

Abstract
Objective: To probe into the role and regulatory mechanisms of INSR in pathogenesis of osteoarthritis (OA). Methods: KLF4 and INSR expression was detected in cartilage tissues of 40 OA patients and 10 controls using RT-qPCR. IL-1β-induced OA chondrocytes and anterior cruciate ligament transection (ACLT)-induced OA models were respectively constructed. After overexpressing or silencing KLF4 or INSR, flow cytometry assay was utilized to detect chondrocyte apoptosis. Furthermore, JAK2/STAT3, cartilage markers and OA-related markers were examined by western blot. Dual luciferase report and CHIP assay were carried out to verify the interactions between KLF4 and INSR, followed by functional gain and loss assay. INSR promoter methylation was assessed by MS-PCR. Results: Both KLF4 and INSR were down-regulated both in OA chondrocytes and cartilage tissues. Knockdown of KLF4 or INSR accelerated apoptosis of IL-1β-induced OA chondrocytes. However, overexpression of KLF4 or INSR ameliorated OA progression both in OA chondrocytes and OA mouse models. Moreover, INSR inactivated JAK2/STAT3 pathway in OA chondrocytes. Dual luciferase report and CHIP assay results confirmed that INSR was transcriptionally regulated by KLF4. As shown in MS-PCR results, INSR expression was mediated by DNA methylation in OA. Conclusion: Our findings suggested that INSR, as a key regulator for OA, was regulated by transcription factor KLF4 and DNA methylation, thereby mediating the activation of JAK2/STAT3 signaling, which was considered as an underlying therapeutic target for OA.
Keywords: INSR, KLF4, DNA methylation, osteoarthritis, JAK2/STAT3 signaling
Introduction
Osteoarthritis (OA) is the most common degenerative joint disease, which has been the main cause of disability in the elderly, affecting 200 million people worldwide [1]. It is characterized by continued destruction of articular cartilage, as well as inflammation and degeneration [2]. OA puts a heavy burden on the society and economy. So far, the etiology and pathogenesis of OA have not been fully understood.
Accumulated evidence shows that inflammatory factors (such as IL-1β) stimulate the synthesis of matrix-degrading enzymes (such as MMP3/13), resulting in the degradation and destruction of articular cartilage tissues [3-5]. As an example, IL-1β stimulation induces the activation of MAPK and NF-kB signaling pathways, thereby mediating the expression of chondrocyte markers (such as Col2a1 Aggrecan) and OA markers (such as MMP13, ADAMTS-4 and ADAMTS-5) [6,7].
Kruppel-like factor (KLF)-4 (KLF4), a family membrane of zinc finger protein regulates various cellular processes like proliferation, differentiation and apoptosis [11]. Increasing evidence suggests that KLF4 may mediate chondrocyte physiology [12]. Its overexpression stimulates the expression of chondrogenic MMPs and aggrecanase, which contributes to cartilage maintenance [13]. Furthermore, KLF4 blocks IL-1β-induced the activation of NF-kB signaling pathway, which may prevent OA progression [14]. A global gene expression analysis shows that lowly expressed transcription factor KLF4 targets many dysregulated genes in OA cartilage tissues [15]. Although these studies have explicitly confirmed the potential role of KLF4 on OA progression, there is still a lack of molecular regulatory mechanisms concerning KLF4 in the development of OA. In addition to MAPK and NF-kB, JAK2/STAT3 signaling is involved in chondrocyte and OA development. In 2003, Legendre F, et al. has found that JAK2/STAT3 signaling could mediate IL-induced inhibition of the expression of chondrocyte makers and contribute to progression of OA [16]. Thus, JAK2/STAT3 signaling has been considered as a therapeutic target for OA therapy [17].
Abnormal gene expression induced by transcription factors or DNA methylation plays an important role in regulating the pathogenesis of OA [8]. For example, the transcription factor, HIF-1α inhibits the expression of catabolic genes by inhibiting NF-κB signaling, such as MMP13 and Hif2a [9]. DNA methylation participates in the progress of OA through mediation of CtBP expression [10]. As previous studies, INSR is involved in multiple biological processes such as inflammatory response [19] and apoptosis [20]. Its abnormal expression contributes to the progression multiple diseases like cancers [21] and Alzheimer’s disease [22]. It has been reported that insulin receptor (INSR) expression is down-regulated in OA chondrocytes [18]. Nevertheless, the role of INSR in OA remains unclear. Furthermore, the regulatory mechanism of its low expression in OA is also unclear. In this study, we identified INSR as a novel key regulator for OA. INSR could be double regulated by transcription factor KLF4 and DNA methylation, thereby affecting JAK2/STAT3 signaling pathway activation during OA progression.
Materials and methods
Bioinformatics analysis
GSE114007, GSE43269, GSM1364027, GSM2183789, GSM447584 and GSM2183790 datasets were downloaded from the Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/). GSE114007 RNA-seq dataset was composed of 20 OA and 18 healthy human cartilage knee samples on the GPL11154 platform [15], which was used for differential expression analysis under the screening criterion of |log2fold change (FC)| > 1 and FDR < 0.05. GSE43269 methylation profiles included 23 OA and 18 healthy human cartilage knees based on the GPL8490 platform [23]. The average methylation difference of each probe group was calculated. Wilcoxon rank sum test was used to screen probes with p-value < 0.05 as differentially methylated genes. The average methylation difference of each probe in each group was calculated. Wilcoxon rank sum test was used to screen probes with p-value < 0.05 as differentially methylated genes. Target genes of KLF4 were screened using the GSM1364027, GSM2183789, GSM447584 and GSM2183790 datasets. According to the DisGeNET (http://www.disgenet.org/web/DisGeNET/menu) database, OA-related genes were identified, which were validated as reported studies. Interested genes related to OA were imported into the STRING (version 11.0; https://string-db.org/) database [24]. A protein-protein interaction (PPI) network was visualized via Cytoscape 3.6.1 [25].
Clinical specimens
Cartilage tissues were collected from 40 OA patients who underwent synovectomy or arthroplasty and 10 normal controls in the Sun Yat-sen Memorial Hospital, Sun Yat-sen University between 2016 and 2018. All procedures performed in this study involving human participants were in line with the Helsinki Declaration. All participants signed written informed consent. This study got the approval of the Ethics Committee of Sun Yat-sen Memorial Hospital, Sun Yat-sen University (2016082).
OA model construction
A total of 40 eight-week-old male C57BL/6J mice were purchased from Hujingda Laboratory Animal Co., Ltd. (Hunan, China). All animals were kept in an environment of 23±2°C, humidity of 50±10%, and 12:12 light cycle. In this study, anterior cruciate ligament transection (ACLT) was adopted to induce OA in mice [26]. All mice were randomly separated into sham operation group (n=10) and ACLT operation group (n=30). In brief, after anesthetizing, the right knee joint was exposed by a medial parapatellar method. The ACL was transected using micro-scissors. Complete transection was confirmed via a positive anterior drawer sign. The mice in the control group experienced arthrotomy without ACLT. Following surgery, all mice exercised freely for 20 min a day. After 2 weeks, the success of the model was determined by immunohistochemistry. The successful rate was 93.3% (28/30). Then, pcDNA3.1-KLF4 lentivirus was injected into the joint. After the experiment was over, all mice were euthanized through intraperitoneal injection of an overdose of pentobarbital sodium (200 mg/kg). The study was approved by the Animal Ethics Committee of Sun Yat-sen Memorial Hospital, Sun Yat-sen University (2016082).
