

Abstract
Osteoarthritis (OA) is the most common skeletal disease and the leading cause of pain and disability in the aged population (>65 years). However, the underlying factors involved in OA pathogenesis remain elusive which has resulted in failure to identify disease-modifying OA drugs. Altered metabolism has been shown to be a prominent pathological change in OA. As a critical bioenergy sensor, AMP-activated protein kinase (AMPK) mediates not only energy homeostasis but also redox balance in chondrocytes to counter various cell stress. Dysfunction of AMPK activity has been associated with reduced autophagy, impaired mitochondrial function, excessive reactive oxygen species generation, and inflammation in joint tissue. These abnormalities ultimately trigger articular cartilage degeneration, synovial inflammation, and abnormal subchondral bone remodeling. This review focuses on recent findings describing the central role of AMPK in joint homeostasis and OA development. We also highlight current advances that target AMPK as a novel therapeutic strategy for OA prevention.
Keywords: AMPK, Osteoarthritis, metabolism, autophagy, inflammation
Introduction
Pathogenesis and development of OA
Osteoarthritis (OA) is the most common skeletal disease and is viewed as the leading cause of joint pain and disability in the aged population (>65 years). The prevalence of OA reached 240 million cases around the world in 2015 [1]. The overall burden of OA has become a challenge to the modern healthcare system as it results in loss of mobility and severe pain in its advanced stage. Currently, OA is viewed as a highly heterogeneous disorder with a number of different phenotypes, all of which share distinct underlying pathogeneses and functional consequences. Aging, mechanical loading, obesity, metabolic syndromes, and genetic mutations appear to increase the risk of OA development. Prominent features of OA pathology involve the whole synovial joint in the form of cartilage degeneration, subchondral bone sclerosis, and osteophyte formation which result in progressive pain and disability of the joint [2]. Unfortunately, there are no disease-modifying drugs available to reverse OA progression due to its elusive etiology and lack of specific biological markers for early OA-associated changes.
The role of metabolic dysfunction in OA
Recently, emerging evidence has shown that metabolic alterations play a key role in OA pathogenesis as evidenced by an increased risk of incidence and severity of OA in patients with metabolic syndromes such as obesity, insulin resistance, and hyperlipidemia [3]. Interestingly, obesity was associated with both weight-bearing joint OA and elevated incidence of non-weight bearing joint OA (hands and wrists) which suggests that it not only increases the risk of OA as a mechanical factor as is conventionally believed, but also as a biological element. The biological effect of obesity on OA development seems to be associated with the elevated levels of adipokines which have been demonstrated to drive catabolic response in chondrocytes [4]. Since metabolic stress appears to be an underlying component in the pathogenesis of OA, understanding the mechanism and signaling pathway involved would yield a novel therapeutic strategy to slow the destruction of joint tissues and halt functional impairment. AMP-activated protein kinase (AMPK), a master energy sensor, has been implicated in multiple aging-related diseases correlated with cellular energy imbalance such as type 2 diabetes mellitus, cancer, cardiovascular disease, Alzheimer’s disease as well as OA [5]. Additionally, dysfunction of AMPK is correlated with impaired autophagy, abnormal endoplasmic reticulum (ER) stress, and increased oxidative stress. Moreover, AMPK activation has an anti-inflammatory impact on chondrocytes in response to cellular stress by modulating the NF-κB pathway [6]. Recently, the critical roles of AMPK in chondrocyte energy homeostasis and inflammation regulation have made it a potential target for OA disease-modifying drugs. To improve understanding of how abnormal energy metabolism contributes to OA development, we highlight new insights into the roles of the AMPK regulatory network in joint tissue homeostasis and OA pathogenesis. An overview of therapeutic approaches by targeting AMPK and its regulatory network are also discussed.
Structure and molecular regulation of AMPK
Structure of AMPK
AMPK, a serine/threonine-protein kinase, is a highly conserved energy status sensor that responds to cellular ATP level changes in eukaryotic cells. Emerging as a heterotrimeric complex, AMPK is composed of a catalytic α subunit and two regulatory β- and γ-subunits encoded by distinct genes. In mammals, expression of α1, α2, β1, β2, γ1, γ2 and γ3 isoforms of AMPK has been revealed and they can form up to 12 different αβγ complexes. Articular cartilage expresses α1, α2, β1, β2, and γ1 subunits [7]. In general, AMPK activity is modulated by three mechanisms: (i) activation by upstream kinase-induced phosphorylation, (ii) dephosphorylation and inactivation by protein phosphatase, and (iii) allosteric kinase-dependent activation.
Regulation and output of AMPK activation
Phosphorylation of threonine 172 (T172) at the catalytic α subunit by several upstream proteins such as calcium/calmodulin-dependent protein kinase kinase (CaMKK) β, transforming growth factor-β-activated kinase 1 (TAK1), and liver kinase B1 (LKB1) is necessary for activation of AMPK [8]. AMPK is highly sensitive to rises in the cellular ratio of AMP to ATP resulting from metabolic stress. In this scenario, AMP and ADP binding to the γ subunit triggers an allosteric change in AMPK which induces the phosphorylation of T172 in the kinase activation domain. This mechanism dominates in mild energetic stress to restore energy homeostasis by promoting ATP-producing pathways and suppressing ATP-consuming processes. In addition, increased intracellular calcium serves as a second message to phosphorylate T172 and thus activate AMPK, which is dependent on CaMKK β activation (Figure 1). To avoid persistent activation, AMPK activity is inhibited by protein phosphatases including PP2A, PP2Cα, and Ppm1E which dephosphorylate the T172 site [9]. In addition to activation by an intracellular AMP/ADP ratio change, AMPK also responds to physical exercise, caloric restriction, local extracellular cues and circulating hormones. Recently, several AMPK agonists such as 5-aminoimidzole-4-carboxamide riboside (AICAR) and A-769662 have been identified to specifically bind to the β1 subunit [8]. The main outputs of AMPK activation are to (1) promote the activity of rate-limiting metabolic enzymes that increase the uptake and metabolism of glucose and fatty acids and (2) suppress the efficacy of anabolic enzymes associated with cholesterol, protein and glycogen synthesis, restoring energy homeostasis [8].
Figure 1.
