

Abstract
Background
Gitelman syndrome (GS) is an autosomal recessive inherited salt-losing tubulopathy (SLT). Here, we describe, for the first time, a case of GS without Gitelman-like features and with concomitant kidney stones, cysts and diabetic nephropathy (DN).
Case presentation
We described a male patient had a 19-year history of recurrent fatigue. From childhood, he had polydipsia and polyuria, paroxysmal tetany and palpitation. Serum biochemistry revealed chronic hypokalemia, metabolic alkalosis, normomagnesemia, mildly elevated Cr. Concomitant 24 h urine collection showed inappropriate renal potassium wasting, borderline hypercalciuria, moderate proteinuria consisting of major glomerular. Ultrasound of urinary tract showed bilateral and multiple kidney stones and cysts. Whole exome sequencing (WES) identified compound heterozygous mutations of SLC12A3. The unusual association of SLTs and glomerular proteinuria prompted us to perform a renal biopsy. Renal pathology showed renal involvement consistent with GS and early stage of diabetic nephropathy (DN). After treatment with KCl, magnesium oxide, perindopril and acarbose, the patient had been cured. The fatigue didn’t relapse.
Conclusion
GS had high variability of phenotype, GS may have no Gitelman-like features, kidney stones are not the exclusion criteria of GS. Renal biopsy should be warranted for GS patients with moderate to massive glomerular proteinuria.
Keywords: Gitelman syndrome, Case report, Diabetic nephropathy, Kidney stones
Background
Gitelman syndrome (GS; OMIM 263800) is an autosomal recessive inherited salt-losing tubulopathy (SLT) characterized by hypokalemic metabolic alkalosis, hypomagnesemia, and hypocalciuria. GS is caused by biallelic inactivating mutations in the SLC12A3 gene encoding the thiazide-sensitive sodium-chloride co-transporter (NCCT) in the distal convoluted tubule (DCT) [1, 2]. Previously, hypomagnesemia (< 0.7 mmol/L) with in appropriaterenal magnesium wasting and hypocalciuria were regarded as diagnostic criteria for GS. On the contrast, nephrolithiasis was considered as the one of the features against a diagnosis of GS due to hypocalciuria [3]. Additionally, SLTs including GS are not classically associated with proteinuria, especially glomerular proteinuria [4]. To date, GS without Gitelman-like features and with concomitant nephrolithiasis and glomerular proteinuria hasn’t been reported.
Case presentation
A 40-year-old, malepatient, was admitted to the hospital with a 19-year history of recurrent fatigue was hospitalized in The First People's Hospital of Huzhou on September 27, 2019.Generalized fatigue and dyspnea developed following upper respiratory infection 19 years ago, laboratory test revealed profound hypokalemia (serum potassium1.8 mmol/L), he was intubated and mechanic ventilation was applied due to respiratory failure resulting from respiratory muscle weakness. After intravenous potassium chloride (KCl) was administered, serum potassium was normalized, the fatigue and dyspnea was subsequently completely resolved. After he was discharged, he wasn’t on KCl supplement though generalized fatigue relapsed occasionally. Fatigue resolved by potassium-rich food intake. From 10 years ago, fatigue failed to be resolved by potassium-rich food intake. Therefore, he was on potassium supplement (KCl 2 ~ 3 g daily). Ten years ago, mild proteinuria developed, the amount of 24 h protein excretion was about 2 g, increased postprandial glucose level was revealed 2 years ago, but he did not take any glucose-lowering drug. From childhood, he had polydipsia and polyuria, paroxysmal tetany and palpitation, but had no salt craving, constipation, physical and mental retardation. His parents were not consanguineous, he had a younger sister and a son. His relatives are all healthy. Physical examination revealed a sitting blood pressure of 118/83 mmHg. His height was 170 cm, body weight 59.3 Kg, BMI 20.52 kg/m2.Serum biochemistry showed hypokalemia, hypochloridemia, normomagnesemia, mildly elevated creatinine (Cr). Concomitant 24 h urine collection showed inappropriate renal potassium wasting, borderline hypercalciuria, moderate proteinuria (2793 mg), and high oxalic acid and low citric acid. Arterial blood gas analysis revealed decompensated metabolic alkalosis. Urinalysis showed elevated microalbuminuria which indicated glomerular injury and elevated β2- microglobulinuria which indicated proximal tubular injury. Fasting blood glucose level was 7.33 mmol/L, the glucose level at 2nd hour of Oral glucose tolerance test (OGTT) was 11.27 mmol/L, both glucose levels confirmed the diagnosis of type2 diabetes mellitus (DM) (Table 1). Ultrasound of urinary tract showed bilateral and multiple kidney stones (Fig. 1a, b) and cysts (Fig. 1c, d).
