

Abstract
Autosomal dominant polycystic kidney disease (ADPKD) is the common hereditary kidney disease, resulting from mutations in polycystic kidney disease 1 (PKD1) and polycystic kidney disease 2 (PKD2). Clinical data and genetic features of six Chinese families including ADPKD patients were analyzed via Next generation sequencing (NGS), Sanger sequencing, and multiplex ligation-dependent probe amplification. In family A, the proband (II5) with polycystic kidney (PK), hypertension, left ventricular hypertrophy, and valvular heart disease exhibited a heterozygous nonsense mutation, c.5086C>T (p.Gln1696Ter), in PKD1 (NM_001009944). In family B, the proband (II3) with PK, polycystic liver (PL), hypertension, hypertrophy of the left ventricle and septum, valvular heart disease, chronic kidney disease (CKD) stage 5, bilateral renal calculi, and right inguinal hernia exhibited a heterozygous missense mutation, c.6695T>C (p.Phe2232Ser), in PKD1. In family C, the proband (III1) with PK, PL, seminal vesicle cyst, hypertension, CKD stage 3, hypertrophy of the left ventricle and septum, and valvular heart disease harbored a heterozygous nonsense mutation, c.662T>G (p.Leu221Ter), in PKD2 (NM_000297). In family D, the proband (III3) with PK, hypertension, and CKD stage 5 harbored a heterozygous missense mutation, c.8311G>A (p.Glu2771Lys), in PKD1. In family E, the proband (II1) with PK, PL, hypertension, and CKD stage 5 exhibited a heterozygous deletion mutation, exon15-22, in PKD1. In family F, the proband (II2) with PK, PL, CKD stage 3, hypertension, thickened interventricular septum, and valvular heart disease carried a heterozygous missense mutation, c.1649A>G (p.His550Arg), in PKD2. Thus, three novel mutation sites which are responsible for ADPKD were discovered in this study.
Keywords: Autosomal dominant polycystic kidney disease, family research, polycystic kidney disease 1, polycystic kidney disease 2, gene mutation
Introduction
Autosomal dominant polycystic kidney disease (ADPKD) is the most common clinical monogenetic kidney disease, with a prevalence of 1/400-1/1000 [1]. The characteristic manifestation of ADPKD is the development and progressive expansion of multiple cysts that diffuse in both kidneys. Cyst formation is caused by cell proliferation, abnormal fluid secretion, and excessive extracellular matrix (ECM) deposition, which lead to increased kidney volume and interstitial fibrosis. Causing irreversible damage to renal structure and function, the illness progresses to end-stage renal disease (ESRD) in approximately half of all patients at 60 years of age, and renal replacement therapy is required to maintain their lives [2-4]. ADPKD ranks fourth among the causes of ESRD, accounting for 5%-10% [5]. ADPKD is a systemic disease involving many systems and develops in adults and infants. Its renal manifestations include renal insufficiency, flank plain, urinary tract infection, hematuria, renal calculi, etc., and its extrarenal manifestations include cysts in epithelial organs such as the liver and pancreas, as well as abnormalities in connective tissues, such as intracranial aneurysms, cardiac valvular abnormalities, aortic dissection, colonic diverticulosis, and ventral hernia [6].
ADPKD is highly genetically heterogeneous. Two genes are predominantly related to the onset of ADPKD: polycystic kidney disease 1 (PKD1) and polycystic kidney disease 2 (PKD2), which are located on chromosomes 16p13.3 and 4q21 and have a mutation probability of 85% and 15% in disease pedigrees, respectively [7,8]. No obvious mutational hotspots have been found in the PKD1 and PKD2 genes, implying that mutations can occur at any site in the gene [9]. Current therapeutic methods are only able to delay the malignant progression of renal function on a limited basis, but it is impossible to reverse the malignant tendency. Consequently, for families with ADPKD patients who have a demand for children, prenatal or pre-implantational genetic diagnosis of high-risk fetuses or embryos as early as possible and provision of reasonable fertility advice are capable of markedly reducing the occurrence of ADPKD and ultimately decreasing the prevalence of ESRD.
Materials and methods
Clinical information
The probands in this study were all patients of Han nationality at the Fujian Provincial Hospital. The diagnostic criteria for ADPKD were as follows: patients aged <30 years with more than two renal cysts on one or both sides; patients aged 30 to 59 years with at least two cysts on both renal sides; patients aged ≥60 years with at least four cysts on both renal sides. However, the diagnostic criteria can be modified if the patient has a family history, extrarenal manifestations, or positive genetic tests for diseases such as polycystic liver, intracranial aneurysms, or cardiac valvular abnormalities [10].
Clinical investigations suggested that there were six families. In family A (Figure 1A), there were four patients; the proband (II5) was a female aged 33 years with manifestations of lumbodynia, hypertension, urinary tract infection, and inguinal hernia. In family B (Figure 1B), there were seven patients; the proband (II3) was a woman aged 61 years with the following manifestations: lumbodynia, upper abdominal discomfort, hypertension, urinary tract infection, multiple renal calculi, gross hematuria, and foam in urine. In family C (Figure 1C), there were nine patients; the proband (III1) was a male aged 48 years, with the manifestations being lumbodynia, upper abdominal discomfort, hypertension, urinary tract infection, foam in urine, and seminal vesicle cysts. In family D (Figure 1D), there were five patients; the proband (III3) was a female aged 36 years, with the manifestations being anemia, hypertension, and foam in urine. In family E (Figure 1E), there were two patients; the proband (II1) was a male aged 48 years with manifestations of lumbodynia, upper abdominal discomfort, hypertension, urinary tract infection, and foam in urine. In family F (Figure 1F), there were two patients; the proband (II2) was a female aged 58 years with manifestations of hypertension, and foam in urine. This study was approved by the Ethics Committee of Fujian Provincial Hospital and all patients.
Figure 1.