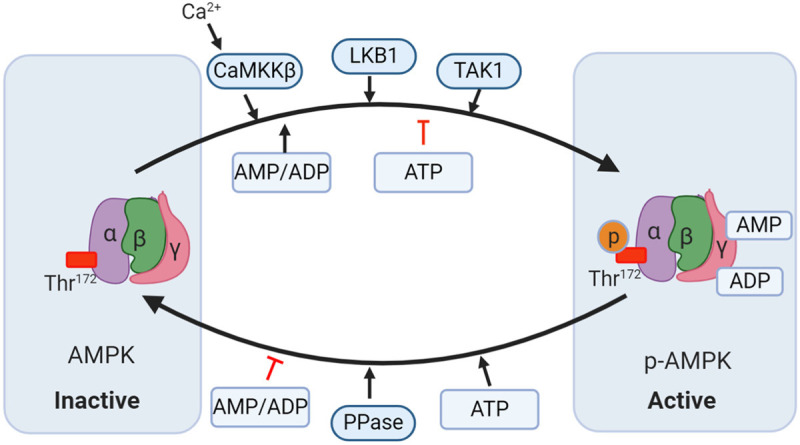
Mechanisms modulating AMP-activated protein kinase (AMPK) activity. AMPK consists of α, β, and γ subunits. There are two distinct ways to trigger AMPK activation. i): AMP and ADP bind to the γ subunits and allosterically activate AMPK; ii) Phosphorylation of Thr172 in the α subunit by upstream kinases such as liver kinase B1 (LKB1), calcium-calmodulin-dependent kinase 2 (CaMKK2), and transforming growth factor-β-activated protein kinase-1 (TAK1) induces the activation of AMPK. AMPK is dephosphorylated by protein phosphatase (PP) thereby being inhibited. Moreover, ATP can also oppose the activation of AMPK by inhibiting the γ subunits that are dependent on allosteric activation and phosphorylating the LKB1-induced α subunit.
Apart from the maintenance of energy homeostasis, AMPK also contributes to several housekeeping processes. For instance, activation of AMPK alleviates inflammatory and oxidative stress by impacting several signaling pathways such as the NF-κB signaling pathway and the nuclear factor erythroid-derived 2 (Nrf2) signaling pathway [10,11]. Moreover, AMPK activation mitigates ER stress and favors DNA damage repair and autophagosome formation which protects cells from stress-induced phenotype changes and apoptosis [12,13]. As AMPK is integrated with essential signaling networks associated with the aging process and metabolism dysfunction for cell survival, it has been targeted by a variety of drugs prescribed for diabetes mellitus and other disorders (e.g. metformin, statins, methotrexate, phytochemicals, and sodium salicylate). Collectively, targeting AMPK would be a novel approach to prevent aging-related disorders and metabolic syndromes as well as OA.
AMPK dysfunction occurs during OA
Recent studies have confirmed that aberrant AMPK activity is implicated in OA. Robust phosphorylation of AMPKα was identified in normal articular chondrocytes. However, compared with normal control tissue, the articular cartilage from an OA human knee and joint tissue from surgically induced and aging-related OA mouse knees exhibited a striking decrease in phosphorylation of AMPKα at T172 [14]. In addition, AMPK activity was dramatically inhibited in chondrocytes treated with inflammatory cytokines and mechanical injury. Using siRNA silencing, Terkeltaub et al reported that depletion of AMPK in chondrocytes aggravated a catabolic response to IL-1β and TNF-α [15]. These data suggest that AMPK activity is indispensable to maintain cartilage homeostasis and chondrocyte phenotype preservation. Additionally, attempts to reverse AMPK dysfunction diminished inflammation and prevented OA progression. For example, chondrocyte catabolic responses to inflammatory cytokines were dramatically suppressed by AICAR, a selective AMPK agonist [15]. In vivo, Li et al demonstrated intra-articular injection of metformin significantly ameliorated the severity of knee joint destruction in mice with destabilization of the medial meniscus. Moreover, this group found that protective effect of metformin against OA progression was blunted by transgenic deletion of AMPKα, indicating AMPKα was the target for the metformin-mediated protection against OA [16].
It has been demonstrated that biomechanical injury, inflammatory cytokines, and aging, the key elements of OA development, are able to inhibit the phosphorylation of AMPK. Elevated inflammatory cytokines (e.g. TNF-α, IL-1β, and IL-6) in OA have been shown to increase the activity of protein phosphatases (e.g. PP2A, PP1, PP2B, and PP2C) leading to dephosphorylation and inhibition of AMPK in chondrocytes [15]. Moreover, the phosphorylation of LKB1 was reduced in aging-related and surgically-induced OA cartilage resulting in a decrease in AMPK activity [14]. Sirtuin 1 (SIRT1), an NAD+ dependent protein deacetylase downstream of AMPK signaling, is able to increase AMPK activity through LKB1 deacetylation. The positive feedback loop between AMPK and SIRT1 is disrupted in human and mouse knee OA chondrocytes due to reduced expression and activity of SIRT1, which leads to a decline in AMPK phosphorylation indirectly through decreased LKB1 activity [17]. Taken together, these studies indicate that reduced responsiveness of AMPK in articular cartilage caused by aging, mechanical injury, and chronic inflammation compromises the joint tissue’s ability to repair and regenerate itself, accelerating OA progression. Notably, the function of AMPK is not restricted to energy metabolism homeostasis control - it can also govern several critical cellular house-keeping processes (Figure 2). Therefore, understanding the underlying mechanisms and outputs of AMPK dysfunction will yield a promising approach to repress OA development.
Figure 2.
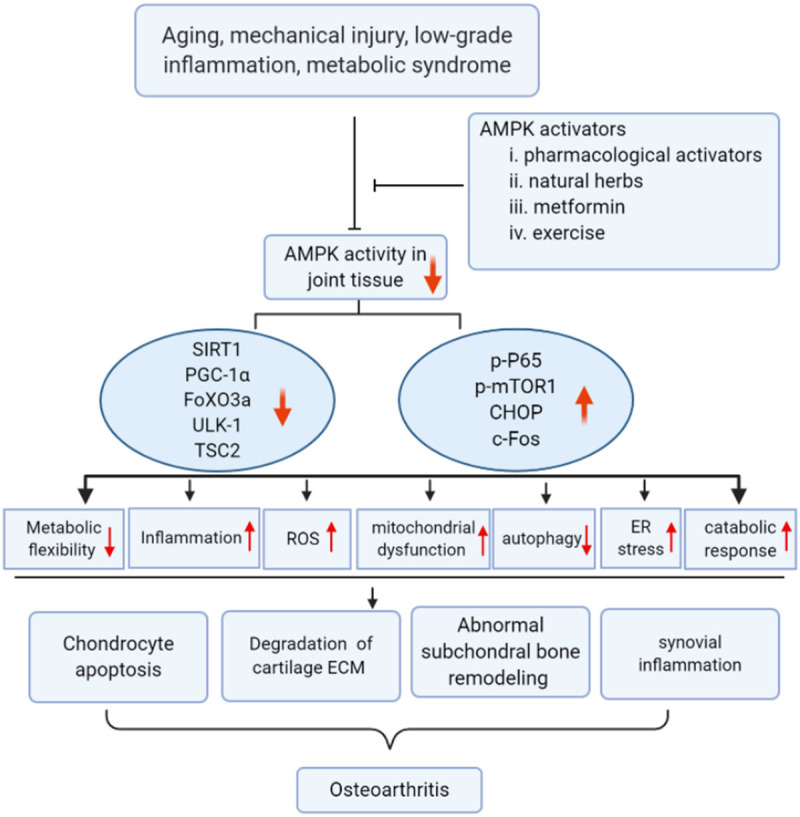
Dysfunction of AMPK activity disrupts cartilage, synovium and subchondral bone homeostasis, thus contributing to OA development. Aging, low-grade inflammation, mechanical injury, and metabolic syndromes result in compromised AMPK activity. Impaired AMPK activity decreases the level and activity of SIRT1, PGC-1α, FoXO3a, TSC2, and ULK-1 which causes mitochondrial dysfunction, aggravates oxidative stress, attenuates autophagy, and increases inflammation-mediated cartilage catabolism. On the contrary, expression of p-P65, p-mTOR1, CHOP, and c-Fos are up-regulated which promotes several signaling pathways, including NF-κB, ER stress, and osteoclastogenesis, resulting in chondrocyte apoptosis, synovial inflammation, and abnormal subchondral bone remodeling. Targeting impaired AMPK activity (e.g. AMPK pharmacological activators, natural herbs, metformin, and appropriate exercise) would be a potential approach to prevent OA.
