

Abstract
β thalassemia major or Cooley’s Anemia (CA) has been difficult to model in mice due to their lack of a fetal hemoglobin gene equivalent. This summary describes novel preclinical humanized mouse models of CA that survive on human fetal hemoglobin at birth and are blood-transfusion dependent for life upon completion of their human fetal-to-adult hemoglobin switch after birth. These CA models are the first to recapitulate the temporal onset of the disease in human patients. These novel humanized CA disease models are useful for the study of the regulation of globin gene expression, synthesis, and switching; examining the onset of disease pathology; development of transfusion and iron chelation therapies; induction of fetal hemoglobin synthesis; and the testing of novel genetic and cell-based therapies for the correction of thalassemia.
Keywords: thalassemia, HPFH, transfusion, mouse model, hemoglobin switching
Introduction
Hemoglobin is a tetramer consisting of two α globin chains and two β globin chains. Thalassemia is caused by an imbalance in α and β globin chain production. The most severe form of β thalassemia, β-thalassemia major or Cooley’s Anemia (CA), is caused by complete absence of the adult β globin chains. Newborn CA patients are initially healthy due to high levels of fetal hemoglobin but develop severe anemia upon completion of their fetal to adult hemoglobin switch at about 1 year of age.1,2 In a futile response to correct their anemia, CA patients expand the production of short-lived thalassemic erythroid progenitors in their bone marrow in a process termed ineffective erythropoiesis.3 For survival into adulthood, CA patients require lifelong regular blood transfusion.4 A preclinical CA animal model that more closely mimics the developmental onset and disease pathology of thalassemia in humans will greatly facilitate the study of CA.
Modeling human hemoglobin disorders in mice is complicated by the fact that mice do not have a true fetal hemoglobin. Both humans and mice change the type of α and β globin chains produced in the blood during development.2,5,6 This hemoglobin switching occurs in two distinct cell types. The first type, the primitive erythroid cell, arises early in development in the yolk sac blood islands. These primitive erythroid cells are large, nucleated, and synthesize embryonic globin chains. The second type termed definitive erythroid cell arises when erythropoiesis shifts to the fetal liver. Definitive erythroid cells are smaller, enucleated, and synthesize predominantly adult (mouse) or fetal (humans) globin chains. When definitive erythropoiesis shifts to the bone marrow after birth, mice continue to express the same adult globin genes synthesized during fetal development, but human definitive erythroid cells undergo a fetal to adult hemoglobin switch.
Murine models of β thalassemia have been made by deleting the adult mouse β goblin genes or replacing the adult mouse β globin genes with nonfunctional human β globin transgenes.7–12 Because murine fetal liver definitive erythropoiesis requires adult β globin gene expression, all of these mice die in utero when homozygous for the adult mouse β globin genes deletion. Mice have no fetal hemoglobin equivalent to sustain definitive erythropoiesis in the absence of their adult globin genes. Furthermore, attempts to produce mice that express high levels of human fetal hemoglobin in definitive erythroid cells by introducing human cosmid, BAC, or YAC transgenes that encompass the entire human β globin locus has been largely unsuccessful.13–17 The human γ globin transgene expression in these models is predominantly confined to the primitive erythroid cells.18–20 Modified human transgenes that contain the locus control region linked directly to human γ and β globin genes with much of the intergenic sequence deleted have been successful in producing human β globin gene expression in definitive erythroid cells.21,22 Indeed, such a delayed fetal to adult hemoglobin-switching transgene was important for the generation of a mouse model of sickle cell disease.21 Recently, our group has used a delayed fetal to adult hemoglobin-switching cassette to generate murine models of CA that more faithfully recapitulate the hemoglobin switch that occurs in humans.18,19
The ideal mouse model of CA should have the following characteristics: express only human hemoglobin in their red blood cells (RBCs), complete the human γ to β globin gene switch after birth, synthesize no functional adult β globin chains in RBCs, require regular blood transfusions for life, and be genetically heritable. Recently, our group has made a series of humanized CA mouse models that have all of these qualities by targeted gene replacement of the adult mouse α and β globin genes with human α and γβ° globin genes in mouse embryonic stem (ES) cells.18,19 Each model synthesizes high levels of human fetal hemoglobin at birth, but becomes severely anemic upon completion of their hemoglobin switch to a nonfunctional human β° globin allele after birth. Humanized CA mice exhibit increasing ineffective erythropoiesis and expire in the absence of life sustaining blood transfusions. These humanized CA mouse models will be useful for the study of the mechanism of human hemoglobin switching, examining the onset of disease pathology, developing novel methods for regulating iron toxicity, and for the testing of novel cellular and genetic therapies for the cure of CA.