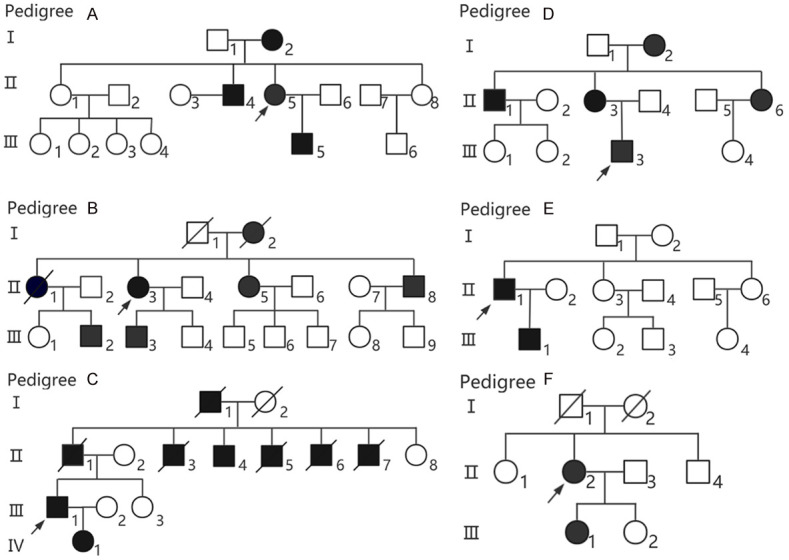
Pedigree chart of each family with ADPKD patients. Males and females are indicated by squares and circles, respectively, and members presenting with ADPKD are represented as squares/circles filled in black. The arrow indicates the proband. Diagonal lines indicate deceased individuals.
Clinical phenotype detection
The clinical manifestations and examination results of related biochemistry and imaging manifestations of the probands and family members were collected, such as Doppler ultrasound, computed tomography (CT), and nuclear magnetic resonance (MRI).
Gene detection
Genomic DNA extraction
For genomic DNA extraction, 10 mL of peripheral blood from the probands and 2 mL of peripheral blood from the other family members were obtained. Genomic DNA was extracted using an Omega E.Z.N.A® Tissue DNA Kit (QIAGEN) according to the manufacturer’s instructions.
Illumina sequencing
(1) Sample detection: The concentrations of the DNA samples from the probands were detected using Nanodrop 2000. DNA samples with concentrations over 3 μg were used for DNA fragmentation. (2) DNA fragmentation: DNA samples were cut into fragments of approximately 250 bp via CovarisAFA technology and then recycled. (3) Construction of the Illumina sequencing library: The recycled DNA fragments were used for the end repair reaction. The sequencing adapter was connected with end repair products, after which the products were amplified according to the binding sites of the universal primers on the adapter. Thereafter, the products were recycled and purified. (4) Target-gene capturing and enrichment: A DNA-capture array that can capture multiple genes was used to enrich various gene fragments, including target genes PKD1 (NM_001009944) and PKD2 (NM_000297). Sequencing of the enriched products was performed using Illumina2000 (Hiseq2000 sequencer) [11,12].
Sanger sequencing
Primers were designed using Primer 5.0 and synthesized by Beijing Liuhe BGI Technology Co., Ltd. (Table 1). The PCR annealing temperature was 53-60°C. PCR products were purified using the E.Z.N.A.™ Gel Extraction Kit (Omega) according to the manufacturer’s instructions. Sanger sequencing was performed using the BigDye Terminator v1.1 Kit according to standard procedures. The sequencing results were compared with the normal sequences using DNAMAN Version 5.2.2. When loss of heterozygosity or insertion was suspected, the PCR products were ligated onto PGEM-T Easy (Promega) vectors to select subclones for sequencing.
Table 1.
The primers design of the fragments with suspected responsibility point mutation of each ADPKD pedigree
| Pedigree | Gene | Transcript accession no. | Exons covered | Forward primer | Reverse primer | Length (bp) |
|---|---|---|---|---|---|---|
| A | PKD1 | NM_001009944 | EX15 | GCCCCTCCTCCAAGGACCAA | GGGTCCTACCATCTCTTACACCTT | 494 |
| B | PKD1 | NM_001009944 | EX15 | GTGGCAAGCTGGGTGTTCTCT | ATCACAGCGCAACTACTTGGAG | 505 |
| C | PKD2 | NM_000297 | EX2 | AAGATATTGCTAATGGGCTTGGGA | AGTTGCCCCTCTGGTGCATAC | 494 |
| D | PKD1 | NM_001009944 | EX23 | GAATGCCATCGAGGCCACCTT | TCTCTGCACTGACCTCACGCA | 428 |
| F | PKD2 | NM_000297 | Ex7 | CCGAACTAAGGAAATGCAAGCAA | GCTTTGGCTGGTCACTTGAATT | 498 |
Multiplex ligation-dependent probe amplification (MLPA)
Large intragenic gene deletions were detected via MLPA. Detection of PKD1/PKD2 exons was performed using the MLPA kit (SALSA MLPA KIT P351-B2/P352-C1 PKD1-PKD2; MRC-Holland, Amsterdam, The Netherlands) following the standard process [13].
Bioinformatic analysis
The original image file was read by the Illumina base calling software 1.7, and paired-end sequences (reads) of 90 bp were obtained. Purified data were used for sequence alignment analysis after removing low-quality and contaminated reads and adapter sequences. The coy number, single nucleotide polymorphism (SNP), and insertion/deletion (INDEL) were analyzed and annotated to screen for suspected pathogenic mutations using the SOAP software. The data were compared using the HGMD database (http://www.hgmd.cf.ac.uk), NCBI ClinVar database, and Mayo clinic PKD gene mutation database (http://pkdb.mayo.edu). The Polyphen (http://genetics.bwh.harvard.edu/pph/) and SIFT (http://sift.jcvi.org/) software were used to predict protein function to evaluate the pathogenic potential, and DNA parental separation analysis was further performed to verify pathogenicity [14,15].
Results
Clinical phenotype analysis
In family A, the proband (II5) visited our hospital for “recurrent bilateral lower back pain for two months and worsening for half a month” and was diagnosed with hypertension and polycystic kidney. The CT results showed that multiple cysts had developed in the renal parenchyma with a maximum diameter of approximately 4.5 cm. Some cystic walls had spotted and nodular calcifications after enhanced CT (Figure 2A). Patient II4 had “polycystic kidney, polycystic liver, hypertension, and post-cyst fenestration and decortication in both kidneys” (Table 2).
Figure 2.

CT or MRI images of the probands in six families (A-F) associated with polycystic kidney disease and/or polycystic liver disease.
Table 2.
