

Abstract
Background
Atrial fibrillation (AF) driver mechanisms are obscured to clinical multielectrode mapping approaches that provide partial, surface‐only visualization of unstable 3‐dimensional atrial conduction. We hypothesized that transient modulation of refractoriness by pharmacologic challenge during multielectrode mapping improves visualization of hidden paths of reentrant AF drivers for targeted ablation.
Methods and Results
Pharmacologic challenge with adenosine was tested in ex vivo human hearts with a history of AF and cardiac diseases by multielectrode and high‐resolution subsurface near‐infrared optical mapping, integrated with 3‐dimensional structural imaging and heart‐specific computational simulations. Adenosine challenge was also studied on acutely terminated AF drivers in 10 patients with persistent AF. Ex vivo, adenosine stabilized reentrant driver paths within arrhythmogenic fibrotic hubs and improved visualization of reentrant paths, previously seen as focal or unstable breakthrough activation pattern, for targeted AF ablation. Computational simulations suggested that shortening of atrial refractoriness by adenosine may (1) improve driver stability by annihilating spatially unstable functional blocks and tightening reentrant circuits around fibrotic substrates, thus unmasking the common reentrant path; and (2) destabilize already stable reentrant drivers along fibrotic substrates by accelerating competing fibrillatory wavelets or secondary drivers. In patients with persistent AF, adenosine challenge unmasked hidden common reentry paths (9/15 AF drivers, 41±26% to 68±25% visualization), but worsened visualization of previously visible reentry paths (6/15, 74±14% to 34±12%). AF driver ablation led to acute termination of AF.
Conclusions
Our ex vivo to in vivo human translational study suggests that transiently altering atrial refractoriness can stabilize reentrant paths and unmask arrhythmogenic hubs to guide targeted AF driver ablation treatment.
Keywords: atrial fibrillation, fibrosis, magnetic resonance imaging, multielectrode mapping, near‐infrared optical mapping
Subject Categories: Translational Studies, Atrial Fibrillation, Magnetic Resonance Imaging (MRI)
Nonstandard Abbreviations and Acronyms
- 3D
3‐ dimensional
- AFCL
atrial fibrillation cycle length
- CE‐MRI
contrast‐enhanced magnetic resonance imaging
- DE‐CMR
delayed enhancement cardiac magnetic resonance
- MEM
multielectrode mapping
- NIOM
near‐infrared optical mapping
- PVI
pulmonary vein isolation
Clinical Perspective
What Is New?
This study demonstrates that persistent atrial fibrillation can be maintained by reentrant drivers within a fibrotic arrhythmogenic hub composed of multiple pathways, allowing beat‐to‐beat variability to obscure activation mapping.
What Are the Clinical Implications
Adenosine challenge can be used in patients with persistent atrial fibrillation to transiently stabilize reentrant drivers, unmask associated arrhythmogenic hubs and improve driver identification via multielectrode mapping.
Structural and electrophysiological features of arrhythmogenic hubs are potential targets for efficient driver ablation in patients with persistent atrial fibrillation.
Atrial fibrillation (AF) continues to pose a serious and worsening health burden. 1 Conventional cardiac ablation treatment of persistent AF suffers from low success rates, partly because of unresolved AF mechanisms. 1 Recent clinical studies suggest that ablation of persistent AF could be improved by targeting spatially stable, extrapulmonary sources of repetitive, rotational activity (reentrant AF drivers) identified by multielectrode mapping (MEM). 2 , 3 , 4 , 5 Typically, AF driver mechanisms are identified by their activation patterns and stability over time. Current clinical mapping, however, is restricted to surface‐only electrode recordings and struggles to define the intramural activity of reentrant AF drivers that span the complex 3‐dimensional (3D) human atrial wall. 6 , 7 The temporal stabilities of AF driver patterns can also be highly variable, which further impair driver visualization by clinical mapping limited by low spatial resolution and/or coverage. 4 , 5 , 8 These challenges hinder proper identification of reentrant AF drivers in patients and significantly impair the outcomes of ablation treatment. 7 , 8 , 9 , 10 Successful treatment of persistent AF is dependent on accurate identification of drivers; therefore, it is crucial to improve our understanding of AF driver mechanisms as well as translate this mechanistic understanding to enhance clinical driver ablation.
Our previous studies have demonstrated that these clinical limitations in the visualization of complete reentrant driver tracks can be overcome in the ex vivo human heart by a 3D integrated, submillimeter‐resolution approach 6 , 11 using near‐infrared optical mapping (NIOM) and 3D contrast‐enhanced magnetic resonance imaging (CE‐MRI). Data from our NIOM and ex vivo CE‐MRI studies have suggested that AF driver reentrant paths may not always follow a single track. Instead, AF driver reentrant paths might be composed of a network of several fibrotically insulated myobundles that create arrhythmogenic hubs, which contain multiple tracks. The tracks may include a primary common path for preferential conduction in addition to several possible return paths to complete the reentry. Although such common arrhythmogenic paths have the potential to serve as better targets to terminate AF, identifying them in vivo remains a significant challenge because of their variability and fibrillatory conduction. We rationalized that heterogeneous local atrial refractoriness, a highly dynamic feature in AF, may cause these return path variations. Therefore, we propose a conceptually novel approach to improve driver visualization through appropriate pharmacological interventions that can stabilize hidden native driver paths and minimize regions of functional block.
Hence, we chose adenosine, an endogenous metabolite of the heart, 12 as a pharmacological challenge of refractoriness to reduce beat‐to‐beat variability. We have previously shown that AF induced during adenosine perfusion is driven by stable intramural reentries along consistent common micro‐anatomic tracks. 13 Moreover, compared to other antiarrhythmic drugs with slow metabolism, adenosine challenge has a transient, recoverable effect on atrial electrophysiology and can be efficiently used to rapidly scan through refractory periods 12 and wavelengths of reentry to challenge the stability of reentrant driver paths. Adenosine bolus was used previously to augment AF frequency and inducibility 12 , 14 and to unmask dormant conduction across pulmonary vein isolation lesion sets. 15 , 16 However, the utility of adenosine in unmasking extrapulmonary AF driver activation patterns during multielectrode mapping to improve AF driver ablation has not been reported previously.
Based on these findings, we hypothesize that modulating atrial refractoriness with adenosine can temporarily stabilize native reentry paths within their fibrotic arrhythmogenic hubs and improve visualization of hidden drivers in patients with persistent AF. In this study, we utilized a novel integrated approach to evaluate ex vivo human hearts with histories of AF and cardiac disease. We used MEM, high‐resolution subsurface NIOM approaches, integrated 3D structural imaging, and heart‐specific 3D computational simulations to gain mechanistic insights as to how adenosine might affect AF driver path visualization. Computational simulations allow the manipulation of driver structural substrate, which cannot be done experimentally. We show that AF driver reentrant paths may not always follow a single track but instead are composed of a network of several fibrotically insulated myobundles creating arrhythmogenic hubs. These hubs may be composed of a primary common path for preferential conduction and several possible return paths to complete the reentry. Our novel approach provides mechanistic support to the use of pharmacologic challenge to aid clinical MEM visualization of critical common paths by stabilizing the return path and improving reentry stability within arrhythmogenic hubs.
METHODS
Data that support the findings of this study are available from the corresponding author upon reasonable request. Extended methods can be found in Data S1. Figure S1 shows a general flowchart for the ex vivo and in vivo experimental protocols.
Ex Vivo Functionally and Structurally Mapped Human Atria and Subsequent Image‐Based 3D Computational Modeling
Human hearts (n=7) were obtained from the Ohio State University Cardiac Transplant Team and LifeLine of Ohio Organ Procurement Organization in accordance with the Ohio State University Institutional Review Board. The characteristics and experimental protocols for the ex vivo hearts are shown in Tables S1 through S4. Hearts were coded with 6‐digit case numbers that blinded investigators to the clinical etiology of the hearts until data analysis was complete. Although some AF mapping data from 2 hearts were included in previous publications (Table S1), we emphasize that the analyses and all data from other ex vivo hearts presented in the current manuscript have never been published before, including the new computer models used for the in‐silico simulations.
Intact human biatrial preparations were isolated, coronary perfused, immobilized with blebbistatin, and stained with the voltage sensitive dye Di‐4‐ANDBQBS, as previously described. 11 , 13 Induction of sustained AF by burst pacing was attempted in all hearts at baseline conditions in the absence of all pharmacologic stimulation. If AF was uninducible or spontaneously terminated, consistent perfusion of isoproterenol was titrated from 1 to 10 nmol/L, and AF induction was tested at each concentration (Table S3). To avoid any recovery of contractile motion at higher doses, isoproterenol did not exceed 10 nmol/L, and pinacidil perfusion was then titrated from 3 to 10 μmol/L, as previously described. 11 , 13 , 17 Pinacidil perfusion did not exceed 10 μmol/L to avoid overtly shortened action potential durations. 17 Importantly, the action potential durations of all hearts under isoproterenol and/or pinacidil perfusion remained within ranges recorded in patients with persistent AF. 18 , 19 Nonintact atria or intact atria in which AF could not be sustained were excluded from the study. Seven ex vivo hearts met the inclusion criteria and sustained AF was mapped by NIOM (0.33–0.9 mm2 resolution). Simultaneously, clinically relevant MEM (9×9 mm2 resolution, customized flattened FIRMap catheter) with 64 electrodes was used in 4 hearts, as previously described (Figure 1). 6 , 11 , 13 Optical activation times were defined as the maximum positive derivative of the optical action potential or using 50% of the optical action potential amplitude. Initial conditions in which sustained AF was induced served as the control group for subsequent adenosine challenge (1 mL bolus, 10–100 μmol/L, n=5) or adenosine perfusion (100 μmol/L, n=1). Ex vivo drivers were defined by NIOM as localized sites with the most repetitive and recurrent reentrant or focal patterns that led neighboring regions and had a minimum temporal stability of 30%. AF driver pattern and AF cycle length (AFCL) were measured by NIOM. Targeted radiofrequency ablation at the NIOM‐defined driver site was used to confirm AF driver patterns as the sustaining AF mechanism (Figure 2).
