

Abstract
Background
Activated vascular cells produce submicron prothrombotic and proinflammatory microparticle vesicles. Atherosclerotic plaques contain high levels of microparticles. Plasma microparticle levels increase during acute coronary syndromes and the thrombotic consequences of plaque rupture likely involve macrophage‐derived microparticles (MΦMPs). The activation pathways that promote MΦMP production remain poorly defined. This study tested the hypothesis that signals implicated in atherogenesis also stimulate MΦMP production.
Methods and Results
We stimulated human primary MΦs with proinflammatory cytokines and atherogenic lipids, and measured MΦMP production by flow cytometry. Oxidized low‐density lipoprotein (oxLDL; 25 µg/mL) induced MΦMP production in a concentration‐dependent manner (293% increase; P<0.001), and these oxLDL MΦMP stimulatory effects were mediated by CD36. OxLDL stimulation increased MΦMP tissue factor content by 78% (P<0.05), and oxLDL‐induced MΦMP production correlated with activation of caspase 3/7 signaling pathways. Salvionolic acid B, a CD36 inhibitor and a CD36 inhibitor antibody reduced oxLDL‐induced MΦMP by 67% and 60%, respectively. Caspase 3/7 inhibition reduced MΦMP release by 52% (P<0.01) and caspase 3/7 activation increased MΦMP production by 208% (P<0.01). Mevastatin pretreatment (10 µM) decreased oxLDL‐induced caspase 3/7 activation and attenuated oxLDL‐stimulated MΦMP production and tissue factor content by 60% (P<0.01) and 43% (P<0.05), respectively.
Conclusions
OxLDL induces the production of prothrombotic microparticles in macrophages. This process depends on caspases 3 and 7 and CD36 and is inhibited by mevastatin pretreatment. These findings link atherogenic signaling pathways, inflammation, and plaque thrombogenicity and identify a novel potential mechanism for antithrombotic effects of statins independent of LDL lowering.
Keywords: caspase, lipids, microparticles, oxidized, signal transduction, tissue factor
Subject Categories: Inflammation, Atherosclerosis, Thrombosis
Nonstandard Abbreviations and Acronyms
- LDL
low‐density lipoprotein
- MΦMP
macrophage‐derived microparticle
- oxLDL
oxidized low‐density lipoprotein
- SMC
smooth muscle cell
- TF
tissue factor
- zDEVD‐FMK
N‐[(phenylmethoxy)carbonyl]‐L‐α‐aspartyl‐L‐α‐glutamyl‐N‐[(1S)‐3‐fluoro‐1‐(2‐methoxy‐2‐oxoethyl)‐2‐oxopropyl]‐L‐valinamide, 1,2‐dimethyl ester
Clinical Perspective
What Is New?
Low‐density lipoprotein, in relevant in vivo concentrations and copper oxidation, promotes microparticle formation in primary macrophages.
Microparticles include phosphatidylserine and tissue factor on their surface and induce thrombin generation.
Microparticle generation is CD36 receptor and caspase 3/7 dependent, is attenuated by statins via the rho/rho‐associated protein kinase pathway, but do not activate apoptotic pathways as shown by the absence of change in the ratio of B‐cell lymphoma 2–associated X protein to B‐cell lymphoma 2 expression.
What Are the Clinical Implications?
Prothrombotic microparticle generation, relevant in atherosclerotic plaque, which contains both oxidized low‐density lipoprotein and macrophages, may play a role in acute coronary syndrome upon plaque rupture or erosion.
Reduction of microparticle formation could be an additional pleiotropic effect of statin therapy related to reducing atherosclerotic complications.
Early in atherogenesis, blood monocytes enter the arterial intima. 1 Within the nascent plaque, monocytes mature into macrophages and can proliferate in the intima. Plaque macrophages internalize oxidized lipids through scavenger receptors and differentiate into foam cells. 2 As the atheroma progresses, a lipid‐rich core develops that contains thrombogenic material, separated from arterial blood by an extracellular matrix that comprises the fibrous cap and the overlying endothelial monolayer. 3 Atherothrombotic complications of atherosclerosis commonly occur when plaque inflammation leads to thinning and fissure of the fibrous cap, known as plaque rupture. 4 This type of plaque disruption exposes the atheroma’s thrombogenic core to the blood and can trigger clot formation and tissue ischemia. 5 Even when plaque rupture does not cause occlusive thrombi, mural fibrin and platelet deposition can drive plaque progression and narrowing of the blood vessel lumen. 6 , 7
Inflammatory and thrombotic signaling pathways intertwine in the pathobiology of atherosclerosis because inflammatory processes stimulate thrombosis, and thrombosis in turn promotes inflammation through bidirectional amplification loops that perpetuate atherogenic and atherothrombotic responses. 8 Inflammatory cytokines and atherogenic lipids directly regulate plaque thrombogenicity by enhancing macrophage expression of procoagulant tissue factor (TF). 9 Within the plaque microenvironment, macrophage lipid uptake associates with increased plaque TF expression and enhanced arterial thrombosis. 10 Lipid overload also inhibits macrophage clearance of apoptotic cells (efferocytosis) and thereby accelerates necrotic core development. 11
In addition to TF, advanced atherosclerotic plaques contain prothrombotic submicron vesicles known as microparticles that carry TF on their surface. 12 Activated vascular cells produce microparticles, and microparticle vesicles carry surface proteins and carry regulatory molecules derived from their source cells. Microparticles function as signaling intermediates that regulate inflammation and thrombosis. 13 Plaque‐associated macrophage‐derived microparticles (MΦMPs) can prove particularly thrombogenic, because they contain TF and phosphatidylserine‐rich plasma membranes. Phosphatidylserine‐containing cell and vesicle membranes promote thrombosis by supporting the assembly and activation of coagulation complexes that generate thrombin. 13 Multiple reports show an association between microparticle levels, severity of cardiovascular disease, and risk of cardiovascular events, 14 , 15 , 16 , 17 and patients with acute coronary syndromes have elevated levels of endothelial‐, platelet‐, and monocyte‐derived microparticles compared with healthy controls. 15 , 18
Stimuli for microparticle production seem to vary by vascular cell type and pathophysiological context. Calcium, phosphate, and acetylated low‐density lipoprotein (LDL) promote smooth‐muscle cell (SMC) microparticle generation. 19 , 20 Tobacco smoke extract 21 and unesterified cholesterol 22 induce monocyte microparticle release, and a previous investigation demonstrated that oxidized low‐density lipoprotein (oxLDL) stimulates immortalized human acute monocytic leukemia cell line monocytic cell microparticle production without affecting cell viability. 23 Notably, several reports have demonstrated that 3‐hydroxy‐3‐methyl‐glutaryl–coenzyme A reductase inhibitors (statins) reduce monocyte and macrophage TF production 23 , 24 , 25 , 26 and statins inhibit activation of rho/rho‐associated protein kinase pathways that regulate endothelial cell microparticle generation. 27
Despite the importance of MΦMPs in mechanisms that underlie thrombotic complications of plaque rupture, the signaling pathways and cellular responses that promote MΦMP production remain poorly defined. This study tested the hypothesis that signals implicated in atherogenesis stimulate MΦMP production and demonstrated that oxLDL potently stimulates MΦMP release. The results implicate caspase 3/7 activation in MΦMP generation and demonstrate that statin treatment limits oxLDL‐induced macrophage caspase activation and caspase‐dependent MΦMP production.
Methods
All supporting materials needed to reproduce or replicate our procedures have been provided in this article and are commercially available except for the inhibitory TF antibody TF8‐5G9. This antibody was generously provided by Dr. James Morrissey, and inquiries for this antibody should be addressed to Dr. James Morrissey, University of Michigan Medical School. Data are available on request from the authors.