Clinical data of the probant in every family associated with ADPKD
| Item | Proband in Pedigree A | Proband in Pedigree B | Proband in Pedigree C | Proband in Pedigree D | Proband in Pedigree E | Proband in Pedigree F |
|---|---|---|---|---|---|---|
| Sex | Male | Male | Female | Male | Female | Male |
| Age (years) | 38 | 61 | 50 | 36 | 48 | 58 |
| First discovery (age, years) | 33 | 53 | 48 | 29 | 39 | 38 |
| Total Kidney Volume (TKV, cm3) | 755.03 | 1154.53 | 111.50 | 1022.59 | 479.06 | 91.93 |
| Staging of chronic kidney disease (CKD) | nomal | CKD5 | CKD3 | CKD5 | CKD5 | CKD3 |
| Blood pressure (SBP/DBP, mmHg) | ≥160/90 | ≥140/90 | ≥160/110 | ≥160/100 | ≥160/90 | ≥180/90 |
| Blood urea nitrogen (BUN, mmol/L) | 4.2 | 14.7 | 5.7 | 43.6 | 20.6 | 8.1 |
| Creatinine (CREA, μmol/L) | 62 | 582 | 139 | 1724 | 425 | 145 |
| Uric acid (URIC, μmol/L) | 280 | 255 | 444 | 444 | 571 | 375 |
| eGFR [ml•min-1•(1.73 m2)] | 99.362 | 7.005 | 50.286 | 2.261 | 14.442 | 37.262 |
| Triglyceride (TG, mmol/L) | 2.13 | 2.09 | 1.38 | 0.96 | 3.00 | 2.07 |
| Cholesterol (CHOL, mmol/L) | 5.13 | 5.89 | 4.59 | 4.40 | 4.80 | 6.01 |
| High-density lipoprotein (HDL, mmol/L) | 0.78 | 1.09 | 1.37 | 1.85 | 0.80 | 1.64 |
| Low-density lipoprotein (LDL, mmol/L) | 3.64 | 4.19 | 2.98 | 2.27 | 2.40 | 4.38 |
| Red blood cell (RBC, ×1012/L) | 3.64 | 3.95 | 12.2 | 2.35 | 3.90 | 4.07 |
| White blood cell (WBC×109/L) | 5.35 | 8.20 | 5.60 | 6.20 | 8.90 | 6.40 |
| Hemoglobin (HGB, g/L) | 149 | 121 | 155 | 72 | 124 | 116 |
| Hematocrit value (HCT, %) | 0.450 | 0.368 | 0.466 | 0.206 | 0.360 | 0.350 |
| Platelet (PLT, ×109/L) | 391 | 167 | 207 | 203 | 150 | 255 |
| Pain of renal origin | Y | Y | Y | N | Y | N |
| urinary system infection | Y | Y | Y | N | Y | N |
| Haematuria (microscopic/gross) | N | Y | N | N | N | N |
| Albuminuria | trace amount | 3+ | trace amount | 2+ | trace amount | 1+ |
| Nephrolithiasis | N | Y | N | N | N | N |
| Extrarenal manifestations | LVH, VDH | PCLD, LVH, VDH, IH | PCLD, SVC, LVH, VDH | N | PCLD | PCLD, LVH, VDH |
| Clinical features of other patients | II4: PKD, PCLD, HBP | II5: PKD, PCLD, HBP, CKD5, LVH, VDH; II8: PKD, Nephtolithiasis, HBP, CKD1; III3: PKD, PCLD, HBP, Nephtolithiasis, CKD1 | II1: PKD, PCLD, HBP, HHD, IA, CAV, IH; II4: PKD, PCLD, HBP, AA, HHD | II1: PKD, PCLD, HBP, CKD3; II3: PKD, HBP, Nephtolithiasis, AA, CKD1 | III1: PKD, PCLD, CKD1 | III1: PKD |
Note: PL, polycystic liver disease; SVC, cyst of seminal vesicle; PC, pancreatic cyst; IAC, intracranial arachnoid cyst; LVH, left ventricular hypertrophy; VDH, valvular disease-heart; IA, intracranial aneurysm; SAH, subarachnoid hemorrhage; CAV, cerebrovascular accident; AA, aortic aneurysm; DD, diverticular disease; AH, abdominal hernia; IH, inguinal hernia; HHD, hypertension heart disease. Y, Yes; N, None.
In family B, the proband (II3) visited our hospital for “lower left back pain with gross hematuria for four days”. She was diagnosed with “polycystic kidney, polycystic liver, hypertension, and left renal calculi” ten years ago, had undergone surgery for a right inguinal hernia three years ago, was diagnosed with chronic kidney disease (CKD) stage 5 two months ago, and had started blood dialysis treatment. The CT urography (CTU) results showed that multiple cysts had developed in both kidneys with a maximum diameter of 8.4 cm. In both kidneys, multiple increased ground-glass opacities were observed, and multiple cysts were seen in the liver, with a maximum diameter of approximately 3.5 cm (Figure 2B). Most members of this family exhibited extrarenal manifestations such as polycystic liver and urinary stones. Patient II5 suffered from “polycystic kidney, polycystic liver, hypertension, left ventricular hypertrophy, heart valve disease, and CKD stage 5 and was undergoing long-term peritoneal dialysis treatment”. Patient II8 had “polycystic kidney, hypertension, bilateral renal calculi, and CKD stage 1”. Patient III3 was diagnosed with “polycystic kidney, polycystic liver, hypertension, bilateral renal calculi, and CKD stage 1” (Table 2).
In family C, the proband (III1) visited our hospital for “recurrent lower left back pain for over ten years”. He was diagnosed with “polycystic kidney, polycystic liver, and urinary tract infections” 12 years ago, had “hypertension” ten years ago, and suffered from “cyst of seminal vesicle” one year ago. The MR urography (MRU) results showed multiple cysts in both kidneys, with a maximum diameter of about 8 cm, and polycystic liver (Figure 2C). Patient II1 died of “stroke” and suffered from “polycystic kidney, polycystic liver, hypertension, hypertensive heart disease, intracranial aneurysm, and inguinal hernia” when alive. Patient II4 had “polycystic kidney, polycystic liver, hypertension, hypertensive heart disease, uremia, long-term hemodialysis, and post-thoracic aortic aneurysm surgery” (Table 2).
In family D, the proband (III3) visited our hospital for “foamy urine for seven consequent years”. She had “polycystic kidney, hypertension, CKD stage 5, and long-term peritoneal dialysis” for four years. The CT urography results showed that multiple cystic lesions had developed in the kidney, along with partially compressed stenosis of the renal pelvis and calyces (Figure 2D). Patient II1 suffered from “polycystic kidney, polycystic liver, hypertension, post right renal cyst fenestration and decortication, and CKD stage 3”. Patient II3 had “polycystic kidney, right renal calculi, hypertension, post thoracoabdominal aortic aneurysm surgery, and CKD stage 1” (Table 2).