Figure 1. Integrated functional mapping of adenosine challenge on AF driver visualization in the ex vivo human atria.
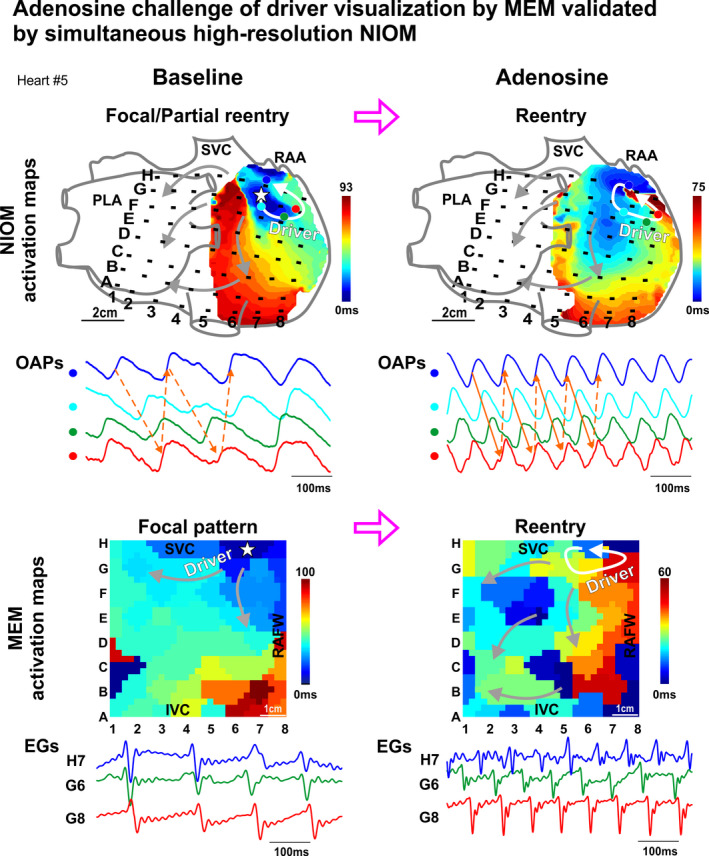
Activation maps, optical action potentials (OAPs), and electrograms (EG) recorded simultaneously from intramural near‐infrared optical mapping (NIOM) and multielectrode mapping (MEM) of the ex vivo human atria Heart #5. Stars represent focal pattern origin, white arrows represent reentry circuits, and gray arrows represent fibrillary conduction. See also Videos [Link], [Link], [Link], [Link], [Link], [Link]. AF indicates atrial fibrillation; EGs, electrograms; I/SVC, inferior/superior vena cava; OAP, optical action potential; PLA, posterior left atrium; RAA, right atrial appendage; and RAFW‐right atrial free wall.
Figure 2. Adenosine challenge during sustained AF unmasks native drivers confirmed by targeted ablation.
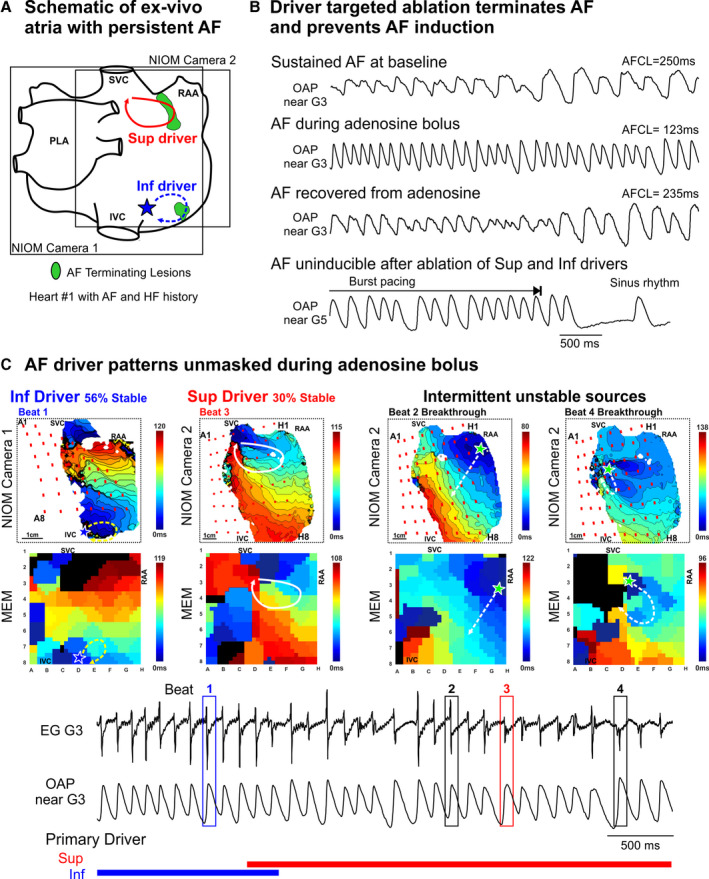
A, Schematic of intact atrial preparation Heart #1 showing optical fields of view and driver and ablation locations. Photos of ablation lesions at the conclusion of the experiment and conduction block caused by ablation lesions are shown in Figure S7. B, Optical action potentials (OAPs) during experimental protocol of AF driver identification and subsequent ablation until atrial fibrillation (AF) was uninducible. C, Top, representative near‐infrared optical mapping (NIOM) and multielectrode mapping (MEM) activation maps depicting each driver activation pattern. Bottom, Electrogram (EG) and OAP during adenosine challenge with active driver patterns across time denoted below. Numbered boxes (1–4) on electrograms denote beats used for activation maps above. HF indicates heart failure; I/SVC, inferior/superior vena cava; LA, left atrium; PLA, posterior left atrium; and RAA, right atrial appendage.
Optically mapped hearts (n=7) were scanned with high‐resolution gadolinium‐based CE‐MRI (155–184 μm3) to define detailed human atrial anatomy, 3D fibrosis distribution, myofiber orientation, and wall thickness (Figure 3A and Figure S2). 20 CE‐MRI‐detected fibrosis was defined as gadolinium‐enhanced voxels with signal intensity exceeding the threshold, found by comparing 2‐dimensional CE‐MRI sections with corresponding Masson's trichrome histology sections. Data from Heart #4 and Heart #5 were used to create biophysically accurate 3D heart‐specific computational models. The human atrial Courtemanche‐Ramirez‐Nattel model 21 with chronic AF was adapted 22 to recreate NIOM‐defined parameters for Heart #5 at baseline and during adenosine challenge (Figure 3B, Figure S3, and Tables S5, S6). 20 , 23 The effect of adenosine in the left atrium was set to be 50% of that in the right atrium based on our previous study. 13 The cellular model was then populated into heart‐specific 3D geometry with fibrosis and fiber orientation. Fibrotic tissue was considered unexcitable, 20 and the total percentage of fibrosis in the computer model was artificially increased or decreased by adjusting the CE‐MRI threshold to study the effect on fibrosis architecture on AF driver stability.
Figure 3. Heart‐specific 3D computational human atrial model of Heart #5.
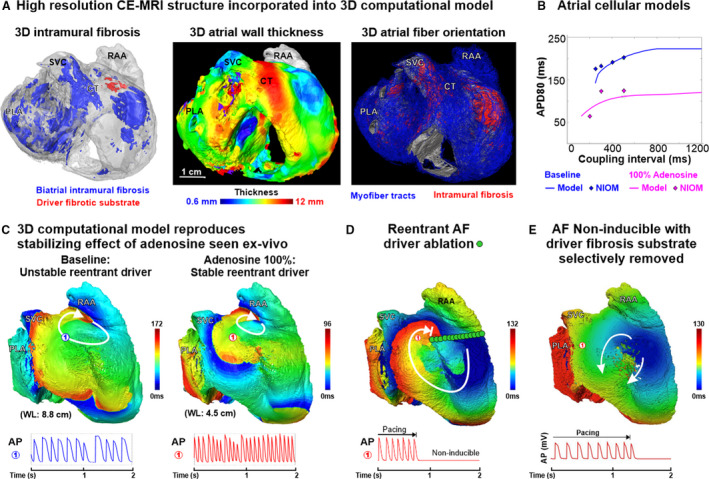
A, Contrast‐enhanced magnetic resonance imaging (CE‐MRI) showing, Left: 3‐dimensional (3D) distribution of intramural fibrosis in the driver region (red) and the whole atria (blue); middle: 3D atrial wall thickness; Right: myofiber orientation (blue) and intramural fibrosis (red), which were included in the heart‐specific computer model for ex vivo Heart #5. B, Courtemanche‐Ramirez‐Nattel atrial fibrillation (AF) cellular model 21 , 22 adapted to match near‐infrared optical mapping (NIOM) action potential duration restitution curve. C, 3D computational model based on Heart #5 shows a large reentrant driver was unstable at baseline, and adenosine shortened the wavelength and stabilized the reentrant AF driver at the same location. D, Applying an unexcitable ablation lesion through the driver arrhythmogenic hub prevents AF reinduction at 100% adenosine. E, Selectively removing the local arrhythmogenic hub fibrosis renders the reentrant AF driver not inducible. AP indicates action potential; APD80, 80% of the action potential duration; CT, crista terminalis; PLA, posterior left atrium; RAA, right atrial appendage; SVC, superior vena cava; and WL, wavelength for reentry.
Population of Patients With Persistent AF
The clinical study was approved by the Ohio State University Wexner Medical Center Institutional Review Board and the Committee on Quality and Safety. Informed consent was obtained from patients at the Ohio State University Wexner Medical Center undergoing first‐time or redo ablation of persistent AF with basket‐catheter MEM‐guided ablation. Ten patients between November 2016 and December 2018 at the Ohio State University Wexner Medical Center (n=3 first time and n=7 redo ablation) had ablation‐confirmed AF drivers, as explained later, during the procedure and were included in the current analyses. See Table S7 for patient demographics. Four of these patients consented to delayed‐enhancement cardiac magnetic resonance (DE‐CMR) before AF ablation and were enrolled in our parallel trial on AF structural substrate (NCT03444337).