Production of humanized mouse models of Cooley’s Anemia
β thalassemia major in mouse models generated by deletion of the adult β globin genes results in early fetal death because the mouse does not have a fetal globin gene equivalent. We hypothesized that replacement of the mouse adult β globin genes with a human fetal to adult hemoglobin-switching cassette would produce animals with high levels of human fetal hemoglobin during fetal life. Using this strategy, we recently reported the production of two humanized mouse models of CA.18,19 In this summary, we compare these models with an additional CA model. Each of these humanized models was produced by replacing the endogenous adult mouse α and β globin genes with human α and γβ or γδβ globin genes, respectively. Material and methods may be found online in the Supporting Information.
Figure 1 illustrates the gene-targeting strategy whereby human β-like globin genes were knocked into (KI) the murine β globin locus simultaneously deleting the adult murine β globin genes in ES cells. All three CA models have a nonfunctional human β° globin KI allele that contains a splice donor site mutation in the first base of intron 1 (IVS 1.1 G to A) that results in the production of no functional β globin chains.19,23 The γβ° KI model contains a wild-type human γ globin promoter, whereas the γHPFHβ° KI and γHPFHδβ° KI models have the Greek-type hereditary persistence of fetal hemoglobin (HPFH) mutation in the γ globin gene promoter (−117 G to A).24 The minor human adult δ globin gene is included in the γHPFHδβ° KI model. Mice produced from each of these KI ES cells were bred to human α2α1 globin KI mice (T.M.R., unpublished data) generated by similar methodology to produce humanized CA mice that synthesize solely human hemoglobin in their RBCs.18,19,25
Figure 1.
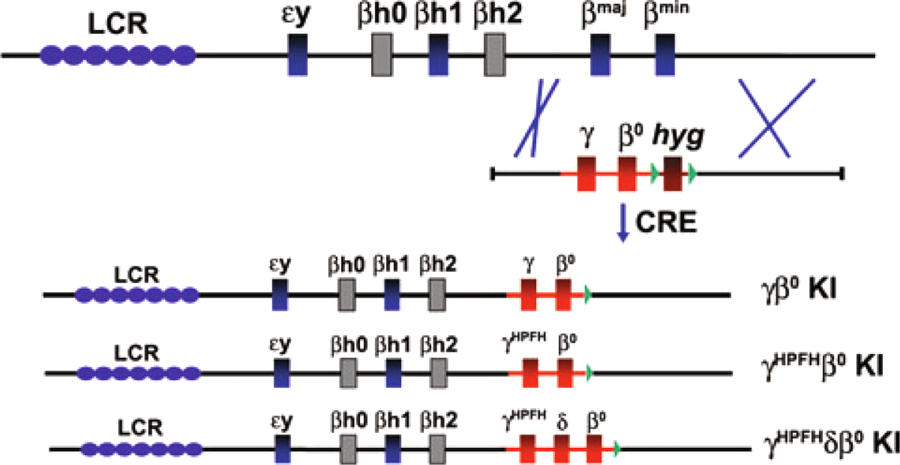
Gene-targeting strategy in ES cells used to replace the adult mouse β globin genes with human globin genes in the mouse β globin locus. Top line represents the wild-type mouse β globin locus. A single-targeting vector is drawn below the locus illustrating homologous recombination between 5′ and 3′ mouse homology regions. After removal of the hygromycin marker gene (hyg) by recombinase (CRE), the resulting human globin gene KI alleles are shown. Locus control region (LCR) represented by blue ovals, functional mouse embryonic genes (εy and βh1), and adult genes (βmaj and βmin) represented by blue boxes, mouse pseudogenes drawn as gray boxes, human genes are red boxes, hygromycin marker gene is brown box, loxP sites are green triangles.