In family E, the proband (II1) visited our hospital for “recurrent bilateral lower back pain for more than eight years”. He suffered from “polycystic kidney and polycystic liver” for nine years and had “CKD stage 5, hypertension, and long-term peritoneal dialysis” for six years. The CT results showed that multiple cysts with a maximum diameter of about 4.6 cm had developed, multiple patchy and cord-like high-density shadows were present in the parenchyma of both kidneys, and polycystic liver had developed with a maximum cyst diameter of approximately 2.7 cm (Figure 2E). Patient III1 was diagnosed with “polycystic kidney, polycystic liver, and CKD stage 1” (Table 2).
In family F, the proband (II2) visited our hospital for “recurring dizziness for over a year and recurrence for a week”. She had “polycystic kidney, polycystic liver, post left renal cyst fenestration and decortication, and hypertension” for ten years. The CT results showed that the liver was slightly deformed with multiple cystic lesions of a maximum diameter of 5.7 cm, multiple cystic lesions had developed in the kidney, with a maximum diameter of 3.7 cm, and residual renal parenchyma was scattered, leading to several nodular dense shadows (Figure 2F). Patient III1 had “polycystic kidney with normal renal function” (Table 2).
Gene detection analysis results
In family A, the sequencing data analysis of PKD1 and PKD2 exons showed that the proband (II5) was found to have no large deletions or duplications in either gene. A heterozygous nonsense mutation, c.5086C>T (p.Gln1696Ter), was found in the coding region of the PKD1 gene (Figure 3A, 3B); this mutation terminated protein synthesis in advance and induced production of truncated proteins or gene degradation, thus affecting protein function. This site has an extremely low probability of being mutated in healthy people. A relevant study has been reported on the pathogenicity of this site [16]. Additionally, two heterozygous missense mutations were found in the coding region of the PKD1 gene: c.10678G>A (p.Gly3560Arg) and c.2813C>T (p.Thr938Met). The two mutations were predicted to be harmless to protein function, using the SIFT and Polyphen software (Table 3). Sanger sequencing confirmed that patients I2, II4, II5, and III5 in family A carried p.Gln1696Ter, while healthy individuals-I1, II1, II2, II3, II6, II7, II8, III1, III2, III3, III4, and III6-tested negative. In this family, the genotype and phenotype were separated. Therefore, the nonsense mutation p.Gln1696Ter was considered to be a pathogenic mutation in this family.
Figure 3.

Sanger sequencing diagram or MLPA test. (A) A heterozygous nonsense mutation, c.5086C>T (p.Gln1696Ter), was found in the coding region of the PKD1 gene in family A; (C) A heterozygous missense mutation, c.6695T>C (p.Phe2232Ser), was found in the coding region of the PKD1 gene in family B; (E) A heterozygous nonsense mutation, c.662T>G (p.Leu221Ter), was found in the coding region of the PKD2 gene in family C; (G) A heterozygous missense mutation, c.8311G>A (p.Glu2771Lys), was found in the coding region of the PKD1 gene in family D; (K) NGS and MLPA suggest that a heterozygous deletion mutation, exon15-22, was found in the coding region of the PKD1 gene in family E; (I) A heterozygous missense mutation, c.1649A>G(p.His550Arg), was found in the coding region of the PKD2 gene in family F; (B, D, F, H, and J) correspond to wild-type forms.
Table 3.
Mutations detected of the proband in every family with ADPKD
| Pedigree | Gene | Transcript accession no. | Coding | Protein | Exon/intron | Het | Chromo-somal location | dbSNP | MAF | Prediction of protein function |
|---|---|---|---|---|---|---|---|---|---|---|
| A | PKD1 | NM_001009944 | c.10678G>A | p.Gly3560Arg | Exon36 | Het | chr16:2143955 | rs79000340 | 0.0156 | - |
| PKD1 | NM_001009944 | c.5086C>T | p.Gln1696Ter | Exon15 | Het | chr16:2160082 | - | 0 | Pathogenic | |
| PKD1 | NM_001009944 | c.2813C>T | p.Thr938Met | Exon11 | Het | chr16:2164211 | rs148709380 | 0.0925 | - | |
| B | PKD1 | NM_001009944 | c.6695T>C | p.Phe2232Ser | Exon15 | Het | chr16:2158473 | - | 0 | VUS |
| C | PKD1 | NM_001009944 | c.11333C>A | p.Thr3778Asn | Exon40 | Het | chr16:2142126 | rs114656915 | 0 | - |
| PKD1 | NM_001009944 | c.10379C>T | p.Ser3460Leu | Exon33 | Het | chr16:2147346 | rs373743750 | 0 | - | |
| PKD2 | NM_000297 | c.662T>G | p.Leu221Ter | Exon2 | Het | chr4:88940676 | - | 0 | Likelypathogenic | |
| D | PKD1 | NM_001009944 | c.2813C>T | p.Thr938Met | Exon11 | Het | chr16:2164211 | rs148709380 | 0.0925 | - |
| PKD1 | NM_001009944 | c.8311G>A | p.Glu2771Lys | Exon23 | Het | chr16:2153747 | - | 0 | Pathogenic | |
| E | PKD1 | NM_001009944 | EX15_22DEL | - | Exon15_22 | Het | - | - | 0 | Likelypathogenic |
| PKD1 | NM_001009944 | c.9067A>G | p.Met3023Val | Exon25 | Het | chr16:2152516 | - | 0 | VUS | |
| F | PKD2 | NM_000297 | c.1649A>G | p.His550Arg | Exon7 | Het | chr4:88973243 | - | 0 |
Note: The mutated sites with MAF >1% from dbSNP 1000 Genomes (population frequency information from the 1000 genomes project) were removed.
In family B, a heterozygous missense mutation, c.6695T>C (p.Phe2232Ser) (Figure 3C, 3D), was found in the coding region of the PKD1 gene of the proband (II3), contributing to the substitution of glutamic acid by serine that may affect protein phosphorylation. This site rarely occurs in normal people, and there have been no reports of its pathogenicity. The SIFT and Polyphen values were 0 and 0.994, respectively, and the mutation was predicted to be harmful to protein function (Table 3). Sanger sequencing confirmed that patients II3, II5, II8, and III3 harbored p.Phe2232Ser, while healthy individuals-II2, II4, II6, II7, III1, III4, III5, III6, III7, III8, and III9-did not. In this family, the genotype and phenotype were separated. Consequently, the missense mutation p.Phe2232Ser was considered to be a newly discovered type of pathogenic mutation in this family.
