

Abstract
Adora2B (adenosine receptor 2B) has been reported as one of the key modulators during cardiac remodeling after acute myocardial infarction (AMI). However, the molecular mechanism involved has not been well investigated. Thus, our study aims to investigate whether Adora2B contributes to cardiac remodeling after AMI and its underlying mechanisms. Adenovirus harboring Adora2B or shAdora2B was injected in the border zone in a mouse model of AMI experimentally produced by permanent ligation of left anterior descending (LAD) coronary artery. Decreased Adora2B expression protected the cardiomyocytes from MI-induced autophagic flux obstacle, improved cardiac function, and reduced fibrosis after MI. Adora2B downregulation attenuated the accumulation of LC3-II and p62, which are autophagy substrate proteins. An adenovirus containing mRFP-GFP-LC3 showed that decreased expression of Adora2B restored the autophagic flux by enhancing autophagosome conversion to autophagolysosome. Also, Adora2B knockdown improved cardiomyocytes’ survival and protected mitochondrial function of cardiomyocytes insulted with hypoxia. Notably, the effect of Adora2B on autophagy flux and cardiomyocyte protection could be mitigated by autophagy inhibitor chloroquine. Our results demonstrate that decreased expression of Adora2B protected cardiomyocytes from impaired autophagy flux induced by MI. Modulation Adora2B expression plays a significant role in blunting the worsening of heart function and reducing scar formation, suggesting therapeutic potential by targeting Adora2B in AMI for the infarct healing.
Keywords: Acute myocardial infarction, Adora2B, autophagy flux, apoptosis
Introduction
Acute myocardial infarction (AMI) is one of the most common heart diseases that cause morbidity and mortality worldwide [1,2]. Heart failure (HF) characterized by adverse cardiac remodeling and diminished cardiac performance is a common outcome after AMI. Despite progress in cardiovascular research and evidence-based therapies, there is no effective therapy available to reverse the progression of left ventricle (LV) adverse remodeling. Therefore, developing new therapeutic strategies against AMI and HF is critical for patients’ better survival.
Autophagy is an evolutionarily conserved self-eating-recycling process involved in the digestion of proteins and organelles and is important for maintaining cellular homeostasis [3]. Many evidence has concluded that autophagy homeostasis in the myocardium is vital for preventing the accumulation of macromolecular aggregates and dysfunctional organelles and thus preserving cardiac function, under normal and stress conditions [4]. Disorganized autophagy in cardiomyocytes especially impaired autophagy flux has been reported in the cardiac ischemia-reperfusion injury [5,6], hypertrophic cardiomyopathy [7], dilated cardiomyopathy [8] and HF [9]. The previous data shows that autophagy was rapidly activated after AMI and impaired autophagy flux worsened heart dysfunction after AMI [10]. However, the underlying mechanism and potential of targeting the impaired autophagy flux after AMI have not been fully understood.
Adenosine is a nucleoside that modulates various physiological functions by interacting with its receptors on the cell surface. Within the cardiovascular system, it plays important roles in various physiological processes and in regulating the myocardial remodeling process in response to various stimuli. Adora2B is the most adenosine-insensitive receptor and has been implicated in tissue adaptation to hypoxic or ischemic conditions [11-13]. Activation of Adora2B is coupled to the protein Gs, which subsequently stimulates the activity of adenylate cyclase and increases intracellular cAMP levels [14,15]. Adora2B is the only one of the four adenosine receptors whose cardiac expression is induced by ischemia in both mice and human [16]. In ischemic mice and human hearts, the induction of Adora2B is associated with cardioprotection against ischemic heart damage, but the mechanism underlying this association remains unclear.
In this study, we aimed to see whether Adora2B regulate the autophagy flux after AMI. We observed that Adora2B is increased after AMI and overexpressed of Adora2B aggravate the OGD injury of cardiomyocytes in vitro. Decreased expression of Adora2B confers cardioprotective effects against AMI while Chloroquine (CQ) abolished these effects, which means that Adora2B regulates the autophagy flux and protected hearts after AMI.
Methods
Recombinant adenovirus construction
Adenovirus containing Adora2B, shAdora2B, negative control, and scramble were prepared by the Genechem (Shanghai, China).