Completion of the fetal-to-adult hemoglobin switch after birth extends the postnatal lifespan of humanized CA mice
Humanized γβ KI mice synthesize high levels of fetal hemoglobin during fetal life that persists after birth. In Figure 2A, the human γ, δ, and β globin chain levels in hemolysates of humanized wild-type control (γβA/γβA) and compound heterozygous mice (γHPFHβ°/γβA and γHPFHδβ°/γβA) are plotted over time. At birth, there are high levels of fetal hemoglobin in all three types of humanized mice illustrating the delayed hemoglobin switch inherent in the γβ KI cassette. In addition, the humanized mice all complete their fetal to adult hemoglobin switch after birth. Although the γ globin chains in humanized control mice (γβA/γβA) decrease to background levels by 3 weeks of age, the fetal chain levels in both the γHPFH KI mice persist into adulthood. Finally, there is also a fetal (HbF) to minor adult (HbA2) hemoglobin switch that occurs over time in humanized γHPFHδβ°/γβA KI mice.18
Figure 2.
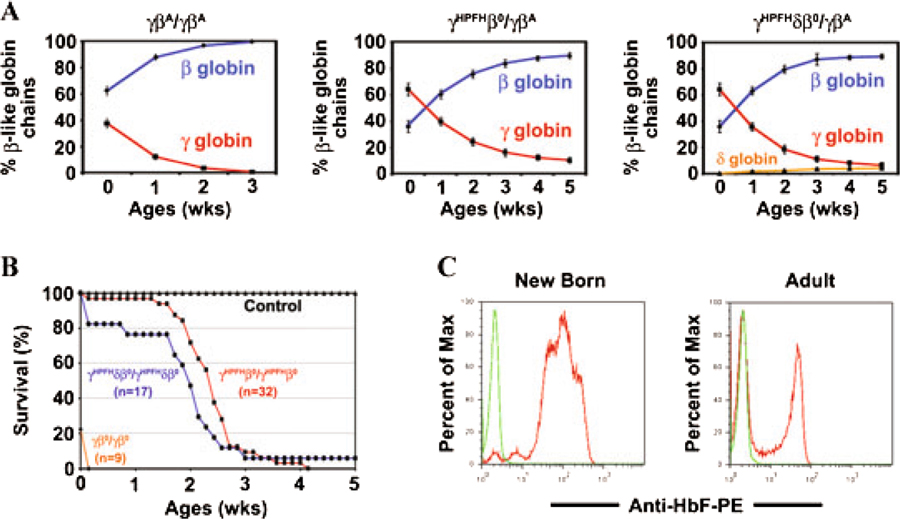
HPFH mutations modulate the human γ globin levels in CA mice. (A) Total β-like globin chains in peripheral blood hemolysates of humanized newborn to 5-week-old mice were separated by HPLC and quantified. High fetal γ globin levels at birth are replaced by adult β globin as the hemoglobin switch is completed after birth. Genotype of each humanized KI mouse shown at the top of each figure. Fetal γ globin chains shown in red, adult β globin in blue, and adult δ globin in orange. Values represent mean ± SEM. n ≥ 9 at each time point. (B) Survival curves of humanized CA mice. The percentage of mice alive for each genotype is plotted with age. The few humanized γβ° KI CA mice that survive to birth die within the first 24 h. Incorporation of the HPFH mutation in the human γ globin gene promoter in CA mice results in 100% survival to birth and an average lifespan of 14 and 15 daysforthe γHPFHδβ° and γHPFHβ° CA mice, respectively. (C) F-cell distribution in newborn and adult heterozygous γHPFHβ°/γβA KI mice. Peripheral mouse blood was fixed and stained with anti-human fetal hemoglobin monoclonal antibody and analyzed by flow cytometry. HbF is pancellularly distributed in newborn humanized mice, whereas adult mice have a heterocellular distribution. Green line is wild-type mouse control.
The level of fetal globin chain production and the completion of the fetal to adult hemoglobin switch greatly influence the severity of anemia and the lifespan of humanized CA mice. Figure 2B is a plot of the survival curves for homozygous CA mice. Only 20% of humanized γβ°/γβ° mice survive to birth. These CA mice are extremely pale and die within the first 24 h after birth.19 In contrast, all humanized CA mice containing the HPFH mutation in the human γ globin gene promoter are alive at birth. As the high fetal hemoglobin levels in these CA mice declines over the first few weeks of life, the majority of the animals die around 2 weeks of age. At birth, virtually 100% of the RBCs in γβ KI mice contain fetal hemoglobin. This pancellular distribution of HbF at birth is evident by flow cytometry in compound heterozygous γHPFHβ°/γβA KI mice after staining with an anti-HbF antibody in Figure 2C. In adult mice, HbF is distributed heterocellularly being present in less than 50% of the circulating RBCs. All the above data suggest that γβ globin KI mice synthesize high levels of fetal hemoglobin throughout fetal life and complete their fetal to adult hemoglobin switch after birth similar to humans.
