

Abstract
Carotid artery stenosis is a leading cause of ischemic stroke, but the underlying mechanism remains unclear. We aimed to determine the molecular mechanisms of carotid plaque progression. We analyzed the molecular and morphometric characteristics of carotid plaque samples obtained from 30 patients who underwent carotid endarterectomy. Additionally, we established a mouse model of carotid atherosclerosis by partially ligating the left common carotid arteries of male ClockΔ19/Δ19 (Clk) and wild-type (WT) C57BL/6J mice fed a high-fat diet. Clk and WT primary mouse aortic endothelial cells (pMAECs) were exposed to disturbed flow (DF) or undisturbed flow (UF) with or without treatment with the IRE-1α inhibitor STF-083010 or the PERK inhibitor GSK2606414. In human carotid artery plaques, CLOCK expression was lower in the lipid-rich necrotic core than in transitional regions, especially in the endothelium. Decreased CLOCK mRNA levels were associated with more extensive stenosis, intraplaque hemorrhage, and complex plaque in human carotid plaques. In mice, the ClockΔ19/Δ19 mutation significantly increased neointima formation and neovascularization but decreased collagen content and lumen area in partially ligated carotid arteries. In addition, ClockΔ19/Δ19 mutants exhibited significantly decreased Cdh5 expression and increased expression of endothelial-mesenchymal transition (EndMT) and endoplasmic reticulum (ER) stress markers in mice with partially ligated carotid arteries and pMAECs exposed to DF. Notably, inhibition of the IRE1α-XBP1 axis abrogated the increased EndMT caused by ClockΔ19/Δ19 mutation and DF in pMAECs. In conclusion, the disruption of CLOCK function aggravates EndMT via the IRE1α-XBP1 axis, contributing to carotid artery stenosis.
Keywords: Carotid artery stenosis, circadian locomotor output cycles kaput, endothelial-mesenchymal transition, endoplasmic reticulum stress, disturbed flow
Introduction
Carotid artery stenosis contributes to the occurrence of ischemic stroke [1]. Therefore, determining the molecular mechanisms of carotid artery stenosis may help in preventing stroke. Carotid artery stenosis is a multistep cardiovascular disease promoted by aging, disturbed blood flow, hypercholesterolemia, hyperglycemia, and smoking [2]. Emerging evidence suggests that endothelial-mesenchymal transition (EndMT) in endothelial dysfunction is a crucial component and an emerging concept in the pathogenesis of vascular diseases [2]. Endothelial dysfunction is closely related to the excessive unfolded protein response (UPR) activated by endoplasmic reticulum (ER) stress [3-5]. However, the upstream regulator of ER stress and the mechanism through which ER stress induces EndMT during carotid artery stenosis remains unclear and needs to be clarified [6].
Proteins involved in UPR and ER chaperones are under the control of biological rhythms [7]. The circadian clock consists of positive and negative feedback control loops of approximately 24 h, which are regulated by core clock genes such as CLOCK and BMAL1 [8]. The disruption of both central and peripheral CLOCK genes is closely associated with the aging process and aging-related diseases such as atherosclerosis [9]. Thus, we hypothesized that the disruption of CLOCK expression in the carotid artery (CA) may lead to excessive ER stress and subsequent carotid artery stenosis.
Here, we investigated CLOCK expression in the carotid plaque tissues of patients with internal carotid artery stenosis (ICAS) and analyzed the correlation of CLOCK expression levels with the pathological features of carotid plaques. We also identified CLOCK as a suppressor of carotid plaque progression and explored the underlying mechanisms by which this may occur. Our data demonstrate that inhibiting the IRE1α-XBP1 axis significantly attenuates the endothelial-to-mesenchymal transition (EndMT) induced by disturbed flow (DF) and the disturbance of CLOCK expression, thereby inhibiting carotid plaque progression.
Materials and methods
Human tissue samples and ethics statement
Experiments involving human samples were approved (IRB approval No. Y2019-219) and supervised by the Ethics Committee of Zhongshan Hospital (Shanghai, China). Clinical carotid plaque specimens were used in accordance with the Declaration of Helsinki, and written informed consent was obtained from all patients. Samples were consecutively obtained from 30 patients who underwent carotid endarterectomy for ICAS at 8:00-10:00 AM following Clinical Practice Guidelines between February 1, 2018, and January 31, 2019 [10]. Samples were stored at -80°C or formalin-fixed and paraffin-embedded until further use.
Animal studies with partial ligation
The in vivo studies were performed at Institute of Vascular Surgery, Zhongshan Hospital, Fudan University. All animal experiments were approved by the Institutional Animal Care and Use Committee of Zhongshan Hospital, Fudan University (Shanghai, China). Male ClockΔ19/Δ19 (mutant) C57BL/6J mice, which showed reduced CLOCK mRNA levels and CLOCK function [11], and wild-type (WT) C57BL/6J mice were obtained from the Model Animal Research Center of Nanjing University (Jiangsu, China). All mice were fed a chow diet and water ad libitum until 6 weeks of age. Mice were then fed the Paigen high-fat diet (1.25% cholesterol, 15% fat, 0.5% cholic acid) and water ad libitum [12]. At 12 weeks, all mice were anesthetized in a closed chamber with 1-2% isoflurane in oxygen for 2 to 5 min until immobile and then underwent partial ligation of the left common CA and mock treatment of the right common CA at 8:00-10:00 AM [12]. All mice were sacrificed at week 18 by pentobarbital overdose. CA tissues were harvested, snap-frozen in liquid nitrogen, and stored at -80°C at 8:00-10:00 AM.
Endothelial cell (EC) isolation and culture
WT or ClockΔ19/Δ19 primary mouse aortic ECs (pMAECs) were isolated and cultured as previously described [13]. Briefly, after adequate anesthesia, mouse aortas were separated and dissected into 2 mm sections that were then placed on Matrigel pre-coated plates. After culture in endothelial cell medium at 37°C for 7-14 days, pMAECs were passaged. At 8:00-10:00 AM, pMAECs at passage 5-6 were exposed to DF and undisturbed flow (UF) using a 10 mm-radius orbital shaking system at 210 rpm and dimethyl sulfoxide in the presence or absence of 15 or 30 µΜ STF-083010 (IRE-1α inhibitor; Selleckchem, Shanghai, China) or 0.015 or 0.03 μM GSK2606414 (PERK inhibitor; Selleckchem) for a further 72 h [14].
Western blotting
In pMAECs and the partially ligated CA, the relative protein expression levels of p-PERK, p-IRE1α, ATF6, p-eIF2α, XBP-1, and GAPDH were measured by western blotting using standard methods. Anti-ATF6 (ab37149) and anti-GAPDH (ab8245) antibodies were obtained from Abcam (Cambridge, MA, USA), anti-Xbp-1 (24385) was from Signalway Antibody (Baltimore, USA), anti-p-IRE1α (Ser724; ARG40603) and anti-p-eIF2α (Ser51; ARG57722) were from arigo Biolaboratories Corp (Taiwan), and anti-p-PERK (Thr980; 3179s) was from Cell Signaling Technology (Danvers, MA, USA). Band intensities were quantified using ImageJ software (https://imagej.nih.gov/ij/) and normalized to the levels of GAPDH.