Table 1.
Laboratory investigations
| Examination item | Test value | Reference value |
|---|---|---|
| Serum biochemistry | ||
| Potassium (mmol/L) | 2.0 | 3.50–5.30 |
| Sodium (mmol/L) | 139.0 | 137.0–147.0 |
| Chloride (mmol/L) | 98.0 | 99.0–110.0 |
| Calcium (mmol/L) | 2.15 | 2.08–2.60 |
| Magnesium (mmol/L) | 0.92 | 0.70–1.10 |
| Phosphate (mmol/L) | 0.86 | 0.85–1.51 |
| Creatinine (μmol/L) | 102.0 | 57.0–97.0 |
| Uric acid (μmol/L) | 438 | 208–428 |
| Arterial blood gas analysis | ||
| PH value | 7.49 | 7.35–7.45 |
| PaO2 (mmHg) | 102.1 | 83.0–108.0 |
| PaCO2 (mmHg) | 37.0 | 35.0–45.0 |
| HCO3−(mmol/L) | 28 | 22–26 |
| Base excess (mmol/L) | 5.0 | −3.0-3.0 |
| Urine analysis | ||
| Urine specific gravity | 1.015 | 1.003–1.030 |
| PH value | 8.0 | 4.5–8.0 |
| Albumin to creatinine ratio (mg/g.Cr) | 1133.33 | < 25 |
| α1- microglobulin to creatinine ratio (mg/g.Cr) | 44.6 | < 15 |
| 24-h Urine tests | ||
| Potassium (mmol/24 h) | 253.10 | 25.00–100.00 |
| Calcium (mmol/24 h) | 5.34 | 0.25–7.5 |
| Creatinine (mg/24 h) | 1250.2 | 700.0–2300.0 |
| Uric acid (mmol/24 h) | 4666.0 | 1488.0–4463.0 |
| Oxalic acid (mg/24 h) | 93.05 | 20–40 |
| Citric acid (mg/24 h) | 80.18 | > 450 |
| Urinay calcium/creatinine (mmol/mmol) | 0.48 | |
| Renin-angiotensin-aldosterone system (upright) | ||
| Plasma renin (μIU/ml) | > 500 | 4.4–46.1 |
| Plasma aldosterone (pg / ml) | 688 | 30–353 |
| Glycosylated hemoglobin A1C (%) | 6.0 | 4.0–6.0 |
| OGTT tests | ||
| Fasting blood glucose (mmol/L) | 7.33 | 3.89–6.11 |
| Postprandial blood glucose (2 h) (mmol/L) | 11.27 | 3.89–7.78 |
Fig. 1.