Chondrocyte isolation and culture
Chondrocytes were isolated from articular cartilage tissue of mice. Briefly, following repeatedly washing cartilage tissues with PBS under sterile conditions, the synovium and fibrous tissues attached to cartilage tissues were carefully peeled off, and then tissues were cut to a thickness of 1 mm3. The pieces were digested with 0.05% type II collagenase for 30 min, followed by centrifugation at 1000 r/min. Then, samples were digested with 0.1% collagenase type II and 0.25% trypsinase at 37°C for 60-100 min. Finally, the isolated chondrocytes were cultured in DMEM medium plus 20% FBS at an atmosphere of 37°C and 5% CO2. The purity of chondrocytes was determined by flow cytometry. Briefly, chondrocytes in log phase were collected. After digestion with 0.25% trypsin, the number of cells were counted. After resuspension, the samples were centrifuged at 1,000 r/min for 5 min. After discarding the supernatant, cells were incubated with anti-col2A1 (1:1000; ab188570, abcam, USA) and FITC-labeled flow cytometry antibodies. Following fixation, flow cytometry analysis was presented.
Transfection
pCDNA3.1-KLF4, PCDNA3.1-INSR, shKLF4, shINSR and their corresponding vectors from Shanghai Generay Biotech Co., Ltd. (Shanghai, China) were transfected into chondrocytes via Lipofectamine 3000 (Invitrogen, Carlsbad, California, USA). The sequences of three siRNAs targeting KLF4 were as follows: 5’-GATGGCTGTGGGTGGAAATTT-3’; 5’-UGAGAUGGGAACUCUUUGUGUAGGU-3’; 5’-CCAUUAUCAAGAGCUCAUGCCACCG-3’. The sequence of pCDNA3.1-KLF4 was as follows: 5’-CCCCGGATGAGTGAGGGGGCTGGAGTGAGTCAC-3’. After 24 h, transfection efficiency was assessed by RT-qPCR and western blot. 48 h after transfection, chondrocytes were stimulated with 10 ng/ml IL-1β [27].
RT-qPCR
RNA extraction from tissues or chondrocytes was presented utilizing TRIzol reagent (Invitrogen). First strand cDNA was synthesized with 1 μg RNA by PrimerScript™ RT Reagent Kit (Takara, Beijing, China). RT-qPCR was carried out through miScript SYBR-Green PCR Kit (Qiagen, Nasdaq, New York, USA) on the ABI7500 Sequence Detection System (Applied Biosystems, Shanghai, China). The PCR conditions were as follows: one cycle for 2 min at 50°C (initial denaturation); one cycle for 10 min at 95°C (denaturation); 40 cycles for 15 sec at 95°C (annealing and elongation) and 40 cycles for 1 min at 60°C (final extension). Relative expression levels were calculated using the 2-ΔΔCT method. The primer sequences are in Table 1. β-actin was utilized as the internal reference.
Table 1.
The primer sequences for RT-qPCR
| Gene | Sequence (5’-3’) |
|---|---|
| KLF4 | GTCCCGGGGATTTGTAGCTC (forward) |
| TGTAGTGCTTTCTGGCTGGG (reverse) | |
| Aggrecan | CAGACCATGACAACTCGCTG (forward) |
| GCAGCACTACCTCCTTCTCC (reverse) | |
| MMP3 | TGAGGACACCAGCATGAACC (forward) |
| ACTTCGGGATGCCAGGAAAG (reverse) | |
| Col2a1 | GCAGGATGGGCAGAGGTATAA (forward) |
| CGAGGTCAGTTGGGCAGATG (reverse) | |
| MMP13 | CAGTTTGCAGAGCGCTAC (forward) |
| TCAAGTTTGCCAGTCACC (reverse) | |
| ADAMTS5 | CTCGGGAGGATTTATGTG (forward) |
| ATGCTGGTAAGGATGGAA (reverse) | |
| ADAMTS-4 | GGAAATTCAGATGTGGTACTGCC (forward) |
| GCCACTAGGACTTGCAGTGT (reverse) | |
| INSR | GATCCGCGCCGCCTTTT (forward) |
| GGGACTGTCTCTCGGCTCTC (reverse) | |
| β-actin | AGGGGCCGGACTCGTCATACT (forward) |
| GGCGGCACCACCATGTACCCT (reverse) |
Western blot
The tissue or cells were lysed through RIPA lysate. The BCA protein kit (Pierce, Shanghai, China) was utilized to measure protein concentration. Protein samples were separated by 12% SDS-PAGE and transferred to PVDF membrane. The membrane was blocked with 5% skim milk and incubated with primary antibodies (KLF4 (1:1000; ab215036, abcam, USA), INSR (1:1000; ab227831, abcam, USA), MMP3 (1:1000; ab52915, abcam, USA), MMP13 (1:1000; ab51072, abcam, USA), ADAMTS-4 (1:200; ab84792, abcam, USA), col2A1 (1:1000; ab188570, abcam, USA), JAK2 (1:2000; ab245303, abcam, USA), STAT3 (1:5000; ab119352, abcam, USA), p-JAK2 (1:1000; ab195055, abcam, USA), p-STAT3 (1:2000; ab76315, abcam, USA) and GAPDH (1:1000; ab8245, abcam, USA)) and HRP-conjugated goat anti-mouse (1:5000; ab97040, abcam, USA) or anti-rabbit (1:5000; ab7090, abcam, USA) secondary antibodies. GAPDH was used as the internal reference. Enhanced chemiluminescence (ECL) was used to visualize the protein blots. ImageJ software was used to quantify grayscale values of each protein.
Flow cytometry
Cell apoptosis was detected via an Annexin V-fluorescein Isothiocyanate (FITC) Apoptosis Kit (Biovision, Milpitas, CA). In brief, after washing using phosphate-buffered saline (PBS) for twice, cells (2×105) were incubated with 200 μl binding buffer and 5 μl Annexin-V FITC (20 mg/ ml) in the dark on ice for 15 min. Then, cells were treated with 10 μl propidium iodide (PI). Apoptosis was analyzed by flow cytometry BD FACSCalibur (BD Biosciences, San Jose, CA).
Immunohistochemistry and Safranin O and fast green staining
Cartilage tissues were fixed in 4% paraformaldehyde and embedded into paraffin. The section was at a thickness of 5 μm. After deparaffinizing in xylene and hydrating with graded ethanol, the sections were stained with Safranin O and fast green. Then, the sections were stained with hematoxylin for 1 min, 0.02% fast green for 5 min, and 0.1% Safranin O for 30 min. The results were investigated under a microscope.
Immunohistochemistry
Cartilage tissues were fixed in 4% paraformaldehyde for paraffin embedding. The slices are cut into 4 μm thick. The sections were dried, deparaffinized, rehydrated, and then placed on the slides for immunohistochemistry analysis. Then sections were incubated with a primary antibody against KLF4 (ab106629, Abcam, Cambridge, United Kingdom), followed by HRP-conjugated secondary antibody.
