

Chemotherapy‐induced cardiotoxicity is an emerging and significant clinical problem with no preventative therapies. 1 Although chemotherapy‐induced cardiac dysfunction is well recognized, 1 our understanding of how specific chemotherapies (eg, doxorubicin) affect cardiac energetics remains limited. Furthermore, the role of the proapoptotic transcription factor p53 (which is directly involved in chemotherapy‐induced cardiotoxicity pathogenesis 2 ) in cardiac energy metabolism is unclear, especially in the setting of chemotherapy‐induced cardiotoxicity. To identify potential metabolic pathways that may be altered by certain chemotherapies, we treated human lung cancer–xenotransplanted mice with doxorubicin for several weeks. This treatment induced p53 and caused cardiac dysfunction (shown in Saleme et al 2 ). We then isolated intact cardiomyocytes from the myocardium and performed unbiased RNA sequencing (Figure—Panel A). As predicted, a number of DNA‐damage and apoptosis‐related genes (many of which are p53 regulated) were significantly induced, whereas cardiomyocyte survival and maintenance genes were predictably suppressed in doxorubicin‐treated mice compared with controls (Figure—Panel B). Intriguingly, our sequencing analysis identified only 1 metabolic pathway that was different between the 2 groups, and this was suppression of APLNR (apelin receptor) signaling pathway, which promotes fatty acid oxidation (FAO), 3 in doxorubicin‐treated mice (Figure—Panel B).
Figure 1. p53‐Mediated repression of the PGC1A (PPARG coactivator 1α) and APLNR (apelin receptor) signaling pathways limit fatty acid oxidation (FAO) energetics, and this can be therapeutically prevented in doxorubicin (DXR)–induced cardiotoxicity by the tetrameric PKM2 –stabilizing drug TEPP‐46.
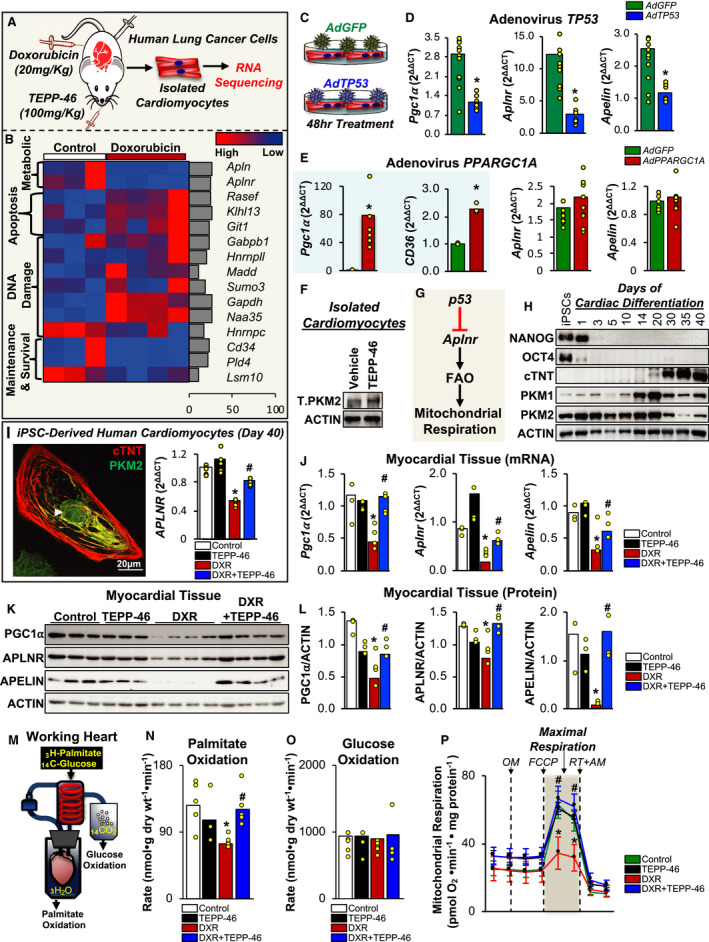
Statistical analysis was performed on SPSS v24.0 (IBM Corp; Somers). Kruskal–Wallis test was used to compare ≥2 independent samples, and the Mann–Whitney U test was used for comparisons between 2 groups, unless otherwise specified. Significance was considered at P<0.05. A, Experimental design for unbiased RNA sequencing platform. Athymic male nude mice were xenotransplanted with human lung tumors, as described previously. 2 Mice were then allocated to receive either vehicle or a cumulative dose of 20 mg/kg of DXR (weekly, intraperitoneally) and/or concurrent daily gavage of TEPP‐46 at 100 mg/kg for 5 weeks. Once cardiac dysfunction was confirmed (shown in Saleme et al 2 ), cardiomyocytes were isolated, RNA was extracted, and RNA sequencing was performed. B, Heat map showing the RNA‐sequencing results from isolated cardiomyocytes from control‐ and DXR‐treated mice. Libraries were prepared using TrueSeq V2 and sequenced on HiSeq 2500 and aligned to GRCm38 using Kallisto. Differential expression analysis was conducted with the DESeq2 R package, and transcript abundance differences with corrected P<0.05 were considered differentially expressed (n=3 for control and n=4 for