mice (Figure 3H). Furthermore, the levels of lipids, proteins, and nucleic acids peroxidation markers, 4-hydroxynonenal (4-HNE), 3-nitrotyrosine (3-NT), and 8-hydroxy-2′-deoxyguanosine (8-OHdG), were significantly downregulated in the hippocampus of Lv-miR-204-treated APP/PS1 mice compared with those in Lv-con-treated mice (Figures 3I–3K).
Given that the activation of glia cells and interaction of glia cells and neurons play an important role in the pathogenesis of AD,16 we assessed whether miR-204-3p overexpression regulated the activation of microglia and astrocytes in APP/PS1 mice. The results of immunofluorescence showed that the levels of Iba-1 were significantly decreased, while the level of GFAP was not changed after Lv-miR-204 treatment (Figure S3). These results demonstrated that miR-204-3p overexpression decreased the level of Aβ, oxidative stress, and microglia activation in the hippocampus of APP/PS1 mice.
Nox4 Was a Target of miR-204-3p
TargetScan was used to predict the potential targets of miR-204-3p, and it was found that there was a potential binding site for miR-204-3p in the 3′ UTR of Nox4 (Figure 4A). The luciferase reporter assay was performed to validate whether miR-204-3p binds to the 3′ UTR of Nox4. The relative luciferase activity was significantly reduced when cells were cotransfected with miR-204-3p and pGL3-Nox4-3′ UTR. However, the inhibitory effects were rescued when cells were cotransfected with miR-204-3p and pGL3-Nox4-3′ UTR mutant (Figure 4B). The mRNA level of Nox4 was not changed in the hippocampus of Lv-miR-204-treated APP/PS1 mice (Figure 4C). However, the protein level of Nox4 was reduced in the hippocampus of Lv-miR-204-treated APP/PS1 mice (Figure 4D). Moreover, the protein level of Nox4 was increased in the hippocampus of 6-month-old and 9-month-old APP/PS1 mice (Figure S4). All of these results indicated that Nox4 was a target of miR-204-3p.
Figure 4.

Nox4 Was a Target of miR-204-3p
(A) Predicted binding sites of miR-204-3p, Nox4 3′ UTR, and Nox4 3′ UTR mutant. (B) N2a cells were cotransfected with miR-204-3p and Nox4 3′-UTR or Nox4 3′-UTR mutant for 48 h, and the relative luciferase activity was assayed. n = 6. (C) The mRNA level of Nox4 was measured in the Lv-con and Lv-miR-204 group. n = 5. (D) The protein level of Nox4 was measured in the hippocampus of Lv-miR-204-treated APP/PS1 mice by western blotting (left panel), and quantification of the intensities normalized to β-actin was shown (right panel). n = 3. (E) Cell viability of neurons treated with Lv-miR-204 or GLX351322 followed by Aβ42 was examined by CCK-8 assays. n = 6. (F) Cell viability of neurons treated with Lv-miR-204 with or without Lv-Nox4 followed by Aβ42 was examined by CCK-8 assays. n = 6. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. ns, no significance.
To verify whether miR-204-3p exerted protective effects against Aβ through Nox4, we overexpressed miR-204-3p and/or Nox4 in primary cortical neurons followed by Aβ1–42 treatment. miR-204-3p overexpression reduced neuronal death induced by Aβ1–42, and GLX351322, a selective Nox4 inhibitor, increased the cell viability in Aβ1–42-treated neurons (Figure 4E). In addition, Nox4 overexpression reversed the protective effects of miR-204-3p (Figure 4F), suggesting that miR-204-3p protected against Aβ1–42-induced neurotoxicity at least partially by inhibition of Nox4.
Nox4 Inhibition Protected against Synaptic and Memory Dysfunctions in APP/PS1 Mice
To examine the effects of Nox4 on memory functions in APP/PS1, we performed the behavior tests after GLX351322 treatment. GLX351322 treatment at the dosage of 5 mg/kg/day did not affect the body weight of APP/PS1 mice (Figure S5A), but decreased the mRNA and protein levels of p22phox (Figures 5A and 5B), which was essential for the activity of Nox4. The open field test showed no significant difference in the traveling distance or time spent in the center and corner area among these three groups (Figures S5B–S5D), indicating that GLX351322 treatment did not affect the motor ability and exploration behavior of APP/PS1 mice. In the NOR tests, GLX351322 treatment significantly increased the discrimination index in APP/PS1 mice (Figure 5C). In the FC tests, the freezing times of GLX351322-treated APP/PS1 mice were significantly increased in the contextual and cued tests (Figures 5D and 5E). In the MWM tests, the escape latency was significantly reduced after GLX351322 treatment during the training trial (Figure 5F). There was no significant difference in the total travel distance and swimming speed (Figures S5E and S5F), while the crossing platform times (Figure 5G) and time in target quadrant (Figure 5H) were increased, and the latency to target quadrant (Figure 5I) was decreased after GLX351322 treatment during the probe trial. These results strongly suggested that GLX351322 treatment ameliorated memory impairment in APP/PS1 mice.
Figure 5.