Animal model of AMI and adenoviral vector delivery
Male mice aged 10-12 weeks were randomly selected for the Sham group (without ligation of left anterior descending coronary artery (LAD) and induction of MI by permanent ligation of the LAD coronary artery as previously described [17,18]. The mice were anesthetized via intraperitoneal injection of 50 mg/kg sodium pentobarbital and ventilated with the use of a volume-regulated respirator and the body temperature was maintained at 37°C throughout the surgical procedure, and the animals remained in a supervised setting until becoming fully conscious. MI was produced through ligation of the LAD coronary artery with an 8-0 Prolene suture. Immediately after LAD ligation, mice were randomized to be injected with either Ad-shAdora2B or Ad-Scramble. A total of 20 μL Ad-shAdora2B (1×109 v.g./mL) or Ad-Scramble (1×109 v.g./mL) was injected into 3 sites in the peri-infarct zone with the use of a syringe attached to a 31-gauge needle. Chloroquine (50 mg/kg/day) was injected intraperitoneally for 10 consecutive days after MI 5 days.
Echocardiography
Transthoracic echocardiography (Vevo 2100, Visual Sonics) with a 25-MHz imaging transducer was performed on anesthetized animals as previously described [18] with measurement of left ventricular (LV) ejection fraction (LVEF), LV fractional shortening (LVFS), and LV systolic dimension (LVDs). The average values were collected from each of three consecutive cardiac cycles.
Determination of infarct size
Hearts were quickly removed from anesthetized mice and washed with 5 ml cold cardiac arresting buffer (10% KCl), PBS, and 4% PFA solution. Ventricular tissues were embedded in OCT (SAKURA) compound for histological analysis. Transversal sections (5 μm) were prepared at 500 μm intervals. The Masson’s Trichrome staining was performed as previously described [19-21]. Fibrous infarct area was measured as described previously [20,21] and analyzed by a Zeiss inverted microscope (Zeiss, Japan). The Infarcted scar size was calculated as a total scar area divided by the left ventricular area.
NRCMs culture and oxygen-glucose deprivation (OGD)
The neonatal rat cardiomyocytes (NRCMs) were isolated from the neonatal (postnatal day 1) rat hearts and cultured as report [22]. The NRCMs were cultured and subjected to OGD as described previously [22]. Briefly, the cells were rinsed twice with serum-free, glucose and sodium pyruvate free DMEM (Cat#D5030, Sigma-Aldrich, St. Louis, MO) and cultured in the same medium at 37°C in an anoxic chamber (In Vivo 500, Ruskin Life Science) saturated with 94% N2/5% CO2/1% O2 for 12 hours. Incubation Chloroquine (20 μM, Cat#C6628, Sigma-Aldrich, St. Louis, MO) was 2 hours before OGD treatment.
Adenovirus, siRNAs, and transfection
The NRCMs cells were plated in a 60 mm dish 24 hours before transfection. Cells were then transfected with Ad-shAdora2B or Ad-Scramble for 48 hours. The NRCMs were plated in 60 mm plates 24 hours prior to transfection.
CCK8 assay
As described previously [23], the cells were incubated with CCK8 reagent (KeyGEN, China) for 5 hours, followed by measurement of absorbance at 570 nm using a spectrophotometer.
mRFP-GFP-LC3 adenovirus transduction
The adenovirus encoding mRFP-GFP-LC3 was purchased from HanBio Inc (Shanghai, China). Cells were infected with adenoviral particles at 50 MOI. 24 hours after adenovirus transduction, the cells were washed and fixed with 4% paraformaldehyde. Histological images were blindly measured using a Zeiss inverted microscope and processed using ZEN software. Three microscopic fields were quantified for each slice by two independent observers. Image-processing software (Image J) was used as the image quantification software. The number of GFP and mRFP dots was determined by manual counting of fluorescent puncta from at least 4 different myocyte preparations with a 60× objective. At least 40 cells were scored in each experiment.
Quantitative RT-PCR (RT-qPCR)
RT-qPCR was performed as described previously [24]. Briefly, Total RNA was extracted from the LV tissue using an RNeasy Plus Mini Kit (QIAGEN) following the manufacturer’s instructions. cDNA was generated by reverse-transcribed total RNA using ReverTra Ace reverse transcriptase (Toyobo). RT-qPCR was performed using the ABI PRISM 7900 system (Applied Biosystems) with the SYBR Green Real-time PCR Master Mix plus (TOYOBO) for relative quantification of the indicated genes. The transcript of GAPDH was used for internal normalization.
Western blotting
The border zone of the infarct hearts or the cell lysate was separated and homogenized. Equal amounts of protein (20 μg) were separated on SDS-PAGE and electro-transferred to polyvinylidene difluoride membranes (Roche, Switzerland). The membranes were incubated with rabbit anti-Adora2B (Cat#bs-5900R), rabbit anti-p62 (Cat#5114), rabbit anti-LC3 (Cat#3868), rabbit anti-Cleaved Caspase 3 (Cat#9664), goat anti-GAPDH (Cat# ab181602) at 1:1,000 dilution at 4°C overnight, and incubated with either goat anti-rabbit (1:4000 dilution; Abcam) for 1 hour at room temperature. Blots were developed using a chemiluminescent substrate and molecular band intensity was determined by densitometry.