Quantitative reverse transcription PCR (qRT-PCR)
TRIzol (Thermo Fisher Scientific, Waltham, MA, USA) was used to isolate total RNA from pMAECs. An Omniscript RT kit (Qiagen, Hilden, Germany) was used to synthesize first-strand complementary DNA. qRT-PCR using SYBR Green was used to quantify changes in mRNA levels. Expression levels were calculated using the 2-ΔCT method. All primers used are listed in Table 1.
Table 1.
Primer sequences for the genes targeted in quantitative reverse transcription PCR
| Target | GenBank accession no. | Primer sequence (5’-3’) | Category |
|---|---|---|---|
| CLOCK | NM_004898 | F: CAGGCAGCATTTACCAGCTCATG | Human Primer |
| R: GTAGCTTGAGACATCACTGGCTG | |||
| GAPDH | NM_002046 | F: GTCTCCTCTGACTTCAACAGCG | Human Primer |
| R: ACCACCCTGTTGCTGTAGCCAA | |||
| S100a4 | NM_011311 | F: AGCTCAAGGAGCTACTGACCAG | Mouse Primer |
| R: GCTGTCCAAGTTGCTCATCACC | |||
| Acta2 | NM_007392 | F: TGCTGACAGAGGCACCACTGAA | Mouse Primer |
| R: CAGTTGTACGTCCAGAGGCATAG | |||
| Vimentin | NM_011701 | F: CGGAAAGTGGAATCCTTGCAGG | Mouse Primer |
| R: AGCAGTGAGGTCAGGCTTGGAA | |||
| Snai1 | NM_011427 | F: TGTCTGCACGACCTGTGGAAAG | Mouse Primer |
| R: CTTCACATCCGAGTGGGTTTGG | |||
| Cdh5 | NM_009868 | F: GAACGAGGACAGCAACTTCACC | Mouse Primer |
| R: GTTAGCGTGCTGGTTCCAGTCA | |||
| Mmp2 | NM_008610 | F: CAAGGATGGACTCCTGGCACAT | Mouse Primer |
| R: TACTCGCCATCAGCGTTCCCAT | |||
| Mmp9 | NM_013599 | F: GCTGACTACGATAAGGACGGCA | Mouse Primer |
| R: TAGTGGTGCAGGCAGAGTAGGA | |||
| Gapdh | NM_008084 | F: CATCACTGCCACCCAGAAGACTG | Mouse Primer |
| R: ATGCCAGTGAGCTTCCCGTTCAG |
Histological and morphometric analysis
Histological analysis of carotid arterial segments was performed as previously reported [12]. Briefly, samples were formalin-fixed and paraffin-embedded. The blocks were sectioned at 5 μm intervals using a microtome. The sections were then deparaffinized in xylene and rehydrated with graded ethanol before staining with hematoxylin-eosin or Masson’s trichrome stain. The luminal, intimal, and medial areas of the vessel and percent area of collagen deposition were measured by two independent investigators blinded to the experimental design using ImageJ software.
Immunofluorescence and confocal microscopy
The carotid arterial segments were formalin-fixed and paraffin-embedded. The blocks were sectioned at 5 μm intervals using a microtome. The resulting sections were deparaffinized in xylene and dehydrated in an ethanol series (100%, 90%, 80%, 70%, and 50% ethanol), followed by rinsing with phosphate-buffered saline (PBS). Antigen retrieval was performed in citrate buffer (10 mM, pH 6.0) at 95°C. After cooling, the tissue slices were blocked for 1 h in 5% bovine serum albumin (BSA) and 20% donkey serum in PBS and then incubated with primary antibodies in 3% BSA overnight at 4°C in a humidified chamber. Primary antibodies used for mice immunofluorescence were anti-CD31 (550274; BD Biosciences), anti-S100A4 (ab27957; Abcam), anti-vimentin (ab24525; Abcam), and anti-XBP-1 (sc-8015; Santa Cruz Biotechnology). Anti-CLOCK (ab3517; Abcam) antibody was used for human IF. After incubation with the primary antibodies, tissue sections were washed with Tris-buffered saline and incubated with cyanine-3 (Cy3) or fluorescein isothiocyanate-conjugated secondary antibodies for 1 h at 20°C. The sections were then observed and photographed using the Axiovert 200 M microscopy system (Carl Zeiss, Jena, Germany). The number of vimentin-, XBP-1-, or S100A4-positive ECs and neovessels (CD31-positive tubules) was quantified by two independent investigators blinded to the experimental design using ImageJ software.
Statistical analyses
Data are expressed as the mean ± standard deviation. All experiments were performed at least three times. Statistical comparisons between two groups were performed using Student’s t-test. For multiple comparison tests, one-way analysis of variance with Bonferroni’s correction was employed. Comparisons of non-parametric data between two groups were performed using the two-tailed Mann-Whitney test. Differences were considered significant at P < 0.05. All these statistical analysis were performed using SPSS version 20.0 (SPSS Inc., Chicago, IL, USA).
Results
Expression of CLOCK in the lipid-rich necrotic core (LRNC) and transitional region (TR) tissues of carotid atherosclerotic plaques
LRNC and TR of carotid atherosclerotic plaque tissues were collected from 30 patients with ICAS (Figure 1A). CLOCK mRNA levels in LRNC tissues were much lower than those in the TR of the carotid atherosclerotic plaque tissues (Figure 1B). The potential cut-off values of the CLOCK mRNA level predicting LRNC formation were sequentially measured by receiver-operating characteristic curve analysis (Figure 1C). Using the maximum Youden index, the optimal cut-off value for dividing high and low levels of CLOCK mRNA was 5.56 × 10-4 (Figure 1C, 1D). We thus defined a CLOCK mRNA level ≤ 5.56 × 10-4 as low CLOCK mRNA expression and a CLOCK mRNA level > 5.56 × 10-4 as high CLOCK mRNA expression (sensitivity = 86.70%, specificity = 70.00%). Low levels of CLOCK mRNA were found in 70% of LRNC specimens and 13.33% of TR tissue samples (P < 0.001; Table 2). Immunostaining demonstrated that CLOCK expression of the LRNC was significantly lower than that of the TR, especially in the endothelium (Figure 1E-H). Hence, CLOCK expression was low in the carotid atherosclerotic plaque tissues of patients with ICAS.
Figure 1.
