

Abstract
CLN8 disease is a rare form of neuronal ceroid lipofuscinosis caused by biallelic mutations in the CLN8 gene, which encodes a transmembrane endoplasmic reticulum protein involved in trafficking of lysosomal enzymes. CLN8 disease patients present with myoclonus, tonic-clonic seizures, and progressive declines in cognitive and motor function, with many cases resulting in premature death early in life. There are currently no treatments that can cure the disease or substantially slow disease progression. Using a mouse model of CLN8 disease, we tested the safety and efficacy of an intracerebroventricularly (i.c.v.) delivered self-complementary adeno-associated virus serotype 9 (scAAV9) gene therapy vector driving expression of human CLN8. A single neonatal injection was safe and well tolerated, resulting in robust transgene expression throughout the CNS from 4 to 24 months, reducing histopathological and behavioral hallmarks of the disease and restoring lifespan from 10 months in untreated animals to beyond 24 months of age in treated animals. While it is unclear whether some of these behavioral improvements relate to preserved visual function, improvements in learning/memory, or other central or peripheral benefits, these results demonstrate, by far, the most successful degree of rescue reported in an animal model of CLN8 disease, and they support further development of gene therapy for this disorder.
Keywords: Batten disease, neurodegeneration, AAV9, AAV, gene therapy, CLN8
Graphical Abstract
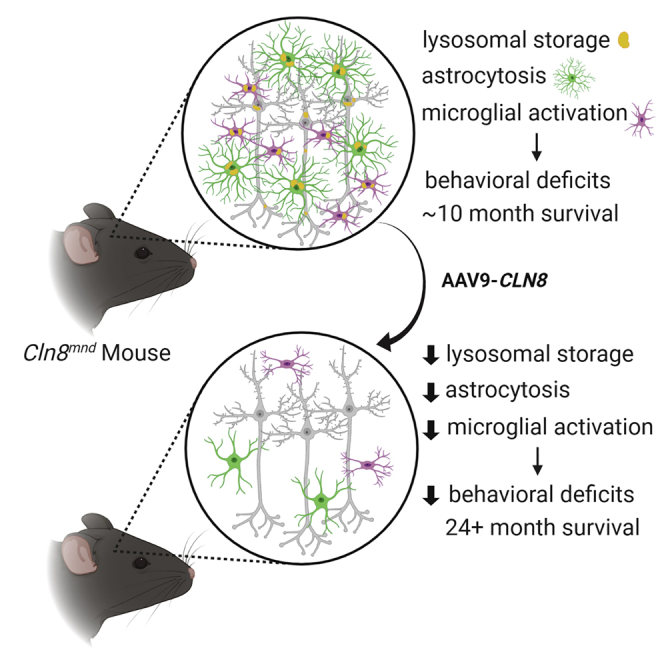
CLN8 disease is a rare form of neuronal ceroid lipofuscinosis characterized by neurological deficits and premature death. In this study, Johnson et al. demonstrate that CNS-delivered AAV9-CLN8 gene therapy reduces histopathological and behavioral hallmarks of the disease, while restoring lifespan from 10 months to beyond 24 months in treated animals.
Introduction
The neuronal ceroid lipofuscinoses (NCLs, also known as Batten disease) are a phenotypically similar group of rare lysosomal storage disorders caused by mutations in 1 of at least 13 identified ceroid-lipofuscinosis neuronal (CLN)-related genes,1 and they are collectively the most prevalent neurodegenerative disease in children (incidence 2–4/100,000 births).2,3 Patients typically present with CNS-related symptoms, including mental and motor deficits, blindness, and epilepsy in the first decade of life, with variable symptomatology, onset, and progression depending on the genetic subtype. All forms of NCL are progressive, most are fatal, and none yet has a cure.1
At the cellular level, NCLs are characterized by the lysosomal accumulation of autofluorescent storage material (ASM) consisting of lipid-based lipofuscin and other waste products.4 Some CLN genes encode soluble lysosomal enzymes and are thus linked directly to lysosomal activity (CLN1, CLN2, CLN10, CLN13), while others have more elusive function.1 Recently, CLN8, a transmembrane endoplasmic reticulum (ER) protein, was shown to regulate the ER to Golgi trafficking of catabolic enzymes destined for the lysosome.5 In the absence of CLN8, lysosomal enzymes are depleted, leading to classic NCL CNS pathology, including ASM accumulation, glial activation, and neurodegeneration throughout the CNS.6
Mutations in CLN8 can lead to an aggressive form of variant late infantile NCL in human patients that typically presents in childhood with myoclonus, tonic-clonic seizures, and progressive motor decline and dementia. Visual deficits or blindness can develop throughout the disease course,7, 8, 9, 10, 11, 12 or not at all.12, 13, 14 Phenotypic severity varies from milder forms, such as the “Northern epilepsy” caused by missense mutations in the Finnish population,12 to more severe forms caused by different sets of mutations, including null alleles.15,16 Various small molecule treatments have shown some efficacy in animal models of CLN8 disease,17, 18, 19 yet none has been adopted in the clinic, resulting in an unmet need to halt or reverse disease progression.
Recently, gene therapy has shown promise for treating a variety of genetic disorders, including the NCLs. Adeno-associated virus (AAV)-based therapies have shown some of the greatest potential, with low immunogenicity and toxicity, persistent gene expression, and favorable tropism patterns.20 AAV-based therapies have been tested in animal models for CLN1, CLN2, CLN3, CLN6, and CLN10 and are in clinical trials for human CLN2, CLN3, and CLN6 patients.1 While AAV-based therapies have been shown to be generally safe and well tolerated, they have not yet been explored for CLN8 disease. In this study, we use the Cln8 motor neuron degeneration (Cln8mnd) mouse model to test the efficacy of AAV serotype 9 (AAV9) gene therapy for CLN8 disease.
The Cln8mnd mouse, a naturally occurring mutant that has been extensively characterized,21 exhibits a disease pattern with strong similarity to human CLN8 disease patients. Cln8mnd homozygotes display early and progressive neuroinflammation and motor neuron degeneration culminating in paralysis and early death around 1 year of age.21, 22, 23 The causative mutation is a single base pair insertion (267–268insC) resulting in a frameshift and premature stop codon.12 Microglial activation is apparent at the early symptomatic stage of 3 months and is followed shortly thereafter by progressive astrocytosis and thalamic and cortical neurodegeneration beginning at 5 months of age.6 Associative learning deficits are detectable as early as 14–16 weeks of age, along with reductions in electroretinogram (ERG) responses and pupillary light reflexes.24 Retinal photoreceptor death begins as early as postnatal day 21 (P21).25
We utilized an intracerebroventricularly (i.c.v.) delivered self-complementary AAV9 (scAAV9) gene therapy vector, based on our prior success with this treatment in a mouse model of CLN6 disease.26 Our vector (scAAV9.pT-MecP2.CLN8) utilizes an AAV9 capsid combined with truncated MecP2 promoter (pT-MecP2). This combination achieves widespread and persistent transduction throughout the CNS, including in the cortex and thalamus, following a single injection. We quantified outcomes by measuring well-characterized disease correlates, including cortical and thalamic lysosomal storage and glial activation, behavioral assays of motor performance and learning/memory, and survival.