Statistical analysis
All data were analyzed with the statistical software GraphPad Prism 5.0 and were expressed as means ± SEM. The differences between the two groups were analyzed using Student’s unpaired t-test, and differences between three or more groups were evaluated via one-way ANOVA with Bonferroni correction. P<0.05 was considered statistically significant.
Results
Adora2B was induced in the heart infarct border after LAD ligation and in the neonatal rat cardiomyocytes (NRCMs) after oxygen-glucose deprivation (OGD) injury
To illustrate the role of Adora2B in the heart, we examined whether Adora2B is involved in cardiac ischemia. Interestingly, in a mice model of MI, Adora2B protein and mRNA were prominently increased in the infarcted hearts compared with that of the sham-treated hearts. In parallel, the protein of p62 was accumulated (Figure 1A and 1C). However, p62 mRNA was relatively stable (Figure 1C). Additionally, Adora2B protein and mRNA were increased in an in vitro model of ischemic myocardial injury induced by OGD treatment, paralleled with the deposition of p62 protein (Figure 1B and 1D).
Figure 1.

Adora2B was induced in the heart infarct border after LAD ligation and in the neonatal rat cardiomyocytes (NRCMs) after oxygen-glucose deprivation (OGD) injury. A. Representative immunoblotting images and analysis result depicting p62 and Adora2B in the heart infarct border at different time points after LAD ligation. B. Representative western blotting image and analysis depicting p62 and Adora2B in the NRCMs for different time points after OGD treatment. C. Quantitative PCR analysis of the relative abundance of p62 and Adora2B in the heart infarct border at different time points after MI. D. Quantitative PCR analysis of the relative abundance of p62 and Adora2B in the NRCMs for different time points after OGD treatment.
Adora2B critically regulates cardiomyocytes’ survival during OGD
To better understand the role of Adora2B in MI hearts function, we transfected the NRCMs with adenoviral vectors encoding Adora2B (Ad-Adora2B) or the short hairpin RNA targeting Adora2B (Ad-shAdora2B). The western blotting assay was performed to confirm that the Adora2B expression was successfully increased in Ad-Adora2B and Adora2B was knocked down in Ad-shAdora2B (Figure 2A). Adora2B overexpression decreased cardiomyocytes viability after OGD treatment, whereas knockdown of Adora2B depressed OGD-induced cardiomyocytes injury (Figure 2B). Annexin V and propidium iodide (PI) staining assay by flow cytometry showed that Ad-Adora2B cardiomyocytes were more susceptible to apoptosis and necrosis under OGD compare with Ad-NC control (Figure 2C). In contrast, Ad-shAdora2B cardiomyocytes exhibited resistance not only to cell apoptosis but also to cell necrosis under OGD (Figure 2D).
Figure 2.

Adora2B critically regulates cardiomyocytes’ survival during OGD. (A) Western blot assay was performed to confirm that the Adora2B expression. (B) The cell viability was analyzed by CCK8 assay with or without OGD treatment on NRCMs transfected with adenovirus carrying Adora2B or shRNA (*P < 0.05 vs. Ad-Null; #P < 0.05 vs. Si-Scramble, n = 5). (C) The representative image and analysis of flow cytometry showing that Adora2B overexpression attenuated cardiomyocytes injury, whereas Adora2B knockdown (D) aggravated cardiomyocytes injury after OGD treatment. *P < 0.05 vs. Ad-Null; #P < 0.05 vs. Si-Scramble, n = 5.
Adora2B critically regulates cardiomyocytes’ mitochondrial function during OGD
We sought to evaluate whether Adora2B regulates mitochondrial function. Tetramethylrhodamine ethyl ester (TMRE), and MitoSOX red was performed to assess mitochondrial membrane potential and mitochondrial matrix oxidative burden in NRCMs suffered from OGD injury. Relative to Ad-NC, Ad-Adora2B increased mitochondrial oxidant burden (Figure 3A), and reduced mitochondrial membrane potential (Figure 3B). Interestingly, Ad-shAdora2B decreased mitochondrial oxidant burden (Figure 3A), and increased mitochondrial membrane potential compared with Ad-NC (Figure 3B). These data suggest that Adora2B leads to mild mitochondrial respiratory chain dysfunction and an increase in mitochondrial oxidative stress, while decreased expression of Adora2B protected the mitochondrial function of cardiomyocytes after ischemic stress.