In family C, a heterozygous nonsense mutation, c.662T>G (p.Leu221Ter) (Figure 3E, 3F), was found in the coding region of the PKD2 gene of the proband (III1); this mutation terminated protein synthesis in advance and induced the production of truncated proteins, leading to a serious effect on protein structure and function. The site rarely occurs in normal people, and there have been no reports of its pathogenicity. Additionally, two heterozygous missense mutations were found in the coding region of the PKD1 gene: c.11333C>A (p.Thr3778Asn) and c.10379C>T (p.Ser3460Leu). These two mutations were predicted to be harmless to protein function by the SIFT and Polyphen software (Table 3). Sanger sequencing confirmed that patients II4, III1, and IV1 carried the c.662T>G (p.Leu221Ter) mutation, but normal individuals-II2, II8, III2, and III3-did not. In this family, the genotype and phenotype were separated. Accordingly, the nonsense mutation Leu221Ter was considered to be a newly discovered type of pathogenic mutation in this family.
In family D, a heterozygous missense mutation, c.8311G>A (p.Glu2771Lys), was found in the coding region of PKD1 (Figure 3G, 3H); this mutation causes glutamic acid to be replaced by lysine and might influence the structure and function of the protein. This site rarely occurs in normal people. The SIFT and Polyphen values were 0.011 and 0.998, respectively, and the mutation was predicted to be harmful to protein function. Relevant studies have been conducted on the pathogenicity of this site [17-19]. Additionally, a heterozygous missense mutation, c.2813C>T (p.Thr938Met), was found in the PKD1 gene and was predicted to be harmless to protein function via the SIFT and Polyphen software (Table 3). Sanger sequencing verified that patients II1, II3, II6, and III3 carried the p.Glu2771Lys mutation, but normal individuals-II2, II8, III2, and III3-did not. In this family, the genotype and phenotype were separated. As a result, the missense mutation p.Glu2771Lys was considered to be a pathogenic mutation in this family.
In family E, NGS and MLPA suggested that a heterozygous deletion mutation, exon15-22, was found in the coding region of PKD1 in the proband (II1) (Figure 3K). This mutation causes a large deletion in the protein-coding region and thus might affect protein structure and function. In addition, a heterozygous deletion mutation, c.9067A>G (p.Met3023Val), was found in the coding region of PKD1 and was predicted to be harmless to the protein function via the SIFT and Polyphen software (Table 3). MLPA results indicated that patients II1 and III1 both carried exon15-22 deletion mutation, whereas normal individuals-I1, I2, II2, II3, II4, II5, II6, III2, III3, and III4-did not. In this family, the genotype and phenotype were separated. As a result, the deletion mutation exon15-22 was considered to be a newly discovered pathogenic mutation in this family.
In family F, a heterozygous missense mutation, c.1649A>G (p.His550Arg) (Figure 3I, 3J), was found in the coding region of the PKD2 gene of the proband (II2); this mutation caused histidine to be replaced by arginine. This site rarely occurs in normal people, and there have been no reports of its pathogenicity so far (Table 3). Sanger sequencing demonstrated that patients II2 and III1 carried the p.His550Arg mutation, while normal individuals-II1, II3, II4, and III2-did not. Co-segregation of genotypes and phenotypes was present in this family. Therefore, the missense mutation p.His550Arg was considered to be a pathogenic mutation in this family.
Discussion
The PKD1 gene, approximately 52 kb in length, contains 46 exons and encodes 4,303 amino acids (aa). Polycystin1 (PC1), its encoding product, has a complete membrane protein structure with a large extracellular N-terminal region (3,074 aa), 11 transmembrane regions (1,032 aa), and a short intracellular C-terminal region (197 aa) (Figure 4) [20]. As a cell membrane receptor, PC1 is distributed in the primary cilia of plasma membranes and in some cell junctions such as focal adhesions, desmosomes, tight junctions, and adhering junctions [21-23]. It is involved in multiple cell signal transduction pathways, including the Wnt, JAK2/signal transducer and activator of transcription (JAK-STAT), JNK-AP1, Ca2+-NFAT, IGF-1, and HDAC6/EGFR pathways, as a regulator of cell proliferation and apoptosis through cell-cell or cell-matrix interactions [24-29]. The PKD2 gene is approximately 68 kb in length and contains 15 exons. Polycystin2 (PC2), encoded by PKD2, is also a membrane-associated protein, with 25% and 50% of its sequence identical and similar to PC1, respectively. Additionally, it has six transmembrane regions and cellular N-terminal and C-terminal regions, and both the amino and carboxyl terminals are located in the cytoplasm (Figure 4). PC2 is detected in the endoplasmic reticulum (ER), primary cilia, and apical and basolateral membranes of the kidney. As a member of the transient receptor potential family of ion channels, PC2 is considered to be a nonselective cation channel and a member of a third class of Ca2+ release channels apart from ryanodine receptors and IP3 receptors [23,30]. In addition, PC2 is a voltage-gated Ca2+ channel protein that stimulates the opening and closing of the Ca2+ channel and regulates the concentration of Ca2+ in renal tubular epithelial cells by receiving the signal transmitted in the baroreceptor; additionally, it is involved in the proliferation and differentiation of renal tubular epithelial cells via the Ca2+ phospholipid-dependent protein kinase. PC1 and PC2 interact with each other in kidney primary cilia to form a spiral polycystic protein complex in the C-terminus that hypothetically functions as a cell surface signal receptor and mechanical sensor to promote tubular epithelial cell proliferation and differentiation [31]. As a cell membrane receptor, PC1 senses extracellular mechanical or chemical stimulation to control the influx of Ca2+ through the PC2 channel to regulate the intracellular Ca2+ concentration, thereby combining with the intracellular signaling pathway to participate in cyst formation. However, when mutations occur in PKD1 and PKD2, they trigger the dysfunction of the polycystic complex, leading to decreased intracellular Ca2+ concentrations and increased intracellular cAMP concentrations. The fluid movement driving cyst formation is stimulated by cAMP and involves the interaction between apical cystic fibrosis transmembrane regulator and the basolateral Na+-K+-2Cl- cotransporter NKCC1, which drives the flow of Cl- into the lumen [24]. Mutations in PKD1 and PKD2 also affect the Wnt pathway, Hippo pathway, mammalian target of rapamycin, mitogen-activated protein kinase, PI3K-AKT signaling, AMP-activated protein kinase, and JAK-STAT pathway, sparking abnormal tubular cell cycle regulation and metabolism along with aberrant proliferation and apoptosis of epithelial cells and ultimately inducing cyst formation [4,32-35].
Figure 4.
