

Abstract
Traumatic brain injury (TBI) is one of the most common causes of death and disability worldwide. We investigated whether inhibition of p53 using pifithrin (PFT)-α or PFT-μ provides neuroprotective effects via p53 transcriptional dependent or -independent mechanisms, respectively. Sprague Dawley rats were subjected to controlled cortical impact TBI followed by the administration of PFTα or PFT-μ (2 mg/kg, i.v.) at 5 h after TBI. Brain contusion volume, as well as sensory and motor functions were evaluated at 24 h after TBI. TBI-induced impairments were mitigated by both PFT-α and PFT-μ. Fluoro-Jade C staining was used to label degenerating neurons within the TBI-induced cortical contusion region that, together with Annexin V positive neurons, were reduced by PFT-μ. Double immunofluorescence staining similarly demonstrated that PFT-μ significantly increased HO-1 positive neurons and mRNA expression in the cortical contusion region as well as decreased numbers of 4-hydroxynonenal (4HNE)-positive cells. Levels of mRNA encoding for p53, autophagy, mitophagy, anti-oxidant, anti-inflammatory related genes and proteins were measured by RT-qPCR and immunohistochemical staining, respectively. PFT-α, but not PFT-μ, significantly lowered p53 mRNA expression. Both PFT-α and PFT-μ lowered TBI-induced pro-inflammatory cytokines (IL-1β and IL-6) mRNA levels as well as TBI-induced autophagic marker localization (LC3 and p62). Finally, treatment with PFT-μ mitigated TBI-induced declines in mRNA levels of PINK-1 and SOD2. Our data suggest that both PFT-μ and PFT-α provide neuroprotective actions through regulation of oxidative stress, neuroinflammation, autophagy, and mitophagy mechanisms, and that PFT-μ, in particular, holds promise as a TBI treatment strategy.
Keywords: Traumatic brain injury (TBI), p53, Pifithrin analogs, PFT-α, PFT-μ, Autophagy, Neuroinflammation, Mitophagy
1. Introduction
Traumatic brain injury (TBI) is one of the most common causes of death and disability worldwide, and its incidence continues to rise. According to the Centers for Disease Control (CDC), rates of TBI-related emergency department visits have risen by 70% in the US over the past decade. Pathological consequences of TBI include alterations in biochemical and metabolic pathways, excessive glutamate release, oxidative stress, inflammation, autophagy and mitophagy. Therapeutic options to stop or reverse these processes are not available yet, in part, because the underlying molecular mechanisms are poorly understood. TBI-associated brain damage can be classified into two distinct phases. First, an initial primary damage phase occurs at the moment of insult and includes direct mechanical injury leading to immediate (necrotic) cell death (Greig et al., 2014; LaPlaca et al., 2007). This is followed by an extended second phase involving cascades of biological processes initiated at the time of injury that persist over much longer times, leading to chronic ischemia, neuroinflammation, glutamate toxicity, astrocyte reactivity, neuronal dysfunction and, ultimately, apoptosis (Baratz et al., 2015; Barkhoudarian et al., 2011; Prins et al., 2013; Morganti-Kossmann et al., 2002; Schmidt et al., 2005).
In the subacute period after TBI, the transcription factor p53 is activated in neurons and results in delayed programmed (apoptotic) cell death. At 6 h after TBI, a robust increase in p53-labeled cells is evident at the site of maximal injury (Plesnila et al., 2007; Yang et al., 2015), and genetic deletion of p53 confers protection against TBI (Tomasevic et al., 2010). A lipophilic (ClogD value 1.75 (Zhu et al., 2002)) small molecular weight p53 inactivator, Pifithrin-α (PFT-α), minimizes apoptotic cell death by inhibiting p53 transcriptional activity and preventing p53-dependent activation of apoptotic pathways (Gudkov and Komarova, 2010). PFT-α analogs have been evaluated across rodent species (mice, rats and hamsters) in a broad number of studies, with pharmacologically effective doses ranging between 0.2 and 8 mg/kg. In addition to transactivation of multiple apoptotic gene pathways within the nucleus, p53 also directly mediates apoptosis by binding and inactivating the anti-apoptotic proteins Bcl-xL and Bcl-2 on the mitochondrial surface. Early mitochondrial p53 translocation induces mitochondrial outer membrane permeabilization (MOMP), release of mitochondrial proapoptotic factors (e.g., cytochrome c and Smac/DIABLO) into the cytosol, and triggers subsequent activation of caspase 3–dependent apoptosis (Yang et al., 2015; Gudkov and Komarova, 2010; Filichia et al., 2015). Subtypes of the small molecule Pifithrin, PFT-α and Pifithrin-μ (PFT-μ) are able to differentially block transcriptional and mitochondrial p53 effects, respectively. PFT-α reversibly inactivates p53-dependent transactivation of p53-responsive genes (such as cyclin G, p21/waf1, and mdm2), whereas PFT-μ directly inhibits p53 binding to mitochondria as well as to the Bcl-xL and Bcl-2 proteins (Gudkov and Komarova, 2010; Sparfel et al., 2006). PFT-μ inhibits the interaction of p53 with mitochondria, and thereby mitigates subsequent release of cytochrome c, caspase-3 activation, and apoptosis. By contrast PFT-α inhibits transcriptionally mediated p53 dependent apoptosis, as detailed above.
Our previous work focused on PFTα and its more potent oxygen analog (Yang et al., 2015; Wang et al., 2016). Our primary objective here was to test the efficacy of PFT-μ in a ‘side by side” comparison with PFTα in the same animal TBI model. From a therapeutic approach, the selectivity of PFT-μ is appealing since it inactivates only p53 mitochondrial pathways without affecting other functions of p53 (including p53-mediated expression of genes unrelated to apoptosis) that may not be involved in the pathology of TBI. In the present study, we compared the effect of PFT-μ with PFT-α on functional recovery and neuronal damage after controlled cortical impact (CCI) TBI. Our aim was to examine whether p53 inhibitors using PFT-α or PFT- μ would be a potential therapeutic strategy in a rat model of TBI, particularly if there was a biological overlap between the effects of these two molecules.
2. Methods
2.1. Pifihrin analogues
PFT-α [1-(4-methyl-phenyl)-2-(4,5,6,7-tetrahydro-2-imino-3(2H) benzothiazolyl) ethanone) was generated as its HBr salt (Fig. 1A), following a synthetic route detailed by Zhu et al. (Zhu et al., 2002). Chemical characterization confirmed the structure of the desired agent in high purity (> 99%). PFT-μ (2-phenylethynesulfonamide) was obtained from Millipore Sigma (St. Louis, MO). Chemical characterization confirmed the structure with > 97% purity. Both agents were prepared freshly in 100% DMSO (Millipore Sigma) immediately prior to use and diluted with physiological saline.
Fig. 1.
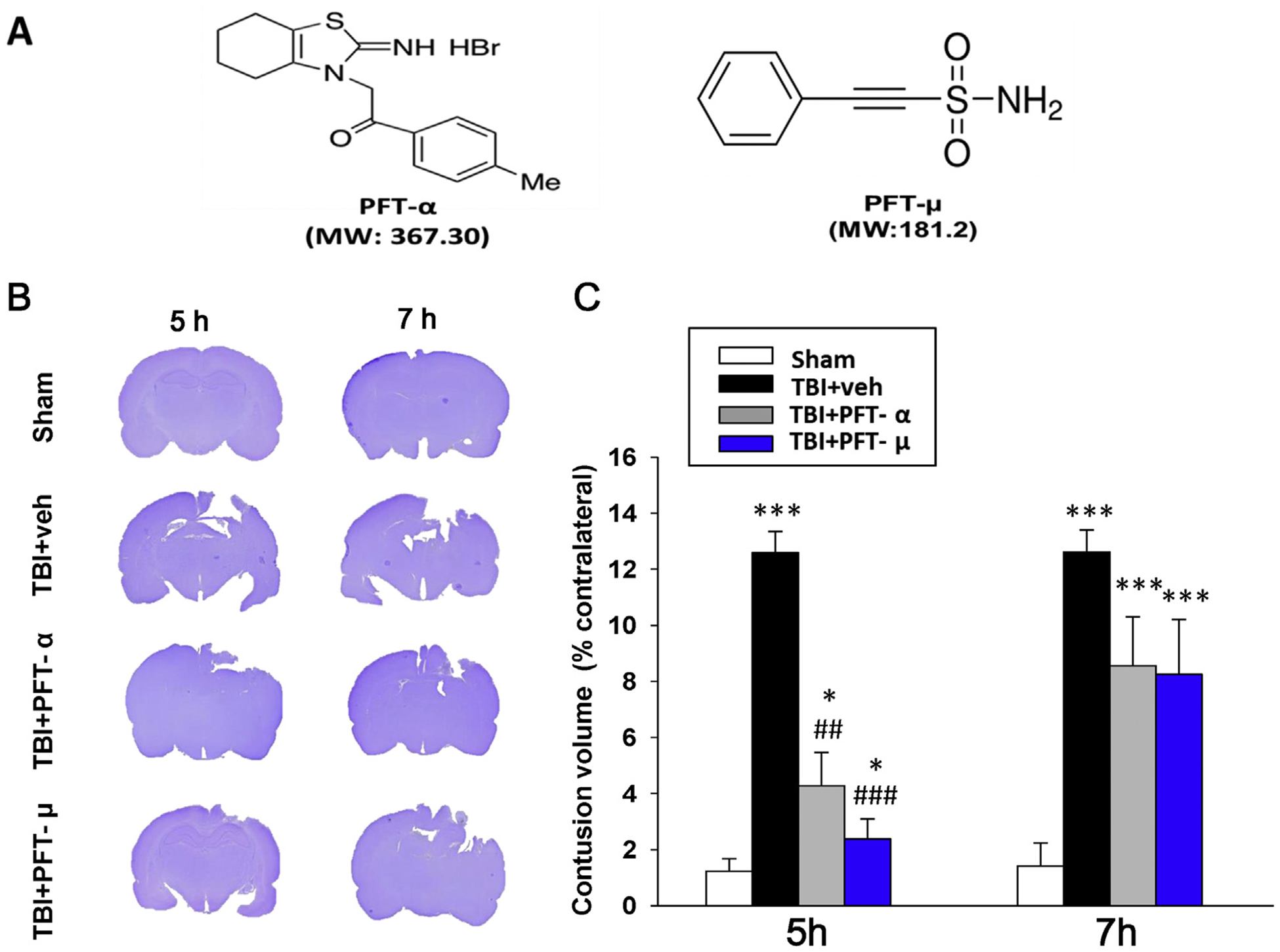
Post-injury administration of PFT-α or PFT-μ at 5 h after TBI significantly reduced contusion volume at 24 h. (A) Structure and molecular weight (MW) of PFT-α and PFT-μ. (B) Coronal sections of sham, vehicle, and PFT-α/ PFT-μ 5 h and 7 h treated TBI rats were obtained and stained with cresyl violet at 24 h post injury. (C) The contusion volumes of TBI groups were not different from one another at 7 h, but at 5 h were significantly reduced by PFT-α or PFT-μ treatment compared to the TBI vehicle (veh) group. Data are expressed as means ± SEM. * P < .05, *** P < .001 versus sham group; ## P < .01, ### P < .001, versus TBI + veh group (n = 8 in TBI + veh and TBI + PFT-α group, n = 5 in sham and TBI + PFT-μ group).