Results
scAAV9.pT-MecP2.CLN8 Drives Widespread Expression of hCLN8 through 24 Months of age in Cln8mnd Mice
To express full-length human CLN8 (hCLN8) in the CNS of treated animals, we designed an AAV9 viral vector expressing hCLN8 under the control of a truncated version of the MecP2 promoter: scAAV9.pT-MecP2.CLN8. The AAV9 capsid has been shown to be translatable between animal models of neurodegenerative disease and human subjects,27,28 and it is currently being used in clinical trials for CLN3 and CLN6 diseases (ClinicalTrials.gov: NCT03770572 and NCT02725580, respectively) following successful preclinical studies.26,29 scAAV9.pT-MecP2.CLN8 drives expression with a truncated version of the MecP2 promoter (pT-MecP2), which has been shown to achieve physiologically appropriate expression levels for this gene, which is endogenously expressed with low abundance.29 For this study, we delivered scAAV9.pT-MecP2.CLN8 via a single i.c.v. injection at postnatal day 1 (P1) in male and female Cln8mnd mice at a dose of 5 × 1010 viral genomes (vg) (the same dose used in our preclinical CLN6 studies).26 PBS was delivered to Cln8mnd controls.
To characterize spatiotemporal patterns of transgene expression, we examined the hCLN8 transcript using RNAscope, a modified in situ hybridization assay. The hCLN8 transcript was present in scAAV9.pT-MecP2.CLN8-treated Cln8mnd mice in all brain regions examined from 4 to 24 months, with the most robust levels in the cerebral cortex (motor, somatosensory, and visual cortices), and lower levels in the thalamus and hindbrain (Figure 1). Transcript levels and regional patterns were sustained during the 24-month period. Transcription throughout the somatosensory thalamocortical pathway was of particular interest, as these areas have been shown to be particularly vulnerable in Cln8mnd mice.6 scAAV9.pT-MecP2.CLN8 also resulted in transcription of hCLN8 in the cervical, thoracic, and lumbar spinal cord, as well as in the kidney, as measured by qRT-PCR with hCLN8-specific primers (Figures 2A–2E; Figure S1).
Figure 1.

scAAV9.pT-MecP2.CLN8 Produces Sustained hCLN8 Transcription in the Brain of Cln8mnd Mice
A single P1 i.c.v. injection of scAAV9.pT-MecP2.CLN8 results in transcription of hCLN8 in multiple areas of the brain until 24 months of age. The highest transcription levels were observed in the motor cortex, S1BF, and visual cortex, with lower levels of transcription in the thalamus, pons, cerebellum, and medulla.
Figure 2.

scAAV9.pT-MecP2.CLN8 Produces Sustained hCLN8 Transcription in the CNS, Resulting in Enhanced Cln8mnd Survival
(A–E) As measured by qRT-PCR, P1 i.c.v. administration of scAAV9.pT-MecP2.CLN8 produces increased hCLN8 transcript in the cerebral cortex (A), cervical spinal cord (B), thoracic spinal cord (C), lumbar spinal cord (D), and kidney (E). (F) Importantly, hCLN8 expression is sustained through 24 months after injection. scAAV9.pT-MecP2.CLN8 extends median lifespan in Cln8mnd mice from 10 months to beyond 24 months of age. Mean ± SEM. Each time point was run independently and analyzed as either a one-way ANOVA, Fisher’s LSD test (2–8 months), or unpaired t test (10–24 months). Samples from each tissue region from each mouse were analyzed using technical triplicates for both hCLN8 and Gapdh. The survival curve was analyzed using a Mantel-Cox log-rank test (n = 30–31). Detailed sample Ns are described in Table S2. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
scAAV9.pT-MecP2.CLN8 Restores Lifespan in Cln8mnd Mice
Cln8mnd mice have a drastically shortened lifespan, with premature death occurring at 7–12 months of age (depending on genetic background), presumably from seizures.23,30 In our study, PBS-treated Cln8mnd mice (C57BL/6J) died between 7 and 12 months of age, with a median survival of 10 months, while scAAV9.pT-MecP2.CLN8-treated Cln8mnd mice had a greatly lengthened lifespan, with a median survival beyond 24 months (Figure 2F). scAAV9.pT-MecP2.CLN8-treated Cln8mnd mice lived significantly longer than did PBS-treated Cln8mnd mice, and they were indistinguishable from wild-type (WT) mice through the duration of the study (24 months) in terms of survival curves. This survival benefit far surpasses what was previously the greatest survival increase reported for a therapeutic in Cln8mnd mice, i.e., a 16% increase in mean lifespan with carnitine supplementation.17
Additionally, gross necropsy was performed on each mouse at the time of sacrifice. In general, no abnormalities were observed in scAAV9.pT-MecP2.CLN8-treated Cln8mnd mice in any of the organs examined, including CNS, heart, lung, liver, spleen, kidney, small intestine, and skeletal muscles. Some animals were euthanized due to issues commonly observed in C57BL/6 mouse colonies (Table S1), but none of these appeared to be related to the genetic mutation or the therapy. Overall, we conclude that the treatment was safe and well tolerated, with no signs of adverse effects.
scAAV9.pT-MecP2.CLN8 Improves Lysosome Pathology in Cln8mnd Mice
Intracellular ASM accumulates in all forms of NCL, including CLN8, and it is frequently used as a correlate for cellular disease burden.23,31 While ASM is apparent in many tissues and cell types, it is most evident in neurons.31 We examined ASM in WT mice and scAAV9.pT-MecP2.CLN8-treated and PBS-treated Cln8mnd mice from 2 to 24 months. ASM was evident in the ventral posteromedial and ventral posterolateral nuclei of the thalamus (VPM/VPL) and in the primary somatosensory cortex barrel field (S1BF) at 2 months of age in PBS-treated Cln8mnd mice, and it accumulated rapidly until premature death between 8 and 10 months of age (Figures 3A and 3B). scAAV9.pT-MecP2.CLN8 greatly reduced ASM in the VPM/VPL and S1BF at 2, 4, 6, and 8 months of age. At 4 and 6 months of age, scAAV9.pT-MecP2.CLN8-treated Cln8mnd mice had significantly more ASM in the VPM/VPL than did WT mice, but levels were still far below those of PBS-treated Cln8mnd mice. At 2 and 8 months of age in the VPM/VPL, and at all time points from 2 through 8 months of age in the S1BF, ASM levels in WT and scAAV9.pT-MecP2.CLN8-treated Cln8mnd mice were indistinguishable.
Figure 3.