Figure 3.
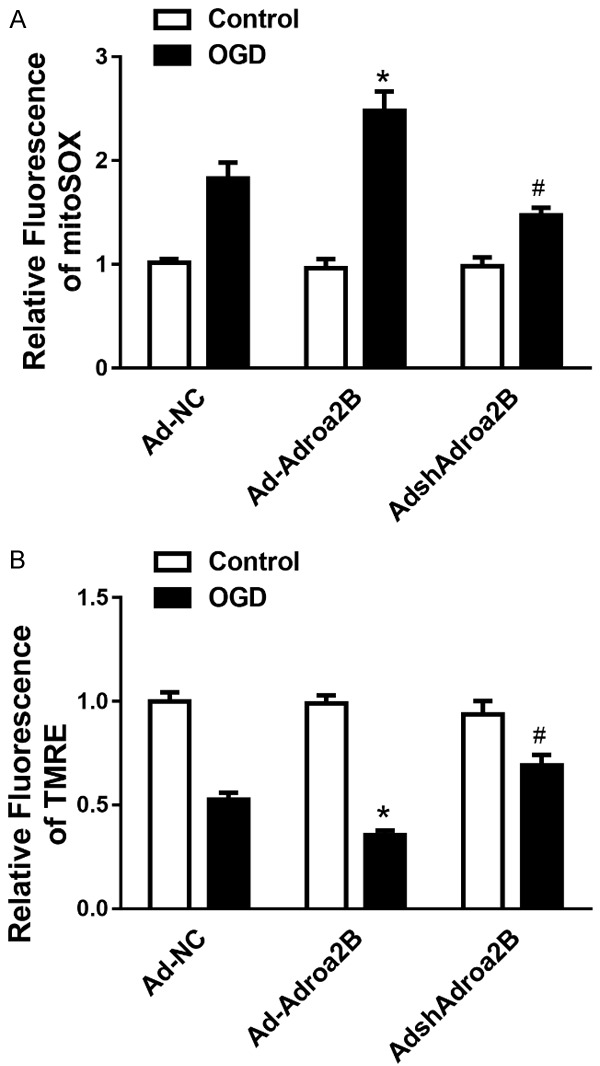
Adora2B critically regulates cardiomyocyte’s mitochondrial function during OGD. Cumulative data of assessing mitochondrial ROS generation (A) and mitochondrial membrane potential (B) in NRCMs suffered from OGD for 12 hours. *P < 0.05 vs. control; #P < 0.05 vs. Ad-scramble-OGD group.
Decreased Adora2B attenuated reverse LV remodeling after MI in mice
To further explore the role of Adora2B in the post-infarct remodeling after AMI, we use adenovirus containing shAdora2B and corresponding scramble sequence were locally injected in the border zone of heart after MI. The echocardiography assessment was performed 4 weeks after MI. Interestingly, the ejection fraction and fractional shortening were significantly increased in the Ad-shAdora2B group compared with Ad-Scramble group. Besides, the LV anterior wall thickness at both systole and diastole was increased in the Ad-shAdora2B group compared with the Ad-Scramble group (Figure 4A, 4B). Massons trichrome staining analysis of the heart infarct size revealed that attenuated cardiac fibrosis in the Ad-shAdora2B group compared with that of the Ad-Scramble group (Figure 4C).
Figure 4.

Decreased Adora2B attenuated reverse LV remodeling after MI in mice. A, B. The representative images and analysis results of echocardiographic assessment of hearts 28 days after MI. C. The representative images and analysis results of Masson staining assessment of the hearts 28 days after MI. *P < 0.05 vs. control; #P < 0.05 vs. Ad-NC-CQ group.
Decreased Adora2B confers myocardial protection by restoring autophagy flux in vitro and in vivo
To view whether decreased Adora2B conferred-cardioprotection was mediated by autophagy regulation, we investigated the expression profile of key protein markers for autophagy, LC3, and p62, in NRCMs after OGD injury. Interestingly, the accumulation of the p62 and LC3II in the NRCMs were alleviated in the Ad-shAdora2B mice compared with the Ad-Scramble group. Remarkably, the lysosomal inhibitor, Chloroquine (CQ) blocked the effect of AdshAdora2B (Figure 5A). Additionally, p62 and LC3II accumulation due to OGD treatment was also alleviated by shAdora2B, which was blunted by CQ (Figure 5A). Furthermore, the autophagy flux rate was measured with live-cell imaging using an adenovirus harboring tandem fluorescent mRFP-GFP-LC3 (Figure 5B). Importantly, shAdora2B restored the autophagy flux after OGD by enhancing autophagosome conversion to autophagolysosome, as indicated by the decrease of yellow puncta (indicating autophagosome) and significant increase of free red puncta (indicating autophagolysosome). Also, the effect of the Adora2B knockdown was mitigated by CQ.