In Vivo Electrophysiology Study
Pulmonary vein isolation was first established or reestablished. One‐minute unipolar recordings were taken with FIRMap™ 64‐electrode MEM catheters (50–60 mm, Abbott EP) and analyzed intraoperatively on RhythmView™ (Abbott EP). Adenosine sufficient to achieve atrioventricular block (0.15–0.2 mg/kg) 14 was injected via intravenous bolus at the onset of recording so that the first 8 to 16 seconds recorded baseline conditions (Figure S4). One‐minute recordings were divided into 15 4 seconds segments. The operator targeted both reentrant and focal patterns of AF drivers based on a subjective determination of the pattern with the highest temporal stability. Driver ablation was limited to a 1×1 to 2×1 interelectrode distance at the portion of the driver activation pattern with highest stability. If ablation led to sinus rhythm after acute termination of AF, then reinduction of AF was attempted by burst pacing. In addition to extrapulmonary driver ablation, 9 patients had roof/floor lines and/or tricuspid‐caval isthmus ablation to prevent flutter per operator's discretion. Ablation time for each patient is reported in Table S7.
Following the ablation procedure, patients were followed during a 3‐month blanking period, and AF and atrial tachycardia burdens were assessed at 12 months (n=10). No repeat ablation procedures were performed during follow‐up. The need for continuing antiarrhythmic medication was discussed between patient and physician. AF recurrence was defined as AF lasting longer than 30 seconds following the 3‐month blanking period on implanted loop recorder (n=4), event monitor (n=5), and/or 30‐day Holter monitor (n=1) due to unclear event monitor.
Postprocedure Analysis of In Vivo AF Drivers
Because MEM can also detect false‐positive drivers, 6 we did not use phase analysis and restricted in vivo AF driver definition to locations only where ablation caused acute termination to sinus rhythm, conversion to atrial tachycardia, or slowing of AFCL by ≥10% (Table S8). 5 AFCL slowing has been shown ex vivo to result from primary driver substrate destruction, yet a secondary driver present in the atria may take over at a slower rate. 11 Driver activation patterns and AFCLs were analyzed after offline annotation based on −dV/dtmax criteria of nonprocessed unipolar electrograms with a customized Matlab program. AF driver visualization patterns, including reentrant (>270° rotation), partial reentrant (>180° rotation), focal (centrifugal activation), or disordered, were defined for each AFCL by the agreement of 2 operators (Figure S5). 24 Disordered activity in the driver region entails inconsistent propagation of activation waves with questionable directionality or intermittent activation by waves originating outside the driver area. Here we report driver visualization as the percentage of time within a 4‐second segment that each driver activation pattern is visualized, rather than reporting driver stability, as any in vivo measurement of driver stability would be affected by limitations of the mapping system. Reentrant visualization stability was calculated as the sum of reentrant and partial reentrant patterns and total driver temporal stability being the sum of reentrant, partial reentrant, and focal patterns.
In Vivo Delayed‐Enhancement Cardiac Magnetic Resonance
Patients #1, 7, 8, and 10 underwent DE‐CMR using 0.2 mmol/kg gadolinium contrast agent and a 3T MAGNETOM Tim Trio or Prisma (Siemens Healthineers) at 0.625×0.625×1.25 mm3 or 0.885 mm3 (Patient #1) resolution. Visualization of the endocardial wall was aided by magnetic resonance angiography, a preceding scan with gadolinium enhanced blood pool. The epicardial wall was then segmented by hand in Amira (Thermo Fisher Scientific). Atrial fibrosis was defined as exceeding an atrial wall/blood pool intensity ratio of >1.0 to 1.2, with atrial fibrosis exceeding a patient‐specific ratio in accordance with previously published atrial fibrosis quantification studies using a similar method. 25 , 26 A 1x1cm2 region, spanning the thickness of the wall, at the driver site and the surrounding 1×1 cm2 regions composing a 3×3 cm2 grid were individually segmented and fibrosis content quantified. These 9 driver regions were compared with 111 randomly selected 1×1 cm2 regions outside the driver region within the same atrial chamber for each patient.
Statistical Analysis
Data are presented as mean±SD unless otherwise stated in figure legend. Analysis was done in R 3.4.4. Pairwise tests between condition levels were adjusted using Tukey's method. Statistical analysis was done using a general linear mixed model with heart/patient as a factor to control for multiple drivers being observed in the same heart or patient. Normality assumption was verified using Shapiro‐Wilk test. Nonparametric data were analyzed with 2‐sided Wilcoxon test. P<0.05 were considered significant. The statistical power and sample size calculations for post hoc (achieved power) and a priori (prospective studies) were done with G‐power 3.1.9.2 (University of Dusseldorf) using F‐test option at the 95% confidence level. The minimal adequate statistical power was assumed at 80% level. Specific statistical analysis can be found in Data S1.
Results
Common Paths of Reentrant AF Drivers Revealed Ex Vivo During Adenosine Challenge
Figure 1 shows NIOM and MEM activation maps of an ex vivo human atrium (Heart #5) in which adenosine improved visualization of the reentrant driver track. At baseline, without any pharmacological stimulation, NIOM revealed an intramural reentrant driver in the superior right atrium with partial reentrant and focal surface activation patterns. Activation at the driver site recurred through a common superior path, whereas returning activation took different paths each cycle (Figure 1 and related Video S1). In contrast, simultaneous MEM recordings revealed only a focal pattern (Videos S2 and S3). During 100 μmol/L adenosine perfusion, atrial refractoriness shortening stabilized a smaller full reentry path, detected by both NIOM (Video S4) and MEM (Videos S5 and S6), which incorporated the baseline common path. Even though both NIOM and MEM visualized the reentrant driver, the full AF driver path length differed from ≈7.1 cm by MEM to ≈3.7 cm by NIOM (Figure S6).
The utility of adenosine challenge in unmasking critical AF drivers in ex vivo human Heart #1 (with clinical history of AF and heart failure) and the complex hierarchical interaction between multiple drivers are shown in Figure 2. This heart had sustained AF that initiated spontaneously and at baseline had complex temporal and spatial activation patterns from 2 primary drivers in the RA interspersed with intermittent unstable sources in both atria. During adenosine challenge effect, these 2 primary drivers became more temporally stable (from 19% to 30% and from 25% to 56%, respectively). Ablation of the superior driver terminated AF, but AF was reinducible by burst pacing. Finally, ablation of the inferior driver terminated the burst‐pacing induced AF and prevented its reinduction (Figure S7). The temporal stability of intermittent unstable sources may also be affected by adenosine challenge, however, low baseline stability prevented them from sustaining AF in the absence of the primary drivers.
Overall, for 9 drivers in 6 hearts (1–2 drivers per heart), adenosine decreased AFCL from 167±50 ms to 122±41 ms (P<0.05, Table S4) but did not significantly change AFCL spatial standard deviation. The response to adenosine for each ex vivo driver is presented in Table S4. Adenosine unmasked native pathways of reentry at 6 AF driver sites with NIOM driver patterns becoming more temporally stable (adenosine‐improved group, 38±23% versus 62±27%, P<0.01). Four of these AF drivers with improved NIOM visualization also had improved MEM visualization. However, 3 stable NIOM driver patterns trended towards less temporally stablility during adenosine (adenosine‐obscured group, 77±21% versus 18±31%; statistical power was <0.8), with one also becoming less stable by MEM. Furthermore, AF drivers returned to baseline patterns after recovery from adenosine challenge. Localized radiofrequency ablation (n=5) was used to validate AF drivers as mechanisms of AF maintenance. Targeting the driver common paths terminated AF to atrial tachycardia around the lesion (n=4) or to sinus rhythm (n=1).
In an analysis done by separating our limited number of mapped hearts, we did not find a difference in driver stability response to adenosine between ex vivo human hearts with documented histories of AF (n=4) and other hearts (n=3) (P=0.83). Hearts with AF history had a slower average conduction velocity (67±11 versus 100±14 cm/s, P=0.0004), but no difference in action potential duration in the driver region (185±24 versus 177±54 ms, P=0.8) during constant atrial pacing at 300 to 500 ms cycle length under conditions when AF was sustained.
Human Heart‐Specific 3D CE‐MRI and In‐Silico Analysis of AF Drivers
High‐resolution ex vivo MRI were able to properly visualize intramural fibrosis strands >100 to 200 μm that were found to be one of the arrhythmogenic “fingerprints” of reentrant driver structural substrates by our previous work 6 , 11 , 27 and the work of others with animal models of persistent AF. 28 , 29 Total atrial fibrosis did not differ between ex vivo hearts with and without AF history (34±8 versus 29±5%, P=0.36 for the left atrium and 26±10 versus 22±5%, P=0.54 for the right atrium). The 3D CE‐MRI fibrosis analysis showed arrhythmogenic hubs of all NIOM‐defined AF drivers were composed of islands of intramural fibrotic tissue, as seen in Figures 3A, 4, and Figure S8.
Figure 4. Modulating atrial refractoriness stabilizes reentrant drivers on micro‐anatomic fibrotic substrate critical for AF maintenance.
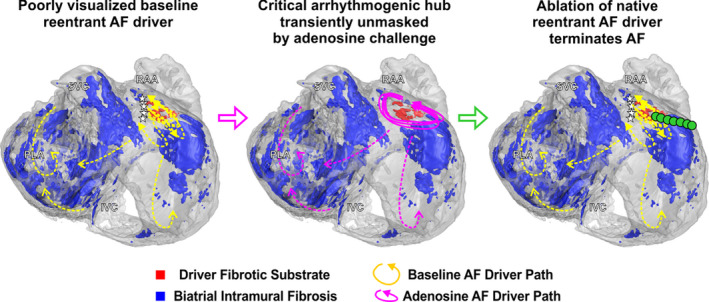
3D human atrial structure showing intramural fibrosis in the atrial fibrillation (AF) driver arrhythmogenic hub (red) and surrounding intramural fibrosis (blue) seen by contrast‐enhanced magnetic resonance imaging (CE‐MRI) in Heart #5. Consecutive reentrant paths observed ex vivo show the reentrant driver paths vary at baseline (yellow, left) and merge along a common path within the arrhythmogenic hub when beat‐to‐beat unstable functional blocks present at baseline are extinguished by adenosine (pink, middle). Stabilizing the unstable baseline driver with adenosine allows planning targeted ablation to terminate the native reentrant AF driver (right). I/SVC indicates inferior/superior vena cava; PLA, posterior left atrium; and RAA, right atrial appendage.
