

Abstract
Background
Hydrogen sulfide (H2S) is an important endogenous physiological signaling molecule and exerts protective properties in the cardiovascular system. Cystathionine γ‐lyase (CSE), 1 of 3 H2S producing enzyme, is predominantly localized in the vascular endothelium. However, the regulation of CSE in vascular endothelium remains incompletely understood.
Methods and Results
We generated inducible endothelial cell‐specific CSE overexpressed transgenic mice (EC‐CSE Tg) and endothelial cell‐specific CSE knockout mice (EC‐CSE KO), and investigated vascular function in isolated thoracic aorta, treadmill exercise capacity, and myocardial injury following ischemia‐reperfusion in these mice. Overexpression of CSE in endothelial cells resulted in increased circulating and myocardial H2S and NO, augmented endothelial‐dependent vasorelaxation response in thoracic aorta, improved exercise capacity, and reduced myocardial‐reperfusion injury. In contrast, genetic deletion of CSE in endothelial cells led to decreased circulating H2S and cardiac NO production, impaired endothelial dependent vasorelaxation response and reduced exercise capacity. However, myocardial‐reperfusion injury was not affected by genetic deletion of endothelial cell CSE.
Conclusions
CSE‐derived H2S production in endothelial cells is critical in maintaining endothelial function, exercise capacity, and protecting against myocardial ischemia/reperfusion injury. Our data suggest that the endothelial NO synthase—NO pathway is likely involved in the beneficial effects of overexpression of CSE in the endothelium.
Keywords: cardioprotection, cystathionine γ‐lyase, endothelial function, hydrogen sulfide, nitric oxide
Subject Categories: Basic Science Research, Endothelium/Vascular Type/Nitric Oxide, Ischemia, Vascular Biology, Physiology
Nonstandard Abbreviations and Acronyms
- CSE
cystathionine γ‐lyase
- EC‐CSE KO
endothelial cell‐specific CSE knockout
- EC‐CSE Tg
endothelial cell‐specific CSE overexpressed transgenic
- eNOS
endothelial NO synthase
- H2S
hydrogen sulfide
- INF
infarct size
- tTA
tetracycline‐controlled trans‐activator
Clinical Perspective
What Is New?
Cystathionine γ‐lyase is a critical enzyme responsible for generating hydrogen sulfide in endothelial cells.
Hydrogen sulfide production in endothelial cells plays important role in maintaining cardiovascular function and protecting against myocardial ischemia/reperfusion injury.
What Are the Clinical Implications?
Modulation of endogenous endothelial hydrogen sulfide production could be a potential therapeutic strategy in the treatment of cardiovascular disease.
Hydrogen sulfide (H2S), a highly toxic gas, is now recognized as an important endogenous physiological signaling molecule. At physiological levels, H2S exerts many protective properties in the cardiovascular system, including vasodilation, antioxidant, and anti‐inflammation. 1 , 2 , 3 , 4
The endogenous production of H2S is catalyzed by 3 enzymes: cystathionine β‐synthase, cystathionine γ‐lyase (CSE), and 3‐mercaptopyruvate sulfurtransferase. 5 While, all 3 enzymes have been found in the cardiovascular system, CSE is believed to be expressed primarily in the heart and vasculature and responsible for the majority endogenous H2S production in the cardiovascular system. 6 Therefore, it appeared that CSE is a critical enzyme that regulates H2S generation, and ultimately modulates cardiovascular function and health. Indeed, decreased CSE expression results in a global decrease in H2S bioavailability and is associated with multiple cardiovascular diseases. 7 , 8 Genetic deletion of CSE globally leads to age‐dependent hypertension, impaired vascular relaxation, early development of atherosclerosis, and exacerbated myocardial and renal ischemia/reperfusion injury. 6 , 9 , 10 , 11 Conversely, genetic overexpression of CSE in cardiomyocytes improves cardiac function and attenuates myocardial infarction following myocardial ischemia reperfusion injury, 12 , 13 suggesting the important role of CSE‐derived H2S in cardioprotection and cardiovascular homeostasis.
Endothelial cells are a critical component of blood vessels that are responsible for regulating vascular tone and downregulating oxidative stress, inflammation, and smooth muscle cell proliferation. 14 Endothelial dysfunction is strongly associated with cardiovascular diseases including hypertension, peripheral vascular disease, coronary artery disease, and heart failure. 2 , 15 , 16 Previous studies have shown that H2S protects endothelial function through various mechanisms, and augmentation of NO signaling is 1 of the major mechanisms. 2 , 16 CSE is predominantly localized to the endothelial layer of blood vessels where it has been shown to produce H2S and mediate smooth muscle relaxation and subsequent vasodilation. 6 , 17 However, the regulation of CSE in vascular endothelium remains incompletely understood.
The purpose of this study was to investigate the impact of endothelial CSE‐derived H2S on cardiovascular function. We first generated inducible endothelial cell‐specific CSE overexpressed transgenic (EC‐CSE Tg) mice and investigated the effect of endothelial CSE overexpression on vascular function, exercise and myocardial protection following myocardial‐reperfusion injury. We then similarly investigated the effect of endothelial CSE deficiency on vascular function, exercise, and myocardial injury following ischemia‐reperfusion using endothelial cell‐specific CSE knockout (EC‐CSE KO) mice.
METHODS
An expanded methods section is available in Data S1.
Data Sharing
The authors declare that all supporting data are available within the article and its online supplementary files.
Experimental Animals
EC‐CSE Tg mice were generated by breeding tetracycline response element (TRE)‐CSE mice as shown in Figure 1A with Cdh5‐tTA (tetracycline‐controlled trans‐activator) mice. Tre‐CSE was developed using human CSE transgene under the control of a tetracycline operator sequence, which contains a minimal promoter, TRE. Cdh5‐tTA mice, which express tTA under the control of Cdh5 promoter, were purchased from Jackson Laboratory. As a result, EC‐CSE Tg mice overexpress CSE selectively in endothelial cells.
Figure 1. Increased cardiac cystathionine γ‐lyase mRNA and protein expression in endothelial cell‐specific cystathionine γ‐lyase overexpressed transgenic mice.
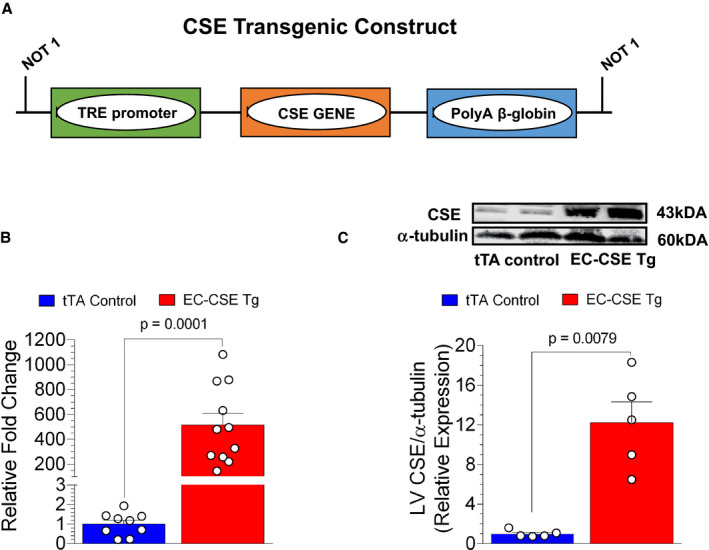
A, Cystathionine γ‐lyase (CSE) transgenic construct. CSE gene is under the control of the tetracycline response element (TRE). In the presence of tTA (tetracycline trans‐activator), CSE transgene expression is induced. B, Endothelial cell‐specific CSE transgenic mice (n=11) had a 500‐fold increase in cardiac CSE mRNA expression compared with tTA controls (n=9). C, CSE protein expression in the left ventricle was increased by 12‐fold in endothelial cell‐specific CSE transgenic mice (n=5) compared with tTA controls (n=5), as shown in the immunoblot and calculated optical density. Data are presented as mean±SEM, Student t‐test (B), Mann–Whitney U test (C). CSE indicates cystathionine γ‐lyase; EC‐CSE Tg, endothelial cell‐specific CSE transgenic mice; LV, left ventricle; TRE, tetracycline response element; and tTA, tetracycline trans‐activator.
EC‐CSE KO mice were generated by breeding CSEflox+/fox+ mice 18 with Cdh5‐Cre mice. CSE flox mice were developed by GenOway (Lyon, France), where CSE gene was loxp‐flanked. The Cdh5‐Cre mice (Jackson Laboratory) express Cre recombinase under the control of Cdh5 promoter. By breeding with Cdh5‐Cre, the loxp‐flanked CSE gene in CSE flox mice was deleted in endothelial cells specifically.
EC‐CSE Tg and EC‐CSE KO mice and age‐matched control littermates (C57Bl/6J, male, 10–12 weeks old) were used in the following studies. Cdh5‐tTA mice were used as controls for EC‐CSE Tg, and CSEflox+/flox+ mice were used as controls for EC‐CSE KO mice. All experimental protocols were approved by the Institute for Animal Care and Use Committee at Louisiana State University Health Sciences Center‐New Orleans and conformed to the Guide for the Care and Use of Laboratory Animals. All experimental animals received humane care in accordance with National Society of Medical Research and National Institutes of Health tenets.