DXR‐treated mice). Gene variability shown as a percentage of standard deviation over fold change for each gene is shown (right). Gene names for the 4 different pathways are shown to the right of each representative row. C, Experimental design for TP53 (tumor protein p53) overexpression using adenovirus infection of isolated adult mouse cardiomyocytes. The TP53 adenovirus coexpresses GFP. Adenovirus for GFP was used as a control. D, Quantitative reverse transcriptase polymerase chain reaction (qRT‐PCR) for the TP53 overexpression experiment shows the mRNA levels for Pgc1A, Aplnr, and Apln (apelin). In addition, 18S was used as a loading control. Mean data are shown, and each individual experiment (n=9) is shown as an individual yellow circle. *P<0.05 compared with adenovirus (Ad) GFP (green fluorescent protein). E, qRT‐PCR for the PPARGC1A (also known as PGC1A) overexpression experiment shows the mRNA levels for Pgc1A, CD36, Aplnr, and Apln. In addition, 18S was used as a loading control. Mean data are shown, and each individual experiment for Aplnr and Apln (n=9) and for Pgc1A and CD36 (n=6) is shown as an individual yellow circle. *P<0.05 compared with adenovirus (Ad) GFP. F, Disuccinimidyl suberate cross‐linking of isolated adult mouse cardiomyocytes treated with TEPP‐46 shows the levels of tetrameric PKM2 (T.PKM2), using immunoblots. Actin (ACTB) was used as a loading control. G, Schematic showing that p53 activation decreases Aplnr mRNA levels, which subsequently suppresses FAO rates, and mitochondrial respiration. H, Cardiac differentiation of induced pluripotent stem cells (iPSCs) is shown by the loss of stem cell markers (NANOG, OCT4) and the acquisition of cardiac markers (cardiac troponin T [cTNT]). Furthermore, similar to adult cardiomyocytes, which have a high PKM1/PKM2 ratio, 2 PKM1 levels continue to increase, whereas PKM2 levels correspondingly decrease during the differentiation time frame from 1 to 40 days, using immunoblots. ACTB was used as a loading control. I, Immunofluorescence staining of a human iPSC‐derived cardiomyocyte (day 40) showing cTNT (cytosolic) in red and PKM2 (cytosolic and nuclear; shown by white arrow) in green (left). The mRNA levels are shown for APLNR on day 40 of human iPSC‐derived cardiomyocytes treated with DXR (150 nmol/L) and TEPP‐46 (50 µmol/L) for 24 hours, using qRT‐PCR. In addition,18S was used as a loading control. Mean data and individual experiments (yellow circles) are shown. *P<0.05 compared with control, # P<0.05 compared with DXR (right). J through L, qRT‐PCR or immunoblots show the mRNA expression (J) or protein levels (K and L), respectively, of PGC1A, APLN, and APLNR from the myocardial tissue of mice treated with TEPP‐46 (100 mg/kg, daily gavage), DXR (20 mg/kg cumulative dose), or DXR+TEPP‐46, as described previously. 