Human heart‐specific 3D computational models were constructed 6 , 13 , 20 incorporating all data acquired by CE‐MRI of atrial preparations Heart #4 and Heart #5 (Figure 3A). Three‐dimensional heart‐specific computational simulations were tailored to reproduce optically defined action potential durations and conduction patterns measured in Heart #5 during baseline and 100 μmol/L adenosine perfusion (Figure 3B, Figure S3, and Table S5). Baseline computational simulations of Heart #5 showed that unstable AF lasting ≈2 seconds was led mainly (75% temporal stability) by a driver at the same superior right atrial location seen by NIOM (Figure 3C). Furthermore, targeted ablation through the common reentry path in arrhythmogenic hub and connected to the tricuspid valve prevented the reinduction of arrhythmia, at both baseline and 100% adenosine conditions (Figure 3D).
Integration with CE‐MRI revealed that, in both computational simulation and NIOM, adenosine alleviated spatially unstable functional block such that reentrant driver paths fluctuating at baseline stabilized along an underlying fibrotic arrhythmogenic hub (Figure 4, Figure S9, and Videos S1, S4). Heart‐specific computational models also allowed us to test the role of local fibrotic structure on the formation of the arrhythmogenic hub through the replacement of arrhythmogenic hub fibrosis with excitable atrial tissue, which cannot be assessed experimentally. Importantly, the driver was no longer inducible when unexcitable voxels representing fibrotic substrate in the driver region were replaced by excitable atrial tissue, highlighting the possible importance of fibrosis in the maintenance of AF drivers (Figure 3E). AF driver stability was also tested in models with 70%, 100%, and 130% total fibrosis and varying concentrations of adenosine from 50% to 200% (Table S6 and Figure S10). A 30% reduction in fibrosis led to AF being noninducible in this model, because of a significant reduction of substrate at the driver region specifically. Spatially stable or unstable reentry was inducible at 100% and 130% fibrosis, while the concentration of adenosine modulated the temporal stability and cycle length. For example, cycle length was reduced from ≈133 to ≈96 ms with changes from 0% to 200% adenosine at 100% fibrosis.
Simulating adenosine challenge 100% reduced atrial refractoriness and shortened the wavelength (8.8 to 4.5 cm, similar to ex vivo changes from 8.2 to 4.6 cm), while stabilizing the reentrant AF driver at the fibrotic arrhythmogenic hub (Figure 3C). These computational simulations suggest that transient refractoriness‐shortening by adenosine can augment wavelengths of reentry until a common path stabilizes within the arrhythmogenic fibrotic hub, thus enabling detection by MEM and targeted AF ablation. However, a different response could be observed when adenosine challenged reentrant driver already stable at baseline. In this case adenosine‐induced wavelength shortening can destabilize the stable reentry, as seen in another heart‐specific computer model (Heart #4). Figure S8 shows the effect of adenosine challenge during sustained AF, maintained by a reentrant driver within fibrotic substrate in the posterior left atrium. Adding adenosine caused acute shortening of refractoriness and allowed neighboring fibrillatory wavelets to destabilize the reentrant driver, leading to further maintenance of AF by several short‐lived secondary drivers.
Challenging AF Driver Physiology in Patients With Persistent AF
Next, we explored the effectiveness of adenosine to improve AF driver detection by MEM for targeted ablation in patients with persistent AF (Table S7). Among the 10 patients included in this study, 15 AF recordings with adenosine challenge resulted in an ablation‐confirmed driver (1.5±0.7 drivers/patient, range 1–3). Because MEM can also detect false‐positive drivers, 6 in vivo AF drivers (n=15) in these 10 patients were defined only as locations where limited ablation caused acute termination to sinus rhythm (10/15 drivers), conversion to atrial tachycardia (3/15), or slowing of AFCL by ≥10% (2/15) (Table S8). 5 Patient‐specific locations of AF drivers are shown in Figure 5A. Thirteen were in the left and 2 in the right atria.
Figure 5. Clinical translation of adenosine challenge during multielectrode mapping in patients with persistent AF.
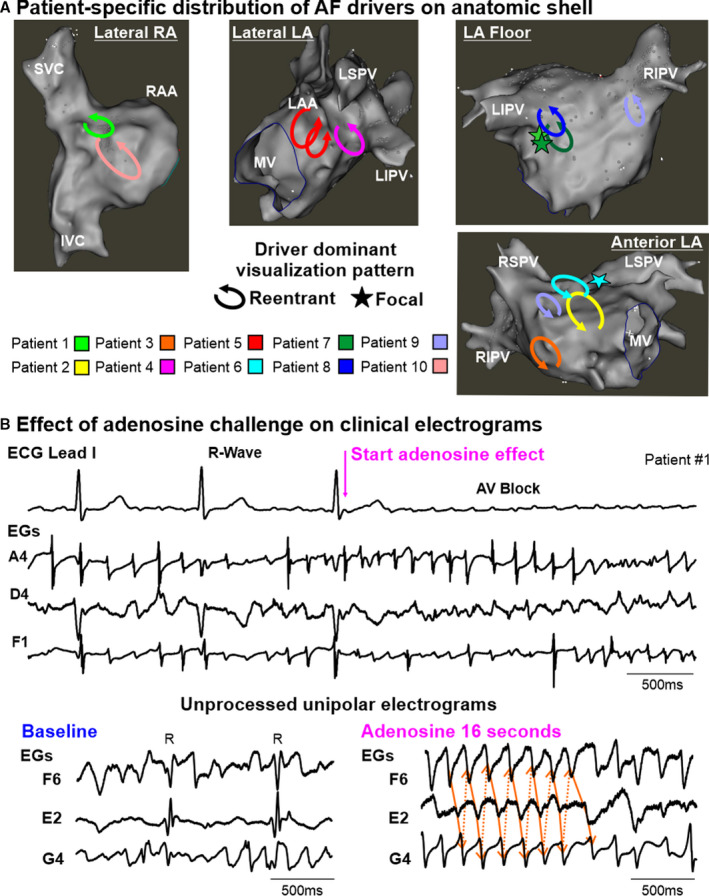
A, Distribution of atrial fibrillation (AF) drivers from all patients marked on representative electroanatomic left (LA) and right atrial (RA) shells. B, ECG lead I and unipolar electrograms (EGs) from driver and nondriver regions of Patient #1, recorded by endocardial 64‐electrode basket catheter. AV block indicates atrioventricular block; ‐dV/dtmax, point of maximum negative derivative; I/SVC, inferior/superior vena cava; LAA, left atrial appendage; LIPV/LSPV/RIPV/RSPV, left/right inferior/superior pulmonary veins; MV, mitral valve; R, ventricular R‐wave; and RAA, right atrial appendage.
Fifteen adenosine challenges improved electrogram annotation by inducing atrioventricular block lasting 7.2±6.1 seconds and reducing ventricular far‐field influence (Figure 5B). The 1‐minute AF recordings included 15 4s segments sequentially covering baseline, the full adenosine effect, and recovery from adenosine. Figures 5C and 6 show examples in the right and left atria, respectively, when temporally stable reentry was observed during adenosine. Although analysis of the same driver site during baseline showed fluctuating activation patterns with each beat. However, a repeating, common path of each baseline cycle represented a portion of the stable reentrant circuit seen during adenosine (Figure 6 and related Videos [Link], [Link], [Link], [Link], [Link]).
Figure 6. Common path of reentrant AF driver in patient with persistent AF unmasked by adenosine challenge.
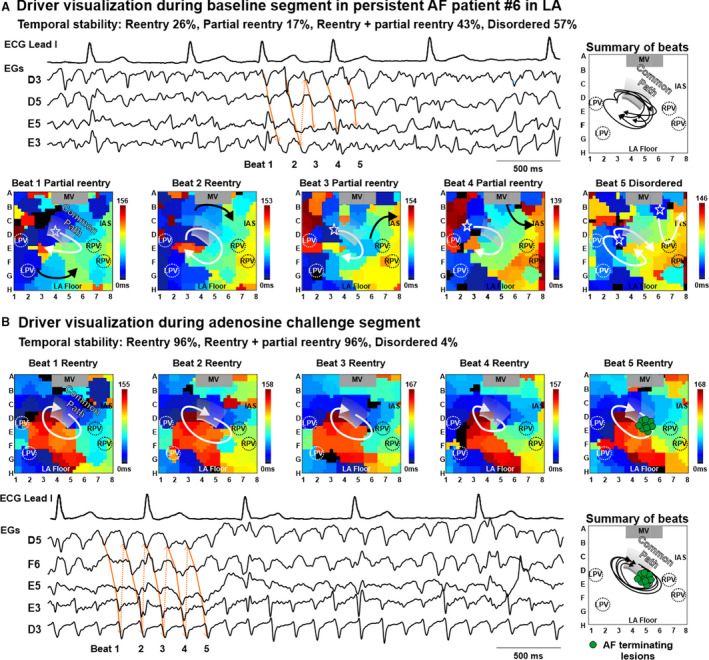
Five consecutive basket‐catheter activation maps and 4 second electrograms (EGs) showing activation patterns at the same driver location during baseline (A) and adenosine (B) in Patient #6. The adenosine segment, with 96% reentrant driver visualization was chosen to guide targeted ablation over the baseline segment when disordered activity was seen 57% of the time. See also Videos [Link], [Link], [Link], [Link], [Link]. AF indicates atrial fibrillation; IAS, interatrial septum; L/RPV, left/right pulmonary veins; LA, left atrium; LA, left atrium; and MV, mitral valve.