H2S, Nitrite, and Cyclic Guanosine Monophosphate Measurements
Plasma and myocardial free H2S and sulfane sulfur were measured using gas chromatography‐chemiluminescence (7890 Series GC, G660XA Series Chemiluminescence Detector; Agilent, Santa Clara, CA, USA), as previously described. 19 , 20
Nitrite content of plasma and myocardial specimens were measured by an automated ion chromatography system (ENO30 Analyzer; Eicom San Diego, CA, USA), as previously described. 20 , 21 Tissue levels of H2S and nitrite were adjusted based on protein concentration.
Cardiac cyclic guanosine monophosphate (cGMP) levels were measured by radio‐immunoassay, as previously described. 9
Real Time‐Polymerase Chain Reaction for mRNA Expression
RNA isolation was performed by using TRIzol reagent (Thermo Fisher 15596018). RNA was transcribed using I‐script cDNA synthesis kit (Bio‐Rad 1708891). Real‐time quantitative polymerase chain reaction were performed on a Thermocycler (StepOnePlus, Applied Biosystem) by using TaqMan Gene Expression assay generated for 18S or CSE (Thermo Fisher 4331182).
Western Blot Analysis
Flash‐frozen tissue was homogenized in radio‐immunoprecipitation assay buffer supplemented with Halt protease and phosphatase inhibitor cocktail (Thermo Fisher). Protein concentration was determined using a bicinchoninic acid protein assay (Thermo Fisher). Immunoblotting used specific antibodies for the N terminus of CSE (Proteintech 60231‐4‐lg) and α‐tubulin (Abcam ab7291).
Vascular Reactivity Studies
Vascular function was measured in isolated segments (3 mm in length) of thoracic aorta as previously described. 16 , 21 Briefly, vascular rings were equilibrated in oxygenated Krebs‐Henseleit solution (118 mmol/L NaCl, 4.7 mmol/L KCl, 1.2 mmol/L MgSO4, 1.25 mmol/L CaCl2, 1.2 mmol/L KH2PO4, 25 mmol/L NaHCO3, and 11 mmol/L Dextrose) at 0.5 g tension for 60 minutes. And then the rings were pre‐contracted with phenylephrine (1 μmol/L) then treated with increasing concentrations of acetylcholine (10−9 to 10−5 mol/L), calcium ionophore A23187 (10−10 to 10−7 mol/L), and sodium nitroprusside (SNP, 10−11 to 10−7 mol/L). Data are reported as percent of relaxation from maximum contraction to phenylephrine.
Exercise Capacity Assessment
Treadmill exercise capacity was measured using a rodent treadmill (IITC, Woodland Hills, California) as previously described. 16 Briefly, immediately before the test, mice were placed on the treadmill to acclimate for 15 minutes. Then, the mice were subjected to a slow walk/run period to be familiarized with treadmill running. Finally, in the test phase, the treadmill was set to a 30°‐incline. The starting speed was set to 10 m/min and programed to increase to 18 m/min in 3 minutes, then treadmill maintained at its speed of 18 m/min. All mice were run at this setting until they reached a state of exhaustion. Distance and duration of the running and shocks per minute were recorded. Shocks per minute is a measurement of endurance. As mice fell behind the speed of the treadmill, they received an electrical shock by the shocking grid. Higher shocks per minute indicated lower endurance. In a blinded fashion, a mouse was deemed to be exhausted when it spent >5 consecutive seconds at the back of the treadmill despite receiving electrical shocks (1.5 mA) repeatedly.
Myocardial Ischemia/Reperfusion Protocol and Myocardial INF Determination
Surgical ligation of the left coronary artery and myocardial INF determination were performed as previously described. 13 , 22 Briefly, mice were fully anesthetized with ketamine (60 mg/kg) and pentobarbital sodium (50 mg/kg), intubated, and connected to rodent ventilator (model 845, MiniVent, Hugo Sachs). A median sternotomy was performed, and the left coronary artery was ligated with 7‐0 silk suture and a small piece of polyethylene‐10 tubing. Mice were subjected to 45 minutes of left coronary artery ischemia. After 24 hours of reperfusion, the left ventricle area‐at‐risk and INF were determined by Evan's blue and 2,3,5‐triphenyltetrazolium chloride staining at 37°C.
Troponin I Measurement
Plasma was collected at 4 hours following reperfusion from the tail vein. Plasma levels of the cardiac‐specific isoform of troponin‐I were assessed using an ELISA kit from Life Diagnostics (West Chester, PA), as previously described. 13
Statistical Analysis
Data were analyzed in a blinded manner to the genetic background of the animals in each study until all measurements were complete. All data in this study are graphically represented as mean±SEM. Prism 6 (GraphPad Software Inc) was used for data analysis. Normal distribution was tested with the D'Agostino‐Pearson test. For 2‐group comparisons, we used a Student unpaired 2‐tailed t‐test, or a Mann–Whitney test when the data did not follow a normal distribution. For multiple group comparison, data were analyzed using ordinary 1‐way ANOVA analysis, or Kruskal–Wallis test when the data did not follow a normal distribution. Two‐way ANOVA analysis followed by a Bonferroni multiple comparison test was used when appropriate. A P value of <0.05 was considered statistically significant. Presented data may have different numbers of animals per end point because of procedural complications, limited sample collection (ie, plasma volume), or lack of participation in involuntary treadmill running. Before conducting statistical analysis, an outlier test was performed using “robust regression and outlier removal" method developed by Prism 6 with 1% aggressiveness to identify and remove any outlier in the data set.
RESULTS
Cardiac CSE mRNA and Protein Expression in EC‐CSE Tg
Real time‐quantitative polymerase chain reaction analysis revealed a 500‐fold increase in CSE mRNA expression in the heart of EC‐CSE Tg mice compared with tTA control mice (Figure 1B). This increase in CSE RNA translated to a 12‐fold increase in protein expression as quantified by western‐blot (Figure 1C).
Increased Circulating and Myocardial H2S Production in EC‐CSE Tg Mice
Figure 2 depicts plasma and myocardial levels of free H2S and sulfane sulfur. Free H2S levels were (P=0.01) increased in the plasma as shown in Figure 2A but not significantly changed in the heart as shown in Figure 2B when comparing EC‐CSE Tg mice with tTA controls. There was no statistically significant difference in free myocardial H2S levels between the 2 groups (P=NS). However, sulfane sulfur, the storage form of H2S, was increased in both plasma (P<0.0001) as shown in Figure 2C and heart tissue (P=0.0028, Figure 2D) of the EC‐CESE Tg mice compared with control. These data indicate that overexpression of CSE specifically in endothelial cells increases H2S levels and storage both locally and systemically.
Figure 2. Overexpression of cystathionine γ‐lyase in endothelial cells increased circulating and myocardial hydrogen sulfide production.
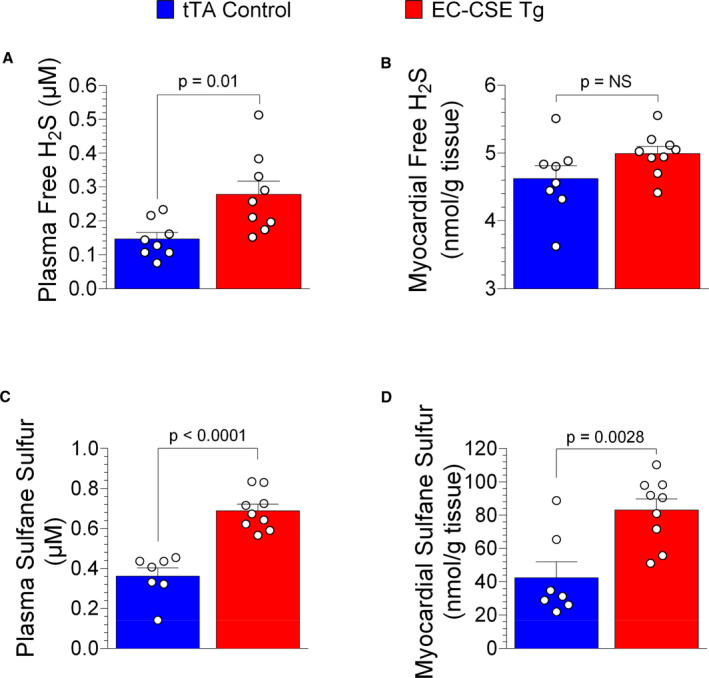
Free hydrogen sulfide levels (μmol/L) were higher in plasma (A) but not significantly changed in cardiac tissue (nmol/g) (B) of endothelial cell‐specific cystathionine γ‐lyase transgenic mice (n=9) compared with tTA (tetracycline trans‐activator) controls (n=8). Sulfane sulfur levels (μmol/L) were elevated in both circulating (C) and cardiac tissue (nmol/mg) (D) of endothelial cell‐specific cystathionine γ‐lyase transgenic mice (n=9) compared with tTA controls (n=7). Data are presented as mean±SEM, Student t‐test (A and B), Mann–Whitney U test (C and D). EC‐CSE Tg indicates endothelial cell‐specific cystathionine γ‐lyase transgenic mice; H2S, hydrogen sulfide; and tTA, tetracycline trans‐activator.