The results of AMPK dysfunction in OA chondrocytes
Mitochondrial function, altered metabolism, and oxidative stress
As the powerhouse of the cell, the mitochondria is responsible for producing ATP by oxidative phosphorylation for a number of activities including proliferation, differentiation, signaling regulation, and movement [18]. During oxidative phosphorylation, reactive oxygen species are generated by the mitochondria as byproducts. Mitochondrial dysfunction results in a deficient supply of energy but excessive production of reactive oxygen species (ROS) which is viewed as a hallmark in aging-related diseases including OA. Augmented ROS production in turn aggravates mitochondrial dysfunction to form a vicious feedback loop, inciting an inflammatory response and degeneration. Impaired mitochondrial function has been linked to pathological changes in cartilage damage such as enhanced apoptosis, defective ECM synthesis, elevated catabolic response, and vulnerability to stress [19]. Moreover, chondrocytes with compromised mitochondrial function were more permissive of inflammation.
It has been demonstrated that OA chondrocytes can switch their metabolism from oxidative phosphorylation to glycolysis, accompanied by an increase in the production of inflammatory mediators and catabolic factors [20]. Mitochondrial dysfunction has been linked to aberrant AMPK activity during OA progression [14]. In healthy chondrocytes, AMPK activation promotes glycolysis by phosphorylation of PFK-2 and enhances glucose uptake by increasing expression of glucose transporter 1, which provides energy and metabolites to repair and regenerate the damaged joint tissue. However, in the absence of AMPK activation, OA chondrocytes compromise their adaptive ability to tolerate these stresses [21]. Yun et al uncovered that the impaired mitochondrial biogenesis capacity in OA chondrocytes was associated with declined AMPK phosphorylation and downstream mediators such as SIRT1 and peroxisome proliferator-activated receptor γ co-activator 1α (PGC1α). Moreover, that group also found A-769662, a pharmacological AMPK activator, strikingly promoted the expression of PGC1α and rescued the deficient mitochondrial biogenesis in OA chondrocytes by SIRT1 signaling [22].
ROS, another notorious element of OA development, has also been connected with AMPK signaling. Activation of the NF-κB pathway plays a critical role in ROS-induced cartilage matrix degradation, hypertrophic shift, inflammatory response, and chondrocyte apoptosis [23]. Observations have reported that AMPK has an inhibitory role in NF-κB activation via SIRT1 deacetylation of the p65 subunit, thereby suppressing oxidative stress in articular chondrocytes. Furthermore, using a ROS inducing model, a recent report showed that an AMPK pharmacological activator enhanced antioxidant enzymes like superoxide dismutase 2 (SOD2) and catalase to limit the production of superoxide in human chondrocytes challenged with menadione through PGC1α and forkhead box O 3 (FoxO3) [24]. Similarly, an increase in AMPK activity by berberine chloride treatment inhibited sodium nitroprusside-induced iNOS expression and protected chondrocytes from apoptosis in vitro [25]. Additionally, occurrence of a mitochondrial DNA (mtDNA) 4977-bp deletion mutant that compromised the ability of mtDNA to encode proteins for the respiratory chain was detected in OA chondrocytes. Moreover, a pharmacological AMPK agonist significantly eradicated the mtDNA 4977-bp deletion by promoting SIRT3 and oxoguanine glycosylase activity, greatly improving mitochondrial function [26]. Using an age-related spontaneous OA mouse model, Chen et al found that systematic administration of berberine chloride, an AMPK agonist, substantially alleviated the severity of knee destruction by restoring aging-associated reduction of the AMPK-SIRT3 axis [26]. These data suggested that decreased AMPK activity is associated with mitochondrial dysfunction, metabolism shift, and redox imbalance which, in turn, disrupts homeostasis of articular cartilage and contributes to OA.
Inflammation and catabolic response
Recent studies have shifted the paradigm of OA from a “wear and tear” disorder to a low-grade inflammatory disease. The link between synovitis and cartilage damage has been identified in many reports. During OA progression, increased levels of pro-inflammatory cytokines such as IL-1β, TNF-α, and IL-6 are secreted from chondrocytes and synovial cells, provoking chondrocyte apoptosis and catabolic response which accelerate joint destruction and cause pain [27]. Production of pro-inflammatory cytokines is governed by the NF-κB pathway in chondrocytes. AMPK has been demonstrated to negatively regulate the NF-κB pathway directly or indirectly.
SIRT1, a primary downstream effector of AMPK, can deacetylate the p65 subunit and then induce proteasome degradation of p65, ultimately inactivating NF-κB signaling [28]. Activation of AMPK is caused by Protectin DX inhibited IL-1β-stimulated expression of NO, PGE2, and iNOS in rat primary chondrocytes via attenuation of the NF-κB pathway [29]. Consistently, using an adjuvant-induced rat arthritis model, Wang et al showed that activation of AMPK by glycolysis reduced synovial inflammation and alleviated joint damage, which mainly relied on inhibition of the NF-κB pathway. Furthermore, depletion of AMPK or SIRT1 via siRNA silencing in chondrocytes aggravated catabolic response to IL-1β and TNF-α treatment [30]. Compared to wild wild-type controls, mice with deletion of AMPKα developed more severe surgically induced and aging-related OA lesion-like changes, evidenced by enhanced expression of MMPs, phosphorylated p65, and apoptosis markers. In vitro, chondrocytes with an AMPKα deficiency demonstrated an increase in IL-1β-induced MMP13 expression [31]. A similar result was also observed after the administration of advanced glycation end products (AGEs). Loss of AMPK and SIRT1 activity was identified in primary human chondrocytes treated with advanced AGEs. In this study, Pioglitazone rescued AMPK activity and significantly abolished AGEs-induced inflammatory cytokine expression [32]. These data suggested that lack of AMPK activity in chondrocytes amplifies inflammatory cytokine production and catabolic response, thus triggering OA development.