2.2. Animal model of TBI using a controlled cortical impactor (CCI)
All animals were treated in accordance with international guidelines for animal research, and the study design was approved by the animal ethics committee of Taipei Medical University (protocol # LAC-2015–0051). Animals were housed in groups in a temperature- (21–25 °C) and humidity (45%~50%)-controlled room with a 12-h light/dark cycle and ad libitum access to pellet chow and water. A CCI was used to produce TBI in rats as previously described (Yang et al., 2015; Wang et al., 2016). All animals were randomized into four groups (sham, TBI + veh (veh, 0.1% DMSO in saline), TBI + PFTμ, and TBI + PFT-α). Male Sprague-Dawley rats (250–300 g body weight) were anesthetized (2.5% isoflurane) and placed in a stereotaxic frame. A 5-mm craniotomy was performed over the left parietal cortex, centered on the coronal suture and 3.5 mm lateral to the sagittal suture. Injury was made using a CCI instrument with a rounded metal tip (5 mm in diameter). A velocity of 4 m/s and a deformation depth of 2 mm below the dura were used as we previously described (Chen et al., 2007). Sham animals received anesthesia and a craniotomy but no TBI. The body temperature was monitored throughout the surgery with a rectal probe; it was maintained at 37 °C using a heating pad. Rats were placed in a heated cage to maintain their body temperature while recovering from anesthesia. Our selected treatment time of 5 h and dosage (PFTα or PFTμ 2 mg/kg intravenous injection) were based on our prior study indicating that the secondary phase of neuronal injury following TBI can be attenuated if appropriate treatment is initiated within this time window (Yang et al., 2015). The body temperature was monitored in TBI animals following vehicle, PFT-α and PFT-μ group treatment over a period of 3 h, as well as in our previous study (Yang et al., 2015).
2.3. Behavioral evaluation of neurological outcome
Beam walking test, modified neurological severity scores (mNSS), swing test and tactile adhesive-removal test were performed to compare functional outcomes in sham or TBI rats treated with vehicle or PFT-α or PFT-μ (2 mg/kg i.v.) (Hsueh et al., 2019). All behavioral tests were performed by an observer blinded to the experimental groups before CCI and at 24 h after CCI, as described in our previous publications. Overall motor activity was measured in automated chambers, as in our prior studies (Yang et al., 2015). To compare neurological deficit severity in TBI rats, a mNSS (a composite of motor, sensory, reflex and balance tests) was performed (Chen et al., 2008). One point was scored for inability to perform the test or for the lack of a tested reflex; thus, the higher the score, the more severe the injury. Neurological function was graded on a scale of 0–18 (normal score: 0; maximal deficit score: 18) (Alder et al., 2011). The tactile removal test was used to evaluate somatosensory function. Two small adhesive-backed stickers are used as bilateral tactile stimuli that are placed on the distal–radial region on the wrist of each forelimb (Chen et al., 2008). Rats were pre-trained daily for 3 days before CCI. The time required (not to exceed 180 s) for the rats to remove the sticker from the forelimb was recorded.
2.4. Fluoro Jade C (FJC) staining
FJC, a derivative of polyanionic fluorescein, selectively binds to degenerating neurons. We used an FJC ready-to-dilute staining kit (TR-100-FJ, Biosensis) to identify degenerating neuronal cells according to the manufacturer’s protocol with some modifications. After behavioral evaluation, rats were deeply anesthetized by Zoletil (20–40 mg/kg, i.p.) and perfused through the left ventricle with cold saline, followed by 4% paraformaldehyde fixative. The brains were immediately removed, and postfixed for 24 h in the same fixative at 4 °C for paraffin sections. The 4 μm coronal sections were prepared for staining (cresyl violet, immunohistochemical, immunofluorescent or FJC staining). Other sets of animals were guillotine decapitated after deep anesthesia for brain dissections for biochemical measurement. Brain sections from the different treatment groups were deparaffinized, rehydrated, and incubated in distilled water for 2 min. These sections were then incubated in a solution of potassium permanganate (1:15) for 10 min, rinsed in distilled water for 2 min, and then incubated in the FJC solution (1:25) for 30 min. After incubation, sections were washed and mounted on coverslips with Vecta-shield mounting medium (Vector Laboratories, Burlingame, CA, USA). All sections were observed and photographed using a fluorescent inverted microscope (IX70, Olympus, Japan). The numbers of FJC-positive cells were thereafter counted in three randomly selected fields per slide by means of SPOT image analysis software (Diagnostic Instruments). Finally, the numbers of FJC-positive cells observed on the slides from different treatment groups were counted and used to generate a mean number per treatment group.
2.5. Quantification of contusion volume
Contusion volume measurement was performed as previously described (Chen et al., 2007). Specifically, cresyl violet was used to measure contusion volume, by staining the sections followed by digitization and analysis using a 1× objective and a computer image analysis system (ImageJ 1.8 software, National Institutes of Health, Bethesda, MD, USA). Contusion area was calculated from all images of cresyl violet-stained sections that contained contused brain. Hemisphere tissue loss was expressed as a percent calculated by (contralateral minus ipsilateral hemispheric volumes) / (contralateral hemispheric volume) × 100%, as previously reported (Zhang et al., 1998).
2.6. Measurement of mRNA levels
Levels of mRNA encoding for p53, HO-1, SOD1, SOD2, IL-1β, IL-6 and PINK1 genes were measured by reverse transcription followed by real time-PCR from TBI without or with PFT-α / PFT-μ treatment.
Total RNA was extracted from brain tissues using the TRIzol® reagent (Invitrogen Life Technologies, Paisley, Scotland). Total RNA (3 μg) was reverse-transcribed into complimentary (c)DNA using the ReverTra Ace-α First-strand cDNA Synthesis Kit (Toyobo Life Science, Japan). The resulting cDNA was incubated with a Rotor-Gene SYBR Green kit (Applied Biosystems, Foster City, CA, USA) and appropriate primers.
HO-1, 5′- GAGATAGAGCGAAACAAGCAGAAC-3′ (forward) and.
5′-CATACCAGAAGGCCAATGTCCTG-3′ (reverse);
p53, 5′- CCATGGCCATCTACAAGAAGT-3′ (forward) and.
5′-TGTCGTCCAGATACTCAGCATA-3′ (reverse);
PINK1, 5′- CTGTCAGGAGATCCAGGCAATT −3′ (forward) and.
5′- GCATGGTGGCTTCATACACAGC-3′ (reverse);
IL-6, 5′-TTCTTGGGACTGATGTTGTTGAC-3′ (forward) and.
5′-AATTAAGCCTCCGACTTGTGAAG-3′ (reverse);
IL-1β, 5′-GTTTGAGTCTGCACAGTTCCC-3′ (forward) and.
5′-CAACTATGTCCCGACCATTGC-3′ (reverse);
SOD1, 5′-CGGCTTCTGTCGTCTCCTTGC-3′ (forward) and.
5′-GTTCACCGCTTGCCTTCTGC-3′ (reverse);
SOD2, 5′-ACGCGACCTACGTGAACAATCT-3′ (forward) and.
5′-CAGTGCAGGCTGAAGAGCAA-3′ (reverse);
β-actin, 5′-GACCCAGATCATGTTTGAGACCTTC-3′ (forward) and.
5′-GGTGACCGTAACACTACCTGAG-3′ (reverse). The comparative threshold cycle (Ct) value of the β-actin gene was used as reference. Relative transcript expression of mRNA levels was calculated by the Ct method.
2.7. Immunofluorescence: double immunofluorescence staining with antibodies against NeuN, HO-1, Annexin V, p62, 4-HNE, and LC3
Coronal sections (10 μm) were obtained from the anterior area of the left hemisphere. Sections were dried, rehydrated in phosphate-buffered saline (PBS), and rinsed in PBS. Sections were blocked for 60 min in 5% bovine serum albumin (BSA, PBS containing 5% BSA and 0.2% Triton X-100; Sigma, St. Louis, MO, USA) and incubated with the appropriate primary antibodies. These were either mouse monoclonal anti-NeuN antibody (Millipore; 1:500, NeuN is a neuronal marker)/rabbit polyclonal anti-HO-1 antibody (Enzo Life Sciences; 1:200)/rabbit polyclonal anti- annexin V (Abcam; 1:500), / rabbit polyclonal anti-4-HNE (Abcam; 1:250), / rabbit polyclonal anti-p62 (Abgent; 1:200), / mouse monoclonal anti-LC3 (Genetex; 1:200) at 4 °C overnight. Thereafter, incubation with a secondary antibody (Alexa Fluor® 488 goat anti-rabbit/anti-mouse immunoglobulin G (IgG) or Alexa Fluor® 594 goat anti-rabbit/anti-mouse IgG; Jackson ImmunoResearch, West Grove, PA, USA; 1:250) was undertaken for 1 h at room temperature. Sections were mounted with mounting medium H-1000 (Vector Laboratories). Immunofluorescence images were viewed using an inverted Olympus IX 70 microscope equipped with a cooled CCD camera and SPOT advanced software (Diagnostic Instruments, Sterling Heights, MI, USA). Images (in three randomly selected fields within the calibrated area which is the same as indicated by the black square box in Fig. 3C) were captured as digital micrographs and counted. Background control sections were evaluated in which primary antibody was omitted. Notably, sections from the four animal groups were incubated with antibodies in the same chamber or well.
Fig. 3.
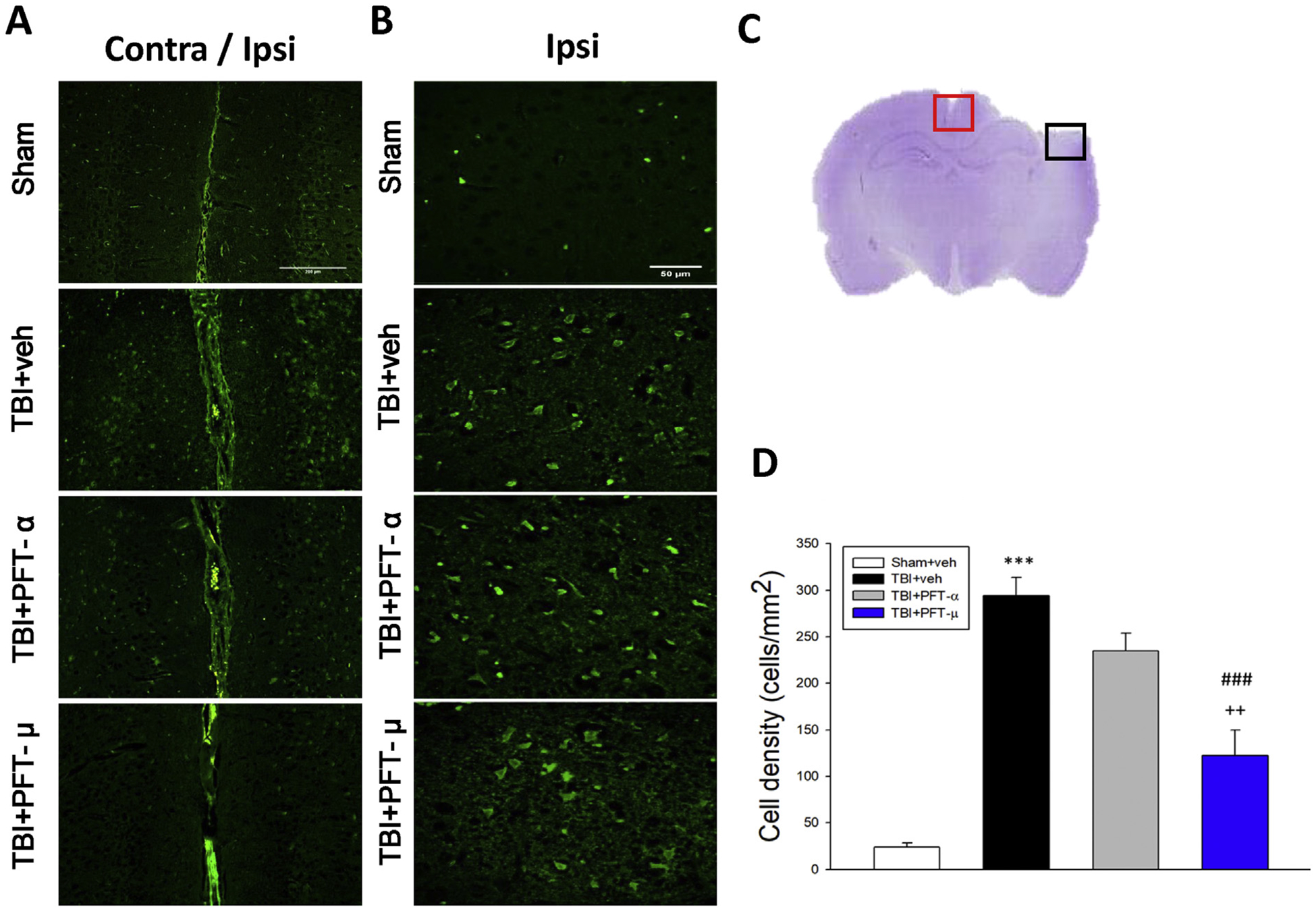
Post-injury administration of PFT-μ at 5 h after TBI significantly decreased FJC-positive cells at 24 h. FJC-stained (A) contralateral and ipsilateral (Bar = 200 μm) (B) ipsilateral cortex (Bar = 50 μm) regions of interest in sham, TBI + veh, TBI + PFT-α or TBI + PFT-μ group. (C) A representative HE-stained coronal section showing the area as indicated by the red (for both contralateral and ipsilateral cortex) and black (only ipsilateral cortex) square box to compare the fluorescent signals across the 4 groups of rats. (D) There was a significant decrease in the number of FJC-positive cells in the TBI + PFT-μ group. Data are expressed as means ± SEM. The total number of FJC-positive cells was expressed as the mean number per field of view (n = 5 per group). ***P < .001 versus sham group; ###P < .001 versus TBI + vehicle (veh) group; ++P < .01 versus TBI + PFT- α group.