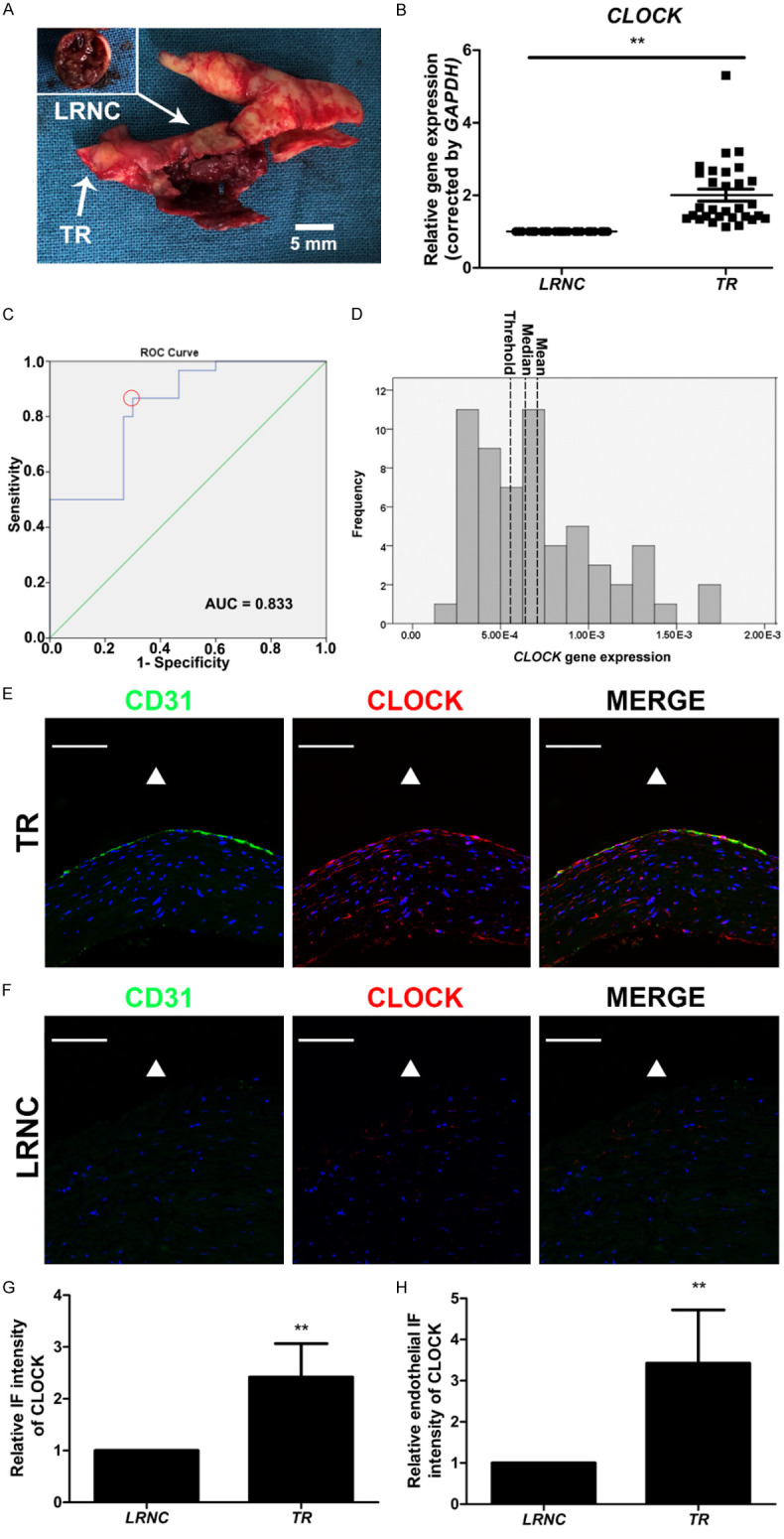
Decreased CLOCK mRNA levels were associated with the development of carotid atherosclerotic plaques. A. Morphology of the lipid-rich necrotic core (LRNC) with intraplaque hemorrhage and transitional region (TR) of carotid atherosclerotic plaque. B. CLOCK mRNA levels were detected in LRNC (n = 30) and TR (n = 30) of tissues from patients with internal carotid artery stenosis. C. Receiver-operating characteristic curve of CLOCK mRNA levels and TR (red dotted circle, optimal cut-off point). D. Histogram of CLOCK mRNA levels (left dotted line, threshold, 5.56 × 10-4; middle dotted line, median, 6.38 × 10-4; right dotted line, mean, 7.09 × 10-4). AUC, area under curve. E, F. Immunofluorescence staining for CLOCK (red) and CD31 (green) of TR and LRNC. Nuclei were stained with 4’,6-diamidino-2-phenylindole (DAPI; blue). The triangle indicates the lumen side. G. Immunofluorescence intensity of CLOCK in the TR and LRNC (n = 5). H. Endothelial immunofluorescence intensity of CLOCK in the TR and LRNC (n = 5). Scale bar: 100 μm. **P < 0.01.
Table 2.
CLOCK mRNA levels in the lipid-rich necrotic core and transitional region
| Sample | n | Low CLOCK mRNA expression (% of samples) |
|---|---|---|
| Lipid-rich necrotic core | 30 | 21 (70.00%) |
| Transitional region | 30 | 4 (13.33%)* |
P < 0.01 in comparisons between tissue samples.
Correlation between CLOCK mRNA expression and clinicopathological plaque features
As shown in Table 3, LRNCs with low CLOCK mRNA levels were significantly and positively associated with the status of complex (AHA type VI) plaques, the extent of stenosis, and intraplaque hemorrhage (IPH).
Table 3.
Association of low CLOCK mRNA expression in the lipid-rich necrotic cores (LRNCs) with clinicopathological plaque features
| Parameter | n | Low CLOCK mRNA expression (% of samples) | P |
|---|---|---|---|
| Sex | |||
| Male | 26 | 69.23% | 0.822 |
| Female | 4 | 75.00% | |
| Age (years) | |||
| 55-75 | 16 | 81.25% | 0.169 |
| < 55 | 14 | 57.14% | |
| Hypertension | |||
| Yes | 15 | 73.33% | 0.702 |
| No | 15 | 66.67% | |
| Diabetes mellitus | |||
| Yes | 18 | 77.78% | 0.270 |
| No | 12 | 58.33% | |
| Hyperlipidemia | |||
| Yes | 17 | 76.47% | 0.394 |
| No | 13 | 61.54% | |
| Smoking | |||
| Yes | 10 | 80.00% | 0.416 |
| No | 20 | 65.00% | |
| Statin use | |||
| Yes | 26 | 69.23% | 0.822 |
| No | 4 | 75.00% | |
| Extent of stenosis (NASCET) | |||
| 50-69% | 7 | 28.57% | 0.005 |
| 70-99% | 23 | 82.61% | |
| Intraplaque hemorrhage | |||
| Yes | 15 | 93.33% | 0.005 |
| No | 15 | 46.67% | |
| Surface defect | |||
| Yes | 7 | 85.71% | 0.264 |
| No | 23 | 65.22% | |
| Complex (AHA type VI) plaque | |||
| Yes | 18 | 88.89% | 0.012 |
| No | 12 | 41.67% |
Abbreviations: AHA, American Heart Association; NASCET, North American Symptomatic Carotid Endarterectomy.