Increased Circulating and Myocardial NO Production and cGMP Levels in EC‐CSE Tg Mice
Several previous studies have shown that H2S promotes endothelial NO synthase (eNOS) activity and increases NO bioavailability. 9 , 23 We investigated whether NO levels were affected by overexpression of CSE in endothelial cells. Nitrite, a stable breakdown metabolite of NO, was measured in plasma and heart tissues. EC‐CSE Tg mice displayed increased nitrite levels in both plasma (P=0.013, Figure 3A) and heart tissue (P=0.026, Figure 3B), suggesting increased NO production in mice with endothelial cell overexpression of CSE.
Figure 3. Overexpression of cystathionine γ‐lyase in endothelial cells increased circulating and myocardial NO production.
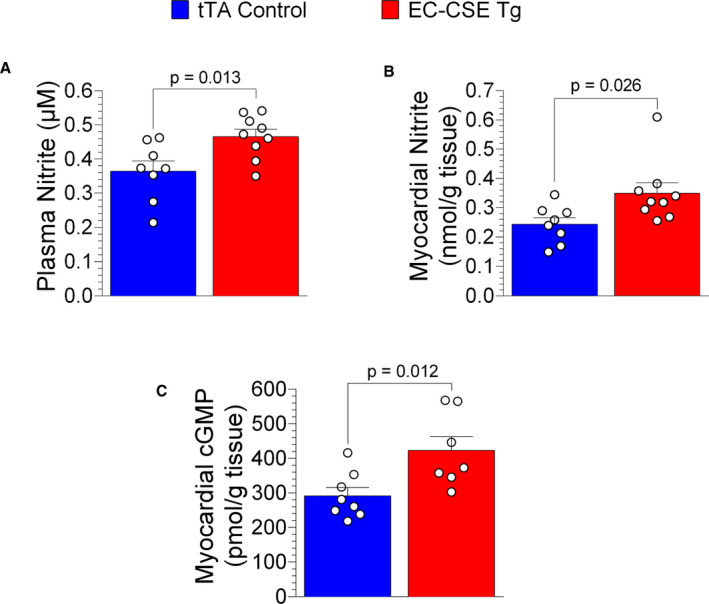
Nitrite levels (μmol/L) were increased in both plasma (A) and cardiac tissue (B) of endothelial cell‐specific cystathionine γ‐lyase transgenic mice (n=9) compared with tTA (tetracycline trans‐activator) controls (n=8). In addition, cardiac cyclic guanosine monophosphate levels (pmol/g) were higher in endothelial cell‐specific cystathionine γ‐lyase transgenic mice (n=7) than in tTA controls (n=8) (C). Data are presented as mean±SEM, Student t‐test (A), Mann–Whitney U test (B and C). cGMP, cyclic guanosine monophosphate; EC‐CSE Tg, endothelial cell‐specific cystathionine γ‐lyase transgenic mice; and tTA, tetracycline trans‐activator.
NO activates soluble guanylyl cyclase to form cGMP. Myocardial cGMP levels were also increased in EC‐CSE Tg mice compared with control (P=0.012, Figure 3C), confirming increased myocardial NO signaling in these mice.
Improved Endothelium‐Dependent Vasorelaxation Responses in EC‐CSE Tg Mice
We evaluated vascular reactivity of isolated thoracic aorta vascular rings to acetylcholine, A23187, and SNP and these data are presented in Figure 4. Aortic rings from EC‐CSE Tg mice displayed augmented vasorelaxation responses to acetylcholine as shown in concentration‐response curve and half maximal effective concentration values (Figure 4A). Aortic vascular rings from EC‐CSE Tg mice also displayed enhanced responses to the eNOS‐activating calcium ionophore, A23187 as shown in Figure 4B. In contrast, the endothelium‐independent vasorelaxation responses to SNP in thoracic aortas were not significantly different in EC‐CSE Tg mice and tTA controls (Figure 4C). These data suggest that overexpression of CSE in endothelial cells increases endothelium‐dependent vasodilation responses, possibly through activation of eNOS.
Figure 4. Overexpression of cystathionine γ‐lyase in endothelial cells augmented endothelium‐dependent vasorelaxation responses in isolated thoracic aortic rings.
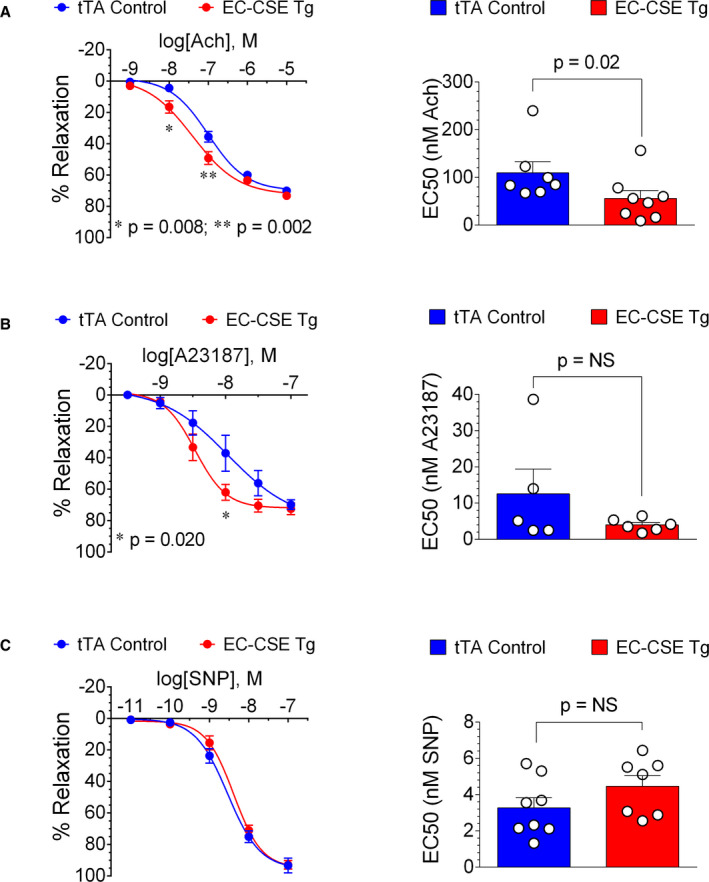
A, Concentration‐response curve shows that acetylcholine induced augmented relaxation in aortic rings from endothelial cell‐specific cystathionine γ‐lyase transgenic mice (EC‐CSE Tg) (n=8) at concentration of 10−8 and 10−7 mol/L (LEFT). In addition, half maximal effective concentration value of acetylcholine was lower in aortic rings from EC‐CSE Tg (n=8) compared with tTA (tetracycline trans‐activator) controls (n=7) (RIGHT). B, Similarly, A23187‐induced vasorelaxation was augmented in EC‐CSE Tg mice (n=5) at concentration of 10−8 mol/L (LEFT), although half maximal effective concentration value of A23187 was not significantly different in EC‐CSE Tg (n=5) compared with tTA controls (n=6) (RIGHT). C, However, neither concentration‐response curve (LEFT) nor half maximal effective concentration values (RIGHT) showed statistically significant differences in vasorelaxation response to sodium nitroprusside between EC‐CSE Tg (n=7) and tTA control (n=8) mice. Data are presented as mean±SEM, 2‐way ANOVA analysis with Bonferroni multiple comparison test was used to compare the concentration—response curves, Mann–Whitney U test was used to compare half maximal effective concentration. EC50 indicates half maximal effective concentration; EC‐CSE Tg, endothelial cell‐specific cystathionine γ‐lyase transgenic mice; SNP, sodium nitroprusside; and tTA, tetracycline trans‐activator.
Increased Exercise Capacity in EC‐CSE Tg Mice
Exercise capacity reveals important information regarding the integration of cardiac and vascular function and is important to consider as an index of overall quality‐of‐life under baseline conditions and during heart failure. We measured treadmill exercise capacity in EC‐CSE Tg mice and tTA controls under baseline conditions and these data are presented in Figure 5. EC‐CSE Tg mice displayed longer distance of running (P=0.004) in Figure 5A and time to exhaustion (P=0.0004) in Figure 5B. Furthermore, the EC‐CSE Tg mice also exhibited lower numbers of shocks per minute (P=0.003) compared with controls (Figure 5C). Body weight was not significantly different between EC‐CSE Tg and tTA controls (Figure S1A). Taken together, these data suggest augmented exercise capacity in the EC‐CSE Tg mice.
Figure 5. Overexpression of cystathionine γ‐lyase in endothelial cells increased exercise capacity.