2.8. Statistics
Data are presented as mean ± standard error of the mean (SEM). Between-group comparisons were evaluated by one-way analysis of ANOVA with a post hoc test (Bonferroni), when required, to determine individual group differences. Differences between means were calculated at the probability level of P ≤ .05, .01, and .001. All analyses and generation of bar graphs were undertaken using Sigma Plot and Stat version 2.0.
3. Results
3.1. Post-injury administration of PFT-α and PFT-μ at 5 h after TBI significantly reduced contusion volume and improved functional outcomes
No temperature changes were observed after PFT-α or PFT-μ administration; additionally, the physiological parameters of blood pressure, heart rate and oxygen saturation were monitored and there were no changes for up to 1 h. After functional evaluation, animals were anesthetized, and brains were removed and sectioned to measure brain contusion volume by cresyl violet staining at 24 h after CCI. As illustrated in Fig. 1, CCI resulted in a loss of cortical tissue in the ipsilateral parietal cortex, as revealed by reductions in cresyl violet staining intensity. By comparison, the structural integrity of the contralateral cortex remained normal (Fig. 1B). The volume of contusion, quantified from cresyl violet-stained sections, in vehicle-treated CCI rats was significantly greater than the equivalent volume in sham animals. PFT-α or PFT-μ post-injury treatment significantly reduced contusion volume compared to the TBI ± veh group at 24 h after CCI when administered at 5 h (Fig. 1C; P ≤ .01 and P ≤ .001, respectively) but not at 7 h. Lesion volumes in groups treated with PFT-α or PFT-μ at 5 h did not return to sham levels. Moreover, there was no significant difference in contusion volume between the PFT-α and PFT-μ treatments.
To evaluate whether or not changes in contusion volume across the CCI groups were of physiological relevance, behavioral studies were performed to quantitatively challenge a broad range of largely motor functions at 24 h following injury (Fig. 2). Specifically, motor coordination impairment, evaluated by the beam walking test, was evident in CCI vehicle-treated animals (Fig. 2A). Notably, 5 h post-injury administration of PFT-μ but not PFT-α significantly ameliorated (P < .01) this beam walking impairment. This benefit was lost when PFT-μ treatment was delayed to 7 h post injury. Neurological function was measured by composite mNSS scores across sham and CCI groups, which revealed an overall decline in functional outcome induced by CCI in the vehicle-treated group (signified by an elevation in the mNSS score, P < .01). Post-injury treatment with PFT-α and, in particular, PFT-μ given at 5 h (P < .001) and to a lesser extent at 7 h (P < .01 and P < .001, respectively) improved functional deficits after CCI (Fig. 2B). Functional evaluation assessed by the elevated body swing test (EBST) in sham and CCI rats indicated impairment, as measured by motor asymmetry, in the vehicle-treated CCI group (Fig. 2C). Both PFT-α and PFT-μ treated rats at 5 h demonstrated improvements in functional deficits after CCI (both P < .05, Fig. 2C), as revealed by a reduction in contralateral swings. This benefit was lost on delaying treatment to 7 h. Finally, functional evaluation appraised by tactile adhesive-removal in sham and CCI rats indicated a deficit caused by CCI in the vehicle-treated group (P < .001; Fig. 2D). Post-injury treatment with PFT-α or PFT-μ given at 5 h equally improved functional deficits after CCI (P < .01), but delaying PFT treatment until 7 h post injury resulted in loss of its effectiveness.
Fig. 2.
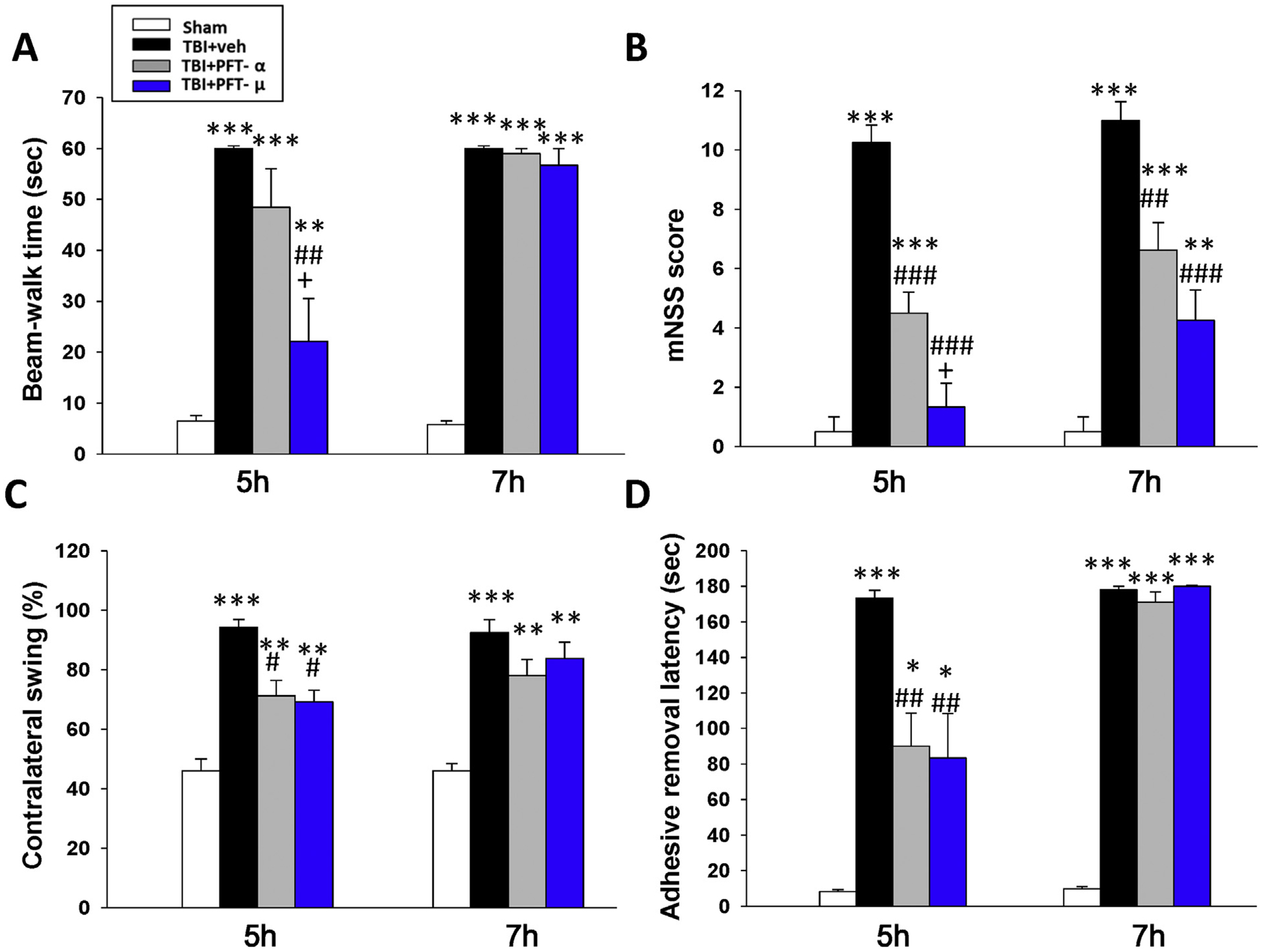
Post-injury administration of PFT-α or PFT-μ given at 5 h improved functional outcomes, as revealed by multiple behavioral evaluations undertaken at 24 h post CCI or sham challenge. (A) Motor coordination measured by beam walking test. (B) Neurological function measured by mNSS. (C) Motor asymmetry measured by elevated body swing test (EBST). (D) Sensory-motor function measured by tactile adhesive removal test. Data represent the means ± SEM. *P < .05, **P < .01, ***P < .001 versus sham group; #P < .05, ##P < .01, ###P < .001 versus TBI + vehicle (veh) group; +P < .05 versus TBI + PFT-α group. (n = 8 in TBI + veh and TBI + PFT-α group, n = 5 in sham and TBI + PFT-μ group). Note that when PFT treatment was administered at 7 h post injury, it proved much less effective.
3.2. Post-injury administration of PFT-μ given at 5 h, but not PFT-α, decreased degenerating neuronal cells induced by TBI at 24 h
FJC-stained contralateral and ipsilateral cortex regions (Fig. 3A) and FJC-positive cells possessing a neuronal morphology (Fig. 3B) were evident 24 h after CCI in the cortical contusion margin (Fig. 3C), but not within the contralateral hemisphere. As illustrated in Fig. 3D following quantification of FJC-positive cells, CCI induced a dramatic rise in degenerating cell counts in the vehicle-treated group (P < .001). Notably, PFT-μ significantly reduced (~58%) the number of FJC-positive cells compared with vehicle (P < .001) as well as PFT-α treatment (P < .01). In contrast, PFT-α demonstrated a trend to lower FJC-positive cells induced by TBI that did not reach statistical significance.
3.3. Post-injury administration of PFT-μ given at 5 h, but not PFT-α, decreased annexin V positive neurons in the cortical contusion region at 24 h
In the light of our results with FJC, to gain a greater insight into apoptosis, we evaluated annexin V staining as it is commonly used to detect apoptotic cells by its ability to bind to phosphatidylserine, a marker of apoptosis when present on the outer leaflet of a cell’s plasma membrane. Co-localization with annexin V and the widely used neuronal marker, NeuN, allowed the quantification of neuronal cells undergoing apoptosis (Fig. 4A), which was dramatically elevated in TBI vehicle-treated animals (P ≤ .001). In accordance with the FJC data, PFT-μ significantly reduced annexin V-positive neuron number (~68%) compared with vehicle (P < .001). In contrast, PFT-α demonstrated a modest trend to lower annexin V-positive neurons induced by TBI that did not reach statistical significance.
Fig. 4.