Ultrasound imaging of the urinary tract. a multiple kidney stones in the left kidney (arrow). b multiple kidney stones in the right kidney (arrow). c multiple kidney cysts in the left kidney (arrow). d multiple kidney cysts in the right kidney (arrow)
Chronic hypokalemia resulting from urine potassium wasting, metabolic alkalosis, normal blood pressure despite elevated renin and aldosterone indicated SLTs. Therefore, whole exome sequencing (WES) was performed to identify the exact type of SLTs. Genomic DNA was extracted from blood peripheral leukocytes. Paired-end sequencing libraries then were prepared using a DNA sample prep reagent set 1 (NEBNext). The amplified DNA was captured use GenCap WES capture kit. The enrichment libraries were sequenced on IlluminaHiSeq X ten sequencer for paired read 150 bp.After sequencing, the bioinformatics analysis was performed. After quality control, the clean reads were mapped to the UCSC hg19 human reference genome using BWA. Variants were filtered using GATK VariantFiltration and further annotated by ANNOVAR and associated with multiple databases. The potential pathogenic mutations were selected. Candidate mutations were confirmed by Sanger sequencing. The mutations of family members were confirmed by the same procedure. WES revealed two heterozygous mutations in SLC12A3 gene: one splicing mutation (c.2038-1delG), potentially leading to loss of function of the gene, one missense mutation (c.2012 T > G) resulting in arginine substitution for leucine at codon 671 (p.Leu671Arg). According to the American College of Medical Genetics (ACMG) mutation guidelines, the splicing mutation (c.2038-1delG) was classified as “pathogenic” and the missense mutation (c.2012 T > G) was classified as “likely pathogenic”.p.Leu671Arg was a novel mutation. His father carries a heterozygous mutation of c.2038-1delG (Fig. 2a), his mother carried a heterozygous mutation of p.Leu671Arg (Fig. 2b).
Fig. 2.

a. The patient and his father carried a c.2038-1delG (arrow) heterozygous mutation in SLC12A3 gene, and his mother had no mutation inSLC12A3 gene at this site. b. The patient and his mother’s carried a c.2012 T > G (arrow) (p.Leu671Arg) heterozygous mutationinSLC12A3 gene, and his father had no mutation in SLC12A3 gene at this site
The unusual association of SLTs and glomerular proteinuria prompted us to perform a renal biopsy. Light microscopy (LM) showed chronic tubulointerstitial nephritis and hypertrophy of the juxtaglomerular apparatus which are consistent with renal involvement resulting from GS; LM also revealed thickened basement membrane, mild mesangial cells and mesangial matrix proliferation, thickening arteriole wall and narrowing lumen. Immunofluorescence demonstrated no deposition of immunoglobulins or complements. Electron microscopy shows foot process effacement (50–75%) and thickening GBM with no electron dense deposits. The renal pathology also indicated early stage of diabetic nephropathy (DN) (Fig. 3).
Fig. 3.
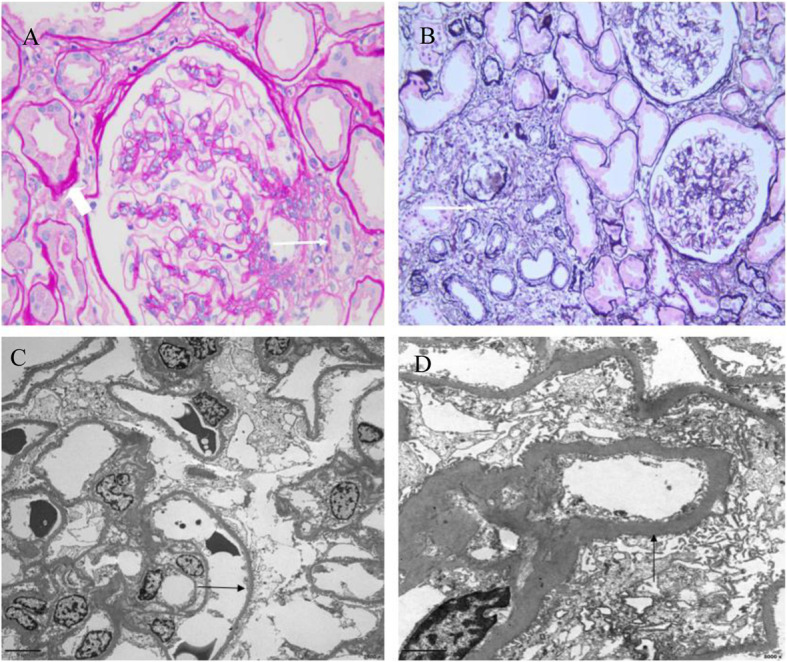
Renal pathology. a Light microscopic findingsof HE staining (magnification × 400) showedjuxtaglomerular apparatus proliferation (arrow) and granular and vacuolar degeneration of tubular epithelial cells (arrowhead). b Light microscopy findings of PASM staining (magnification × 200) showed multifocal atrophy of renal tubular, multifocal infiltration of inflammatory cells and fibrosis (arrow). c Electron microscopy (magnification × 2000) showed foot process effacement (50–75%) (arrow) and no electron dense deposits. d Electron microscopy (magnification × 8000) showed thickening GBM (744 nm)
Eventually, the diagnosis of GS without Gitelman-like features and with kidney stones, cysts and DN was established. KCl at the dose of 3 g three times daily, magnesium oxide 188 mg once daily and perindopril 4 mg once every night are administered to correct hypokalemia, acarbose100 mg three times to correct hyperglycemia. On Dec 20th, 2019, serum Cr was 104 μmol/L, serum potassium3.57 mmol/L and fasting blood glucose was 6.89 mmol/L.The fatigue didn’t relapse.