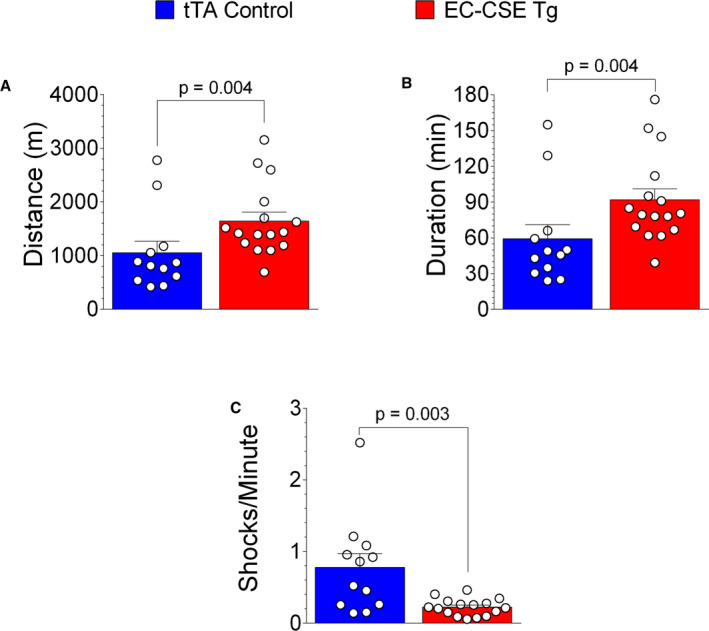
Treadmill exercise testing shows that the distance of running (meters) (A) and time to exhaustion (minutes) (B) were higher, while numbers of shocks per minute (C) were lower in endothelial cell‐specific cystathionine γ‐lyase transgenic mice (n=16) compared with tTA (tetracycline trans‐activator) controls (n=12). Data are presented as mean±SEM, Mann–Whitney U test. EC‐CSE Tg indicates endothelial cell‐specific cystathionine γ‐lyase transgenic mice; and tTA, tetracycline trans‐activator.
Myocardial Ischemia‐Reperfusion Injury is Attenuated in EC‐CSE Tg Mice
We have previously reported that H2S therapy with H2S donors significantly attenuates myocardial and hepatic ischemia‐reperfusion injury. 9 , 13 , 24 To investigate whether selective genetic overexpression of CSE in endothelial cells protects the ischemic‐reperfused myocardium, we subjected EC‐CSE Tg mice to transient coronary artery ligation and reperfusion. Mice were subjected to 45 minutes of myocardial ischemia and 24 hours of reperfusion (Figure 6A). We evaluated the influence of the promoter cdh5‐tTA with a tTA control group and also included a group of wild‐type mice as another control. Neither myocardial infarct size (INF) per area‐at‐risk nor troponin‐I levels were significantly different between cdh5‐tTA and wild‐type mice, suggesting the activator protein tTA has no influence on myocardial ischemia‐reperfusion injury. However, the EC‐CSE Tg mice displayed decrease in INF/area‐at‐risk (P<0.0001) compared with wild type or tTA controls (Figure 6B and 6C). Likewise, circulating troponin‐I levels were reduced in EC‐CSE Tg mice versus wild type (P=0.045 Figure 6D). The area of left ventricle at risk was not significantly different among all groups (P=NS). These data suggest that increased CSE expression in vascular endothelial cells protects against myocardial ischemia‐reperfusion injury, likely through increased endothelial H2S and NO production.
Figure 6. Overexpression of cystathionine γ‐lyase in endothelial cells reduced myocardial ischemia‐reperfusion injury.
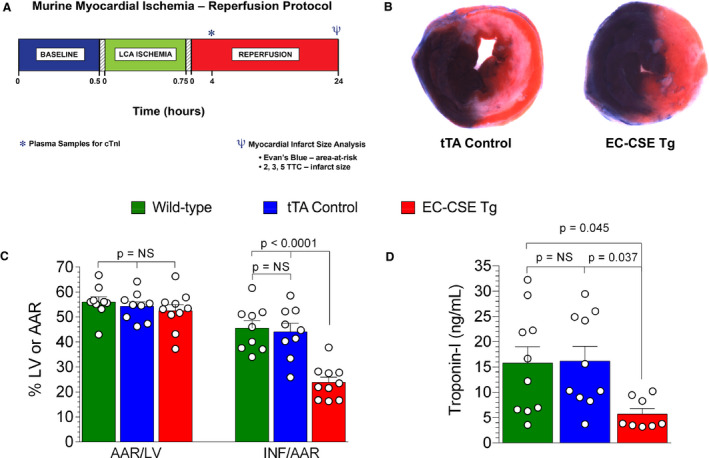
A, Myocardial ischemia‐reperfusion protocol. Mice were subjected to 45 minutes of left coronary artery (LCA) ischemia followed by 24 hours of reperfusion. Plasma samples were collected at 4 hours of reperfusion for troponin‐I measurement. Evan's blue and 2,3,5‐triphenyltetrazolium chloride staining were performed at 24 hours of reperfusion to determine the left ventricle area‐at‐risk (AAR) and infarct size. B, Representative images for left ventricle AAR and infarct size in tTA (tetracycline trans‐activator) and endothelial cell‐specific cystathionine γ‐lyase transgenic mice following myocardial ischemia‐reperfusion. C, AAR/left ventricle was not significantly different among all groups. Infarct size/AAR showed no statistically significant differences between cdh5‐tTA (n=9) and wild‐type mice (n=9), suggesting the activator protein tTA has no influence on myocardial ischemia‐reperfusion injury. However, the endothelial cell‐specific cystathionine γ‐lyase transgenic mice (n=10) displayed a significant decrease in infarct size/AAR compared with wild type. D, Plasma troponin‐1 level (a marker for myocardial injury) was not significantly changed in tTA (n=10) but reduced in endothelial cell‐specific cystathionine γ‐lyase transgenic mice (n=8) compared with wild type (n=10). Data are presented as mean±SEM, ordinary 1‐way ANOVA with Tukey multiple comparison test. AAR indicates area‐at‐risk; EC‐CSE Tg, endothelial cell‐specific cystathionine γ‐lyase transgenic mice; LCA, left coronary artery; LV, left ventricular; and tTA, tetracycline trans‐activator.
Reduced Endothelium‐Dependent Vasodilation Responses and Treadmill Exercise Capacity in EC‐CSE KO Mice
To evaluate the role of constitutive endothelial cell CSE on cardiovascular physiology and pathology, we generated conditional knockout mice with endogenous CSE deleted specifically in endothelial cells. We measured H2S, NO, vascular function, and exercise capacity in EC‐CSE KO mice and CSEflox+/flxo+ controls. As shown in Figure 7A, plasma free H2S levels were not significantly decreased in EC‐CSE KO mice compared with CSEflox+/flxo+ (P=0.15). In contrast, we observed decreased circulating sulfane sulfur levels (Figure 7B) in EC‐CSE KO mice compared with control (P=0.01). Interestingly, we also observed a trend of decreased circulating nitrite level (P=0.06. Figure 7C) in the KO compared with CSEflox+/flxo+ controls. In heart tissue, neither free H2S (Figure 8A) nor sulfane sulfur levels (Figure 8B) were significantly changed in EC‐CSE KO mice compared with control. However, cardiac nitrite levels were decreased in the knockout compared with CSEflox+/flxo+ controls (P=0.002, Figure 8C).
Figure 7. Genetic deletion of cystathionine γ‐lyase in endothelial cells decreased circulating hydrogen sulfide storage and tended to decrease circulating NO production.
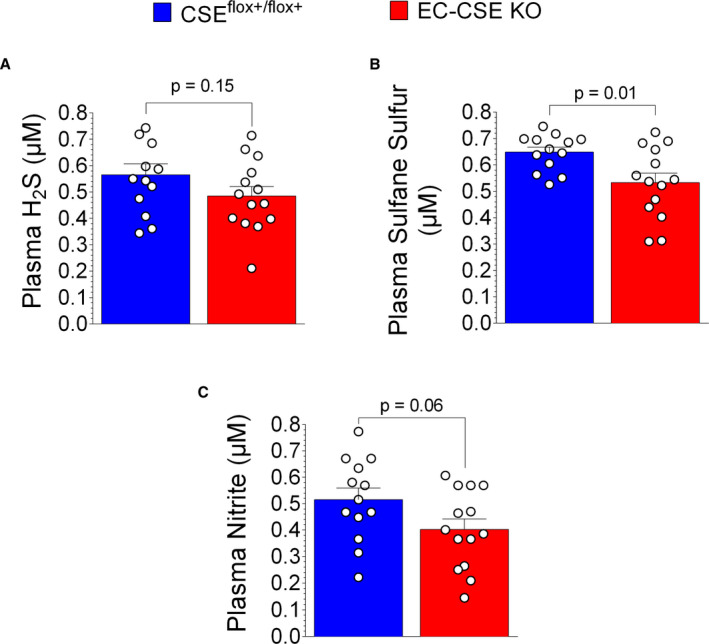
A, Plasma free hydrogen sulfide levels (μmol/L) were not significantly different between endothelial cell‐specific cystathionine γ‐lyase knockout (EC‐CSE KO, n=14) and CSEflox+/flox+ mice (n=13). B, Plasma sulfane sulfur levels (μmol/L) were decreased in EC‐CSE KO mice (n=14) compared with CSEflox+/flox+ (n=13). C, A trend of decreased circulating nitrite levels (μmol/L) were also observed in EC‐CSE KO mice (n=14) compared with CSEflox+/flox+ (n=13). Data are presented as mean±SEM, Student t‐test. CSE indicates cystathionine γ‐lyase; EC‐CSE KO, endothelial cell‐specific CSE knockout mice; and H2S, hydrogen sulfide.