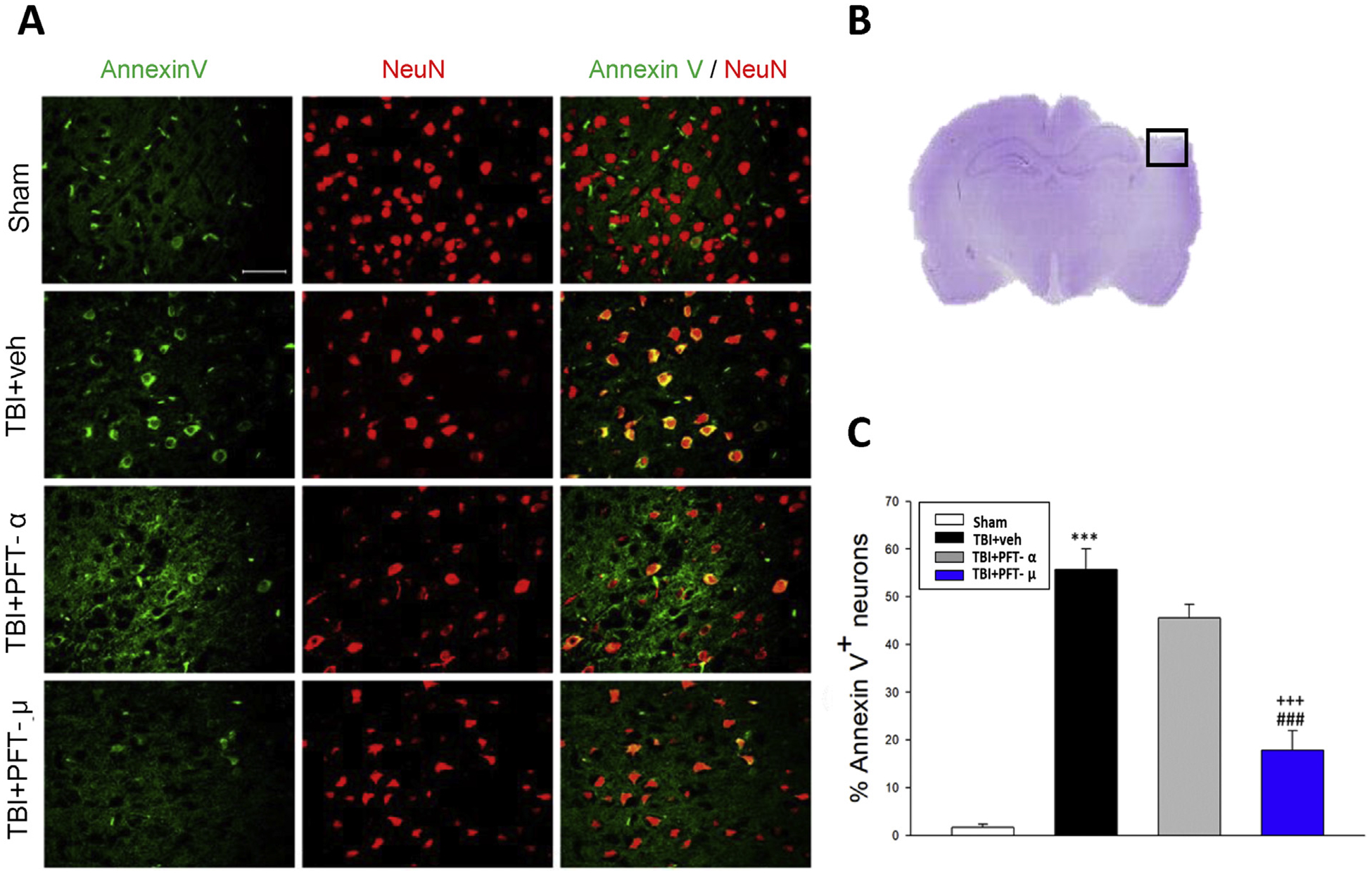
Post-injury administration of PFT-μ at 5 h after TBI decreased annexin V positive neurons in the cortical contusion region at 24 h. (A) Co-immunohistochemistry of annexin V and NeuN in the cortical region. Annexin V (a marker of apoptosis) immunoreactivity is shown in green, and NeuN (a cell marker for neurons) is shown in red. Yellow labelling indicates colocalization. (B) A representative HE-stained coronal section showing the area as indicated by the black (ipsilateral cortex) square box to compare the fluorescent signals across the 4 groups of rats. (C) There was a significant decrease in the number of Annexin V positive neurons in the TBI + PFT-μ group. Data are expressed as means ± SEM (n = 5 per group). ***P < .001, compared with the sham group; ###P < .001, versus the TBI + veh group; +++P < .001 versus the TBI + PFT-α group. Scale bar = 50 um.
3.4. Post-injury PFT-μ treatment induces HO-1 expression and mitigates lipid peroxidation in the cortical contusion region of rats with TBI at 24 h
Oxidative stress is a primary mediator that drives neuronal damage and contributes to the pathogenesis of TBI. Endogenous neuroprotective mechanisms to counter oxidative stress include heme oxygenase-1 (HO-1, an inducible 32-kDa heat shock protein that is rapidly upregulated by numerous pro-oxidant and inflammatory signals (Neis et al., 2018). As illustrated in Fig. 5, levels of HO-1 mRNA expression and neurons expressing HO-1 protein were significantly elevated in the TBI + veh-treated group (8.8 ± 1.3-fold and 13.7 ± 3.5% of neurons expressing HO-1, respectively) compared to the sham group (1.00 ± 0.1-fold and 3.2 ± 1.2% of neurons expressing HO-1, respectively). These effects were significantly enhanced in the TBI + PFT-α-treated group (16.2 ± 4.0-fold, P < .01; and 23.4 ± 1.6%, P < .05, respectively) and, particularly in the TBI + PFT-μ -treated group (20.1 ± 3.4-fold, P < .001; and 44.5 ± 8.0%, P < .001, respectively) (Fig. 5B and C). These results suggest that TBI-induced HO-1 expression is, in part, dependent on p53.
Fig. 5.
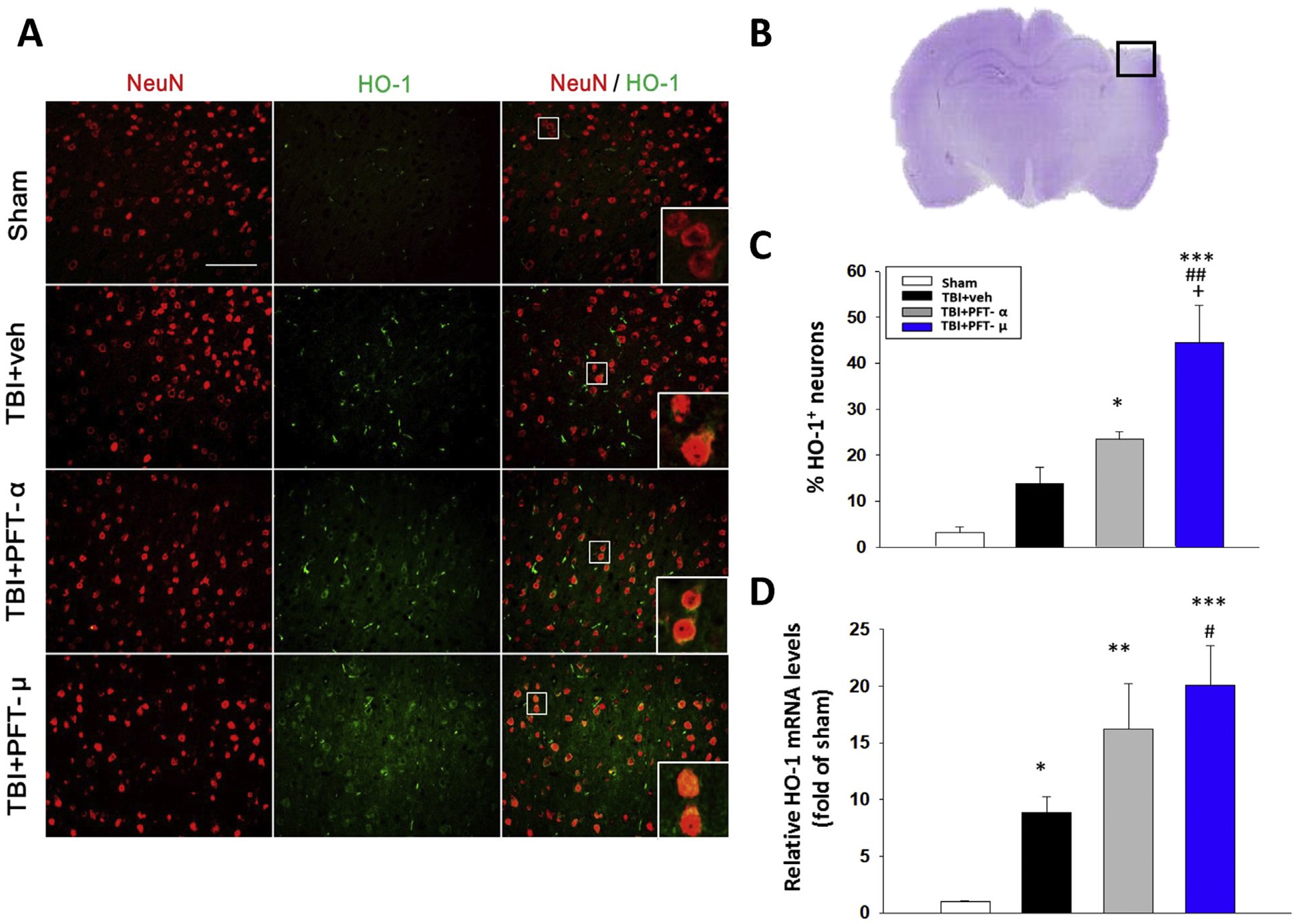
Post-injury administration of PFT-α and, in particular, PFT-μ at 5 h after TBI elevated HO-1 positive neurons and mRNA expression in the cortical contusion region at 24 h. (A) Co-immunohistochemistry of HO-1 and NeuN in cortical brain tissue. (B) A representative HE-stained coronal section showing the area as indicated by the black (ipsilateral cortex) square box to compare the fluorescent signals across the 4 groups of rats. (C) There was a significant increase of HO-1 positive neurons in the TBI + PFT-μ group beyond that induced in the TBI + vehicle group. (D) The mRNA levels of HO-1 were analyzed by RT-PCR. Data are expressed as means ± SEM (n = 5 per group). *P < .05, **P < .01, ***P < .001, compared with the sham group; #P < .05, ##P < .01, compared with the TBI + veh group; +P < .05, compared with the TBI + PFT-α group. Scale bar = 100 μm.
As HO-1 mediates cellular responses to oxidative stress and autophagy, we additionally examined the effect of PFT-α and PFT-μ on oxidative stress induced by TBI, specifically focusing on the expression of the lipid peroxidation marker 4-hydroxynonenal (4-HNE) (Fig. 6). Whereas TBI induced a significant 4.7-fold rise (P < .001) in 4-HNE-positive neurons in the cortical contusion area, PFT-μ, but not PFT-α, significantly mitigated this effect (P < .01).
Fig. 6.