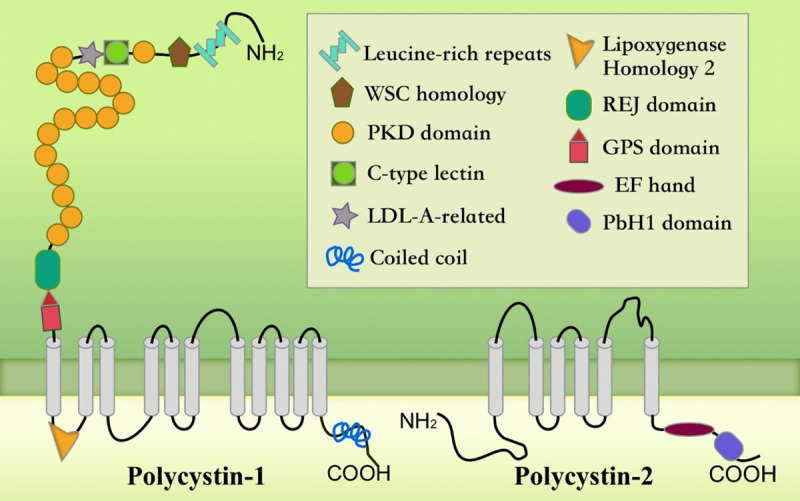
Structure of polycystins. PC1 has a large extracellular domain, 11-transmembrane domain, and short C tail. PC2 has a six-transmembrane domain and cytoplasmic N- and C-termini. The coiled-coil domain in the C-terminal end of PC1 interacts with the C-terminal tail of PC2.
Currently, the clinical diagnosis of ADPKD chiefly involves a renal imaging technique based on the age-associated cyst number [36]. For ADPKD sufferers with a family history, ultrasound could be the first choice, whose detection rate can be as high as 96% [37]. However, for patients who have no family history, are unable to be diagnosed by imaging, or the current diagnostic criteria are unable to provide a definitive diagnosis for patients who have highly similar clinical manifestations caused by mutations in other genes such as TSC, HNF-1β, and PKHD; in such cases, genetic diagnosis is the gold standard. The ADPKD mutation database (Version 3.0, http://pkdb.mayo.edu/) indicates that until December 15, 2019, the PKD1 gene had 2,323 mutation sites, including 868 clear pathogenic mutations, while PKD2 had 278 mutation sites, including 162 pathogenic mutations. The database is continuously updated. PKD1 is a complex gene in composition and structure [38]. There are six highly homologous pseudogenes in the proximal 5’ exon regions 1-33 [39] and a number of GC base pairs in exon region 1 [17]; additionally, there is a polypyrimidine repeat sequence (PyRE) of approximately 2.5 kb in the intron region 21, which may interfere with PKD1 replication, transcription, and RNA processing [32]. These unique circumstances make the analysis and screening of mutations in PKD1 extremely challenging. NGS application significantly increases the positive detection rate of responsible mutations in the coding, multiple copy, and single copy regions of the PKD1 and PKD2 genes [40]; however, NGS lacks sensitivity to large copy number changes, whereas MLPA can be used to detect copy number alterations in target gene fragments. In the six Chinese families with ADPKD in this study, three novel co-segregated mutations were discovered. The p.Leu221Ter mutation in PKD2 forms a stop codon at exon221, leading to the truncation of PC2. This truncated protein contains the intracellular N-terminal of PC2 but lacks the intracellular C-terminal. The lack of proper C-terminal-mediated signal transduction cascades contributes to improper regulation of gene transcription and aberrant renal development [31]. The mRNA chain transcribed upon the exon15-22del mutation in PKD1 is incomplete and would eventually produce a pathogenic effect and lead to genetic phenotype changes. Nonetheless, large deletions and nonsense mutations have been clearly regarded as pathogenic mutations, and DNA parental separation can be demonstrated by family linkage analysis. The p.Phe2232Ser mutation in PKD1 is located in the extracellular N-terminal domain of PC1. Glutamic acid is a nonpolar and hydrophobic amino acid, while serine is a polar and neutral amino acid. The substitution of serine for glutamic acid is likely to affect the phosphorylation process.
In conclusion, the analysis of a number of families with ADPKD patients indicated that patients with PKD1 mutations had suffered from ESRD about 20 years earlier than those with PKD2 mutations. Patients with truncated mutations were more likely to develop ESRD than were those with nontruncated mutations, and patients with mutations in the 5’ region had more severe clinical manifestations than did those with mutations in the 3’ region [41-43]. However, this study found that patients from the same family had different clinical phenotypes even though they carried the same mutant genes and genetic background. Consequently, it is difficult to explain the “one-on-one” relationship between the genetic mutation and clinical phenotype of ADPKD, possibly due to the high degree of heterogeneity in the inheritance of ADPKD and the effects of environment and genetic modification [44].
In recent years, the number of newly identified disease-associated genes has grown exponentially in all fields of ADPKD since the emergence of NGS technology. However, different genetic mutation modes have different sensitivities to detection techniques. For example, NGS lacks sensitivity to large deletions of gene, and thus, a combined MLPA detection method is often required. Therefore, in order to reduce false negative or false positive results, the selection of genetic testing techniques should be performed carefully. This study found that patients with considerable exon-deletion mutations did not present severe clinical manifestations; this is contrary to traditional cognition and reveals that the degree of clinical manifestation is not related to the degree of genetic mutation. In addition, NGS has opened the door not only for molecular diagnosis but also for the discovery of treatments for diseases that were considered death sentences in the past. Nevertheless, for many years, gene therapy has been associated with very few success stories. There are reasons to believe that the journey has only begun, and much work remains to overcome many unknown challenges.
Acknowledgements
This work was supported in part by grants from the National Natural Science Foundation of China (No. 81874379), the Fujian Province Medical Innovation Foundation (No. 2018-CXR-2, 2019-CXB-3, 2019-CXB-4), and the Special Research Foundation of Fujian Provincial Department of Finance (No. 2018 643#), China.
Disclosure of conflict of interest
None.