scAAV9.pT-MecP2.CLN8 Treatment Prevents Storage Material Accumulation in Cln8mnd Mice
(A–D) As measured by immunohistochemistry, scAAV9.pT-MecP2.CLN8 treatment significantly prevents autofluorescent storage material (ASM; A and B) and mitochondrial ATP synthase subunit C accumulation (SubC; C and D) in the somatosensory cortex (S1BF; A and C) and thalamus (VPM/VPL; B and D) from 2 through 24 months of age. Images are representative of 8 months of age. Mean ± SEM. Each time point was analyzed independently by either a one-way ANOVA, Fisher’s LSD test (2–8 months), or unpaired t test (10–24 months) (n = 8–72 images/treatment group, biological n = 4/sex/genotype). Detailed sample Ns are described in Table S2. ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
Due to the premature death of many PBS-treated Cln8mnd mice, only WT and scAAV9.pT-MecP2.CLN8-treated Cln8mnd mice could be examined from 10 to 24 months of age. During these time points, scAAV9.pT-MecP2.CLN8-treated Cln8mnd mice had more ASM in the VPM/VPL and S1BF than did their WT counterparts, but ASM levels never approached those observed in moribund (8-month-old) PBS-treated Cln8mnd mice, even at 24 months of age. These reductions in ASM are far greater than those documented for any other therapy in Cln8mnd mice.17
Mitochondrial ATP synthase subunit C (SubC) is a major constituent of the storage material in NCLs.4 SubC was elevated in the VPM/VPL and S1BF in PBS-treated Cln8mnd mice at 2, 4, 6, and 8 months of age, with large increases at the latest time point (Figures 3C and 3D). At all of these time points, VPM/VPL and S1BF SubC levels in WT and scAAV9.pT-MecP2.CLN8-treated Cln8mnd mice were indistinguishable. As with ASM, only WT and scAAV9.pT-MecP2.CLN8-treated Cln8mnd mice could be analyzed from 10 to 24 months of age. While scAAV9.pT-MecP2.CLN8-treated Cln8mnd mice showed some elevation of SubC in the VPM/VPL and S1BF at these late time points, SubC levels never approached those observed in moribund PBS-treated Cln8mnd mice. These drastic reductions we observed in cortical and thalamic ASM and SubC suggest that cellular disease burden in the brain is greatly reduced in scAAV9.pT-MecP2.CLN8-treated Cln8mnd mice.
Gliosis Is Reduced in scAAV9.pT-MecP2.CLN8-Treated Cln8mnd Mice
In neurodegenerative disorders including NCLs, glial cells respond to neuronal damage with a predictable cascade of morphological and molecular changes known as reactive gliosis.32 In the Cln8mnd mouse, brain gliosis has been reported to precede neuron loss, with microglial activation appearing prior to 3 months of age, followed shortly thereafter by astrocytosis.6 We observed the first signs of reactive microglia, as evidenced by increased immunolabeling for cluster of differentiation 68 (CD68) in the VPM/VPL and S1BF of PBS-treated Cln8mnd mice at 2 months of age (Figures 4A and 4B). In the subsequent months, CD68 immunoreactivity increased dramatically, with particularly large increases at the moribund time point of 8 months of age. CD68-positive microglia in PBS-treated Cln8mnd mice displayed morphology consistent with a reactive state, with hypertrophic soma and little ramification.
Figure 4.

scAAV9.pT-MecP2.CLN8 Treatment Reduces Glial Activation in Cln8mnd Mice
(A–D) As measured by immunohistochemistry, scAAV9.pT-MecP2.CLN8 treatment significantly prevents microglial activation (CD68; A and B) and astrocyte activation (GFAP; C and D) in the S1BF (A and C) and VPM/VPL (B and D) of 24-month-old Cln8mnd mice. Images are representative of 8 months of age. Mean ± SEM. Each time point was analyzed independently by either a one-way ANOVA, Fisher’s LSD test (2–8 months), or unpaired t test (10–24 months) (n = 24–72 images/treatment group, biological n = 4/sex/genotype. Detailed sample Ns are described in Table S2. ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001.
scAAV9.pT-MecP2.CLN8-treated Cln8mnd mice had significantly less microglial activation than did PBS-treated Cln8mnd mice at all time points examined in the S1BF, and from 4 months of age onward in the VPM/VPL. CD68 levels were indistinguishable from WT levels at several early (2- to 8-month) time points in scAAV9.pT-MecP2.CLN8-treated Cln8mnd mice, although slight elevations were present at 4 and 8 months of age. CD68 levels remained elevated in scAAV9.pT-MecP2.CLN8-treated Cln8mnd mice through 24 months of age, but they generally remained lower than the levels observed in moribund PBS-treated Cln8mnd mice.
We observed the first signs of astrocytosis, as evidenced by slight increases in glial fibrillary acidic protein (GFAP) immunoreactivity, in PBS-treated Cln8mnd mice at 2 months of age in the VPM/VPL and S1BF (Figures 4C and 4D). This was followed at 4 months of age by dramatic increases in GFAP immunoreactivity in both areas, indicating profound astrocytosis. Astrocytes in PBS-treated Cln8mnd mice appeared hypertrophic and intensely GFAP-positive, typical of the reactive responses observed in neurodegenerative diseases. GFAP immunoreactivity remained highly elevated from 4 through 8 months.
Treatment with scAAV9.pT-MecP2.CLN8 greatly reduced or eliminated astrocytosis, resulting in GFAP immunoreactivity indistinguishable from WT levels at 2 and 6 months of age, and far lower than PBS-treated Cln8mnd mice at 4 and 8 months of age, both in the VPM/VPL and S1BF. Moderate astrocytosis became evident from 10 through 24 months in the VPM/VPL and S1BF in scAAV9.pT-MecP2.CLN8-treated Cln8mnd mice, but it never exceeded that observed in moribund PBS-treated Cln8mnd mice. Taken together, these results demonstrate that scAAV9.pT-MecP2.CLN8 administration greatly attenuates, but does not completely eliminate, glial pathology in Cln8mnd mice.
scAAV9.pT-MecP2.CLN8 Reduces Motor and Behavioral Abnormalities in Cln8mnd Mice
Given the profound improvements we observed in lifespan and brain histopathological parameters in scAAV9.pT-MecP2.CLN8-treated Cln8mnd mice, we anticipated improvements in behavioral measures of CNS function. We performed a battery of behavioral tests to comprehensively assess performance in motor and learning/memory-related tasks, focusing on behavioral parameters that have been shown to be disrupted in the Cln8mnd model.18,19,24,33 Since the Cln8mnd model exhibits a vast range of neurophysiological and behavioral deficits, it can sometimes be difficult to ascribe improvements to specific facets of the disease. Still, we hypothesized that an effective therapy should elicit improvements in complex behavioral outcomes.
To quantify motor coordination-related behaviors, we measured rotarod performance from 2 to 24 months of age. Rotarod performance declined rapidly in PBS-treated Cln8mnd mice, with significant performance deficits evident at 8 and 10 months of age (Figure 5A). scAAV9.pT-MecP2.CLN8 administration substantially restored rotarod performance at all time points, with scAAV9.pT-MecP2.CLN8-treated Cln8mnd mice performing similarly to WT mice from 2 to 24 months of age. Similar deficits were also evident in the vertical pole test, where PBS-treated Cln8mnd mice exhibited increased descent time, time to turn down the pole, and fall frequency at 8 and 10 months of age (Figures 5B–5D). Treatment with scAAV9.pT-MecP2.CLN8 reduced these three deficits at both time points, with scAAV9.pT-MecP2.CLN8-treated animals maintaining WT levels of performance until 24 months of age.
Figure 5.

scAAV9.pT-MecP2.CLN8 Prevents Motor Abnormalities in Cln8mnd mice
(A–H) Neonatal administration of scAAV9.pT-MecP2.CLN8 leads to prevention of motor abnormalities as measured by the accelerating rotarod (A); vertical pole climb descent time (B), turn down time (C) and fall number (D); and coordination tasks (E–H). Mean ± SEM; two-way ANOVA, Fisher’s LSD test. Detailed Ns are described in Table S2. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, for differences from WT group. #p < 0.05, ##p < 0.01, ###p < 0.001, ####p < 0.0001, for differences from all other groups at that time point.