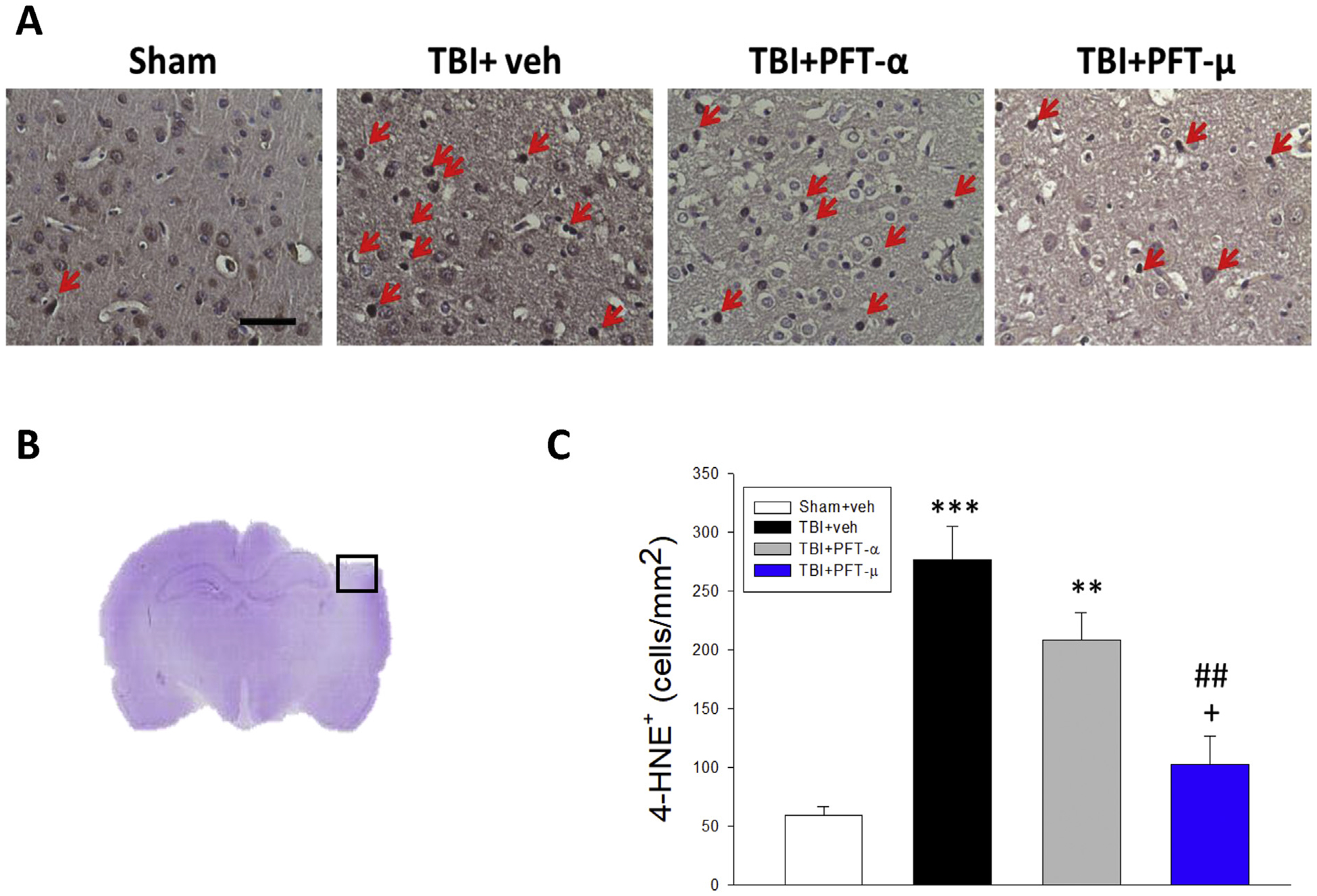
Post-injury administration of PFT-μ, but not PFT-α, at 5 h after TBI decreased 4-HNE positive cells in the cortical contusion region at 24 h. (A) Representative photomicrographs showing the results of immunohistochemical staining with anti-4HNE in 4 groups. Red arrows indicate 4-HNE-positive cells (brown colour). (B) A representative HE-stained coronal section showing the area as indicated by the black (ipsilateral cortex) square box to compare the fluorescent signals across the 4 groups of rats. (C) There was a significant decrease in the number of 4-HNE-positive cells in the TBI + PFT-μ group. The total number of 4-HNE-positive cells was expressed as the mean number of cells/mm2. Mean ± SEM. **P < .01, ***P < .001, compared with the sham group. ##P < .01, compared with the TBI + veh group. +P < .05, compared with the TBI + PFT-α group. (Scale bar = 50 μm; n = 5 per group).
3.5. Post-injury PFT-μ treatment attenuates TBI-induced elevated expression levels of autophagic markers LC3 and p62 in the cortical contusion region of TBI rats at 24 h
In the light of prior research demonstrating that the autophagy pathway is involved in pathophysiologic responses induced by TBI, and inhibition of this pathway attenuates neuronal damage and functional outcome deficits (Luo et al., 2011), we evaluated the actions of PFT-α and PFT-μ on TBI-induced autophagy by focusing on two key autophagic markers. Fig. 7 illustrates the immunostaining of the autophagic proteins, microtubule-associated protein 1 light chain 3 (LC3) and p62 (the sequestosome 1 (SQSTM1)/A170 protein), which were elevated in response to TBI. PFT-μ substantially mitigated these TBI-induced changes (inducing 78% inhibition, P < .01), more so than did PFT-α (inducing 28% inhibition, P < .05), and indicating the involvement of p53 transcription-independent signaling in TBI-induced autophagy in rats.
Fig. 7.
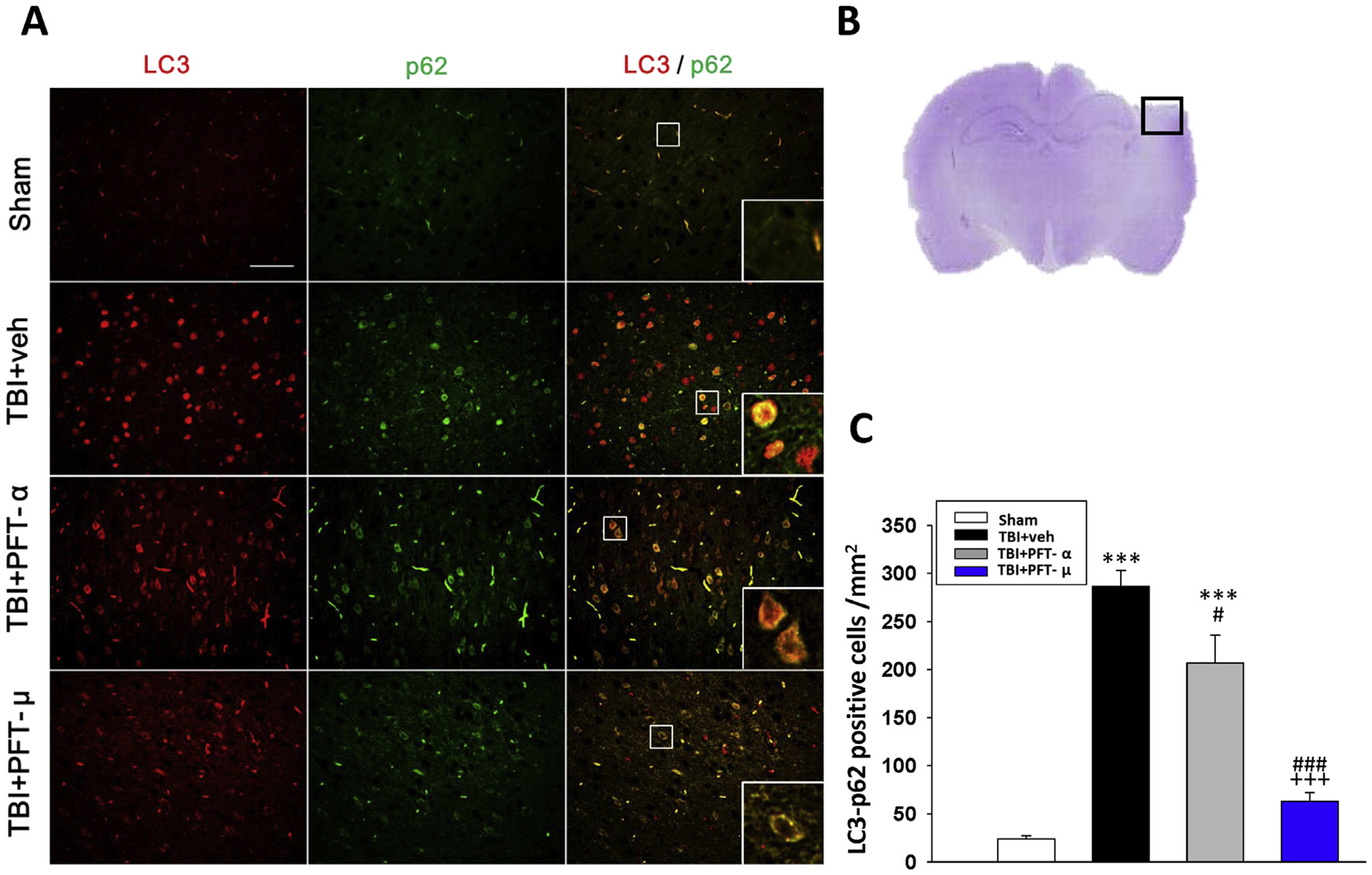
Post-injury administration of PFT-μ at 5 h after TBI decreases LC3/p62 positive cells in the cortical contusion region at 24 h. (A) Co-immunohistochemistry of LC3 and p62 in cortical brain tissue. LC3: green, p62: red, and yellow labelling indicating colocalization (Bar = 100 μm, 0.259 mm2). Inserts: Magnified colocalization of LC3 (red) and p62 (green) cells at a greater magnification. (B) A representative HE-stained coronal section showing the area as indicated by the black (ipsilateral cortex) square box to compare the fluorescent signals across the 4 groups of rats. (C) A significant decrease in LC3/p62 positive cells was evident in the TBI + PFT-μ group. The total number of LC3/p62-positive cells was expressed as the mean number per field of view in cortex. Data represent mean ± S.E.M. (n = 5 per group). **P < .01, ***P < .001 compared to the sham group; #P < .05, ### P < .001 compared to the TBI + veh group; +++ P < .001 compared to the TBI + PFT-α group.
3.6. Post-injury PFT-μ treatment significantly increased PINK-1, SOD2 and decreased IL-1β, IL-6 mRNA expression at 24 h
To further define mechanisms underpinning how PFT-μ and PFT-α mitigate TBI-induced neuronal cell loss and behavioral impairments, RNA expression levels of key proteins involved in apoptosis (p53), mitophagy (PINK1), neuroinflammation (IL-1β and IL-6) and oxidative stress (SOD1 and SOD2) were quantified (Fig. 8). Notably, TBI induced an increase in p53 mRNA expression (1.4 ± 0.1-fold., P < .05) versus the sham group. As expected, and confirming its transcription-dependent p53 action (Gudkov and Komarova, 2010), PFT-α inhibited this elevation. In contrast, PFT-μ had no effect on TBI-elevated p53 mRNA expression, thereby, substantiating its transcription-independent mitochondrial action (Fig. 8A). In response to TBI, mRNA levels of PINK1, a molecular sensor of damaged mitochondria, declined by 71% (P < .01), which was equally attenuated by PFT-μ and -α (P < .05; Fig. 8B). In contrast, mRNA levels of the proinflammatory markers IL-1β and IL-6 were dramatically elevated by 13.6- and 4.8-fold, respectively (P < .001). These increments also were equally mitigated by PFT-μ and -α (P < .001 and .01; Fig. 8C). Finally, in response to TBI, the levels of the critical endogenous anti-oxidant proteins cytoplasmic SOD1 and mitochondrial SOD2 were reduced by 66% and 50%, respectively (P < .001). As illustrated in Fig. 8D, neither PFT-μ nor PFT-α eliminated this TBI-induced SOD1 decline; however, PFT-μ attenuated the SOD2 decrease.
Fig. 8.