Nox4 Inhibition Protected against Synaptic and Memory Dysfunctions in APP/PS1 Mice
(A) The mRNA level of Nox4 subunit p22phox in the hippocampus of APP/PS1 mice treated with GLX351322 was determined by qPCR. n = 4. (B) The protein level of p22phox in the hippocampus of APP/PS1 mice treated with GLX351322 by western blotting. n = 3. (C) In NOR tests, the percentage of time to explore new objects was recorded. n = 16. (D and E) In FC tests, the percentage of freezing time in contextual (D) and cued (E) tests was recorded. n = 15. (F) In the acquisition trial, the escape latency was analyzed in the MWM tests. (G–I) The number of target platform crossings (G), time in the target quadrant (H), and latency to the target quadrant (I) were recorded in the probe trial. n = 14–15. (J) The I-O curve of hippocampal CA1 of WT-vehicle mice, APP-vehicle mice, and APP-GLX35132 mice. n = 8–9 slices from three mice in each group. (K and L) LTP induced by high-frequency stimulation was evaluated in hippocampal CA1. n = 8–9 slices from three mice in each group. (M–O) The levels of synaptic proteins in the hippocampus were determined by western blotting, and quantification of the intensities normalized to β-actin was shown. n = 3. (P and Q) Representative images of dendrite morphology and quantitative analysis of dendritic spine density in each group. n = 6 slices from three mice in each group. Scale bars: 10 μm. (R) The percentage of mushroom spines per 10 μm dendrite length in each group. n = 10 slices from three mice in each group. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. ns, no significance.
Similarly, we detected the effects of GLX351322 on synaptic transmission, synaptic proteins, and morphology in the hippocampus of APP/PS1 mice. GLX351322 treatment resulted in significantly facilitated synaptic transmission and LTP induction (Figures 5J–5L), and increased the protein levels of NMDAR2A, GluA1, GluA2, and PSD-95 (Figures 5M–5O). Furthermore, GLX351322 treatment promoted dendrite branching and increased the percentage of mushroom spines in the CA1 region of APP/PS1 mice (Figures 5P–5R). These data suggest that Nox4 inhibition by GLX351322 improved synaptic and memory functions in APP/PS1 mice.
Nox4 Inhibition Ameliorated Amyloid Levels and Oxidative Stress in the Hippocampus of APP/PS1 Mice
To explore whether Nox4 inhibition regulated the amyloid deposition and Aβ levels in APP/PS1 mice, we determined the amyloid levels by immunofluorescence and ELISA. As shown in Figures 6A and 6B, the amyloid deposition in the hippocampus of APP/PS1 mice was reduced after GLX351322 treatment. The levels of TBS, TBS-T, and FA soluble Aβ1–40 and Aβ1–42 were also significantly decreased in the hippocampus of GLX351322-treated APP/PS1 mice (Figures 6C and 6D). Furthermore, GLX351322 treatment significantly downregulated the level of PSEN1, while the levels of APP, ADAM10, ADAM17, and BACE1 were not changed (Figures 6E and 6F).
Figure 6.

Nox4 Inhibition Decreased Amyloid Levels and Oxidative Stress in the Hippocampus of APP/PS1 Mice
(A and B) Representative image of Aβ staining in the hippocampus of GLX351322-treated APP/PS1 mice and quantification of the area percentage of Aβ plaque load. n = 10 slices from three mice in each group. Scale bars: 250 μm. (C and D) The levels of TBS-soluble, TBS-T-soluble, and FA-soluble Aβ1–40 (C) and Aβ1–42 (D) were determined by ELISA in the hippocampus of GLX351322-treated APP/PS1 mice. n = 3. (E and F) The levels of APP and secretases in the hippocampus were examined by western blotting, and quantification of the intensities normalized to GAPDH as a loading control was shown. n = 3. (G) The level of ROS in the hippocampus of GLX351322-treated APP/PS1 mice was detected by fluorescence. Scale bars: 250 μm. (H) The level of H2O2 in the hippocampus of GLX351322-treated APP/PS1 mice was examined by spectrophotometry. n = 5. (I–K) The levels of 4-HNE (I), 3-NT (J), and 8-OHdG (K) in the hippocampus of GLX351322-treated APP/PS1 mice were measured by ELISA. n = 5–6. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. ns, no significance.
Because Nox4 is a main source of superoxide and H2O2,17 we next examined whether Nox4 inhibition could rescue the oxidative stress in the APP/PS1 mice. The levels of ROS and H2O2 were decreased after GLX351322 treatment (Figures 6G and 6H), and the levels of 4-HNE, 3-NT, and 8-OHdG were significantly reduced after GLX351322 treatment (Figures 6I–6K). These results demonstrated that Nox4 inhibition could reverse the oxidative stress in the hippocampus of APP/PS1 mice.
We also investigated the effects of Nox4 inhibition by GLX351322 on glia cell activation in the APP/PS1 mice. The level of Iba-1 in the hippocampus of APP/PS1 mice was significantly decreased after GLX351322 treatment, while the expression of GFAP was not changed (Figure S6). These results showed that GLX351322 treatment ameliorated Aβ levels, oxidative stress, and microglia activation in APP/PS1 mice.
Discussion
Our study has uncovered a novel role of miR-204-3p in the pathogenesis of AD. miR-204-3p was downregulated in the hippocampus and plasma of 6-month-old APP/PS1 mice. Lv-mediated overexpression of miR-204-3p in the hippocampus improved synaptic plasticity and attenuated memory deficits of APP/PS1 mice. In addition, miR-204-3p overexpression decreased the burdens of both Aβ1–40 and Aβ1–42, and inhibited oxidative stress and the activation of microglia. We also identified Nox4 as a target of miR-204-3p, and Nox4 inhibition showed a protective effect against oxidative stress and synaptic and memory dysfunction in APP/PS1 mice. Thus, our results revealed that miR-204-3p/Nox4 played an important role in the AD pathogenesis, indicating that miR-204-3p overexpression and/or Nox4 inhibition might be a potential therapy for the treatment of AD.