To further examine parameters related to motor performance, we deployed a three-component assay that has previously shown utility in mouse models of neurological disease.26,34 At 8 and 10 months of age, PBS-treated Cln8mnd mice showed impairments in hind limb clasping frequency, ledge descent ability, and overall gait, and they had significantly worse combined scores (Figures 5E–5H). scAAV9.pT-MecP2.CLN8-treated Cln8mnd mice, however, performed at or near WT levels in all three tasks at both time points, with combined scores indistinguishable from WT at 8 and 10 months of age. In all measures related to motor performance (rotarod, pole climb, and three-component test), the behavioral improvements noted with scAAV9.pT-MecP2.CLN8 administration persisted well beyond the moribund time point of 8 months for Cln8mnd mice, with performance remaining at or near WT levels for scAAV9.pT-MecP2.CLN8-treated Cln8mnd mice through 24 months of age.
We also measured performance in the Morris water maze as a complex assessment of visual, motor, and cognitive performance. During “training” trials, 6- and 8-month-old PBS-treated Cln8mnd mice took longer to swim to a platform marked by a visual cue despite having similar swim speed (Figures 6A and 6B), likely reflecting visual deficits. scAAV9.pT-MecP2.CLN8 treatment improved time to reach the platform to intermediate levels at both time points, although treated animals ultimately performed similarly to moribund untreated animals as they aged. In “memory” trials in which the visual cue was removed from the platform, PBS-treated Cln8mnd mice took longer to swim to the platform at 6 and 8 months of age, but these results were confounded by differences in swim speed during these trials (Figures 6C and 6D), and by the likely visual deficits noted in training trials. scAAV9.pT-MecP2.CLN8 treatment improved 8-month swim speed to WT levels, and 6- and 8-month platform finding latency to intermediate levels. Finally, during intermittent “learning” (also known as “reversal”) trials, in which the unmarked platform was moved to a novel location, PBS-treated Cln8mnd mice, which were only tested at 6 months of age, as they perished by later trials, took longer to swim to the platform at 6 months of age, with no apparent improvement in the scAAV9.pT-MecP2.CLN8-treated group (Figures 6E and 6F). Taken together, while the data suggest that some impairments may have been alleviated with scAAV9.pT-MecP2.CLN8 treatment, it is difficult to interpret whether these improvements relate to visual, motor, or cognitive function due to differences in overall motor performance and vision (as demonstrated by deficits in swim speed in the memory tests and deficits in time to the platform in the training trials).
Figure 6.

scAAV9.pT-MecP2.CLN8 Does Not Prevent Morris Water Maze Deficits in Cln8mnd Mice
(A–F) Memory and learning abnormalities are partially, but not entirely, corrected as measured by the Morris water maze “training” time to platform (A), “training” swim speed (B), “memory” time to platform (C), “memory” swim speed (D), “learning” time to platform (E), and “learning” swim speed (F). Swim speeds are shown as a control, showing that motor deficits may play a role in water maze deficits. Mean ± SEM; two-way ANOVA, Fisher’s LSD test. Detailed Ns are described in Table S2. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗∗p < 0.0001, for differences from WT group. ##p < 0.01, ###p < 0.001, ####p < 0.0001, for differences from all other groups at that time point.
While caring for these mice, we were surprised to observe tremors in many moribund PBS-treated Cln8mnd mice, a phenotype that mirrors symptoms observed in some human CLN8 disease patients,35 but which has not been reported in the Cln8mnd literature. To quantify the phenotype, we measured tremor scores in a force plate actimeter. Force plate data showed that scAAV9.pT-MecP2.CLN8-treated mice maintained a healthy weight through 24 months of age, and they showed no consistent differences in force plate parameters of total distance traveled, bouts of low mobility, or total area covered, but they exhibited slightly less focused stereotypy as they aged (Figures 7A–7F). PBS-treated Cln8mnd mice exhibited progressive elevations in tremor scores beginning at 2 months of age, which culminated in large elevations in tremor scores at 10 months of age (Figures 7G–7J). Treatment with scAAV9.pT-MecP2.CLN8 prevented the development of increased tremor scores at most time points and frequencies. Notably, even at the last time point examined (24 months), scAAV9.pT-MecP2.CLN8-treated Cln8mnd mice failed to exhibit the large elevations in tremor scores evident in moribund PBS-treated Cln8mnd mice. Taken together, these results demonstrate marked improvements in survival, brain pathology, and behavioral dysfunction in Cln8mnd mice following a single postnatal i.c.v. injection of scAAV9.pT-MecP2.CLN8.
Figure 7.

scAAV9.pT-MecP2.CLN8 Prevents Tremor Presentations in Cln8mnd Mice
(A–E) scAAV9.pT-MecP2.CLN8-treated mice show slight differences in weight, but they maintain a healthy weight through 24 months of age (A, males; B, females) and show no consistent differences in force plate parameters of total distance traveled (C), total area covered (D), and bouts of low mobility (E). (F) scAAV9.pT-MecP2.CLN8-treated mice show slight, but significantly fewer stereotypic movements as they age. (G–J) Lastly, AAV9 treatment prevents tremor presentation in Cln8mnd mice at various frequencies through 24 months of age, although AAV9 treated mice show slightly more tremors than do their wild-type counterparts as they age. Mean ± SEM; two-way ANOVA, Fisher’s LSD test. Detailed Ns are described in Table S2. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001, for differences from WT group. #p < 0.05, ##p < 0.01, ###p < 0.001, ####p < 0.0001, for differences from all other groups at that time point.
Discussion
CLN8 disease is a phenotypically variable, but often severe form of NCL with no known cure or treatment. While some preclinical studies have shown positive effects following the administration of small molecule therapies, behavioral and pathological improvements have been modest,18,19 and lifespan extension has been minimal.17 To date, the paucity of effective treatments is reflected in the clinic, with no clinical trials registered at ClinicalTrials.gov to test any therapy in CLN8 disease patients.
As is the case with many other NCLs, the lack of therapies can be attributed to a poor understanding of CLN8 function. Recent studies have made great progress toward this end, demonstrating that CLN8 binds directly to both soluble lysosomal enzymes and COPI/COPII machinery, facilitating the ER to Golgi trafficking of lysosomal enzymes.5 Still, while this knowledge provides valuable insight into potential drug targets, it is only a first step in an often long and arduous pathway toward the development of a small molecule therapy. Treatments that can directly resolve CLN8 deficiency are desperately needed.
Recent clinical trials for NCLs have focused on two primary treatment modalities, i.e., enzyme replacement therapy (ERT) and gene therapy, both of which aim to restore functional CLN proteins in the most affected cell populations (i.e., neurons). ERT has been used to deliver recombinant proteins directly into the brain through i.c.v. injection. This approach has been very successful in some cases; for example, cerliponase alfa (trade name Brineura) was US Food and Drug Administration (FDA) approved in 2017 for the treatment of CLN2 disease, which is caused by a deficiency in the soluble lysosomal enzyme tripeptidyl peptidase-1 (TPP1). Cerliponase alfa is a recombinant proenzyme form of endogenous TPP1, and while the therapy slows disease progression, it must be administered biweekly for a patient’s entire remaining lifespan, and adverse events such as hypersensitivity reactions are common.36 ERT is even less appealing for some other NCLs, particularly those caused by deficiencies in larger transmembrane proteins that would be more difficult to target to the correct subcellular locations.