Autophagy
Autophagy is a conservative housekeeping mechanism that targets dysfunctional cellular organelles and removes damaged molecular aggregates, which is an essential component of cellular homeostasis [33]. As a survival signaling pathway, autophagy exists in articular cartilage and is associated with normal chondrocyte phenotype preservation. It has been demonstrated that expression of key autophagy-related proteins are substantially reduced in degenerated cartilage [34]. There is consensus that compromised autophagy results in enhanced generation of catabolic proteases and increased risk of OA development. Moreover, activation of autophagy by inhibiting mammalian target of rapamycin complex 1 (mTORC1) alleviated the severity of articular cartilage damage and synovitis in a surgically-induced knee OA mouse model [35].
It has been demonstrated that AMPK serves as a potent activator of autophagy to protect chondrocytes from cellular stress. For instance, in the nutrient-poor context, active AMPK phosphorylates raptor, an essential component for mTORC1 activation, and then directly inhibits mTORC1, which induces autophagy [36]. Furthermore, AMPK can also phosphorylate tuberous sclerosis protein 2 (TSC2) which, in turn, inactivates small GTPases like Rheb and represses mTORC1 signaling, stimulating autophagy [37]. Moreover, ULK1, an essential initiator of autophagy, is phosphorylated and activated by AMPK [38]. In addition, SIRT1 and FoxO3a, signaling molecules downstream of AMPK, have been demonstrated to trigger autophagosome formation either by deacetylating autophagy-related proteins (e.g., Atg5, Atg7, and Atg8) or by inducing the expression of autophagy-associated proteins (e.g., LC3B, Gabarapl1, and Beclin1) [39,40]. Recently, chondrocytes with depletion of AMPK by RNA interference showed reduced autophagy in response to increased intracellular calcium influx [41]. Using the destabilized medial meniscus (DMM)-induced knee OA mouse model, Qin et al showed restoration of AMPK activity by intra-articular injection of resveratrol promoted autophagy through suppression of mTOR which inhibited catabolic signals and ameliorated joint destruction [42]. Collectively, these results indicate that alteration of AMPK activity in OA chondrocytes can suppress the autophagy response to cellular stress which leads to chondrocyte apoptosis and OA initiation.
ER stress
The endoplasmic reticulum (ER) governs the correct folding and post-translational modification of all secretory and integral membrane proteins, which is essential for protein quality control and normal cellular function. When ER homeostasis is disrupted by various cellular stresses such as an imbalanced redox state, a perturbation in calcium homeostasis, a depletion of energy stores or an inflammatory stimulus, the aggregation of misfolded or unfolded proteins in the ER lumen triggers an unfolded protein response (UPR) [43]. UPR aims to restore the equilibrium of stressed ER through (1) promoting chaperone proteins to facilitate correct folding; (2) attenuating new protein translation; and (3) inducing apoptosis when ER stress is too prolonged to be resolved [44]. In the UPR, three-armed signaling cascades are initiated by ER transmembrane proteins protein kinase R (PKR)-like ER kinase (PERK), activating transcription factor 6 (ATF6), and dissociation of the chaperone protein glucose-regulated protein (GRP78) from ER stress sensor inositol-requiring enzyme 1 (IRE1) [45].
Increasing evidence has demonstrated that ER stress is involved in OA development. For instance, the activation of ER stress was confirmed in OA cartilage with a substantial increase in expression of X-box binding protein 1 (XBP1), GRP78, and C/EBP (CHOP). Mice with CHOP deletion exhibited more severe cartilage damage and chondrocyte apoptosis in a surgically-induced knee OA mouse model suggesting ER stress-induced expression of CHOP contributed to OA pathogenesis [46]. The expression of p-PERK and CHOP, markers of ER stress, were positively associated with the severity of cartilage degeneration in human knee OA [47]. In addition, prolonged ER stress triggered by a biomechanical injury, nitric oxide, and advanced glycation end products (AGEs) resulted in substantially increased chondrocyte apoptosis [48,49]. ER stress also induced cartilage extracellular matrix degradation which appeared to be mediated through increasing the expression of MMP13 [50].
It has been demonstrated that AMPK plays a pivotal role in ER stress regulation to slow OA progression. For instance, AICAR, an AMPK pharmacological activator, was shown to attenuate ER stress-mediated apoptotic signals via repression of CHOP expression in chondrocytes challenged by IL-1β or mechanical injury [13]. Moreover, Kai et al described that treatment with quercetin, a member of the flavonoid family, can increase the level of active AMPK and suppress ER stress-induced apoptosis in chondrocytes treated by tert-butyl hydroperoxide. In vivo, intra-articular injection of quercetin markedly promoted phosphorylated AMPK level in knee cartilage but inhibited ER stress marker expression thereby ameliorating cartilage damage and reducing chondrocyte apoptosis in a DMM-induced knee OA rat model [51]. Collectively, these studies suggested that AMPK can target the ER stress-induced apoptotic signal to exert its chondroprotective effect.
The role of dysfunctional AMPK in synovitis and subchondral bone sclerosis
Since OA pathogenesis not only involves articular cartilage destruction but also includes synovial inflammation and subchondral bone sclerosis, understanding the association between AMPK dysfunction and subchondral bone sclerosis as well as synovial inflammation would yield an effective connection for developing disease-modifying OA drugs. It has been reported that abnormal AMPK activity was linked to synovial pathological changes in OA. For example, mechanical stretching of synovial fibroblasts (SF) can trigger phosphorylation of AMPKα at the T172 site to counter the TNF-α-induced inflammatory response, suggesting that AMPK activation alleviates synovitis in an exercise that was generally prescribed for OA treatment [52]. Similarly, the increase in AMPK activity induced by chitosan oligosaccharide, an oligomer of D-glucosamine, has been shown to attenuate TNF-α-mediated NF-κB signaling activation and pro-inflammatory cytokine generation in both primary rabbit and human synoviocytes. Using an anterior cruciate ligament transection-induced knee OA rabbit model, the same group also demonstrated that systematic administration of chitosan oligosaccharide significantly ameliorated the degree of synovitis in vivo [53]. In addition, transforming growth factor-beta 1 (TGF-β1), a well-known driving force in OA development, can activate AMPK to promote FoXO3 expression which, in turn, suppresses synthesis of inflammatory mediators in human knee OA synovial fibroblasts [54]. Therefore, these data indicated that AMPK activation had an anti-inflammatory effect on synovium, but the detailed mechanism still remains elusive. Further research should be carried out to investigate how dysfunctional AMPK activity contributes to synovitis during OA development.