Dual luciferase activity
A luciferase assay was performed to detect the effects of KLF4 on transcriptional regulation of INSR. An upstream promoter region of INSR was cloned into the specific luciferase reporter pGL3 basic vector (Promega, Madison, Wisconsin, USA). Then, 0.5 mg of the luciferase was constructed and 100 ng cytomegalovirus-Renilla vector were co-transfected into cells via lipofectamine 2000 (Life Technologies). After 24 h, cells were transfected with the KLF4 lentiviral vector and incubated for 24 h. A commercial dual luciferase detection kit (Promega) was used to analyze the luciferase activity.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (CHIP) assay was utilized to evaluate whether KLF4 could bind to the INSR promoter. Briefly, chondrocytes were transfected with KLF4 lentiviral vector. Cells were cross-linked with 1% formaldehyde for 10 min. The reaction was stopped with 100 mM glycine. Then, intracellular DNA was sheared into 0.3- to 3-kb fragments and incubated with an anti-KLF4 antibody overnight at 4°C. A subsequent PCR analysis was performed with the specific INSR primers. The reaction mixture was analyzed on 1.8% agarose gel electrophoresis.
Methylation-specific polymerase chain reaction (MS-PCR)
Chondrocyte DNA was extracted using a QIAamp DNA Blood Mini Kit (Qiagen, 51104), and genomic DNA was processed using a CpGenome DNA kit (Chemicon International Inc., Temecula, CA). INSR promoter methylation was assessed via a two-step method. Methylation primers were as follows: M-F: GGTCGAGAGTCGAGAGATAGTTTC and M-R: CAAATACTAAACGAAAACCCTTACG. Nonmethylated primers were as follows: U-F: GGGTTGAGAGTTGAGAGATAGTTTT and U-M: AATACTAAACAAAAACCCTTACAAT. The fragments of methylated and nonmethylated PCR products were 128 bp and 127 bp, respectively.
Statistical analysis
Data are presented as the means ± standard deviation (SD). All statistical analysis was carried out using GraphPad Prism 8.0 (GraphPad, San Diego, CA). Comparisons between two groups were assessed by student’s t test, while multiple comparisons were presented using one-way ANOVA followed by Tukey’s post hoc test. P < 0.05 was statistically significant.
Results
Identification of down-regulated and hypermethylated target genes of KLF4
From GSE114007 dataset, a total of 2247 differentially expressed genes were screened between OA and normal cartilage tissues, including 936 down-regulated and 1311 up-regulated genes (Figure 1A). Among them, KLF4 was down-regulated in OA cartilage tissues (Figure 1B). Using GSE43269 dataset, 1381 hypermethylated genes were identified in OA than normal cartilage tissues. Through comprehensively analyzing GSM1364027, GSM2183789, GSM447584 and GSM2183790 dataset, we identified 8130 target genes of KLF4. After overlapping target genes of KLF4, hypermethylated and down-regulated genes, a total of 28 genes were finally identified for further analysis (Figure 1C). Heat map depicted the expression patterns of the 28 genes between OA and normal cartilage tissues (Figure 1D). Using the DisGeNET database, ten genes have been reported to be related to OA, including COL1A1, COL1A1, COL1A2, COL2A1, COL9A2, COL11A2, GDF5, FBN1, FRZB and SMAD3. Then, a PPI network was constructed where INSR could be considered as a hub gene (Figure 1E).
Figure 1.
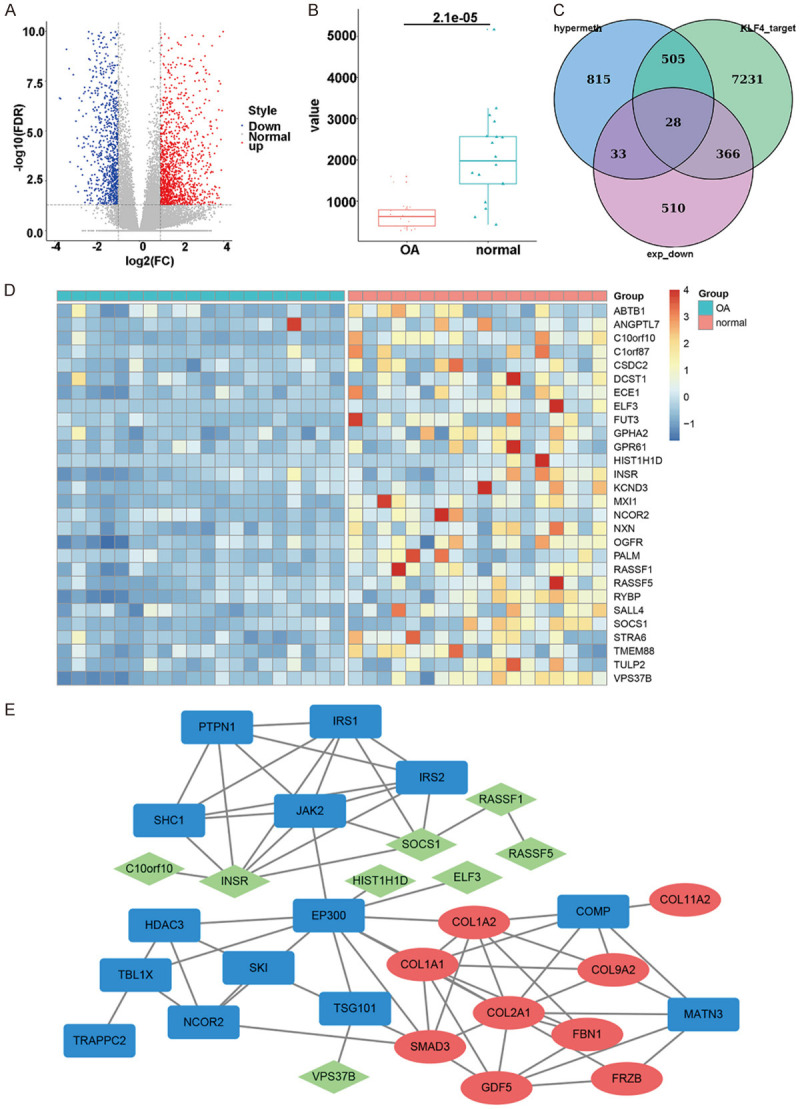
Bioinformatics analysis was utilized to identify down-regulated and hypermethylated target genes of KLF4. A. Volcano plots depicting differentially expressed genes between OA and normal cartilage tissues. Red: up-regulation; blue: down-regulation. B. Box plots showing the expression patterns of KLF4 between OA and normal cartilage tissues. C. Venn diagram was utilized to overlap down-regulated and hypermethylated target genes of KLF4. D. Heat map visualizing the expression patterns of above genes between OA and normal cartilage tissues. E. A PPI network for OA.