Figure 8. Genetic deletion of cystathionine γ‐lyase in endothelial cells did not significantly change cardiac hydrogen sulfide levels but decreased cardiac NO production.
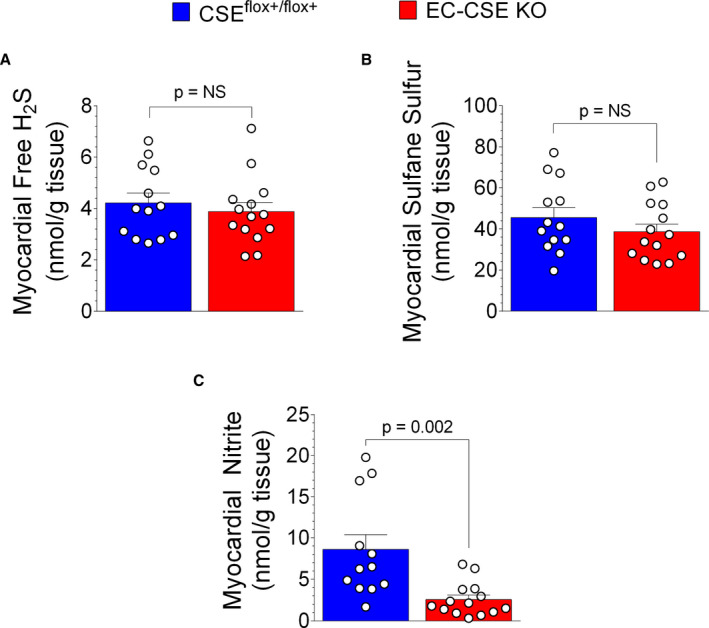
A, Cardiac free hydrogen sulfide levels (nmol/mg) were not significantly different between endothelial cell‐specific cystathionine γ‐lyase CSE knockout mice (n=14) and CSEflox+/flox+ mice (n=13). B, Cardiac sulfane sulfur levels (nmol/mg) were not significantly changed in endothelial cell‐specific CSE knockout (n=14) compared with CSEflox+/flox+ (n=13). C, However, cardiac nitrite levels (nmol/mg) were decreased in endothelial cell‐specific CSE knockout mice (n=14) compared with CSEflox+/flox+ (n=13). Data are presented as mean±SEM, Student t‐test. CSE indicates cystathionine γ‐lyase; EC‐CSE KO, endothelial cell‐specific CSE knockout mice; and H2S, hydrogen sulfide.
Analysis of isolated aortic vascular rings harvested from EC‐CSE KO mice revealed decreased relaxation responses to acetylcholine and A23187 when compared with controls (Figure 9A and 9B). In contrast we failed to observe any statistically significant differences in the vascular relaxation response to SNP as shown in concentration‐response curve and half maximal effective concentration values (Figure 9C).
Figure 9. Genetic deletion of cystathionine γ‐lyase in endothelial cells impaired endothelium‐dependent vasorelaxation responses in isolated thoracic aortic rings.
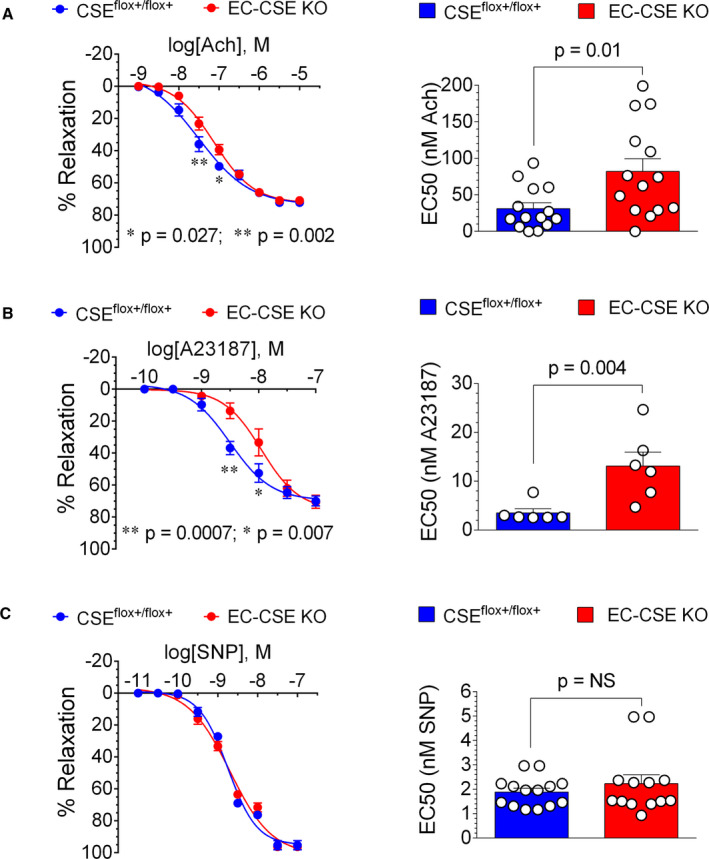
A, Concentration‐response curve shows that acetylcholine induced less relaxation (%) in aortic rings in endothelial cell‐specific cystathionine γ‐lyase knockout mice (EC‐CSE KO) (n=13) than in CSEflox+/flox+ (n=14) mice at concentration of 10−7.5 and 10−7 mol/L (LEFT). Half maximal effective concentration (EC50) value of acetylcholine was higher in aortic rings from EC‐CSE KO (n=13) compared with CSEflox+/flox+ (n=14) (RIGHT). B, A23187‐induced vasorelaxation (%) was reduced in EC‐CSE KO mice (n=6) compared with CSEflox+/flox+ (n=6) at concentration of 10−8.5 and 10−8 mol/L (LEFT). The EC50 value of A23187 was higher in EC‐CSE KO (n=6) compared with controls (n=6). C, Neither concentration‐response curve (LEFT) nor EC50 values (RIGHT) showed statistically significant differences in vasorelaxation response to sodium nitroprusside between endothelial cell‐specific cystathionine γ‐lyase knockout mice (n=13) and CSEflox+/flox+ control (n=14). Data are presented as mean±SEM, 2‐way ANOVA analysis with Bonferroni multiple comparison test was used to compare the concentration—response curves, Student t‐test was used to compare EC50 (A and C), Mann–Whitney U test was used to compare EC50 (B). CSE indicates cystathionine γ‐lyase; EC50, half maximal effective concentration; EC‐CSE KO, endothelial cell‐specific CSE knockout mice; and SNP, sodium nitroprusside.
Moreover, treadmill exercise capacity testing (Figure 10) revealed that EC‐CSE KO mice displayed reduced total distance of running (P=0.012) as shown in Figure 10A and reduced time to exhaustion (P=0.012) in Figure 10B. We also observed higher numbers of shocks per minute (P=0.018) in EC CSE KO mice compared with control (Figure 10C). Body weights were not significantly different in EC‐CSE KO and CSEflox+/flox+ mice (Figure S1B). Our data suggest that deletion of CSE in endothelial cells results in endothelial dysfunction, decreased vascular reactivity, and decreased exercise capacity. This decreased exercise capacity is likely because of the impairment of H2S production and downstream NO signaling.
Figure 10. Genetic deletion of cystathionine γ‐lyase in endothelial cells decreased exercise capacity.
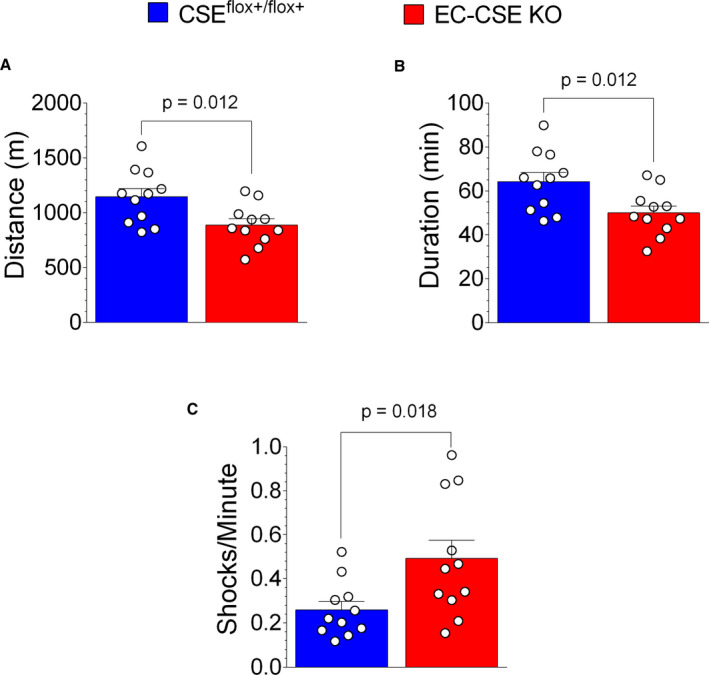
Treadmill exercise testing showed that the distance of running (meters) (A) and time to exhaustion (minutes) (B) were lower, while numbers of shocks per minute (C) were higher in endothelial cell‐specific cystathionine γ‐lyase knockout mice (n=11) compared with CSEflox+/flox+ control (n=11). Data are presented as mean±SEM, Student t‐test. CSE indicates cystathionine γ‐lyase; and EC‐CSE KO, endothelial cell‐specific CSE knockout mice.