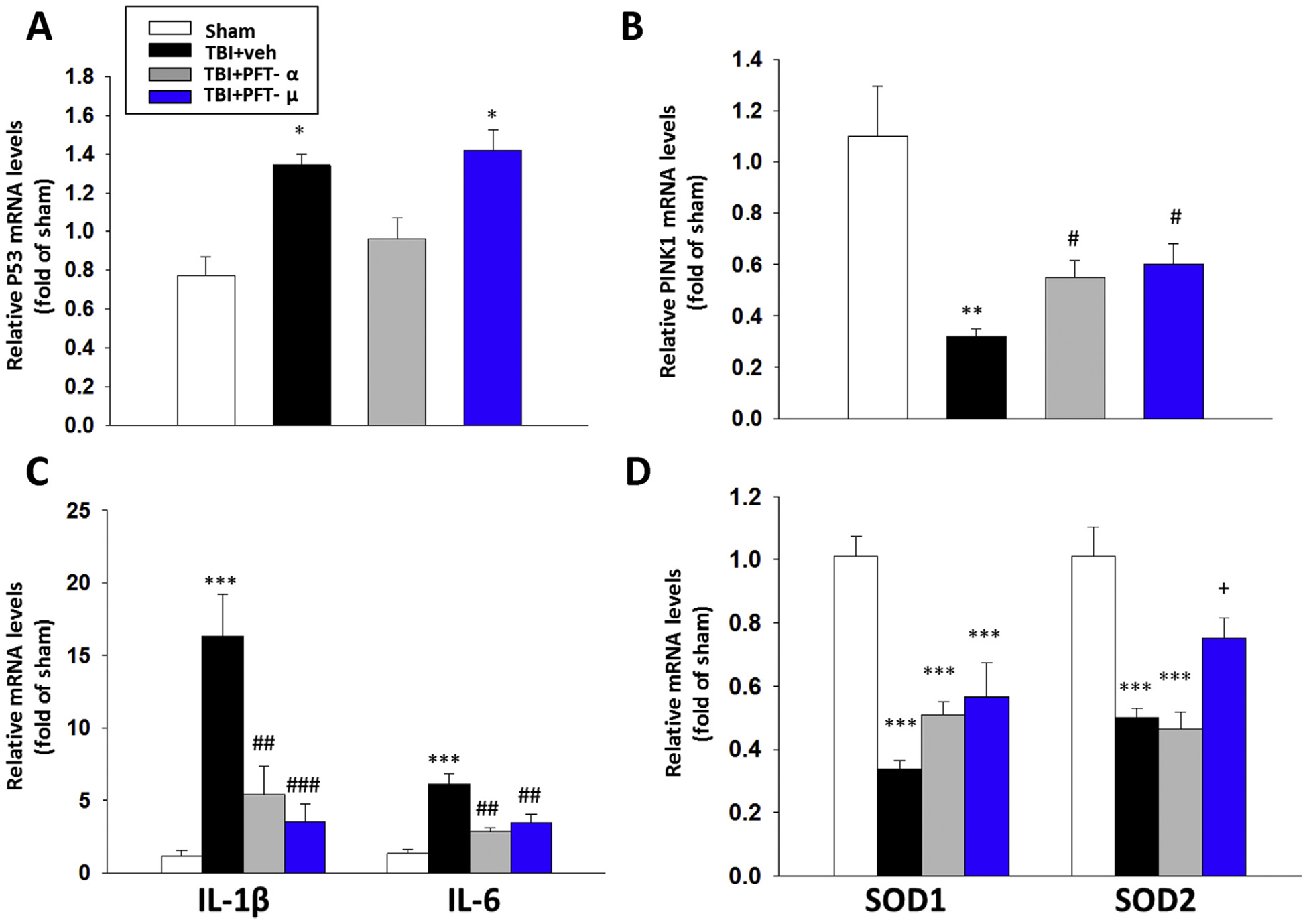
Treatment with PFT-μ at 5 h after TBI significantly mitigated TBI-induced reductions in PINK-1, and SOD2, and inhibited TBI-mediated rises in IL-1β, IL-6 mRNA expression in the cortical contusion region at 24 h. The mRNA levels of (A) p53, (B) PINK1, (C) IL-1β and IL-6, and (D) SOD1 and SOD2 across the four animal groups were evaluated by RT-PCR. PFT-μ significantly increased PINK1 and SOD2 mRNA expression, and decreased IL1β and IL-6 mRNA compared with TBI + vehicle-treated rats. Notably, PFT-μ did not change the level of p53 gene expression in accord with its transcriptional independent mechanism of action. Data are expressed as means ± SEM (n = 5 per group). * P < .05, **P < .01, ***P < .001 vs. sham; #P < .05, ##P < .01, ###P < .001 vs. TBI + veh group. +P < .05 vs. TBI + PFT-α group.
4. Discussion
The present study demonstrates a primary role for p53 in the delayed neuronal cell death that occurs following moderate TBI, as induced by CCI in rats. Multiple previous studies have demonstrated that p53 protein levels are rapidly elevated within the injured brain region as little as 6 to 12h after an experimental brain trauma and remain increased for up to 24h post injury or even later (Plesnila et al., 2007; Napieralski et al., 1999; Wan et al., 2013). The accumulation of p53 correlates largely with later neuronal cell loss in not only TBI (Plesnila et al., 2007; Yang et al., 2015; Wan et al., 2013; Rachmany et al., 2013; Huang et al., 2018) but also other neurological disorders, such as those initiated by ischemia and neurotoxic agents, and the use of both p53 inhibitors and knockout mice can significantly attenuate this process (Filichia et al., 2015; Culmsee et al., 2001; Leker et al., 2004; Nijboer et al., 2011; Lu et al., 2017; Chen et al., 2019). To gain greater insight into how p53 induces neuronal cell loss following TBI, we evaluated the small molecule p53 inactivators, PFT-α and PFT-μ, to differentiate transcriptional and direct mitochondrial p53 effects, respectively, in a ‘side by side” comparison in the same animal TBI model. We have previously shown that intravenous PFT-α administration did not affect body temperature or other physiological parameters (Yang et al., 2015). Similar results were found here with PFT-μ treatment. Notably, a neuroprotective effect, measured at 24h after injury, was achieved when a single dose of either PFT analogue was administered up to 5h after TBI. Although our study was acute, this finding for PFT-α is in line with that of Plesnila and colleagues (Plesnila et al., 2007), who additionally demonstrated that post-injury neuroprotection of brain tissue was still evident 7 days after trauma, as evaluated by improved motor and cognitive outcomes. Specifically, in the current study we demonstrated that 5 h post-injury administration of PFT-α and PFT-μ significantly reduced contusion volume and improved functional outcomes at 24 h, with the therapeutic window largely closing by 7 h (Figs. 1 and 2). While microscopy consistently showed better histological improvement following PFT-μ treatment, better improvement in functional outcome was found only in the TBI + PFT-μ group as compared to the TBI + PFT-α in two (beam-walk and mNSS) of the four behavioral tests at 5 h. We evaluated these 4 different behavioral tests as each test reflects different neurological outcomes involving different neural anatomical pathways. It is not unusual that the results from histology and functional outcome measures may not be perfectly correlated. However, the overall finding of our study is that greater improvement was found in the TBI + PFT-μ group than in the TBI + PFT-α group at 5 h but not 7 h after TBI. Post-injury administration of PFT-μ at 5 h, in particular, significantly decreased degenerating neuronal cells, measured by Fluoro-Jade staining, and by decreased annexin V positive neurons, a marker of apoptosis (Figs. 2 and 3), with PFT-α showing a more modest trend. In evaluating biochemical underpinnings, the mRNA expression level of HO-1 and number of HO-1 positive staining neurons within the cortical contusion area of PFT-μ treated animals were dramatically elevated. HO-1, a key enzyme that protects against oxidative injury, is also anti-apoptotic and anti-inflammatory and is induced by pro-oxidant and inflammatory stimuli. In contrast, 4-HNE positive cells, a marker for lipid peroxidation triggered by oxidative stress, were substantially reduced by PFT-μ more strikingly than by PFT-α. We view elevated HO-1 expression as a homeostatic compensatory response to the oxidative stress induced by TBI, and that this response is augmented by PFT-α and, in particular, by PFT-μ. We speculate that the cytosolic (mitochondrial) inhibition of p53 activity occurs more rapidly than transcriptional inhibition and PFT-μ does not target p53 as a transcriptional factor. In addition, the PFT moieties reduce cell death and thus there are more viable cellular elements in the lesion area (as shown in Fig. 4 A). This would result in elevated levels of HO-1 from the increased cell numbers. Moreover, 5 h post-injury administration of PFT-μ significantly decreased LC3 and p62 positive cells, markers for autophagy, increased mRNA expression of PINK-1, an initiator of mitochondrial mitophagy, and antioxidant mitochondrial-localized SOD2, and also decreased IL-1β and IL-6 mRNA expression, further supporting its neuroprotective activity. We believe there are critical time-dependent changes in pro-apoptotic cascades induced by TBI involving p53 mechanisms during this 5–7 h period, as shown in our prior studies (Yang et al., 2015; Wang et al., 2016; Huang et al., 2018). Once these changes are established, blocking p53 activity would have little efficacy.
The protein p53 is recognized as the ‘guardian of the genome’, as its loss of action consequent to mutation can lead to an increased occurrence of tumor development consequent to a loss of chromosome integrity (Gudkov and Komarova, 2007). Basal cellular levels of p53 are regulated by the binding of p53 to the E3 ubiquitin ligase Mdm2 that promotes breakdown of the protein by targeting both itself and p53 for degradation by the proteasome (Gudkov and Komarova, 2007). As a response to acute stress, p53 becomes post-translationally modified through phosphorylation and acetylation, binding between Mdm2 and p53 declines, and levels of p53 are stabilized. Accrual of p53 occurs within the nucleus where p53 interacts with specific DNA sequences to modify the transcription of responsive genes (Jiang et al., 2010). Multiple p53 responsive genes have been implicated in cell cycle management, the induction of apoptosis, as well as regulation of DNA repair enzymes to ultimately govern the outcome of a cell’s response to a particular stress (Hafner et al., 2019). However, the specific pathway(s) activated largely depends on the target cell type as well as the stress and type of DNA damage induced, and how a specific cell integrates these signals and selects between the opposing outcomes of cell survival versus death remains largely unknown (Hafner et al., 2019).
The cell death-inducing roles of p53 are classically considered tumor suppressive, consequent to p53’s ability to provoke apoptosis and hereby, to eliminate damaged cells from multiplying and potentially adding to the gene pool. While valuable to curb tumor development, the same molecular machinery is present in end-differentiated neuronal cells that are incapable of dividing and adding to the gene pool and in which stress-induced p53-dependent cell death is often disadvantageous. Apoptosis is instigated by the permeabilization of the outer mitochondrial membrane, mediated by members of BCL-2 family. A critical function of p53 in this regard, is as a nuclear transcription factor that can upregulate the transcription of key pro-apoptotic BCL-2 family members, comprising BAX, PUMA, NOXA and BID, and can lower the transcription of anti-apoptotic members that include BCL-2 and BCL-XL (Hafner et al., 2019; Danial, 2007; Kale et al., 2018). Via alternate mechanisms, however, p53 can likewise provoke apoptosis separate from its transcriptional actions. Depending on the cellular stress, cytoplasmic accumulation of p53 can occur and detrimentally directly impact mitochondria activity, providing a transcription-independent function of p53 in the induction of apoptosis. p53 has been reported to operate via direct interactions to activate BCL-2 family proteins at the mitochondria level (Hafner et al., 2019; Speidel, 2010).
Strategies that allow a short term inactivation of p53 function in normal cells have been developed to potentially mitigate the adverse effects of cancer therapies or of physiological and pathological insults (Golubovskaya and Cance, 2013), and the resulting two classes of small molecule p53 inhibitors target either the transcriptional or the mitochondrial activity of p53 (Zhu et al., 2002; Komarov et al., 1999; Strom et al., 2006). PFT-α and analogs are widely considered to prevent cell death by reversibly inhibiting p53-transcriptional activity (confirmed here in Fig. 8A), inhibiting p53-induced apoptosis, cell cycle arrest and DNA-synthesis block (Komarov et al., 1999; Bassi et al., 2002; Arango et al., 2001; Havre et al., 2002; Qin et al., 2002; Toillon et al., 2002). PFT-α and analogs have been successfully used in vitro and in vivo to protect normal cells from otherwise lethal doses of chemotherapy and radiotherapy (Gudkov and Komarova, 2007), as well as to protect neuronal cells from a host of generally fatal challenges (Filichia et al., 2015; Culmsee et al., 2001; Leker et al., 2004; Checler and Alves Da Costa, 2014; Szybińska and Leśniakx, 2019; Jazvinšćak Jembrek et al., 2018). PFT-α and analogs have hence proven to be a valuable pharmacological tool for the identification of genes under the control of p53, and mechanisms underpinning cell death (Gudkov and Komarova, 2007). In light of extensive reports of PFT-α mediated protection of cell death across cell culture and animal models, the mechanism(s) of action of PFT-α and analogs warrant further characterization, although they are widely considered to disrupt the nuclear transport of p53 (Gudkov and Komarova, 2007). PFT-α is considered to cyclize to PFT-β (originally synthesized by Zhu and colleagues (Greig et al., 2002)) for biological action, and to mediate its primary actions through inhibiting the transcriptional activity of p53, leading to the suppressed transactivation of p53 responsive genes (for example, lowering p21 and Bax, and elevating Bcl-2 levels (Gudkov and Komarova, 2007)). Support for this hypothesis derives from multiple studies both undertaken and reviewed by Gudkov and Komarova (Gudkov and Komarova, 2007) and from Charlot et al. (2006) in studies combining staurosporine-induced apoptosis and PFT-α to significantly decrease the apoptosis of cells with transcriptionally active p53, whereas no effect was found in mutant p53 or p53 deficient cells. Of note, multiple downstream targets are impacted by p53 transcriptional activation and its inhibition by PFT-α and analogs can thereby result in multiple protein changes even in the light of a relatively selective action. In addition to these p53 transcription-dependent actions, PFT-α has been reported at high concentrations to interact with the aryl hydrocarbon receptor (Hoagland, 2005) to mediate its actions on p53, and to suppress heat shock and glucocorticoid receptor signaling (Komarova et al., 2003).