Observation has demonstrated that AMPK is essential to control the balance of bone metabolism. Activation of AMPK favored human mesenchymal stem cell (MSC) differentiation into osteoblasts at the expense of adipogenic differentiation whereas silencing AMPKα attenuated the osteogenic differentiation of human MSCs [55]. Both AICAR and metformin, two well-known AMPK agonists, have been shown to improve osteogenic differentiation of MC3T3-E1 which appeared to be mediated by activation of the ERK pathway [56,57]. Consistently, mice with double deletion of AMPKα1 and α2 developed enhanced bone resorption and a high bone turnover phenotype which resulted in decreased bone mass and density compared with wild-type controls [58]. In addition to osteogenic differentiation, evidence also has indicated that AMPK is inversely associated with osteoclastogenesis. Inhibition of AMPKα in bone marrow macrophages augmented osteoclastogenesis and enhanced receptor activator of nuclear factor kappa-B ligand (RANKL) signaling, suggesting a critical role of AMPK in osteoclast differentiation [59]. Furthermore, AICAR treatment was found to inhibit the RANKL-mediated osteoclast differentiation by induction of autophagy [60].
Mechanistically, AMPK served as a negative regulator in the expression of c-Fos and NFAc1 thereby inhibiting osteoclast differentiation [61]. Similarly, Peng et al found that Carnosic acid functioned as an AMPK agonist and substantially suppressed expression of osteoclastogenesis related genes in osteoclast precursors in vitro. Moreover, the inhibitory effect of AMPK on osteoclast formation was further identified using a lipopolysaccharides-induced calvarial osteolysis mouse model [62]. Taken together, AMPK emerged as a key mechanism to regulate bone homeostasis. However, the effect of AMPK on subchondral bone remodeling has not been elucidated yet which would be a promising topic to identify an effective therapeutic approach for OA.
Therapeutic prospects targeting AMPK for osteoarthritis
To date, the main goal of OA management remains pain control and symptom relief. There are no effective disease-modifying OA drugs available to halt or even delay OA progression. Lack of understanding of the molecular events involved in OA pathogenesis may be one of the factors that accounts for the disappointing outcomes of the latest clinical trials. As irreversible joint structural damage occurs in end-stage OA, surgical arthroplasty, a costly procedure associated with several complications, is mandated for patients. Therefore, it is of great importance to elucidate the underlying key molecules and signaling pathways associated with the pathogenesis of OA. Since the dysregulation of AMPK is linked to various aging-related diseases, including OA, targeting it may yield a promising approach for OA treatment.
Herb and small molecule
Recently, a number of drugs and natural plant compounds from traditional medicine acted as AMPK activators to exert protective effects against OA. For instance, medicinal herbs such as berberine, quercetin, and resveratrol attenuated oxidative stress and mitochondrial dysfunction by activation of AMPK signaling in chondrocytes, thereby ameliorating joint damage in a surgically-induced knee OA mouse model [25,42,63]. Interestingly, metformin, a first-line drug in the clinic for combating diabetes, has demonstrated that it can limit OA development through activation of AMPK in an experimental OA model [64]. In addition, several selective AMPK activators (e.g. A-769962, AICAR) were shown to restore impaired mitochondrial function and diminish IL-1β-induced catabolic response in chondrocytes [22]. Protectin DX, a fatty acid metabolite, has demonstrated that it can suppress the generation of pro-catabolic effectors in IL-1β-treated chondrocytes and alleviate cartilage damage in a chemically-induced knee OA rat model in an AMPK dependent way [29].
MicroRNA
MicroRNAs, small non-coding RNA molecules, also play a role in regulating AMPK signaling and OA development. For instance, Kuo et al revealed the miR-92a is implicated in TGF-β1-induced FoXO3 expression in human OA synovial fibroblasts and contributes to OA development. FoXO3, a main downstream effector of AMPK, was shown to increase the expression of genes related to anti-oxidization, DNA repair, and autophagic/lysosomal pathways which benefitted chondrocyte homeostasis. Transfection of OA synovial fibroblasts with miR-92a mimics mitigated TGF-β1-induced FoXO3 expression and compromised chondrocyte stress resistance. Interestingly, AMPKα phosphorylation suppressed miR-92a expression, thereby facilitating FoXO3 activity and preventing OA progression [54]. Similarly, miR-449a was viewed as a novel agent of cartilage destruction due to its role in targeting the SIRT1 mRNA, a main downstream effector of AMPK signaling. Chondrocytes transfected with miR-449a exhibited a hypertrophic phenotype with high levels of catabolic gene expression and decreased anabolic signals in response to IL-1β, whereas miR-449a’s impact can be effectively reversed by forced overexpression of SIRT1 [65]. These data indicate that manipulation of AMPK signaling by targeting microRNAs could be a potential therapeutic approach to delay OA development.
Others
In addition, some strategies correlated with AMPK activation have also exhibited potential protective effects against OA. For instance, inhibition of glycolysis and stabilization of HIF-1α inhibited catabolic response and ameliorated joint degeneration by activating AMPK signaling [30,66]. Similarly, mechanical stretching induced antioxidant enzyme generation and promoted osteogenic differentiation of human bone marrow mesenchymal stem cells by activation of AMPK, indicating that AMPK is associated with the benefits of exercise [67] which is highly recommended by the Osteoarthritis Research Society to alleviate pain and improve function in knee OA [68]. Based on the current data, targeting AMPK to restore altered energy metabolism and impaired mitochondrial function in OA joint tissues could be a potential strategy for OA. However, as OA is a heterogeneous disorder with various subtypes and different etiologies, further studies are needed to define the role of AMPK in different types of OA pathogeneses and identify how AMPK is involved in the interplay between mechanical loading, inflammation, and energy metabolism.
Conclusion
Recently, disruption of energy balance in articular joint tissue has been considered one of the most essential pathogeneses in OA development. This new paradigm is strongly supported by the fact that individuals with metabolic syndromes are predisposed to suffer both weight-bearing and non-weight-bearing joint OA. Reduced AMPK activity compromises the capacity of energy flexibility for cellular homeostasis, triggering adverse events such as mitochondrial dysfunction, oxidative stress, impaired autophagy, and prolonged ER stress which ultimately resulted in articular cartilage degeneration, synovial inflammation, and abnormal subchondral bone remodeling. Restoring AMPK activation has been demonstrated to coordinate chondrocyte survival mechanisms and improve joint tissue resistance to OA driven forces. Further insight into the details of how AMPK is connected to mechanical loading, inflammation, and metabolism and the role of AMPK in subchondral bone remodeling, as well as synovial inflammation, could yield a novel therapeutic approach for OA.
Acknowledgements
This study was supported by the Natural Science Foundation of Hunan Province (No. 2018JJ3766), China.
Disclosure of conflict of interest
None.