Analysis of all driver sites confirmed by acute ablation effect showed that 9 drivers had improved reentrant visualization during adenosine effect (adenosine‐improved group), whereas 6 drivers had better reentrant visualization during baseline conditions (adenosine‐obscured group). In the adenosine‐improved group, adenosine increased reentrant pattern stability in driver regions (from 41±26% to 68±25%, P<0.01 during baseline and adenosine, respectively), and adenosine worsened driver reentrant pattern stability in the adenosine‐obscured group (from 74±14% to 34±12%, P<0.05) (Figure 7). Logistic regression analysis of patient‐specific changes showed that a baseline reentrant visualization stability <43.5% predicted an increase by adenosine with sensitivity 89% and specificity 83% (Figure S11). Four of the 15 drivers had predominantly focal activation patterns (42±14% focal versus 25±21% reentrant) at baseline conditions, and all these drivers were visualized more often as reentry, rather than focal or disordered, during adenosine effect (48±7% reentrant versus 5±9% focal).
Figure 7. AF driver‐specific effect of adenosine challenge on driver visualization.
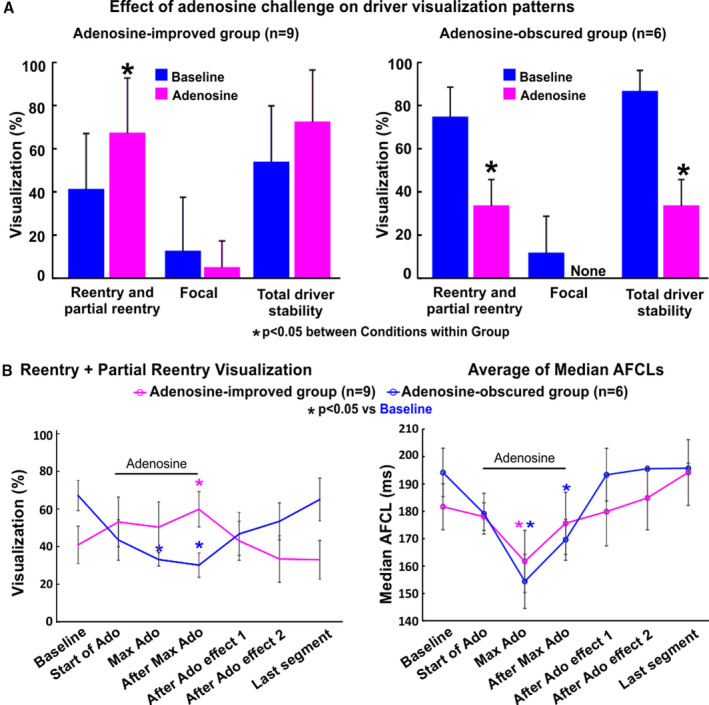
A, Driver‐specific effect of adenosine (Ado) on driver visualization pattern shows 2 distinct groups, unstable at baseline and adenosine‐improved, or stable at baseline and adenosine‐obscured. *P<0.05 between Conditions within Group. Statistical analysis was done using a general linear mixed model. Driver level responses are shown in Figure S11. B: Left, Driver visualization of reentrant and partial reentrant patterns across the 1‐minute recordings for adenosine‐improved and adenosine‐obscured groups. Right, Full‐basket average of median atrial fibrillation cycle lengths (AFCL) across the 1‐minute recording for adenosine‐improved and adenosine‐obscured groups. To normalize the time course of adenosine challenge across patients, we set start of Ado at the first 4s segment with increase in ventricular R‐R interval. Max Ado effect was by definition the segment with shortest median AFCL. After restoration of ventricular R‐R intervals to baseline values, measurements were taken at 4 seconds (After Ado Effect 1) and 8 seconds (After Ado Effect 2). Minimum time steps were 4 seconds, maximum steps did not exceed 16 seconds and the time between Baseline and Last segment did not exceed 56 seconds. Atrial fibrillation (AF) reentry visualization data are presented as average±SE. AFCL data are presented as average of medians±SE. *P<0.05 compared with baseline. Statistical analysis was done using general linear mixed model.
Figure 7B shows that time‐dependent changes in full‐basket AFCL were also associated with changes in driver reentrant visualization during adenosine challenge. The highest temporal stability of reentrant driver pattern was seen during recovery from maximum AFCL shortening, as was common for the adenosine‐improved group (Figure S4A). AF driver visualization was also seen to return to baseline values toward the end of the recording. Moreover, differentiation of adenosine‐improved or adenosine‐obscured groups did not correlate with maximum magnitude of adenosine‐induced AFCL shortening, a surrogate of refractoriness shortening (Figure 7B). Figure S4B shows that adenosine‐induced AFCL shortening was accompanied by decreasing AFCL spatial heterogeneity. This reduction in spatial heterogeneity was likely because of a narrowing of the range of AFCL measured at all electrodes toward the shorter values as baseline AFCL had a positive correlation to the reduction in AFCL due to adenosine (P<0.001). AFCL analysis across a 1‐minute recording revealed that adenosine challenge transiently allows evaluation of a range of refractoriness‐shortening (up to 35% from baseline) over 12 to 36 seconds. Adenosine decreased AFCL 38±12% and 34±10% for the 2 right atrial drivers and 10±11% for the left atrial drivers (n=13). Additional results from AFCL analysis can be found in Figure S4C.
Total radiofrequency ablation time was 37.8±19.5 minutes (Table S7). This time included 23±14.8 minutes for first time pulmonary vein isolation (n=3) and redo pulmonary vein isolation (n=4). Driver ablation (n=15) required 4.6±2.3 minutes of radiofrequency ablation, and an additional 6.2±3 minutes of ablation were used to connect driver ablations to unexcitable obstacles to prevent macro‐reentrant tachycardia (n=6), together covering 0.9±0.5% (range 0.2–1.7%) of the endocardial biatrial surface. Other linear ablations took 4.9±3.4 minutes to prevent atrial flutter.
Long‐term follow‐up after driver ablation in this cohort is presented in Table S9. At 1 year, 8 out of 10 patients did not have recurrences of atrial fibrillation. As per mutual patient and physician decision, all patients remained on antiarrhythmic medication throughout follow‐up (Table S9). Further randomized controlled trials with a larger population of patients with persistent AF are necessary to assess the clinical benefit of adenosine challenge for AF driver mapping. In this cohort, maximum driver visualization, regardless of adenosine condition, was higher in patients who were free from AF and atrial tachycardia at 1 year (86±13% versus 72±28%, P<0.001). Moreover, an ablated reentrant driver with temporal stability of >70.1% at either baseline or during adenosine challenge predicted freedom from AF at 1 year with an F1‐score of 0.89.
AF Driver Substrate Visualized in Patients With Persistent AF
DE‐CMR was performed before the ablation procedure in 4 patients to assess structural substrates underlying AF drivers defined by MEM and confirmed by ablation. All 4 clinical DE‐CMR were of sufficient quality to segment atrial walls and differentiate delayed‐enhancement voxels, a surrogate for fibrosis. Figure 8 shows a driver found in the base of the left inferior pulmonary vein with a high level of enhancement, targeted ablation through the fibrotic substrates terminated AF in this case. To prevent subsequent reentrant tachycardias in this and 12 other cases, the targeted lesion set was connected to an unexcitable obstacle, 11 such as pulmonary vein isolation lesions (Figure 8) or the superior vena cava. Retrospective analysis revealed that all driver sites contained islands of DE‐CMR fibrosis. Clusters/islands of enhancement found in driver regions were 2.4±1.4 cm2 (n=4). The percentage of enhancement at driver regions was higher than surrounding regions in the left atria (25.1±16.2% versus 14.0±21.8%, P<0.05), but no difference was found in the right atria (9.2±8.8% versus 11.2±12.8%, P=0.5). Upon further analysis, a threshold of 8.34% regional fibrosis was found to have the best f1‐score (65) and bore a modest specificity of 62.2% and sensitivity of 67.6%.
Figure 8. Targeted ablation of patient‐specific fibrotic reentrant driver substrate terminates AF.
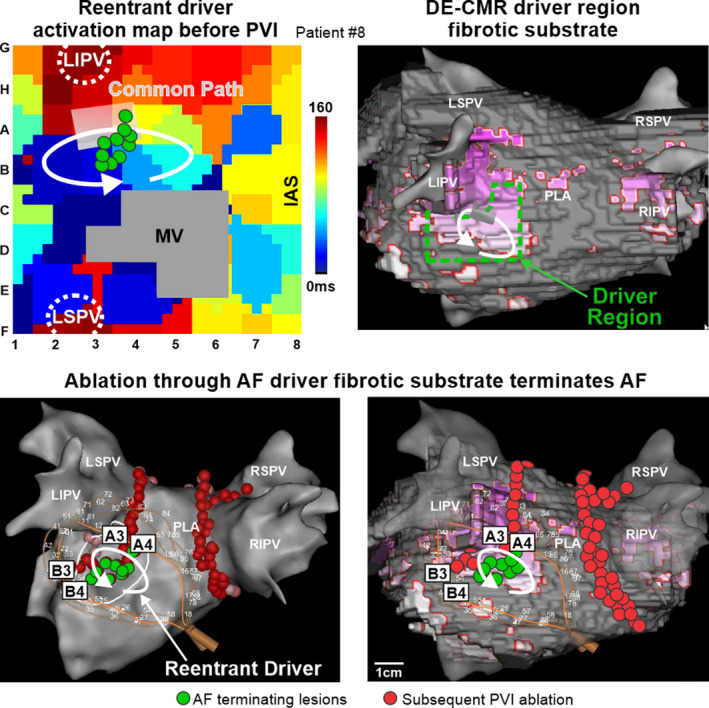
Top Left, basket‐catheter activation map of a reentrant driver in the left atria (LA) of Patient #8. Top Right, delayed‐enhancement cardiac magnetic resonance (DE‐CMR) 3D reconstructions integrated with electroanatomic maps show the relationship between enhanced voxels (purple) and the analyzed driver region (dashed green box). Bottom, 3D electroanatomic map showing location of reentrant atrial fibrillation (AF) driver, AF terminating ablation lesion placement (green dots) and basket‐catheter position (left) and with integrated DE‐CMR reconstruction (right). Red dots denote subsequent pulmonary vein isolation. IAS indicates interatrial septum; LIPV/LSPV/RIPV/RSPV, left/right inferior/superior pulmonary veins; MV, mitral valve; PLA, posterior left atrium; and PVI, pulmonary vein isolation.
