

Abstract
Background
Perturbations in myocardial substrate utilization have been proposed to contribute to the pathogenesis of cardiac dysfunction in diabetic subjects. The failing heart in nondiabetics tends to decrease reliance on fatty acid and glucose oxidation, and increases reliance on ketone body oxidation. In contrast, little is known regarding the mechanisms mediating this shift among all 3 substrates in diabetes mellitus. Therefore, we tested the hypothesis that changes in myocardial glucose utilization directly influence ketone body catabolism.
Methods and Results
We examined ventricular‐cardiac tissue from the following murine models: (1) streptozotocin‐induced type 1 diabetes mellitus; (2) high‐fat‐diet–induced glucose intolerance; and transgenic inducible cardiac‐restricted expression of (3) glucose transporter 4 (transgenic inducible cardiac restricted expression of glucose transporter 4); or (4) dominant negative O‐GlcNAcase. Elevated blood glucose (type 1 diabetes mellitus and high‐fat diet mice) was associated with reduced cardiac expression of β‐hydroxybutyrate‐dehydrogenase and succinyl‐CoA:3‐oxoacid CoA transferase. Increased myocardial β‐hydroxybutyrate levels were also observed in type 1 diabetes mellitus mice, suggesting a mismatch between ketone body availability and utilization. Increased cellular glucose delivery in transgenic inducible cardiac restricted expression of glucose transporter 4 mice attenuated cardiac expression of both Bdh1 and Oxct1 and reduced rates of myocardial BDH1 activity and β‐hydroxybutyrate oxidation. Moreover, elevated cardiac protein O‐GlcNAcylation (a glucose‐derived posttranslational modification) by dominant negative O‐GlcNAcase suppressed β‐hydroxybutyrate dehydrogenase expression. Consistent with the mouse models, transcriptomic analysis confirmed suppression of BDH1 and OXCT1 in patients with type 2 diabetes mellitus and heart failure compared with nondiabetic patients.
Conclusions
Our results provide evidence that increased glucose leads to suppression of cardiac ketolytic capacity through multiple mechanisms and identifies a potential crosstalk between glucose and ketone body metabolism in the diabetic myocardium.
Keywords: cardiomyopathy, diabetes mellitus, ketone metabolism, O‐GlcNAcylation, diabetic cardiomyopathy, glucose, mitochondria
Subject Categories: Basic Science Research, Metabolism, Mechanisms, Metabolic Syndrome, Cellular Reprogramming
Nonstandard Abbreviations and Acronyms
- BDH1
β‐hydroxybutyrate dehydrogenase
- dnOGAh
transgenic inducible cardiac‐restricted expression of dominant negative O‐GlcNAcase
- HFD
high‐fat diet
- HMGCS2
3‐hydroxy‐3‐methylglutaryl‐CoA synthase 2
- KB
ketone body
- mG4H
transgenic inducible cardiac‐restricted expression of glucose transporter 4
- SCOT
succinyl‐CoA:3‐oxoacid CoA transferase
- T1D
type 1 diabetes mellitus
- T2D
type 2 diabetes mellitus
Clinical Perspective
What Is New?
We demonstrate transcriptional suppression of ketone body utilizing enzymes (Bdh1 and Oxct1) in hearts of 2 independent mouse models of diabetes mellitus and insulin resistance (type 1 diabetes mellitus and high‐fat diet), coupled with elevated myocardial β‐hydroxybutyrate in type 1 diabetes mellitus mice.
Identification of reduced β‐hydroxybutyrate utilization pathways in hearts of patients with type 2 diabetes mellitus and end‐stage heart failure.
We found consistent suppression of Bdh1 gene expression in 4 distinct mouse models of altered myocardial glucose handling, and high myocardial glucose availability is sufficient to attenuate myocardial ketone body utilization via mechanisms that may be further altered in diabetes mellitus.
What Are the Clinical Implications?
Our findings provide a snapshot of attenuated cardiac expression of ketolytic enzymes in multiple mouse models associated with impaired cardiac glucose metabolism and identify that increased myocardial glucose availability can independently modulate myocardial ketone body metabolism.
These findings may improve our understanding of the complex relationship between myocardial substrate utilization and development of diabetic cardiomyopathy.
Diabetes mellitus increases the risk of heart failure independently of underlying coronary artery disease. 1 Though diabetic heart failure is a multifactorial disease, an established body of evidence suggests that impaired cardiac energy metabolism is a leading mechanism that contributes to development of cardiac dysfunction. For example, increased substrate availability during diabetes mellitus is associated with cardiac metabolic inefficiency and accumulation of signaling/cytotoxic metabolites. 2 However, the precise mechanisms by which impaired myocardial substrate metabolism could be linked to pathology in the diabetic heart is incompletely understood.
The majority of the work investigating myocardial substrate selection in the past 60+ years has focused primarily on the relationship between altered fatty acid and glucose metabolism in the diabetic heart. 3 For example, increased fatty acid uptake and oxidation (as occurs during diabetes mellitus) inhibits glucose oxidation in a number of ways, including both allosteric inhibition and posttranslational modification of the pyruvate dehydrogenase complex. 4 Conversely, increased glucose uptake and oxidation inhibits fatty acid oxidation through malonyl‐CoA‐mediated inhibition of carnitine palmitoyltransferase I. 5 However, cardiomyocytes also readily utilize lactate, pyruvate, amino acids, and ketone bodies (KB) as substrates. 6 , 7 Acetoacetate and β‐hydroxybutyrate, the 2 primary KB, are synthesized from fatty acids by the liver, particularly during starvation, type 1 diabetes mellitus (T1D), and following consumption of ketogenic diets. Once generated, KB are released into the circulation and subsequently utilized by multiple oxidative tissues, including the heart. Two important enzymes, mitochondrial BDH1 (D‐β‐hydroxybutyrate (BHB)‐dehydrogenase) (encoded by Bdh1) and succinyl‐CoA:3‐oxoacid CoA transferase (SCOT, encoded by Oxct1), produce acetyl CoA by catabolizing β‐hydroxybutyrate and acetoacetate, respectively. 7 A growing number of pharmacologic, 8 dietary, 9 , 10 and genetic manipulation 11 studies suggest important roles of ketone bodies in cardiac health and disease. Recently 2 studies examining a mouse model of cardiac hypertrophy, as well as human heart failure patients, demonstrated increased reliance on KB utilization by the failing heart (in the absence of diabetes mellitus). 12 , 13 However, the interrelationship between myocardial glucose, fatty acid, and KB oxidation, or whether this relationship is disrupted during diabetes mellitus, remains unclear. In the present study, we sought to investigate whether increased glucose availability may directly influence myocardial KB metabolism. Our data show that genetic augmentation of glucose influx into the heart (glucose transporter 4 [GLUT4] overexpression; mG4H) decreased the rate of KB oxidation, concomitant with decreased expression of both Bdh1 and Oxct1. Furthermore, we observed decreased Bdh1 (but not Oxct1) in transgenic inducible cardiac‐restricted expression of dominant negative O‐GlcNAcase (dnOGAh) hearts, suggesting that the Bdh1 gene could be regulated by protein O‐GlcNAcylation, while SCOT protein levels were not changed but potentially directly regulated by O‐GlcNAcylation. Both T1D and high‐fat diet (HFD)‐induced hyperglycemia decreased cardiac Bdh1 and Oxct1 expression. BDH1 and OXCT1 were similarly repressed in failing hearts of diabetic but not from nondiabetic patients. Collectively these observations suggest that increased levels of glucose‐derived metabolites suppress cardiac ketolytic machinery via overlapping transcriptional and posttranslational mechanisms.
Methods
The data, analytic methods, and study materials are presented here following the Transparency and Openness Promotion (TOP) Guidelines, and any additional information will be made available upon request to facilitate other researchers for purposes of reproducing the results or replicating the procedures.
Ethics statement
The human study protocol was approved by the University of Alabama at Birmingham Institutional Review Board. Informed consent was granted to the UAB Cardiovascular Tissue Bank for the procurement of left ventricular assist device core biopsies and de‐identified patient health history with demographics were obtained to maintain confidentiality. All animal experimental protocols were approved by the Institutional Animal Care and Use Committee at the University of Alabama at Birmingham.
Animals
Inducible cardiomyocyte‐specific overexpression of glucose transporter 4
To dissect a glucose‐specific contribution, a double transgenic mouse for a myc‐tagged rat GLUT4 cDNA under the control of tetracycline response element promoter and the codon optimized tetracycline transactivator under control of the α‐myosin heavy chain promoter (tON) was used. The mice generated have been described previously, 14 and extensively phenotyped recently. 15 Briefly, transgene induction is initiated by a single injection of 100 μg of doxycycline and mice were maintained on a doxycycline‐chow (1 g/kg doxycycline; Bio‐Serv, Frenchtown, NJ).
Inducible cardiomyocyte‐specific overexpression of OGA‐inactive splice variant of O‐GlcNAcase gene (dnOGAh)
To determine an O‐GlcNAc‐specific role in regulating KB metabolism, we generated a second mouse model overexpressing an OGA‐inactive splice variant of the O‐GlcNAcase gene (dnOGA), 16 specifically in cardiac muscle using the same α‐myosin heavy chain tON mouse as above to drive the cardiomyocyte‐specific expression of dnOGA (dnOGAh). Generation of the GFP‐tagged dnOGA construct was previously described. 17 This mouse was bred with the tON mouse described above and backcrossed more than 6 times onto the FVB background. The resulting transgene is a dominant‐negative form of OGA lacking OGA enzyme activity. Single transgenic littermates were used as controls (Con). Both Con and transgenic mice (transgenic inducible cardiac‐restricted expression of glucose transporter 4 [mG4H] or dnOGAh) were injected with doxycycline and then kept on doxycycline‐chow for 2 weeks. The animals were housed at 22°C with a 12‐hour light, 12‐hour dark cycle with free access to water and chow. All experiments were performed in male mice and cardiac tissues were collected between 06:00 and 08:00 hours.
Diabetes mellitus induction and HFD treatment
Diabetes mellitus was induced in FVB mice for 4 weeks using the Animal Models of Diabetic Complications Consortium low‐dose (55 mg/kg bw) streptozotocin protocol, which includes injections over 5 consecutive days. Mice were euthanized 4 weeks after the initial injection of streptozotocin or control vehicle. HFD studies in C57BL/6J mice were performed as described previously. 18 Briefly, mice were fed HFD (58.0 kcal% fat, D12331i Research Diets, New Brunswick, NJ) or low‐fat diet (Con, 10.5 kcal% fat, D12329i Research Diets, New Brunswick, NJ) for 12 weeks. Cardiac tissues were collected after 2‐hour fasting (starting at 06:00).