miRNAs have been shown to play critical roles in synaptic plasticity, which regulates the pathogenesis of neurodegenerative disorders, including AD.18,19 Fragile X mental retardation protein (FMRP)-associated microRNAs miR-125b and miR-132 have opposing effects on dendritic spine morphology in hippocampal neurons by regulating different target synaptic proteins.20 miR-181a is significantly upregulated in AD mice, while inhibition of miR-181a attenuates memory deficits and increases the expression of its target GluA2 in AD mice.21 miR-101b has been shown to regulate the expression of APP,22 and miR-101b mimics rescue histone deacetylase 2 (HDAC2)-induced tau pathology and dendritic and memory impairment in hippocampal neurons and AD mice.23 Here, we demonstrated that miR-204-3p overexpression or Nox4 inhibition facilitated synaptic transmission and increased synaptic proteins and dendritic spine density in the hippocampus of APP/PS1 mice, which was associated with improved memory functions. Interestingly, miR-204-5p is shown to suppress the expression of EphB2 and surface NR1 in hippocampal neurons in aging,12 which seems to be detrimental to synaptic function. We speculated that the inconsistent results might be because of the different sequences between miR-204-5p and miR-204-3p and the different pathogenesis between aging and AD.
Biomarkers of oxidative stress, including ROS production, mitochondria dysfunction, protein, lipid, and DNA/RNA oxidation, are significantly increased in the human AD brain and AD models.24, 25, 26 Excessive oxidative stress promotes the generation of Aβ, and Aβ overproduction causes lipid peroxidation, protein oxidation, and DNA oxidation, which forms a vicious circle.5,27,28 Nox4, a member of the NOX family, is constitutively active and widely expressed in the brain.29 Nox4 is highly expressed in human brain pericytes, and inhibition of Nox4 decreases the level of ROS, which indicates that Nox4 is a major source of superoxide production.30 The functions of Nox4 in the center nerve system have been excessively studied in stroke and traumatic brain injury (TBI).31 The level of Nox4 is upregulated after TBI, and Nox4 knockout mice show attenuated brain injury and decreased oxidative markers.32 The level of Nox4 is increased mainly in the pericytes of ischemic stroke brains, and transgenic mice overexpressing Nox4 in pericytes demonstrate greater infarct volume and more severe blood-brain barrier damage.33 However, the role of Nox4 in the pathogenesis of AD has not been fully understood. Aβ1–42 treatment significantly increases the activity of Nox and upregulates the mRNA level of Nox4.34,35 In humanized APP × PS1 knockin mice, the level of Nox4 is increased in the frontal cortex and is correlated with the increase of Aβ1–42 and cognitive dysfunction.36 In this study, we demonstrated that Nox4 inhibition by miR-204-3p or GLX351322 decreased oxidative stress and attenuated memory deficits and synaptic dysfunction in APP/PS1 mice. Moreover, Nox4 seemed to mediate Aβ1–42-induced neuronal death, because inhibition of Nox4 by miR-204-3p or GLX351322 could partially rescue the detrimental effects. Both apoptotic and necrotic neurons are significantly decreased in neuronal Nox4 knockout mice of ischemic stroke, indicating the proapoptotic effects of Nox4.37 Nox4 interacts with p-STAT3 and promotes neuronal apoptosis by regulating Bcl-2, Bax, and p-Akt, which is a target of leonurine in ischemic stroke.38 NOX4-mediated ROS production induces cancer cell apoptosis by downregulation of c-FLIP and Mcl-1 expression.39 The imbalance of Nox4 and Nrf2 modulates the apoptosis of fibroblasts in aging.40 In addition, Nox4 and Nox4-mediated ROS increase cardiomyocyte autophagy via the PERK/eIF-2α/ATF4 pathway.41 Here we have shown that Nox4 inhibition by miR-204-3p or GLX351322 reversed Aβ1–42-induced neuronal death and rescued synaptic and memory deficits in APP/PS1 mice. The mechanisms underlying the detrimental effects of Nox4 will be investigated in future research.
PSEN1 is an essential subunit of the γ-secretase complex, and more than 200 mutations of PSEN1 have been identified in familial AD,42 which mostly increases the ratio of Aβ42/Aβ40 and promotes the progress of AD.43 A PSEN1 mutation (S169del) enhances Aβ generation and memory deficits in AD mice without altering the Notch signaling pathway.44 In addition, PSEN1 exerts several biological functions independent of the γ-secretase activity. PSEN1 activates the NF-κB signaling through interaction with breakpoint cluster region (BCR) and casein kinase II (CK2a), which is not affected by a γ-secretase inhibitor.45 The PSEN1 ΔE9 mutant-induced pluripotent stem cell (iPSC)-derived astrocytes show increased secretion of Aβ42, disturbance of Ca2+ signaling, extensive ROS production, and mitochondrial dysfunction, which contributes to the phenotype of AD.46 Therefore, PSEN1 is a potential therapeutic target for AD treatment. Here we showed that both miR-204-3p overexpression and Nox4 inhibition decreased the expression of PSEN1 and subsequently mitigated Aβ generation. Additionally, adenovirus-associated virus (AAV)-mediated Nox4 knockdown in the hippocampus reverses memory impairment and inhibits the expression of Aβ in the brains of 6-hydroxydopamine (6-OHDA)-induced Parkinson’s disease with dementia (PDD) mice,47 which is consistent with our results in AD mice. Notably, the level of ADAM10 was increased in the hippocampus of Lv-miR204-treated APP/PS1 mice, while inhibition of Nox4 by GLX351322 did not affect the expression of ADAM10, which indicated that Nox4 was not the only target of miR-204-3p in the pathogenesis of AD and further investigation is needed in future studies. Taken together, our results indicated that miR-204-3p overexpression or Nox4 inhibition not only decreased the production of Aβ but also inhibited Aβ-induced oxidative stress and synaptic deficits in APP/PS1 mice, which contributed to memory improvement.