Severe anemia, pathology, and death of humanized CA mice are prevented by serial blood transfusion
Humanized CA mice become severely anemic after birth. Compared to wild-type humanized controls, homozygous γHPFHδβ° KI CA mice have marked reductions in their RBC, hemoglobin, and hematocrit levels (Fig. 3A). Homozygous CA mice have a massive reticulocytosis exceeding 70% of peripheral blood cells. Peripheral blood smears show numerous erythroblasts and nucleated RBCs, hypochromic RBCs, and anisopoikilocytosis (Fig. 3B). Heterozygous γHPFHδβ°/γβA KI mice also show significant changes in their RBC indices, but are only mildly anemic. The persistent expression of fetal hemoglobin from the HPFH allele results in a much milder anemia compared to heterozygous β globin knockout or human γβ° globin KI mice.8,18,19
Figure 3.
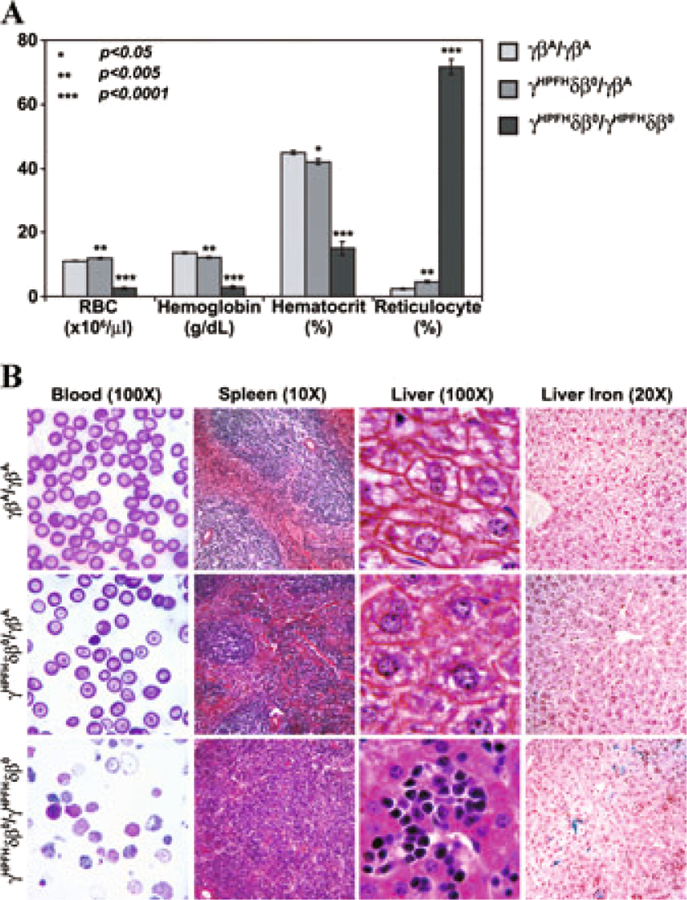
Hematological RBC indices and histopathology of humanized control, heterozygous, and homozygous CA mice. (A) RBC indices of peripheral blood from humanized mice showed a slight but significant anemia in heterozygous CA mice. Homozygous CA mice are severely anemic with marked reductions in RBC, hemoglobin, and hematocrit levels and an extreme reticulocytosis compared to control mice. Humanized control and heterozygous mice were analyzed eight weeks after birth. Moribund homozygous CA mice were analyzed prior to their euthanasia around two weeks of age. Values represent mean ± SEM. n ≥ 9 for each mouse line. Statistical significances were determined compared to the γβA/γβA control mice. P values were calculated by two-tailed unpaired Student’s t-test. (B) Peripheral blood smear, spleen, and liver sections of humanized control (γβA/γβA), heterozygous (γHPFHδβ°/γβA), and homozygous (γHPFHδβ°/γHPFHδβ°) CA mice. Control mice have normocytic RBCs and typical splenic and hepatic structure. Heterozygous CA mice are slightly thalassemic with frequent RBC targeting, but little erythroid hyperplasia in the spleen or extramedullary hematopoiesis in the liver. Homozygous CA mice have numerous nucleated RBCs and erythroblasts in the peripheral blood, marked erythroid hyperplasia in the spleen and extramedullary hematopoiesis, and extensive iron overload in the liver.