Gene therapy offers another method to restore functional NCL proteins in vulnerable cell populations. With these therapies, viral vectors are used to drive expression of the gene of interest, with AAVs being the vector of choice in recent preclinical NCL studies.26,37, 38, 39, 40, 41 Different AAV serotypes have varying tropism and distribution patterns, allowing for specific tissues and cell populations to be targeted. AAV9 is particularly useful for CNS disorders, as it crosses the blood-brain barrier following intravenous administration to target both neurons and glia with high efficiency.27, 28, 29,42 Despite this useful property, other more direct routes of administration to the CNS are often still preferred since doses can be decreased, minimizing the potential for hepatotoxicity and other peripheral side effects.43 The use of i.c.v. delivery facilitates much smaller doses while still transducing CNS neurons with high efficiency, and while our study utilized i.c.v. delivery for practicality in mice, the results are supportive of other CSF-mediated delivery methods that are currently used in patients, such as intrathecal delivery.26
In this study, we demonstrate that a single P1 injection of an AAV9 viral vector driving expression of human CLN8 ameliorates many of the pathological and behavioral hallmarks of the disease in a mouse model of CLN8 disease. Our vector, scAAV9.pT-MecP2.CLN8, utilizes a truncated form of the MecP2 promoter in order to achieve physiologically relevant expression levels29 in transduced cells. We administered the virus at P1, since this timing in mice best recapitulates the transduction patterns observed in non-human primates and anticipated in human patients.44, 45, 46 This dosing scheme would be most representative of early postnatal treatment in humans rather than symptomatic stages, and future adoption of newborn screening for CLN8 mutations could make this type of treatment both realistic and feasible.
This single P1 i.c.v. injection of scAAV9.pT-MecP2.CLN8 achieved widespread and persistent expression of hCLN8 in the CNS from 4 to 24 months of age. Restoration of functional CLN8 greatly attenuated, but did not entirely eliminate, histopathological hallmarks of the disease, including ASM, SubC accumulation, microglial activation, and astrocytosis, providing protection through 24 months of age (the final time point assessed). The therapy also elicited improvements in a number of behavioral parameters, including motor performance in rotarod, pole climb, and three-component tests. Morris water maze test performance was partially improved in scAAV9.pT-MecP2.CLN8-treated Cln8mnd mice, although likely differences in overall vision and motor performance precluded any definitive assessment of learning or memory.
We also documented, for the first time, the presence of tremors in Cln8mnd mice. scAAV9.pT-MecP2.CLN8 treatment reduced tremor scores from 2 through 18 months, with scores in scAAV9.pT-MecP2.CLN8-treated Cln8mnd mice indistinguishable from WT at most frequencies and time points. Finally, and perhaps the most encouraging result in our study, scAAV9.pT-MecP2.CLN8 treatment conferred a significant survival benefit to Cln8mnd mice. While PBS-treated Cln8mnd mice all died by 12 months of age, most scAAV9.pT-MecP2.CLN8-treated animals survived for the duration of the study (24 months), with survival curves indistinguishable from WT mice.
While these results are encouraging, several important questions remain. At later time points beyond 8 months, increased lysosomal storage material and gliosis were evident in the brains of scAAV9.pT-MecP2.CLN8-treated Cln8mnd mice. While levels never approached those observed in PBS-treated Cln8mnd mice, this suggests that the rescue obtained by a single P1 i.c.v. injection of scAAV9.pT-MecP2.CLN8 is incomplete. Whether these minor tissue-level defects reflect incomplete rescue in transduced cells or are the consequences of cellular dysfunction in untransduced cells such as microglia is currently unknown. It is also unknown how potential systemic, rather than CNS, improvements may have contributed toward the improvements we observed in lifespan and behavioral parameters. Finally, a number of studies have demonstrated that in mice, the tropism of AAV9 viruses switches from predominately neuronal at P1–P7 to many more glial and fewer neurons after P7.42 This tropism switch does not occur in higher mammals (pigs, nonhuman primates). This caveat in tropism switch prevents us from testing the outcome of dosing at different ages in the mice.
Further experiments will be necessary to answer these questions, to optimize dose levels and timing, and to further enhance the efficacy of the therapy. Collectively, the improvements in survival, pathology, and behavior obtained from scAAV9.pT-MecP2.CLN8 administration in this study far surpass those reported for any other therapy in Cln8mnd mice, suggesting great potential for a similar strategy in human CLN8 disease patients.
Materials and Methods
Ethics Statement/Animals
WT and homozygous Cln8 mutant mice (Cln8mnd) on C57BL/6J backgrounds were used for all studies. Cln8mnd mice harbor a single nucleotide insertion (267–268C, codon 90), predicting a frameshift and premature stop codon. All mice were housed under identical conditions in an AAALAC International-accredited facility with Institutional Animal Care and Use Committee (IACUC) approval (US Department of Agriculture [USDA] license 46-R-0009; protocol #156-01-22D, Sanford Research, Sioux Falls, SD, USA).
Vector and AAV9 Delivery
A human CLN8 cDNA clone was subcloned into an AAV vector under the control of a truncated MecP2 (pT-MecP2) promoter. Self-complementary AAV9.pT-MecP2.CLN8 was produced as previously described, using transient transfection procedures with a double-stranded AAV2-ITR-based pT-MecP2.CLN8 vector, with a plasmid encoding Rep2Cap9 sequence along with an adenoviral helper plasmid pHelper (Stratagene, Santa Clara, CA, USA) in HEK293 cells.42 Vectors were purified by double cesium chloride gradient centrifugation. Silver staining analysis was used to measure the purity and titer of the vector.
At P0 or P1, mouse pups were sexed and then randomly assigned to a treatment group of either PBS or AAV9.pT-MecP2.CLN8 treated. Pups were randomly assigned until all treatment groups were filled. Mice received a single i.c.v. injection of either PBS or scAAV9.pT-MecP2.CLN8 at a dose of 5.0 × 1010 vg/animal. Animals were anesthetized with hypothermia at P1, injected with 4 μL of solution, and monitored until fully recovered. All animals were monitored daily by trained animal technicians and genotyped as previously described.26
qRT-PCR
Mice were CO2 euthanized and a 3-mm lateral section of the outer right hemisphere was collected and frozen for RNA isolation. Total RNA was extracted according to the manufacturer’s protocol for the Maxwell 16 LEV simplyRNA tissue kit (AS1280), using a Maxwell 16 MDx machine (Promega). RNA quality and concentration were assayed using a BioTek Epoch microplate spectrophotometer, with all samples having concentrations between 200 and 1,000 ng/μL and A260/A280 >2. Using the Promega GoScript reverse transcription system (A5001) and the manufacturer’s protocol, cDNA synthesis was performed on 1 μg of total RNA. The following primers were used from Integrated DNA Technologies: hCLN8 forward, 5′-GGACTGGCTCTGCTTACGCTAA-3′, reverse, 5′-GCTCTTGGCTTCTGGCTGTG-3′; Gapdh forward, 5′-ACC ACA GTC CAT GCC ATC AC-3′, reverse, 5′-TCC ACC ACC CTG TTG CTG TA-3′. Reactions were run on an Applied Biosystems ABI 7500 fast real-time PCR system, and PCR products were run on 3% Tris/borate/EDTA (TBE) gels at 125 V for 90 min. Bands were visualized under ultraviolet light using a Gel-Doc Imager (UVP). Relative levels of hCLN8 transcript were normalized against Gapdh by 2−ΔΔCt calculation from averages of triplicate Ct values for both transcripts.