Discussion
Results from our integrated ex vivo and in‐silico to in vivo studies demonstrate the existence of patient‐specific arrhythmogenic hubs created by a network of several fibrotically insulated myobundles that form a common segment of AF driver circuits with varying return paths. Mechanistically, our studies suggest that modulating refractoriness with adenosine could stabilize these return paths by decreasing beat‐to‐beat variability and thereby improve visualization of these hidden native AF driver paths (Figure 4). Furthermore, these ex vivo findings from intact human atria were translated directly to patients with persistent AF, where adenosine challenge significantly improved the visualization of native AF drivers that were previously hidden to MEM (Figure 6). Moreover, adenosine challenge also revealed the reentrant nature of AF drivers that appeared with focal surface activation patterns at baseline conditions. Notably, ablating these common reentrant paths under ex vivo, in‐silico, and in vivo conditions terminated AF, indicating that modulating driver physiology is a feasible tool to help guide catheter ablation. These findings expand our fundamental understanding of the structural physiology of human AF drivers, by identifying fibrotic arrhythmogenic hubs that sustain AF and highlight the importance of translating ex vivo mechanisms of extrapulmonary vein drivers to their in vivo assessment and localization for clinical ablation.
Clinical applications of adenosine challenge for AF driver identification could include recording panoramic AF activation movies that include baseline conditions, adenosine effect, and post‐adenosine recovery. The site with the most stable driver pattern, regardless of condition, can then be targeted with limited ablation to the driver path. If AF persists after ablation application, then a second recording that again includes all 3 conditions can be collected and the most stable driver pattern in this new recording would be targeted for ablation. This process would be repeated until acute termination of AF, conversion to atrial tachycardia, or until no more drivers are identifiable.
Clinical studies, 2 , 3 , 4 , 5 , 7 , 8 , 9 , 30 limited to surface only mapping, have reported diverse activation patterns of AF drivers, and ablation of these surface driver patterns did not always improve treatment. Currently, there is no clear consensus regarding the importance of extrapulmonary vein targets, mainly because of the inability of clinical mapping systems to reliably produce meaningful results when employed by different operators. Difficulty discriminating true AF drivers from disordered wavelets, because of multiple limitations in the clinical setting, is likely one of the causes of disparate results. 6 , 31 The challenges discussed next include the inability of current MEM to map intramural activity, the low resolution of MEM, the presence of multiple, competing AF drivers, and the beat‐to‐beat variability within AF driver patterns caused by heterogeneous refractoriness.
First, clinical surface MEM alone often misrepresents intramural reentry activation as spatially unstable fibrillatory wavelets, whereas activation patterns elsewhere may be erroneously identified as false‐positive drivers. 6 , 31 Importantly, our recent ex vivo study 6 directly validated that each of these diverse driver activation types (Figure S5), including full reentry, partial reentry, and focal patterns, seen by clinical MEM may all result from intramural reentry. Moreover, in this study all 4 in vivo drivers with predominantly focal activation patterns at baseline conditions were revealed to be reentrant patterns during adenosine effect. This finding is important for 2 reasons: (1) it reconciles a unifying mechanism underlying the discrepant results from prior clinical MEM studies that showed either reentrant or focal activation patterns; and (2) because our previous ex vivo studies showed focal breakthrough sites at the atrial surface could be 1 cm away from the intramural reentrant track, 6 , 11 , 13 the common reentrant path within the arrhythmogenic hub revealed by adenosine could represent a more efficient target. Thus, understanding that these focal MEM surface patterns represent underlying native reentrant AF drivers will allow clinical operators to plan ablation lesion sets that result in acute termination of AF.
Second, the resolution of mapping may also affect how reentrant AF drivers are visualized because of the size of the reentrant tracks. The current ex vivo results, supported by our previous ex vivo human atria studies, 6 , 11 , 13 , 20 suggest that common 3D reentrant driver tracks are on average ≈1.5×0.6 cm, with a 0.3 cm depth. Thus, a minimum resolution of 0.6 cm would be necessary to record full reentry from at least 4 points on the driver track, which was not available with the current MEM basket design and may need to be supplemented by catheters with higher resolution.
Third, we also found that the temporal stability of an AF driver can be negatively affected by the presence of other competing drivers and intermittent unstable sources. 6 , 13 The temporal stability of AF driver visualization also varies among mapping approaches 4 , 5 , 8 and baseline stability of native drivers averaged 64±22%. Although multiple drivers may be active simultaneously, it was common for one driver to induce another and then be subsequently terminated by it (Figure 2). AF drivers may trade off in this fashion reducing the total stability calculated for each, whereas AF remains sustained because of the presence of at least one driver. This finding may render the threshold of 100% temporal stability in defining a driver both futile and counterproductive, as Figure 2 shows ablation of multiple drivers may be necessary for AF termination and prevention. However, any improvement in the visualization of driver temporal stability may aid in the clinical identification of AF drivers. The intermittent visualization of reentrant drivers in patients with AF suggests that longer recording times and panoramic mapping may be necessary to ensure driver identification, which is difficult to achieve in the clinical setting. 5
Fourth, as explored in this study, beat‐to‐beat variability of the reentry path, because of a dynamic relationship between local electrophysiology and the structural arrhythmogenic hub, may decrease the perceived AF driver stability. This variability has a negative impact on AF driver identification and promotes unnecessarily extensive ablation of the entire driver pattern, whereas only a critical portion of the hub, commonly used by many reentrant paths, could represent a better target for effective ablation.
The pharmacologic challenge of AF driver refractoriness used in this study was adenosine, an endogenous metabolite of the heart, which opens G protein‐coupled inwardly rectifying potassium GIRK channels to shorten atrial refractoriness 13 , 32 and can accelerate AF activation frequency in patients. 14 , 33 Adenosine also has a well‐known safety profile, 15 , 16 , 34 and hyperpolarizing properties of adenosine have been used to restore dormant conduction across stunned atrial tissue in the gaps between ablation lesions. Ishihara et al's case study 35 found that adenosine triphosphate injection induces extra beats from the right atrial appendage of 1 patient, which induced but did not sustain AF. It is likely their adenosine triphosphate injection dropped the blood pressure in their patient, which resulted in a compensatory sympathetic release and in extra beats. 36 In contrast, our study is the first to propose arrhythmogenic hubs as the mechanistic substrate behind stable AF drivers and leverage that the repolarization shortening effect of adenosine can be used to stabilize AF drivers within these hubs. As adenosine is metabolized quickly in the blood, AF patterns return to native conditions, so native driver patterns can be ablated without delaying the procedure, and if more than one driver is present in the patient (Table S8), adenosine challenge can be repeated. Previously, we showed a heterogeneous intra‐atrial distribution of adenosine A1 receptors (A1R) and their G‐protein‐coupled potassium channels GIRK1/4 proteins across the human atria as well as diversity among heart histories, which regulate the local amplitude of adenosine‐induced repolarization shortening. 13 Thus, we propose AF driver activation patterns and their response to pharmacologic challenge depend on a complex network of structural (fibrosis, wall thickness, myofiber orientation), baseline electrophysiological, and molecular (A1R‐GIRK1/4, etc) substrates culminating in localized patient‐specific AF drivers.
Results from our translational study of AF driver dynamics provide insights into several possible mechanisms of adenosine's ability to alter temporal stability/visualization of ablation‐confirmed AF drivers. Based on our ex vivo human atrial and computational studies, it appears that transient refractoriness‐shortening by adenosine scans through a range of wavelengths until a path of reentry is stabilized within the arrhythmogenic hub, thus enabling detection by MEM and targeted AF ablation (Figure 4). Another possible mechanism includes adenosine‐induced hyperpolarization, which may also relieve unstable functional blocks and synchronize surface activation with an intramural reentrant driver previously disconnected from the atrial surface, and therefore, poorly visualized at baseline by MEM. 34 Additional improvement of in vivo driver visualization could be related to adenosine‐induced atrioventricular block that diminishes far‐field ventricular influence on atrial electrograms and aid annotation of local activation times; however, this was also observed in the adenosine‐obscured group, so it is unlikely to be the major effect of adenosine on driver visualization. Because the effects of adenosine challenge are transient, 14 the acute effect of ablation was invariably observed during baseline, suggesting that the common paths unmasked by adenosine were indeed native driver tracks and responsible, in part, for persistent AF maintenance.