Abbreviations
- ADPKD
Autosomal dominant polycystic kidney disease
- PKD1
polycystic kidney disease1
- PKD2
polycystic kidney disease2
- NGS
Next generation sequencing
- MLPA
multiplex ligation-dependent probe amplification
- CKD
chronic kidney disease
- PK
polycystic kidney
- PL
polycystic liver
References
- 1.Simms RJ. Autosomal dominant polycystic kidney disease. BMJ. 2016;352:i679. doi: 10.1136/bmj.i679. [DOI] [PubMed] [Google Scholar]
- 2.He J, Zhou H, Meng J, Zhang S, Li X, Wang S, Shao G, Jin W, Geng X, Zhu S, Yang B. Cardamonin retards progression of autosomal dominant polycystic kidney disease via inhibiting renal cyst growth and interstitial fibrosis. Pharmacol Res. 2020;155:104751. doi: 10.1016/j.phrs.2020.104751. [DOI] [PubMed] [Google Scholar]
- 3.Shaw C, Simms RJ, Pitcher D, Sandford R. Epidemiology of patients in England and Wales with autosomal dominant polycystic kidney disease and end-stage renal failure. Nephrol Dial Transplant. 2014;29:1910–1918. doi: 10.1093/ndt/gfu087. [DOI] [PubMed] [Google Scholar]
- 4.Bergmann C, Guay-Woodford LM, Harris PC, Horie S, Peters DJM, Torres VE. Polycystic kidney disease. Nat Rev Dis Primers. 2018;4:50. doi: 10.1038/s41572-018-0047-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Testa F, Magistroni R. ADPKD current management and ongoing trials. J Nephrol. 2020;33:223–237. doi: 10.1007/s40620-019-00679-y. [DOI] [PubMed] [Google Scholar]
- 6.Muller RU, Benzing T. Management of autosomal-dominant polycystic kidney disease-state-of-the-art. Clin Kidney J. 2018;11:i2–i13. doi: 10.1093/ckj/sfy103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Riella C, Czarnecki PG, Steinman TI. Therapeutic advances in the treatment of polycystic kidney disease. Nephron Clin Pract. 2014;128:297–302. doi: 10.1159/000368244. [DOI] [PubMed] [Google Scholar]
- 8.Li A, Tian X, Zhang X, Huang S, Ma Y, Wu D, Moeckel G, Somlo S, Wu G. Human polycystin-2 transgene dose-dependently rescues ADPKD phenotypes in Pkd2 mutant mice. Am J Pathol. 2015;185:2843–2860. doi: 10.1016/j.ajpath.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bergmann C. Recent advances in the molecular diagnosis of polycystic kidney disease. Expert Rev Mol Diagn. 2017;17:1037–1054. doi: 10.1080/14737159.2017.1386099. [DOI] [PubMed] [Google Scholar]
- 10.Pei Y, Obaji J, Dupuis A, Paterson AD, Magistroni R, Dicks E, Parfrey P, Cramer B, Coto E, Torra R, San Millan JL, Gibson R, Breuning M, Peters D, Ravine D. Unified criteria for ultrasonographic diagnosis of ADPKD. J Am Soc Nephrol. 2009;20:205–212. doi: 10.1681/ASN.2008050507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.He J, Wu J, Jiao Y, Wagner-Johnston N, Ambinder RF, Diaz LA Jr, Kinzler KW, Vogelstein B, Papadopoulos N. IgH gene rearrangements as plasma biomarkers in Non-Hodgkin’s lymphoma patients. Oncotarget. 2011;2:178–185. doi: 10.18632/oncotarget.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu J, Matthaei H, Maitra A, Dal Molin M, Wood LD, Eshleman JR, Goggins M, Canto MI, Schulick RD, Edil BH, Wolfgang CL, Klein AP, Diaz LA Jr, Allen PJ, Schmidt CM, Kinzler KW, Papadopoulos N, Hruban RH, Vogelstein B. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci Transl Med. 2011;3:92ra66. doi: 10.1126/scitranslmed.3002543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harris PC, Torres VE. Polycystic kidney disease, autosomal dominant. In: Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJH, Stephens K, Amemiya A, editors. GeneReviews((R)) Seattle (WA): University of Washington, Seattle; 1993-2020. [Google Scholar]
- 14.Li R, Li Y, Kristiansen K, Wang J. SOAP: short oligonucleotide alignment program. Bioinformatics. 2008;24:713–714. doi: 10.1093/bioinformatics/btn025. [DOI] [PubMed] [Google Scholar]
- 15.Audrezet MP, Cornec-Le Gall E, Chen JM, Redon S, Quere I, Creff J, Benech C, Maestri S, Le Meur Y, Ferec C. Autosomal dominant polycystic kidney disease: comprehensive mutation analysis of PKD1 and PKD2 in 700 unrelated patients. Hum Mutat. 2012;33:1239–1250. doi: 10.1002/humu.22103. [DOI] [PubMed] [Google Scholar]
- 16.Rossetti S, Consugar MB, Chapman AB, Torres VE, Guay-Woodford LM, Grantham JJ, Bennett WM, Meyers CM, Walker DL, Bae K, Zhang QJ, Thompson PA, Miller JP, Harris PC, Consortium C. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 2007;18:2143–2160. doi: 10.1681/ASN.2006121387. [DOI] [PubMed] [Google Scholar]
- 17.Rossetti S, Strmecki L, Gamble V, Burton S, Sneddon V, Peral B, Roy S, Bakkaloglu A, Komel R, Winearls CG, Harris PC. Mutation analysis of the entire PKD1 gene: genetic and diagnostic implications. Am J Hum Genet. 2001;68:46–63. doi: 10.1086/316939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Obeidova L, Elisakova V, Stekrova J, Reiterova J, Merta M, Tesar V, Losan F, Kohoutova M. Novel mutations of PKD genes in the Czech population with autosomal dominant polycystic kidney disease. BMC Med Genet. 2014;15:41. doi: 10.1186/1471-2350-15-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Trujillano D, Bullich G, Ossowski S, Ballarin J, Torra R, Estivill X, Ars E. Diagnosis of autosomal dominant polycystic kidney disease using efficient PKD1 and PKD2 targeted next-generation sequencing. Mol Genet Genomic Med. 2014;2:412–421. doi: 10.1002/mgg3.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Torres VE, Harris PC. Autosomal dominant polycystic kidney disease: the last 3 years. Kidney Int. 2009;76:149–168. doi: 10.1038/ki.2009.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet. 2007;369:1287–1301. doi: 10.1016/S0140-6736(07)60601-1. [DOI] [PubMed] [Google Scholar]
- 22.Igarashi P, Somlo S. Genetics and pathogenesis of polycystic kidney disease. J Am Soc Nephrol. 2002;13:2384–2398. doi: 10.1097/01.asn.0000028643.17901.42. [DOI] [PubMed] [Google Scholar]