RNAscope
hCln8 transcript localization was visualized with RNAscope as previously described.26 Briefly, brains were collected from CO2-euthanized mice and placed on a 1-mm sagittal brain block. Tissue blocks from 0 to 3 mm right of the midline were flash-frozen. Brain sections were sliced on a cryostat at 16 μm, series dehydrated, placed on slides, and processed for RNAscope according to the manufacturer’s protocols. Sections were labeled with a human CLN8 probe (ACDBio catalog no. 516851), counterstained with DAPI, and mounted on slides using antifade mounting media (Dako Faramount, Agilent Technologies).
Immunohistochemistry
The remaining left hemisphere of the brain was fixed in 4% paraformaldehyde and sectioned into 50-μm slices with selected slices placed into a 24-well plate. Immunohistochemistry was performed on free-floating sections as previously described using anti-ATP synthase SubC (Abcam, ab181243), anti-GFAP (Dako, Z0334), and anti-CD68 (AbD Serotec, MCA1957) antibodies.26 Secondary antibodies included anti-rat and anti-rabbit biotinylated (Vector Laboratories, BA-9400). Sections were placed on slides and mounted using xylene and xylene-based mounting media (DPX, VWR International). Sections were imaged and analyzed using an Aperio digital pathology slide scanner (VERSA) and associated software. Multiple images of each animal were taken in the VPM/VPL of the thalamus and the S1BF, and images were quantified using ImageJ.
Neurobehavior Testing
Rotarod
Animals participated in an accelerated rotarod protocol as previously described to assess motor coordination (Columbus Instruments, Columbus, OH, USA).26 The machine was set to accelerate 0.3 rpm every 2 s, with a starting speed of 0.3 rpm and a maximum speed of 36 rpm. Mice were trained for three consecutive trials, given a 30-min rest period, trained for three consecutive trials, given a second 30-min rest period, and trained for three final consecutive trials. After a 4-h rest period, mice were tested using the same paradigm as the training session. The latency time to fall from the rod was averaged from each of the nine afternoon testing sessions to produce one value per mouse.
Pole Climb
The pole climb descent test was performed as previously described.47 Mice were placed downward on a metal pole and given 60 s to descend the pole. Mice were placed upward on a metal pole and given 60 s to turn downward on the pole. Lastly, the number of falls made by each mouse during the two tests was recorded.
Water Maze
Mice were tested in a 4-ft-diameter Morris water maze apparatus as previously described.26 The apparatus was filled with water to ∼26 in and the goal platform submerged by 0.5 cm at 315°. The tub was aligned with four distinct visual cues at 0°, 90°, 180°, and 270° to aid in spatial memory. Mice were first trained in a clear pool with a flagged platform to assess confounding factors such as visual, motor, and anxious difficulties. Mice were given 60 s to find the platform each trial, with four trials in the morning, followed by a 3-h rest period, and four additional trials in the afternoon. Mice that could not locate the platform with 50% accuracy in the time allotted were eliminated from further testing. Mice were then tested in water colored with white, non-toxic tempura paint and an unflagged platform. Mice were given 60 s to complete each trial, with four trials in the morning, followed by a 3-h rest period, and four additional trials in the afternoon. Mice were tested for 4 consecutive days, each day starting at a different visual cue. Mice were recorded using ANY-maze video tracking software (Stoelting, Wood Dale, IL, USA). Test duration and swim speed were represented as the average from the 16 afternoon trials per mouse. At 6, 12, 18, and 24 months of age, an additional 4 days of reversal testing were introduced, with the hidden platform moved from 315° to 45°.
Clasping, Ledge, and Gait Tests
Tests were performed as previously described.26,34 For hind limb clasping measurements, animals were scored on the extent to which their limbs clasped into their abdomen when held by the base of their tail (score 0–3). For ledge-lowering measurements, animals were scored on their ability to climb down from the edge of their home cage (score 0–3). For gait measurements, animals were scored on their overall ease of walking, including whether their abdomen dragged on the ground and whether their limbs were splayed out while walking (score 0–3). The same blinded experimenter determined all scores.
Force Plate
A force plate actimeter was used to measure tremors as previously described.47 Animals were recorded in a sound-proof chamber for 20 min, and data were processed using FPA analysis software (BASi, West Lafayette, IN, USA).
Statistical Analysis
Statistical analyses were performed as previously described26 using GraphPad Prism (v6.04+), and details are noted in the figure legends. In general, one-way ANOVA was used with Fisher’s least significant difference (LSD) test, and outliers were removed with the ROUT method (Q = 0.1%). When appropriate, an unpaired t test was used. For the survival curve analysis, the log-rank (Mantel-Cox) test was used. ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001, ∗∗∗∗p < 0.0001. Detailed sample Ns are described in Table S2. For RT-qPCR, individual samples from each tissue region from each mouse were analyzed using technical triplicates in 96-well plates for both hCLN8 and Gapdh quantitation/normalization. For histopathological quantitation, three brain slices from each animal were used for each assay with four images/region/tissue taken for 12 technical replicates per/region/mouse. During the course of analysis, statistical outliers and any obvious technical failures (e.g., tissue folding, loss of desired region during processing) were removed from analysis, which accounts for the variability in technical replicate Ns in Table S2.
Author Contributions
Conceptualization, T.B.J., J.T.C., K.M., and J.M.W.; Methodology, T.B.J., J.T.C., M.A.P., and C.D.B.; Formal Analysis, T.B.J., K.A.W., J.T.C., L.L., and M.A.P.; Investigation, T.B.J., L.L., M.A.P., C.D.B., D.J.T., S.S.D., B.M., and S.L.; Resources, K.M.; Writing – Original Draft, J.B.; Writing – Review and Editing, T.B.J., K.A.W., J.B., K.M., and J.M.W.; Visualization, T.B.J. and K.A.W.; Supervision, T.B.J., J.T.C., K.M., and J.M.W.; Project Administration, T.B.J., K.A.W., and J.M.W.; Funding Acquisition, J.M.W.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work received support from the Cure Batten CLN8 Velona Foundation, Amicus Therapeutics, the Sanford Research Imaging Core within the Sanford Research Center for Pediatric Research (NIH P20GM103620), and the Sanford Research Molecular Pathology Core within the Sanford Research Center for Cancer Biology (NIH P20GM103548). The graphical abstract was created with BioRender.