RNA Sequencing Analysis
Left ventricular tissue was obtained from patients with end‐stage heart failure before left‐ventricular assist device implantation, as described previously. 19 Briefly, RNA was isolated using the RNeasy Fibrous Tissue Mini Kit (Qiagen Inc., Valencia, CA), following the manufacturer’s protocol. Extracted RNA was measured for quality to ensure RNA quality, with RNA Integrity Numbers >7. Next‐generation RNA sequencing was then performed using Illumina HiSeq2000 at the Heflin Genomics Core at the University of Alabama at Birmingham. Sequenced reads were aligned to the human genome (hg19) using TopHat (version 2.0.12) and Bowtie2 (version 2.2.3). Cufflinks (version 2.2.1) was then used to assemble transcripts from the aligned reads, estimate their abundances, and test for differential expression. Cuffcompare (version 2.2.1), a component of Cufflinks, related the assembled transcripts to a reference annotation and tracked Cufflinks transcripts across multiple experiments. Finally, Cuffdiff (version 2.2.1) generated statistical comparison between type 2 diabetes mellitus (T2D) and nondiabetic used to identify significant changes in transcript expression.
RNA Extraction and Quantitative Reverse Transcription Polymerase Chain Reaction Analysis
Quantitative polymerase chain reaction (qPCR) was performed as described previously. 20 In brief, total RNA was extracted with TRIzol reagent (Invitrogen, Carlsbad, CA) and reverse transcribed to cDNA using MultiScribe High Capacity cDNA Reverse Transcription Kit as per manufacturer’s instruction (Applied Biosystems, Carlsbad, CA). Reverse transcription quantitative polymerase chain reactions were performed using SYBR Green (Fermentas, Thermo Scientific, St. Leon‐Rot, Germany) in CFX96 Touch Real‐Time PCR Detection System according to the manufacturer’s recommended protocol. Relative mRNA expression was quantified using the comparative ΔCT method, normalized to β‐actin. The following primers were used for qPCR analysis: (Ppia) 3 peptidylprolyl isomerase; cyclophilin, 5′‐ AGCACTGGAGAGAAAGGATTTGG ‐3′, and reverse, 5′‐TCTTCTTGCTGGTCTTGCCATT ‐3′; (Bdh1) 3‐hydroxybutyrate dehydrogenase, type 1 forward, 5′‐AGCGTCTTACAACTTGGTGCCA‐3′, and reverse, 5′‐TTTGCCACTAGCCGCATCTG‐3′; (Oxct1) 3‐oxoacid CoA transferase 1 forward, 5′‐AAGCCATCACGGGAGATTTT‐3′, and reverse, 5′‐CCACGGTAGTTTCTGCAG‐3′; (Hmgcs2) 3‐hydroxy‐3‐methylglutaryl‐coenzyme A synthase 2 forward, 5′‐GGCCTTCAGGGGTCTAAAGC‐3′, and reverse, 5′‐AGGCAGCCATAGAGGGAAGA‐3′; (Slc16a1) solute carrier family 16 member 1, forward, 5′‐ TGCAACGACCAGTGAAGTATC‐3′, and reverse, 5′‐ GACAACCACCAGCGATCATTA ‐3′; (Slc16a7) solute carrier family 16 member 7, forward, 5′‐ATACTTGCAGGTCCTCTCATTC‐3′, and reverse, 5′‐GGAAGAGGCAGACAACGATAA‐3′.
Western Blot Analysis and Immunoprecipitation
Quantitative analysis of protein levels and O‐GlcNAcylation status was performed via standard Western blotting procedures as described previously. 21 Briefly, total protein lysates were isolated in RIPA buffer (150 mmol/L NaCl, 1 mmol/L EDTA, 50 mmol/L Tris/HCl, 0.5% sodium dodecyl sulfate, 1% NP‐40, plus Halt protease and phosphatase inhibitor cocktail [Thermo Scientific]). Protein was quantified by BCA protein assay kit (Pierce 23225). Protein lysate (25–50 μg) was separated on polyacrylamide gels and transferred to a 0.45 μM polyvinylidene difluoride membrane (Bio‐Rad, Hercules, CA). Membranes were immunoblotted with primary antibodies for BDH1 (Catalog No. 15417‐1‐AP), SCOT (Catalog No. 12175‐1‐AP), HMGCS2 (3‐hydroxy‐3‐methylglutaryl‐CoA synthase 2) (Catalog No. ab137043), actin (Catalog No. GTX110564; GTX629630), O‐GlcNAc (CTD110.6; UAB Epitope Recognition and Immunoreagent Core), GAPDH (Catalog No. ab8245) or OGA (Catalog No. 14711‐1‐AP) in casein blocking buffer (Sigma). Proteins were detected using goat anti‐rabbit IgG (A10043) and a goat anti‐mouse IgG (926‐32212) secondary antibody labeled with NIR fluorochrome dyes (800 and 700 nm) followed by signal visualization using an Odyssey CLx infrared imaging system (model 9120) (LI‐COR Biosciences‐US, Lincoln, NE). O‐GlcNAc blots were developed using horseradish peroxidase–conjugated secondary (anti‐mouse IgM) (Calbiochem, 401225), and chemiluminescent substrate (Perkin Elmer). The immunoprecipitation was conducted as modified method described previously. 22 Briefly, 2 µg of anti‐O‐GlcNAc (RL2; Catalog No. MA1‐072, Thermo Fisher) was added to 2 mg of cell lysates. The antibody and lysate complex were incubated 4 hours at 4°C with gentle agitation and then magnetic beads were added (Dynabeads 10004D, Thermo Fisher Scientific) and incubated overnight at 4°C. Beads were washed 5 times with RIPA buffer and protein was eluted with 0.1 M citrate buffer (pH 2–3), and then NuPAGE LDS sample buffer was added (#NP0008, Thermo Fisher) and immuno‐blotted (IB) with the anti‐SCOT antibody.
Metabolomic Analysis of Heart Tissue
Metabolomic analysis was performed in nonfasted mice as described previously. 23 Specifically, left‐ventricle tissue was quickly dissected away in ice‐cold saline solution and freeze‐clamped in liquid nitrogen. Between 58 and 114.2 mg of tissue was carefully transferred into a bead mill tube containing 1.4 mm ceramic beads (MoBio Laboratories, Carlsbad, CA). To this tube 900 μL of pre‐chilled 90% MeOH was added. Using an Omni Bead Ruptor 24 bead mill (Omni‐Inc, Kennesaw, GA) the tissue was homogenized for 30 s at 6.5 m/s. The lysate was incubated for 1 hour at −20°C to precipitate protein. Following incubation, cell debris was removed by centrifugation, 14 000 g for 5 minutes at 4°C. The supernatant was then transferred to fresh microfuge tubes. A second extraction was performed using 60% MeOH in the same manner as the first extraction. The 2 supernatants were combined, split equally into 2 portions, 1 for liquid chromatography (LC) and the other for gas chromatography mass spectroscopy, and the solvent was removed en vacuo.
Catalytic Activity of Ketolytic Enzymes
3‐Hydroxybutyrate dehydrogenase activity was determined by measuring the reduction of NAD+ to NADH in a spectrophotometric unit as previously described with slight modification. 24 , 25 Briefly, ventricular tissue was homogenized in buffer (300 mmol/L sucrose, 20 mmol/L Tris, and 2 mmol/L EGTA) using a glass homogenizer and the mitochondria pellet was suspended using the same buffer. Enzyme activity is expressed as nmol of NAD+ reduced/min per mg of protein. CoA transferase activity assay was measured by using the assay procedure described previously. 26 Briefly, ventricle pieces were homogenized with phosphate buffer saline (pH 7.2) containing with protease inhibitors in glass homogenizer followed by centrifugation at 20 000 g at 4°C for 20 minutes. The supernatant was used as source for CoA transferase. The incubation mixture contained 50 mmol/L Tris·HCl, pH 8.5, 10 mmol/L MgCl2, 4 mmol/L iodoacetamide, 1 mmol/L succinyl‐CoA, 10 mmol/L lithium acetoacetate, and 100 μg of the lysate. SCOT catalytic activity was measured spectrophotometrically by following the formation of acetoacetyl‐CoA at 313 nm for 2 minutes. SCOT catalytic activity was normalized to the total protein in high‐speed supernatants. Enzyme activity was expressed as nmol of acetoacetyl‐CoA formed per min per mg of protein. Isolated ventricle mitochondrial HMGCS2 activity was measured as previously described with modification. 27 Briefly, mitochondria sonicated supernatants were added to a reaction mixture that had a final concentration of 50 mmol/L Tris (pH 8.0), 0.1 mmol/L acetoacetyl‐CoA (Sigma), and 10 mmol/L acetyl‐CoA (Sigma) in a total volume of 150 µL at room temperature. After 10, 20, and 30 minutes, 27.5 µL of reaction mixtures were removed and 21 µL of 3% perchloric acid deproteinization and 7 µL of 2 M KOH were rapidly added to achieve pH 6.5–8 and terminate the reaction. The deproteinized reaction mixtures were briefly centrifuged and 20 µL of supernatant was used for total KB quantification using a colorimetric assay kit (#415‐73301, Fujifilm Wako Pure Chemical Corporation, Osaka, Japan).
Ketone Utilization ex vivo Using Isolated Working Hearts
Myocardial substrate utilization was measured ex vivo in isolated perfused working mouse hearts, as described previously. 28 Male 16‐ to 17‐week‐old mG4H mice were subjected to perfusion experiments 2 weeks after transgene induction. Mice were euthanized following chloral hydrate injection, and cardiac excision was performed. All hearts were perfused in the working mode (nonrecirculating manner) for 40 minutes with a preload of 12.5 mm Hg and an afterload of 50 mm Hg. Standard Krebs‐Henseleit buffer was supplemented with 8 mmol/L glucose, 0.4 mmol/L oleate conjugated to 3% bovine serum albumin (fatty acid–free), 10 μU/mL insulin (basal/fasting concentration), 2 mmol/L D‐β‐hydroxybutyrate (BHB), 0.2 mmol/L acetoacetate, 0.05 mmol/L l‐carnitine, and 0.13 mmol/L glycerol. Metabolic flux was assessed through the use of radiolabeled tracers: [U‐14C]‐BHB (0.04 mCi/L; BHB oxidation), and d‐[2‐5H]‐glucose (0.04 mCi/L; glycolysis). Measures of cardiac metabolism (i.e., β‐hydroxybutyrate oxidation) and function were determined as described previously. 25 Data are presented as steady‐state values during the last 10 minutes of the perfusion protocol. At the end of the perfusion protocol, the left ventricles were snap‐frozen with liquid nitrogen and stored at −80°C.
Biochemical parameters analysis
Blood glucose was measured using a commercial glucometer. Blood was collected from the facial vein of mice and plasma was isolated for subsequent biochemical parameters. Plasma β‐hydroxybutyrate (ab83390, Abcam, Cambridge, United Kingdom) and acetoacetate (ab180875, Abcam) concentrations were measured with commercially available kits. For cardiac ventricle tissue, the samples were deproteinated with 1 M perchloric acid according to the manufacturer’s instruction.