2 β2M and ACTB were used as housekeeping genes for qRT‐PCR and immunoblots, respectively. Mean data and individual animal data (yellow circles) are shown. *P<0.05 compared with control, # P<0.05 compared with DXR. Immunoblots of analyzed data in (K) are shown in (L). M, The working heart model allows for the measurement of glucose and palmitate oxidation rates. Isolated hearts are perfused with radiolabeled glucose or palmitate, and radiolabeled H2O or CO2 provides a relative rate for palmitate oxidation or glucose oxidation, respectively. N and O, DXR treatment in vivo decreases palmitate oxidation (N), but not glucose oxidation (O), compared with control‐treated animals, and this is prevented by cotreatment with TEPP‐46, using the isolated working heart system (n=5 animals per group for all groups except TEPP‐46 [n=3 animals]). *P<0.05 compared with control‐treated animals, # P<0.05 compared with DXR‐treated animals). P, TEPP‐46 prevents a DXR‐induced decrease in oxygen consumption rates in isolated mouse cardiomyocytes, as assessed by Seahorse (n=3 different experiments; * P<0.05 compared with baseline respiration, # P<0.05 compared with DXR; values are expressed as mean±SEM). AM indicates antimycin A; FCCP, carbonyl cyanide‐4‐(trifluoromethoxy)phenylhydrazone; OM, oligomycin; and RT, rotenone.
A recent study showed that p53 can bind and repress the promoter for the transcriptional coactivator promoter of PGC1A (PPARG coactivator 1α; approved as PPARGC1A), 4 suggesting that p53 can have a repressive effect on the promoter of specific genes. Thus, we sought to determine whether the decrease in Aplnr (apelin receptor) and Apln (apelin) by doxorubicin was directly mediated by p53. To investigate, we overexpressed p53 in adult cardiomyocytes (Figure—Panel C). We not only observed the predictable decrease in the expression of Pgc1A (as previously described 4 ) but also found a significant decrease with Aplnr and Apln, compared with GFP (green fluorescent protein) control‐treated cardiomyocytes (Figure—Panel D). Because PGC1A is a prominent inducer of FAO, 5 we wanted to determine whether the decrease in Aplnr and Apln by p53 was in part due to a p53‐mediated repression of Pgc1A. To assess this, we overexpressed PGC1A in adult cardiomyocytes. Although we observed a predictable increase in the expression of CD36 (a target of PGC1A 5 ), we did not observe any differences in the expression of Aplnr or Apln compared with GFP control‐treated cardiomyocytes (Figure—Panel E). These data suggest that suppression of the APLNR signaling pathway could be mediated by p53.
We speculated that induction of p53 would suppress and myocardial‐specific inhibition of p53 would preserve the APLNR signaling pathway, along with FAO energetics, in doxorubicin‐ versus control‐treated mice. We recently described a myocardial‐specific p53 therapy in which tetrameric PKM2 (pyruvate kinase M2) prevented doxorubicin‐mediated cardiac dysfunction. 2 Thus, we proposed that stabilization of tetrameric PKM2 with TEPP‐46 (Figure—Panel F) might inhibit the repressive function of p53 on the APLNR signaling cascade and preserve FAO rates and mitochondrial respiration (Figure—Panel G).