ClockΔ19/Δ19 mutation aggravated carotid artery stenosis in the partially ligated CA model
To confirm the suppressive effect of CLOCK on carotid artery stenosis, a partially ligated CA mouse model was employed. The luminal area of partially ligated CAs was smaller in ClockΔ19/Δ19 mice than in WT mice, while the intimal area and intima/media ratio of partially ligated CAs was higher in ClockΔ19/Δ19 mice than in WT mice (Figure 2A-F). Masson trichrome staining revealed that the relative collagen fractional area of partially ligated CAs were significantly lower in ClockΔ19/Δ19 mice than in WT mice (Figure 2A, 2G). In contrast, the relative collagen fractional area of CAs were comparable in the mock treatments of both mouse groups (Figure 2A, 2H). Moreover, neovessel density of partially ligated CAs was higher in ClockΔ19/Δ19 mice than in WT mice (Figure 2B, 2I).
Figure 2.

ClockΔ19/Δ19 increased the extent of stenosis and changed the structure of the partially ligated carotid artery (CA) ligation model. A. Masson trichrome staining of mouse CA. Arrows indicate neovessels. B. Immunofluorescence staining for CD31 (green). Nuclei were stained with DAPI (blue). Arrows indicate neovessels. C-F. Mean lumen, intimal, and medial areas, and intima/media ratio (n = 6). G, H. Relative collagen fractional area of right common CAs (mock surgery, n = 6) and left common CAs (partial ligation, n = 6). I. Neovessel density of partially ligated CAs (n = 6). Scale bar: 100 mm. *P < 0.05, **P < 0.01.
ClockΔ19/Δ19 mutation aggravated EndMT in the partially ligated CA
We performed a set of experiments to investigate the effects of CLOCK on EndMT in the partially ligated CA. Double immunostaining demonstrated that the proportion of vimentin (+) or S100A4 (+) ECs was significantly higher in the intima of partially ligated CAs of ClockΔ19/Δ19 mice than in those of WT mice (Figure 3A-F). Moreover, vimentin and S100A4 expression levels were significantly increased in the intima of partially ligated CAs of ClockΔ19/Δ19 mice compared to WT mice (Figure 3A-F). qRT-PCR results demonstrated that the levels of the endothelial marker Cdh5 were lower, while the levels of mesenchymal markers S100a4, Acta2, vimentin, Twist1, Snai1, Mmp2, and Mmp9 were higher in the partially ligated CAs of ClockΔ19/Δ19 mice than in the partially ligated CAs of WT mice (Figure 3G).
Figure 3.
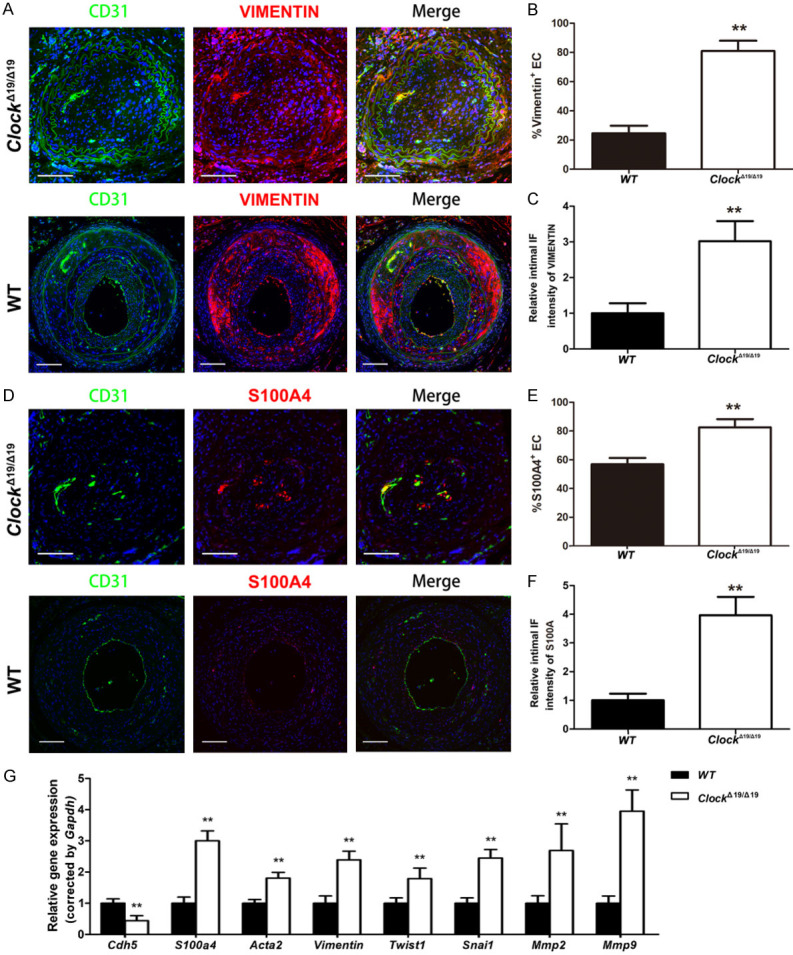
ClockΔ19/Δ19 stimulated EndMT in vivo. A. Immunofluorescence staining for CD31 (green) and vimentin (red). Nuclei were stained with DAPI (blue). B. Immunocytochemical analysis of the percentages of vimentin+ ECs in the intima of partially ligated CAs (n = 6). C. Immunofluorescence intensity of vimentin in the intima of partially ligated CAs (n = 6). D. Immunofluorescence staining for CD31 (green) and S100A4 (red). Nuclei were stained with DAPI (blue). E. Immunocytochemical analysis of the percentage of S100A4+ ECs in the intima of partially ligated CAs (n = 6). F. Immunofluorescence intensity of S100A4 in the intima of partially ligated CAs (n = 6). G. Relative mRNA levels of Cdh5, S100a4, Acta2, Vimentin, Twist1, Snai1, Mmp2, and Mmp9 in partially ligated CAs (n = 6). Scale bar: 100 μm (n = 6). *P < 0.05, **P < 0.01.
ClockΔ19/Δ19 mutation aggravated ER stress and UPR in the partially ligated CA
ER stress and the UPR involve the activation of three transmembrane proteins: IRE1α, protein kinase RNA-like ER kinase (PERK), and activating transcription factor 6 (ATF6). Immunoblotting results showed that after partial ligation, ClockΔ19/Δ19 mutation significantly increased p-PERK and p-IRE1α protein expression of CA, whereas ATF6 showed no significant difference between ClockΔ19/Δ19 and WT mice (Figure 4A). We also found that p-eIF2α and XBP-1, downstream targets of PERK and IRE1α respectively, were significantly upregulated in ClockΔ19/Δ19 mice compared to WT mice (Figure 4A). Furthermore, double immunostaining demonstrated that the proportion of XBP-1 (+) ECs was significantly higher in the intima of partially ligated CAs of ClockΔ19/Δ19 mice than in those of WT mice (Figure 4B, 4C). XBP-1 expression was also significantly increased in the intima of partially ligated CAs of ClockΔ19/Δ19 mice compared to that in WT mice (Figure 4B, 4D).