Statistical Analysis
Analyses were performed with GraphPad Prism 8.4 software (Prism, San Diego, CA). All the results are expressed as mean±SEM. Mann‐Whitney test was performed to examine sensitivity and results are reported as unpaired Student t tests (2‐tailed) when comparisons are made between 2 groups. Statistical analysis was performed using the criterion for significance of P<0.05 for all comparisons. For the RNA‐sequencing analysis of human heart failure (HF), a 2‐way comparison was made between diabetic and nondiabetic patients with HF, with data presented as means±SEM. When comparing normalized gene expression between groups, statistical significance was determined using a Student t test with Bonferroni posttest adjustment.
Results
Differential expression of KB utilization enzymes in the heart of T1D mice
Recent findings from HF patients without diabetes mellitus suggest that myocardial KB oxidation is increased in failing hearts. 13 However, the effect of T1D on myocardial KB metabolism of failing hearts is not well known. In the present study, we used a low‐dose streptozotocin‐induced T1D mouse model to study KB metabolism. Seven days following the initial streptozotocin injection, mice with blood glucose levels > 200 mg/dL were considered hyperglycemic and were followed an additional 3 weeks before analysis. Previously, we have shown that these mice develop cardiac dysfunction at this time point. 29 To determine the effect of T1D on the expression of enzymes involved in KB metabolism in the heart, we examined gene expression levels of Bdh1 and Oxct1, which encode for 2 important enzymes responsible for KB oxidation. Cardiac expression of both genes (Bdh1 and Oxct1) was significantly reduced (Figure 1A and 1B) in T1D mice. Protein levels of cardiac BDH1 were also significantly lower in hearts of diabetic mice (Figure 1C). However, cardiac SCOT protein abundance was unchanged in hearts of diabetic mice at this time point (Figure 1D). This finding is in alignment with a previous report showing that cardiac SCOT protein expression is unchanged after 4 weeks of streptozotocin‐induced diabetes mellitus in rats. 30
Figure 1. Regulation of ketone body utilizing genes, protein, and activity by streptozotocin (STZ)‐induced hyperglycemia.
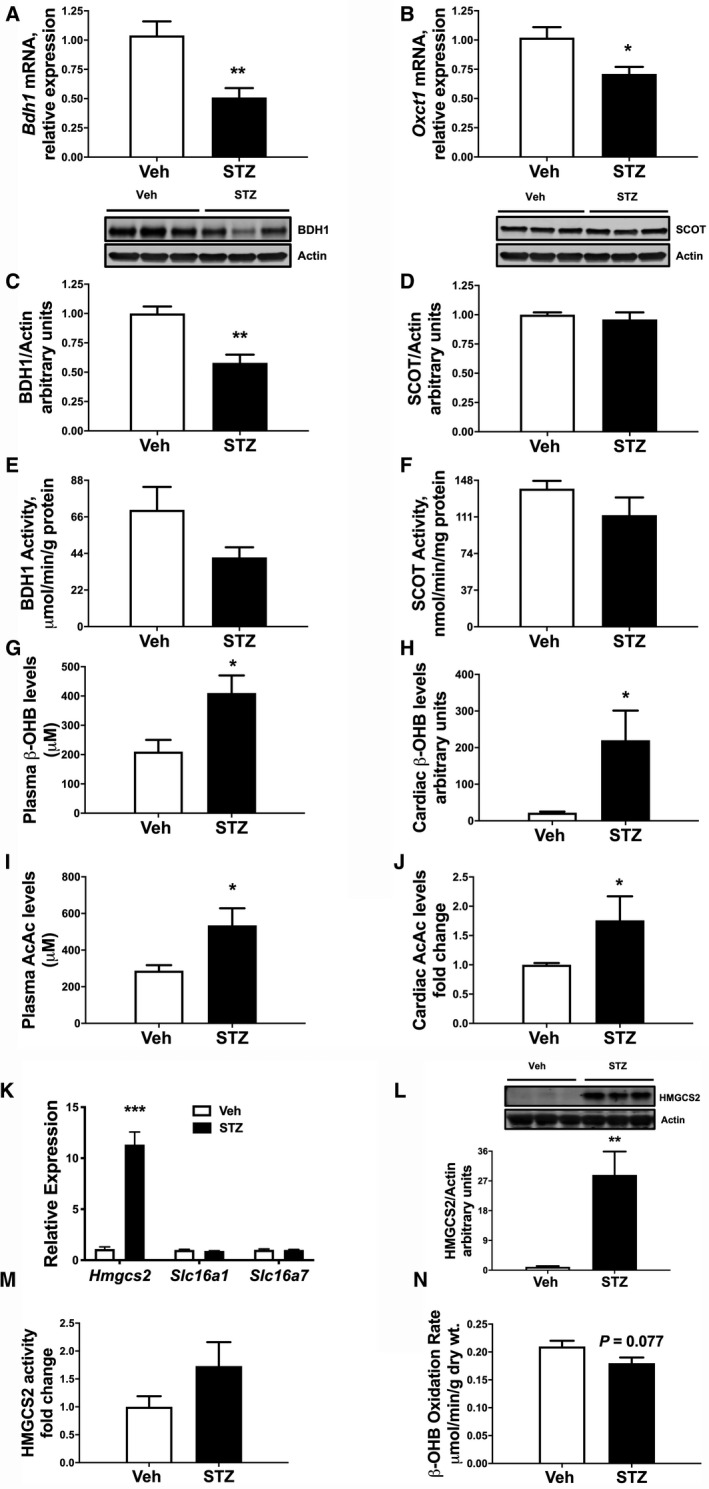
Gene expression from qPCR analysis showing expression of Bdh1 (A) and Oxct1 (B) in mice 4 weeks after STZ treatment. Western analysis of whole cell extracts from left ventricular heart tissue for BDH1 and SCOT. C, Upper panel: Immunoblot illustrating BDH1 levels in ventricular tissue of STZ mice compared with vehicle‐treated mice. Lower panel: Densitometric analysis of immunoblots shown in the upper panel. D, Upper panel: Immunoblot illustrating SCOT levels in ventricular tissue of STZ mice compared with vehicle‐treated mice. Lower panel: Densitometric analysis of immunoblots shown in upper panel. E, Catalytic activity of BDH1 in mitochondrial fraction prepared from left ventricle in STZ mice. F, SCOT enzymatic activity was measured in the Co‐A transferase enriched fractions from the ventricular tissue of Vehicle (Veh) and STZ mice. G, Plasma β‐hydroxybutyrate (β‐OHB) and metabolomics data showing cardiac β‐OHB levels (H) in Veh and STZ mice. I, Plasma acetoacetate (AcAc) and direct measure of cardiac AcAc levels (J) in Veh and STZ mice. K, mRNA expression of Hmgcs2, Slc16a1, and Slc16a7 in ventricular tissue of Veh and STZ mice. L, Upper panel: immunoblot illustrating HMGCS2 expression in ventricular tissue of STZ mice compared with Veh mice. Lower panel: Densitometric analysis of immunoblots shown in upper panel, n≥6. M, HMGCS2 activity in tissue lysates from ventricular tissue of STZ mice compared to Veh mice, n≥5. N, Hearts from STZ mice were isolated and perfused ex vivo in presence of [U‐14C]‐βOHB for oxidation, n=4. Means±SEM; *P<0.05; **P<0.01; ***P. qPCR indicates quantitative polymerase chain reaction. <0.001; vs. Veh.
Functional Consequences of Reduced KB Utilization Gene Expression in T1D Mouse Hearts
We next sought to determine whether observed changes in Bdh1 mRNA and protein expression influence enzymatic activity. Previous studies found that BDH1 enzyme activity is reduced in mitochondria isolated from hearts of T1D rats. 24 To this end, we observed a trend for reduced cardiac catalytic activity of BDH1 (−27%; P=0.084; Figure 1E) in diabetic hearts. However, SCOT activity was not altered (−19%; Figure 1F). In line with our prior studies, 29 plasma β‐hydroxybutyrate levels were increased in T1D mice (1.9‐fold; Figure 1G). Additionally, we found that there was a striking 10‐fold increase in cardiac β‐hydroxybutyrate levels (Figure 1H; P=0.034), compared with hearts of nondiabetic mice. Furthermore, both circulating and cardiac tissue acetoacetate levels were also significantly increased (Figure 1I and Figure 1J).
To further explore potential mechanisms for the elevated tissue levels of myocardial β‐hydroxybutyrate, we also examined expression of genes involved in KB synthesis and uptake. Interestingly, there was a robust 10‐fold elevation of mRNA levels of Hmgcs2, which encodes for HMGCS2, a mitochondrial protein that controls ketone bodies synthesis, in T1D myocardium (Figure 1K; P<0.001). Moreover, while cardiac HMGCS2 protein was weakly detected in nondiabetic mice, cardiac HMGCS2 was significantly induced (P=0.003) in diabetic mice (Figure 1L). These findings support recent reports showing similar induction of cardiac Hmgcs2 expression in streptozotocin‐induced diabetes mellitus in mice and rats. 31 , 32 KBs are transported to extrahepatic tissues by a family of monocarboxylate transporters belonging to the solute carrier 16A family. Therefore, we measured expression of the genes encoding the KB transporters (Slc16a1 [MCT1] and Slc16a7 [MCT2]). 33 mRNA expression of both cardiac Slc16a1 and Slc16a7 were unaltered in T1D hearts (Figure 1K), suggesting that accumulation of cardiac β‐hydroxybutyrate could be a consequence of decreased KB utilization and increased ketogenesis. Despite the marked induction of HMGCS2 protein, total activity of the enzyme was not significantly increased (Figure 1M). However, consistent with the decreased BDH1 protein (Figure 1C) and trend in decreased BDH1 enzyme activity (Figure 1E), there was a strong trend for decreased KB utilization in isolated working hearts (Figure 1N, P=0.077). Taken together, these data suggest that T1D alters the regulation of genes that encode enzymes and proteins involved in KB metabolism.
Reduced Expression of Cardiac Bdh1 in an Established Mouse Model of Glucose Intolerance
Our findings from the T1D mouse model stimulated our interest to understand whether the observed expression changes in KB utilization are present in other models of disrupted cardiac metabolism. Diet‐induced obesity is a major risk factor for developing glucose intolerance and T2D, and it is well established that obesity greatly increases the risk for heart failure. 34 Therefore, we fed mice a HFD for 12 weeks to induce obesity and insulin resistance, which has been shown by some groups to induce diabetic cardiomyopathy. 35 We performed qPCR and immunoblot analysis to measure cardiac expression of KB metabolic enzymes. Interestingly, mRNA levels of both Bdh1 (−47%) and Oxct1 (−24%) were reduced following HFD feeding (Figure 2A and 2B). In contrast, no differences were observed for Hmgcs2, Slc16a1, and Slc16a7 expression (Figure 2C and 2D). At the protein level, BDH1, but not SCOT, was significantly reduced in hearts of HFD‐fed mice compared with Con mice (Figure 2E and 2F). Assessment of enzymatic activities revealed a trend for reduced myocardial BDH1 activity in HFD mice compared with Con mice (−37.1%; P=0.149), with no significant changes in SCOT activity (Figure 2G and 2H). Collectively, these observations suggest that cardiac KB utilization may be impaired in obese mice, and for both T1D and HFD models there may be overlapping but distinct regulation of ketolytic machinery in diabetic insulin‐dependent versus insulin‐resistant states.