In conclusion, our data have demonstrated a key role for miR-204-3p/Nox4 in the pathogenesis of AD, and miR-204-3p overexpression and/or Nox4 inhibition appeared to be a potential therapeutic strategy for AD treatment.
Materials and Methods
Animals and Treatment
Male APPswe/PS1dE9 (APP/PS1) transgenic mice and WT C57BL/6 (B6) littermates were obtained from the Model Animal Research Center of Nanjing University. The Lv overexpressing miR-204-3p (Lv-miR-204) and control (Lv-con) and the Lv overexpressing Nox4 (Lv-Nox4) and control (Lv-con2) were purchased from Shanghai Genechem (China). Lv-miR-204 (4 × 105 transduction units [TU]) or Lv-con (4 × 105 TU) was slowly injected into bilateral hippocampus of 6-month-old APP/PS1 mice (Lv-miR-204 group and Lv-con group) as previously described.48,49 Meanwhile, Lv-con (4 × 105 TU) was slowly injected into the bilateral hippocampus of 6-month-old WT mice (Lv-WT group). The behavior tests or electrophysiology experiments were performed 30 days after the injection, and then the mice were sacrificed for the other experiments. To access the effects of Nox4 on memory functions, a selective Nox4 inhibitor, GLX351322 (5 mg/kg/day; MedChemExpress, Monmouth Junction, NJ, USA), was intraperitoneally injected into 6-month-old APP/PS1 mice for 4 weeks followed by the behavior tests or electrophysiology experiments. All animal experiments were approved by the Animal Care Committee of Nanjing University.
Cell Culture and Treatment
Primary cortical neurons were prepared from embryonic day (E) 15–17 embryos of B6 mice as previously described,50 and cultured in Neurobasal medium with B27 (Invitrogen, Carlsbad, CA, USA) and 25 nM glutamine at 37°C in a humidified 5% CO2 incubator. The cells were infected by Lv-miR-204 (MOI = 5) with or without Lv-Nox4 (MOI = 5) at day 4 and then treated with synthetic Aβ1–42 (2 μM; Millipore, Boston, MA, USA) at day 7 for 24 h followed by the cell viability tests. Alternatively, the neuronal cells were pretreated with GLX351322 (10 μM) for 2 h at day 7 and then treated with Aβ1–42 for 24 h followed by the cell viability tests. Mouse neuroblastoma N2a cells were obtained from American Type Culture Collection (ATCC) and maintained in Dulbecco’s modified Eagle’s medium (DMEM, Invitrogen, Carlsbad, CA, USA) containing 10% fetal bovine serum (FBS; Invitrogen, Carlsbad, CA, USA).
Behavior Tests
Open field, NOR, FC, and MWM tests were performed as previously described.48,51 Detailed information can be found in the Supplemental Materials and Methods.
Microarray and Quantitative Real-Time PCR
The hippocampus was isolated and homogenized using a tissue homogenizer (Servicebio, Wuhan, China), and the RNA in tissues or plasma was extracted and purified using RNeasy Mini Kit (QIAGEN, Hilden, Germany) or miRNeasy Serum/Plasma Kit (QIAGEN, Hilden, Germany). Mouse miRNA microarray (Release 21.0; Shanghai Aksomics, China) with 1,881 unique miRNA probes was used to analyze the miRNA expression profile. miRNAs with differential expressions in the hippocampus of APP/PS1 mice were picked out with the fold change >2 and adjusted p <0.05. Quantification of mRNA or miRNA expression was performed using TaqMan Advanced miRNA Assays or SYBR Advantage qPCR Premix (Thermo Fisher, Carlsbad, CA, USA) according to the manufacturer’s instructions. The reactions were performed using an ABI 7500 PCR instrument (Applied Biosystems, Carlsbad, CA, USA). U6 small nuclear RNA (snRNA) and cel-miR-39 (Thermo Fisher, Carlsbad, CA, USA) were used as controls for tissue and plasma, respectively. Gapdh was used as an internal control for mRNA. The primer sequences were shown in the Supplemental Materials and Methods.
Electrophysiology
The hippocampal slices (300 μm) were prepared as previously described.52 The acute slices were transferred to the microelectrode array, continuously perfused with oxygenated artificial cerebrospinal fluid (ACSF) (2 mL/min), and maintained at 32°C for recording. field excitatory postsynaptic potentials (fEPSPs) in the CA1 stratum radiatum were recorded by using the MEA-2100-60-System (Multi Channel Systems, Reutlingen, Germany). To evaluate the input-output relationships of synapses, we measured the slope of fEPSPs. The stimulation intensity was half of the maximum evoked response in LTP experiments. The LTP was induced by high-frequency stimulation (HFS; 100 Hz, three trains, 1-s duration, 10-s interval time). Initial fEPSP slopes were normalized by the averaged slope value during the control period. Data were acquired by LTP-Director software, and data analysis was performed using LTP-Analyzer software.
Golgi Staining
Golgi staining was performed according to the manufacturer’s instructions of a fast Golgi staining kit (FD Neurotechnologies, Columbia, USA) as previously described.52 Detailed information could be found in the Supplemental Materials and Methods.
Luciferase Activity Assay
The relative luciferase activity was examined by using the Promega Bright-N-Glo system (Promega, Madison, WI, USA) as previously described.53 Detailed information can be found in the Supplemental Materials and Methods.