Discussion and conclusion
We described a male patient had a 19-year history of recurrent fatigue. From childhood, he had polydipsia and polyuria, paroxysmal tetany and palpitation. Serum biochemistry revealed chronic hypokalemia, metabolic alkalosis, normomagnesemia, mildly elevated Cr. Concomitant 24 h urine collection showed inappropriate renal potassium wasting, borderline hypercalciuria, moderate proteinuria consisting of major glomerular. Ultrasound of urinary tract showed bilateral and multiple kidney stones and cysts. WES identified compound heterozygous mutations of SLC12A3.Renal pathology showed renal involvement consistent with GS and early stage of DN. To our best knowledge, this is the first case of GS without Gitelman-like features and with concomitant kidney stones, cysts and DN.
GS is an autosomal recessive, salt-losing tubulopathy. The prevalence of GS is approximately 1–10 per 40,000 persons in Western countries, and potentially higher in Asia [3]. Undoubtedly, identification of biallelic inactivating mutations in SLC12A3 is the indispensable criteria for establishing a diagnosis of GS. In this case, WES revealed two heterozygous mutations in SLC12A3 gene: one splicing mutation (c.2038-1delG), one missense mutation (c.2012 T > G) resulting in arginine substitution forleucine at codon 671 (p.Leu671Arg). p.Leu671Arg was a novel mutation. Therefore, it is indisputable that the patient was diagnosed with GS. Up to now, more than 400 SLC12A3 gene mutation sites have been reported to be associated with GS, of which mutations in exons 1 and 10 are more common [5].
As is well known, GS was characterized by hypomagnesemia (serum magnesium < 0.7 mmol/L) and hypocalciuria (a spot urine calcium to creatinine ratio < 0.2 mmol/mmol), Kidney Disease: Improving Global Outcomes (KDIGO) guidance recommended both hypomagnesemia and hypocalciuria as diagnostic criteria for GS. On the contrast, nephrolithiasis was considered as the one of the features against a diagnosis of GS due to hypocalciuria [3]. However, a recent study from China showed that both hypomagnesemia and hypocalciuria were not sensitive enough to diagnose GS. The diagnostic sensitivity and specificity of hypomagnesemia for GS were 72.41 and 87.50%, respectively, the diagnostic sensitivity and specificity of hypocalciuriafor GS were 44.83 and 75.00%, respectively [6]. The present patient had normomagnesemia, borderline hypercalciuria, bilateral and multiple kidney stones and cysts which were all not consistent with typical GS. Eventually, gene sequencing identified the diagnosis of GS. Accordingly, GS had highly variable phenotype and gene sequencing was indispensable to diagnose correctly. Therefore, kidney stones and cysts are not exclusion criteria of GS. The mechanism of nephrolithiasis of the present case are unclear now, no relevant report was found in the Chinese and foreign literatures. In this case, the 24-h Urine tests showed high oxalic acid and low citric acid, indicating that the formation of kidney stones probably was a result of the increase in the urinary excretion rate of oxalic acid and the decrease in the urinary excretion rate of citric acid, which correlated with the increase in nephrolithiasis, on the other side, the polydipsia indicates insufficient capacity, which may further increase the risk of stone formation. The elevated β2- microglobulinuria which indicated proximal tubular injury, it may be associated with the increased reabsorption of sodium and calcium from proximal convoluted tubules. The connection between the kidney stones and cysts is unclear, the WES also excluded other 500 hereditary disease, including polycystic kidney.