Supplemental Information
References
- 1.Johnson T.B., Cain J.T., White K.A., Ramirez-Montealegre D., Pearce D.A., Weimer J.M. Therapeutic landscape for Batten disease: current treatments and future prospects. Nat. Rev. Neurol. 2019;15:161–178. doi: 10.1038/s41582-019-0138-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Santavuori P. Neuronal ceroid-lipofuscinoses in childhood. Brain Dev. 1988;10:80–83. doi: 10.1016/s0387-7604(88)80075-5. [DOI] [PubMed] [Google Scholar]
- 3.Rider J.A., Rider D.L. Batten disease: past, present, and future. Am. J. Med. Genet. Suppl. 1988;5:21–26. doi: 10.1002/ajmg.1320310606. [DOI] [PubMed] [Google Scholar]
- 4.Palmer D.N., Fearnley I.M., Walker J.E., Hall N.A., Lake B.D., Wolfe L.S., Haltia M., Martinus R.D., Jolly R.D. Mitochondrial ATP synthase subunit c storage in the ceroid-lipofuscinoses (Batten disease) Am. J. Med. Genet. 1992;42:561–567. doi: 10.1002/ajmg.1320420428. [DOI] [PubMed] [Google Scholar]
- 5.di Ronza A., Bajaj L., Sharma J., Sanagasetti D., Lotfi P., Adamski C.J., Collette J., Palmieri M., Amawi A., Popp L. CLN8 is an endoplasmic reticulum cargo receptor that regulates lysosome biogenesis. Nat. Cell Biol. 2018;20:1370–1377. doi: 10.1038/s41556-018-0228-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kuronen M., Lehesjoki A.E., Jalanko A., Cooper J.D., Kopra O. Selective spatiotemporal patterns of glial activation and neuron loss in the sensory thalamocortical pathways of neuronal ceroid lipofuscinosis 8 mice. Neurobiol. Dis. 2012;47:444–457. doi: 10.1016/j.nbd.2012.04.018. [DOI] [PubMed] [Google Scholar]
- 7.Cannelli N., Cassandrini D., Bertini E., Striano P., Fusco L., Gaggero R., Specchio N., Biancheri R., Vigevano F., Bruno C. Novel mutations in CLN8 in Italian variant late infantile neuronal ceroid lipofuscinosis: another genetic hit in the Mediterranean. Neurogenetics. 2006;7:111–117. doi: 10.1007/s10048-005-0024-y. [DOI] [PubMed] [Google Scholar]
- 8.Reinhardt K., Grapp M., Schlachter K., Brück W., Gärtner J., Steinfeld R. Novel CLN8 mutations confirm the clinical and ethnic diversity of late infantile neuronal ceroid lipofuscinosis. Clin. Genet. 2010;77:79–85. doi: 10.1111/j.1399-0004.2009.01285.x. [DOI] [PubMed] [Google Scholar]
- 9.Topçu M., Tan H., Yalnizoğlu D., Usubütün A., Saatçi I., Aynaci M., Anlar B., Topaloğlu H., Turanli G., Köse G., Aysun S. Evaluation of 36 patients from Turkey with neuronal ceroid lipofuscinosis: clinical, neurophysiological, neuroradiological and histopathologic studies. Turk. J. Pediatr. 2004;46:1–10. [PubMed] [Google Scholar]
- 10.Vantaggiato C., Redaelli F., Falcone S., Perrotta C., Tonelli A., Bondioni S., Morbin M., Riva D., Saletti V., Bonaglia M.C. A novel CLN8 mutation in late-infantile-onset neuronal ceroid lipofuscinosis (LINCL) reveals aspects of CLN8 neurobiological function. Hum. Mutat. 2009;30:1104–1116. doi: 10.1002/humu.21012. [DOI] [PubMed] [Google Scholar]
- 11.Zelnik N., Mahajna M., Iancu T.C., Sharony R., Zeigler M. A novel mutation of the CLN8 gene: is there a Mediterranean phenotype? Pediatr. Neurol. 2007;36:411–413. doi: 10.1016/j.pediatrneurol.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 12.Ranta S., Zhang Y., Ross B., Lonka L., Takkunen E., Messer A., Sharp J., Wheeler R., Kusumi K., Mole S. The neuronal ceroid lipofuscinoses in human EPMR and mnd mutant mice are associated with mutations in CLN8. Nat. Genet. 1999;23:233–236. doi: 10.1038/13868. [DOI] [PubMed] [Google Scholar]
- 13.Herva R., Tyynelä J., Hirvasniemi A., Syrjäkallio-Ylitalo M., Haltia M. Northern epilepsy: a novel form of neuronal ceroid-lipofuscinosis. Brain Pathol. 2000;10:215–222. doi: 10.1111/j.1750-3639.2000.tb00255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hirvasniemi A., Herrala P., Leisti J. Northern epilepsy syndrome: clinical course and the effect of medication on seizures. Epilepsia. 1995;36:792–797. doi: 10.1111/j.1528-1157.1995.tb01616.x. [DOI] [PubMed] [Google Scholar]
- 15.Gao Z., Xie H., Jiang Q., Wu N., Chen X., Chen Q. Identification of two novel null variants in CLN8 by targeted next-generation sequencing: first report of a Chinese patient with neuronal ceroid lipofuscinosis due to CLN8 variants. BMC Med. Genet. 2018;19:21. doi: 10.1186/s12881-018-0535-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ranta S., Topcu M., Tegelberg S., Tan H., Ustübütün A., Saatci I., Dufke A., Enders H., Pohl K., Alembik Y. Variant late infantile neuronal ceroid lipofuscinosis in a subset of Turkish patients is allelic to Northern epilepsy. Hum. Mutat. 2004;23:300–305. doi: 10.1002/humu.20018. [DOI] [PubMed] [Google Scholar]
- 17.Katz M.L., Rice L.M., Gao C.L. Dietary carnitine supplements slow disease progression in a putative mouse model for hereditary ceroid-lipofuscinosis. J. Neurosci. Res. 1997;50:123–132. doi: 10.1002/(SICI)1097-4547(19971001)50:1<1::AID-JNR1>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 18.Elger B., Schneider M., Winter E., Carvelli L., Bonomi M., Fracasso C., Guiso G., Colovic M., Caccia S., Mennini T. Optimized synthesis of AMPA receptor antagonist ZK 187638 and neurobehavioral activity in a mouse model of neuronal ceroid lipofuscinosis. ChemMedChem. 2006;1:1142–1148. doi: 10.1002/cmdc.200600144. [DOI] [PubMed] [Google Scholar]
- 19.Zeman R.J., Peng H., Etlinger J.D. Clenbuterol retards loss of motor function in motor neuron degeneration mice. Exp. Neurol. 2004;187:460–467. doi: 10.1016/j.expneurol.2004.03.006. [DOI] [PubMed] [Google Scholar]
- 20.Wang D., Tai P.W.L., Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat. Rev. Drug Discov. 2019;18:358–378. doi: 10.1038/s41573-019-0012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Messer A., Flaherty L. Autosomal dominance in a late-onset motor neuron disease in the mouse. J. Neurogenet. 1986;3:345–355. doi: 10.3109/01677068609106858. [DOI] [PubMed] [Google Scholar]
- 22.Messer A., Plummer J., Wong V., Lavail M.M. Retinal degeneration in motor neuron degeneration (mnd) mutant mice. Exp. Eye Res. 1993;57:637–641. doi: 10.1006/exer.1993.1169. [DOI] [PubMed] [Google Scholar]
- 23.Bronson R.T., Lake B.D., Cook S., Taylor S., Davisson M.T. Motor neuron degeneration of mice is a model of neuronal ceroid lipofuscinosis (Batten’s disease) Ann. Neurol. 1993;33:381–385. doi: 10.1002/ana.410330408. [DOI] [PubMed] [Google Scholar]
- 24.Wendt K.D., Lei B., Schachtman T.R., Tullis G.E., Ibe M.E., Katz M.L. Behavioral assessment in mouse models of neuronal ceroid lipofuscinosis using a light-cued T-maze. Behav. Brain Res. 2005;161:175–182. doi: 10.1016/j.bbr.2005.02.024. [DOI] [PubMed] [Google Scholar]