- 23.Zhou J. Polycystins and primary cilia: primers for cell cycle progression. Annu Rev Physiol. 2009;71:83–113. doi: 10.1146/annurev.physiol.70.113006.100621. [DOI] [PubMed] [Google Scholar]
- 24.Chapin HC, Caplan MJ. The cell biology of polycystic kidney disease. J Cell Biol. 2010;191:701–710. doi: 10.1083/jcb.201006173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kashyap S, Hein KZ, Chini CC, Lika J, Warner GM, Bale LK, Torres VE, Harris PC, Oxvig C, Conover CA, Chini EN. Metalloproteinase PAPP-A regulation of IGF-1 contributes to polycystic kidney disease pathogenesis. JCI Insight. 2020;5:e135700. doi: 10.1172/jci.insight.135700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ke B, Chen Y, Tu W, Ye T, Fang X, Yang L. Inhibition of HDAC6 activity in kidney diseases: a new perspective. Mol Med. 2018;24:33. doi: 10.1186/s10020-018-0027-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rastogi A, Ameen KM, Al-Baghdadi M, Shaffer K, Nobakht N, Kamgar M, Lerma EV. Autosomal dominant polycystic kidney disease: updated perspectives. Ther Clin Risk Manag. 2019;15:1041–1052. doi: 10.2147/TCRM.S196244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tang Y, Shi G, Yang J, Zheng W, Tang J, Chen XZ, Yang J, Wang Z. Role of PKR in the inhibition of proliferation and translation by polycystin-1. Biomed Res Int. 2019;2019:5320747. doi: 10.1155/2019/5320747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang Y, Yang J, Zheng W, Tang J, Chen XZ, Yang J, Wang Z. Polycystin-1 inhibits cell proliferation through phosphatase PP2A/B56alpha. Biomed Res Int. 2019;2019:2582401. doi: 10.1155/2019/2582401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Semmo M, Kottgen M, Hofherr A. The TRPP subfamily and polycystin-1 proteins. Handb Exp Pharmacol. 2014;222:675–711. doi: 10.1007/978-3-642-54215-2_27. [DOI] [PubMed] [Google Scholar]
- 31.Xu P, Huang S, Li J, Zou Y, Gao M, Kang R, Yan J, Gao X, Gao Y. A novel splicing mutation in the PKD1 gene causes autosomal dominant polycystic kidney disease in a Chinese family: a case report. BMC Med Genet. 2018;19:198. doi: 10.1186/s12881-018-0706-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parker MI, Nikonova AS, Sun D, Golemis EA. Proliferative signaling by ERBB proteins and RAF/MEK/ERK effectors in polycystic kidney disease. Cell Signal. 2020;67:109497. doi: 10.1016/j.cellsig.2019.109497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Margaria JP, Campa CC, De Santis MC, Hirsch E, Franco I. The PI3K/Akt/mTOR pathway in polycystic kidney disease: a complex interaction with polycystins and primary cilium. Cell Signal. 2020;66:109468. doi: 10.1016/j.cellsig.2019.109468. [DOI] [PubMed] [Google Scholar]
- 34.Merrick D, Bertuccio CA, Chapin HC, Lal M, Chauvet V, Caplan MJ. Polycystin-1 cleavage and the regulation of transcriptional pathways. Pediatr Nephrol. 2014;29:505–511. doi: 10.1007/s00467-013-2548-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Paul BM, Vanden Heuvel GB. Kidney: polycystic kidney disease. Wiley Interdiscip Rev Dev Biol. 2014;3:465–87. doi: 10.1002/wdev.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu D, Ma Y, Gu X, Bian R, Lu Y, Xing X, Mei C. Novel mutations in the PKD1 and PKD2 genes of chinese patients with autosomal dominant polycystic kidney disease. Kidney Blood Press Res. 2018;43:297–309. doi: 10.1159/000487899. [DOI] [PubMed] [Google Scholar]
- 37.Bhutani H, Smith V, Rahbari-Oskoui F, Mittal A, Grantham JJ, Torres VE, Mrug M, Bae KT, Wu Z, Ge Y, Landslittel D, Gibbs P, O’Neill WC, Chapman AB CRISP Investigators. A comparison of ultrasound and magnetic resonance imaging shows that kidney length predicts chronic kidney disease in autosomal dominant polycystic kidney disease. Kidney Int. 2015;88:146–151. doi: 10.1038/ki.2015.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cornec-Le Gall E, Audrezet MP, Le Meur Y, Chen JM, Ferec C. Genetics and pathogenesis of autosomal dominant polycystic kidney disease: 20 years on. Hum Mutat. 2014;35:1393–1406. doi: 10.1002/humu.22708. [DOI] [PubMed] [Google Scholar]
- 39.Song X, Haghighi A, Iliuta IA, Pei Y. Molecular diagnosis of autosomal dominant polycystic kidney disease. Expert Rev Mol Diagn. 2017;17:885–895. doi: 10.1080/14737159.2017.1358088. [DOI] [PubMed] [Google Scholar]
- 40.Cheung CY, Chan HW, Liu YL, Chau KF, Li CS. Long-term graft function with tacrolimus and cyclosporine in renal transplantation: paired kidney analysis. Nephrology (Carlton) 2009;14:758–763. doi: 10.1111/j.1440-1797.2009.01155.x. [DOI] [PubMed] [Google Scholar]
- 41.Rossetti S, Burton S, Strmecki L, Pond GR, San Millan JL, Zerres K, Barratt TM, Ozen S, Torres VE, Bergstralh EJ, Winearls CG, Harris PC. The position of the polycystic kidney disease 1 (PKD1) gene mutation correlates with the severity of renal disease. J Am Soc Nephrol. 2002;13:1230–1237. doi: 10.1097/01.asn.0000013300.11876.37. [DOI] [PubMed] [Google Scholar]
- 42.Cornec-Le Gall E, Audrezet MP, Chen JM, Hourmant M, Morin MP, Perrichot R, Charasse C, Whebe B, Renaudineau E, Jousset P, Guillodo MP, Grall-Jezequel A, Saliou P, Ferec C, Le Meur Y. Type of PKD1 mutation influences renal outcome in ADPKD. J Am Soc Nephrol. 2013;24:1006–1013. doi: 10.1681/ASN.2012070650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ong AC, Harris PC. A polycystin-centric view of cyst formation and disease: the polycystins revisited. Kidney Int. 2015;88:699–710. doi: 10.1038/ki.2015.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Al-Muhanna FA, Al-Rubaish AM, Vatte C, Mohiuddin SS, Cyrus C, Ahmad A, Shakil Akhtar M, Albezra MA, Alali RA, Almuhanna AF, Huang K, Wang L, Al-Kuwaiti F, Elsalamouni TSA, Al Hwiesh A, Huang X, Keating B, Li J, Lanktree MB, Al-Ali AK. Exome sequencing of Saudi Arabian patients with ADPKD. Ren Fail. 2019;41:842–849. doi: 10.1080/0886022X.2019.1655453. [DOI] [PMC free article] [PubMed] [Google Scholar]