Figure 2. Regulation of ketone body utilizing genes, protein, and activity by high‐fat diet (HFD)‐induced obesity.
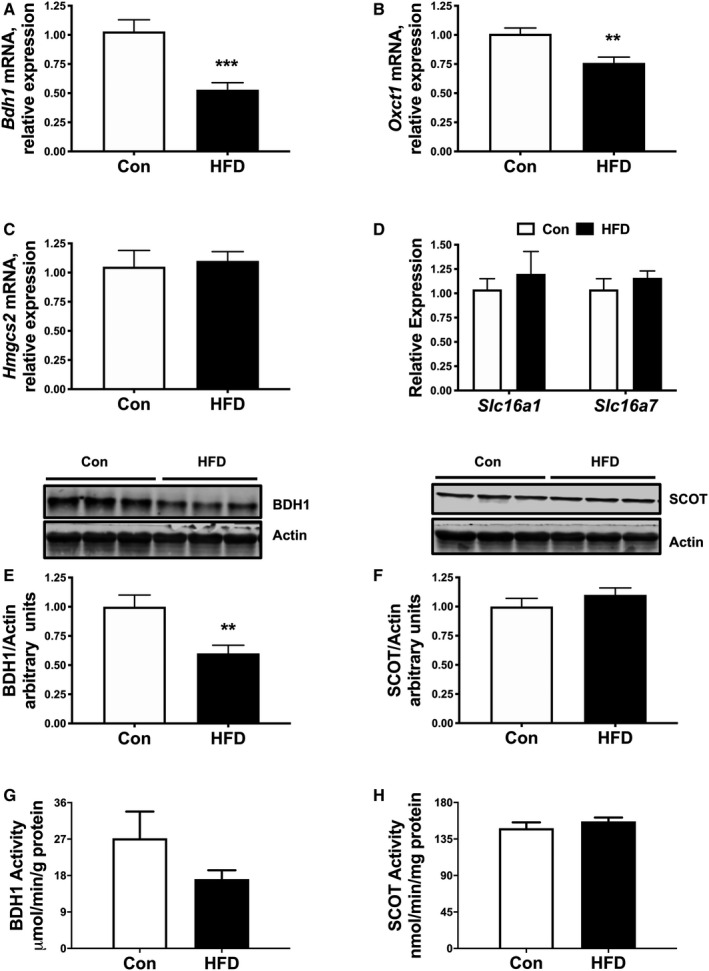
qPCR analysis showing expression of Bdh1 (A) and Oxct1 (B) in mice after 12 weeks of HFD or control diet (Con) feeding. mRNA expression of Hmgcs2 (C), and ketone transport genes, Slc16a1 and Slc16a7 (D) in ventricular tissue. Western analysis of whole cell extracts prepared from left ventricular heart tissue for β‐hydroxybutyrate dehydrogenase (BDH1) and succinyl‐CoA:3‐oxoacid CoA transferase (SCOT). E, Upper panel: Immunoblot illustrating BDH1 levels in ventricular tissue of HFD mice compared with Con mice. Lower panel: Densitometric analysis of immunoblots shown in upper panel. F, Upper panel: Immunoblot illustrating SCOT levels in ventricular tissue of HFD mice compared with Con mice. Lower panel: Densitometric analysis of immunoblots shown in upper panel. Cardiac BDH1 (G) and SCOT (H) enzyme activities in Con and HFD mice. n=6; Means±SEM. qPCR indicates quantitative polymerase chain reaction. **P<0.01; ***P<0.001; vs. Con.
Cardiac GLUT4 Overexpression Results in Reduced Myocardial KBs Utilization
Hyperglycemia may independently mediate the adverse cardiovascular effects of diabetes mellitus through multiple mechanisms including formation of advanced glycation end products and protein posttranslational modification of proteins via O‐linked attachment of β‐N‐acetyl‐glucosamine, O‐GlcNAcylation. 36 Therefore, we asked whether glucose alone is sufficient to alter cardiac KB metabolism. To determine this, we used a transgenic mouse model with inducible cardiac‐specific overexpression of a myc‐tagged glucose transporter 4 (GLUT4; mG4H) with single transgenic littermates as controls (Con). Similar to the gene expression changes observed in T1D and HFD mouse hearts, enhanced cellular glucose delivery led to a robust reduction in Bdh1 and Oxct1 expression in mG4H mice (Figure 3A and 3B). Furthermore, BDH1 protein was also reduced in this model (Figure 3C). Unlike observations in T1D and HFD mice, enhanced glucose delivery alone repressed cardiac SCOT protein levels (Figure 3D). These findings suggest that cardiac SCOT protein levels in T1D and HFD mice are not regulated via glucose‐mediated mechanisms (comparing Figures 1D, 2F, and 3D). In contrast to the T1D mouse model (Figure 1K and 1L), mRNA levels of Hmgcs2 were unchanged in mG4H mice (Figure 3E). Similarly to the other models, expression of the KB transporters Slc16a1 and Slc16a7 was unchanged (Figure 3F). Despite the marked reduction in ketone metabolic enzymes, there was no measurable change in cardiac β‐hydroxybutyrate levels (Figure 3G). Taken together, these findings support that induction of cardiac Hmgcs2 expression in T1D is independent of direct glucose‐mediated regulation of gene expression.
Figure 3. Glucose specific regulation of ketone body utilizing genes and protein by cardiac specific glucose transporter 4 (GLUT4)‐induced glucose delivery.
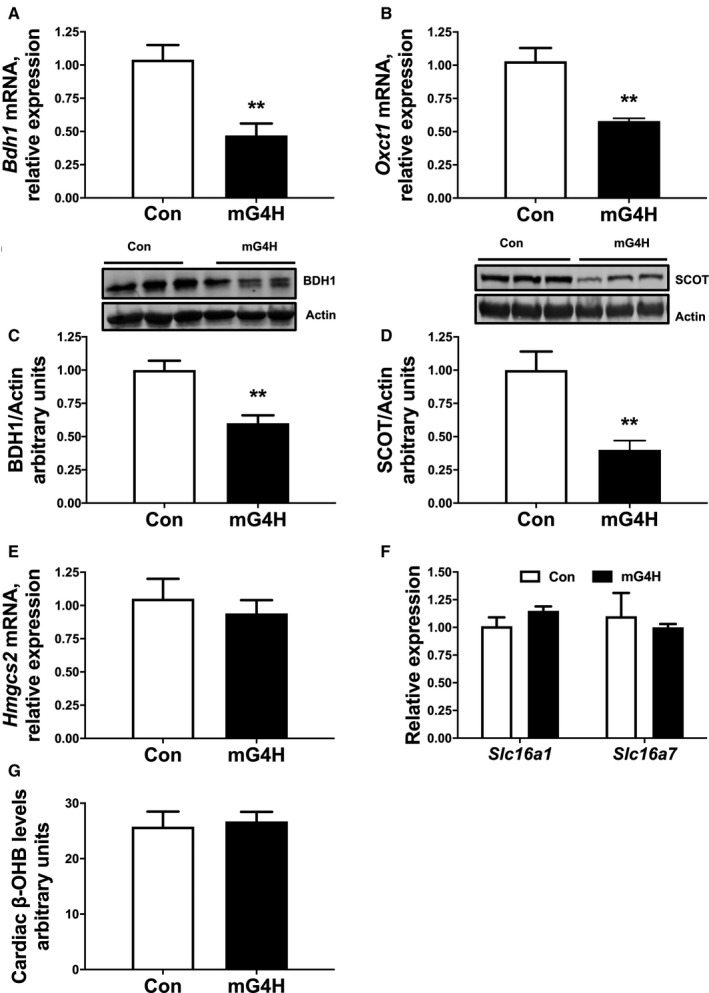
Gene expression from qPCR analysis showing expression of Bdh1 (A) and Oxct1 (B) in mice 4 weeks after GLUT4 induction in the heart (mG4H). Western analysis of whole cell extracts from ventricular heart tissue for BDH1 and SCOT. C, Upper panel: Immunoblot illustrating BDH1 levels in ventricular tissue of mG4H mice compared with Control (Con) mice. Lower panel: Densitometric analysis of immunoblots shown in upper panel. D, Upper panel: Immunoblot illustrating SCOT levels in ventricular tissue of mG4H mice compared with Con mice. mRNA expression of Hmgcs2 (E), and ketone transport genes, Slc16a1 and Slc16a7 (F) in ventricular tissue. G, Tissue metabolomics data of cardiac β‐hydroxybutyrate (β‐OHB) levels. n≥6; Means±SEM; BDH1 indicates β‐hydroxybutyrate dehydrogenase; MG4H, transgenic inducible cardiac‐restricted expression of glucose transporter 4; qPCR, quantitative polymerase chain reaction; SCOT, succinyl‐CoA:3‐oxoacid CoA transferase. **P<0.01; vs. Con.
We next asked whether reduced abundance of ketolytic proteins in mG4H mice translates to reduced catalytic activities of enzymes involved in KB utilization. We measured specific activities of BDH1 in myocardial mitochondria isolated from Con or mG4H mice following transgene induction. mG4H mice demonstrated a significant reduction in BDH1 enzyme activity compared with Con mice (Figure 4A). However, catalytic activity of SCOT was similar in both groups (Figure 4B). Next, KB utilization was determined in isolated working hearts. GLUT4 overexpression in mG4H mice increased glycolysis relative to Con mice (Figure 4C). Concomitant with reduced BDH1 expression and enzyme activity results, GLUT4 induction resulted in a significant reduction in ex vivo β‐hydroxybutyrate oxidation rates compared with Con hearts (Figure 4D). Taken together, these findings strongly suggest that glucose alone is sufficient to attenuate cardiac KB utilization, potentially through attenuation of BDH1 expression/activity.
Figure 4. Glucose specific regulation of ketone body utilizing protein activity and cardiac metabolic rates.
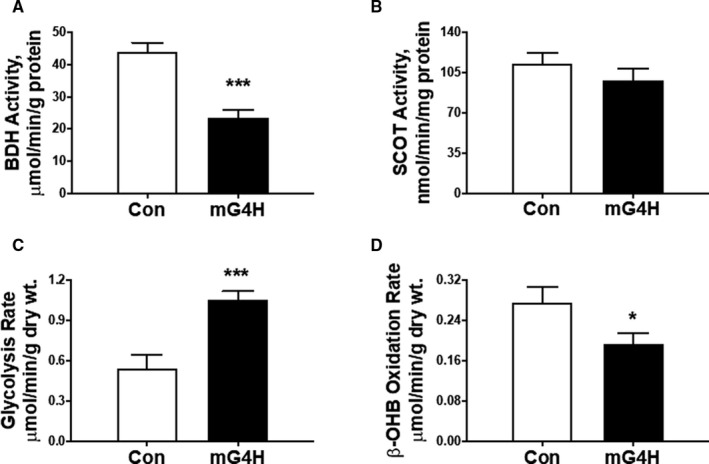
A, Catalytic activity of BDH1 in mitochondrial fractions prepared from the hearts of mG4H mice. B, SCOT enzymatic activity was measured in the CoA transferase–enriched fractions from the heart. C, After 2 weeks of transgene induction, hearts from mG4H mice were isolated and perfused ex vivo in presence of D‐[5‐3H]‐glucose for glycolysis (C) and [U‐14C]‐βOHB for oxidation (D). Values are expressed as means±SEM (n=5–9; sample size range varies dependent on the parameter investigated); *P<0.05; ***P<0.001; vs. Con. BDH1 indicates β‐hydroxybutyrate dehydrogenase; MG4H, transgenic inducible cardiac‐restricted expression of glucose transporter 4; SCOT, succinyl‐CoA:3‐oxoacid CoA transferase.