Myocardial Ischemia‐Reperfusion Injury is Unchanged in EC‐CSE KO Mice
Global deletion of CSE has been shown to exacerbate cardiac ischemia‐reperfusion injury. 9 To examine the impact of CSE deletion specifically in endothelial cells, EC‐CSE KO mice were subjected to the same myocardial ischemia‐reperfusion injury protocol. Interestingly, no statistically significant differences in myocardial INF/area‐at‐risk were observed between EC‐CSE KO and CSEflox+/flox+ mice, as shown in Figure 11A and 11B.
Figure 11. Genetic deletion of cystathionine γ‐lyase in endothelial cells did not significantly affect myocardial ischemia‐reperfusion injury.
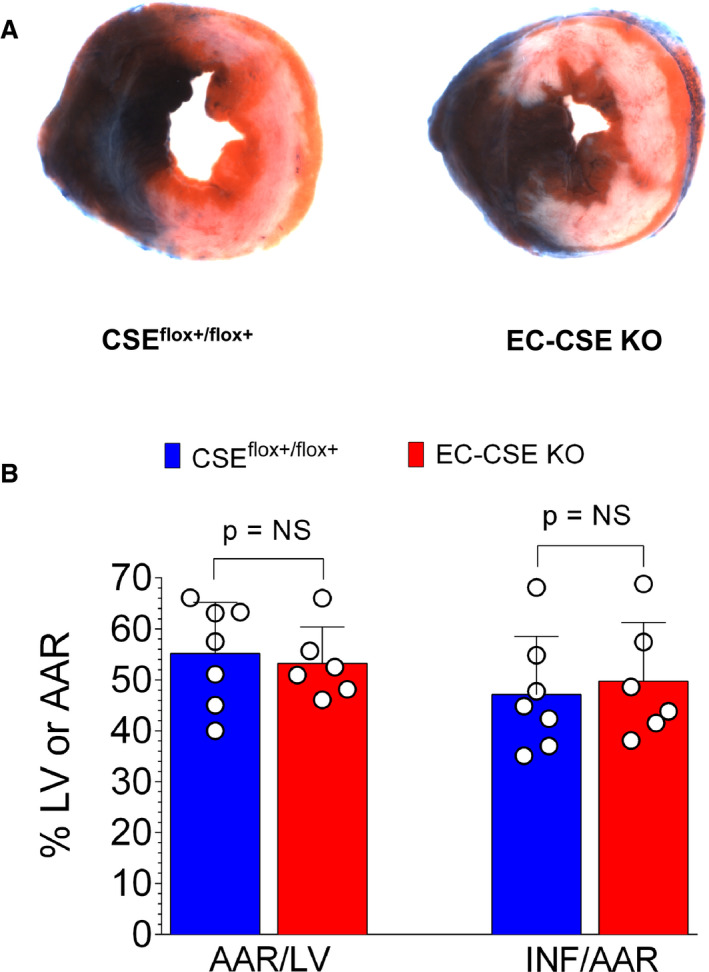
Representative heart slice images taken at the mid‐ventricular level (A) and quantified data (B) showed no statistically significant differences in area‐at‐risk/left ventricle (AAR/LV) or infarct size/area‐at‐risk (INF/AAR) between cystathionine γ‐lyase knockout mice (n=6) and CSEflox+/flox+ (n=7). Data are presented as mean±SEM, Mann–Whitney U test. AAR indicates area‐at‐risk; EC‐CSE KO, endothelial cell‐specific cystathionine γ‐lyase knockout mice; INF, infarct size; and LV, left ventricular.
DISCUSSION
Since the discovery of endogenously produced H2S in mammalian brain, 25 , 26 , 27 numerous studies have demonstrated that H2S is an important mediator of diverse physiological processes. 28 , 29 H2S is a potent gaseous signaling molecule that works in concert with NO and carbon monoxide. 30 In the cardiovascular system, H2S modulates vascular tone, promotes angiogenesis, reduces inflammation, attenuates oxidative stress, downregulates apoptosis, protects against multiple cardiovascular disease states, and rescue compromised organs secondary to cardiovascular system such as kidney in a similar manner as NO. 16 , 31 , 32
To date, the physiological functions of H2S in the cardiovascular system have been investigated primarily by using H2S donors and inhibitors. However, these studies have limitations because of the lack of cell selectivity of the compounds. Genetic modulation of H2S producing enzyme levels in a tissue specific manner is an effective means to more precisely investigate the cardiovascular actions of endogenous H2S. Using a global CSE deletion mouse model, studies from Yang et al and our group have demonstrated the critical role of endogenous H2S derived from CSE for maintaining normal cardiovascular function and protecting against cardiac injury. 6 , 9 To evaluate the protective effects of cardiac specific H2S, we later generated cardiomyocyte specific CSE overexpressed mice and demonstrated the cardioprotective role of endogenous H2S produced in cardiomyocytes. 12 , 13 In the current study, we sought to identify the effects of endothelial cell‐specific H2S production on cardiovascular function and the response to ischemic injury.
The adult mammalian heart is composed of several cell types, including cardiomyocytes, fibroblasts, endothelial cells, smooth muscle cells, and immune cells. In addition to cardiomyocytes, non‐cardiomyocytes are critical for cardiac homeostasis, providing the extracellular matrix, intercellular network, and other needs for efficient cardiomyocyte architecture and metabolic function. Cardiomyocytes occupy 70% to 85% of the volume of the mammalian heart and account for ≈25% to 35% of cardiac cells. 33 , 34 Using combined analyses, a recent study from Pinto et al demonstrated that endothelial cells are the most abundant cell population in human and mouse hearts (≈50% versus ≈30% cardiomyocytes). 35 The increase in CSE mRNA (≈500‐fold) and protein levels (≈12‐fold) in total heart tissue samples resulting from overexpression in endothelial cells in the current study confirm that endothelial cells comprise a significant proportion of total cardiac cells. Importantly, increased CSE protein expression resulted in increased H2S production, as evidenced by increased free H2S in the circulation and increased sulfane sulfur levels in the heart and circulation. Although CSE protein expression in isolated vascular tissue was not measured, we can speculate that increased circulating H2S levels in EC‐CSE Tg mice are also attributed to increased CSE expression in the endothelium of peripheral vessels. Conversely, deletion of CSE in endothelial cells resulted in decreased circulating sulfane sulfur levels in EC‐CSE mice, confirming that endothelial CSE is critical for endogenous H2S production. However, cardiac H2S and sulfane sulfur levels were not significantly affected by CSE deletion in endothelial cells, suggesting that the sources of cardiac H2S generation are not limited to endothelial CSE. CSE expression in other cells, such as cardiomyocytes, and the other H2S producing enzymes cystathionine β‐synthase and 3‐MST, also contribute to H2S production in the heart.
We also observed increased NO bioavailability following CSE overexpression in endothelial cells. H2S and NO share a wide range of physical properties and physiological functions. Growing evidence has shown that these 2 gaseous signaling molecules interact to regulate the biosynthesis of NO and H2S as well cellular function. Both exogenously and endogenously derived H2S have been shown to promote NO bioavailability and signaling. 9 , 23 , 36 On the other hand, NO donors increase CSE expression and H2S production. 37 In endothelial cells, NO is synthesized from L‐arginine by the endothelial isoform of NO synthase. NO diffuses through the endothelial cell membrane into smooth muscle cells where it activates soluble guanylyl cyclase and produces the second messenger cGMP, which regulates vascular tone, blood flow, and platelet function. 38 In the current study, overexpression of CSE in endothelial cells led to increased circulating and myocardial nitrite levels as well as myocardial cGMP in EC‐CSE Tg mice. This finding suggests that an increase in endogenous H2S production, specifically in endothelial cells, augments NO bioavailability in the cardiovascular system. These results also suggest that H2S generated in endothelial cells could provide beneficial effects in the cardiovascular system via NO signaling. However, deletion of CSE in endothelial cells only resulted in a trend of decreased circulating nitrite level. The mild change in circulating nitrite level could result from a mild decrease in circulating H2S production (no significant change in free H2S and 18% decrease in sulfane sulfur levels in EC‐CSE KO mice). In addition, NO is not only generated in endothelium. Besides eNOS, other NO Synthases also contribute to NO production in circulation. Deletion of CSE in endothelial cells might impair eNOS function, but might not affect neuronal NO synthase function. As a result, the total circulating NO levels were not dramatically decreased following EC‐CSE deletion. Interestingly, despite no significant changes in cardiac free H2S and sulfane sulfur levels, cardiac nitrite levels were reduced by CSE deletion in endothelial cells. These data suggest that cardiac NO is mainly generated in endothelial cells. Deletion of CSE in endothelial cells impaired eNOS function in the heart therefore decreasing local NO production.