Cell Counting Kit-8 (CCK-8) Assay
Cell viability was determined using the CCK-8 assay (Dojindo, Kumamoto, Japan) as previously described.54 Detailed information could be found in the Supplemental Materials and Methods.
Western Blotting
Tissue lysate was prepared and subjected to western blotting as previously described.50 Detailed information can be found in the Supplemental Materials and Methods.
Immunofluorescence
Immunofluorescence was performed in brain sections or primary neurons as previously described.48 Detailed information can be found in the Supplemental Materials and Methods.
Aβ ELISA
Soluble and insoluble Aβ in the hippocampus were sequentially extracted as previously reported.48 Detailed information can be found in the Supplemental Materials and Methods.
Oxidative Stress Measurement
The markers of lipid peroxidation 4-HNE, protein oxidation 3-NT, and DNA oxidation 8-OHdG were detected using the corresponding ELISA kits (CUSABIO, Wuhan, PR China). In brief, the supernatants of hippocampus tissue were added to a plate pre-coated with mouse 4-HNE, 3-NT, or 8-OHdG antibody and incubated for the indicated times at 37°C. After adding the TMB solution and stop solution, the absorbance was recorded at 450 nm. The concentrations of 4-HNE, 3-NT, and 8-OHdG were calculated according to the standard curve.
The level of H2O2 was measured according to the manufacturer’s instructions (Elabscience, Wuhan, PR China). The hippocampus was homogenized with 10 vol (w/v) iced PBS buffer containing a mixture of phosphatase and protease inhibitor (Sigma, St. Louis, MO, USA) and centrifuged at 3,000 rpm for 15 min. The supernatant was collected for H2O2 detection at 240 nm.
ROS production in the hippocampus was measured using a ROS assay kit (GENMED, Shanghai, PR China). Fresh brain tissue was isolated and immediately cut into 20-μm-thick cryosections (Leica, Wetzlar, Germany). According to the manufacturer’s instructions, 200 μL of dihydroethidium (DHE) working solution was added to the slices and incubated in the dark at 37°C for 20 min. DHE fluorescence was measured using a microscope (Olympus, Tokyo, Japan).
Statistical Analysis
All data were presented as mean ± standard error of the mean (SEM). Statistical analysis was performed using SPSS 17.0 (SPSS, USA). The difference between two groups was analyzed using unpaired Student’s t test, and the difference among multiple groups was analyzed by one- or two-way analysis of variance (ANOVA) with repeated measures followed by Bonferroni post hoc test. A statistically significant difference was set at p <0.05.
Data Accessibility
The microarray data are deposited in the Gene Expression Omnibus (GEO) database with the accession number GEO: GSE138382. All other data are available from the corresponding author upon reasonable request.
Author Contributions
X.Z. and Y.X. designed research; W.T., L.Y., S.S., Y.L., Z.Z., S.X., X.B., and Y.G. performed experiments; W.T. and L.Y. analyzed data; W.T. and X.Z. wrote the manuscript; F.C. and W.S. provided valuable comments and revised the manuscript. All authors read and approved the final version of the manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This study was supported by the National Natural Science Foundation of China (grants 81671055 and 81971009 to X.Z. and 81901091 to S.S.); the Key Research and Development Program of Jiangsu Province of China (grant BE2016610 to Y.X.); Jiangsu Province Key Medical Discipline Grant (ZDXKA2016020 to Y.X.); Jiangsu Province Medical Youth Talent (grant QNRC2016024 to X.Z.); Young Talent Support Program from Jiangsu Association for Science and Technology (to X.Z.); and Canadian Institutes of Health Research Project (grant PJT-166127 to W.S). W.S. was the holder of the Tier 1 Canada Research Chair in Alzheimer’s Disease.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2020.09.006.
Contributor Information
Yun Xu, Email: xuyun20042001@aliyun.com.
Xiaolei Zhu, Email: zhuquelee@126.com.