Protein O‐GlcNAcylation Influences Cardiac BDH1 Expression
Hyperglycemia increases glucose flux through the hexosamine biosynthetic pathway and leads to posttranslational modification of nuclear, cytoplasmic, and mitochondrial proteins via O‐GlcNAcylation. Moreover, aberrant cardiac O‐GlcNAc signaling appears to play a causal role in the adverse effects of diabetes mellitus on the heart. 37 , 38 Consistent with that prior work, total cellular levels of protein O‐GlcNAcylation in streptozotocin and HFD mouse hearts were increased (Figure 5A and 5B). A smaller number of proteins also showed increased O‐GlcNAcylation in mG4H mouse hearts, but the overall change to total protein did not reach significance (Figure 5C). To directly test a mechanistic role for O‐GlcNAcylation in the molecular changes of KB utilization machinery, we generated a novel mouse model with inducible cardiomyocyte‐specific overexpression of a dominant negative OGA gene (dnOGAh), similar to that generated previously in skeletal muscle. 17 After 2 weeks of doxycycline induction, Western blot analysis revealed 2 OGA bands in dnOGAh hearts and not in other tissues examined (Figure 6A, and data not shown), representing the endogenous OGA protein (130 kDa) and the GFP‐tagged OGA transgene (apparent size, 150 kDa), which contrasts with the presence of the wild‐type protein in control mice (Figure 6A). Transgene induction increased protein O‐GlcNAcylation in the heart (Figure 6B and 6C). Consistent with all models tested (Figure 1A, 2A, and 3A), mRNA expression of cardiac Bdh1 was also reduced in dnOGAh mice (Figure 6D), while levels of Oxct1, Hmgcs2, Slc16a1, and Slc16a7 were unchanged (Figure 6E and 6G). Also, similar to the mG4H model, tissue KB levels were unchanged for either β‐hydroxybutyrate or acetoacetate (Figure 6H and 6I, respectively). Furthermore, protein levels of BDH1 or SCOT were not significantly altered (Figure 6J and 6K). Owing to the role of O‐GlcNAcylation to directly regulate enzymatic function, we further assessed whether either SCOT or BDH1 were directly modified by GlcNAc. Immunoprecipitation for O‐GlcNAc followed by Western blot revealed that BDH1 was not modified under these conditions but SCOT was modified, and this was significantly increased by dnOGAh (Figure 6L) Taken together, these results suggest that protein O‐GlcNAcylation may regulate Bdh1 gene expression and directly regulate SCOT protein in the heart.
Figure 5. Protein posttranslational regulation via O‐GlcNAcylation is enhanced by STZ, HFD, and glucose.
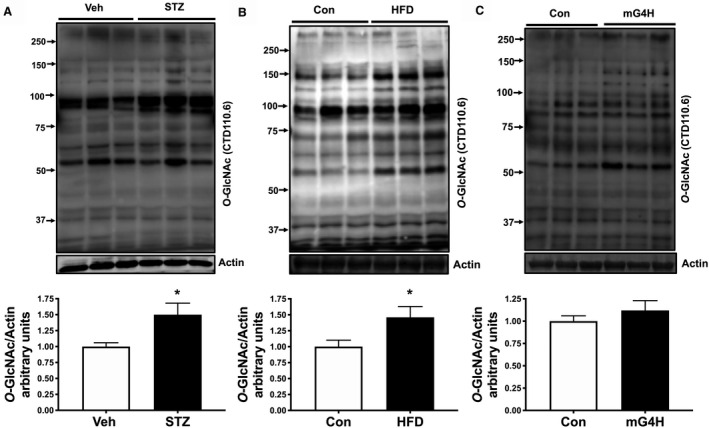
Cardiac protein O‐GlcNAcylation was measured in ventricular lysates prepared from 4 weeks STZ‐induced diabetic mice, 12‐week HFD, or Con diet fed mice and mG4H mice after 2 weeks of transgene induction. Fifty micrograms of lysates was resolved and probed using CTD110.6 antibody. A, Upper panel: immunoblot illustrating O‐GlcNAc detection in ventricular tissue of STZ mice compared with vehicle‐treated mice. Lower panel: Densitometric analysis of immunoblots shown in upper panel. B, Upper panel: immunoblot illustrating O‐GlcNAc detection in ventricular tissue of HFD mice compared with vehicle‐treated mice. Lower panel: Densitometric analysis of immunoblots shown in upper panel. (C) Upper panel: immunoblot illustrating O‐GlcNAc detection in ventricular tissue of mG4H mice compared with vehicle‐treated mice. Lower panel: Densitometric analysis of immunoblots shown in upper panel. *P<0.05. Con indicates Control; HFD, high‐fat diet; MG4H, transgenic inducible cardiac‐restricted expression of glucose transporter 4; STZ, streptozotocin; Veh, Vehicle.
Figure 6. O‐GlcNAcylation specific regulation of ketone body utilizing genes and protein is altered by cardiac‐specific dominant negative O‐GlcNAcase (dnOGA) transgene induction.
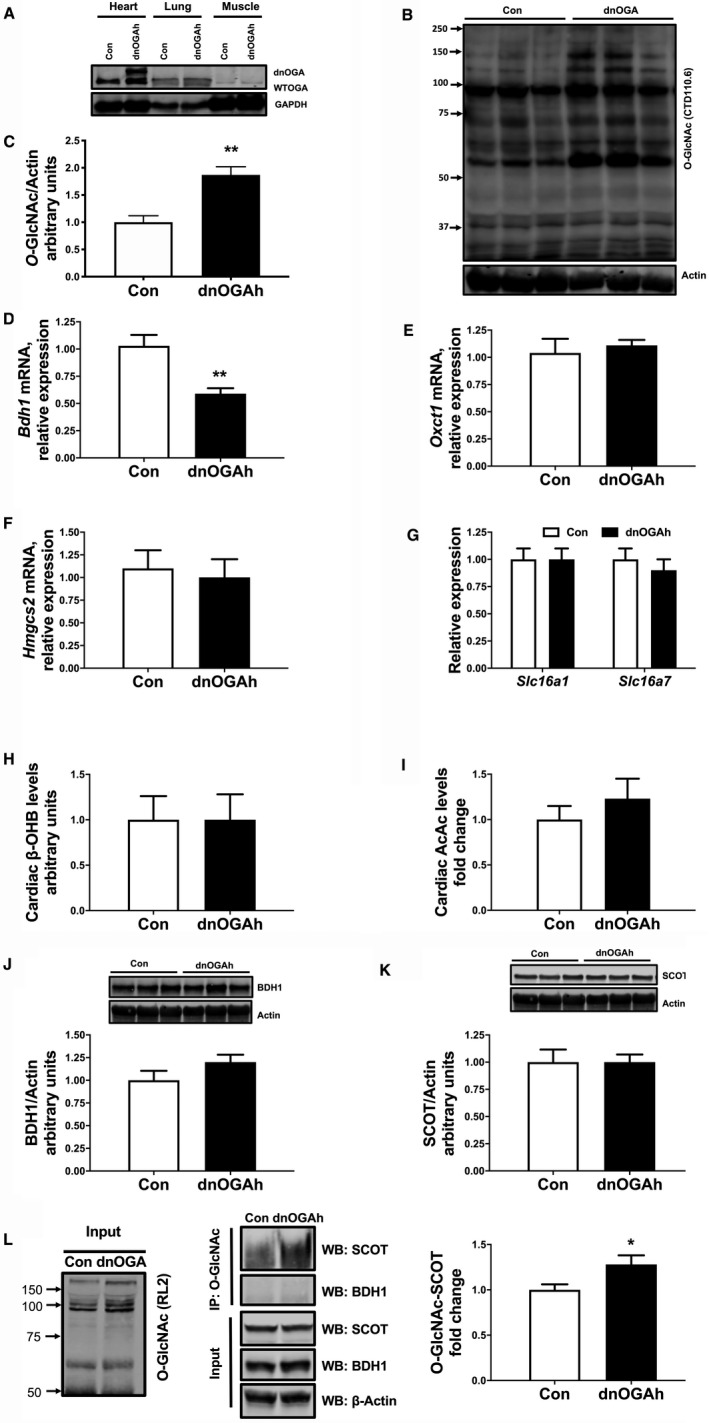
Inducible cardiomyocyte‐specific overexpression of OGA‐inactive splice variant of O‐GlcNAcase gene. Following transgene induction, protein expression of endogenous OGA and dnOGA in ventricular tissue (A) and levels of O‐GlcNAcylated proteins (B and C). qPCR analysis showing mRNA levels of Bdh1 (D) and Oxct1 (E) in mice 2 weeks after transgene induction in dnOGAh mouse hearts. mRNA expression of Hmgcs2 (F), and ketone transport genes, Slc16a1 and Slc16a7 (G). Western analysis of whole cell extracts from left ventricular heart tissue for BDH1 and SCOT. (H) Tissue metabolomics data of cardiac β‐hydroxybutyrate (β‐OHB) levels, n=6. (I) Cardiac tissue acetoacetate (AcAc) levels in Con and dnOGAh mice. J, Upper panel: Immunoblot illustrating BDH1 levels in ventricular tissue of dnOGAh mice compared with Con mice. Lower panel: Densitometric analysis of immunoblots shown in upper panel. K, Upper panel: Immunoblot illustrating SCOT levels in ventricular tissue of dnOGAh mice compared with Con mice, n≥6. L, Left panel: Immunoblot illustrating RL2 antibody detection of total O‐GlcNAcylation of cardiac proteins in input from Con and dnOGAh hearts. Central Panel: Immunoblot illustrating input of BDH1, SCOT, and loading control β‐actin as well as immunoprecipitation (IP) for O‐GlcNAcylated proteins followed by Western blot (WB) for SCOT and BDH1. Right panel: Densitometric analysis of immunoblots, n≥3. Means±SEM; *P<0.05; **P<0.01; vs. Con‐dnOGAh. AcAc indicates acetoacetate; Con, Control; BDH1, β‐hydroxybutyrate dehydrogenase; O‐GlcNAc, β‐linked N‐acetylglucosamine posttranslational modification; qPCR, quantitative polymerase chain reaction; SCOT, succinyl‐CoA:3‐oxoacid CoA transferase.