Reagents
Reagents include the following: tumor necrosis factor‐α, and monocyte chemotactic protein 1 (PeproTech, Rocky Hill, NJ); human interferon‐γ, and interleukin‐1 beta (R&D Systems, Minneapolis, MN); 2‐(5‐oxovaleryl)phosphatidylcholine (POV‐PC) and 1‐palmitoyl‐2‐glutaryl phosphatidylcholine (Cayman Chemicals, Ann Arbor, MI); Phe‐Pro‐Arg‐chloromethylketone, Glu‐Gly‐Arg‐chloromethylketone coagulation protease inhibitors and Lactadherin (BLAC 1200; Haematologic Technologies Inc., Essex Junction, VT); lipopolysaccharide, mevastatin, salvianolic acid B, and polymyxin B (Sigma‐Aldrich, St. Louis, MO); Ly294002 (Cell Signaling Technology, Inc., Danvers, MA); N‐[(phenylmethoxy)carbonyl]‐L‐α‐aspartyl‐L‐α‐glutamyl‐N‐[(1S)‐3‐fluoro‐1‐(2‐methoxy‐2‐oxoethyl)‐2‐oxopropyl]‐L‐valinamide, 1,2‐dimethyl ester (zDEVD; BD Biosciences, San Jose, CA); human native LDL (0.03–0.1 nmol/L malondialdehyde/mg protein) and oxLDL (endotoxin tested, <0.5 EU/mg) (Biomedical Technologies Inc., Stoughton, MA); and CD36 monoclonal functional antibody, FA6‐152 (HM2122‐FS; Hycult Biotech, Plymouth Meeting, PA). The LDL preparations used in this study were purified to homogeneity via ultra‐centrifugation (1.019–1.063 g/cc) before oxidation with 20 µmol/L CuSO4 in phosphate‐buffered saline at 37°C for 2 hours (thiobarbituric acid reactive substances: 6‐17 nmol/L malondialdehyde/mg), 4 hours (11 nmol/L malondialdehyde/mg), or 24 hours (29–31 nmol/L malondialdehyde/mg). Oxidation was terminated by adding an excess of 0.3 mmol/L EDTA to chelate the oxidizing copper. Following oxidation, agarose gel electrophoresis demonstrated indiscernible migration compared with native LDL.
Macrophage Culture
Peripheral blood mononuclear cells were isolated from freshly prepared leukocyte concentrates (Research Blood Components, Boston, MA) by density gradient centrifugation employing lymphocyte separation medium (MP Biomedicals, LLC, Solon, OH). To prepare macrophages, we plated the mononuclear cell fraction on Primaria cell culture plates (Becton Dickinson, Franklin Lakes, NJ), as previously described. 28 Adherent monocytes were cultured for 9 days in RPMI 1640 (Lonza, Walkersville, MD) with 1% penicillin/streptomycin (Lonza) and 5% human serum (Gemini BioProducts, West Sacramento, CA) to promote macrophage differentiation. To test the effect of cytokines and lipids under quiescent conditions, we cultured macrophages in low serum (1%) for 18 hours prior to stimulation. During inhibitor experiments, we pretreated cells with mevastatin (10µM) and geranylgeraniol (10µM) for 48 hours, zDEVD (20 µmol/L) for 1 hour. Mevastatin, LY294002, geranylgeraniol and zDEVD were resuspended in dimethyl sulfoxide for a final concentration of 0.16%, 0.05%, 0.012% and 0.1% of dimethyl sulfoxide, respectively. Vehicle treatment of macrophages with 0.2% of dimethyl sulfoxide did not alter microparticle production (not shown). For microparticle analysis, we centrifuged conditioned culture medium at 13 000g for 2 minutes at room temperature to pellet cellular debris, and the microparticle‐containing supernatant was aliquoted and stored at −81°C. Experiments were performed with multiple donors and multiple (2‐4) biological (well) replicates (minimum of n = 3 donors for each experiment).
Microparticle Flow Cytometry Analysis
The microparticle flow cytometry protocol combined 20 µL of macrophage cultured medium, 42.5 µL of filtered (0.22 µm) annexin V binding buffer (1X Tris Buffered Saline with 2.5 mmol/L CaCl2), and 2.5 µL of annexin V‐fluorescein isothiocyanate (BMS306FI, eBioscience, San Diego, CA) to enable quantification of phosphatidylserine‐positive microparticles. Before flow cytometry, annexin V–labeled microparticles were combined with 385 µL of annexin V–binding buffer and 50 µL of fluorescent counting beads, which enabled determination of flow rate and microparticle concentration (Flow‐Count Fluorospheres, Beckman Coulter, Brea, CA). Sample analysis was performed on a FACS Calibur flow cytometer (Becton Dickinson, Franklin Lake, NJ), and we analyzed the flow cytometry data with FCS Express software 3.0 (DeNovo Software, Los Angeles, CA). Microparticles were identified by side‐scatter size compared with sizing beads (fluorescent green silica beads 200 nm, #141114‐10; Corpuscular, Cold Spring, NY, and Megamix, Biocytex 7801, France), and by annexin V binding as described previously. 29 We defined the microparticle gate as annexin V–positive events sized approximately 1 μm or smaller. Annexin V binding to phosphatidylserine‐containing plasma membranes is calcium dependent; thus, samples treated with the calcium‐chelating agent EDTA (20 mmol/L) or lactadherin (28µM) served as a negative control for annexin V gating. The threshold of annexin V–positive microparticle events was set above the 99.99th percentile of the EDTA‐treated negative control sample.
Tissue Factor Assays
To obtain washed microparticles, we centrifuged 250 µL of MP‐containing macrophage cultured medium at 100 000g for 1 hour at 4°C and washed the microparticle pellet twice with 250 µL of TF ELISA assay buffer. Flow cytometry analysis of the supernatant and microparticle pellet confirmed effective centrifugation of >99% of microparticles (data not shown). We used two methods for TF determination. In the first method we measured TF concentration in the washed microparticle fraction with the Imubind TF ELISA kit per manufacturer’s protocol (Sekisui, previously American Diagnostica, Stamford, CT). In the second method, we measured TF activity (Assaysense Human Tissue Factor chromogenic activity kit; Assaypro, St. Charles, MO) per the manufacturer’s protocol. Specificity of TF activity was tested using the inhibitory TF antibody TF8‐5G9 (generously provided by Dr. James Morrissey).
Microparticle Thrombin Generation
Microparticle prothrombotic activity was measured in a microparticle capture assay using published methods. 30 We treated MΦMP samples with the coagulation factor inhibitors Phe‐Pro‐Arg‐chloromethylketone (50 µmol/L) and Glu‐Gly‐Arg‐chloromethylketone (50 µmol/L), and a 50‐µL microparticle aliquot was added to wells of an annexin V–coated 96‐well plate (StreptaWell plate; Roche, San Francisco, CA; biotinylated annexin V, 0.36 ng/µL coating for 30 minutes; Biovision, Milpitas, CA). After 30‐minute incubation and 3 wash steps, we added prothrombin (1.3 µmol/L), factor Va (2.5 nmol/L) and factor Xa (2.5 nmol/L) (Haematologic Technologies, Inc, Essex Junction, VT) in calcium‐containing Tris buffer (25 mmol/L Tris, 2.5 mmol/L calcium) to the microparticle‐containing wells. Following 30‐minute incubation at 37°C, EDTA addition (0.1M) halted the prothrombinase reaction, and we added Chromozym TH chromogenic thrombin substrate (0.57 mmol/L, Roche) to quantify thrombin activity. The assay measured microparticle‐stimulated thrombin production in reference to a standard curve in a multiplate reader (405 nm optic diameter at 1 minute).
Impedance Flow‐Cytometry Analysis of Tissue Factor Microparticles
We measured TF‐positive microparticles with an SC MPL Quanta flow cytometer (Beckman Coulter) using published methods. 31 The Quanta flow cytometer uses impedance to determine particle size, and fluorescence to detect TF. Fluorescent microspheres (0.78‐μm; Bangs Laboratories, Fishers, IN) functioned to calibrate particle size. Before quantification, MP samples were stained with Alexa Fluor 488–labeled monoclonal antibody (clone cH36) against human TF, or with Alexa‐labeled IgG antibody control (I4506, Sigma Aldrich). TF microparticles from human pancreas adenocarcinoma ascites metastasis‐1 pancreatic cancer cells served as a positive control.