- 25.Seigel G.M., Wagner J., Wronska A., Campbell L., Ju W., Zhong N. Progression of early postnatal retinal pathology in a mouse model of neuronal ceroid lipofuscinosis. Eye (Lond.) 2005;19:1306–1312. doi: 10.1038/sj.eye.6701770. [DOI] [PubMed] [Google Scholar]
- 26.Cain J.T., Likhite S., White K.A., Timm D.J., Davis S.S., Johnson T.B., Dennys-Rivers C.N., Rinaldi F., Motti D., Corcoran S. Gene therapy corrects brain and behavioral pathologies in CLN6-Batten disease. Mol. Ther. 2019;27:1836–1847. doi: 10.1016/j.ymthe.2019.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Foust K.D., Wang X., McGovern V.L., Braun L., Bevan A.K., Haidet A.M., Le T.T., Morales P.R., Rich M.M., Burghes A.H., Kaspar B.K. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat. Biotechnol. 2010;28:271–274. doi: 10.1038/nbt.1610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 28.Mendell J.R., Al-Zaidy S., Shell R., Arnold W.D., Rodino-Klapac L.R., Prior T.W., Lowes L., Alfano L., Berry K., Church K. Single-dose gene-replacement therapy for spinal muscular atrophy. N. Engl. J. Med. 2017;377:1713–1722. doi: 10.1056/NEJMoa1706198. [DOI] [PubMed] [Google Scholar]
- 29.Bosch M.E., Aldrich A., Fallet R., Odvody J., Burkovetskaya M., Schuberth K., Fitzgerald J.A., Foust K.D., Kielian T. Self-complementary AAV9 gene delivery partially corrects pathology associated with juvenile neuronal ceroid lipofuscinosis (CLN3) J. Neurosci. 2016;36:9669–9682. doi: 10.1523/JNEUROSCI.1635-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Messer A., Plummer J., MacMillen M.C., Frankel W.N. Genetics of primary and timing effects in the mnd mouse. Am. J. Med. Genet. 1995;57:361–364. doi: 10.1002/ajmg.1320570251. [DOI] [PubMed] [Google Scholar]
- 31.Haltia M. The neuronal ceroid-lipofuscinoses. J. Neuropathol. Exp. Neurol. 2003;62:1–13. doi: 10.1093/jnen/62.1.1. [DOI] [PubMed] [Google Scholar]
- 32.Pekny M., Pekna M. Reactive gliosis in the pathogenesis of CNS diseases. Biochim. Biophys. Acta. 2016;1862:483–491. doi: 10.1016/j.bbadis.2015.11.014. [DOI] [PubMed] [Google Scholar]
- 33.Bolivar V.J., Scott Ganus J., Messer A. The development of behavioral abnormalities in the motor neuron degeneration (mnd) mouse. Brain Res. 2002;937:74–82. doi: 10.1016/s0006-8993(02)02470-8. [DOI] [PubMed] [Google Scholar]
- 34.Guyenet S.J., Furrer S.A., Damian V.M., Baughan T.D., La Spada A.R., Garden G.A. A simple composite phenotype scoring system for evaluating mouse models of cerebellar ataxia. J. Vis. Exp. 2010;(39):1787. doi: 10.3791/1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pesaola F., Kohan R., Cismondi I.A., Guelbert N., Pons P., Oller-Ramirez A.M., Noher de Halac I. Congenital CLN8 disease of neuronal ceroid lipofuscinosis: a novel phenotype. Rev. Neurol. 2019;68:155–159. [PubMed] [Google Scholar]
- 36.Schulz A., Ajayi T., Specchio N., de Los Reyes E., Gissen P., Ballon D., Dyke J.P., Cahan H., Slasor P., Jacoby D., Kohlschütter A., CLN2 Study Group Study of intraventricular cerliponase alfa for CLN2 disease. N. Engl. J. Med. 2018;378:1898–1907. doi: 10.1056/NEJMoa1712649. [DOI] [PubMed] [Google Scholar]
- 37.Arrant A.E., Onyilo V.C., Unger D.E., Roberson E.D. Progranulin gene therapy improves lysosomal dysfunction and microglial pathology associated with frontotemporal dementia and neuronal ceroid lipofuscinosis. J. Neurosci. 2018;38:2341–2358. doi: 10.1523/JNEUROSCI.3081-17.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Katz M.L., Tecedor L., Chen Y., Williamson B.G., Lysenko E., Wininger F.A., Young W.M., Johnson G.C., Whiting R.E., Coates J.R., Davidson B.L. AAV gene transfer delays disease onset in a TPP1-deficient canine model of the late infantile form of Batten disease. Sci. Transl. Med. 2015;7:313ra180. doi: 10.1126/scitranslmed.aac6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roberts M.S., Macauley S.L., Wong A.M., Yilmas D., Hohm S., Cooper J.D., Sands M.S. Combination small molecule PPT1 mimetic and CNS-directed gene therapy as a treatment for infantile neuronal ceroid lipofuscinosis. J. Inherit. Metab. Dis. 2012;35:847–857. doi: 10.1007/s10545-011-9446-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sondhi D., Johnson L., Purpura K., Monette S., Souweidane M.M., Kaplitt M.G., Kosofsky B., Yohay K., Ballon D., Dyke J. Long-term expression and safety of administration of AAVrh.10hCLN2 to the brain of rats and nonhuman primates for the treatment of late infantile neuronal ceroid lipofuscinosis. Hum. Gene Ther. Methods. 2012;23:324–335. doi: 10.1089/hgtb.2012.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Macauley S.L., Roberts M.S., Wong A.M., McSloy F., Reddy A.S., Cooper J.D., Sands M.S. Synergistic effects of central nervous system-directed gene therapy and bone marrow transplantation in the murine model of infantile neuronal ceroid lipofuscinosis. Ann. Neurol. 2012;71:797–804. doi: 10.1002/ana.23545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Foust K.D., Nurre E., Montgomery C.L., Hernandez A., Chan C.M., Kaspar B.K. Intravascular AAV9 preferentially targets neonatal neurons and adult astrocytes. Nat. Biotechnol. 2009;27:59–65. doi: 10.1038/nbt.1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hinderer C., Katz N., Buza E.L., Dyer C., Goode T., Bell P., Richman L.K., Wilson J.M. Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human SMN. Hum. Gene Ther. 2018;29:285–298. doi: 10.1089/hum.2018.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Schuster D.J., Dykstra J.A., Riedl M.S., Kitto K.F., Belur L.R., McIvor R.S., Elde R.P., Fairbanks C.A., Vulchanova L. Biodistribution of adeno-associated virus serotype 9 (AAV9) vector after intrathecal and intravenous delivery in mouse. Front. Neuroanat. 2014;8:42. doi: 10.3389/fnana.2014.00042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chakrabarty P., Rosario A., Cruz P., Siemienski Z., Ceballos-Diaz C., Crosby K., Jansen K., Borchelt D.R., Kim J.Y., Jankowsky J.L. Capsid serotype and timing of injection determines AAV transduction in the neonatal mice brain. PLoS ONE. 2013;8:e67680. doi: 10.1371/journal.pone.0067680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Green F., Samaranch L., Zhang H.S., Manning-Bog A., Meyer K., Forsayeth J., Bankiewicz K.S. Axonal transport of AAV9 in nonhuman primate brain. Gene Ther. 2016;23:520–526. doi: 10.1038/gt.2016.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Geraets R.D., Langin L.M., Cain J.T., Parker C.M., Beraldi R., Kovacs A.D., Weimer J.M., Pearce D.A. A tailored mouse model of CLN2 disease: a nonsense mutant for testing personalized therapies. PLoS ONE. 2017;12:e0176526. doi: 10.1371/journal.pone.0176526. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.