Knockdown of KLF4 accelerates apoptosis of IL-1β-induced OA chondrocytes
To verify bioinformatics analysis results, we presented RT-qPCR assay to detect the expression of KLF4 between OA and normal cartilage tissues. As expected, down-regulated KLF4 was found in OA cartilage tissues (Figure 2A). We then further probed into the function of KLF4 in OA progression. Chondrocytes were isolated from normal cartilage tissues. The flow cytometry results confirmed that the purity of chondrocytes was up to 95% (Figure 2B). We designed and synthesized three shRNAs targeting KLF4 as well as pcDNA3.1-KLF4. As shown in Figure 2C, shKLF4-3 had the most stable transfection effect in chondrocytes, which was chosen for further analysis. Also, KLF4 was successfully overexpressed in chondrocytes following transfection with pcDNA3.1-KLF4 (Figure 2D). Transfection effect was also confirmed by western blot (Figure 2E, 2F). Intriguingly, we found that the apoptotic rate of IL-1β-induced OA chondrocytes was elevated by shKLF4 and decreased by pcDNA3.1-KLF4 (Figure 2G, 2H).
Figure 2.

Knockdown of KLF4 accelerates apoptosis of IL-1β-induced OA chondrocytes. A. Low KLF4 expression in OA than control cartilage tissues via RT-qPCR. B. The purity of isolated chondrocytes was determined by flow cytometry. C-F. Transfection efficiency of shKLF4 and pcDNA3.1-KLF4 was assessed through RT-qPCR and western blot. G, H. Apoptosis rate was determined after transfection with shKLF4 and pcDNA3.1-KLF4 in IL-1β-induced OA chondrocytes. I-K. MMP3/13, COL2A1, Aggrecan and ADAMTS-4/5 expression was quantified using RT-qPCR. *P < 0.05; ***P < 0.001; ****P < 0.0001; ns: no statistical significance.
KLF4 ameliorates OA progression both in IL-1β-induced OA chondrocytes and ACLT-induced OA mouse model
Our RT-qPCR results showed that shKLF4 distinctly promoted MMP3 and MM13 expression, while pcDNA3.1-KLF4 inhibited their expression in IL-1β-induced OA chondrocytes (Figure 2I). shKLF4 suppressed COL2A1 and Aggrecan expression and promoted ADAMTS-4/5 expression in IL-1β-induced OA chondrocytes, and converse results were detected after overexpressing KLF4 (Figure 2J, 2K). We established an ACLT-induced OA mouse model. Safranin O and fast green staining results suggested that the OA model was successfully constructed (Figure 3A). Our immunohistochemistry results showed that KLF4 was primarily expressed in the nucleus of chondrocytes, and its expression was decreased as OA severity increased (Figure 3B). In Figure 3C, after injection with pcDNA3.1-KLF4, OA progression was obviously suppressed. Furthermore, western blot results showed that COL2A1 and Aggrecan expression was increased, whereas MMP3, MMP13 and ADAMTS-4 expression was decreased (Figure 3D, 3E) in OA model after overexpression of KLF4. These results indicated that KLF4 could inhibit the progression of OA.
Figure 3.
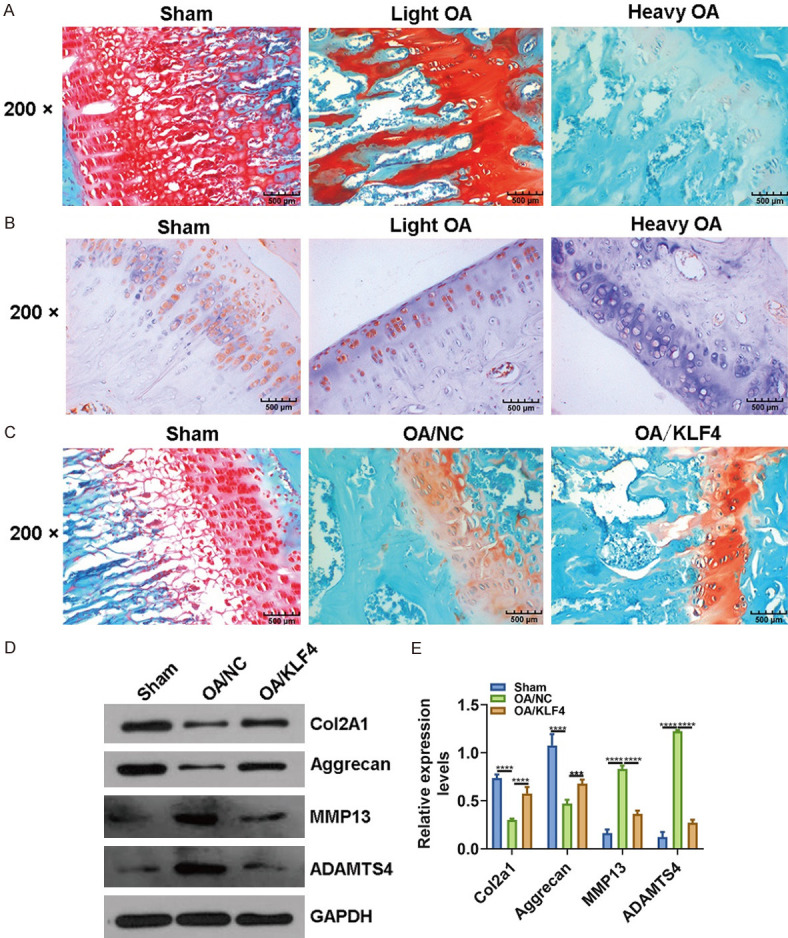
Overexpression of KLF4 suppresses the progression of OA in an OA mouse model. A. Safranin O and fast green staining was used to assess the pathological changes of cartilage tissues in an ACLT-induced OA mouse model. B. Immunohistochemistry was presented to detect the expression of KLF4 in an OA mouse model. C. Safranin O and fast green staining was presented to detect the pathological changes of cartilage tissues in an OA mouse model following injection with pcDNA3.1-KLF4. Magnification: 200×. D, E. COL2A1, Aggrecan, MMP3, MMP13 and ADAMTS-4 expression was examined in OA cartilage tissues using RT-qPCR. ***P < 0.001; ****P < 0.0001.
INSR promotes cell apoptosis and inactivates JAK2/STAT3 pathway in IL-1β-induced OA chondrocytes
Our RT-qPCR results revealed that INSR was lowly expressed in OA cartilage tissues, which was consistent with bioinformatics results (Figure 4A). As shown in Figure 4B, 4C, western blot results confirmed that INSR was successfully overexpressed and silenced in chondrocytes transfected with pcDNA3.1-INSR and shINSR, respectively. After overexpression of INSR, the apoptosis of IL-1β-induced OA chondrocytes was distinctly suppressed (Figure 4D, 4E). Furthermore, western blot results showed that p-JAK2 and p-STAT3 levels not JAK2 and STAT3 were prominently elevated in IL-1β-induced OA chondrocytes transfected with pcDNA3.1-INSR (Figure 4F, 4G).
Figure 4.
