

Hadders and Lens preview work from the Zachos laboratory describing a new pathway activating the abscission checkpoint during cytokinesis.
Abstract
How chromatin bridges are relayed to the chromosomal passenger complex (CPC) during mammalian cell division is unknown. In this issue, Petsalaki and Zachos (2020. J. Cell Biol. https://doi.org/10.1083/jcb.202008029) show that the DNA damage checkpoint kinases ATM and Chk2 signal to the CPC to associate with a pool of cytoskeletal regulators, MKLP2–Cep55, in the midbody center and to delay abscission.
The birth of two daughter cells from one mother cell is the outcome of the intricate process of cell division. During cell division, the duplicated chromosomes are first equally separated and segregated in mitosis, after which cytoplasmic division or cytokinesis begins to physically separate two new daughter cells with identical copies of the genome. Cytokinesis starts in anaphase with processive ingression of the cell membrane at the cell equator (aka furrow ingression) caused by constriction of an actomyosin-based ring structure. It eventually ends in late telophase with abscission, the process during which the two daughter cells are pinched off from each other at a structure called the midbody. The midbody is part of a narrow, microtubule-filled intercellular canal that connects the two daughter cells until abscission and serves as a landing platform for the abscission machinery (Fig. 1; 1). To avoid chromosome damage by the act of cytokinesis (2, 3), cytokinesis onset and progression has to be coordinated with prior chromosome segregation to ensure that all chromatin is cleared from the area where the furrow ingresses and the intercellular canal is eventually formed. In line with this notion, the presence of unsegregated chromatin in the division plane delays the final phase of cytokinesis, and this abscission delay is referred to as the “abscission checkpoint.” Midbody-localized Aurora B kinase, the enzymatic subunit of the chromosomal passenger complex (CPC), is a central and evolutionary-conserved component of this checkpoint (3, 4). Over the years, several downstream targets of Aurora B have been identified that explain how Aurora B activity can delay the final cut (5). Interestingly, many of these Aurora B targets accumulate in a narrow, ring-shaped structure in the center of the midbody, known as the Flemming body, suggesting this specific part of the midbody might act as a signaling hub for the abscission checkpoint (5). Yet, it has remained elusive how an active pool of Aurora B is recruited to and retained in the Flemming body, particularly in response to missegregated chromatin in the division plane. In this issue, Petsalaki and Zachos shed new light on this mechanism (6).
Figure 1.
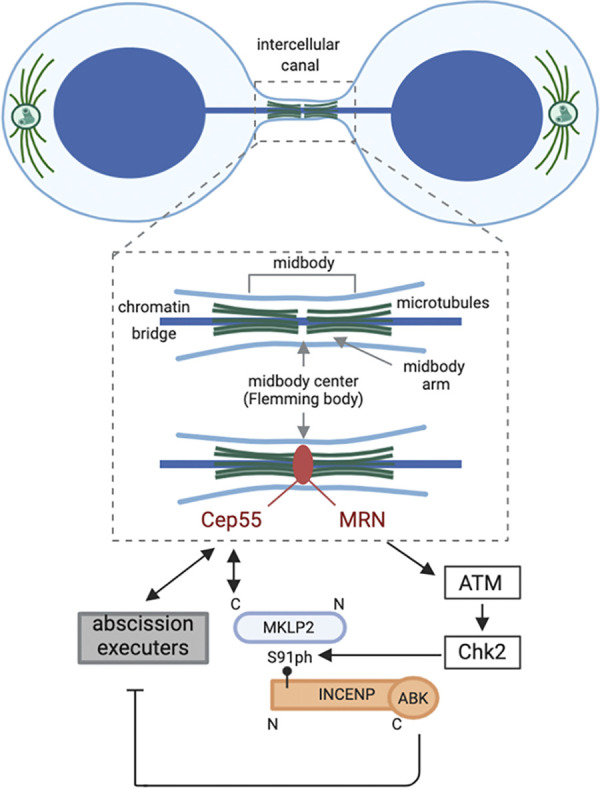
Checkpoint kinase encounter at the midbody. Cartoon of a cell undergoing cytokinesis while a chromatin bridge links the two daughter nuclei. The future daughter cells are connected by an intercellular canal through which the chromatin bridge passes. An enlargement of the intercellular canal is drawn to highlight the central part of the canal where the midbody is positioned. The antiparallel-oriented microtubules that form the midbody arms overlap in the very center of the midbody (indicated as a gap between the microtubule arms). A large number of proteins is recruited to the midbody during cytokinesis. These proteins either localize on the midbody arms (not indicated), the Flemming body (such as Cep55, indicated in red), or dynamically exchange between these locations. An example of the latter is the CPC, which localizes on the midbody arms in early telophase but also becomes detectable in the Flemming body in late telophase (12). In the present study, Petsalaki and Zachos reveal the mechanism of this Flemming body localization of the CPC. They find that MRN, ATM, and Chk2 are required to drive the association between S91-phosphorylated INCENP and Cep55-bound MKLP2 in late telophase and demonstrate that CPC recruitment to the Flemming body is essential to delay abscission. ABK, Aurora B kinase; S91ph, phosphorylated S91; N, N-terminus; C, C-terminus. Created with Biorender.com.
Petsalaki and Zachos identified the DNA double-strand break (DSB) signaling kinase ataxia-telangiectasia mutated (ATM) and its downstream target checkpoint kinase 2 (Chk2) as regulators of Aurora B recruitment to the Flemming body. The researchers found that active ATM and Chk2 colocalize with Aurora B and its CPC interaction partner, INCENP, at Flemming bodies in late telophase in cancer cell lines. Inhibition of these DNA damage checkpoint kinases accelerated the onset of abscission, both in cells without or with chromatin bridges. In case of the latter, ATM or Chk2 inhibition caused the chromatin bridge to break, resulting in DNA damage.