T2D Alters Expression of KB Enzymes in Failing Human Hearts
To determine whether the observed changes in ketolytic gene expression in T1D and HFD mouse models in mice corresponded with biologically conserved findings in humans, we applied an integrated transcriptomic analysis of both human and mouse. Left ventricular biopsies were obtained from diabetic (T2D, n=6) and nondiabetic (n=5) patients with end‐stage heart failure (HF) who underwent left‐ventricular assist device implantation. Clinical characteristics of the patients are shown in Figure 7A. Tissues were then analyzed via RNA‐sequencing analysis (GSE109097), 19 and compared with array‐based gene expression analyses of streptozotocin‐induced diabetic and mG4H mice (GSE123975), 15 and separate previously published HFD (GSE106180) 39 models (Figure 7B). We identified 14 genes that were regulated in all groups. We found that within this small set of genes there was an enrichment for KB metabolism (Figure 7C). Inspection of all 14 genes regulated in each the systems demonstrated suppression of BDH1 (51.7%, P<0.001) and OXCT1 (34.7%, P=0.005) in humans with similar decreases in each of the mouse models (Figure 7D). Taken together, these observations suggest that diabetes mellitus influences cardiac expression of KB metabolism enzymes in a comparable manner between mice and humans.
Figure 7. Gene expression pathway analysis reveals that glucose, diabetes mellitus, and HFD alter ketone body gene expression similarly between mice and diabetic human patients.
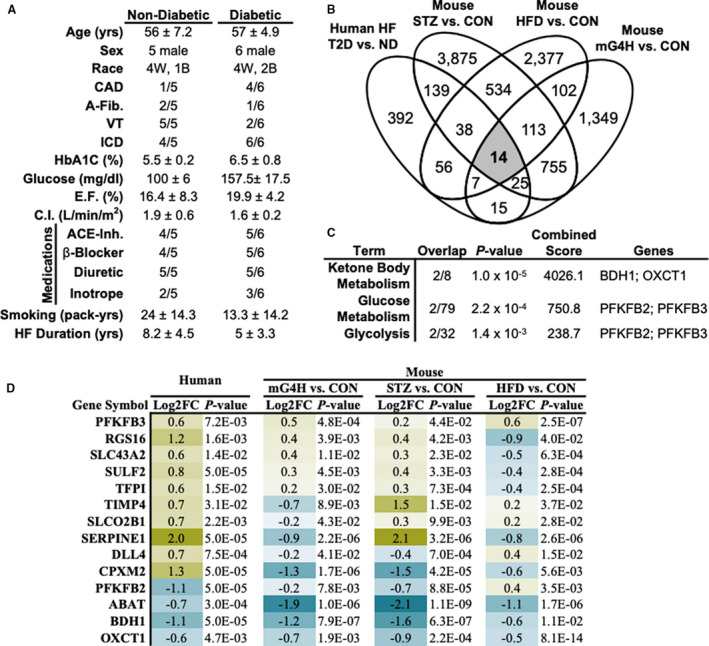
Gene expression analysis of human HF and mouse models demonstrate conserved suppression of ketone metabolic pathways. A, Table summarizing patient health information at time of left ventricular assist device implantation. B, Venn diagram comparison of differentially expressed genes (DEGs)* based on diabetic relative to nondiabetic HF, STZ‐induced diabetes mellitus relative to VEH‐treated mice (n=6), mG4H relative to littermate controls (n=6), and HFD relative to CON (GSE106180). 39 C, Gene set enrichment of co‐regulated DEGs, ranked by P‐value. D, Relative expression of co‐regulated gene expression among all 4 comparisons. *Differential gene expression was assessed via RNA sequencing analysis (human) or Agilent 4×40k array (mouse) and was considered significant at the P<0.05 level and >1.5‐fold change. A‐Fib indicates atrial fibrillation; ACE‐Inh, angiotensin‐converting enzyme inhibitor; CAD, coronary artery disease; CON, Control; E.F., ejection fraction; HbA1C, hemoglobin A1C; HF, heart failure; HFD, high‐fat diet; ICD, implantable cardioverter defibrillator; mG4H, transgenic inducible cardiac‐restricted expression of glucose transporter 4; ND, nondiabetic; STZ, streptozotocin; VEH, Vehicle; VT, ventricular tachycardia.
Discussion
Cardiovascular disease is the major cause of morbidity and mortality in diabetic patients. It has been established that diabetic cardiomyopathy is a distinct phenomenon independent of coronary artery disease and hypertension. 1 Recently published findings collectively demonstrate that the failing heart preferentially switches to KB utilization in the absence of diabetes mellitus. 12 , 13 In contrast, in the present study we show a suppression of genes encoding proteins involved in KB utilization in the diabetic heart. Using a combination of gene expression, protein, and metabolomic analyses, we found that (1) diabetes mellitus and insulin resistance (T1D and HFD) are associated with changes in the expression of genes involved in myocardial KB metabolism; (2) increased myocardial glucose delivery alone is sufficient to attenuate KB utilization, and the hexosamine biosynthetic pathway could represent an accessory glucose metabolic pathway involved in this effect; (3) T2D is associated with transcriptional suppression of BDH1 and OXCT1 in patients with end‐stage HF compared with nondiabetic patients with HF; and (4) transcriptional suppression of cardiac Bdh1 was consistently observed in all 4 mouse models (T1D, HFD, mG4H, and dnOGAh), suggesting that Bdh1 is the predominant KB metabolic pathway gene that is transcriptionally regulated by glucose. While expression of ketolytic enzymes was suppressed, we did not measure KB utilization in T1D or HFD hearts. However, a recent study showed that the rate of myocardial KB oxidation in db/db mice is markedly reduced compared with C57BL/6J mice, 40 suggesting that the intrinsic ability to utilize KB may be suppressed in diabetic heart despite increased circulating ketones. Thus, in contrast to nondiabetic HF, the systemic changes in metabolism associated with diabetes mellitus induce an overall suppression of ketolytic pathways.
Our metabolite analysis showed a significant increase in the myocardial content of both β‐hydroxybutyrate and acetoacetate in T1D mice, indicating possible mismatch between available KBs and their utilization in T1D mice. However, consistent with recent findings, 31 , 32 we noted an interesting observation that both mRNA and protein levels of cardiac Hmgcs2 were induced in our T1D mice (Figure 1K and 1L). Hmgcs2 encodes a mitochondrial protein that controls KB synthesis, by which acetoacetate and β‐hydroxybutyrate are generated. It is well established that KBs are synthesized primarily in the liver via the hydroxymethylglutaryl‐CoA pathway and transported through blood to peripheral tissues where they are readily oxidized. Therefore, elevated cardiac β‐hydroxybutyrate levels in T1D mice raise the possibility that myocardial KB synthesis and/or uptake is activated in the cardiac myocyte. Our qPCR analysis did not show changes in expression of genes encoding KB transporters, ruling out the possibility of increased ketone transport levels as the mechanism of myocardial β‐hydroxybutyrate elevation in T1D mice. Several possibilities arise from our studies in the context of induction of cardiac Hmgcs2 expression and β‐hydroxybutyrate in T1D mice. First, as the metabolic phenotype of a diabetic myocardium is primarily characterized by increased fatty acid oxidation, it may be possible that as a compensatory response, the heart reprograms its metabolic machinery to flux excess fatty acid–derived acetyl Co‐A to ketogenic pathways by inducing Hmgcs2 expression. Previously, in work from another group, Wentz et al. reported similar cardiac Hmgcs2 induction (mRNA and protein) in response to 8 weeks of ketogenic diet. However, HMGCS2 catalytic activity in isolated mitochondria and isolated working heart perfusions showed no change in de novo synthesis of ketone bodies. 27 Indeed, we observed that myocardial HMGCS2 induction was associated with a nonsignificant but modest trend toward increased HMGCS2 activity in T1D mice (Figure 1M). Therefore, it is possible that cardiac Hmgcs2 induction in our T1D mice might represent a response of the diabetic myocardium to acetyl CoA spillover from the citric acid cycle. Second, increased peroxisome proliferator‐activated receptor signaling has been suggested to be an important regulator of fatty acid metabolism 41 and that cardiac Hmgcs2 has been shown to be regulated by peroxisome proliferator‐activated receptor‐α. 27 , 42 Therefore, it would be reasonable to conclude that Hmgcs2 induction in T1D results from peroxisome proliferator‐activated receptor‐α‐mediated induction. Nevertheless, whether cardiac Hmgcs2 induction in T1D mice is sufficient to produce measurable levels of de novo ketones that can impact cardiac metabolism needs further investigation. Cardiac Hmgcs2 expression was unchanged in mG4H mice, suggesting that diabetes mellitus–induced expression of Hmgcs2 is independent of increased glucose availability. Collectively, our Hmgcs2 expression data with other published findings suggest that diabetes mellitus–induced perturbations in systemic metabolism, which are potentially independent of glycemia, can activate the expression of ketogenic enzymes in the heart.
Diabetes mellitus is a progressive disease that develops over time, and is associated with an array of metabolic abnormalities including hyperglycemia, hyperlipidemia, and impaired insulin signaling. Data presented here strongly suggest that metabolic abnormalities associated with diabetes mellitus and insulin resistance significantly alter the transcriptional signature of KB metabolic enzymes in both rodents and humans. However, to our knowledge, mechanisms that may regulate myocardial ketone metabolic gene expression have not been shown before. Hyperglycemia alone has been shown to contribute to impaired cardiac performance, which may be reversed by restoring euglycemic conditions. 43 Our isolated working heart results in mG4H mice strongly suggest that high cardiac glucose availability via GLUT4 transgene induction diminishes KB utilization. This is consistent with a previous finding showing reduction of KB oxidation in a transgenic mouse model that constitutively overexpressed insulin‐independent GLUT1 in the heart. 5 In fact, this previous study also found that Oxct1 expression is suppressed in the context of GLUT1 overexpression, 5 similar to what we report here on GLUT4 overexpression. Cumulatively, the prior GLUT1 transgenic mouse results are consistent with our new findings from GLUT4 transgenic mice, both of which suggest that glucose alone is sufficient to suppress myocardial ketolysis.
Protein O‐GlcNAcylation has been recognized as a key glucose‐induced posttranslational modification directly regulating the activity of several molecular and cellular pathways in the diabetic heart by multiple mechanisms. 44 Given the simple association of hyperglycemia with diabetes mellitus and elevated O‐GlcNAcylation, we reasoned that the reduced ketolytic capacity in the mG4H mice could be mediated via increased cardiac protein O‐GlcNAcylation. In this study, we show for the first time that increased cardiac protein O‐GlcNAcylation induced similar suppression of cardiac Bdh1 mRNA levels in dnOGAh mice as observed in mouse models of diabetes mellitus or GLUT4 overexpression, suggesting that high glucose availability may inhibit KB oxidation via increased O‐GlcNAcylation. However, protein abundance of BDH1 was unchanged at this early time point. While these findings from 2 independent mouse models (mG4H and dnOGAh) indicate enhanced protein, O‐GlcNAcylation may be a potential mechanistic link connecting glucose and KB metabolism. We now provide preliminary data that in the dnOGAh mouse heart, elevated protein O‐GlcNAcylation occurs specifically on SCOT (Figure 6L) but was not detected on BDH1. However, additional mechanistic work is required to define the direct mechanisms linking glucose‐induced alterations in cellular proteins with the regulation of KB metabolism.