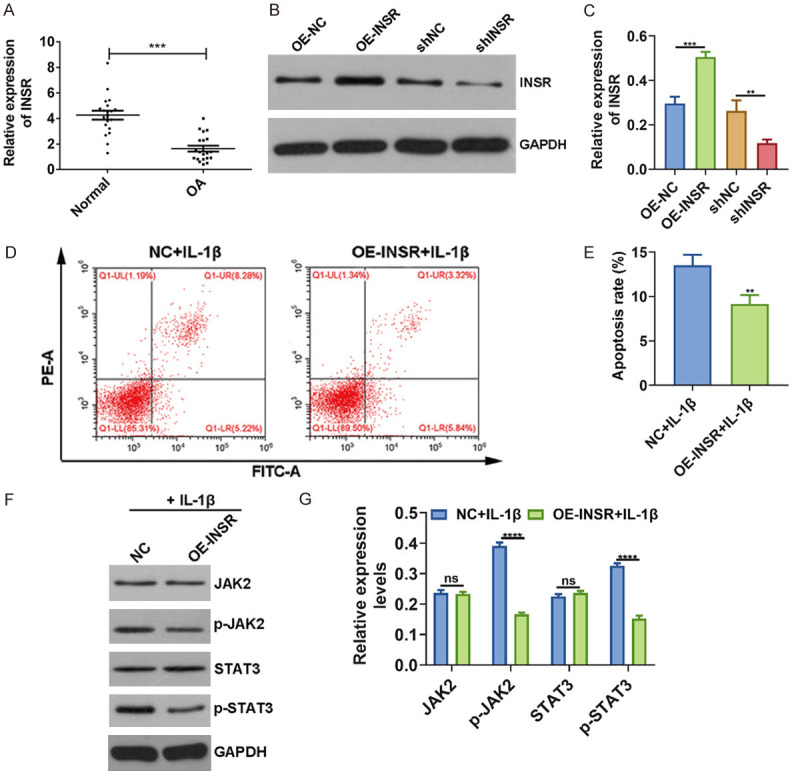
INSR promotes cell apoptosis and inactivates JAK2/STAT3 pathway in IL-1β-induced OA chondrocytes. A. Low INSR expression in OA cartilage tissues according to RT-qPCR. B, C. Transfection efficiency of pcDNA3.1-INSR and shINSR was evaluated in chondrocytes. D, E. Flow Cytometry showing lower apoptosis rate of IL-1β-induced OA chondrocytes transfected with pcDNA3.1-INSR. F, G. JAK2, p-JAK2, STAT3 and p-STAT3 levels were determined in chondrocytes using western blot. **P < 0.01; ***P < 0.001; ****P < 0.0001; ns: no statistical significance.
KLF4 ameliorates OA progression via activation of INSR transcription
We found that INSR mRNA and protein expression was distinctly decreased after transfection with shKLF4 and was significantly increased following overexpressing KLF4 in chondrocytes (Figure 5A-C). Further analysis results showed that shINSR elevated the expression of MMP3/13 and ADAMTS-4/5 and decreased the expression of COL2A1 and Aggrecan in IL-1β-induced OA chondrocytes, which was reversed after overexpressing KLF4 (Figure 5D, 5E), indicating that KLF4 could mediate the expression of these proteins by targeting INSR. Furthermore, western blot results suggested that KLF4 overexpression could reverse the activation of JAK2 and STAT3 induced by shINSR in IL-1β-induced OA chondrocytes (Figure 5F, 5G). To further probe into the mechanism by which KLF4 could regulate INSR, the LASAGNA-Search 2.0 (http://biogrid-lasagna.engr.uconn.edu/lasagna_search/) online software was utilized to predict the binding sites of KLF4 and INSR (Figure 5H). Dual luciferase report results revealed that KLF4 promoted INSR promoter activity (Figure 5I). Furthermore, Chip-qPCR results showed that KLF4 bound to the INSR promoter (Figure 5J), indicating that KLF4 could promote INSR expression by activating its promoter. Above results demonstrated that KLF4 could mediate OA progression via the activation of INSR transcription.
Figure 5.
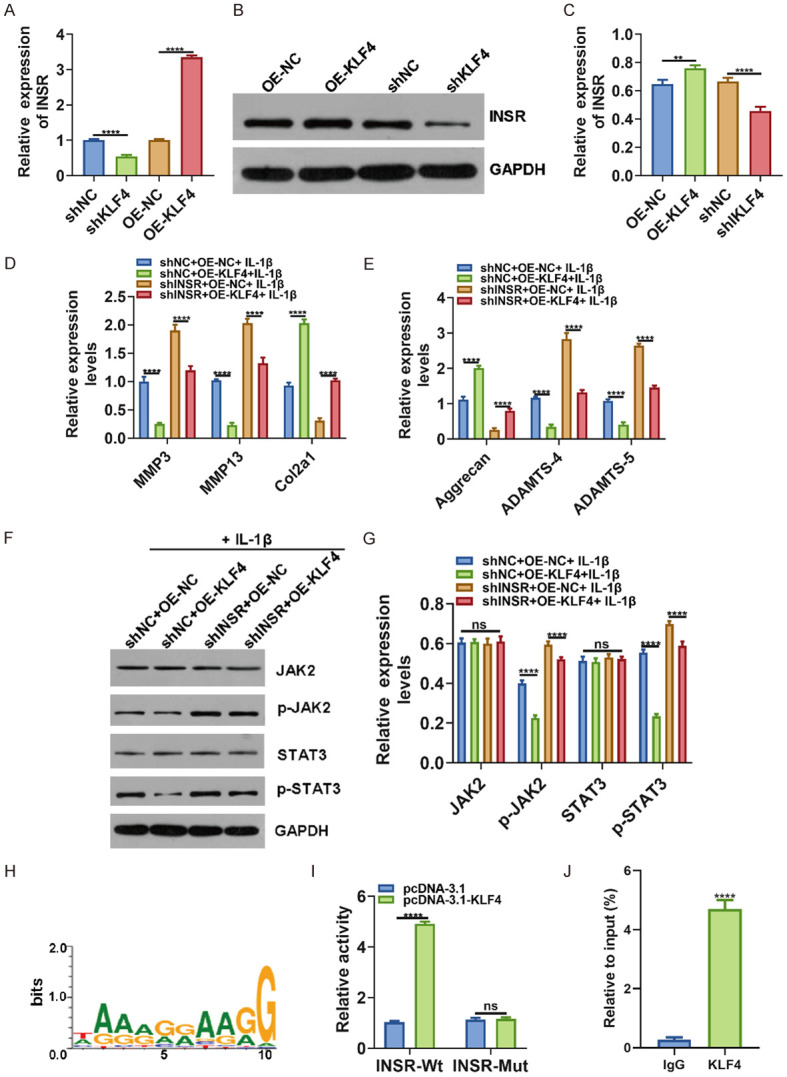
KLF4 ameliorates OA progression via targeting INSR. (A) RT-qPCR was utilized to determine INSR expression in chondrocytes transfected with shKLF4. (B, C) Western blot was presented to examine INSR expression in chondrocytes transfected with pcDNA3.1-KLF4. (D, E) MMP3/13, COL2A1, Aggrecan and ADAMTS-4/5 expression was quantified in IL-1β-induced OA chondrocytes transfected with shINSR and/or KLF4 using RT-qPCR. (F, G) Western blot was utilized to examine the expression of JAK2/p-JAK2 and STAT3/p-STAT3. (H) Prediction of binding sites between KLF4 and INSR. (I) INSR promoter activity was detected using dual luciferase report. (J) Chip-PCR confirmed that KLF4 could bind to the INSR promoter. **p < 0.01; ****p < 0.0001; ns: no statistical significance.