Figure 4.

The ClockΔ19/Δ19 mutation exacerbated endoplasmic reticulum (ER) stress and the unfolded protein response (UPR) in vivo. A. p-PERK, p-eIF2α, p-IRE1α, XBP-1, and ATF6 protein levels in partially ligated CAs assayed by western blotting (n = 6). B. Immunofluorescence staining for CD31 (green) and XBP-1 (red). Nuclei were stained with DAPI (blue). Scale bar: 100 μm. C. Immunocytochemical analysis of the percentage of XBP-1+ ECs in the intima of partially ligated CAs (n = 6). D. Immunofluorescence intensity of XBP-1 in the intima of partially ligated CAs (n = 6). *P < 0.05, **P < 0.01.
In vitro
ClockΔ19/Δ19 mutation aggravated DF-induced EndMT and ER stress
qRT-PCR results revealed that ClockΔ19/Δ19 mutation significantly aggravated DF-induced increase in expression levels of the mesenchymal markers S100a4, Acta2, vimentin, Snai1, Mmp2, and Mmp9 and the decrease in expression levels of the endothelial marker Cdh5 in the pMAECs (Figure 5A).
Figure 5.
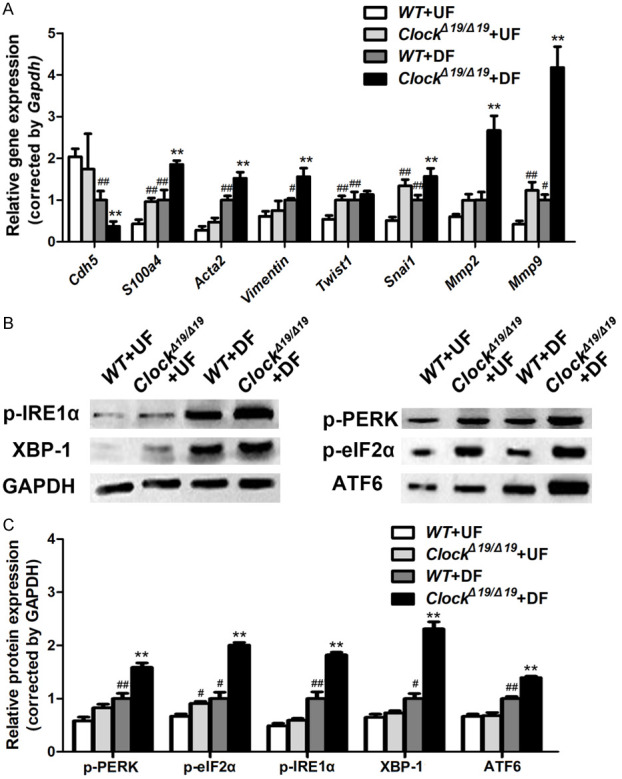
ClockΔ19/Δ19 aggravated EndMT and endoplasmic reticulum (ER) stress induced by disturbed flow (DF) in vitro. A. Relative mRNA levels of Cdh5, S100a4, Acta2, Vimentin, Twist1, Snai1, Mmp2, and Mmp9 in pMAECs exposed to disturbed flow (DF) or undisturbed flow (UF) (n = 3). B, C. p-PERK, p-eIF2α, p-IRE1α, XBP-1, and ATF6 protein levels of pMAECs exposed to disturbed flow (DF) or undisturbed flow (UF) assayed by western blotting (n = 3). *P < 0.05 vs. WT+DF, **P < 0.01 vs. WT+DF, #P < 0.05 vs. WT+UF, ##P < 0.01 vs. WT+UF.
We also investigated the effects of CLOCK on ER stress. Western blot analyses revealed that DF significantly upregulated p-PERK, p-IRE1α, ATF6, p-eIF2α, and XBP-1 expression in the pMACEs, and ClockΔ19/Δ19 mutation significantly aggravated these changes (Figure 5B, 5C).
ClockΔ19/Δ19 mutation induced EndMT by activating the IRE1α-XBP1 axis
To further investigate the effects of the ClockΔ19/Δ19 mutation on EndMT, pMAECs exposed to DF were treated with an IRE-1α inhibitor (STF-083010; 15 or 30 µΜ) and PERK inhibitor (GSK2606414; 0.015 or 0.03 μM). qRT-PCR revealed that STF-083010 significantly attenuated the decreased expression level of Cdh5 (Figure 6A) and the increased expression of S100a4, vimentin, and Snai1 in ClockΔ19/Δ19 pMAECs exposed to DF in a dose-dependent manner (Figure 6B-D). However, GSK2606414 failed to rescue the aggravated EndMT in ClockΔ19/Δ19 pMAECs exposed to DF despite the increased expression level of vimentin (Figure 6A-D).
Figure 6.
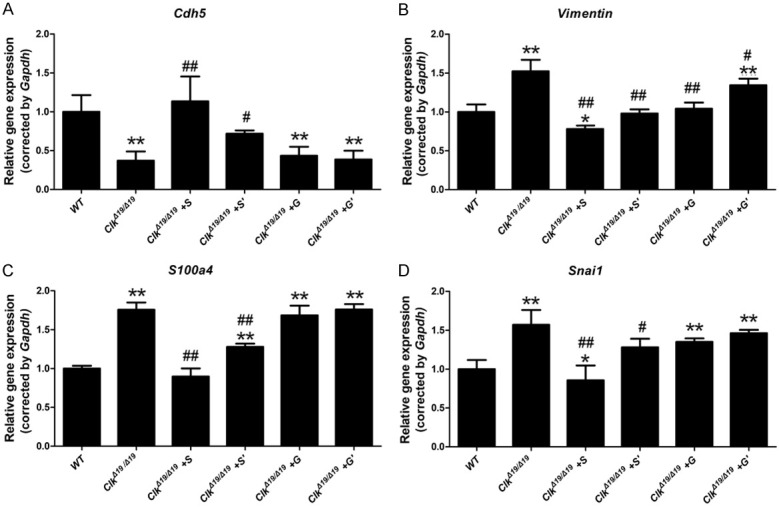
ClockΔ19/Δ19 (Clk) mutation aggravated disturbed flow (DF)-induced EndMT by activating the IRE1α-XBP1 axis. Wild-type (WT) or ClockΔ19/Δ19 pMAECs were exposed to disturbed flow (DF) and DMSO (WT or Clk), 30 µΜ STF-083010 (Clk+S), 15 µΜ STF-083010 (Clk+S’), 0.03 μM GSK2606414 (Clk+G), or 0.015 μM GSK2606414 (Clk+G’) for 72 h. A-D. Cdh5, S100a4, Vimentin, and Snai1 mRNA levels were assayed by quantitative reverse transcription PCR (n = 3). *P < 0.05 vs. WT, **P < 0.01 vs. WT, # P < 0.05 vs. Clk, ## P < 0.01 vs. Clk.