Limitations of the Study
From multiple mouse models of diabetes mellitus and insulin resistance, combined with mouse models of increased cardiac glucose delivery or downstream glucose signaling, our findings collectively illustrate that glucose and its related metabolic pathways suppress ketolytic genes, and that glucose alone is sufficient to modulate KB metabolism. Nevertheless, our study has several limitations. First, in the T1D model we found elevated myocardial β‐hydroxybutyrate levels together with suppression of ketolytic genes (Bdh1 and Oxct1), but HMGCS2 protein abundance was also induced under the same experimental condition. Though the heart is a net consumer of KBs and is not known to be a ketone‐synthesizing tissue, additional studies are required to ascertain the role of myocardial ketogenesis in diabetic heart. Second, our human heart analysis did not include nondiabetic nonfailing patients. Inclusion of this group in future studies will help to clarify the extent of the transcriptional changes in diabetic failing hearts relative to a healthy normoglycemic group. Also, we did not measure cardiac function in our HFD mice. Finally, while data from the mG4H mouse model clearly revealed that high glucose availability can independently attenuate ketone oxidation, direct modulation of glucose availability in the diabetic heart would provide a better understanding of this link in diabetes mellitus.
Conclusions and Perspectives
In summary, our results from 4 independent mouse models of altered substrate metabolism as well as T2D patients with HF support a hypothesis that the diabetic heart undergoes gene expression and catalytic regulatory reprogramming to decrease the intrinsic capacity for oxidation of ketone bodies. However, we do not know whether reduced KB catabolic gene expression is cause or consequence of diabetes mellitus, and whether increasing KB utilization in the diabetic heart would also enhance ATP production. The recent (Empagliflozin) EMPA‐REG OUTCOME (Cardiovascular Outcome Event Trial in Type 2 Diabetes Mellitus Patients) trial using an inhibitor of sodium–glucose cotransporter 2 (SGLT2i) in patients with T2D and high cardiovascular risk has shown reduced cardiovascular mortality. 45 Furthermore, inhibitor of sodium–glucose cotransporter 2 administration elevated circulating KB levels in these patients, and thus it has been proposed that increased cardiac ketone oxidation contributed to the beneficial effects in this trial. 46 , 47 Also, a recent study showed that T2D patients with diabetic ketosis had reduced all‐cause mortality compared with those with no ketosis, suggesting that ketosis may potentially be protective in diabetes mellitus. 48 Thus, our results could be consistent with the hypothesis that downregulation of myocardial ketolytic enzymes could contribute to the progression of diabetic cardiomyopathy. However, it is important to consider several aspects that regulate cardiac energy metabolism in the healthy and diseased heart in order to determine whether changes in the transcriptional signature of ketone metabolic genes in diabetic heart are adaptive or maladaptive. In particular, a unique aspect of cardiac metabolism in diabetes mellitus is that despite being exposed to excess substrate supply (fatty acids, glucose, and/or ketones), the heart is energy‐deprived and progressively fails over time. First, prevailing evidence suggests that glucose and fatty acids compete for contribution to the tricarboxylic acid cycle and subsequent mitochondrial oxidative energy production. On the other hand, both in vitro and in vivo studies have shown that ketones compete with fatty acids, and inhibit their oxidation, 49 , 50 which might be responsible for triglyceride accumulation in the diabetic heart. Hence, it may be detrimental for the diabetic heart to have increased ketone oxidation. Second, it has been demonstrated that chronic exposure to β‐hydroxybutyrate induces insulin resistance in isolated cardiomyocytes and both glucose uptake and oxidation are suppressed by β‐hydroxybutyrate in the failing heart. 51 , 52 , 53 Therefore, it could be expected that in diabetes mellitus, increased KB oxidation would compete for tricarboxylic acid cycle acetyl CoA, resulting in an impairment of fatty acid or carbohydrate utilization. Thus, we speculate that the shift toward less KB utilization in the diabetic heart might be an early adaptive response to maintain contractile function through substrate competition. However, it is possible that over the longer term, low rates of ketone utilization may lead to maladaptive consequences, which warrants future investigation. It is noted that there is growing evidence that the abundance of fatty acids present in the diabetic condition can itself regulate ketone metabolism regulation, 7 thus highlighting the further need for defining changes related to control via glucose or fatty acids. Furthermore, human studies that examined temporary enhancement of KB levels via infusion found that the inverse may also be true. 54 Specifically, the authors of that study conclude that increasing KBs specifically displaces myocardial glucose uptake without affecting palmitate uptake, thus supporting a role for ketone metabolism as an important fuel with therapeutic potential. This combined with the decreases in ketone body metabolic genes and pathways that we observe by either glucose alone or diabetes mellitus highlight that timing of metabolite delivery may play a crucial role in the functional outcomes. Diabetic cardiomyopathy develops slowly over time, and thus, there is a need to replicate the long‐term course of the disease in animals to characterize the changes in KB metabolism that take place in the myocardium in relation to disease duration.
Sources of Funding
The project has been funded by National Institutes of Health (NIH) grants R00 HL111322 and R01 HL133011 to A.R.W., F30 HL137240 to M.E.P., an American Heart Association (AHA) Association‐wide Postdoctoral Fellowship 17POST33630169 to M.K.B, and NIH grants R01 DK092065, R01 HL108379, and U01 HL087947 to E.D.A., who is an Established Investigator of the AHA.
Disclosures
None.
(J Am Heart Assoc. 2020;9:e013039 DOI: 10.1161/JAHA.120.013039.)
This manuscript was sent to Hossein Ardehali, MD, PhD, Guest Editor, for review by expert referees, editorial decision, and final disposition.
For Sources of Funding and Disclosures, see page 16.
References
- 1. Jia G, Hill MA, Sowers JR. Diabetic cardiomyopathy: an update of mechanisms contributing to this clinical entity. Circ Res. 2018;122:624–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bayeva M, Sawicki KT, Ardehali H. Taking diabetes to heart—deregulation of myocardial lipid metabolism in diabetic cardiomyopathy. J Am Heart Assoc. 2013;2:e000433–e000449. DOI: 10.1161/JAHA.113.000433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ungar I, Gilbert M, Siegel A, Blain JM, Bing RJ. Studies on myocardial metabolism: Iv. Myocardial metabolism in diabetes. Am J Med. 1955;18:385–396. [DOI] [PubMed] [Google Scholar]
- 4. Park S, Jeon J‐H, Min B‐K, Ha C‐M, Thoudam T, Park B‐Y, Lee I‐K. Role of the pyruvate dehydrogenase complex in metabolic remodeling: differential pyruvate dehydrogenase complex functions in metabolism. Diabetes Metab J. 2018;42:270–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yan J, Young ME, Cui L, Lopaschuk GD, Liao R, Tian R. Increased glucose uptake and oxidation in mouse hearts prevent high fatty acid oxidation but cause cardiac dysfunction in diet‐induced obesity. Circulation. 2009;119:2818–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stowe KA, Burgess SC, Merritt M, Sherry AD, Malloy CR. Storage and oxidation of long‐chain fatty acids in the c57/bl6 mouse heart as measured by nmr spectroscopy. FEBS Lett. 2006;580:4282–4287. [DOI] [PubMed] [Google Scholar]
- 7. Puchalska P, Crawford PA. Multi‐dimensional roles of ketone bodies in fuel metabolism, signaling, and therapeutics. Cell Metab. 2017;25:262–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Valayannopoulos V, Bajolle F, Arnoux JB, Dubois S, Sannier N, Baussan C, Petit F, Labrune P, Rabier D, Ottolenghi C, et al. Successful treatment of severe cardiomyopathy in glycogen storage disease type iii with d, l‐3‐hydroxybutyrate, ketogenic and high‐protein diet. Pediatr Res. 2011;70:638–641. [DOI] [PubMed] [Google Scholar]
- 9. Liu J, Wang P, Zou L, Qu J, Litovsky S, Umeda P, Zhou L, Chatham J, Marsh SA, Dell'Italia LJ, et al. High‐fat, low‐carbohydrate diet promotes arrhythmic death and increases myocardial ischemia‐reperfusion injury in rats. Am J Physiol Heart Circ Physiol. 2014;307:H598–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Krebs P, Fan W, Chen YH, Tobita K, Downes MR, Wood MR, Sun L, Li X, Xia Y, Ding N, et al. Lethal mitochondrial cardiomyopathy in a hypomorphic med30 mouse mutant is ameliorated by ketogenic diet. Proc Natl Acad Sci USA. 2011;108:19678–19682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Schugar RC, Moll AR, Andre d'Avignon D, Weinheimer CJ, Kovacs A, Crawford PA. Cardiomyocyte‐specific deficiency of ketone body metabolism promotes accelerated pathological remodeling. Mol. Metab. 2014;3:754–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Aubert G, Martin OJ, Horton JL, Lai L, Vega RB, Leone TC, Koves T, Gardell SJ, Kruger M, Hoppel CL, et al. The failing heart relies on ketone bodies as a fuel. Circulation. 2016;133:698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bedi KC Jr, Snyder NW, Brandimarto J, Aziz M, Mesaros C, Worth A, Wang L, Javaheri A, Blair IA, Margulies K, et al. Evidence for intramyocardial disruption of lipid metabolism and increased myocardial ketone utilization in advanced human heart failure. Circulation. 2016;133:706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhu Y, Pereira RO, O'Neill BT, Riehle C, Ilkun O, Wende AR, Rawlings TA, Zhang YC, Zhang Q, Klip A, et al. Cardiac pi3k‐akt impairs insulin‐stimulated glucose uptake independent of mtorc1 and glut4 translocation. Mol. Endocrinol. 2013;27:172–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Wende AR, Schell JC, Ha CM, Pepin ME, Khalimonchuk O, Schwertz H, Pereira RO, Brahma MK, Tuinei J, Contreras‐Ferrat A, et al. Maintaining myocardial glucose utilization in diabetic cardiomyopathy accelerates mitochondrial dysfunction. Diabetes. 2020; May 4 [epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Toleman C, Paterson AJ, Whisenhunt TR, Kudlow JE. Characterization of the histone acetyltransferase (hat) domain of a bifunctional protein with activable o‐glcnacase and hat activities. J Biol Chem. 2004;279:53665–53673. [DOI] [PubMed] [Google Scholar]