DNA methylation mediates INSR expression in OA
It has been known that DNA methylation could regulate the expression of INSR. In this study, the methylation region of the INSR promoter was predicted, as shown in Figure 6A. The methylation level of INSR was examined in OA and normal cartilage tissues by MSP. The results were consistent with the bioinformatics results (Figure 6B). Furthermore, methylation inhibitor 5-Aza could effectively promote the expression of INSR (Figure 6C), suggesting that hypermethylation of INSR promoter could inhibit the expression of INSR in OA.
Figure 6.
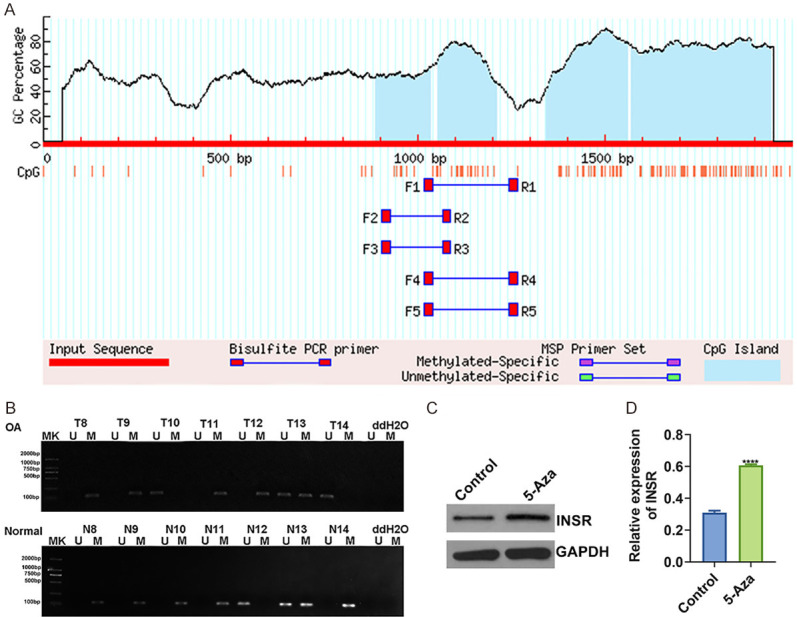
DNA methylation mediates INSR expression in OA. (A) Prediction of methylation sites in the INSR promoter. (B) Methylation levels of INSR were detected in OA and normal cartilage tissues by MSP. (C, D) Western blot showing the promotion effect of methylation inhibitor 5-Aza on INSR expression in OA cartilage tissues. ****p < 0.0001.
Discussion
In the current study, we probed into a novel regulator INSR for OA. INSR was down-regulated in OA cartilage tissues and chondrocytes, induced by KLF4 transcription regulation and DNA methylation. Overexpression of INSR could inactivate JAK2/STAT3 pathway and suppress OA progression, which possessed potential as a therapeutic target for OA.
We employed the GSE114007 dataset to confirm that KLF4 expression was down-regulated in OA cartilage tissues, consistent with previous research results [15,28]. Its down-regulation has been confirmed in OA compared to non-OA samples using RNA-seq analysis [15]. Furthermore, it has been found that Astragalus polysaccharide, a traditional Chinese herbal medicine, could ameliorate lipopolysaccharide-induced cell injury in ATDC5 cells by mediating KLF4 expression [28]. Thus, it is of importance to further explore the regulatory mechanisms of KLF4 in OA. Herein, we further verified the prediction results of bioinformatics. Consistently, the expression of KLF4 was down-regulated in OA cartilage tissues. Its overexpression inhibited IL-1β-induced apoptosis of OA chondrocytes. Additionally, RT-qPCR results showed that overexpression of KLF4 stimulated the expression of cartilage markers, but suppressed the expression of OA-related markers. These results indicated that KLF4, as an inhibitor of OA progression, could inhibit the apoptosis of chondrogenic cells and synthesis of matrix degrading enzymes, and could protect the maintenance of chondrogenic matrix. Also, in an OA mouse model, KLF4 overexpression attenuated the progression of OA. Herein, we concluded that KLF4 could inhibit the progression of OA in vivo and in vitro.
Through comprehensive analysis of multiple datasets, we identified 28 KLF4-targeted hypermethylation and low expression candidate genes in OA cartilage tissues. Among them, INSR was considered as a hub gene in the OA-related PPI network, indicating that it had a strong correlation with OA. According to our bioinformatics prediction, KLF4 may target INSR and regulate its expression in chondrocytes. Thus, our study further studied the role of INSR in OA. A previous finding has demonstrated that INSR expression is reduced in OA chondrocytes [18]. However, the exact role of INSR in the pathogenesis of OA remains unknown. INSR expression was down-regulated in OA cartilage tissues in this study. Overexpression of INSR inhibited apoptosis in IL-1β-induced OA chondrocytes and the expression of matrix-degrading enzymes including MMP3, MMP13 and ADAMTS-4/5, but stimulated the expression of chondrogenic markers including Col2a1 and Aggrecan [6,7]. These results indicated that INSR could exert an inhibitory effect on the OA development. Additionally, we observed that INSR suppressed the activation of JAK2/STAT3 signaling pathway. Previous studies have confirmed that JAK2/STAT3 signaling pathway can contribute to the development of OA [29]. This pathway is activated in IL-1β-induced OA chondrocytes and OA mouse models, and its inactivation can suppress the degradation of articular cartilage of OA [29]. Therefore, INSR may be used as a therapeutic target for OA by inhibiting JAK2/STAT3 signaling pathway. As previous studies, JAK2/STAT3 pathway has been confirmed to be mediated by several factors. For instance, lncRNA DANCR regulates the proliferation and apoptosis of chondrocytes in OA through the JAK2-STAT3 pathway [30]. HIF-1α induces osteocyte-mediated osteoclastogenesis via activation of JAK2/STAT3 pathway [31]. In this study, we discovered the novel role of INSR in regulating the pathogenesis of OA.
In line with our bioinformatic analysis, INSR may be a regulatory target of transcription factor KLF4. The experimental results showed that overexpression of KLF4 stimulated the expression of INSR, while knockdown of KLF4 suppressed its expression. These results suggested that KLF4 may be an upstream factor of INSR and positively regulated its expression. Moreover, overexpression of INSR inhibited the cartilage formation mediated by KLF4 gene silencing, including chondrogenic markers and OA-related markers. Intriguingly, knockdown of KLF4 activated the JAK2/STAT3 signal pathway. However, INSR overexpression inhibited the activation of this pathway. More importantly, our dual luciferase report and ChIP results confirmed that KLF4 may directly bind to the predicted promoter region of INSR. As a transcription factor, KLF4 can activate INSR expression by binding to the promoter region. These results indicated that KLF4 may inhibit the activation of JAK2/STAT3 signaling by inducing INSR expression, thereby negatively affecting OA progression. Interestingly, we also confirmed the epigenetic mechanism that regulated INSR expression. Consistent with bioinformatics predictions, MSP analysis showed that INSR was hypermethylated in OA cartilage tissues. DNA methylation status can suppress gene expression [32]. INSR hypermethylation has been found in various diseases, such as obesity [33], diabetes [34] and endometrial cancer [35]. Therefore, in addition to transcription factor KLF4, DNA hypermethylation status also suppressed the expression of INSR in OA.