Discussion
Substantial evidence over the past 20 years suggests that both circadian rhythms and ER stress participate in atherosclerosis [6,15]. However, the molecular connections between these two signaling pathways in carotid artery stenosis remain unknown. Here, we confirmed the suppressive effects of CLOCK on carotid artery stenosis by controlling endoplasmic reticulum stress-induced endothelial-mesenchymal transition through the IRE1α-XBP1 axis.
CLOCK is a core circadian protein that regulates biological rhythms [9]. Disturbance of the CLOCK-dependent biological rhythm might be directly involved in the deterioration of physiological function and aging-related diseases [9,16]. In this study, we found that carotid plaque LRNCs of low CLOCK level were more closely associated with LRNC size, and IPH and AHA type VI plaque, which might predict poor prognosis for patients with ICAS. To determine whether the mutational disruption of CLOCK can lead to carotid artery stenosis, ClockΔ19/Δ19 mice were employed to establish a partial ligation model to mimic the disrupted blood flow of carotid artery stenosis [12]. Consistent with the results of previous studies [17,18], our in vivo results showed that ClockΔ19/Δ19 mice had significantly increased neointima formation compared to WT mice in the partially ligated CAs, and the ClockΔ19/Δ19 mutation led to decreased collagen content and increased neovascularization in partially ligated CAs. Thus, CLOCK may play an important role in suppressing carotid artery stenosis and plaque vulnerability.
We next sought to determine the mechanism by which CLOCK suppresses carotid artery stenosis. Using chronological in vivo imaging, a recent study showed that endothelial inflammation occurs earlier than neutrophil accumulation and lipid deposition in zebrafish fed a high-fat, cholesterol-rich diet [19], emphasizing the important role of the vascular endothelium in cardiovascular homeostasis. Several studies indicate that the EndMT of ECs plays a key role in atherosclerosis progression [20,21]. EndMT is an extreme form of endothelial cell plasticity [22], which can be promoted by hypoxia, high glucose, and disrupted blood flow [23]. Here, the ClockΔ19/Δ19 mutation resulted in increased EndMT in vitro and in vivo. EndMT is widely known to be associated with angiogenesis and an altered balance of collagen-matrix metalloproteinases [21,24,25], which might explain the decreased collagen content and increased neovascularization observed in partially ligated CAs from ClockΔ19/Δ19 mice.
Hydrodynamic shear stress regulates inflammation in human aortic ECs through ER stress by activating transcription factor x-box binding protein 1 (XBP1) [3], and the regulatory role of CLOCK in ER stress has been demonstrated [4,26]. Thus, CLOCK might be involved in carotid artery stenosis via the regulation of ER stress, as evidenced in the present study. We evaluated the effect of ClockΔ19/Δ19 on ER stress in vivo and in vitro and found that ClockΔ19/Δ19 resulted in the activation of the IRE1α-XBP1 and PERK-eIF2α signaling pathways in vivo and in vitro. The activation of these pathways is known to induce apoptosis and angiogenesis [27,28]. Our results showing increased LRNC size and IPH in human plaques and increased neovascularization in partial ligated CAs of ClockΔ19/Δ19 mice are consistent with this mechanism. Moreover, ER stress has been found to induce the expression of the EndMT transcription factor SNAI1 [29]. SNAI1 activation by ER stress promotes arterial endothelial EndMT of plaques. The present study also showed the regulatory effect of the IRE1α-XBP1 axis on EndMT by investigating the rescue effect of the IRE-1α inhibitor and the PERK inhibitor on DF-induced EndMT in ClockΔ19/Δ19 pMAECs. Only the IRE-1α inhibitor successfully and significantly attenuated the EndMT caused by ClockΔ19/Δ19 mutation, which might be due to the regulatory effects of the IRE1α-XBP1 axis on endothelial apoptosis and migration [30,31]. It is also interesting to note that ClockΔ19/Δ19 increased ATF6 expression in pMAECs but failed to increase ATF6 expression in the partially ligated CAs of the mice. The difference between our in vivo and in vitro results might be attributed to the suppression of ATF6 by a cholesterol-enriched diet and/or the influence of other (non-EC) components within the artery [32].
This study has a few limitations. First, the mouse CA partial ligation model might differ from a traditional mouse model of atherosclerosis induced by a high-fat diet only. However, this CA partial ligation model has been proved effective in studies on the mechanism of atherosclerotic plaque progression [12,33,34]. Second, the molecular mechanisms between CLOCK and the IRE1α-XBP1 axis are complex, warranting additional studies to elucidate the relevant mechanisms.
Nevertheless, loss of CLOCK function activated the IRE1α-XBP1 axis and subsequently increased SNAI1 expression, thereby stimulating the EndMT of ECs under DF. These findings suggest that CLOCK functions as a suppressor of carotid artery stenosis (Figure 7). Further analysis of the molecular mechanisms focusing on the CLOCK-IRE1α-XBP1 axis is needed to develop promising therapies for carotid artery stenosis.
Figure 7.
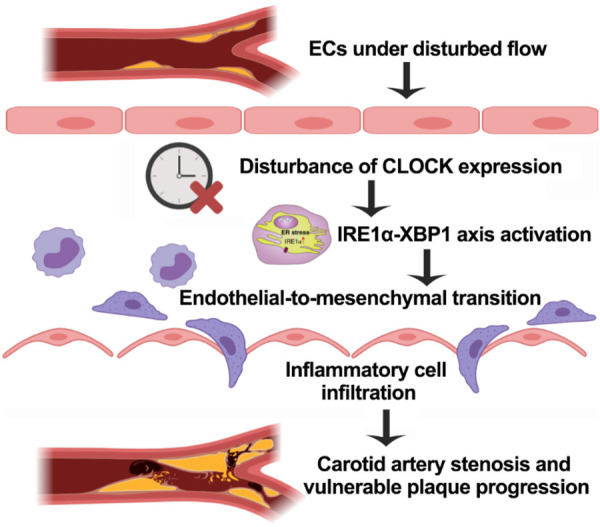
The mechanism for the inhibitory effect of CLOCK as a suppressor of carotid plaque progression. Under disturbed flow, the disturbance of endothelial CLOCK expression activates the IRE1α-XBP1 axis and then leads to endothelial-to-mesenchymal transition; this results in increased inflammation. The results of our research showed that CLOCK attenuated carotid plaque stenosis and vulnerable plaque progression through the signaling pathways above.