Caspase 3/7 Assay
We measured caspase 3/7 activity with a commercial kit, according to the manufacturers’ instructions (Caspase‐Glo 3/7 Assay #G8090, Promega, San Luis Obispo, CA).
Quantitative mRNA Analysis
RNA isolated from cells with a QIAshredder and RNeasy mini kit (QIAGEN, Valencia, CA) was reverse‐transcribed using Superscript First‐Stand Synthesis for real‐time quantitative polymerase chain reaction (Invitrogen, Grand Island, NY). We performed quantitative polymerase chain reaction on a Bio‐RAD MyIQ system using 25‐µL reactions with iQ SYBR Green Supermix (Bio‐RAD, Hercules, CA), and normalized with reference genes as described previously. 32 , 33
Statistical Analysis
We used Prism 4.0 software (Graphpad Software, La Jolla, CA) for all statistical analyses, and P values <0.05 were deemed significant. Microparticle quantification, cytotoxicity percentage, caspase 3/7 activity, and TF ELISA concentrations were analyzed by 1‐way ANOVA and by the Newman‐Keuls multiple comparison test. Data are reported as mean ± SEM, unless noted otherwise.
Results
OxLDL Stimulates Macrophage Prothrombotic Microparticle Production
We measured agonist‐induced MΦMP production by flow cytometry using standard methods, and identified microparticles based on particle size and staining with fluorescent‐labeled annexin V, which binds to phosphatidylserine‐containing plasma membranes (Figure 1). As expected, EDTA or lactadherin (28 µM) treatment abolished calcium‐dependent annexin V microparticle binding (Figure 1 and Figure S1). LDL and oxLDL do not form structures that are recognized as microparticles by fluorescence‐activated cell sorting analysis (Figure S2). Reference sizing beads established that the events measured by fluorescence‐activated cell sorting analysis conform to the definition of microparticles of being smaller than 1 µm (Figure 1 and Figure S3). To determine the effect of atherogenic cytokines and oxLDL on MΦMP production, we stimulated primary human monocyte‐derived macrophages with oxLDL, interferon‐γ, interleukin‐1β, monocyte chemotactic protein 1, tumor necrosis factor‐α, or bacterial lipopolysaccharide. OxLDL — but not interferon‐γ, interleukin‐1β, tumor necrosis factor‐α, monocyte chemotactic protein 1, or lipopolysaccharide — induced MΦMP production in a concentration‐dependent manner, with a peak effect at 16 hours (293% increase at 16 hours, P<0.001)(Figure 2A through 2C). OxLDL induction of MΦMP production was not related to lipopolysaccharide contamination because lipopolysaccharide failed to generate MΦMPs (Figure 2A), and pretreatment with polymyxin B (5 µg/mL for 1 hour) did not attenuate oxLDL‐induced MΦMP generation (not shown).
Figure 1. Flow cytometry quantification of macrophage microparticles.
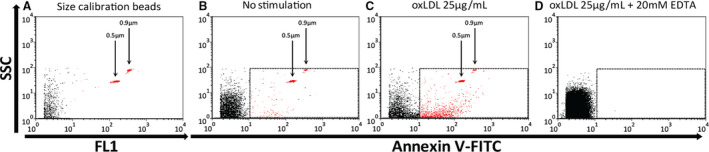
Flow cytometry of fluorescent sizing beads (A through C) demonstrates that the microparticle gate captures microparticle events that are <0.9 µmol/L. Representative flow cytometry of fluorescent sizing beads alone (A), annexin V–stained microparticles from nonstimulated (B), and oxLDL‐stimulated macrophages (C). EDTA treatment abolishes calcium‐dependent annexin V binding to MΦMPs (D). FITC indicates fluorescein isothiocyanate; FL1, fluorescence intensity using 530/30 filter (used for FITC); oxLDL, oxidized low‐density lipoprotein; SSC, side scatter.
Figure 2. Oxidized LDL stimulates macrophage microparticle production.
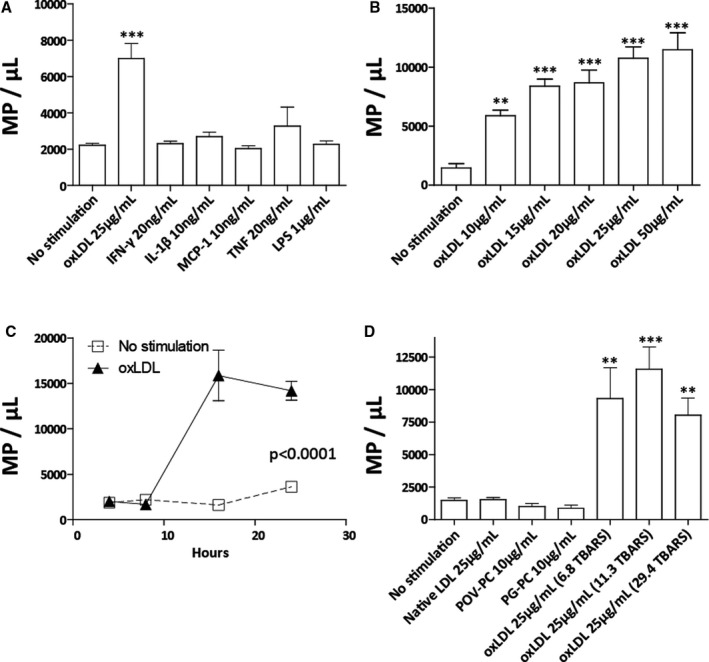
Macrophage stimulation with atherogenic cytokines and oxLDL. OxLDL treatment (30.3 TBARS, 16 hours) led to a 416% increase in macrophage microparticle (MΦMP) production (A). OxLDL‐induced MΦMP production in relation to oxLDL concentration (30.3 TBARS, 16 hours) (B), and hours of stimulation (25 µg/mL, 29.4 TBARS) (C). MΦMP production in response to stimulation with nonoxidized LDL, minimally modified oxLDL components POV‐PC and PG‐PC, or oxLDL with 6.8, 11.3, and 29.4 TBARS oxidation (D). (TBARS in nmol/L of malondialdehyde/mg of LDL protein) (*P<0.05, **P<0.01, and ***P<0.001 vs. no stimulation). IFN‐γ indicates inteferon–γ; Il‐1β, interleukin‐1β; LDL, low‐density lipoprotein; LPS, lipopolysaccharide; MCP‐1, monocyte chemotactic protein 1; oxLDL, oxidized low‐density lipoprotein; PG‐PC, 1‐palmitoyl‐2‐glutaryl phosphatidylcholine; POV‐PC, 2‐(5‐oxovalery) phosphatidylcholine; TBARS, thiobarbituric acid reactive substances; TNF, tumor necrosis factor.
To explore the effect of LDL oxidation on MΦMP production, we stimulated macrophages with nonoxidized LDL, with the minimally modified LDL components 2‐(5‐oxovaleryl) phosphatidylcholine and 1‐palmitoyl‐2‐glutaryl phosphatidylcholine, and with oxLDL that had undergone increasing amounts of copper‐mediated oxidation (6‐31 thiobarbituric acid reactive substances)(Figure 2D). Nonoxidized native LDL and the minimally modified LDL components failed to induce MΦMP production, while oxLDL caused significant production of MΦMPs independent of the degree of oxidation. These experiments demonstrated a requirement for LDL oxidation for LDL‐induced MΦMP production.