Supplemental Information
References
- 1.Alzheimer’s Association 2019 Alzheimer’s disease facts and figures. Alzheimers Dement. 2019;15:321–387. [Google Scholar]
- 2.Selkoe D.J., Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016;8:595–608. doi: 10.15252/emmm.201606210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zhang Y., Song W. Islet amyloid polypeptide: Another key molecule in Alzheimer’s pathogenesis? Prog. Neurobiol. 2017;153:100–120. doi: 10.1016/j.pneurobio.2017.03.001. [DOI] [PubMed] [Google Scholar]
- 4.Butterfield D.A., Boyd-Kimball D. Oxidative Stress, Amyloid-β Peptide, and Altered Key Molecular Pathways in the Pathogenesis and Progression of Alzheimer’s Disease. J. Alzheimers Dis. 2018;62:1345–1367. doi: 10.3233/JAD-170543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheignon C., Tomas M., Bonnefont-Rousselot D., Faller P., Hureau C., Collin F. Oxidative stress and the amyloid beta peptide in Alzheimer’s disease. Redox Biol. 2018;14:450–464. doi: 10.1016/j.redox.2017.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.He L., Hannon G.J. MicroRNAs: small RNAs with a big role in gene regulation. Nat. Rev. Genet. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 7.Maciotta S., Meregalli M., Torrente Y. The involvement of microRNAs in neurodegenerative diseases. Front. Cell. Neurosci. 2013;7:265. doi: 10.3389/fncel.2013.00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Putteeraj M., Fairuz Y.M., Teoh S.L. MicroRNA Dysregulation in Alzheimer’s Disease. CNS Neurol. Disord. Drug Targets. 2017;16:1000–1009. doi: 10.2174/1871527316666170807142311. [DOI] [PubMed] [Google Scholar]
- 9.Hébert S.S., Horré K., Nicolaï L., Papadopoulou A.S., Mandemakers W., Silahtaroglu A.N., Kauppinen S., Delacourte A., De Strooper B. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer’s disease correlates with increased BACE1/beta-secretase expression. Proc. Natl. Acad. Sci. USA. 2008;105:6415–6420. doi: 10.1073/pnas.0710263105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang X., Liu D., Huang H.Z., Wang Z.H., Hou T.Y., Yang X., Pang P., Wei N., Zhou Y.F., Dupras M.J. A Novel MicroRNA-124/PTPN1 Signal Pathway Mediates Synaptic and Memory Deficits in Alzheimer’s Disease. Biol. Psychiatry. 2018;83:395–405. doi: 10.1016/j.biopsych.2017.07.023. [DOI] [PubMed] [Google Scholar]
- 11.Swarbrick S., Wragg N., Ghosh S., Stolzing A. Systematic Review of miRNA as Biomarkers in Alzheimer’s Disease. Mol. Neurobiol. 2019;56:6156–6167. doi: 10.1007/s12035-019-1500-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mohammed C.P., Rhee H., Phee B.K., Kim K., Kim H.J., Lee H., Park J.H., Jung J.H., Kim J.Y., Kim H.C. miR-204 downregulates EphB2 in aging mouse hippocampal neurons. Aging Cell. 2016;15:380–388. doi: 10.1111/acel.12444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schneider R., McKeever P., Kim T., Graff C., van Swieten J.C., Karydas A., Boxer A., Rosen H., Miller B.L., Laforce R., Jr., Genetic FTD Initiative (GENFI) Downregulation of exosomal miR-204-5p and miR-632 as a biomarker for FTD: a GENFI study. J. Neurol. Neurosurg. Psychiatry. 2018;89:851–858. doi: 10.1136/jnnp-2017-317492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sinha M., Mukhopadhyay S., Bhattacharyya N.P. Mechanism(s) of alteration of micro RNA expressions in Huntington’s disease and their possible contributions to the observed cellular and molecular dysfunctions in the disease. Neuromolecular Med. 2012;14:221–243. doi: 10.1007/s12017-012-8183-0. [DOI] [PubMed] [Google Scholar]
- 15.Li K., Wei Q., Liu F.F., Hu F., Xie A.J., Zhu L.Q., Liu D. Synaptic Dysfunction in Alzheimer’s Disease: Aβ, Tau, and Epigenetic Alterations. Mol. Neurobiol. 2018;55:3021–3032. doi: 10.1007/s12035-017-0533-3. [DOI] [PubMed] [Google Scholar]
- 16.Alibhai J.D., Diack A.B., Manson J.C. Unravelling the glial response in the pathogenesis of Alzheimer’s disease. FASEB J. 2018;32:5766–5777. doi: 10.1096/fj.201801360R. [DOI] [PubMed] [Google Scholar]
- 17.Muñoz M., López-Oliva M.E., Rodríguez C., Martínez M.P., Sáenz-Medina J., Sánchez A., Climent B., Benedito S., García-Sacristán A., Rivera L. Differential contribution of Nox1, Nox2 and Nox4 to kidney vascular oxidative stress and endothelial dysfunction in obesity. Redox Biol. 2020;28:101330. doi: 10.1016/j.redox.2019.101330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Juźwik C.A., Drake S.S., Zhang Y., Paradis-Isler N., Sylvester A., Amar-Zifkin A., Douglas C., Morquette B., Moore C.S., Fournier A.E. microRNA dysregulation in neurodegenerative diseases: A systematic review. Prog. Neurobiol. 2019;182:101664. doi: 10.1016/j.pneurobio.2019.101664. [DOI] [PubMed] [Google Scholar]