We also assessed the impact of endothelial generated H2S on vascular function. It is postulated that the primary action of H2S in the vasculature is vasodilation. It has been shown that global deletion of CSE resulted in decreased circulating H2S levels and impaired endothelium dependent vasodilation. 6 However, it is not clear whether endothelial cell‐specific production of H2S is critical for regulating vascular function. It has also been suggested that the vasodilatory action of H2S is mediated by eNOS generated NO. 39 , 40 By performing vascular reactivity measurements in isolated thoracic aorta of EC‐CSE Tg and EC‐CSE KO mice, we found enhanced vascular vasorelaxation responses to the endothelium‐dependent vasodilator acetylcholine and eNOS‐specific vasodilator A23187 in endothelial CSE transgenic mice. In contrast, endothelial‐dependent vasorelaxation responses were impaired by the deletion of CSE in endothelial cells. These findings provide novel evidence that endothelial cell‐specific H2S production is critical for maintaining endothelial function and normal vascular tone. Although some reports have shown that H2S mediates vasodilation responses via an endothelium‐independent mechanism, such as inhibiting cGMP degradation, 41 we did not see significant changes in the direct vascular smooth muscle relaxation response to SNP in our endothelial cells specifically modified mice. In combination, our findings of increased NO production in EC‐CSE Tg and decreased NO levels in EC‐CSE KO mice suggest that CSE‐derived H2S in endothelial cells protects vascular function, at least in part, through the eNOS—NO pathway.
Exercise capacity is influenced by cardiac contractile function, vascular function, oxygen‐carrying capacity, and peripheral muscle mass and function. 42 Systemic administration of an H2S donor has been shown to improve exercise capacity and vascular function in heart failure. 16 In the current study, although cardiac structure and function at rest were not significantly changed by CSE overexpression or deletion in endothelial cells (Figures S2 and S3), treadmill exercise capacity testing revealed that running distance, duration, and endurance were increased in mice with endothelial cell restricted overexpression of CSE and were all reduced by genetic deletion of constitutive endothelial cell CSE. In addition to endothelial function, the augmented exercise capacity in EC‐CSE Tg mice could also reflect better cardiac function, improvement of mitochondrial function, or improved oxygen delivery to skeletal muscle. All of which are previously demonstrated actions of H2S an NO. 2 , 38
It is well established that H2S exerts potent cytoprotective effects in the setting of cardiovascular diseases in various animal model systems. 31 , 43 Using transgenic mouse models, previous studies reported that systemic deficiency of CSE significantly attenuates H2S bioavailability and results in exacerbated myocardial ischemia/reperfusion injury. 6 , 44 In contrast, cardiomyocyte‐specific genetic overexpression of CSE increased endogenous cardiac H2S production and resulted in cardiac protection against acute myocardial ischemia‐reperfusion injury and ischemia‐induced heart failure. 12 , 13 Here, we investigated the effects of endothelial cell‐specific CSE modulation and H2S production on acute myocardial ischemia‐reperfusion injury. Similar to what was shown in cardiomyocyte‐specific overexpression, endothelial cell‐specific CSE overexpression significantly limited myocardial infarct size following acute myocardial ischemia reperfusion, suggesting that endothelial cell‐derived H2S production is critical in myocyte protection following ischemic insult. The mechanisms responsible for this cardiac protection, could involve, but are not limited to, the numerous mechanisms elucidated by previous studies. These include reduction in oxidative stress, 45 inflammation, 46 apoptosis 47 and mitochondrial dysfunction 13 which are associated with ischemia/reperfusion injury. Surprisingly, myocardial injury following ischemia‐reperfusion was not significantly augmented by CSE deletion in endothelial cells. Combining with unchanged cardiac H2S levels in EC‐CSE KO mice, our findings suggest that H2S from other sources also contribute to cardiac protection during ischemia‐reperfusion injury.
In summary, to our knowledge, this is the first study using tissue/cell‐specific gene modulation to address the role of endothelial cell‐specific H2S generation in the cardiovascular system. Our results provide evidence that CSE is a critical enzyme responsible for generating H2S in endothelial cells. Moreover, we demonstrated that H2S produced in endothelial cells is critical in maintaining endothelial function, exercise capacity, and protecting against myocardial ischemia/reperfusion injury. Modulation of endogenous endothelial H2S production may be a potential therapeutic strategy in the treatment of cardiovascular disease.
Sources of Funding
This work was supported by Grants from National Institutes of Health National Heart, Lung, and Blood Institute (1R01 HL146098, 1R01 HL146514, HL137711) to Dr Lefer, American Heart Association Postdoctoral Grant (20POST3520075) to Dr Li, American Heart Association Postdoctoral Grant (18POST34020143) to Dr Sharp III, and the German Research Foundation (SFB 815) to Prof Pfeilschifter.
Disclosures
None.
Supporting information
Data S1
Figures S1–S3
(J Am Heart Assoc. 2020;9:e017544 DOI: 10.1161/JAHA.120.017544.)
Supplementary Materials for this article are available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.120.017544
For Sources of Funding and Disclosures, see page 15.
References
- 1. Kanagy NL, Szabo C, Papapetropoulos A. Vascular biology of hydrogen sulfide. Am J Physiol Cell Physiol. 2017;C537–C549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Polhemus DJ, Lefer DJ. Emergence of hydrogen sulfide as an endogenous gaseous signaling molecule in cardiovascular disease. Circ Res. 2014;730–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kimura Y, Goto Y‐I, Kimura H. Hydrogen sulfide increases glutathione production and suppresses oxidative stress in mitochondria. Antioxid Redox Signal. 2010;1–13. [DOI] [PubMed] [Google Scholar]
- 4. Sarathi M, Ashley U, Lingyun W, Rui W. Hydrogen sulfide and the pathogenesis of atherosclerosis. Antioxid Redox Signal. 2014;805–817. [DOI] [PubMed] [Google Scholar]
- 5. Kabil O, Banerjee R. Enzymology of H(2)S biogenesis, decay and signaling. Antioxid Redox Signal. 2014;770–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yang G, Wu L, Jiang B, Yang W, Qi J, Cao K, Meng Q, Mustafa AK, Mu W, Zhang S, et al. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine γ‐lyase. Science. 2008;587–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Manna P, Gungor N, McVie R, Jain SK. Decreased cystathionine‐γ‐lyase (CSE) activity in livers of type 1 diabetic rats and peripheral blood mononuclear cells (PBMC) of type 1 diabetic patients. J Biol Chem. 2014;11767–11778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang C, Han J, Xiao L, Jin C‐E, Li D‐J, Yang Z. Role of hydrogen sulfide in portal hypertension and esophagogastric junction vascular disease. World J Gastroenterol. 2014;1079–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. King AL, Polhemus DJ, Bhushan S, Otsuka H, Kondo K, Nicholson CK, Bradley JM, Islam KN, Calvert JW, Tao Y‐X, et al. Hydrogen sulfide cytoprotective signaling is endothelial nitric oxide synthase‐nitric oxide dependent. Proc Natl Acad Sci USA. 2014;3182–3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mani S, Li H, Untereiner A, Wu L, Yang G, Austin RC, Dickhout JG, Lhotak S, Meng QH, Wang R. Decreased endogenous production of hydrogen sulfide accelerates atherosclerosis. Circulation. 2013;2523–2534. [DOI] [PubMed] [Google Scholar]
- 11. Bos EM, Wang R, Snijder PM, Boersema M, Damman J, Fu M, Moser J, Hillebrands J‐L, Ploeg RJ, Yang G, et al. Cystathionine γ‐lyase protects against renal ischemia/reperfusion by modulating oxidative stress. J Am Soc Nephrol. 2013;759–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Calvert J, Elston M, Nicholson C, Gundewar S, Jha S, Elrod J, Ramachandran A, Lefer D. Genetic and pharmacologic hydrogen sulfide therapy attenuates ischemia‐induced heart failure in mice. Circulation. 2010;11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Elrod JW, Calvert JW, Morrison J, Doeller JE, Kraus DW, Tao L, Jiao X, Scalia R, Kiss L, Szabo C, et al. Hydrogen sulfide attenuates myocardial ischemia‐reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci USA. 2007;15560–15565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Deanfield JE, Halcox JP, Rabelink TJ. Endothelial function and dysfunction. Circulation. 2007;1285–1295. [DOI] [PubMed] [Google Scholar]