Histopathological sections of spleen and liver of humanized control (γβA/γβA), heterozygous (γHPFHδβ°/γβA), and homozygous (γHPFHδβ°/γHPFHδβ°) CA mice are shown in Figure 3B. The severe anemia in CA mice causes a massive erythroid hyperplasia resulting in splenomegaly characterized by the loss of lymphoid white pulp regions and the expansion of the red pulp with erythroid progenitors. The livers of anemic CA mice have numerous foci of extramedullary erythropoiesis and increased iron deposition. In contrast, the heterozygous γHPFHδβ°/γβA KI mice show little disease pathology consistent with their slight anemia.
The severe anemia and shortened lifespan of humanized CA mice are similar to human patients with β thalassemia major that do not receive treatment. The average age of death of CA mice is approximately equivalent to a 1- to 2-year-old human. To extend the life of CA mice, we transfused them with packed RBCs from GFP transgenic mice so that the donor RBCs could be tracked in vivo (Fig. 4). All the transfused CA mice were rescued from their lethal anemia and survived beyond weaning age. After five weekly transfusions, the endogenous erythropoiesis in the CA mice was effectively suppressed. Over 99% of the peripheral blood was from donor GFP transgenic mice and the hemoglobin level stabilized at 14 g/dL. After eight weeks of transfusion, the experiment was stopped and the CA mice were analyzed. All had marked reductions in spleen size and endogenous erythropoiesis was suppressed.18 Importantly, the transfused CA mice had increased iron deposition in the liver similar to transfused CA patients.18
Figure 4.
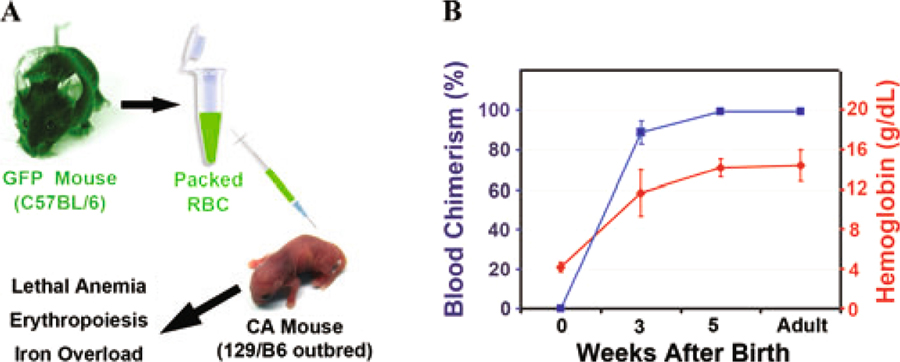
Humanized CA mice can be rescued from lethal anemia by blood transfusion. (A) Blood transfusion scheme for CA mice. Each week fresh peripheral RBCs were collected from GFP transgenic mice, pelleted through Ficoll, washed, resuspended to 70% hematocrit in saline, and transfused into CA mice. (B) Blood chimerism (blue line) and hemoglobin levels (red line) measured over time in transfused CA mice. The donor RBC chimerism was determined by measuring the percentage of GFP+ RBCs in the peripheral blood by flow cytometry. Hemoglobin concentrations maintained at 14 g/dL efficiently suppressed endogenous erythropoiesis indicated by the virtual absence of GFP negative RBCs in the peripheral blood. Transfused CA mice live into adulthood free of anemia but develop severe iron overload.18
Summary of improvements in humanized CA mice over previous β thalassemia major mouse models
Successful generation of mice with true fetal hemoglobin during development was achieved by replacement of the adult mouse β globin genes with delayed switching human γβ globin KI cassettes. The definitive erythroid cells produced in these humanized KI mice switch from fetal to adult hemoglobin similar to humans rather than continuously express the endogenous mouse βmaj and βmin globins throughout both fetal and adult life in wild type mice. Presumably, the high-level expression of the human KI genes is enhanced through interactions with the murine β globin LCR sequences located far upstream (Fig. 1).