OxLDL Increases Macrophage Tissue Factor–Positive Prothrombotic Microparticles
To assess the effect of oxLDL stimulation on MΦMP TF content, microparticles from resting or oxLDL‐stimulated macrophages were ultracentrifuged, washed, and the amount of TF was assessed by ELISA and by impedance flow cytometry. Samples might also contain smaller extracellular vesicles such as exosomes. ELISA quantification demonstrated that oxLDL stimulation increases the production of microparticle‐associated TF by 79.0%, compared with nonstimulated controls (Figure 3A). In addition to standard light‐based flow cytometry, we used impedance flow cytometry as an alternative method to characterize and quantify oxLDL‐induced TF‐positive microparticles. 31 Impedance flow cytometry quantification also demonstrated that oxLDL stimulation caused a 5.6‐fold increase in the production of TF‐positive MΦMPs, which were defined as TF‐positive events <0.78 µm in size (Figure 3B and 3E). In addition to measurement of microparticle‐associated TF antigen by ELISA and impedance flow cytometry, we quantified TF activity using a standard TF assay. oxLDL stimulation increased microparticle‐associated TF activity in washed microparticle samples (Figure 3C). To demonstrate the prothrombotic activity of the TF‐positive MΦMPs measured in our light and impedance flow cytometry experiments, we evaluated microparticles from resting or oxLDL‐stimulated macrophages in a microparticle thrombin generation assay. This assay involves capture of phosphatidylserine containing microparticles on an annexin V–coated plate, and measurement of microparticle‐induced thrombin generation. 30 MΦMPs significantly increased thrombin generation, and oxLDL stimulation resulted in a 2.7‐fold increase in MΦMP prothrombotic activity, compared with nonstimulated controls (Figure 3D).
Figure 3. Oxidized LDL stimulation increases production of macrophage prothrombotic microparticles.
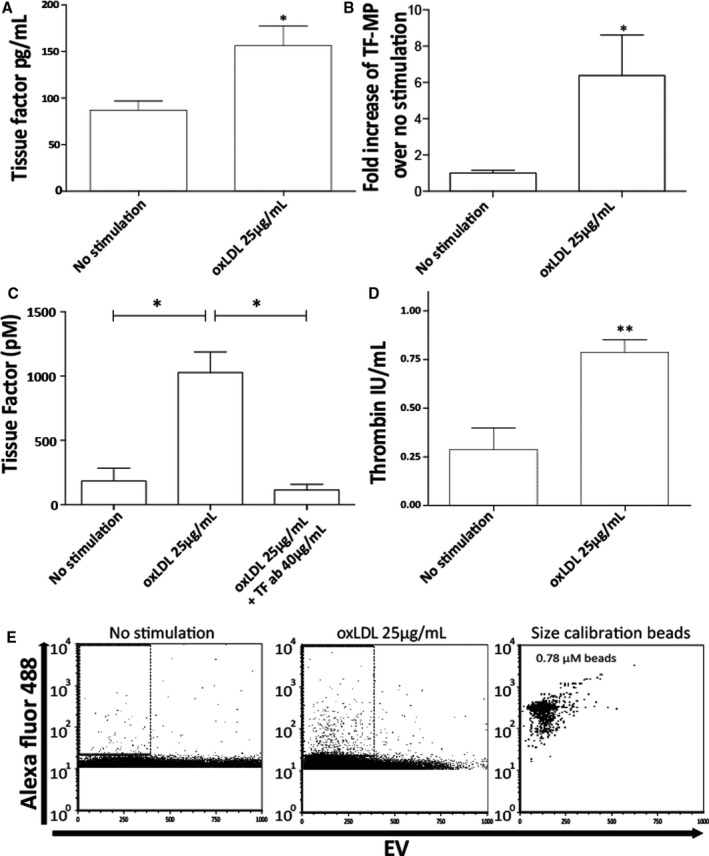
ELISA measurement of TF content in washed microparticles from resting and oxLDL‐treated (25 µg/mL) macrophages (A). Impedance flow cytometry (iFACS) analysis of TF‐positive MΦMPs (B, E). Quantification of TF‐positive MΦMP events measured by iFACS (B). MΦMP TF activity in the absence and presence of inhibitory TF antibody TF8‐5G9 (C). iFACS analysis of MΦ TF‐positive microparticles (E); no stimulation, left panel; oxLDL stimulation (25 µg/mL), middle panel, size calibration beads, right panel. (*P<0.05, **P<0.01). MΦMP from conditioned cell medium (ie, not ultra‐centrifuged) were tested for thrombin production (D). MΦMP indicates macrophage microparticle; oxLDL, oxidized low‐density lipoprotein; and TF, tissue factor.
OxLDL‐Induced Macrophage Microparticle Production Correlates With Caspase 3/7 Activation
Caspase 3/7 activation participates in microparticle production in several cell types. 34 , 35 Calcium phosphate‐induced SMC microparticle production and microparticle‐dependent vascular calcification appear to couple inextricably with SMC caspase activation, 19 and studies that used B‐cell lymphoma 2/B‐cell lymphoma 2–associated X protein expression ratios to quantify SMC apoptotic responses concluded that microparticle production by SMC exposed to lipids does not involve the activation of apoptotic pathways 20 . To determine whether oxLDL‐stimulated MΦMP generation correlates with caspase activation, we treated macrophages with oxLDL and measured caspase 3/7 activity and MΦMP generation. Compared with nonstimulated controls, oxLDL (25 µg/mL) significantly increased caspase 3/7 activity 7‐fold after 16 hours of treatment (Figure 4A), and oxLDL‐stimulated caspase activation correlated with MΦMP production (Figure 4B). LDL‐induced macrophage caspase 3/7 activation required LDL oxidation because, similar to microparticle production (Figure 2D), stimulation with nonoxidized LDL and the minimally modified LDL components 2‐(5‐oxovaleryl)phosphatidylcholine and 1‐palmitoyl‐2‐glutaryl phosphatidylcholine did not increase caspase 3/7 activation (Figure 4C). Consistent with prior reports on the effects of oxLDL‐induced caspase activation on SMC and macrophage apoptosis, 20 , 36 oxLDL treatment and caspase 3/7 activation did not alter activation of transcriptional mechanisms that control apoptotic pathways because there was no change in the mRNA expression ratio of the apoptosis promoter B‐cell lymphoma 2–associated X protein to the apoptosis inhibitor B‐cell lymphoma 2 (Figure 4D).
Figure 4. OLDL‐induced macrophage microparticle production correlates with caspase 3/7 activation.

OxLDL activates caspase 3/7 (oxLDL 25 µg/mL, 31 TBARS, 16 hours) (A). Linear regression of macrophage caspase 3/7 activity and microparticle production (R2 = 0.56) following stimulation with oxLDL (0‐25 µg/mL)(B). Macrophage caspase 3/7 activation in response to stimulation with native LDL, minimally modified oxLDL components POV‐PC and PG‐PC, or oxLDL with 6.8, 11.3, and 29.4 TBARS of copper oxidation (oxLDL 25 µg/mL, 16 hours) (C). Apoptotic index of BAX/BCL2 measured by real‐time quantitative polymerase chain reaction in resting macrophages or macrophages stimulated with oxLDL (25 µg/mL, 16 hours) (D). (NS: P≥0.05, ***P<0.001 vs no stimulation). BAX/BCL2 indicates B‐cell lymphoma 2–associated X protein / B‐cell lymphoma 2; LDL indicates low‐density lipoprotein; oxLDL: oxidized low‐density lipoprotein; PG‐PC: 1‐palmitoyl‐2‐glutaryl phosphatidylcholine; POV‐PC: 2‐(5‐oxovalery) phosphatidylcholine; and TBARS: thiobarbituric acid reactive substances.