- 19.Reza-Zaldivar E.E., Hernández-Sápiens M.A., Minjarez B., Gómez-Pinedo U., Sánchez-González V.J., Márquez-Aguirre A.L., Canales-Aguirre A.A. Dendritic Spine and Synaptic Plasticity in Alzheimer’s Disease: A Focus on MicroRNA. Front. Cell Dev. Biol. 2020;8:255. doi: 10.3389/fcell.2020.00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Edbauer D., Neilson J.R., Foster K.A., Wang C.F., Seeburg D.P., Batterton M.N., Tada T., Dolan B.M., Sharp P.A., Sheng M. Regulation of synaptic structure and function by FMRP-associated microRNAs miR-125b and miR-132. Neuron. 2010;65:373–384. doi: 10.1016/j.neuron.2010.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rodriguez-Ortiz C.J., Prieto G.A., Martini A.C., Forner S., Trujillo-Estrada L., LaFerla F.M., Baglietto-Vargas D., Cotman C.W., Kitazawa M. miR-181a negatively modulates synaptic plasticity in hippocampal cultures and its inhibition rescues memory deficits in a mouse model of Alzheimer’s disease. Aging Cell. 2020;19:e13118. doi: 10.1111/acel.13118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vilardo E., Barbato C., Ciotti M., Cogoni C., Ruberti F. MicroRNA-101 regulates amyloid precursor protein expression in hippocampal neurons. J. Biol. Chem. 2010;285:18344–18351. doi: 10.1074/jbc.M110.112664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu D., Tang H., Li X.Y., Deng M.F., Wei N., Wang X., Zhou Y.F., Wang D.Q., Fu P., Wang J.Z. Targeting the HDAC2/HNF-4A/miR-101b/AMPK Pathway Rescues Tauopathy and Dendritic Abnormalities in Alzheimer’s Disease. Mol. Ther. 2017;25:752–764. doi: 10.1016/j.ymthe.2017.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luo Z., Luo W., Li S., Zhao S., Sho T., Xu X., Zhang J., Xu W., Xu J. Reactive oxygen species mediated placental oxidative stress, mitochondrial content, and cell cycle progression through mitogen-activated protein kinases in intrauterine growth restricted pigs. Reprod. Biol. 2018;18:422–431. doi: 10.1016/j.repbio.2018.09.002. [DOI] [PubMed] [Google Scholar]
- 25.Kamat P.K., Kalani A., Rai S., Swarnkar S., Tota S., Nath C., Tyagi N. Mechanism of Oxidative Stress and Synapse Dysfunction in the Pathogenesis of Alzheimer’s Disease: Understanding the Therapeutics Strategies. Mol. Neurobiol. 2016;53:648–661. doi: 10.1007/s12035-014-9053-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chew H., Solomon V.A., Fonteh A.N. Involvement of Lipids in Alzheimer’s Disease Pathology and Potential Therapies. Front. Physiol. 2020;11:598. doi: 10.3389/fphys.2020.00598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Butterfield D.A., Reed T., Newman S.F., Sultana R. Roles of amyloid beta-peptide-associated oxidative stress and brain protein modifications in the pathogenesis of Alzheimer’s disease and mild cognitive impairment. Free Radic. Biol. Med. 2007;43:658–677. doi: 10.1016/j.freeradbiomed.2007.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sultana R., Perluigi M., Butterfield D.A. Oxidatively modified proteins in Alzheimer’s disease (AD), mild cognitive impairment and animal models of AD: role of Abeta in pathogenesis. Acta Neuropathol. 2009;118:131–150. doi: 10.1007/s00401-009-0517-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo S., Chen X. The human Nox4: gene, structure, physiological function and pathological significance. J. Drug Target. 2015;23:888–896. doi: 10.3109/1061186X.2015.1036276. [DOI] [PubMed] [Google Scholar]
- 30.Kuroda J., Ago T., Nishimura A., Nakamura K., Matsuo R., Wakisaka Y., Kamouchi M., Kitazono T. Nox4 is a major source of superoxide production in human brain pericytes. J. Vasc. Res. 2014;51:429–438. doi: 10.1159/000369930. [DOI] [PubMed] [Google Scholar]
- 31.Ma M.W., Wang J., Zhang Q., Wang R., Dhandapani K.M., Vadlamudi R.K., Brann D.W. NADPH oxidase in brain injury and neurodegenerative disorders. Mol. Neurodegener. 2017;12:7. doi: 10.1186/s13024-017-0150-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma M.W., Wang J., Dhandapani K.M., Brann D.W. Deletion of NADPH oxidase 4 reduces severity of traumatic brain injury. Free Radic. Biol. Med. 2018;117:66–75. doi: 10.1016/j.freeradbiomed.2018.01.031. [DOI] [PubMed] [Google Scholar]
- 33.Nishimura A., Ago T., Kuroda J., Arimura K., Tachibana M., Nakamura K., Wakisaka Y., Sadoshima J., Iihara K., Kitazono T. Detrimental role of pericyte Nox4 in the acute phase of brain ischemia. J. Cereb. Blood Flow Metab. 2016;36:1143–1154. doi: 10.1177/0271678X15606456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oguchi T., Ono R., Tsuji M., Shozawa H., Somei M., Inagaki M., Mori Y., Yasumoto T., Ono K., Kiuchi Y. Cilostazol Suppresses Aβ-induced Neurotoxicity in SH-SY5Y Cells through Inhibition of Oxidative Stress and MAPK Signaling Pathway. Front. Aging Neurosci. 2017;9:337. doi: 10.3389/fnagi.2017.00337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sagy-Bross C., Hadad N., Levy R. Cytosolic phospholipase A2α upregulation mediates apoptotic neuronal death induced by aggregated amyloid-β peptide1-42. Neurochem. Int. 2013;63:541–550. doi: 10.1016/j.neuint.2013.09.007. [DOI] [PubMed] [Google Scholar]