References
- 1.GBD 2015 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016;388:1545–1602. doi: 10.1016/S0140-6736(16)31678-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sherwood J. Osteoarthritis year in review 2018: biology. Osteoarthritis Cartilage. 2019;27:365–370. doi: 10.1016/j.joca.2018.10.005. [DOI] [PubMed] [Google Scholar]
- 3.June RK, Liu-Bryan R, Long F, Griffin TM. Emerging role of metabolic signaling in synovial joint remodeling and osteoarthritis. J Orthop Res. 2016;34:2048–2058. doi: 10.1002/jor.23420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Francisco V, Perez T, Pino J, Lopez V, Franco E, Alonso A, Gonzalez-Gay MA, Mera A, Lago F, Gomez R, Gualillo O. Biomechanics, obesity, and osteoarthritis. The role of adipokines: when the levee breaks. J Orthop Res. 2018;36:594–604. doi: 10.1002/jor.23788. [DOI] [PubMed] [Google Scholar]
- 5.Salminen A, Kaarniranta K. AMP-activated protein kinase (AMPK) controls the aging process via an integrated signaling network. Ageing Res Rev. 2012;11:230–241. doi: 10.1016/j.arr.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 6.Liu-Bryan R. Inflammation and intracellular metabolism: new targets in OA. Osteoarthritis Cartilage. 2015;23:1835–1842. doi: 10.1016/j.joca.2014.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hardie DG, Schaffer BE, Brunet A. AMPK: an energy-sensing pathway with multiple inputs and outputs. Trends Cell Biol. 2016;26:190–201. doi: 10.1016/j.tcb.2015.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Carling D. AMPK signalling in health and disease. Curr Opin Cell Biol. 2017;45:31–37. doi: 10.1016/j.ceb.2017.01.005. [DOI] [PubMed] [Google Scholar]
- 9.Viollet B, Horman S, Leclerc J, Lantier L, Foretz M, Billaud M, Giri S, Andreelli F. AMPK inhibition in health and disease. Crit Rev Biochem Mol Biol. 2010;45:276–295. doi: 10.3109/10409238.2010.488215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Salminen A, Hyttinen JM, Kaarniranta K. AMP-activated protein kinase inhibits NF-kappaB signaling and inflammation: impact on healthspan and lifespan. J Mol Med. 2011;89:667–676. doi: 10.1007/s00109-011-0748-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ni YL, Shen HT, Su CH, Chen WY, Huang-Liu R, Chen CJ, Chen SP, Kuan YH. Nerolidol suppresses the inflammatory response during lipopolysaccharide-induced acute lung injury via the modulation of antioxidant enzymes and the AMPK/Nrf-2/HO-1 pathway. Oxid Med Cell Longev. 2019;2019:9605980. doi: 10.1155/2019/9605980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim J, Guan KL. Regulation of the autophagy initiating kinase ULK1 by nutrients: roles of mTORC1 and AMPK. Cell Cycle. 2011;10:1337–1338. doi: 10.4161/cc.10.9.15291. [DOI] [PubMed] [Google Scholar]
- 13.Husa M, Petursson F, Lotz M, Terkeltaub R, Liu-Bryan R. C/EBP homologous protein drives pro-catabolic responses in chondrocytes. Arthritis Res Ther. 2013;15:R218. doi: 10.1186/ar4415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Petursson F, Husa M, June R, Lotz M, Terkeltaub R, Liu-Bryan R. Linked decreases in liver kinase B1 and AMP-activated protein kinase activity modulate matrix catabolic responses to biomechanical injury in chondrocytes. Arthritis Res Ther. 2013;15:R77. doi: 10.1186/ar4254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Terkeltaub R, Yang B, Lotz M, Liu-Bryan R. Chondrocyte AMP-activated protein kinase activity suppresses matrix degradation responses to proinflammatory cytokines interleukin-1beta and tumor necrosis factor alpha. Arthritis Rheum. 2011;63:1928–1937. doi: 10.1002/art.30333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li J, Zhang B, Liu WX, Lu K, Pan H, Wang T, Oh CD, Yi D, Huang J, Zhao L, Ning G, Xing C, Xiao G, Liu-Bryan R, Feng S, Chen D. Metformin limits osteoarthritis development and progression through activation of AMPK signalling. Ann Rheum Dis. 2020;79:635–645. doi: 10.1136/annrheumdis-2019-216713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ruderman NB, Xu XJ, Nelson L, Cacicedo JM, Saha AK, Lan F, Ido Y. AMPK and SIRT1: a long-standing partnership? Am J Physiol Endocrinol Metab. 2010;298:E751–760. doi: 10.1152/ajpendo.00745.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shutt TE, Shadel GS. A compendium of human mitochondrial gene expression machinery with links to disease. Environ Mol Mutagen. 2010;51:360–379. doi: 10.1002/em.20571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Loeser RF, Collins JA, Diekman BO. Ageing and the pathogenesis of osteoarthritis. Nat Rev Rheumatol. 2016;12:412–420. doi: 10.1038/nrrheum.2016.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lotz M, Loeser RF. Effects of aging on articular cartilage homeostasis. Bone. 2012;51:241–248. doi: 10.1016/j.bone.2012.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Marsin AS, Bertrand L, Rider MH, Deprez J, Beauloye C, Vincent MF, Van den Berghe G, Carling D, Hue L. Phosphorylation and activation of heart PFK-2 by AMPK has a role in the stimulation of glycolysis during ischaemia. Curr Biol. 2000;10:1247–1255. doi: 10.1016/s0960-9822(00)00742-9. [DOI] [PubMed] [Google Scholar]
- 22.Wang Y, Zhao X, Lotz M, Terkeltaub R, Liu-Bryan R. Mitochondrial biogenesis is impaired in osteoarthritis chondrocytes but reversible via peroxisome proliferator-activated receptor gamma coactivator 1alpha. Arthritis Rheumatol. 2015;67:2141–2153. doi: 10.1002/art.39182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bolduc JA, Collins JA, Loeser RF. Reactive oxygen species, aging and articular cartilage homeostasis. Free Radic Biol Med. 2019;132:73–82. doi: 10.1016/j.freeradbiomed.2018.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao X, Petursson F, Viollet B, Lotz M, Terkeltaub R, Liu-Bryan R. Peroxisome proliferator-activated receptor gamma coactivator 1alpha and FoxO3A mediate chondroprotection by AMP-activated protein kinase. Arthritis Rheumatol. 2014;66:3073–3082. doi: 10.1002/art.38791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhou Y, Liu SQ, Yu L, He B, Wu SH, Zhao Q, Xia SQ, Mei HJ. Berberine prevents nitric oxide-induced rat chondrocyte apoptosis and cartilage degeneration in a rat osteoarthritis model via AMPK and p38 MAPK signaling. Apoptosis. 2015;20:1187–1199. doi: 10.1007/s10495-015-1152-y. [DOI] [PubMed] [Google Scholar]