Caspase 3/7 Inhibition Attenuates Macrophage Microparticle Production and Caspase 3/7 Activation Increases Macrophage Microparticle Production
To determine whether caspase activation regulates MΦMP production, we examined oxLDL‐induced MΦMP production in the presence of the selective caspase 3/7 inhibitor zDEVD. 37 Pretreatment of macrophages with zDEVD (20 µmol/L, 1 hour) resulted in caspase 3/7 inhibition and led to a 52% reduction in oxLDL‐induced MΦMP production (Figure 5A and 5B). Monocyte and macrophage quiescence requires constitutive phosphatidylinositol 3‐kinase activation and downstream Akt signaling, which suppresses caspase 3 activity. 38 Phosphatidylinositol 3‐kinase and Akt inhibitors activate monocyte and macrophage caspases. 38 Further demonstration that caspase 3/7 controls MΦMP production used the phosphatidylinositol 3‐kinase inhibitor LY294002 (25 µmol/L) to induce caspase activity and then measured MΦMP generation. Macrophage exposure to LY294002 for 24 hours caused a 3.3‐fold enhancement of caspase 3/7 activity and promoted a 208% increase in MΦMP production (Figure 5C and 5D). Caspase inhibition with zDEVD significantly attenuated LY294002‐induced caspase activation and MΦMP production, which further demonstrates that MΦMP production, requires caspase 3/7 function (Figure 5C and 5D).
Figure 5. Caspase 3/7 inhibition attenuates macrophage microparticle production and caspase 3/7 activation increases macrophage microparticle production.
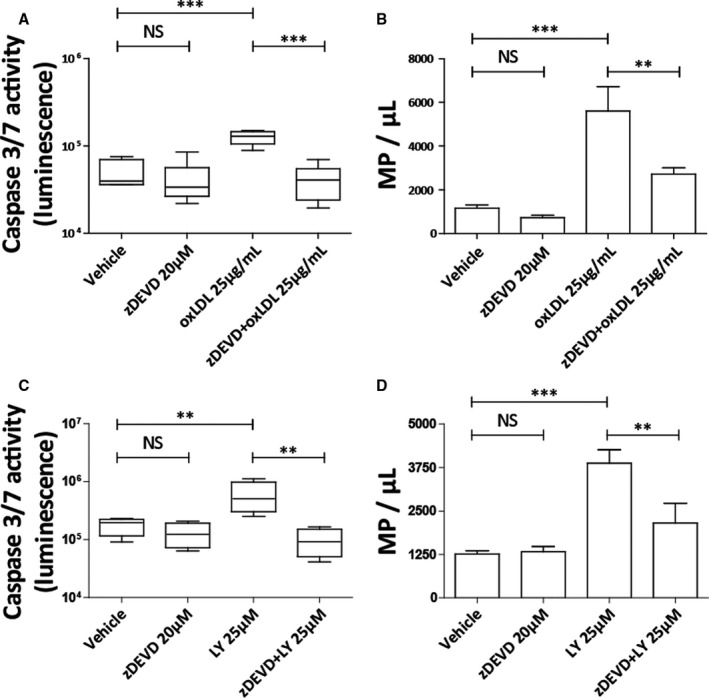
zDEVD‐FMK (20 µmol/L), a caspase 3/7 inhibitor, attenuates oxLDL‐induced (25 µg/mL) caspase 3/7 activation by 67% and macrophage microparticle (MΦMP) production by 52% (A through B). Ly294002 (25 µmol/L), a PI‐3K/Akt pathway inhibitor, increases caspase 3/7 activation by 232% and increases MΦMP production by 208% (C through D). zDEVD‐FMK pretreatment (20 µmol/L) also attenuates Ly294002‐induced caspase 3/7 activation by 84% and MΦMP production by 44% (C through D). Vehicle is 0.2% dimethyl sulfoxide. (NS: P≥0.05, **P<0.01, and ***P<0.001). LY indicates Ly2940002; oxLDL, oxidized low‐density lipoprotein; and zDEVD, N‐[(phenylmethoxy)carbonyl]‐L‐α‐aspartyl‐L‐α‐glutamyl‐N‐[(1S)‐3‐fluoro‐1‐(2‐methoxy‐2‐oxoethyl)‐2‐oxopropyl]‐L‐valinamide, 1,2‐dimethyl ester.
The Scavenger Receptor CD36 Mediates oxLDL‐Induced Macrophage Microparticle Production
OxLDL binding to the macrophage scavenger receptor CD36 results in signal transduction and cellular activation. 23 , 39 To examine whether oxLDL‐induced MΦMP production requires CD36 function, we treated macrophages with a CD36 inhibitor, salvianolic acid B. Salvianolic acid B is an established inhibitor of CD36 dependent cellular activation that in addition to other effects reduces oxLDL stimulated CD36 expression and blocks CD36‐mediated uptake of oxLDL. 40 , 41 Salvianolic acid B pretreatment (100 µmol/L, 12‐hour pretreatment) led to a 33% decrease in caspase 3/7 activity and a 67% decrease in oxLDL‐induced MΦMP production (Figure 6C and 6D). To confirm that this effect results from inhibition of CD36 we treated macrophages with a CD36 inhibitory antibody (20 µg/mL, 1 hour pretreatment). 42 CD36 antibody inhibition caused a 58% decrease in oxLDL‐induced caspase 3/7 activity and a 60% decrease of oxLDL‐induced MΦMP production (Figure 6A and 6B).
Figure 6. The scavenger receptor CD36 mediates oxLDL‐induced macrophage microparticle production.

The CD36 inhibitory antibody FA6‐152 (20 µg/mL) attenuates oxLDL‐induced (25 µg/mL) caspase 3/7 activation by 58% and macrophage microparticle (MΦMP) production by 60% (A through B). The CD36 inhibitor SAB (100 µmol/L) attenuates oxLDL‐induced caspase 3/7 activation by 33% and MΦMP production by 67% (B, D) (NS: P≥0.05, *P<0.05, **P<0.01, and ***P<0.001). oxLDL indicates oxidized low‐density lipoprotein; and SAB, salvianolic acid B.
Statins Inhibit Caspase 3/7 Activation and Macrophage Microparticle Production
The statin class of hypercholesterolemia drugs has clinically important antithrombotic effects that do not depend on LDL lowering. 43 Statins can reduce monocyte and macrophage TF production, 23 , 24 , 25 , 26 but it is unknown whether statins modulate macrophage caspase activation and caspase‐dependent MΦMP production. To test the hypothesis that statin treatment alters caspase 3/7‐dependent oxLDL‐induced MΦMP production, we pretreated macrophages with mevastatin (10 µmol/L) for 48 hours before stimulation with oxLDL. Mevastatin treatment reduced macrophage caspase 3/7 activation by 50% (Figure 7A), attenuated MΦMP production by 51% (Figure 7B), and decreased production of macrophage‐associated TF by 56% (not shown). Treatment with atorvastatin (10 µmol/L) also inhibited oxLDL‐induced MΦMP production by 65% (not shown).
Figure 7. Statin treatment attenuates macrophage caspase 3/7 activation and macrophage microparticle production.

Mevastatin (10 µmol/L) reduces oxLDL‐induced (25 µg/mL) macrophage caspase 3/7 activation by 50% (A). Mevastatin (10 µmol/L) reduces oxLDL‐induced (25 µg/mL) macrophage microparticle (MΦMP) production by 51% (B). Addition of GGO (10 µmol/L) to mevastatin‐treated macrophages (10 µmol/L) partially restores oxLDL‐induced (25 µg/mL) MΦMP production (C). (NS: P≥0.05, *P<0.05, **P<0.01, and ***P<0.001). GGO indicates geranylgeraniol; and oxLDL, oxidized low‐density lipoprotein.
The effects of statins on inflammation depend in part on reduction in the prenylation of key signaling proteins. Statins inhibit inflammatory signaling by reducing the synthesis of intermediates in the 3‐hydroxy‐3‐methyl‐glutaryl–coenzyme A reductase pathway that furnish substrates for prenylation of certain critical proteins. To test whether statins inhibit MΦMP production through a prenylation‐dependent mechanism, we added the prenylation substrate geranylgeraniol to mevastatin‐inhibited macrophages. Geranylgeraniol (10 µmol/L) partially restored MΦMP production, an observation consistent with a mechanism whereby statins inhibit oxLDL‐induced MΦMP generation by reducing prenylation of intermediate signaling molecules that control microparticle production (Figure 7C).