Collectively, the present study focused on the role of KLF4 and INSR and revealed a new mechanism of OA pathogenesis. KLF4 stimulated INSR expression, which led to the suppression of JAK2/STAT3 signaling and ultimately exerted a suppressive role in OA progression. We also identified that the inhibition of INSR expression in OA cartilage was mediated by the downregulation of KLF4 and high levels of INSR gene methylation. Our discoveries could provide a new mechanism of OA pathogenesis and facilitate the development of therapeutic agents for the treatment of OA.
Conclusion
In summary, INSR was identified as a key regulator for OA, which distinctly mediated OA progression. Furthermore, its downregulation could be co-regulated by transcription factor KLF4 and DNA methylation. Thus, the roles of INSR in OA worth further study.
Disclosure of conflict of interest
None.
Abbreviations
- OA
osteoarthritis
- ACLT
anterior cruciate ligament transection
- KLF4
Kruppel-like factor-4
- INSR
insulin receptor
- PBS
phosphate-buffered saline
- CHIP
Chromatin immunoprecipitation
- MS-PCR
Methylation-specific polymerase chain reaction
References
- 1.Liang B, Mamidi MK, Samsa WE, Chen Y, Lee B, Zheng Q, Zhou G. Targeted and sustained Sox9 expression in mouse hypertrophic chondrocytes causes severe and spontaneous osteoarthritis by perturbing cartilage homeostasis. Am J Transl Res. 2020;12:1056–1069. [PMC free article] [PubMed] [Google Scholar]
- 2.Reker D, Siebuhr AS, Thudium CS, Gantzel T, Ladel C, Michaelis M, Aspberg A, Berchtold MW, Karsdal MA, Gigout A, Bay-Jensen AC. Sprifermin (rhFGF18) versus vehicle induces a biphasic process of extracellular matrix remodeling in human knee OA articular cartilage ex vivo. Sci Rep. 2020;10:6011. doi: 10.1038/s41598-020-63216-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu F, Liu X, Yang Y, Sun Z, Deng S, Jiang Z, Li W, Wu F. NEAT1/miR-193a-3p/SOX5 axis regulates cartilage matrix degradation in human osteoarthritis. Cell Biol Int. 2020;44:947–957. doi: 10.1002/cbin.11291. [DOI] [PubMed] [Google Scholar]
- 4.Zuo S, Zou W, Wu RM, Yang J, Fan JN, Zhao XK, Li HY. Icariin alleviates IL-1β-induced matrix degradation by activating the Nrf2/ARE pathway in human chondrocytes. Drug Des Devel Ther. 2019;13:3949–3961. doi: 10.2147/DDDT.S203094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cho H, Bhatti FU, Hasty KA, Yi AK. Nanosome-mediated delivery of protein kinase D inhibitor protects chondrocytes from interleukin-1β-induced stress and apoptotic death. Int J Nanomedicine. 2019;14:8835–8846. doi: 10.2147/IJN.S218901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wu TJ, Lin CY, Tsai CH, Huang YL, Tang CH. Glucose suppresses IL-1beta-induced MMP-1 expression through the FAK, MEK, ERK, and AP-1 signaling pathways. Environ Toxicol. 2018;33:1061–1068. doi: 10.1002/tox.22618. [DOI] [PubMed] [Google Scholar]
- 7.Lee SA, Moon SM, Han SH, Hwang EJ, Park BR, Kim JS, Kim DK, Kim CS. Chondroprotective effects of aqueous extract of Anthriscus sylvestris leaves on osteoarthritis in vitro and in vivo through MAPKs and NF-kappaB signaling inhibition. Biomed Pharmacother. 2018;103:1202–1211. doi: 10.1016/j.biopha.2018.04.183. [DOI] [PubMed] [Google Scholar]
- 8.Nishimura R, Hata K, Takahata Y, Murakami T, Nakamura E, Ohkawa M, Ruengsinpinya L. Role of Signal transduction pathways and transcription factors in cartilage and joint diseases. Int J Mol Sci. 2020;21:1340. doi: 10.3390/ijms21041340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Okada K, Mori D, Makii Y, Nakamoto H, Murahashi Y, Yano F, Chang SH, Taniguchi Y, Kobayashi H, Semba H, Takeda N, Piao W, Hanaoka K, Nagano T, Tanaka S, Saito T. Hypoxia-inducible factor-1 alpha maintains mouse articular cartilage through suppression of NF-κB signaling. Sci Rep. 2020;10:5425. doi: 10.1038/s41598-020-62463-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun X, Xiao L, Chen J, Chen X, Chen X, Yao S, Li H, Zhao G, Ma J. DNA methylation is involved in the pathogenesis of osteoarthritis by regulating CtBP expression and CtBP-mediated signaling. Int J Biol Sci. 2020;16:994–1009. doi: 10.7150/ijbs.39945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Park CS, Shen Y, Lewis A, Lacorazza HD. Role of the reprogramming factor KLF4 in blood formation. J Leukoc Biol. 2016;99:673–685. doi: 10.1189/jlb.1RU1215-539R. [DOI] [PubMed] [Google Scholar]
- 12.Cota P, Helmi SA, Hsu C, Rancourt DE. Cytokine directed chondroblast trans-differentiation: JAK inhibition facilitates direct reprogramming of fibroblasts to chondroblasts. Cells. 2020;9:191. doi: 10.3390/cells9010191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cheng Z, Zou X, Jin Y, Gao S, Lv J, Li B, Cui R. The role of KLF4 in Alzheimer’s disease. Front Cell Neurosci. 2018;12:325. doi: 10.3389/fncel.2018.00325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chang SF, Huang KC, Chang HI, Lee KC, Su YP, Chen CN. 2 dyn/cm(2) shear force upregulates kruppel-like factor 4 expression in human chondrocytes to inhibit the interleukin-1beta-activated nuclear factor-kappaB. J Cell Physiol. 2018;234:958–968. doi: 10.1002/jcp.26924. [DOI] [PubMed] [Google Scholar]
- 15.Fisch KM, Gamini R, Alvarez-Garcia O, Akagi R, Saito M, Muramatsu Y, Sasho T, Koziol JA, Su AI, Lotz MK. Identification of transcription factors responsible for dysregulated networks in human osteoarthritis cartilage by global gene expression analysis. Osteoarthritis Cartilage. 2018;26:1531–1538. doi: 10.1016/j.joca.2018.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Legendre F, Dudhia J, Pujol JP, Bogdanowicz P. JAK/STAT but not ERK1/ERK2 pathway mediates interleukin (IL)-6/soluble IL-6R down-regulation of Type II collagen, aggrecan core, and link protein transcription in articular chondrocytes. Association with a down-regulation of SOX9 expression. J Biol Chem. 2003;278:2903–2912. doi: 10.1074/jbc.M110773200. [DOI] [PubMed] [Google Scholar]