Acknowledgements
We thank Drs. Yong Ding and Yifan Liu for their assistance in human sample curation and Dr. Mengjiao Zhu for the technical advice on immunofluorescence and confocal microscopy analyses. This work was supported by grants from Shanghai Sailing Program (Grant No. 20YF1406700), the National Natural Science Foundation of China (Grant No. 81970408), and Science and Technology Commission of Shanghai Municipality (Grant No. 19411966900).
Disclosure of conflict of interest
None.
References
- 1.Zhou C, Yuan C, Li R, Wang W, Li C, Zhao X CARE-II Study Collaborators. Association between incomplete circle of Willis and carotid vulnerable atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 2018;38:2744–2749. doi: 10.1161/ATVBAHA.118.311797. [DOI] [PubMed] [Google Scholar]
- 2.Tang H, Zhu M, Zhao G, Fu W, Shi Z, Ding Y, Guo D. Loss of CLOCK under high glucose upregulates ROCK1-mediated endothelial to mesenchymal transition and aggravates plaque vulnerability. Atherosclerosis. 2018;275:58–67. doi: 10.1016/j.atherosclerosis.2018.05.046. [DOI] [PubMed] [Google Scholar]
- 3.Bailey KA, Moreno E, Haj FG, Simon SI, Passerini AG. Mechanoregulation of p38 activity enhances endoplasmic reticulum stress-mediated inflammation by arterial endothelium. FASEB J. 2019;33:12888–12899. doi: 10.1096/fj.201900236R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Battson ML, Lee DM, Gentile CL. Endoplasmic reticulum stress and the development of endothelial dysfunction. Am J Physiol Heart Circ Physiol. 2017;312:H355–H367. doi: 10.1152/ajpheart.00437.2016. [DOI] [PubMed] [Google Scholar]
- 5.Hutt DM, Powers ET, Balch WE. The proteostasis boundary in misfolding diseases of membrane traffic. FEBS Lett. 2009;583:2639–2646. doi: 10.1016/j.febslet.2009.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hotamisligil GS. Endoplasmic reticulum stress and atherosclerosis. Nat Med. 2010;16:396–399. doi: 10.1038/nm0410-396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Moore PC, Oakes SA. CPEB4 links the clock and the UPR to protect the liver. Nat Cell Biol. 2017;19:79–81. doi: 10.1038/ncb3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bell-Pedersen D, Cassone VM, Earnest DJ, Golden SS, Hardin PE, Thomas TL, Zoran MJ. Circadian rhythms from multiple oscillators: lessons from diverse organisms. Nat Rev Genet. 2005;6:544–556. doi: 10.1038/nrg1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao J, Warman GR, Cheeseman JF. The functional changes of the circadian system organization in aging. Ageing Res Rev. 2019;52:64–71. doi: 10.1016/j.arr.2019.04.006. [DOI] [PubMed] [Google Scholar]
- 10.Naylor AR, Ricco JB, de Borst GJ, Debus S, de Haro J, Halliday A, Hamilton G, Kakisis J, Kakkos S, Lepidi S, Markus HS, McCabe DJ, Roy J, Sillesen H, van den Berg JC, Vermassen F Esvs Guidelines Committee; Kolh P, Chakfe N, Hinchliffe RJ, Koncar I, Lindholt JS, Vega de Ceniga M, Verzini F Esvs Guideline Reviewers. Archie J, Bellmunt S, Chaudhuri A, Koelemay M, Lindahl AK, Padberg F, Venermo M. Editor’s choice - management of atherosclerotic carotid and vertebral artery disease: 2017 clinical practice guidelines of the European society for vascular surgery (ESVS) Eur J Vasc Endovasc Surg. 2018;55:3–81. doi: 10.1016/j.ejvs.2017.06.021. [DOI] [PubMed] [Google Scholar]
- 11.King DP, Zhao Y, Sangoram AM, Wilsbacher LD, Tanaka M, Antoch MP, Steeves TD, Vitaterna MH, Kornhauser JM, Lowrey PL, Turek FW, Takahashi JS. Positional cloning of the mouse circadian clock gene. Cell. 1997;89:641–653. doi: 10.1016/s0092-8674(00)80245-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nam D, Ni CW, Rezvan A, Suo J, Budzyn K, Llanos A, Harrison D, Giddens D, Jo H. Partial carotid ligation is a model of acutely induced disturbed flow, leading to rapid endothelial dysfunction and atherosclerosis. J Physiol Heart Circ Physiol. 2009;297:H1535–H1543. doi: 10.1152/ajpheart.00510.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.He D, Zhao M, Wu C, Zhang W, Niu C, Yu B, Jin J, Ji L, Willard B, Mathew AV, Chen YE, Pennathur S, Yin H, He Y, Pan B, Zheng L. Apolipoprotein A-1 mimetic peptide 4F promotes endothelial repairing and compromises reendothelialization impaired by oxidized HDL through SR-B1. Redox Biol. 2018;15:228–242. doi: 10.1016/j.redox.2017.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Warboys CM, de Luca A, Amini N, Luong L, Duckles H, Hsiao S, White A, Biswas S, Khamis R, Chong CK, Cheung WM, Sherwin SJ, Bennett MR, Gil J, Mason JC, Haskard DO, Evans PC. Disturbed flow promotes endothelial senescence via a p53-dependent pathway. Arterioscler Thromb Vasc Biol. 2014;34:985–995. doi: 10.1161/ATVBAHA.114.303415. [DOI] [PubMed] [Google Scholar]
- 15.Yang G, Zhang J, Jiang T, Monslow J, Tang SY, Todd L, Puré E, Chen L, FitzGerald GA. Bmal1 deletion in myeloid cells attenuates atherosclerotic lesion development and restrains abdominal aortic aneurysm formation in hyperlipidemic mice. Arterioscler Thromb Vasc Biol. 2020;40:1523–1532. doi: 10.1161/ATVBAHA.120.314318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Anea CB, Zhang M, Stepp DW, Simkins GB, Reed G, Fulton DJ, Rudic RD. Vascular disease in mice with a dysfunctional circadian clock. Circulation. 2009;119:1510–1517. doi: 10.1161/CIRCULATIONAHA.108.827477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang Q, Liu H, Wang S, Wang J, Tang Y, He Z, Wu F, Huang Z, Cong X, Ding R, Liang C. Circadian locomotor output cycles kaput accelerates atherosclerotic plaque formation by upregulating plasminogen activator inhibitor-1 expression. Acta Biochim Biophys Sin (Shanghai) 2018;50:869–879. doi: 10.1093/abbs/gmy087. [DOI] [PubMed] [Google Scholar]