Discussion
This study made the novel discovery that oxLDL induces primary macrophages to produce TF‐expressing prothrombotic microparticles in a CD36‐ and caspase 3/7–dependent manner. The results also establish that statin treatment reduces oxLDL‐induced caspase 3/7–dependent MΦMP production.
Several reports have demonstrated that circulating oxidized lipids predict the presence of atherosclerosis and the risk of ischemic atherothrombotic events. 44 We derived our hypothesis — that atherogenic lipids promote macrophage production of prothrombotic microparticles — on the basis of data that atherosclerotic lesions contain extensively oxLDL, 45 and that oxLDL increases macrophage TF expression. 9 Distinct scavenger receptor signaling mechanisms mediate oxLDL‐induced macrophage activation, and oxLDL specifically increases macrophage TF expression via CD36 scavenger receptor–dependent pathways, 23 , 46 that activate rho guanosine triphosphatases and caspases. 10 Mouse experiments have extensively characterized TF‐positive MΦMP generation 47 . Mouse macrophages primed with interferon‐γ and lipopolysaccharide and activated with ATP exhibit cytoskeletal disorganization (actin remodeling), cell membrane formation into filopodia, and caspase‐1 activation. Mouse MΦMPs are constituted from phosphatidylserine‐rich membrane domains in the filopodia with TF freed from filamin by caspase 1–activated calpain and imported via lipid rafts. Caspase 1 activation and increased actin remodeling are also key factors in the production of human neutrophil microparticles induced by a hyperglycemic medium. 48
Our study used complementary approaches —pharmacologic caspase inhibition attenuated oxLDL‐induced microparticle production, and caspase activation with a phosphatidylinositol 3‐kinase inhibitor increased caspase‐dependent microparticle production — to demonstrate that in nonimmortalized primary human monocyte–derived macrophages, caspase 3/7 regulates oxLDL‐induced MΦMP release. Using inhibitory CD36 antibodies and pharmacologic CD36 inhibition, we also established that the CD36 scavenger receptor mediates oxLDL‐induced caspase 3/7 activation and MΦMP production.
The connection between macrophage caspase 3/7 activity and MΦMP production agree with investigations in endothelial cells, which established that caspases control endothelial MP generation via rho‐associated protein kinase‐II signaling pathways that do not stimulate apoptosis. 27 , 49 We similarly demonstrate that oxLDL activates caspase 3/7–dependent MΦMP production without affecting transcriptional apoptosis programs controlled by the expression ratio of the apoptosis inhibitor B‐cell lymphoma 2 and the apoptosis promoter B‐cell lymphoma 2–associated X protein. Our data — that concentrations of oxLDL relevant in vivo do not activate macrophage apoptosis transcriptional programs — support prior observations that oxLDL activates SMC and macrophage caspase 3/7 independent of apoptosis. 20 , 36 Indeed, Asmis and colleagues 36 specifically demonstrated that caspase 3 activation is neither required nor sufficient to promote human macrophage cell lysis and apoptosis, and caspase 3/7 activity regulates processes such as monocyte differentiation and T‐cell expansion without triggering programmed cell death. 50 , 51 , 52 , 53
Statin medications lower cholesterol and reduce atherothrombotic events in patients with coronary artery disease. As a class, these medications have several beneficial anti‐inflammatory and antithrombotic effects that appear independent of their ability to lower LDL cholesterol. Statins reduce monocyte and macrophage TF expression in vitro and in atherosclerotic plaques, 23 , 24 , 25 , 26 and a large randomized clinical trial demonstrated that statins reduce both arterial and venous thrombotic events in individuals with chronic inflammation and normal LDL cholesterol levels. 43 This study therefore tested whether statins attenuate oxLDL‐stimulated macrophage caspase activation and microparticle production to shed light on the mechanisms by which statins reduce thrombotic events.
In clinical studies, statin use associates with lower levels of circulating endothelial microparticles, 54 , 55 and mechanistic investigations have shown that statins reduce endothelial cell microparticle generation 27 by inhibiting rho/rho‐associated protein kinase signaling pathways that control endothelial cell microparticle production. In addition to supporting the concept that statins inhibit oxLDL‐induced leukocyte microparticle production, our experiments provide the novel insights that in human primary monocyte–derived macrophages, (1) caspase 3/7 inhibition reduces MP production, (2) caspase 3/7 activation with the phosphatidylinositol 3‐kinase inhibitor LY294002 increases microparticle production, and (3) statin‐induced reduction in caspase activation and microparticle production likely depends on alterations in signaling protein prenylation. Future studies will focus on identifying the specific microparticle‐signaling molecules upstream of caspase 3/7 that statin therapy affects.
Our findings support a model where oxLDL activates macrophage CD36 and caspase 3/7–dependent microparticle formation, which increases the thrombogenicity of the atheroma core. oxLDL‐stimulated production of TF‐positive microparticles likely increases plaque thrombogenicity and heightens the risk of ischemic atherothrombotic complications. In this study, statins attenuated macrophage caspase activation and reduced MΦMP production. The ability of statins to reduce MΦMPs may help explain the ability of this class of medications to decrease atherothrombotic events independent of their ability to lower LDL cholesterol.
Sources of Funding
This work was supported by a grant from Capes Foundation–Ministry of Education of Brazil (A.M.); a Gilead Pharmaceuticals Science Foundation Research Award (Dr Croce); a Harris Family Foundation Award (KC); and a grant from the National Institutes of Health/National Heart, Lung and Blood Institute (1K08HL086672; Dr Croce). Dr Marchini was supported by a Lemann Foundation Harvard Medical School Cardiovascular Research Fellowship at Brigham & Women’s Hospital. Dr Libby receives funding support from the National Heart, Lung, and Blood Institute (1R01HL134892), the American Heart Association (18CSA34080399), and the RRM Charitable Fund.
Disclosures
Dr Libby is an unpaid consultant to, or involved in clinical trials for, Amgen, AstraZeneca, Baim Institute, Beren Therapeutics, Esperion, Therapeutics, Genentech, Kancera, Kowa Pharmaceuticals, Medimmune, Merck, Norvo Nordisk, Merck, Novartis, Pfizer, and Sanofi‐Regeneron. Dr Libby is a member of scientific advisory board for Amgen, Corvidia Therapeutics, DalCor Pharmaceuticals, Kowa Pharmaceuticals, Olatec Therapeutics, Medimmune, Novartis, and XBiotech, Inc. Dr Libby’s laboratory has received research funding in the past 2 years from Novartis. Dr Libby is on the Board of Directors of XBiotech, Inc. Dr Libby has a financial interest in Xbiotech, a company developing therapeutic human antibodies. Dr Libby’s interests were reviewed and are managed by Brigham and Women’s Hospital and Partners HealthCare in accordance with their conflict‐of‐interest policies. The remaining authors have no disclosures to report.
Supporting information
Figures S1‐S3
Acknowledgments
We thank Drs Bruce and Barbara Furie (Beth Israel Deaconess Medical Center) for providing access to their impedance flow cytometer, and Drs Jean‐Marie Freyssinet and Fatiha Zobair for consultation on the microparticle thrombin assay. We also thank Dr James Morrissey (University of Illinois) for providing the inhibitory TF antibody (TF8‐5G9), and Ms Yevgenia Tesmenitsky for technical support.
(J Am Heart Assoc. 2020;9:e015878 DOI: 10.1161/JAHA.120.015878.)
For Sources of Funding and Disclosures, see pages 12 and 13.