There are multiple improvements in these humanized KI mice over previous models of CA. For the first time, homozygous CA pups are alive at birth. Furthermore, similar to humans the high level of human α and γ globin chains synthesized during fetal life generates CA mice that survive upon human HbF at birth. Similarly, as the fetal to adult hemoglobin switch is completed after birth in humanized CA mice, the onset of anemia and disease pathology develops postnatally. Incorporation of an HPFH mutation in the human γ globin promoter and the minor adult δ globin gene in the KI allele extends the postnatal lifespan of the CA mice, making them more amenable for experimental manipulation. The disease in humanized CA mice is heritable in contrast to an adult transplant model of β thalassemia major produced by transplantation of early fetal liver cells from homozygous β globin knockout mice into irradiated wild-type mice.26 Furthermore, disease in the transplant model occurs in adults only after senescence of the recipient’s wild-type RBCs and is marked by abnormal erythroblast differentiation of the donor fetal erythroblasts in the absence of any hemolysis or reticulocytosis.27 Anemia in humanized CA KI mice occurs postnatally as a consequence of hemoglobin switching and is marked by ineffective erythropoiesis, hemolysis, and reticulocytosis similar to human patients.18
Future studies
These novel preclinical humanized mouse models of CA are amenable to many diverse areas of study. Because they survive to birth and develop disease postnatally similar to humans, the pathophysiology of disease progression can be analyzed in parallel with transfusion and chelation therapy. Indeed, new transfusion regimens, iron chelators, or novel small molecules designed to make existing iron chelation therapy more efficient can be analyzed in these mice. Cellular and genetic therapies with stem or progenitor cells designed to cure or ameliorate CA are straightforwardly tested in this model. Because human fetal to adult hemoglobin switching is an integral component of this model, studies of the epigenetic changes during the silencing of the human γ globin KI allele or experiments designed to reactivate the γ globin KI allele in the adult could provide important insights into the mechanism of globin gene switching in humans. For example, the role that BCL11A, a modulator of γ globin levels identified in genome-wide association studies in humans,28,29 may play in the regulation of fetal to adult globin gene switching in humanized CA KI mice could help identify important cis-regulatory sequences required for its action.20,30
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants R01 HL072351 (T.M.R.), R01 HL073440 (T.M.R.), and T32 GM008111 (S.C.M.). The authors thank the Cooley’s Anemia Foundation RFA for Translational Research in Adult Thalassemia for support. S.C.M. and T.Z. were supported by Carmichael Scholarships. The authors thank UNICO Foundation, Mr. Joseph Ruisi, and the Thalassemia-Cooley’s Anemia Group at UAB.
Footnotes
Conflicts of interest
The authors declare no conflicts of interest.
Supporting Information
Additional supporting information describing Materials and Methods may be found in the online version of this article:
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Bunn H & Forget B 1986. Hemoglobin: Molecular, Genetic, and Clinical Aspects. W.B. Saunders; Philadelphia. [Google Scholar]
- 2.Nienhuis AW & Stamatoyannopoulos G 1978. Hemoglobin switching. Cell 15: 307–315. [DOI] [PubMed] [Google Scholar]
- 3.Yuan J, Angelucci E, Lucarelli G, et al. 1993. Accelerated programmed cell death (apoptosis) in erythroid precursors of patients with severe beta-thalassemia (Cooley’s anemia). Blood 82: 374–377. [PubMed] [Google Scholar]
- 4.Piomelli S & Loew T 1991. Management of thalassemia major (Cooley’s anemia). Hematol. Oncol. Clin. North Am 5: 557–569. [PubMed] [Google Scholar]
- 5.Kingsley PD, Malik J, Emerson RL, et al. 2006. “Maturational” globin switching in primary primitive erythroid cells. Blood 107: 1665–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Whitelaw E, Tsai SF, Hogben P & Orkin SH 1990. Regulated expression of globin chains and the erythroid transcription factor GATA-1 during erythropoiesis in the developing mouse. Mol. Cell Biol 10: 6596–6606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shehee WR, Oliver P & Smithies O 1993. Lethal thalassemia after insertional disruption of the mouse major adult beta-globin gene. Proc. Natl. Acad. Sci. USA 90: 3177–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ciavatta DJ, Ryan TM, Farmer SC & Townes TM 1995. Mouse model of human beta zero thalassemia: targeted deletion of the mouse beta maj- and beta min-globin genes in embryonic stem cells. Proc. Natl. Acad. Sci. USA 92: 9259–9263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang B, Kirby S, Lewis J, et al. 1995. A mouse model for beta-thalassemia. Proc. Natl. Acad. Sci. USA 92: 11608–11612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jamsai D, Zaibak F, Khongnium W, et al. 2005. A humanized mouse model for a commonbeta-thalassemia mutation. Genomics 85: 453–461. [DOI] [PubMed] [Google Scholar]