- 17. Huang P, Ho SR, Wang K, Roessler BC, Zhang F, Hu Y, Bowe DB, Kudlow JE, Paterson AJ. Muscle‐specific overexpression of ncoatgk, splice variant of o‐glcnacase, induces skeletal muscle atrophy. Am J Physiol Cell Physiol. 2011;300:C456–C465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pepin ME, Padgett LE, McDowell RE, Burg AR, Brahma MK, Holleman C, Kim T, Crossman D, Kutsch O, Tse HM, et al. Antiretroviral therapy potentiates high‐fat diet induced obesity and glucose intolerance. Mol Metab. 2018;12:48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pepin ME, Ha CM, Crossman DK, Litovsky SH, Varambally S, Barchue JP, Pamboukian SV, Diakos NA, Drakos SG, Pogwizd SM, et al. Genome‐wide DNA methylation encodes cardiac transcriptional reprogramming in human ischemic heart failure. Lab Invest. 2019;99:371–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wende AR, Kim J, Holland WL, Wayment BE, O'Neill BT, Tuinei J, Brahma MK, Pepin ME, McCrory MA, Luptak I, et al. Glucose transporter 4‐deficient hearts develop maladaptive hypertrophy in response to physiological or pathological stresses. Am J Physiol Heart Circ Physiol. 2017;313:H1098–H1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wende AR, O'Neill BT, Bugger H, Riehle C, Tuinei J, Buchanan J, Tsushima K, Wang L, Caro P, Guo A, et al. Enhanced cardiac akt/protein kinase b signaling contributes to pathological cardiac hypertrophy in part by impairing mitochondrial function via transcriptional repression of mitochondrion‐targeted nuclear genes. Mol Cell Biol. 2015;35:831–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhang Z, Parker MP, Graw S, Novikova LV, Fedosyuk H, Fontes JD, Koestler DC, Peterson KR, Slawson C. O‐glcnac homeostasis contributes to cell fate decisions during hematopoiesis. J Biol Chem. 2019;294:1363–1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pereira RO, Wende AR, Olsen C, Soto J, Rawlings T, Zhu Y, Anderson SM, Abel ED. Inducible overexpression of glut1 prevents mitochondrial dysfunction and attenuates structural remodeling in pressure overload but does not prevent left ventricular dysfunction. J Am Heart Assoc. 2013;2:e000301–e000317. DOI: 10.1161/JAHA.113.000301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Grinblat L, Pacheco Bolaños LF, Stoppani AO. Decreased rate of ketone‐body oxidation and decreased activity of d‐3‐hydroxybutyrate dehydrogenase and succinyl‐coa:3‐oxo‐acid coa‐transferase in heart mitochondria of diabetic rats. Biochem J. 1986;240:49–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Young ME, Brewer RA, Peliciari‐Garcia RA, Collins HE, He L, Birky TL, Peden BW, Thompson EG, Ammons BJ, Bray MS, et al. Cardiomyocyte‐specific bmal1 plays critical roles in metabolism, signaling, and maintenance of contractile function of the heart. J. Biol. Rhythms. 2014;29:257–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cotter DG, Schugar RC, Wentz AE, André d'Avignon D, Crawford PA. Successful adaptation to ketosis by mice with tissue‐specific deficiency of ketone body oxidation. Am J Physiol Endocrinology and Metabolism. 2012;304:E363–E374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Wentz AE, d'Avignon DA, Weber ML, Cotter DG, Doherty JM, Kerns R, Nagarajan R, Reddy N, Sambandam N, Crawford PA. Adaptation of myocardial substrate metabolism to a ketogenic nutrient environment. J Biol Chem. 2010;285:24447–24456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Durgan DJ, Pat BM, Laczy B, Bradley JA, Tsai JY, Grenett MH, Ratcliffe WF, Brewer RA, Nagendran J, Villegas‐Montoya C, et al. O‐glcnacylation, novel post‐translational modification linking myocardial metabolism and cardiomyocyte circadian clock. J Biol Chem. 2011;286:44606–44619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bugger H, Riehle C, Jaishy B, Wende AR, Tuinei J, Chen D, Soto J, Pires KM, Boudina S, Theobald HA, et al. Genetic loss of insulin receptors worsens cardiac efficiency in diabetes. J Mol Cell Cardiol. 2012;52:1019–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Turko IV, Marcondes S, Murad F. Diabetes‐associated nitration of tyrosine and inactivation of succinyl‐coa:3‐oxoacid coa‐transferase. Am J Physiol Heart Circ Physiol. 2001;281:H2289–H2294. [DOI] [PubMed] [Google Scholar]
- 31. Shukla SK, Liu W, Sikder K, Addya S, Sarkar A, Wei Y, Rafiq K. Hmgcs2 is a key ketogenic enzyme potentially involved in type 1 diabetes with high cardiovascular risk. Sci Rep. 2017;7:4590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Cook GA, Lavrentyev EN, Pham K, Park EA. Streptozotocin diabetes increases mrna expression of ketogenic enzymes in the rat heart. Biochim Biophys Acta. 2017;1861:307–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Halestrap AP, Wilson MC. The monocarboxylate transporter family: role and regulation. IUBMB Life. 2012;64:109–119. [DOI] [PubMed] [Google Scholar]
- 34. Kenchaiah S, Evans JC, Levy D, Wilson PWF, Benjamin EJ, Larson MG, Kannel WB, Vasan RS. Obesity and the risk of heart failure. N Engl J Med. 2002;347:305–313. [DOI] [PubMed] [Google Scholar]
- 35. Roberts NW, González‐Vega M, Berhanu TK, Mull A, García J, Heydemann A. Successful metabolic adaptations leading to the prevention of high fat diet‐induced murine cardiac remodeling. Cardiovasc Diabetol. 2015;14:127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Brahma MK, Pepin ME, Wende AR. My sweetheart is broken: role of glucose in diabetic cardiomyopathy. Diabetes Metab J. 2017;41:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Banerjee PS, Ma J, Hart GW. Diabetes‐associated dysregulation of o‐glcnacylation in rat cardiac mitochondria. Proc Natl Acad Sci USA. 2015;112:6050–6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Ramirez‐Correa G, Ma J, Slawson C, Zeidan Q, Lugo‐Fagundo NS, Xu M, Shen X, Gao WD, Caceres V, Chakir K, et al. Removal of abnormal myofilament o‐glcnacylation restores ca2+ sensitivity in diabetic cardiac muscle. Diabetes. 2015;64:3573–3587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Spallotta F, Cencioni C, Atlante S, Garella D, Cocco M, Mori M, Mastrocola R, Kuenne C, Guenther S, Nanni S, et al. Stable oxidative cytosine modifications accumulate in cardiac mesenchymal cells from type 2 diabetes patients. Circ. Res. 2018;122:31. [DOI] [PubMed] [Google Scholar]
- 40. Verma S, Rawat S, Ho KL, Wagg CS, Zhang L, Teoh H, Dyck JE, Uddin GM, Oudit GY, Mayoux E, et al. Empagliflozin increases cardiac energy production in diabetes: novel translational insights into the heart failure benefits of sglt2 inhibitors. JACC Basic Transl Sci. 2018;3:575–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD, et al. The cardiac phenotype induced by pparα overexpression mimics that caused by diabetes mellitus. J Clin Invest. 2002;109:121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rodríguez JC, Gil‐Gómez G, Hegardt FG, Haro D. Peroxisome proliferator‐activated receptor mediates induction of the mitochondrial 3‐hydroxy‐3‐methylglutaryl‐coa synthase gene by fatty acids. J Biol Chem. 1994;269:18767–18772. [PubMed] [Google Scholar]
- 43. Rohm M, Savic D, Ball V, Curtis MK, Bonham S, Fischer R, Legrave N, MacRae JI, Tyler DJ, Ashcroft FM. Cardiac dysfunction and metabolic inflexibility in a mouse model of diabetes without dyslipidemia. Diabetes. 2018;67:1057–1067. [DOI] [PubMed] [Google Scholar]
- 44. Wende AR. Post‐translational modifications of the cardiac proteome in diabetes and heart failure. Proteomics Clin Appl. 2016;10:25–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ, et al. Empagliflozin, cardiovascular outcomes, and mortality in type 2 diabetes. N. Engl. J. Med. 2015;373:2117–2128. [DOI] [PubMed] [Google Scholar]
- 46. Ferrannini E, Mark M, Mayoux E. Cv protection in the empa‐reg outcome trial: A “thrifty substrate” hypothesis. Diabetes Care. 2016;39:1108. [DOI] [PubMed] [Google Scholar]
- 47. Henry RR, Rosenstock J, Edelman S, Mudaliar S, Chalamandaris A‐G, Kasichayanula S, Bogle A, Iqbal N, List J, Griffen SC. Exploring the potential of the sglt2 inhibitor dapagliflozin in type 1 diabetes: A randomized, double‐blind, placebo‐controlled pilot study. Diabetes Care. 2014;38:412–419. [DOI] [PubMed] [Google Scholar]
- 48. Kruljac I, Ćaćić M, Ćaćić P, Ostojić V, Štefanović M, Šikić A, Vrkljan M. Diabetic ketosis during hyperglycemic crisis is associated with decreased all‐cause mortality in patients with type 2 diabetes mellitus. Endocrine. 2017;55:139–143. [DOI] [PubMed] [Google Scholar]
- 49. Hasselbaink DM, Glatz JFC, Luiken JJFP, Roemen THM, Van der Vusse GJ. Ketone bodies disturb fatty acid handling in isolated cardiomyocytes derived from control and diabetic rats. Biochem J. 2003;371:753–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stanley WC, Meadows SR, Kivilo KM, Roth BA, Lopaschuk GD. Β‐hydroxybutyrate inhibits myocardial fatty acid oxidation in vivo independent of changes in malonyl‐coa content. Am J Physiol Heart Circ Physiol. 2003;285:H1626–H1631. [DOI] [PubMed] [Google Scholar]
- 51. Pelletier A, Tardif A, Gingras MH, Chiasson JL, Coderre L. Chronic exposure to ketone bodies impairs glucose uptake in adult cardiomyocytes in response to insulin but not vanadate: the role of pi3‐k. Mol Cell Biochem. 2007;296:97–108. [DOI] [PubMed] [Google Scholar]
- 52. Tardif A, Julien N, Pelletier A, Thibault G, Srivastava AK, Chiasson J‐L, Coderre L. Chronic exposure to β‐hydroxybutyrate impairs insulin action in primary cultures of adult cardiomyocytes. Am J Physiol Endocrinol Metab. 2001;281:E1205–E1212. [DOI] [PubMed] [Google Scholar]
- 53. Horton JL, Davidson MT, Kurishima C, Vega RB, Powers JC, Matsuura TR, Petucci C, Lewandowski ED, Crawford PA, Muoio DM, et al. The failing heart utilizes 3‐hydroxybutyrate as a metabolic stress defense. JCI Insight. 2019;4:e124079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Gormsen LC, Svart M, Thomsen HH, Sondergaard E, Vendelbo MH, Christensen N, Tolbod LP, Harms HJ, Nielsen R, Wiggers H, et al. Ketone body infusion with 3‐hydroxybutyrate reduces myocardial glucose uptake and increases blood flow in humans: A positron emission tomography study. J Am Heart Assoc. 2017;6:e005066–e005076. DOI: 10.1161/JAHA.116.005066. [DOI] [PMC free article] [PubMed] [Google Scholar]