Conversely, we also found that modulating refractoriness may obscure the visualization of a reentrant driver path already stable at baseline, possibly mediated through several mechanisms. An optimal relationship may exist between the micro‐anatomic fibrotic driver track and local refractoriness because adenosine's stabilizing effects were no longer observed when global fibrosis content was altered in the computer models (Figure S10 and Table S6). Thus, modulating driver physiology could disrupt this optimal relationship and obscure identification of an already stable driver. For example, adenosine challenge could accelerate short‐lived secondary drivers that are inactive during baseline conditions, which could transiently compete with/or overdrive the primary baseline driver (Figure S8). Alternately, the reentrant driver may have remained in the same location but was obscured to low‐resolution MEM because of the path length shortening and filtered out by mapping algorithms. Moreover, low‐resolution phase‐based MEM can detect false‐positive phase singularities 37 and mislead phase singularity‐based driver algorithms, but this did not occur in the current in vivo or ex vivo study, which used activation mapping to detect micro‐anatomic reentrant drivers, not phase singularities. 6 Ultimately, the combination of panoramic MEM with high‐resolution MEM may be necessary to rule out false positives and accurately guide in vivo AF driver ablation given the smaller reentrant paths in which the NIOM visualized drivers are localized in ex vivo heart studies (Figure S6). 38
Clinical and experimental studies have shown that reentrant drivers are present in regions with increased fibrotic content. 6 , 10 , 11 Our current study suggests that AF drivers are localized within heart‐specific fibrotic arrhythmogenic hubs (Figure 4). The importance of fibrosis at these hubs was suggested in‐silico where the removal of only driver fibrosis prevented the induction of sustained AF. Our data also suggest that driver stability is dictated by the composition of electrophysiologic (refractoriness and conduction velocity) and structural (fibrosis, wall thickness, myofiber orientation) 20 substrates for each driver arrhythmogenic hub (Table S6). Biatrial structural remodeling can create several arrhythmogenic hubs, which would allow persistent AF to be sustained even during fluctuations in atrial refractoriness. However, the percentage of regional fibrosis may not be sufficient for identifying arrhythmogenic hubs in the persistent AF remodeled atria for targeted ablation. Therefore, multiple components contribute to the 3D structural architecture of the hub, including fibrosis pattern, myofiber orientations, and wall thickness and may all be necessary to define these ablation targets by structural analysis. 20 At the same time, assessing the 3D architecture of arrhythmogenic hubs in human atria may prove difficult for clinical CMR 39 because of its limited resolution of 1.25 mm3. Because of the current clinical limitations, we suggest that driver prediction based on structural imaging should be complemented with functional multielectrode mapping. 6 , 27 The effect of driver ablation on atrial fibrosis is currently under investigation by our group and will be included in a future study. Our study, by improving the accuracy of AF driver detection, would also mitigate extensive ablation caused by false positive driver ablation. Accurate driver ablation that covers only 1% to 2% of the total atrial surface, as reported here and previously, 6 may mitigate atrial dysfunction possibly caused by extensive ablation.
Our data show that modulating AF driver physiology, not only refractoriness but conduction as well, by pharmacologic challenge could be a novel approach to overcome the limitations of clinical multielectrode AF driver mapping in patients. In addition to adenosine, other pharmacologic challenges, such as antiarrhythmic drugs (ibutilide 40 ) or autonomic blockers (atropine and propranolol 41 ) may be used to modulate AF driver wavelengths through atrial refractoriness or conduction to improve AF driver substrate identification. Future studies integrating DE‐CMR and computational simulation should explore how micro‐anatomic fibrotic substrates could be effectively utilized in determining which patients would benefit from which specific pharmacologic modulation of driver physiology for targeted ablation of extrapulmonary vein AF drivers in order to prevent extensive ablation procedure and preserve atrial function.
Limitations
Acute findings from this small sample of patients with persistent AF may not represent commonalities among all patients with persistent AF or mapping techniques. Only locations where limited ablation acutely affected AF pattern were considered as AF drivers. Although acute ablation success may predict long‐term outcome of treatment, 42 the long‐term clinical utility of ablating drivers that were unmasked by adenosine challenge should be rigorously tested in large randomized clinical trials. In ex vivo human hearts at baseline‐sustained AF conditions, AFCL (116 and 250 ms, ≈6Hz) was in the range reported in clinical studies 43 , 44 as well as in the clinical cohort of this study (117–225 ms). Moreover, ex vivo human action potential duration (at 500 ms cycle length pacing), ex vivo 80% repolarization=193±22 ms matches the clinical 90% repolarization ≈225±25 ms reported in patients with persistent AF by Franz et al 18 and in this study and others, which supports that our ex vivo results may represent clinical phenomena of AF and its utility in preclinical testing. Clinical relevance of AF mechanisms observed in the ex vivo human atria model is also validated by the structural similarities between the exvivo hearts used (32±7% left atrial and 24±8% right atrial fibrosis) and fibrotic remodeling shown in patients with persistent AF by histology (30–35%). 45 The physiological relevance of ex vivo conditions were also recently validated by in vivo and ex vivo NIOM of the same canine heart. 38 The effect of adenosine was not assessed twice on the same driver before ablation. Further studies on a larger number of heart‐specific computational models are necessary to establish a causative role of fibrotic architecture on arrhythmogenic hub formation. Nonstructural atrial features, such as patient‐specific vagal/sympathetic innervation and heterogeneities in adenosine receptor expression and ion channels, 13 may potentially play a role in adenosine‐induced AF changes, which will be incorporated in future computational studies.
Conclusions
In summary, our data translated from ex vivo human atria to patients with persistent AF, suggest that micro‐anatomic reentrant drivers through fibrotic arrhythmogenic hubs are one of the critical mechanisms responsible for AF maintenance. Our results show that pharmacological modulation of AF driver physiology can unmask common reentrant tracks of hidden native drivers, which could be targeted to improve ablation treatment in patients with persistent AF.
Sources of Funding
This work was supported by National Institutes of Health HL115580 and HL135109 to VVF, T32HL134616 and F30HL142179 to BJH. American Heart Association #16GRNT31010036 to VVF. Health Research Council of New Zealand to JZ. The Bob and Corrine Frick Center for Heart Failure and Arrhythmia.
Disclosures
Dr Fedorov has received research support from Abbott. Dr Hummel is a consultant to Abbott. Dr Simonetti receives research support from Siemens. Dr Mokadam is a consultant and investigator for Abbott, Medtronic, and SynCardia. The remaining authors have no disclosures to report.
Supporting information
Videos S1
Videos S2
Videos S3
Videos S4
Videos S5
Videos S6
Videos S7
Videos S8
Videos S9
Videos S10
Videos S11
Acknowledgments
We would like to thank LifeLine of Ohio Organ Procurement for assisting in donor heart collection and Abbott engineers Carey Briggs and Brian Pederson for supplying custom MatLab programs for electrogram signal annotation. Special thanks to Galina Rozenberg, Suhaib Abudulwahed, Aleksei Mikhailov, Benjamin Buck, Shane Scott, Alex Arreguin, and Annie Huang for their help in editing the manuscript. We would like to thank the Small Animal Imaging Core at OSU for the help with ex vivo CE‐MRI.
(J Am Heart Assoc. 2020;9:e017789 DOI: 10.1161/JAHA.120.017789.)
Supplementary Materials for this article are available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.120.017789
For Sources of Funding and Disclosures, see page 17.
REFERENCES
- 1. Calkins H, Hindricks G, Cappato R, Kim YH, Saad EB, Aguinaga L, Akar JG, Badhwar V, Brugada J, Camm J, et al. 2017 HRS/EHRA/ECAS/APHRS/SOLAECE expert consensus statement on catheter and surgical ablation of atrial fibrillation. Europace. 2018;20:e1–e160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Miller JM, Kalra V, Das MK, Jain R, Garlie JB, Brewster JA, Dandamudi G. Clinical benefit of ablating localized sources for human atrial fibrillation: the Indiana University FIRM Registry. J Am Coll Cardiol. 2017;69:1247–1256. [DOI] [PubMed] [Google Scholar]
- 3. Narayan SM, Krummen DE, Shivkumar K, Clopton P, Rappel WJ, Miller JM. Treatment of atrial fibrillation by the ablation of localized sources: CONFIRM (Conventional Ablation for Atrial Fibrillation With or Without Focal Impulse and Rotor Modulation) trial. J Am Coll Cardiol. 2012;60:628–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Haissaguerre M, Hocini M, Denis A, Shah AJ, Komatsu Y, Yamashita S, Daly M, Amraoui S, Zellerhoff S, Picat MQ, et al. Driver domains in persistent atrial fibrillation. Circulation. 2014;130:530–538. [DOI] [PubMed] [Google Scholar]
- 5. Kowalewski CAB, Shenasa F, Rodrigo M, Clopton P, Meckler G, Alhusseini MI, Swerdlow MA, Joshi V, Hossainy S, Zaman JAB, et al. Interaction of localized drivers and disorganized activation in persistent atrial fibrillation: reconciling putative mechanisms using multiple mapping techniques. Circ Arrhythm Electrophysiol. 2018;11:e005846 DOI: 10.1161/CIRCEP.117.005846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hansen BJ, Zhao J, Li N, Zolotarev A, Zakharkin S, Wang Y, Atwal J, Kalyanasundaram A, Abudulwahed SH, Helfrich KM, et al. Human atrial fibrillation drivers resolved with integrated functional and structural imaging to benefit clinical mapping. JACC Clin Electrophysiol. 2018;4:1501–1515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. de Groot N, van der Does L, Yaksh A, Lanters E, Teuwen C, Knops P, van de Woestijne P, Bekkers J, Kik C, Bogers A, et al. Direct proof of endo‐epicardial asynchrony of the atrial wall during atrial fibrillation in humans. Circ Arrhythm Electrophysiol. 2016;9:e003648 DOI: 10.1161/CIRCEP.115.003648. [DOI] [PubMed] [Google Scholar]