Despite the numerous beneficial actions of PFT-α to protect host cells from the toxicities of radio- and chemotherapy in cancer treatment (Gudkov and Komarova, 2010; Gudkov and Komarova, 2007; Komarov et al., 1999), to mitigate neuronal loss and behavioral impairments in acute and chronic neurological disorders (Plesnila et al., 2007; Yang et al., 2015; Zhu et al., 2002; Rachmany et al., 2013; Huang et al., 2018; Culmsee et al., 2001; Leker et al., 2004; Nijboer et al., 2011; Chen et al., 2019; Checler and Alves Da Costa, 2014; Luo et al., 2009; Duan et al., 2002), as in the current study, and to provide myocardial protection following ischemia (Liu et al., 2006), no compound on this backbone has yet translated into human clinical trials (Parrales and Iwakuma, 2015). The key element in this paper is the efficacy of PFT-μ in many parameters of reducing TBI pathophysiology – at the behavioral, histochemical and biochemical levels. A critical issue with the translational impact of the PFT-α moieties is that they block both transcriptional (nuclear) and non-transcriptional p53 activity, leading to many potential side effects. As a consequence, no agents on the backbone of PFT-α have moved from the bench to the bedside (despite their original discovery almost 20 years ago). As PFT-μ would block only non-transcriptional p53 activity and is on a different chemical scaffold, it could potentially have much more translational value.
Whereas PFT-α prevents p53 mediated transcriptional activation and subsequent apoptosis (Gudkov and Komarova, 2007), PFT-μ (2-phenylethynesulfonamide), albeit shown to have effects on HSP70 at high concentrations (Leu et al., 2009), has been demonstrated to selectively inhibit the transcription-independent mitochondrial arm of the p53 cascade by decreasing p53 binding affinity to BCL-2 and BCL-XL (Strom et al., 2006). In this distinct p53 mitochondrial pathway (Mossalam et al., 2012), selective cellular stresses provoke interaction of p53 with mitochondria by binding with Bcl-2 family protein members (Mossalam et al., 2012; Geng et al., 2010) to cause mitochondrial outer membrane permeabilization, cytochrome c release into the cytosol, and ultimately, caspase-3-induced apoptosis (Vaseva and Moll, 2009). PFT-μ appears to effectively protect against a range of insults that mediate their pro-apoptotic actions via this direct and transcriptionally-independent mitochondrial pathway (as reviewed by Maj et al., 2017) (Maj et al., 2017). The evaluation of PFT-μ and PFT-α, side-by-side in the same models, support the relative differential pathway selectivity of these p53 inactivators (Nijboer et al., 2011; Maj et al., 2017; Jamil et al., 2015; Jebelli et al., 2014). However, studies such as the current one on TBI as well as others on cerebral ischemia (Nijboer et al., 2011) suggest that selective stressors induce neuronal cell death by triggering both p53 transcriptional dependent and independent cascades; each of which can be inhibited by PFT-α and PFT-μ respectively, to mitigate injury-induced neuronal loss and associated behavioral impairments. For both PFT-μ and PFT-α, their mitigation of neuronal cell dysfunction and death at 5 h and reduced efficacy at 7 h suggest that key p53-dependent pro-apoptotic biochemical cascades can be time-dependently inhibited and the processes leading to cell death stalled up to a certain point, beyond which they become irrevocable.
We consider that both PFT-α and PFT-μ primarily have central rather than peripheral actions to provide the neuroprotection evident in our TBI study, as there is a notable and early loss of blood-brain barrier (BBB) integrity following TBI (Alluri et al., 2015). This is particularly evident in CCI TBI in which maximal BBB breakdown occurs after 4 h (Başkaya et al., 1997). In addition, PFT-α and PFT-μ have lipophilicities in line with high distribution into the brain (octanol/water partition coefficient log D values, PFT-α: 1.75 (Zhu et al., 2002), PFT-μ: 0.97). From a therapeutic perspective, the selectivity of PFT-μ is a positive feature as it inactivates only p53 mitochondrial pathways without affecting other functions of p53 (including p53-mediated expression of genes unrelated to apoptosis) that may not be implicated in the pathology of TBI, but underpin the wider spectrum of pharmacological actions (both on and off target) associated with PFT-α.
5. Conclusion
Our data suggest that mechanisms underlying the neuroprotective effects of PFT-μ are manifested, in part, through amelioration of neurological functional deficits, as well as by attenuating neuroinflammation, oxidative stress, autophagy, and mitophagy following an experimental TBI in rats. The positive actions of PFT-μ or PFT-α (2 mg/kg) in mitigating TBI indicate the involvement of both mitochondrial and transcriptional pathways in TBI-induced p53 mediated apoptosis and support the potential of PFT-μ as a new TBI treatment strategy. In the light of our studies, further preclinical research appears warranted on PFT-μ in relation to longer term outcome measures following TBI, as well as dose-dependent, pharmacokinetic and toxicological studies to define doses that could be extrapolated to human equivalency to support potential clinical translation.
Acknowledgements
This research was supported in part by (i) the Intramural Research Program, National Institute on Aging, National Institutes of Health, USA, and (ii) grants from (a) the Ministry of Science and Technology (MOST104-2923-B-038-001-MY3 and MOST 108-2321-B-038-008 to JYW and YHC, MOST 108-2811-B-002-637 to LYY), Taiwan. (b) DP2-107-21121-01-N-05 to JYW, Taipei Medical University, Taipei, Taiwan, and (c) National Institutes of Health R56 AG057028 to BJH, USA.
Footnotes
Declaration of Competing Interest
The authors declare no conflicts of Interest.
References
- Alder J, Fujioka W, Lifshitz J, Crockett DP, Thakker-Varia S, 2011. Lateral fluid percussion: model of traumatic brain injury in mice. J. Vis. Exp 3063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alluri H, Wiggins-Dohlvik K, Davis ML, Huang JH, Tharakan B, 2015. October Blood-brain barrier dysfunction following traumatic brain injury. Metab. Brain Dis 30 (5), 1093–1104. 10.1007/s11011-015-9651-7. [DOI] [PubMed] [Google Scholar]
- Arango D, Corner GA, Wadler S, Catalano PJ, 2001. Augenlicht, L.H. C-myc/p53 interaction determines sensitivity of human colon carcinoma cells to 5-fluorouracil in vitro and in vivo. Cancer Res. 61, 4910–4915. [PubMed] [Google Scholar]
- Baratz R, Tweedie D, Wang JY, Rubovitch V, Luo W, Hoffer BJ, Greig NH, Pick CG, 2015. Transiently lowering tumor necrosis factor-α synthesis ameliorates neuronal cell loss and cognitive impairments induced by minimal traumatic brain injury in mice. J. Neuroinflammation 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkhoudarian G, Hovda DA, Giza CC, 2011. The molecular pathophysiology of concussive brain injury. Clin. Sports Med 30, 33–48. [DOI] [PubMed] [Google Scholar]
- Başkaya MK, Rao AM, Doğan A, Donaldson D, Dempsey RJ, 1997. April 18 The biphasic opening of the blood-brain barrier in the cortex and hippocampus after traumatic brain injury in rats. Neurosci. Lett 226 (1), 33–36. [DOI] [PubMed] [Google Scholar]
- Bassi L, Carloni M, Fonti E, Palma De La Peña N, Meschini R, Palitti F, 2002. Pifithrin-α, an inhibitor of p53, enhances the genetic instability induced by etoposide (VP16) in human lymphoblastoid cells treated in vitro. Mutat. Res. - Fundam. Mol. Mech. Mutagen 499, 163–176. [DOI] [PubMed] [Google Scholar]
- Charlot JF, Nicolier M, Prétet JL, Mougin C, 2006. Modulation of p53 transcriptional activity by PRIMA-1 and Pifithrin-α on staurosporine-induced apoptosis of wild-type and mutated p53 epithelial cells. Apoptosis 11, 813–827. [DOI] [PubMed] [Google Scholar]
- Checler F, Alves Da Costa C, 2014. P53 in neurodegenerative diseases and brain cancers. Pharmacol. Ther 142, 99–113. [DOI] [PubMed] [Google Scholar]
- Chen SF, Hung TH, Chen CC, Lin KH, Huang YN, Tsai HC, Wang JY, 2007. Lovastatin improves histological and functional outcomes and reduces inflammation after experimental traumatic brain injury. Life Sci. 81, 288–298. [DOI] [PubMed] [Google Scholar]
- Chen SF, Hsu CW, Huang WH, Wang JY, 2008. Post-injury baicalein improves histological and functional outcomes and reduces inflammatory cytokines after experimental traumatic brain injury. Br. J. Pharmacol 155, 1279–1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YH, Bae E, Chen H, Yu SJ, Harvey BK, Greig NH, Wang Y, 2019. Pifithrin-alpha reduces methamphetamine neurotoxicity in cultured dopaminergic neurons. Neurotox. Res 36 (2), 347–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culmsee C, Zhu X, Yu QS, Chan SL, Camandola S, Guo Z, Greig NH, Mattson MP, 2001. A synthetic inhibitor of p53 protects neurons against death induced by ischemic and excitotoxic insults, and amyloid β-peptide. J. Neurochem 77, 220–228. [DOI] [PubMed] [Google Scholar]
- Danial NN, 2007. BCL-2 family proteins: critical checkpoints of apoptotic cell death. Clin. Cancer Res 13, 7254–7263. [DOI] [PubMed] [Google Scholar]
- Duan W, Zhu X, Ladenheim B, Yu QS, Guo Z, Oyler J, Cutler RG, Cadet JL, Greig NH, Mattson MP, 2002. P53 inhibitors preserve dopamine neurons and motor function in experimental parkinsonism. Ann. Neurol 52, 597–606. [DOI] [PubMed] [Google Scholar]
- Filichia E, Shen H, Zhou X, Qi X, Jin K, Greig N, Hoffer B, Luo Y, 2015. Forebrain neuronal specific ablation of p53 gene provides protection in a cortical ischemic stroke model. Neuroscience 295, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng Y, Walls KC, Ghosh AP, Akhtar RS, Klocke BJ, Roth KA, 2010. Cytoplasmic p53 and activated bax regulate p53-dependent, transcription-independent neural precursor cell apoptosis. J. Histochem. Cytochem 58, 265–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golubovskaya VM, Cance WG, 2013. Targeting the p53 pathway. Surg. Oncol. Clin. N. Am 22, 747–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greig N, Mattson M, Zhu X, Holloway H, 2002. Tetrahydrobenzothiazole Analogues as Neuroprotective Agents (compound Z2–035II). International Patent Application. WO 02/004409. . [Google Scholar]