- 26.Chen LY, Wang Y, Terkeltaub R, Liu-Bryan R. Activation of AMPK-SIRT3 signaling is chondroprotective by preserving mitochondrial DNA integrity and function. Osteoarthritis Cartilage. 2018;26:1539–1550. doi: 10.1016/j.joca.2018.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Woodell-May JE, Sommerfeld SD. Role of inflammation and the immune system in the progression of osteoarthritis. J Orthop Res. 2020;38:253–257. doi: 10.1002/jor.24457. [DOI] [PubMed] [Google Scholar]
- 28.Kauppinen A, Suuronen T, Ojala J, Kaarniranta K, Salminen A. Antagonistic crosstalk between NF-kappaB and SIRT1 in the regulation of inflammation and metabolic disorders. Cell Signal. 2013;25:1939–1948. doi: 10.1016/j.cellsig.2013.06.007. [DOI] [PubMed] [Google Scholar]
- 29.Piao S, Du W, Wei Y, Yang Y, Feng X, Bai L. Protectin DX attenuates IL-1beta-induced inflammation via the AMPK/NF-kappaB pathway in chondrocytes and ameliorates osteoarthritis progression in a rat model. Int Immunopharmacol. 2020;78:106043. doi: 10.1016/j.intimp.2019.106043. [DOI] [PubMed] [Google Scholar]
- 30.Wang Y, Xian H, Qi J, Wei F, Cheng X, Li S, Wang Q, Liu Z, Yu Y, Zhou J, Sun X, Liu H, Wei Y. Inhibition of glycolysis ameliorate arthritis in adjuvant arthritis rats by inhibiting synoviocyte activation through AMPK/NF-small ka, CyrillicB pathway. Inflamm Res. 2020;69:569–578. doi: 10.1007/s00011-020-01332-2. [DOI] [PubMed] [Google Scholar]
- 31.Zhou S, Lu W, Chen L, Ge Q, Chen D, Xu Z, Shi D, Dai J, Li J, Ju H, Cao Y, Qin J, Chen S, Teng H, Jiang Q. AMPK deficiency in chondrocytes accelerated the progression of instability-induced and ageing-associated osteoarthritis in adult mice. Sci Rep. 2017;7:43245. doi: 10.1038/srep43245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huang H, Wang ZJ, Zhang HB, Liang JX, Cao WD, Wu Q, He CP, Chen C. The function of PPARgamma/AMPK/SIRT-1 pathway in inflammatory response of human articular chondrocytes stimulated by advanced glycation end products. Biol Pharm Bull. 2019;42:1303–1309. doi: 10.1248/bpb.b19-00036. [DOI] [PubMed] [Google Scholar]
- 33.Lamark T, Svenning S, Johansen T. Regulation of selective autophagy: the p62/SQSTM1 paradigm. Essays Biochem. 2017;61:609–624. doi: 10.1042/EBC20170035. [DOI] [PubMed] [Google Scholar]
- 34.Meckes JK, Carames B, Olmer M, Kiosses WB, Grogan SP, Lotz MK, D’Lima DD. Compromised autophagy precedes meniscus degeneration and cartilage damage in mice. Osteoarthritis Cartilage. 2017;25:1880–1889. doi: 10.1016/j.joca.2017.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carames B, Hasegawa A, Taniguchi N, Miyaki S, Blanco FJ, Lotz M. Autophagy activation by rapamycin reduces severity of experimental osteoarthritis. Ann Rheum Dis. 2012;71:575–581. doi: 10.1136/annrheumdis-2011-200557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Holczer M, Hajdu B, Lorincz T, Szarka A, Banhegyi G, Kapuy O. A double negative feedback loop between mTORC1 and AMPK kinases guarantees precise autophagy induction upon cellular stress. Int J Mol Sci. 2019;20:5543. doi: 10.3390/ijms20225543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Alers S, Loffler AS, Wesselborg S, Stork B. Role of AMPK-mTOR-Ulk1/2 in the regulation of autophagy: cross talk, shortcuts, and feedbacks. Mol Cell Biol. 2012;32:2–11. doi: 10.1128/MCB.06159-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shang L, Wang X. AMPK and mTOR coordinate the regulation of Ulk1 and mammalian autophagy initiation. Autophagy. 2011;7:924–926. doi: 10.4161/auto.7.8.15860. [DOI] [PubMed] [Google Scholar]
- 39.Dai B, Zhu F, Chen Y, Zhou R, Wang Z, Xie Y, Wu X, Zu S, Li G, Ge J, Chen F. ASIC1a promotes acid-induced autophagy in rat articular chondrocytes through the AMPK/FoxO3a pathway. Int J Mol Sci. 2017;18:2125. doi: 10.3390/ijms18102125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee IH, Cao L, Mostoslavsky R, Lombard DB, Liu J, Bruns NE, Tsokos M, Alt FW, Finkel T. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A. 2008;105:3374–3379. doi: 10.1073/pnas.0712145105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bohensky J, Leshinsky S, Srinivas V, Shapiro IM. Chondrocyte autophagy is stimulated by HIF-1 dependent AMPK activation and mTOR suppression. Pediatr Nephrol. 2010;25:633–642. doi: 10.1007/s00467-009-1310-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qin N, Wei L, Li W, Yang W, Cai L, Qian Z, Wu S. Local intra-articular injection of resveratrol delays cartilage degeneration in C57BL/6 mice by inducing autophagy via AMPK/mTOR pathway. J Pharmacol Sci. 2017;134:166–174. doi: 10.1016/j.jphs.2017.06.002. [DOI] [PubMed] [Google Scholar]
- 43.Oakes SA, Papa FR. The role of endoplasmic reticulum stress in human pathology. Annu Rev Pathol. 2015;10:173–194. doi: 10.1146/annurev-pathol-012513-104649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell. 2010;140:900–917. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li YH, Tardif G, Hum D, Kapoor M, Fahmi H, Pelletier JP, Martel-Pelletier J. The unfolded protein response genes in human osteoarthritic chondrocytes: PERK emerges as a potential therapeutic target. Arthritis Res Ther. 2016;18:172. doi: 10.1186/s13075-016-1070-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Uehara Y, Hirose J, Yamabe S, Okamoto N, Okada T, Oyadomari S, Mizuta H. Endoplasmic reticulum stress-induced apoptosis contributes to articular cartilage degeneration via C/EBP homologous protein. Osteoarthritis Cartilage. 2014;22:1007–1017. doi: 10.1016/j.joca.2014.04.025. [DOI] [PubMed] [Google Scholar]