SLTs including GS are not classically associated with proteinuria, especially glomerular proteinuria. However, in this case, the 24-h urinary protein excretion was 2793 mg, while most of the previously reported GS patients had normal or mildly elevated urinary protein [7, 8]. The findings of renal pathology were consistent with renal involvements of GS and early stage of DN. Therefore, for the GS patients with moderate to massive glomerular proteinuria, renal biopsy should to be warranted. Possible mechanisms between GS and glomerular proteinuria include angiotensin II or renin-induced podocyte lesions, as well as chronic hypokalaemia. One reason is that the chronic activation of the renin angiotensin-aldosterone pathway, leading to increased systemic and local levels of angiotensin-II and renin, may in turn cause podocyte lesions. Presently, the mechanism of GS with DN is still unclear, and some scholars have proposed that the dysfunction of NCC may be one of the main reasons for the risk of DN due to chronic hyperglycemia caused by insulin resistance in individuals with Type 2 Diabetes [9, 10]. In addition, Arg913Gln variation of SLC12A3 gene is associated with DN in type 2 diabetes and GS [11]. A patient with GS and nephrotic syndrome was reported, the renal pathology revealed minimal lesions [7]. Maybe DN is coincidental to the GS, however, when glomerular proteinuria occurred in patients with GS, renal biopsy should be warranted in order to identify overlapping kidney diseases.
GS had high variability of phenotype, GS may have no Gitelman-like features, kidney stones are not the exclusion criteria of GS. Renal biopsy should be warranted for GS patients with moderate to massive glomerular proteinuria.
Acknowledgements
Not applicable.
Abbreviations
- GS
Gitelman syndrome
- SLT
Salt-losing tubulopathy
- NCCT
Thiazide-sensitive sodium-chloride co-transporter
- DCT
Distal convoluted tubule
- WES
Whole exome sequencing
- DM
Diabetes mellitus
- ACMG
American College of Medical Genetics
- LM
Light microscopy
- DN
Diabetic nephropathy
- GBM
Glomerular basement membrane
Authors’ contributions
QC was responsible for designing and data analysis and was a major contributor in writing the manuscript. JM assisted in editing the manuscript. XW participated in the treatment as primary physician and contributed to the collection and analysis of patient’s data. LM drafted the manuscript, and analyzed pathological and mutational data. LW performed the pathological interpretation of the renal tissue. All authors have read and approved the manuscript.
Funding
This work is financially supported by grants of Zhejiang Province Public Welfare Technology Application Research Project (2017C37106) and Zhejiang Medical and Health Science and Technology Project (2018KY080,2020RC118) and Huzhou Municipal Science and Technology Bureau Public Welfare Application Research Project (2016GYB25), mainly for the gene sequencedetection.
Availability of data and materials
Data regarding this study were obtained from clinical charts stored in the physician office records of The First People's Hospital of Huzhou therefore, cannot be shared. Any reasonable request to access the data must be approved before the data can be released.
Ethics approval and consent to participate
Ethical approval was obtained from the ethics committee of The First People's Hospital of Huzhou. All procedures in this study involving consent to participate were performed in accordance with the ethical standards of the institutional ethics and research committee. Written informed consent was obtained from the patient and his family members for participate in this study.