Figure 5.

Decreased Adora2B confers myocardial protection by restoring autophagy flux in vitro. A. The autophagy associated protein marker p62 and LC3 I/II was examined by Western blotting in the heart infarct border 28 days after MI with or without CQ. B. The representative fluorescent image and analysis of the autophagosome and autophagolysosome in the presence or absence of CQ after Ad-shAdora2B transfection in the NRCMs with OGD treatment. (*P < 0.05 vs. OGD + Ad-Null; #P < 0.05 vs. OGD + Ad-shAdora2B, n = 5).
To determine whether the myocardial protective effect of Adora2B efficiency is mediated by improving autophagy flux in vivo, we injected the autophagy flux inhibitor CQ intraperitoneally together with local injection of Ad-shAdora2B in the infracted hearts (Figure 3A, 3B). Both the cardiac dysfunction and infarct size were significantly attenuated in the Ad-shAdora2B group; however, these effects were significantly blunted by CQ treatment.
Decreased Adora2B promotes cardiomyocytes survival against OGD is mediated by restoring autophagy flux
To determine whether decreased Adora2B promotes cardiomyocytes survival against OGD is mediated by restoring autophagy flux, we measured the apoptosis of NRCMs suffered from OGD injury through Annexin V and PI staining and Caspase 3 activation by flow cytometry and WB assays. The analysis of flow cytometry showing that cardio-protection of decreased Adora2B was ameliorated after CQ administration (Figure 6A). The WB analysis of cleaved Caspase-3 expression showing that promoting cardiomyocyte survival of decreased Adora2B was ameliorated after CQ administration (Figure 6B). The above results indicate that decreased Adora2B promotes cardiomyocytes survival against OGD is mediated by restoring autophagy flux.
Figure 6.

Decreased Adora2B promotes cardiomyocytes survival against OGD is mediated by restoring autophagy flux. A. The representative image and analysis of flow cytometry showing that cardio-protection of decreased Adora2B expression was ameliorated after CQ administration. B. The representative immunoblotting image and analysis of cleaved-Caspase 3 expressions showing that cardio-protection of decreased Adora2B expression was ameliorated after CQ administration. (*P < 0.05 vs. CTL; #P < 0.05 vs. OGD + Ad-Null; &P < 0.05 vs. OGD + Ad-shAdora2B, n = 6).
Discussion
It is generally considered that activation of autophagy is cardioprotective, while over-activation of autophagy increased accumulation of autophagic vacuoles which cannot be degraded leads to cell death [25]. After AMI, autophagy was acutely activated, however, sustained ischemia impaired autophagy flux, which aggravated cardiac dysfunction in the latter phase of AMI [10]. To assess the pathophysiological role of autophagy in heart diseases, autophagy flux is much more important than autophagy itself [5]. The autophagy flux rate depends on both autophagosome formation and autophagolysosome degradation [26]. However, the underlying mechanism and potential of targeting the impaired autophagy flux after AMI need to be elucidated.
Based on our analysis of Adora2B in regulating the autophagy flux after AMI, we found that Adora2B is increased after AMI and it aggravates the ischemic injury of cardiomyocytes, while decreased expression of Adora2B confers cardioprotective effects against AMI. CQ abolished the cardioprotective effects of decreased expression of Adora2B, which means that Adora2B regulates the autophagy flux after AMI and protected hearts. This study is the first time to find that decreased expression of adenosine receptor 2B confers cardiac protection against ischemia via restoring autophagy flux.
Adenosine is commonly recognized as playing a crucial role in the synthesis of ATP [27]. In contrast to its intracellular activities, adenosine in the extracellular compartment functions as a signaling factor through activating GPCRs [28]. Importantly, to hearts, adenosine has cardioprotective effects during ischemic cardiac disease [29,30]. Four distinct adenosine receptors have been found, Adora1, -2A, -2B, and -3, which differ in their affinity for adenosine and the biological effects [31,32]. Among these 4 receptors, Adora2B is the most adenosine-insensitive receptor and has been implicated in tissue adaptation to ischemic stimulations [11,33,34]. Activation of Adora2B is coupled to the protein Gs, which subsequently stimulates the activity of adenylate cyclase and increases intracellular cAMP levels [35].