- 47.Takada K, Hirose J, Senba K, Yamabe S, Oike Y, Gotoh T, Mizuta H. Enhanced apoptotic and reduced protective response in chondrocytes following endoplasmic reticulum stress in osteoarthritic cartilage. Int J Exp Pathol. 2011;92:232–242. doi: 10.1111/j.1365-2613.2010.00758.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Takada K, Hirose J, Yamabe S, Uehara Y, Mizuta H. Endoplasmic reticulum stress mediates nitric oxide-induced chondrocyte apoptosis. Biomed Rep. 2013;1:315–319. doi: 10.3892/br.2013.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yamabe S, Hirose J, Uehara Y, Okada T, Okamoto N, Oka K, Taniwaki T, Mizuta H. Intracellular accumulation of advanced glycation end products induces apoptosis via endoplasmic reticulum stress in chondrocytes. FEBS J. 2013;280:1617–1629. doi: 10.1111/febs.12170. [DOI] [PubMed] [Google Scholar]
- 50.Hamamura K, Lin CC, Yokota H. Salubrinal reduces expression and activity of MMP13 in chondrocytes. Osteoarthritis Cartilage. 2013;21:764–72. doi: 10.1016/j.joca.2013.02.657. [DOI] [PubMed] [Google Scholar]
- 51.Feng K, Chen Z, Pengcheng L, Zhang S, Wang X. Quercetin attenuates oxidative stress-induced apoptosis via SIRT1/AMPK-mediated inhibition of ER stress in rat chondrocytes and prevents the progression of osteoarthritis in a rat model. J Cell Physiol. 2019;234:18192–18205. doi: 10.1002/jcp.28452. [DOI] [PubMed] [Google Scholar]
- 52.Kunanusornchai W, Muanprasat C, Chatsudthipong V. Adenosine monophosphate-activated protein kinase activation and suppression of inflammatory response by cell stretching in rabbit synovial fibroblasts. Mol Cell Biochem. 2016;423:175–185. doi: 10.1007/s11010-016-2835-6. [DOI] [PubMed] [Google Scholar]
- 53.Kunanusornchai W, Witoonpanich B, Tawonsawatruk T, Pichyangkura R, Chatsudthipong V, Muanprasat C. Chitosan oligosaccharide suppresses synovial inflammation via AMPK activation: an in vitro and in vivo study. Pharmacol Res. 2016;113:458–467. doi: 10.1016/j.phrs.2016.09.016. [DOI] [PubMed] [Google Scholar]
- 54.Kuo SJ, Liu SC, Huang YL, Tsai CH, Fong YC, Hsu HC, Tang CH. TGF-beta1 enhances FOXO3 expression in human synovial fibroblasts by inhibiting miR-92a through AMPK and p38 pathways. Aging. 2019;11:4075–4089. doi: 10.18632/aging.102038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim EK, Lim S, Park JM, Seo JK, Kim JH, Kim KT, Ryu SH, Suh PG. Human mesenchymal stem cell differentiation to the osteogenic or adipogenic lineage is regulated by AMP-activated protein kinase. J Cell Physiol. 2012;227:1680–1687. doi: 10.1002/jcp.22892. [DOI] [PubMed] [Google Scholar]
- 56.Hou CH, Tan TW, Tang CH. AMP-activated protein kinase is involved in COX-2 expression in response to ultrasound in cultured osteoblasts. Cell Signal. 2008;20:978–988. doi: 10.1016/j.cellsig.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 57.Kanazawa I, Yamaguchi T, Yano S, Yamauchi M, Sugimoto T. Metformin enhances the differentiation and mineralization of osteoblastic MC3T3-E1 cells via AMP kinase activation as well as eNOS and BMP-2 expression. Biochem Biophys Res Commun. 2008;375:414–419. doi: 10.1016/j.bbrc.2008.08.034. [DOI] [PubMed] [Google Scholar]
- 58.Kang H, Viollet B, Wu D. Genetic deletion of catalytic subunits of AMP-activated protein kinase increases osteoclasts and reduces bone mass in young adult mice. J Biol Chem. 2013;288:12187–12196. doi: 10.1074/jbc.M112.430389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lee YS, Kim YS, Lee SY, Kim GH, Kim BJ, Lee SH, Lee KU, Kim GS, Kim SW, Koh JM. AMP kinase acts as a negative regulator of RANKL in the differentiation of osteoclasts. Bone. 2010;47:926–937. doi: 10.1016/j.bone.2010.08.001. [DOI] [PubMed] [Google Scholar]
- 60.Tong X, Zhang C, Wang D, Song R, Ma Y, Cao Y, Zhao H, Bian J, Gu J, Liu Z. Suppression of AMP-activated protein kinase reverses osteoprotegerin-induced inhibition of osteoclast differentiation by reducing autophagy. Cell Prolif. 2020;53:e12714. doi: 10.1111/cpr.12714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim JY, Min JY, Baek JM, Ahn SJ, Jun HY, Yoon KH, Choi MK, Lee MS, Oh J. CTRP3 acts as a negative regulator of osteoclastogenesis through AMPK-c-Fos-NFATc1 signaling in vitro and RANKL-induced calvarial bone destruction in vivo. Bone. 2015;79:242–251. doi: 10.1016/j.bone.2015.06.011. [DOI] [PubMed] [Google Scholar]
- 62.Peng M, Qiang L, Xu Y, Li C, Li T, Wang J. Inhibition of JNK and activation of the AMPK-Nrf2 axis by corosolic acid suppress osteolysis and oxidative stress. Nitric Oxide. 2019;82:12–24. doi: 10.1016/j.niox.2018.11.002. [DOI] [PubMed] [Google Scholar]
- 63.Qiu L, Luo Y, Chen X. Quercetin attenuates mitochondrial dysfunction and biogenesis via upregulated AMPK/SIRT1 signaling pathway in OA rats. Biomed Pharmacother. 2018;103:1585–1591. doi: 10.1016/j.biopha.2018.05.003. [DOI] [PubMed] [Google Scholar]
- 64.Feng X, Pan J, Li J, Zeng C, Qi W, Shao Y, Liu X, Liu L, Xiao G, Zhang H, Bai X, Cai D. Metformin attenuates cartilage degeneration in an experimental osteoarthritis model by regulating AMPK/mTOR. Aging. 2020;12:1087–1103. doi: 10.18632/aging.102635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Park KW, Lee KM, Yoon DS, Park KH, Choi WJ, Lee JW, Kim SH. Inhibition of microRNA-449a prevents IL-1beta-induced cartilage destruction via SIRT1. Osteoarthritis Cartilage. 2016;24:2153–2161. doi: 10.1016/j.joca.2016.07.002. [DOI] [PubMed] [Google Scholar]
- 66.Tchetina EV, Markova GA, Poole AR, Zukor DJ, Antoniou J, Makarov SA, Kuzin AN. Deferoxamine suppresses collagen cleavage and protease, cytokine, and COL10A1 expression and upregulates AMPK and krebs cycle genes in human osteoarthritic cartilage. Int J Rheumatol. 2016;2016:6432867. doi: 10.1155/2016/6432867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen X, Yan J, He F, Zhong D, Yang H, Pei M, Luo ZP. Mechanical stretch induces antioxidant responses and osteogenic differentiation in human mesenchymal stem cells through activation of the AMPK-SIRT1 signaling pathway. Free Radic Biol Med. 2018;126:187–201. doi: 10.1016/j.freeradbiomed.2018.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Bennell KL, Dobson F, Hinman RS. Exercise in osteoarthritis: moving from prescription to adherence. Best Pract Res Clin Rheumatol. 2014;28:93–117. doi: 10.1016/j.berh.2014.01.009. [DOI] [PubMed] [Google Scholar]