References
- 1. Ley K, Miller YI, Hedrick CC. Monocyte and macrophage dynamics during atherogenesis. Arterioscler Thromb Vasc Biol. 2011;31:1506–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kzhyshkowska J, Neyen C, Gordon S. Role of macrophage scavenger receptors in atherosclerosis. Immunobiology. 2012;217:492–502. [DOI] [PubMed] [Google Scholar]
- 3. Libby P. Molecular and cellular mechanisms of the thrombotic complications of atherosclerosis. J Lipid Res. 2009;50(Suppl):S352–S357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Falk E, Nakano M, Bentzon JF, Finn AV, Virmani R. Update on acute coronary syndromes: the pathologists' view. Eur Heart J. 2013;34:719–728. [DOI] [PubMed] [Google Scholar]
- 5. Libby P. Mechanisms of acute coronary syndromes and their implications for therapy. N Engl J Med. 2013;368:2004–2013. [DOI] [PubMed] [Google Scholar]
- 6. Burke AP, Kolodgie FD, Farb A, Weber DK, Malcom GT, Smialek J, Virmani R. Healed plaque ruptures and sudden coronary death: evidence that subclinical rupture has a role in plaque progression. Circulation. 2001;103:934–940. [DOI] [PubMed] [Google Scholar]
- 7. Kramer MC, Rittersma SZ, de Winter RJ, Ladich ER, Fowler DR, Liang YH, Kutys R, Carter‐Monroe N, Kolodgie FD, van der Wal AC, et al. Relationship of thrombus healing to underlying plaque morphology in sudden coronary death. J Am Coll Cardiol. 2010;55:122–132. [DOI] [PubMed] [Google Scholar]
- 8. Croce K, Libby P. Intertwining of thrombosis and inflammation in atherosclerosis. Curr Opin Hematol. 2007;14:55–61. [DOI] [PubMed] [Google Scholar]
- 9. Meisel SR, Xu XP, Edgington TS, Cercek B, Ong J, Kaul S, Shah PK. Dose‐dependent modulation of tissue factor protein and procoagulant activity in human monocyte‐derived macrophages by oxidized low density lipoprotein. J Atheroscler Thromb. 2011;18:596–603. [DOI] [PubMed] [Google Scholar]
- 10. Wintergerst ES, Jelk J, Rahner C, Asmis R. Apoptosis induced by oxidized low density lipoprotein in human monocyte‐derived macrophages involves CD36 and activation of caspase‐3. Eur J Biochem. 2000;267:6050–6059. [DOI] [PubMed] [Google Scholar]
- 11. Moore KJ, Tabas I. Macrophages in the pathogenesis of atherosclerosis. Cell. 2011;145:341–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Leroyer AS, Isobe H, Leseche G, Castier Y, Wassef M, Mallat Z, Binder BR, Tedgui A, Boulanger CM. Cellular origins and thrombogenic activity of microparticles isolated from human atherosclerotic plaques. J Am Coll Cardiol. 2007;49:772–777. [DOI] [PubMed] [Google Scholar]
- 13. Leroyer AS, Tedgui A, Boulanger CM. Role of microparticles in atherothrombosis. J Intern Med. 2008;263:528–537. [DOI] [PubMed] [Google Scholar]
- 14. Heloire F, Weill B, Weber S, Batteux F. Aggregates of endothelial microparticles and platelets circulate in peripheral blood. Variations during stable coronary disease and acute myocardial infarction. Thromb Res. 2003;110:173–180. [DOI] [PubMed] [Google Scholar]
- 15. Bernal‐Mizrachi L, Jy W, Jimenez JJ, Pastor J, Mauro LM, Horstman LL, de Marchena E, Ahn YS. High levels of circulating endothelial microparticles in patients with acute coronary syndromes. Am Heart J. 2003;145:962–970. [DOI] [PubMed] [Google Scholar]
- 16. Tan KT, Tayebjee MH, Lim HS, Lip GY. Clinically apparent atherosclerotic disease in diabetes is associated with an increase in platelet microparticle levels. Diabetic Med. 2005;22:1657–1662. [DOI] [PubMed] [Google Scholar]
- 17. Porto I, Biasucci LM, De Maria GL, Leone AM, Niccoli G, Burzotta F, Trani C, Tritarelli A, Vergallo R, Liuzzo G, et al. Intracoronary microparticles and microvascular obstruction in patients with ST elevation myocardial infarction undergoing primary percutaneous intervention. Eur Heart J. 2012;33:2928–2938. [DOI] [PubMed] [Google Scholar]
- 18. Morel O, Pereira B, Averous G, Faure A, Jesel L, Germain P, Grunebaum L, Ohlmann P, Freyssinet JM, Bareiss P, et al. Increased levels of procoagulant tissue factor‐bearing microparticles within the occluded coronary artery of patients with ST‐segment elevation myocardial infarction: role of endothelial damage and leukocyte activation. Atherosclerosis. 2009;204:636–641. [DOI] [PubMed] [Google Scholar]
- 19. Reynolds JL, Joannides AJ, Skepper JN, McNair R, Schurgers LJ, Proudfoot D, Jahnen‐Dechent W, Weissberg PL, Shanahan CM. Human vascular smooth muscle cells undergo vesicle‐mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J Am Soc Nephrol. 2004;15:2857–2867. [DOI] [PubMed] [Google Scholar]
- 20. Llorente‐Cortes V, Otero‐Vinas M, Camino‐Lopez S, Llampayas O, Badimon L. Aggregated low‐density lipoprotein uptake induces membrane tissue factor procoagulant activity and microparticle release in human vascular smooth muscle cells. Circulation. 2004;110:452–459. [DOI] [PubMed] [Google Scholar]
- 21. Li M, Yu D, Williams KJ, Liu ML. Tobacco smoke induces the generation of procoagulant microvesicles from human monocytes/macrophages. Arterioscler Thromb Vasc Biol. 2010;30:1818–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu ML, Reilly MP, Casasanto P, McKenzie SE, Williams KJ. Cholesterol enrichment of human monocyte/macrophages induces surface exposure of phosphatidylserine and the release of biologically‐active tissue factor‐positive microvesicles. Arterioscler Thromb Vasc Biol. 2007;27:430–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Owens AP 3rd, Passam FH, Antoniak S, Marshall SM, McDaniel AL, Rudel L, Williams JC, Hubbard BK, Dutton JA, Wang J, et al. Monocyte tissue factor‐dependent activation of coagulation in hypercholesterolemic mice and monkeys is inhibited by simvastatin. J Clin Invest. 2012;122:558–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Aikawa M, Rabkin E, Sugiyama S, Voglic SJ, Fukumoto Y, Furukawa Y, Shiomi M, Schoen FJ, Libby P. An HMG‐CoA reductase inhibitor, cerivastatin, suppresses growth of macrophages expressing matrix metalloproteinases and tissue factor in vivo and in vitro. Circulation. 2001;103:276–283. [DOI] [PubMed] [Google Scholar]
- 25. Baetta R, Camera M, Comparato C, Altana C, Ezekowitz MD, Tremoli E. Fluvastatin reduces tissue factor expression and macrophage accumulation in carotid lesions of cholesterol‐fed rabbits in the absence of lipid lowering. Arterioscler Thromb Vasc Biol. 2002;22:692–698. [DOI] [PubMed] [Google Scholar]
- 26. Colli S, Eligini S, Lalli M, Camera M, Paoletti R, Tremoli E. Vastatins inhibit tissue factor in cultured human macrophages. A novel mechanism of protection against atherothrombosis. Arterioscler Thromb Vasc Biol. 1997;17:265–272. [DOI] [PubMed] [Google Scholar]
- 27. Sapet C, Simoncini S, Loriod B, Puthier D, Sampol J, Nguyen C, Dignat‐George F, Anfosso F. Thrombin‐induced endothelial microparticle generation: identification of a novel pathway involving ROCK‐II activation by caspase‐2. Blood. 2006;108:1868–1876. [DOI] [PubMed] [Google Scholar]