- 11.Lewis J, Yang B, Kim R, et al. 1998. A common human beta globin splicing mutation modeled in mice. Blood 91: 2152–2156. [PubMed] [Google Scholar]
- 12.Vadolas J, Nefedov M, Wardan H, et al. 2006. Humanized beta-thalassemia mouse model containing the common IVSI-110 splicing mutation. J. Biol. Chem 281: 7399–7405. [DOI] [PubMed] [Google Scholar]
- 13.Behringer RR, Ryan TM, Palmiter RD, et al. 1990. Human gamma- to beta-globin gene switching in transgenic mice. Genes Dev 4: 380–389. [DOI] [PubMed] [Google Scholar]
- 14.Gaensler KM, Kitamura M & Kan YW 1993. Germ-line transmission and developmental regulation of a 150-kb yeast artificial chromosome containing the human beta-globin locus in transgenic mice. Proc. Natl. Acad. Sci. USA 90: 11381–11385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaufman RM, Pham CT & Ley TJ 1999. Transgenic analysis of a 100-kb human beta-globin cluster-containing DNA fragment propagated as a bacterial artificial chromosome. Blood 94: 3178–3184. [PubMed] [Google Scholar]
- 16.Peterson KR, Clegg CH, Huxley C, et al. 1993. Transgenic mice containing a 248-kb yeast artificial chromosome carrying the human beta-globin locus display proper developmental control of human globin genes. Proc. Natl. Acad. Sci. USA 90: 7593–7597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Strouboulis J, Dillon N & Grosveld F 1992. Developmental regulation of a complete 70-kb human beta-globin locus in transgenic mice. Genes Dev 6: 1857–1864. [DOI] [PubMed] [Google Scholar]
- 18.Huo Y, C McConnell S & Ryan TM 2009. Preclinical transfusion-dependent humanized mouse model of beta thalassemia major. Blood 113: 4763–4770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huo Y, McConnell SM, Liu S-R, et al. 2008. Humanized mouse model of Cooley’s Anemia. J. Biol. Chem 284: 4889–4896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sankaran VG, Xu J, Ragoczy T, et al. 2009. Developmental and species-divergent globin switching are driven by BCL11A. Nature 460: 1093–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ryan TM, Ciavatta DJ & Townes TM 1997. Knockout-transgenic mouse model of sickle cell disease. Science 278: 873–876. [DOI] [PubMed] [Google Scholar]
- 22.Ryan TM, Sun CW, Ren J & Townes TM 2000. Human gamma-globin gene promoter element regulates human beta-globin gene developmental specificity. Nucleic Acids Res 28: 2736–2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Treisman R, Orkin SH & Maniatis T 1983. Specific transcription and RNA splicing defects in five cloned beta-thalassaemia genes. Nature 302: 591–596. [DOI] [PubMed] [Google Scholar]
- 24.Gelinas R, Endlich B, Pfeiffer C, et al. 1985. G to A sub-stitution in the distal CCAAT box of the A gamma-globin gene in Greek hereditary persistence of fetal haemoglobin. Nature 313: 323–325. [DOI] [PubMed] [Google Scholar]
- 25.Wu LC, Sun CW, Ryan TM, et al. 2006. Correction of sickle cell disease by homologous recombination in embryonic stem cells. Blood 108: 1183–1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rivella S, May C, Chadburn A, et al. 2003. A novel murine model of Cooley anemia and its rescue by lentiviral-mediated human beta-globin gene transfer. Blood 101: 2932–2939. [DOI] [PubMed] [Google Scholar]
- 27.Libani IV, Guy EC, Melchiori L, et al. 2008. Decreased differentiation of erythroid cells exacerbates ineffective erythropoiesis in beta-thalassemia. Blood 112: 875–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Menzel S, Garner C, Gut I, et al. 2007. A QTL influencing F cell production maps to a gene encoding a zincfinger protein on chromosome 2p15. Nat Genet 39: 1197–1199. [DOI] [PubMed] [Google Scholar]
- 29.Uda M, Galanello R, Sanna S, et al. 2008. Genome-wide association study shows BCL11A associated with persistent fetal hemoglobin and amelioration of the phenotype of beta-thalassemia. Proc. Natl. Acad. Sci. USA 105: 1620–1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sankaran VG, Menne TF, Xu J, et al. 2008. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science 322: 1839–1842. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.