Consent for publication
Written informed consent was obtained from the patient and his family members prior to the use of their data for publication.
Competing interests
The authors declared that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Nakhoul F, Nakhoul N, Dorman E, Berger L, Skorecki K, Magen D. Gitelman’s syndrome: a pathophysiological and clinical update. Endocrine. 2012;41(1):53–57. doi: 10.1007/s12020-011-9556-0. [DOI] [PubMed] [Google Scholar]
- 2.Gitelman Syndrome Collaborative Study G Expert consensus for the diagnosis and treatment of patients with Gitelman syndrome. Chinese J Internal Med. 2017;56(9):712. doi: 10.3760/cma.j.issn.0578-1426.2017.09.021. [DOI] [PubMed] [Google Scholar]
- 3.Blanchard A, Bockenhauer D, Bolignano D, Calo LA, Cosyns E, Devuyst O, et al. Gitelman syndrome: consensus and guidance from a kidney disease: improving global outcomes (KDIGO) controversies conference. Kidney Int. 2017;91(1):24–33. doi: 10.1016/j.kint.2016.09.046. [DOI] [PubMed] [Google Scholar]
- 4.Demoulin N, Aydin S, Cosyns J-P, Dahan K, Cornet G, Auberger I, et al. Gitelman syndrome and glomerular proteinuria: a link between loss of sodium-chloride cotransporter and podocyte dysfunction? Nephrol Dial Transplant. 2014;29(suppl_4):iv117–iv120. doi: 10.1093/ndt/gfu075. [DOI] [PubMed] [Google Scholar]
- 5.Takeuchi Y, Mishima E, Shima H, Akiyama Y, Suzuki C, Suzuki T, et al. Exonic mutations in the SLC12A3 gene cause exon skipping and premature termination in Gitelman syndrome. J Am Soc Nephrol. 2015;26(2):271–279. doi: 10.1681/ASN.2013091013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Peng X, Zhao B, Zhang L, Jiang L, Yuan T, Wang Y, et al. Hydrochlorothiazide test as a tool in the diagnosis of Gitelman syndrome in Chinese patients. Front Endocrinol. 2018;559. [DOI] [PMC free article] [PubMed]
- 7.Chen Q, Wu Y, Zhao J, Jia Y, Wang W. A case of hypokalemia and proteinuria with a new mutation in the SLC12A3 gene. BMC Nephrol. 2018;19(1):1–6. doi: 10.1186/s12882-018-1083-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fulchiero R, Seo-Mayer P. Bartter syndrome and Gitelman syndrome. Pediatr Clin. 2019;66(1):121–134. doi: 10.1016/j.pcl.2018.08.010. [DOI] [PubMed] [Google Scholar]
- 9.Ren H, Qin L, Wang W, Ma J, Zhang W, Shen PY, et al. Abnormal glucose metabolism and insulin sensitivity in Chinese patients with Gitelman syndrome. Am J Nephrol. 2013;37(2):152–157. doi: 10.1159/000346708. [DOI] [PubMed] [Google Scholar]
- 10.Yuan T, Jiang L, Chen C, Peng X, Nie M, Li X, et al. Glucose tolerance and insulin responsiveness in Gitelman syndrome patients. Endocrine Connect. 2017;6(4):243–252. doi: 10.1530/EC-17-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De la Cruz-Cano E, CdC J-G, Morales-García V, Pineda-Pérez C, Tejas-Juárez JG, Rendón-Gandarilla FJ, et al. Arg913Gln variation of SLC12A3 gene is associated with diabetic nephropathy in type 2 diabetes and Gitelman syndrome: a systematic review. BMC Nephrol. 2019;20(1):393. doi: 10.1186/s12882-019-1590-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data regarding this study were obtained from clinical charts stored in the physician office records of The First People's Hospital of Huzhou therefore, cannot be shared. Any reasonable request to access the data must be approved before the data can be released.