There is an ongoing debate about Adora2B signaling in cardioprotection from ischemia. Adora2B-elicited circadian rhythm protein period 2 (Per2) stabilization confers protective effects via promoting a HIF-dependent metabolic switch during ischemia. Although a recent study indicates that Adora2B agonist treatment as a novel form of cardioprotection [36-38], several other studies implicate Adora2B signaling also could be detrimental during cardiac remodeling after acute myocardial infarction [39], as our findings in this study. Most likely, these differences are due to differences in the animal models, or the timing of treatment [40,41].
Our results indicated that decreased expression of Adora2B protected cardiomyocytes autophagy flux induced by MI. Modulation Adora2B expression plays a significant role in blunting the worsening of heart function and reducing scar formation, suggesting therapeutic potential by targeting Adora2B in AMI for the infarct healing. Together, using both in vivo and in vitro heart ischemic stimulation, we firstly highlighted Adora2B as a crucial factor involved in autophagy flux. To be highlighted, the detailed molecular mechanism of Adora2B in modulating autophagy flux still needs further research.
Acknowledgements
This work was supported by the Young Innovative Science Research Fund of the Second Hospital Affiliated to the Harbin Medical University (Grand No.: KYCX2018-20) and the National Nature Science Foundation of China (Grant No. 81700234).
Disclosure of conflict of interest
None.
References
- 1.Ruparelia N, Godec J, Lee R, Chai JT, Dall’Armellina E, McAndrew D, Digby JE, Forfar JC, Prendergast BD, Kharbanda RK, Banning AP, Neubauer S, Lygate CA, Channon KM, Haining NW, Choudhury RP. Acute myocardial infarction activates distinct inflammation and proliferation pathways in circulating monocytes, prior to recruitment, and identified through conserved transcriptional responses in mice and humans. Eur Heart J. 2015;36:1923–34. doi: 10.1093/eurheartj/ehv195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Frangogiannis NG. Inflammation in cardiac injury, repair and regeneration. Curr Opin Cardiol. 2015;30:240–5. doi: 10.1097/HCO.0000000000000158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 4.Nishida K, Taneike M, Otsu K. The role of autophagic degradation in the heart. J Mol Cell Cardiol. 2015;78:73–79. doi: 10.1016/j.yjmcc.2014.09.029. [DOI] [PubMed] [Google Scholar]
- 5.Ma X, Liu H, Foyil SR, Godar RJ, Weinheimer CJ, Hill JA, Diwan A. Impaired autophagosome clearance contributes to cardiomyocyte death in ischemia/reperfusion injury. Circulation. 2012;125:3170–3181. doi: 10.1161/CIRCULATIONAHA.111.041814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu Q, Li X, Lu Y, Shen L, Zhang J, Cao S, Huang X, Bin J, Liao Y. Pharmacological modulation of autophagy to protect cardiomyocytes according to the time windows of ischaemia/reperfusion. Br J Pharmacol. 2015;172:3072–3085. doi: 10.1111/bph.13111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Saito T, Asai K, Sato S, Hayashi M, Adachi A, Sasaki Y, Takano H, Mizuno K, Shimizu W. Autophagic vacuoles in cardiomyocytes of dilated cardiomyopathy with initially decompensated heart failure predict improved prognosis. Autophagy. 2016;12:579–587. doi: 10.1080/15548627.2016.1145326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schlossarek S, Mearini G, Carrier L. Cardiac myosin-binding protein C in hypertrophic cardiomyopathy: mechanisms and therapeutic opportunities. J Mol Cell Cardiol. 2011;50:613–620. doi: 10.1016/j.yjmcc.2011.01.014. [DOI] [PubMed] [Google Scholar]
- 9.Shirakabe A, Zhai P, Ikeda Y, Saito T, Maejima Y, Hsu CP, Nomura M, Egashira K, Levine B, Sadoshima J. Drp1-dependent mitochondrial autophagy plays a protective role against pressure overload-induced mitochondrial dysfunction and heart failure. Circulation. 2016;133:1249–1263. doi: 10.1161/CIRCULATIONAHA.115.020502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu X, He L, Chen F, He X, Cai Y, Zhang G, Yi Q, He M, Luo J. Impaired autophagy contributes to adverse cardiac remodeling in acute myocardial infarction. PLoS One. 2014;9:e112891. doi: 10.1371/journal.pone.0112891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kong T, Westerman KA, Faigle M, Eltzschig HK, Colgan SP. HIF-dependent induction of adenosine A2B receptor in hypoxia. FASEB J. 2006;20:2242–2250. doi: 10.1096/fj.06-6419com. [DOI] [PubMed] [Google Scholar]