- 36.Bruce-Keller A.J., Gupta S., Knight A.G., Beckett T.L., McMullen J.M., Davis P.R., Murphy M.P., Van Eldik L.J., St Clair D., Keller J.N. Cognitive impairment in humanized APP×PS1 mice is linked to Aβ(1-42) and NOX activation. Neurobiol. Dis. 2011;44:317–326. doi: 10.1016/j.nbd.2011.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Casas A.I., Geuss E., Kleikers P.W.M., Mencl S., Herrmann A.M., Buendia I., Egea J., Meuth S.G., Lopez M.G., Kleinschnitz C., Schmidt H.H.H.W. NOX4-dependent neuronal autotoxicity and BBB breakdown explain the superior sensitivity of the brain to ischemic damage. Proc. Natl. Acad. Sci. USA. 2017;114:12315–12320. doi: 10.1073/pnas.1705034114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang Q.Y., Wang Z.J., Miao L., Wang Y., Chang L.L., Guo W., Zhu Y.Z. Neuroprotective Effect of SCM-198 through Stabilizing Endothelial Cell Function. Oxid. Med. Cell. Longev. 2019;2019:7850154. doi: 10.1155/2019/7850154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Seo S.U., Kim T.H., Kim D.E., Min K.J., Kwon T.K. NOX4-mediated ROS production induces apoptotic cell death via down-regulation of c-FLIP and Mcl-1 expression in combined treatment with thioridazine and curcumin. Redox Biol. 2017;13:608–622. doi: 10.1016/j.redox.2017.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hecker L., Logsdon N.J., Kurundkar D., Kurundkar A., Bernard K., Hock T., Meldrum E., Sanders Y.Y., Thannickal V.J. Reversal of persistent fibrosis in aging by targeting Nox4-Nrf2 redox imbalance. Sci. Transl. Med. 2014;6:231ra47. doi: 10.1126/scitranslmed.3008182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sciarretta S., Volpe M., Sadoshima J. NOX4 regulates autophagy during energy deprivation. Autophagy. 2014;10:699–701. doi: 10.4161/auto.27955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cacace R., Sleegers K., Van Broeckhoven C. Molecular genetics of early-onset Alzheimer’s disease revisited. Alzheimers Dement. 2016;12:733–748. doi: 10.1016/j.jalz.2016.01.012. [DOI] [PubMed] [Google Scholar]
- 43.Borchelt D.R., Thinakaran G., Eckman C.B., Lee M.K., Davenport F., Ratovitsky T., Prada C.M., Kim G., Seekins S., Yager D. Familial Alzheimer’s disease-linked presenilin 1 variants elevate Abeta1-42/1-40 ratio in vitro and in vivo. Neuron. 1996;17:1005–1013. doi: 10.1016/s0896-6273(00)80230-5. [DOI] [PubMed] [Google Scholar]
- 44.Zhang S., Cai F., Wu Y., Bozorgmehr T., Wang Z., Zhang S., Huang D., Guo J., Shen L., Rankin C. A presenilin-1 mutation causes Alzheimer disease without affecting Notch signaling. Mol. Psychiatry. 2020;25:603–613. doi: 10.1038/s41380-018-0101-x. [DOI] [PubMed] [Google Scholar]
- 45.Tanaka Y., Sabharwal L., Ota M., Nakagawa I., Jiang J.J., Arima Y., Ogura H., Okochi M., Ishii M., Kamimura D., Murakami M. Presenilin 1 Regulates NF-κB Activation via Association with Breakpoint Cluster Region and Casein Kinase II. J. Immunol. 2018;201:2256–2263. doi: 10.4049/jimmunol.1701446. [DOI] [PubMed] [Google Scholar]
- 46.Oksanen M., Petersen A.J., Naumenko N., Puttonen K., Lehtonen Š., Gubert Olivé M., Shakirzyanova A., Leskelä S., Sarajärvi T., Viitanen M. PSEN1 Mutant iPSC-Derived Model Reveals Severe Astrocyte Pathology in Alzheimer’s Disease. Stem Cell Reports. 2017;9:1885–1897. doi: 10.1016/j.stemcr.2017.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Choi D.H., Choi I.A., Lee C.S., Yun J.H., Lee J. The Role of NOX4 in Parkinson’s Disease with Dementia. Int. J. Mol. Sci. 2019;20:696. doi: 10.3390/ijms20030696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu X., Wang S., Yu L., Jin J., Ye X., Liu Y., Xu Y. HDAC3 negatively regulates spatial memory in a mouse model of Alzheimer’s disease. Aging Cell. 2017;16:1073–1082. doi: 10.1111/acel.12642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu L., Liu Y., Jin Y., Cao X., Chen J., Jin J., Gu Y., Bao X., Ren Z., Xu Y., Zhu X. Lentivirus-Mediated HDAC3 Inhibition Attenuates Oxidative Stress in APPswe/PS1dE9 Mice. J. Alzheimers Dis. 2018;61:1411–1424. doi: 10.3233/JAD-170844. [DOI] [PubMed] [Google Scholar]
- 50.Zhu X., Wang S., Yu L., Yang H., Tan R., Yin K. TL-2 attenuates β-amyloid induced neuronal apoptosis through the AKT/GSK-3β/β-catenin pathway. Int. J. Neuropsychopharmacol. 2014;17:1511–1519. doi: 10.1017/S1461145714000315. [DOI] [PubMed] [Google Scholar]
- 51.Yu L., Liu Y., Yang H., Zhu X., Cao X., Gao J., Zhao H., Xu Y. PSD-93 Attenuates Amyloid-β-Mediated Cognitive Dysfunction by Promoting the Catabolism of Amyloid-β. J. Alzheimers Dis. 2017;59:913–927. doi: 10.3233/JAD-170320. [DOI] [PubMed] [Google Scholar]
- 52.Chen J., Shu S., Chen Y., Liu Z., Yu L., Yang L., Xu Y., Zhang M. AIM2 deletion promotes neuroplasticity and spatial memory of mice. Brain Res. Bull. 2019;152:85–94. doi: 10.1016/j.brainresbull.2019.07.011. [DOI] [PubMed] [Google Scholar]
- 53.Zhu X., Chen C., Ye D., Guan D., Ye L., Jin J., Zhao H., Chen Y., Wang Z., Wang X., Xu Y. Diammonium glycyrrhizinate upregulates PGC-1α and protects against Aβ1-42-induced neurotoxicity. PLoS ONE. 2012;7:e35823. doi: 10.1371/journal.pone.0035823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang X., Zhu X.L., Ji B.Y., Cao X., Yu L.J., Zhang Y., Bao X.Y., Xu Y., Jin J.L. LncRNA-1810034E14Rik reduces microglia activation in experimental ischemic stroke. J. Neuroinflammation. 2019;16:75. doi: 10.1186/s12974-019-1464-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The microarray data are deposited in the Gene Expression Omnibus (GEO) database with the accession number GEO: GSE138382. All other data are available from the corresponding author upon reasonable request.