- 15. Yannoutsos A, Levy B, Safar M, Slama G, Blacher J. Pathophysiology of hypertension: interactions between macro and microvascular alterations through endothelial dysfunction. J Hypertens. 2014;216–224. [DOI] [PubMed] [Google Scholar]
- 16. Li Z, Organ CL, Kang J, Polhemus DJ, Trivedi RK, Sharp TE III, Jenkins JS, Tao Y‐X, Xian M, Lefer DJ. Hydrogen sulfide attenuates renin angiotensin and aldosterone pathological signaling to preserve kidney function and improve exercise tolerance in heart failure. JACC Basic Transl Sci. 2018;796–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhao W, Wang R. H2S‐induced vasorelaxation and underlying cellular and molecular mechanisms. Am J Physiol Heart Circ Physiol. 2002;H474–H480. [DOI] [PubMed] [Google Scholar]
- 18. Syhr KMJ, Boosen M, Hohmann SW, Longen S, Köhler Y, Pfeilschifter J, Beck K‐F, Geisslinger G, Schmidtko A, Kallenborn‐Gerhardt W. The H2S‐producing enzyme CSE is dispensable for the processing of inflammatory and neuropathic pain. Brain Res. 2015;380–389. [DOI] [PubMed] [Google Scholar]
- 19. Predmore BL, Kondo K, Bhushan S, Zlatopolsky MA, King AL, Aragon JP, Grinsfelder DB, Condit ME, Lefer DJ. The polysulfide diallyl trisulfide protects the ischemic myocardium by preservation of endogenous hydrogen sulfide and increasing nitric oxide bioavailability. Am J Physiol Heart Circ Physiol. 2012;H2410–H2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Polhemus DJ, Li Z, Pattillo CB, Gojon G Sr, Gojon G Jr, Giordano T, Krum H. A novel hydrogen sulfide prodrug, SG1002, promotes hydrogen sulfide and nitric oxide bioavailability in heart failure patients. Cardiovasc Ther. 2015;216–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Trivedi RK, Polhemus DJ, Li Z, Yoo D, Koiwaya H, Scarborough A, Goodchild TT, Lefer DJ. Combined angiotensin receptor‐neprilysin inhibitors improve cardiac and vascular function via increased NO bioavailability in heart failure. J Am Heart Assoc. 2018;9:e008268 DOI: 10.1161/JAHA.117.008268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Elrod JW, Greer JJM, Bryan NS, Langston W, Szot JF, Gebregzlabher H, Janssens S, Feelisch M, Lefer DJ. Cardiomyocyte‐specific overexpression of NO synthase‐3 protects against myocardial ischemia‐reperfusion injury. Arterioscler Thromb Vasc Biol. 2006;1517–1523. [DOI] [PubMed] [Google Scholar]
- 23. Kondo K, Bhushan S, King AL, Prabhu SD, Hamid T, Koenig S, Murohara T, Predmore BL, Gojon G Sr, Gojon G Jr, et al. H2S protects against pressure overload‐induced heart failure via upregulation of endothelial nitric oxide synthase. Circulation. 2013;1116–1127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Jha S, Calvert JW, Duranski MR, Ramachandran A, Lefer DJ. Hydrogen sulfide attenuates hepatic ischemia‐reperfusion injury: role of antioxidant and antiapoptotic signaling. Am J Physiol Heart Circ Physiol. 2008;H801–H806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Goodwin LR, Francom D, Dieken FP, Taylor JD, Warenycia MW, Reiffenstein RJ, Dowling G. Determination of sulfide in brain tissue by gas dialysis/ion chromatography: postmortem studies and two case reports. J Anal Toxicol. 1989;105–109. [DOI] [PubMed] [Google Scholar]
- 26. Warenycia MW, Goodwin LR, Benishin CG, Reiffenstein RJ, Francom DM, Taylor JD, Dieken FP. Acute hydrogen sulfide poisoning. Demonstration of selective uptake of sulfide by the brainstem by measurement of brain sulfide levels. Biochem Pharmacol. 1989;973–981. [DOI] [PubMed] [Google Scholar]
- 27. Savage JC, Gould DH. Determination of sulfide in brain tissue and rumen fluid by ion‐interaction reversed‐phase high‐performance liquid chromatography. J Chromatogr B Biomed Sci Appl. 1990;540–545. [DOI] [PubMed] [Google Scholar]
- 28. Szabó C. Hydrogen sulphide and its therapeutic potential. Nat Rev Drug Discov. 2007;917–935. [DOI] [PubMed] [Google Scholar]
- 29. Zhang Y, Tang Z‐H, Ren Z, Qu S‐L, Liu M‐H, Liu L‐S, Jiang Z‐S. Hydrogen sulfide, the next potent preventive and therapeutic agent in aging and age‐associated diseases. Mol Cell Biol. 2013;1104–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang R. Two's company, three's a crowd: can H2S be the third endogenous gaseous transmitter? FASEB J. 2002;1792–1798. [DOI] [PubMed] [Google Scholar]
- 31. Polhemus DJ, Calvert JW, Butler J, Lefer DJ. The cardioprotective actions of hydrogen sulfide in acute myocardial infarction and heart failure. Scientifica. 2014;768607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Li Z, Polhemus DJ, Lefer DJ. Evolution of hydrogen sulfide therapeutics to treat cardiovascular disease. Circ Res. 2018;590–600. [DOI] [PubMed] [Google Scholar]
- 33. Nag AC. Study of non‐muscle cells of the adult mammalian heart: a fine structural analysis and distribution. Cytobios. 1980;41–61. [PubMed] [Google Scholar]
- 34. Bergmann O, Zdunek S, Felker A, Salehpour M, Alkass K, Bernard S, Sjostrom SL, Szewczykowska M, Jackowska T, Dos Remedios C, et al. Dynamics of cell generation and turnover in the human heart. Cell. 2015;1566–1575. [DOI] [PubMed] [Google Scholar]
- 35. Pinto AR, Ilinykh A, Ivey MJ, Kuwabara JT, D'Antoni ML, Debuque R, Chandran A, Wang L, Arora K, Rosenthal NA, et al. Revisiting cardiac cellular composition. Circ Res. 2016;400–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sojitra B, Bulani Y, Putcha UK, Kanwal A, Gupta P, Kuncha M, Banerjee SK. Nitric oxide synthase inhibition abrogates hydrogen sulfide‐induced cardioprotection in mice. Mol Cell Biochem. 2012;61–69. [DOI] [PubMed] [Google Scholar]
- 37. Zhao W, Zhang J, Lu Y, Wang R. The vasorelaxant effect of H(2)S as a novel endogenous gaseous K(ATP) channel opener. EMBO J. 2001;6008–6016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Strijdom H, Chamane N, Lochner A. Nitric oxide in the cardiovascular system: a simple molecule with complex actions. Cardiovasc J Afr. 2009;303–310. [PMC free article] [PubMed] [Google Scholar]
- 39. Coletta C, Papapetropoulos A, Erdelyi K, Olah G, Módis K, Panopoulos P, Asimakopoulou A, Gerö D, Sharina I, Martin E, et al. Hydrogen sulfide and nitric oxide are mutually dependent in the regulation of angiogenesis and endothelium‐dependent vasorelaxation. Proc Natl Acad Sci USA. 2012;9161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Hosoki R, Matsuki N, Kimura H. The possible role of hydrogen sulfide as an endogenous smooth muscle relaxant in synergy with nitric oxide. Biochem Biophys Res Commun. 1997;527–531. [DOI] [PubMed] [Google Scholar]
- 41. Bucci M, Papapetropoulos A, Vellecco V, Zhou Z, Zaid A, Giannogonas P, Cantalupo A, Dhayade S, Karalis KP, Wang R, et al. cGMP‐dependent protein kinase contributes to hydrogen sulfide‐stimulated vasorelaxation. PLoS One. 2012;9:e53319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mohammed SF, Borlaug BA, McNulty S, Lewis GD, Lin G, Zakeri R, Semigran MJ, LeWinter M, Hernandez AF, Braunwald E, et al. Resting ventricular‐vascular function and exercise capacity in heart failure with preserved ejection fraction: a RELAX trial ancillary study. Circ Heart Fail. 2014;580–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nicholson CK, Calvert JW. Hydrogen sulfide and ischemia‐reperfusion injury. Pharmacol Res. 2010;289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Calvert JW, Coetzee WA, Lefer DJ. Novel insights into hydrogen sulfide–mediated cytoprotection. Antioxid Redox Signal. 2010;1203–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Azizi F, Seifi B, Kadkhodaee M, Ahghari P. Administration of hydrogen sulfide protects ischemia reperfusion‐induced acute kidney injury by reducing the oxidative stress. Ir J Med Sci. 2016;649–654. [DOI] [PubMed] [Google Scholar]
- 46. Zanardo RCO, Brancaleone V, Distrutti E, Fiorucci S, Cirino G, Wallace JL. Hydrogen sulfide is an endogenous modulator of leukocyte‐mediated inflammation. FASEB J. 2006;2118–2120. [DOI] [PubMed] [Google Scholar]
- 47. Sivarajah A, Collino M, Yasin M, Benetti E, Gallicchio M, Mazzon E, Cuzzocrea S, Fantozzi R, Thiemermann C. Anti‐apoptotic and anti‐inflammatory effects of hydrogen sulfide in a rat model of regional myocardial I/R. Shock. 2009;267–274. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1
Figures S1–S3