- Greig NH, Tweedie D, Rachmany L, Li Y, Rubovitch V, Schreiber S, Chiang YH, Hoffer BJ, Miller J, Lahiri DK, et al. , 2014. Incretin mimetics as pharmacologic tools to elucidate and as a new drug strategy to treat traumatic brain injury. Alzheimers Dement. 10, S62–S75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gudkov AV, Komarova EA, 2007. Dangerous habits of a security guard: the two faces of p53 as a drug target. Hum. Mol. Genet 16. [DOI] [PubMed] [Google Scholar]
- Gudkov AV, Komarova EA, 2010. Pathologies associated with the p53 response. Cold Spring Harb. Perspect. Biol 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hafner A, Bulyk ML, Jambhekar A, Lahav G, 2019. The multiple mechanisms that regulate p53 activity and cell fate. Nat. Rev. Mol. Cell Biol 20, 199–210. [DOI] [PubMed] [Google Scholar]
- Havre PA, O’Reilly S, McCormick JJ, Brash DE, 2002. Transformed and tumor-derived human cells exhibit preferential sensitivity to the thiol antioxidants, N-acetyl cysteine and penicillamine. Cancer Res. 62, 1443–1449. [PubMed] [Google Scholar]
- Hoagland MS, 2005. The p53 inhibitor Pifithrin- is a potent agonist of the aryl hydrocarbon receptor. J. Pharmacol. Exp. Ther 314, 603–610. [DOI] [PubMed] [Google Scholar]
- Hsueh S-C, Lecca D, Greig NH, Wang J-Y, Selman W, Hoffer BJ, Miller JP, Chiang Y-H, 2019. (−)-Phenserine ameliorates contusion volume, neuroinflammation, and behavioral impairments induced by traumatic brain injury in mice. Cell Transplant. 96368971985469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang YN, Yang LY, Greig NH, Wang YC, Lai CC, Wang JY, 2018. Neuroprotective effects of pifithrin-α against traumatic brain injury in the striatum through suppression of neuroinflammation, oxidative stress, autophagy, and apoptosis. Sci. Rep 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamil S, Lam I, Majd M, Tsai SH, Duronio V, 2015. Etoposide induces cell death via mitochondrial-dependent actions of p53. Cancer Cell Int. 15, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazvinšćak Jembrek M, Slade N, Hof PR, Šimić G, 2018. The interactions of p53 with tau and Aß as potential therapeutic targets for Alzheimer’s disease. Prog. Neurobiol 168, 104–127. [DOI] [PubMed] [Google Scholar]
- Jebelli J, Hooper C, Pocock JM, 2014. Microglial p53 activation is detrimental to neuronal synapses during activation-induced inflammation: implications for neurodegeneration. Neurosci. Lett 583, 92–97. [DOI] [PubMed] [Google Scholar]
- Jiang L, Sheikh MS, Huang Y, 2010. Decision Making by p53: Life versus Death. (Vol. 2). [PMC free article] [PubMed] [Google Scholar]
- Kale J, Osterlund EJ, Andrews DW, 2018. BCL-2 family proteins: changing partners in the dance towards death. Cell Death Differ. 25, 65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, Gudkov AV, 1999. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science (80-.) 285, 1733–1737. [DOI] [PubMed] [Google Scholar]
- Komarova EA, Neznanov N, Komarov PG, Chernov MV, Wang K, Gudkov AV, 2003. P53 inhibitor pifithrin A can suppress heat shock and glucocorticoid Signaling pathways. J. Biol. Chem 278, 15465–15468. [DOI] [PubMed] [Google Scholar]
- LaPlaca MC, Simon CM, Prado GR, Cullen DK, 2007. CNS injury biomechanics and experimental models. Prog. Brain Res 161, 13–26. [DOI] [PubMed] [Google Scholar]
- Leker RR, Aharonowiz M, Greig NH, Ovadia H, 2004. The role of p53-induced apoptosis in cerebral ischemia: effects of the p53 inhibitor pifithrin α. Exp. Neurol 187, 478–486. [DOI] [PubMed] [Google Scholar]
- Leu JIJ, Pimkina J, Frank A, Murphy ME, George DL, 2009. A small molecule inhibitor of inducible heat shock protein 70. Mol. Cell 36, 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu P, Xu B, Cavalieri TA, Hock CE, 2006. Pifithrin-α attenuates p53-mediated apoptosis and improves cardiac function in response to myocardial ischemia/reperfusion in aged rats. Shock 26, 608–614. [DOI] [PubMed] [Google Scholar]
- Lu T, Kim PP, Greig NH, Luo Y, 2017. Dopaminergic neuron-specific deletion of p53 gene attenuates methamphetamine neurotoxicity. Neurotox. Res 32, 218–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, Kuo CC, Shen H, Chou J, Greig NH, Hoffer BJ, Wang Y, 2009. Delayed treatment with a p53 inhibitor enhances recovery in stroke brain. Ann. Neurol 65, 520–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo CL, Li BX, Li QQ, Chen XP, Sun YX, Bao HJ, Dai DK, Shen YW, Xu HF, Ni H, et al. , 2011. Autophagy is involved in traumatic brain injury-induced cell death and contributes to functional outcome deficits in mice. Neuroscience 184, 54–63. [DOI] [PubMed] [Google Scholar]
- Maj MA, Ma J, Krukowski KN, Kavelaars A, Heijnen CJ, 2017. Inhibition of mitochondrial p53 accumulation by PFT-μ prevents cisplatin-induced peripheral neuropathy. Front. Mol. Neurosci 10, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morganti-Kossmann MC, Rancan M, Stahel PF, Kossmann T, 2002. Inflammatory response in acute traumatic brain injury: a double-edged sword. Curr. Opin. Crit. Care 8, 101–105. [DOI] [PubMed] [Google Scholar]
- Mossalam M, Matissek KJ, Okal A, Constance JE, Lim CS, 2012. Direct induction of apoptosis using an optimal mitochondrially targeted p53. Mol. Pharm 9, 1449–1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Napieralski JA, Raghupathi R, McIntosh TK, 1999. The tumor-suppressor gene, p53, is induced in injured brain regions following experimental traumatic brain injury. Mol. Brain Res 71, 78–86. [DOI] [PubMed] [Google Scholar]
- Neis VB, Rosa PB, Moretti M, Rodrigues ALS, 2018. Involvement of Heme Oxygenase-1 in neuropsychiatric and neurodegenerative diseases. Curr. Pharm. Des 24, 2283–2302. [DOI] [PubMed] [Google Scholar]
- Nijboer CH, Heijnen CJ, Van Der Kooij MA, Zijlstra J, Van Velthoven CTJ, Culmsee C, Van Bel F, Hagberg H, Kavelaars A, 2011. Targeting the p53 pathway to protect the neonatal ischemic brain. Ann. Neurol 70, 255–264. [DOI] [PubMed] [Google Scholar]
- Parrales A, Iwakuma T, 2015. Targeting oncogenic mutant p53 for Cancer therapy. Front. Oncol 5, 288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plesnila N, von Baumgarten L, Retiounskaia M, Engel D, Ardeshiri A, Zimmermann R, Hoffmann F, Landshamer S, Wagner E, Culmsee C, 2007. Delayed neuronal death after brain trauma involves p53-dependent inhibition of NF-κB transcriptional activity. Cell Death Differ. 14, 1529–1541. [DOI] [PubMed] [Google Scholar]
- Prins M, Greco T, Alexander D, Giza CC, 2013. The pathophysiology of traumatic brain injury at a glance. Dis. Model. Mech 6, 1307–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin JZ, Chaturvedi V, Denning MF, Bacon P, Panella J, Choubey D, Nickoloff BJ, 2002. Regulation of apoptosis by p53 in UV-irradiated human epidermis, psoriatic plaques and senescent keratinocytes. Oncogene 21, 2991–3002. [DOI] [PubMed] [Google Scholar]
- Rachmany L, Tweedie D, Rubovitch V, Yu QS, Li Y, Wang JY, Pick CG, Greig NH, 2013. Cognitive impairments accompanying rodent mild traumatic brain injury involve p53-dependent neuronal cell death and are ameliorated by the tetrahydrobenzothiazole PFT-α. PLoS One 8, 79837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt OI, Heyde CE, Ertel W, Stahel PF, 2005. Closed head injury - an inflammatory disease? Brain Res. Rev 48, 388–399. [DOI] [PubMed] [Google Scholar]
- Sparfel L, Van Grevenynghe J, Le Vee M, Aninat C, Fardel O, 2006. Potent inhibition of carcinogen-bioactivating cytochrome P450 1B1 by the p53 inhibitor pifithrin α. Carcinogenesis 27, 656–663. [DOI] [PubMed] [Google Scholar]
- Speidel D, 2010. Transcription-independent p53 apoptosis: an alternative route to death. Trends Cell Biol. 20, 14–24. [DOI] [PubMed] [Google Scholar]
- Strom E, Sathe S, Komarov PG, Chernova OB, Pavlovska I, Shyshynova I, Bosykh DA, Burdelya LG, Macklis RM, Skaliter R, et al. , 2006. Small-molecule inhibitor of p53 binding to mitochondria protects mice from gamma radiation. Nat. Chem. Biol 2, 474–479. [DOI] [PubMed] [Google Scholar]
- Szybińska A, Leśniakx W, 2019. P53 dysfunction in neurodegenerative diseases - the cause or effect of pathological changes? Aging Dis. 8, 506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toillon RA, Descamps S, Adriaenssens E, Ricort JM, Bernard D, Boilly B, Le Bourhis X, 2002. Normal Breast Epithelial CELLS induce Apoptosis of Breast CANCER cells via Fas Signaling. (Vol. 275). [DOI] [PubMed] [Google Scholar]
- Tomasevic G, Raghupathi R, Scherbel U, Wieloch T, McIntosh TK, 2010. Deletion of the p53 tumor suppressor gene improves neuromotor function but does not attenuate regional neuronal cell loss following experimental brain trauma in mice. J. Neurosci. Res 88, 3414–3423. [DOI] [PubMed] [Google Scholar]
- Vaseva AV, Moll UM, 2009. The mitochondrial p53 pathway. Biochim. Biophys. Acta Bioenerg 1787, 414–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan C, Jiang J, Mao H, Cao J, Wu X, Cui G, 2013. Involvement of upregulated P53-induced death domain protein (PIDD) in neuronal apoptosis after rat traumatic brain injury. J. Mol. Neurosci 51, 695–702. [DOI] [PubMed] [Google Scholar]
- Wang JY, Huang YN, Chiu CC, Tweedie D, Luo W, Pick CG, Chou SY, Luo Y, Hoffer BJ, Greig NH, et al. , 2016. Pomalidomide mitigates neuronal loss, neuroinflammation, and behavioral impairments induced by traumatic brain injury in rat. J. Neuroinflammation 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang LY, Chu YH, Tweedie D, Yu QS, Pick CG, Hoffer BJ, Greig NH, Wang JY, 2015. Post-trauma administration of the pifithrin-α oxygen analog improves histological and functional outcomes after experimental traumatic brain injury. Exp. Neurol 269, 56–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Raghupathi R, Saatman KE, Smith DH, Stutzmann JM, Wahl F, McIntosh TK, 1998. Riluzole attenuates cortical lesion size, but not hippocampal neuronal loss, following traumatic brain injury in the rat. J. Neurosci. Res 52, 342–349. [DOI] [PubMed] [Google Scholar]
- Zhu X, Yu Q. sheng, Cutler RG, Culmsee CW, Holloway HW, Lahiri DK, Mattson MP, Greig NH, 2002. Novel p53 inactivators with neuroprotective action: syntheses and pharmacological evaluation of 2-imino-2,3,4,5,6,7-hexahydrobenzothiazole and 2-imino-2,3,4,5,6,7-hexahydrobenzoxazole derivatives. J. Med. Chem 45, 5090–5097. [DOI] [PubMed] [Google Scholar]