Interestingly, Petsalaki and Zachos identified serine 91 (S91) in INCENP as a substrate of Chk2 and found that this phosphorylation promoted the direct association of INCENP with the kinesin-6 motor protein MKLP2. While a more central part of MKLP2 is known to interact with INCENP (7, 8, 9), Petsalaki and Zachos found that the very C-terminal part of MKLP2 directly bound to Cep55, an established Flemming body protein and recruiter of abscission regulators (5). Deletion of the C-terminal part of MKLP2 specifically disrupted Flemming body localization of the CPC. Similarly, inhibition of Chk2 only affected INCENP-S91 phosphorylation in late Flemming bodies but not in other parts of the late midbody, suggesting that ATM and Chk2 act locally to mediate the binding between the CPC and Cep55-associated MKLP2 (Fig. 1). To further substantiate this, the authors engineered an INCENP fusion protein that directed INCENP and Aurora B specifically toward the Flemming body and not to any other part of the midbody. Expression of this fusion protein restored the abscission delay observed in Chk2-inhibited cells, implying that by recruiting Aurora B to the Flemming body, ATM and Chk2 ensure that a pool of active Aurora B is in close proximity of critical executors of abscission, thereby most likely allowing efficient inhibition of the process.
ATM activity is required at DNA DSB sites to delay cell cycle progression and to facilitate DNA damage repair. ATM recruitment and activation at DSB sites is mediated by the Mre11–Rad50–Nbs1 (MRN) complex, which recognizes and processes the DNA damage site (10). Interestingly, in dividing cells with chromatin bridges, Petsalaki and Zachos found that the MRN proteins localized at late Flemming bodies and were required for ATM and Chk2 activation in, and for CPC recruitment to, these sites (Fig. 1). Similar to ATM and Chk2 inhibition, knockdown of MRN proteins accelerated abscission, causing chromatin bridges to break. The here-described involvement of DNA damage signaling kinases in activation of the abscission checkpoint is not without precedent. DNA lesions resulting from prior replication stress have been described to delay abscission in an Aurora B–dependent manner (11). However, instead of ATM and Chk2, the DNA damage signaling kinases ATR (ataxia-telangiectasia and Rad3–related) and Chk1 relayed the presence of these lesions in the nucleus to Aurora B at the midbody (11). The DNA lesions giving rise to the chromatin bridges in the Petsalaki and Zachos study most likely have a different origin, fueling the idea that, similar to DNA damage sensing and signaling in, for instance, S or G2 phase of the cell cycle, different types of DNA lesions are also detected by different sensors and DNA damage signaling kinases in cells undergoing cytokinesis. In the latter cells, however, these signaling cascades culminate in the retention of active Aurora B in the Flemming body, which inhibits the abscission machinery.
As such, the findings of Petsalaki and Zachos raise the more specific question of how chromatin bridges promote the recruitment of MRN proteins toward the midbody center. It is tempting to speculate that the ring-shaped Flemming body through which the chromatin bridge likely passes causes some sort of DNA stress that is recognized by the MRN complex, which in turn activates the abscission checkpoint and promotes chromatin bridge resolution. It will be interesting to learn what eventually happens to chromatin bridges that recruit the MRN complex in cells with active ATM and Chk2. Is the bridge resolved before abscission proceeds, and if so, what kind of DNA repair mechanism would facilitate chromatin bridge resolution? The discovered role for the MRN–ATM–Chk2 pathway in the abscission checkpoint will guide future efforts in addressing these important questions.
Acknowledgments
The authors apologize to colleagues whose work could not be cited or discussed due to space restrictions.
The Lens laboratory is part of Oncode Institute, which is partly financed by the Dutch Cancer Society.
The authors declare no competing financial interests.
References
- 1.Mierzwa, B., and Gerlich D.W. Dev. Cell. 2014 doi: 10.1016/j.devcel.2014.11.006. [DOI] [PubMed] [Google Scholar]
- 2.Janssen, A., et al. Science. 2011 doi: 10.1126/science.1210214. [DOI] [Google Scholar]
- 3.Norden, C., et al. Cell. 2006 doi: 10.1016/j.cell.2006.01.045. [DOI] [Google Scholar]
- 4.Steigemann, P., et al. Cell. 2009 doi: 10.1016/j.cell.2008.12.020. [DOI] [Google Scholar]
- 5.Nähse, V., et al. Trends Cell Biol. 2017 doi: 10.1016/j.tcb.2016.10.001. [DOI] [PubMed] [Google Scholar]
- 6.Petsalaki, E., and Zachos G. J. Cell Biol. 2020 doi: 10.1083/jcb.202008029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kitagawa, M., et al. Cell Reports. 2014 doi: 10.1016/j.celrep.2014.02.034. [DOI] [Google Scholar]
- 8.Serena, M., et al. J. Cell Biol. 2020 doi: 10.1083/jcb.201910059. [DOI] [Google Scholar]
- 9.Adriaans, I.E., et al. Curr. Biol. 2020 doi: 10.1016/j.cub.2020.04.081. [DOI] [PubMed] [Google Scholar]
- 10.Zabolotnaya, E., et al. Biochem. Soc. Trans. 2020 doi: 10.1042/BST20170168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mackay, D.R., and Ullman K.S. Mol. Biol. Cell. 2015 doi: 10.1091/mbc.E14-11-1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Petsalaki, E., and Zachos G. Nat. Commun. 2016 doi: 10.1038/ncomms11451. [DOI] [PMC free article] [PubMed] [Google Scholar]