- 17.Lim H, Park H, Kim HP. Effects of flavonoids on matrix metalloproteinase-13 expression of interleukin-1beta-treated articular chondrocytes and their cellular mechanisms: inhibition of c-Fos/AP-1 and JAK/STAT signaling pathways. J Pharmacol Sci. 2011;116:221–231. doi: 10.1254/jphs.11014fp. [DOI] [PubMed] [Google Scholar]
- 18.Rosa SC, Rufino AT, Judas F, Tenreiro C, Lopes MC, Mendes AF. Expression and function of the insulin receptor in normal and osteoarthritic human chondrocytes: modulation of anabolic gene expression, glucose transport and GLUT-1 content by insulin. Osteoarthritis Cartilage. 2011;19:719–727. doi: 10.1016/j.joca.2011.02.004. [DOI] [PubMed] [Google Scholar]
- 19.Tsai S, Clemente-Casares X, Zhou AC, Lei H, Ahn JJ, Chan YT, Choi O, Luck H, Woo M, Dunn SE, Engleman EG, Watts TH, Winer S, Winer DA. Insulin receptor-mediated Stimulation Boosts T cell immunity during inflammation and infection. Cell Metab. 2018;28:922–934. e4. doi: 10.1016/j.cmet.2018.08.003. [DOI] [PubMed] [Google Scholar]
- 20.Ma Y, Tang N, Thompson RC, Mobley BC, Clark SW, Sarkaria JN, Wang J. InsR/IGF1R pathway mediates resistance to EGFR inhibitors in glioblastoma. Clin Cancer Res. 2016;22:1767–1776. doi: 10.1158/1078-0432.CCR-15-1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petkevicius V, Salteniene V, Juzenas S, Wex T, Link A, Leja M, Steponaitiene R, Skieceviciene J, Kupcinskas L, Jonaitis L, Kiudelis G, Malfertheiner P, Kupcinskas J. Polymorphisms of microRNA target genes IL12B, INSR, CCND1 and IL10 in gastric cancer. World J Gastroenterol. 2017;23:3480–3487. doi: 10.3748/wjg.v23.i19.3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.da Costa IB, de Labio RW, Rasmussen LT, Viani GA, Chen E, Villares J, Turecki G, Smith MAC, Payao SLM. Change in INSR, APBA2 and IDE gene expressions in brains of Alzheimer’s disease patients. Curr Alzheimer Res. 2017;14:760–765. doi: 10.2174/1567205014666170203100734. [DOI] [PubMed] [Google Scholar]
- 23.Fernández-Tajes J, Soto-Hermida A, Vázquez-Mosquera ME, Cortés-Pereira E, Mosquera A, Fernández-Moreno M, Oreiro N, Fernández-López C, Fernández JL, Rego-Pérez I, Blanco FJ. Genome-wide DNA methylation analysis of articular chondrocytes reveals a cluster of osteoarthritic patients. Ann Rheum Dis. 2014;73:668–677. doi: 10.1136/annrheumdis-2012-202783. [DOI] [PubMed] [Google Scholar]
- 24.Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, Jensen LJ, Mering CV. STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Res. 2019;47:D607–D613. doi: 10.1093/nar/gky1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Doncheva NT, Morris JH, Gorodkin J, Jensen LJ. Cytoscape StringApp: network analysis and visualization of proteomics data. J Proteome Res. 2019;18:623–632. doi: 10.1021/acs.jproteome.8b00702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim BJ, Kim DW, Kim SH, Cho JH, Lee HJ, Park DY, Park SR, Choi BH, Min BH. Establishment of a reliable and reproducible murine osteoarthritis model. Osteoarthritis Cartilage. 2013;21:2013–2020. doi: 10.1016/j.joca.2013.09.012. [DOI] [PubMed] [Google Scholar]
- 27.Hu J, Wang Z, Pan Y, Ma J, Miao X, Qi X, Zhou H, Jia L. MiR-26a and miR-26b mediate osteoarthritis progression by targeting FUT4 via NF-κB signaling pathway. Int J Biochem Cell Biol. 2018;94:79–88. doi: 10.1016/j.biocel.2017.12.003. [DOI] [PubMed] [Google Scholar]
- 28.Fan L, Li M, Cao FY, Zeng ZW, Li XB, Ma C, Ru JT, Wu XJ. Astragalus polysaccharide ameliorates lipopolysaccharide-induced cell injury in ATDC5 cells via miR-92a/KLF4 mediation. Biomed Pharmacother. 2019;118:109180. doi: 10.1016/j.biopha.2019.109180. [DOI] [PubMed] [Google Scholar]
- 29.Lu W, Ding Z, Liu F, Shan W, Cheng C, Xu J, He W, Huang W, Ma J, Yin Z. Dopamine delays articular cartilage degradation in osteoarthritis by negative regulation of the NF-κB and JAK2/STAT3 signaling pathways. Biomed Pharmacother. 2019;119:109419. doi: 10.1016/j.biopha.2019.109419. [DOI] [PubMed] [Google Scholar]
- 30.Zhang L, Zhang P, Sun X, Zhou L, Zhao J. Long non-coding RNA DANCR regulates proliferation and apoptosis of chondrocytes in osteoarthritis via miR-216a-5p-JAK2-STAT3 axis. Biosci Rep. 2018;38:BSR20181228. doi: 10.1042/BSR20181228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhu J, Tang Y, Wu Q, Ji YC, Feng ZF, Kang FW. HIF-1α facilitates osteocyte-mediated osteoclastogenesis by activating JAK2/STAT3 pathway in vitro. J Cell Physiol. 2019;234:21182–21192. doi: 10.1002/jcp.28721. [DOI] [PubMed] [Google Scholar]
- 32.Ham S, Bae JB, Lee S, Kim BJ, Han BG, Kwok SK, Roh TY. Epigenetic analysis in rheumatoid arthritis synoviocytes. Exp Mol Med. 2019;51:1–13. doi: 10.1038/s12276-019-0215-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schellong K, Melchior K, Ziska T, Ott R, Henrich W, Rancourt RC, Plagemann A. Hypothalamic insulin receptor expression and DNA promoter methylation are sex-specifically altered in adult offspring of high-fat diet (HFD)-overfed mother rats. J Nutr Biochem. 2019;67:28–35. doi: 10.1016/j.jnutbio.2019.01.014. [DOI] [PubMed] [Google Scholar]
- 34.Ott R, Melchior K, Stupin JH, Ziska T, Schellong K, Henrich W, Rancourt RC, Plagemann A. Reduced insulin receptor expression and altered DNA methylation in fat tissues and blood of women with gdm and offspring. J Clin Endocrinol Metab. 2019;104:137–149. doi: 10.1210/jc.2018-01659. [DOI] [PubMed] [Google Scholar]
- 35.Wu X, Miao J, Jiang J, Liu F. Analysis of methylation profiling data of hyperplasia and primary and metastatic endometrial cancers. Eur J Obstet Gynecol Reprod Biol. 2017;217:161–166. doi: 10.1016/j.ejogrb.2017.08.036. [DOI] [PubMed] [Google Scholar]