- 12.Eltzschig HK, Thompson LF, Karhausen J, Cotta RJ, Ibla JC, Robson SC, Colgan SP. Endogenous adenosine produced during hypoxia attenuates neutrophil accumulation: coordination by extracellular nucleotide metabolism. Blood. 2004;104:3986–3992. doi: 10.1182/blood-2004-06-2066. [DOI] [PubMed] [Google Scholar]
- 13.Eltzschig HK, Ibla JC, Furuta GT, Leonard MO, Jacobson KA, Enjyoji K, Robson SC, Colgan SP. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J Exp Med. 2003;198:783–796. doi: 10.1084/jem.20030891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grenz A, Homann D, Eltzschig HK. Extracellular adenosine: a safety signal that dampens hypoxia-induced inflammation during ischemia. Antioxid Redox Signal. 2011;15:2221–2234. doi: 10.1089/ars.2010.3665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Eltzschig HK. Adenosine: an old drug newly discovered. Anesthesiology. 2009;111:904–915. doi: 10.1097/ALN.0b013e3181b060f2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yang G, FitzGerald GA. (Almost) everything is illuminated: adenosine shines a light on cardioprotection. Circ Res. 2012;111:965–966. doi: 10.1161/CIRCRESAHA.112.279752. [DOI] [PubMed] [Google Scholar]
- 17.Liu ML, Nagai T, Tokunaga M, Iwanaga K, Matsuura K, Takahashi T, Kanda M, Kondo N, Naito AT, Komuro I, Kobayashi Y. Anti-inflammatory peptides from cardiac progenitors ameliorate dysfunction after myocardial infarction. J Am Heart Assoc. 2014;3:e001101. doi: 10.1161/JAHA.114.001101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dai B, Huang W, Xu M, Millard RW, Gao MH, Hammond HK, Menick DR, Ashraf M, Wang Y. Reduced collagen deposition in infarcted myocardium facilitates induced pluripotent stem cell engraftment and angiomyogenesis for improvement of left ventricular function. J Am Coll Cardiol. 2011;58:2118–27. doi: 10.1016/j.jacc.2011.06.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.de Couto G, Liu W, Tseliou E, Sun B, Makkar N, Kanazawa H, Arditi M, Marbán E. Macrophages mediate cardioprotective cellular postconditioning in acute myocardial infarction. J Clin Invest. 2015;125:3147–62. doi: 10.1172/JCI81321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gálvez-Montón C, Fernandez-Figueras MT, Martí M, Soler-Botija C, Roura S, Perea-Gil I, Prat-Vidal C, Llucià-Valldeperas A, Raya Á, Bayes-Genis A. Neoinnervation and neovascularization of acellular pericardial-derived scaffolds in myocardial infarcts. Stem Cell Res Ther. 2015;6:108. doi: 10.1186/s13287-015-0101-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quijada P, Salunga HT, Hariharan N, Cubillo JD, El-Sayed FG, Moshref M, Bala KM, Emathinger JM, De La Torre A, Ormachea L, Alvarez R Jr, Gude NA, Sussman MA. Cardiac stem cell hybrids enhance myocardial repair. Circ Res. 2015;117:695–706. doi: 10.1161/CIRCRESAHA.115.306838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu FP, Chen MS, Wang YZ, Yi Q, Lin SB, Chen AF, Luo JD. Leptin induces hypertrophy via endothelin-1-reactive oxygen species pathway in cultured neonatal rat cardiomyocytes. Circulation. 2004;110:1269–1275. doi: 10.1161/01.CIR.0000140766.52771.6D. [DOI] [PubMed] [Google Scholar]
- 23.Huang Z, Han Z, Ye B, Dai Z, Shan P, Lu Z, Dai K, Wang C, Huang W. Berberine alleviates cardiac ischemia/reperfusion injury by inhibiting excessive autophagy in cardiomyocytes. Eur J Pharmacol. 2015;762:1–10. doi: 10.1016/j.ejphar.2015.05.028. [DOI] [PubMed] [Google Scholar]