We found that doxorubicin significantly decreased the mRNA expression of APLNR in pluripotent stem cell–derived human cardiomyocytes (which expressed nuclear PKM2), in vitro, and in mRNA, along with protein levels of PGC1A, APLNR, and APLN in myocardial tissue compared with vehicle‐treated cardiomyocytes and mice, respectively, and this was prevented by cotreatment with TEPP‐46 (Figure—Panels H through L). Because PGC1A and the APLNR signaling pathways can promote FAO, 3 , 5 we assessed cardiac energetics by perfusion of isolated hearts with radiolabeled palmitate and glucose and assessed radiolabeled H2O or CO2 as an index of FAO or glucose oxidation rates, respectively (Figure—Panel M). We observed a significant decrease in FAO rates (whereas glucose oxidation rates remained similar) in doxorubicin‐ and control‐treated mice, and this was completely prevented by cotreatment with TEPP‐46 (Figure—Panels N and O). We speculated that the reason why the decrease in FAO did not result in the predicted increase in glucose oxidation (via the Randle cycle) was because glucose oxidation, which was the preferential energetic pathway in these mice (Figure—Panels N and O), was already maximized. However, we predicted that the decrease in FAO would still be meaningful and would be associated with a decrease in mitochondrial respiration (ie, oxidative phosphorylation). Indeed, mitochondrial respiration was significantly decreased in doxorubicin‐treated versus vehicle‐treated cardiomyocytes, and this was prevented by cotreatment with TEPP‐46 (Figure—Panel P).
We identified novel p53‐mediated repression of both the PGC1A and APLNR signaling pathways in the myocardium of doxorubicin‐ versus control‐treated mice; this results in a relatively selective decrease in FAO rates. The decrease in FAO rates may explain, in part, the decrease in contractility commonly associated with doxorubicin. Along with our original study, in which TEPP‐46 prevented doxorubicin‐induced cardiomyocyte apoptosis and cardiac dysfunction, 2 this work confirms that compounds that stabilize the tetrameric form of PKM2 could provide a novel class of therapeutics to prevent not only chemotherapy‐induced cardiotoxicity but also doxorubicin‐induced changes in cardiac energy metabolism.
All animal procedures were performed as described previously 2 and were in compliance with the Canadian Council on Animal Care regulations and the University of Alberta Animal Welfare Committee. All raw data and detailed methods for experiments and analysis are available on request to any researcher for the purposes of reproducing results and replicating the procedure.
Sources of Funding
Saleme is supported by a graduate scholarship from Alberta Innovates. Zhang is supported by a graduate scholarship from the Li Ka Shing Sino‐Exchange Program, and Lorenzana Carrillo is supported by the Government of Mexico CONACYT Scholarship for Doctoral Studies Abroad. Lopaschuk and Michelakis are supported by the Heart and Stroke Foundation of Canada, and Canadian Institutes of Health Research Foundation grants. Sutendra is supported by an Alberta Innovates Translational Health Chair in Cardio‐Oncology, and a National and Alberta New Investigator Award from Heart and Stroke Foundation of Canada, along with the Canadian Institutes of Health Research.
Disclosures
None.
J Am Heart Assoc. 2020;9:e017247 DOI: 10.1161/JAHA.120.017247.
For Sources of Funding and Disclosures, see page 4.
References
- 1. Albini A, Pennesi G, Donatelli F, Cammarota R, De Flora S, Noonan DM. Cardiotoxicity of anticancer drugs: the need for cardio‐oncology and cardio‐oncological prevention. J Natl Cancer Inst. 2010;102:14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Saleme B, Gurtu V, Zhang Y, Kinnaird A, Boukouris AE, Gopal K, Ussher JR, Sutendra G. Tissue‐specific regulation of p53 by PKM2 is redox dependent and provides a therapeutic target for anthracycline‐induced cardiotoxicity. Sci Transl Med. 2019;11:eaau8866. [DOI] [PubMed] [Google Scholar]
- 3. Attane C, Foussal C, Le Gonidec S, Benani A, Daviaud D, Wanecq E, Guzman‐Ruiz R, Dray C, Bezaire V, Rancoule C, et al. Apelin treatment increases complete fatty acid oxidation, mitochondrial oxidative capacity, and biogenesis in muscle of insulin‐resistant mice. Diabetes. 2012;61:310–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mak TW, Hauck L, Grothe D, Billia F. p53 regulates the cardiac transcriptome. Proc Natl Acad Sci USA. 2017;114:2331–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rowe GC, Jiang A, Arany Z. PGC‐1 coactivators in cardiac development and disease. Circ Res. 2010;107:825–838. [DOI] [PMC free article] [PubMed] [Google Scholar]