- 28. Folco EJ, Rocha VZ, Lopez‐Ilasaca M, Libby P. Adiponectin inhibits pro‐inflammatory signaling in human macrophages independent of interleukin‐10. J Biol Chem. 2009;284:25569–25575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tzur A, Moore JK, Jorgensen P, Shapiro HM, Kirschner MW. Optimizing optical flow cytometry for cell volume‐based sorting and analysis. PLoS One. 2011;6:e16053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mallat Z, Hugel B, Ohan J, Leseche G, Freyssinet JM, Tedgui A. Shed membrane microparticles with procoagulant potential in human atherosclerotic plaques: a role for apoptosis in plaque thrombogenicity. Circulation. 1999;99:348–353. [DOI] [PubMed] [Google Scholar]
- 31. Zwicker JI, Liebman HA, Neuberg D, Lacroix R, Bauer KA, Furie BC, Furie B. Tumor‐derived tissue factor‐bearing microparticles are associated with venous thromboembolic events in malignancy. Clin Cancer Res. 2009;15:6830–6840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Vandesompele J, De Preter K, Pattyn F, Poppe B, Van Roy N, De Paepe A, Speleman F. Accurate normalization of real‐time quantitative RT‐PCR data by geometric averaging of multiple internal control genes. Genome Biol. 2002;3:RESEARCH0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hruz T, Laule O, Szabo G, Wessendorp F, Bleuler S, Oertle L, Widmayer P, Gruissem W, Zimmermann P. Genevestigator v3: a reference expression database for the meta‐analysis of transcriptomes. Adv Bioinformatics. 2008;2008:420747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mallat Z, Tedgui A. Current perspective on the role of apoptosis in atherothrombotic disease. Circ Res. 2001;88:998–1003. [DOI] [PubMed] [Google Scholar]
- 35. Schecter AD, Spirn B, Rossikhina M, Giesen PL, Bogdanov V, Fallon JT, Fisher EA, Schnapp LM, Nemerson Y, Taubman MB. Release of active tissue factor by human arterial smooth muscle cells. Circ Res. 2000;87:126–132. [DOI] [PubMed] [Google Scholar]
- 36. Asmis R, Begley JG. Oxidized LDL promotes peroxide‐mediated mitochondrial dysfunction and cell death in human macrophages: a caspase‐3‐independent pathway. Circ Res. 2003;92:e20–e29. [DOI] [PubMed] [Google Scholar]
- 37. Garcia‐Calvo M, Peterson EP, Leiting B, Ruel R, Nicholson DW, Thornberry NA. Inhibition of human caspases by peptide‐based and macromolecular inhibitors. J Biol Chem. 1998;273:32608–32613. [DOI] [PubMed] [Google Scholar]
- 38. Goyal A, Wang Y, Graham MM, Doseff AI, Bhatt NY, Marsh CB. Monocyte survival factors induce Akt activation and suppress caspase‐3. Am J Respir Cell Mol Biol. 2002;26:224–230. [DOI] [PubMed] [Google Scholar]
- 39. Chavez‐Sanchez L, Garza‐Reyes MG, Espinosa‐Luna JE, Chavez‐Rueda K, Legorreta‐Haquet MV, Blanco‐Favela F. The role of TLR2, TLR4 and CD36 in macrophage activation and foam cell formation in response to oxLDL in humans. Hum Immunol. 2014;75:322–329. [DOI] [PubMed] [Google Scholar]
- 40. Ho JH, Hong CY. Salvianolic acids: small compounds with multiple mechanisms for cardiovascular protection. J Biomed Sci. 2011;18:30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bao Y, Wang L, Xu Y, Yang Y, Si S, Cho S, Hong B. Salvianolic acid B inhibits macrophage uptake of modified low density lipoprotein (mLDL) in a scavenger receptor CD36‐dependent manner. Atherosclerosis. 2012;223:152–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Volf I, Moeslinger T, Cooper J, Schmid W, Koller E. Human platelets exclusively bind oxidized low density lipoprotein showing no specificity for acetylated low density lipoprotein. FEBS Lett. 1999;449:141–145. [DOI] [PubMed] [Google Scholar]
- 43. Glynn RJ, Danielson E, Fonseca FA, Genest J, Gotto AM Jr, Kastelein JJ, Koenig W, Libby P, Lorenzatti AJ, MacFadyen JG, et al. A randomized trial of rosuvastatin in the prevention of venous thromboembolism. N Engl J Med. 2009;360:1851–1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kiechl S, Willeit J, Mayr M, Viehweider B, Oberhollenzer M, Kronenberg F, Wiedermann CJ, Oberthaler S, Xu Q, Witztum JL, et al. Oxidized phospholipids, lipoprotein(a), lipoprotein‐associated phospholipase A2 activity, and 10‐year cardiovascular outcomes: prospective results from the Bruneck study. Arterioscler Thromb Vasc Biol. 2007;27:1788–1795. [DOI] [PubMed] [Google Scholar]
- 45. Palinski W, Rosenfeld ME, Yla‐Herttuala S, Gurtner GC, Socher SS, Butler SW, Parthasarathy S, Carew TE, Steinberg D, Witztum JL. Low density lipoprotein undergoes oxidative modification in vivo. Proc Natl Acad Sci USA. 1989;86:1372–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stewart CR, Stuart LM, Wilkinson K, van Gils JM, Deng J, Halle A, Rayner KJ, Boyer L, Zhong R, Frazier WA, et al. CD36 ligands promote sterile inflammation through assembly of a Toll‐like receptor 4 and 6 heterodimer. Nat Immunol. 2010;11:155–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Rothmeier AS, Marchese P, Petrich BG, Furlan‐Freguia C, Ginsberg MH, Ruggeri ZM, Ruf W. Caspase‐1‐mediated pathway promotes generation of thromboinflammatory microparticles. J Clin Invest. 2015;125:1471–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Thom SR, Bhopale VM, Yu K, Huang W, Kane MA, Margolis DJ. Neutrophil microparticle production and inflammasome activation by hyperglycemia due to cytoskeletal instability. J Biol Chem. 2017;292:18312–18324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vion AC, Birukova AA, Boulanger CM, Birukov KG. Mechanical forces stimulate endothelial microparticle generation via caspase‐dependent apoptosis‐independent mechanism. Pulm Circ. 2013;3:95–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Sordet O, Rebe C, Plenchette S, Zermati Y, Hermine O, Vainchenker W, Garrido C, Solary E, Dubrez‐Daloz L. Specific involvement of caspases in the differentiation of monocytes into macrophages. Blood. 2002;100:4446–4453. [DOI] [PubMed] [Google Scholar]
- 51. McComb S, Mulligan R, Sad S. Caspase‐3 is transiently activated without cell death during early antigen driven expansion of CD8(+) T cells in vivo. PLoS One. 2010;5:e15328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Abraham MC, Shaham S. Death without caspases, caspases without death. Trends Cell Biol. 2004;14:184–193. [DOI] [PubMed] [Google Scholar]
- 53. Khalil H, Peltzer N, Walicki J, Yang JY, Dubuis G, Gardiol N, Held W, Bigliardi P, Marsland B, Liaudet L, et al. Caspase‐3 protects stressed organs against cell death. Mol Cell Biol. 2012;32:4523–4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Suades R, Padro T, Alonso R, Mata P, Badimon L. Lipid‐lowering therapy with statins reduces microparticle shedding from endothelium, platelets and inflammatory cells. Thromb Haemost. 2013;110:366–377. [DOI] [PubMed] [Google Scholar]
- 55. Huang B, Cheng Y, Xie Q, Lin G, Wu Y, Feng Y, Gao J, Xu D. Effect of 40 mg versus 10 mg of atorvastatin on oxidized low‐density lipoprotein, high‐sensitivity C‐reactive protein, circulating endothelial‐derived microparticles, and endothelial progenitor cells in patients with ischemic cardiomyopathy. Clin Cardiol. 2012;35:125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1‐S3