- 24.Singh MK, Li Y, Li S, Cobb RM, Zhou D, Lu MM, Epstein JA, Morrisey EE, Gruber PJ. Gata4 and Gata5 cooperatively regulate cardiac myocyte proliferation in mice. J Biol Chem. 2010;285:1765–1772. doi: 10.1074/jbc.M109.038539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schiattarella GG, Hill JA. Therapeutic targeting of autophagy in cardiovascular disease. J Mol Cell Cardiol. 2016;95:86–93. doi: 10.1016/j.yjmcc.2015.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gottlieb RA, Andres AM, Sin J, Taylor DP. Untangling autophagy measurements: all fluxed up. Circ Res. 2015;116:504–14. doi: 10.1161/CIRCRESAHA.116.303787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khakh BS, Burnstock G. The double life of ATP. Sci Am. 2009;301:84–90. 92. doi: 10.1038/scientificamerican1209-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Colgan SP, Eltzschig HK. Adenosine and hypoxia-inducible factor signaling in intestinal injury and recovery. Annu Rev Physiol. 2012;74:153–175. doi: 10.1146/annurev-physiol-020911-153230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hori M, Kitakaze M. Adenosine, the heart, and coronary circulation. Hypertension. 1991;18:565–74. doi: 10.1161/01.hyp.18.5.565. [DOI] [PubMed] [Google Scholar]
- 30.Kitakaze M, Hori M, Morioka T, Minamino T, Takashima S, Sato H, Shinozaki Y, Chujo M, Mori H, Inoue M, et al. Infarct size-limiting effect of ischemic preconditioning is blunted by inhibition of 5’-nucleotidase activity and attenuation of adenosine release. Circulation. 1994;89:1237–1246. doi: 10.1161/01.cir.89.3.1237. [DOI] [PubMed] [Google Scholar]
- 31.Bauerle JD, Grenz A, Kim JH, Lee HT, Eltzschig HK. Adenosine generation and signaling during acute kidney injury. J Am Soc Nephrol. 2011;22:14–20. doi: 10.1681/ASN.2009121217. [DOI] [PubMed] [Google Scholar]
- 32.Eltzschig HK. Adenosine: an old drug newly discovered. Anesthesiology. 2009;111:904–15. doi: 10.1097/ALN.0b013e3181b060f2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eltzschig HK, Thompson LF, Karhausen J, Cotta RJ, Ibla JC, Robson SC, Colgan SP. Endogenous adenosine produced during hypoxia attenuates neutrophil accumulation: coordination by extracellular nucleotide metabolism. Blood. 2004;104:3986–92. doi: 10.1182/blood-2004-06-2066. [DOI] [PubMed] [Google Scholar]
- 34.Synnestvedt K, Furuta GT, Comerford KM, Louis N, Karhausen J, Eltzschig HK, Hansen KR, Thompson LF, Colgan SP. Ecto-5’-nucleotidase (CD73) regulation by hypoxia-inducible factor-1 mediates permeability changes in intestinal epithelia. J Clin Invest. 2002;110:993–1002. doi: 10.1172/JCI15337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Eltzschig HK, Ibla JC, Furuta GT, Leonard MO, Jacobson KA, Enjyoji K, Robson SC, Colgan SP. Coordinated adenine nucleotide phosphohydrolysis and nucleoside signaling in posthypoxic endothelium: role of ectonucleotidases and adenosine A2B receptors. J Exp Med. 2003;198:783–96. doi: 10.1084/jem.20030891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eckle T, Hartmann K, Bonney S, Reithel S, Mittelbronn M, Walker LA, Lowes BD, Han J, Borchers CH, Buttrick PM, Kominsky DJ, Colgan SP, Eltzschig HK. Adora2b-elicited Per2 stabilization promotes a HIF-dependent metabolic switch crucial for myocardial adaptation to ischemia. Nat Med. 2012;18:774–82. doi: 10.1038/nm.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maas JE, Wan TC, Figler RA, Gross GJ, Auchampach JA. Evidence that the acute phase of ischemic preconditioning does not require signaling by the A2B adenosine receptor. J Mol Cell Cardiol. 2010;49:886–893. doi: 10.1016/j.yjmcc.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Koeppen M, Harter PN, Bonney S, Bonney M, Reithel S, Zachskorn C, Mittelbronn M, Eckle T. Adora2b signaling on bone marrow derived cells dampens myocardial ischemia-reperfusion injury. Anesthesiology. 2012;116:1245–1257. doi: 10.1097/ALN.0b013e318255793c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maas JE, Wan TC, Figler RA, Gross GJ, Auchampach JA. Evidence that the acute phase of ischemic preconditioning does not require signaling by the A 2B adenosine receptor. J Mol Cell Cardiol. 2010;49:886–93. doi: 10.1016/j.yjmcc.2010.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Eckle T, Koeppen M, Eltzschig H. Use of a hanging weight system for coronary artery occlusion in mice. J Vis Exp. 2011:2526. doi: 10.3791/2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eckle T, Grenz A, Köhler D, Redel A, Falk M, Rolauffs B, Osswald H, Kehl F, Eltzschig HK. Systematic evaluation of a novel model for cardiac ischemic preconditioning in mice. Am J Physiol Heart Circ Physiol. 2006;291:H2533–H2540. doi: 10.1152/ajpheart.00472.2006. [DOI] [PubMed] [Google Scholar]