- 18.Pan X, Jiang XC, Hussain MM. Impaired cholesterol metabolism and enhanced atherosclerosis in clock mutant mice. Circulation. 2013;128:1758–1769. doi: 10.1161/CIRCULATIONAHA.113.002885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luo H, Li QQ, Wu N, Shen YG, Liao WT, Yang Y, Dong E, Zhang GM, Liu BR, Yue XZ, Tang XQ, Yang HS. Chronological in vivo imaging reveals endothelial inflammation prior to neutrophils accumulation and lipid deposition in HCD-fed zebrafish. Atherosclerosis. 2019;290:125–135. doi: 10.1016/j.atherosclerosis.2019.09.017. [DOI] [PubMed] [Google Scholar]
- 20.Chen PY, Qin L, Baeyens N, Li G, Afolabi T, Budatha M, Tellides G, Schwartz MA, Simons M. Endothelial-to-mesenchymal transition drives atherosclerosis progression. J Clin Invest. 2015;125:4514–4528. doi: 10.1172/JCI82719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Evrard SM, Lecce L, Michelis KC, Nomura-Kitabayashi A, Pandey G, Purushothaman KR, d’Escamard V, Li JR, Hadri L, Fujitani K, Moreno PR, Benard L, Rimmele P, Cohain A, Mecham B, Randolph GJ, Nabel EG, Hajjar R, Fuster V, Boehm M, Kovacic JC. Endothelial to mesenchymal transition is common in atherosclerotic lesions and is associated with plaque instability. Nat Commun. 2016;7:11853. doi: 10.1038/ncomms11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van Meeteren LA, ten Dijke P. Regulation of endothelial cell plasticity by TGF-β. Cell Tissue Res. 2012;347:177–186. doi: 10.1007/s00441-011-1222-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moonen JR, Lee ES, Schmidt M, Maleszewska M, Koerts JA, Brouwer LA, van Kooten TG, van Luyn MJ, Zeebregts CJ, Krenning G, Harmsen MC. Endothelial-to-mesenchymal transition contributes to fibro-proliferative vascular disease and is modulated by fluid shear stress. Cardiovasc Res. 2015;108:377–386. doi: 10.1093/cvr/cvv175. [DOI] [PubMed] [Google Scholar]
- 24.Sun JX, Chang TF, Li MH, Sun LJ, Yan XC, Yang ZY, Liu YS, Xu WQ, Lv Y, Su JB, Liang L, Han H, Dou GR, Wang YS. SNAI1, an endothelial-mesenchymal transition transcription factor, promotes the early phase of ocular neovascularization. Angiogenesis. 2018;21:635–652. doi: 10.1007/s10456-018-9614-9. [DOI] [PubMed] [Google Scholar]
- 25.Tanaka M, Koyama T, Sakurai T, Kamiyoshi A, Ichikawa-Shindo Y, Kawate H, Liu T, Xian X, Imai A, Zhai L, Hirabayashi K, Owa S, Yamauchi A, Igarashi K, Taniguchi S, Shindo T. The endothelial adrenomedullin-RAMP2 system regulates vascular integrity and suppresses tumour metastasis. Cardiovasc Res. 2016;111:398–409. doi: 10.1093/cvr/cvw166. [DOI] [PubMed] [Google Scholar]
- 26.Ohta Y, Taguchi A, Matsumura T, Nakabayashi H, Akiyama M, Yamamoto K, Tanizawa Y. Clock gene dysregulation induced by chronic ER stress disrupts β-cell function. EBioMedicine. 2017;18:146–156. doi: 10.1016/j.ebiom.2017.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Liu C, Liu Y, He J, Mu R, Di Y, Shen N, Liu X, Gao X, Wang J, Chen T, Fang T, Li H, Tian F. Liraglutide increases VEGF expression via CNPY2-PERK pathway induced by hypoxia/reoxygenation injury. Front Pharmacol. 2019;10:789. doi: 10.3389/fphar.2019.00789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang X, Xia HY, Qin HY, Kang XP, Hu HY, Zheng J, Jiang JY, Yao LA, Xu YW, Zhang T, Zhang XL. 20(S)-protopanaxadiol induces apoptosis in human umbilical vein endothelial cells by activating the PERK-eIF2alpha-ATF4 signaling pathway. J Cell Biochem. 2019;120:5085–5096. doi: 10.1002/jcb.27785. [DOI] [PubMed] [Google Scholar]
- 29.Liu J, Wu Z, Han D, Wei C, Liang Y, Jiang T, Chen L, Sha M, Cao Y, Huang F, Geng X, Yu J, Shen Y, Wang H, Feng L, Wang D, Fang S, Wang S, Shen Y. Mesencephalic astrocyte-derived neurotrophic factor inhibits liver cancer through small ubiquitin-related modifier (SUMO)ylation-related suppression of NF-κB/Snail signaling pathway and epithelial-mesenchymal transition. Hepatology. 2020;71:1262–1278. doi: 10.1002/hep.30917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Anspach L, Tsaryk R, Seidmann L, Unger RE, Jayasinghe C, Simiantonaki N, Kirkpatrick CJ, Pröls F. Function and mutual interaction of BiP-, PERK-, and IRE1α-dependent signaling pathways in vascular tumours. J Pathol. 2020;251:123–134. doi: 10.1002/path.5423. [DOI] [PubMed] [Google Scholar]
- 31.Yang J, Xu J, Danniel M, Wang X, Wang W, Zeng L, Shen L. The interaction between XBP1 and eNOS contributes to endothelial cell migration. Exp Cell Res. 2018;363:262–270. doi: 10.1016/j.yexcr.2018.01.016. [DOI] [PubMed] [Google Scholar]
- 32.Tumanovska LV, Swanson RJ, Serebrovska ZO, Portnichenko GV, Goncharov SV, Kysilov BA, Moibenko OO, Dosenko VE. Cholesterol enriched diet suppresses ATF6 and PERK and upregulates the IRE1 pathways of the unfolded protein response in spontaneously hypertensive rats: relevance to pathophysiology of atherosclerosis in the setting of hypertension. Pathophysiology. 2019;26:219–226. doi: 10.1016/j.pathophys.2019.05.005. [DOI] [PubMed] [Google Scholar]
- 33.Schürmann C, Dienst FL, Pálfi K, Vasconez AE, Oo JA, Wang S, Buchmann GK, Offermanns S, van de Sluis B, Leisegang MS, Günther S, Humbert PO, Lee E, Zhu J, Weigert A, Mathoor P, Wittig I, Kruse C, Brandes RP. The polarity protein Scrib limits atherosclerosis development in mice. Cardiovasc Res. 2019;115:1963–1974. doi: 10.1093/cvr/cvz093. [DOI] [PubMed] [Google Scholar]
- 34.Seo Y, Park J, Choi W, Ju Son D, Sung Kim Y, Kim MK, Yoon BE, Pyee J, Tae Hong J, Go YM, Park H. Antiatherogenic effect of resveratrol attributed to decreased expression of ICAM-1 (intercellular adhesion molecule-1) Arterioscler Thromb Vasc Biol. 2019;39:675–684. doi: 10.1161/ATVBAHA.118.312201. [DOI] [PubMed] [Google Scholar]