- 8. Lee S, Sahadevan J, Khrestian CM, Cakulev I, Markowitz A, Waldo AL. Simultaneous biatrial high‐density (510–512 electrodes) epicardial mapping of persistent and long‐standing persistent atrial fibrillation in patients: new insights into the mechanism of its maintenance. Circulation. 2015;132:2108–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lee G, Kumar S, Teh A, Madry A, Spence S, Larobina M, Goldblatt J, Brown R, Atkinson V, Moten S, et al. Epicardial wave mapping in human long‐lasting persistent atrial fibrillation: transient rotational circuits, complex wavefronts, and disorganized activity. Eur Heart J. 2014;35:86–97. [DOI] [PubMed] [Google Scholar]
- 10. Cochet H, Dubois R, Yamashita S, Al JN, Berte B, Sellal JM, Hooks D, Frontera A, Amraoui S, Zemoura A, et al. Relationship between fibrosis detected on late gadolinium‐enhanced cardiac magnetic resonance and re‐entrant activity assessed with electrocardiographic imaging in human persistent atrial fibrillation. JACC Clin Electrophysiol. 2018;4:17–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Hansen BJ, Zhao J, Csepe TA, Moore BT, Li N, Jayne LA, Kalyanasundaram A, Lim P, Bratasz A, Powell KA, et al. Atrial fibrillation driven by micro‐anatomic intramural re‐entry revealed by simultaneous sub‐epicardial and sub‐endocardial optical mapping in explanted human hearts. Eur Heart J. 2015;36:2390–2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Botteron GW, Smith JM. Spatial and temporal inhomogeneity of adenosine's effect on atrial refractoriness in humans: using atrial fibrillation to probe atrial refractoriness. J Cardiovasc Electrophysiol. 1994;5:477–484. [DOI] [PubMed] [Google Scholar]
- 13. Li N, Csepe TA, Hansen BJ, Sul LV, Kalyanasundaram A, Zakharkin SO, Zhao J, Guha A, Van Wagoner DR, Kilic A, et al. Adenosine‐induced atrial fibrillation: localized reentrant drivers in lateral right atria due to heterogeneous expression of adenosine A1 receptors and GIRK4 subunits in the human heart. Circulation. 2016;134:486–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Atienza F, Almendral J, Moreno J, Vaidyanathan R, Talkachou A, Kalifa J, Arenal A, Villacastin JP, Torrecilla EG, Sanchez A, et al. Activation of inward rectifier potassium channels accelerates atrial fibrillation in humans: evidence for a reentrant mechanism. Circulation. 2006;114:2434–2442. [DOI] [PubMed] [Google Scholar]
- 15. Andrade JG, Deyell MW, Nattel S, Khairy P, Dubuc M, Champagne J, Leong‐Sit P, Jolly U, Badra‐Verdu M, Sapp J, et al. Prevalence and clinical impact of spontaneous and adenosine‐induced pulmonary vein reconduction in the Contact‐Force vs. Cryoballoon Atrial Fibrillation Ablation (CIRCA‐DOSE) study. Heart Rhythm. 2020;17:897–904. [DOI] [PubMed] [Google Scholar]
- 16. van Rosmalen F, Delhaas T, Dagres N, Arbelo E, Blomstrom‐Lundqvist C, Crijns H, Da Costa A, Pytkowski M, Sharikov N, Laroche C, et al. Adenosine usage during AF ablation in Europe and selected long‐term findings from the ESC‐EHRA EORP Atrial Fibrillation Ablation Long‐Term registry. J Interv Card Electrophysiol. 2020. April 30 [epub ahead of print]. 10.1007/s10840-020-00744-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fedorov VV, Glukhov AV, Ambrosi CM, Kostecki G, Chang R, Janks D, Schuessler RB, Moazami N, Nichols CG, Efimov IR. Effects of KATP channel openers diazoxide and pinacidil in coronary‐perfused atria and ventricles from failing and non‐failing human hearts. J Mol Cell Cardiol. 2011;51:215–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Franz MR, Karasik PL, Li C, Moubarak J, Chavez M. Electrical remodeling of the human atrium: similar effects in patients with chronic atrial fibrillation and atrial flutter. J Am Coll Cardiol. 1997;30:1785–1792. [DOI] [PubMed] [Google Scholar]
- 19. Narayan SM, Kazi D, Krummen DE, Rappel WJ. Repolarization and activation restitution near human pulmonary veins and atrial fibrillation initiation: a mechanism for the initiation of atrial fibrillation by premature beats. J Am Coll Cardiol. 2008;52:1222–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhao J, Hansen BJ, Wang Y, Csepe TA, Sul LV, Tang A, Yuan Y, Li N, Bratasz A, Powell KA, et al. Three‐dimensional integrated functional, structural, and computational mapping to define the structural "fingerprints" of heart‐specific atrial fibrillation drivers in human heart ex vivo. J Am Heart Assoc. 2017;6:e005922 DOI: 10.1161/JAHA.117.005922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Courtemanche M, Ramirez RJ, Nattel S. Ionic mechanisms underlying human atrial action potential properties: insights from a mathematical model. Am J Physiol. 1998;275:H301–H321. [DOI] [PubMed] [Google Scholar]
- 22. Loewe A, Lutz Y, Wilhelms M, Sinnecker D, Barthel P, Scholz EP, Dossel O, Schmidt G, Seemann G. In‐silico assessment of the dynamic effects of amiodarone and dronedarone on human atrial patho‐electrophysiology. Europace. 2014;16(suppl_4):iv30–iv38. [DOI] [PubMed] [Google Scholar]
- 23. Krummen DE, Bayer JD, Ho J, Ho G, Smetak MR, Clopton P, Trayanova NA, Narayan SM. Mechanisms of human atrial fibrillation initiation: clinical and computational studies of repolarization restitution and activation latency. Circ Arrhythm Electrophysiol. 2012;5:1149–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zaman JAB, Sauer WH, Alhusseini MI, Baykaner T, Borne RT, Kowalewski CAB, Busch S, Zei PC, Park S, Viswanathan MN, et al. Identification and characterization of sites where persistent atrial fibrillation is terminated by localized ablation. Circ Arrhythm Electrophysiol. 2018;11:e005258 DOI: 10.1161/CIRCEP.117.005258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Benito EM, Cabanelas N, Nunez‐Garcia M, Alarcon F, Figueras I Ventura RM, Soto‐Iglesias D, Guasch E, Prat‐Gonzalez S, Perea RJ, Borras R, et al. Preferential regional distribution of atrial fibrosis in posterior wall around left inferior pulmonary vein as identified by late gadolinium enhancement cardiac magnetic resonance in patients with atrial fibrillation. Europace. 2018;20:1959–1965. [DOI] [PubMed] [Google Scholar]
- 26. Csepe TA, Zhao J, Sul LV, Wang Y, Hansen BJ, Li N, Ignozzi AJ, Bratasz A, Powell KA, Kilic A, et al. Novel application of 3D contrast‐enhanced CMR to define fibrotic structure of the human sinoatrial node in vivo. Eur Heart J Cardiovasc Imaging. 2017;18:862–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hansen BJ, Zhao J, Fedorov VV. Fibrosis and atrial fibrillation: computerized and optical mapping; a view into the human atria at submillimeter resolution. JACC Clin Electrophysiol. 2017;3:531–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Angel N, Li LI, MacLeod RS, Marrouche N, Ranjan R, Dosdall DJ. Diverse fibrosis architecture and premature stimulation facilitate initiation of reentrant activity following chronic atrial fibrillation. J Cardiovasc Electrophysiol. 2015;26:1352–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Verheule S, Tuyls E, Gharaviri A, Hulsmans S, van Hunnik A, Kuiper M, Serroyen J, Zeemering S, Kuijpers NH, Schotten U. Loss of continuity in the thin epicardial layer because of endomysial fibrosis increases the complexity of atrial fibrillatory conduction. Circ Arrhythm Electrophysiol. 2013;6:202–211. [DOI] [PubMed] [Google Scholar]
- 30. Honarbakhsh S, Schilling RJ, Dhillon G, Ullah W, Keating E, Providencia R, Chow A, Earley MJ, Hunter RJ. A novel mapping system for panoramic mapping of the left atrium: application to detect and characterize localized sources maintaining atrial fibrillation. JACC Clin Electrophysiol. 2018;4:124–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Podziemski P, Zeemering S, Kuklik P, van Hunnik A, Maesen B, Maessen J, Crijns HJ, Verheule S, Schotten U. Rotors detected by phase analysis of filtered, epicardial atrial fibrillation electrograms colocalize with regions of conduction block. Circ Arrhythm Electrophysiol. 2018;11:e005858 DOI: 10.1161/CIRCEP.117.005858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Tebbenjohanns J, Schumacher B, Pfeiffer D, Jung W, Luderitz B. Dose and rate‐dependent effects of adenosine on atrial action potential duration in humans. J Interv Card Electrophysiol. 1997;1:33–37. [DOI] [PubMed] [Google Scholar]
- 33. Nakai T, Watanabe I, Kunimoto S, Kojima T, Kondo K, Saito S, Ozawa Y, Kanmatsuse K. Electrophysiological effect of adenosine triphosphate and adenosine on atrial and ventricular action potential duration in humans. Jpn Circ J. 2000;64:430–435. [DOI] [PubMed] [Google Scholar]
- 34. Strickberger SA, Man KC, Daoud EG, Goyal R, Brinkman K, Knight BP, Weiss R, Bahu M, Morady F. Adenosine‐induced atrial arrhythmia: a prospective analysis. Ann Intern Med. 1997;127:417–422. [DOI] [PubMed] [Google Scholar]
- 35. Ishihara T, Takasugi N, Naruse G, Okura H. Adenosine‐induced atrial fibrillation arising from arrhythmogenic right atrial appendage. J Cardiovasc Electrophysiol. 2020;31:234–235. [DOI] [PubMed] [Google Scholar]
- 36. Belhassen B, Pelleg A. Electrophysiologic effects of adenosine triphosphate and adenosine on the mammalian heart: clinical and experimental aspects. J Am Coll Cardiol. 1984;4:414–424. [DOI] [PubMed] [Google Scholar]
- 37. Roney CH, Cantwell CD, Bayer JD, Qureshi NA, Lim PB, Tweedy JH, Kanagaratnam P, Peters NS, Vigmond EJ, Ng FS. Spatial resolution requirements for accurate identification of drivers of atrial fibrillation. Circ Arrhythm Electrophysiol. 2017;10:e004899 DOI: 10.1161/CIRCEP.116.004899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hansen BJ, Li N, Helfrich KM, Abudulwahed SH, Artiga EJ, Joseph ME, Mohler PJ, Hummel JD, Fedorov VV. First in vivo use of high‐resolution near‐infrared optical mapping to assess atrial activation during sinus rhythm and atrial fibrillation in a large animal model. Circ Arrhythm Electrophysiol. 2018;11:e006870 DOI: 10.1161/CIRCEP.118.006870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Marrouche NF, Wilber D, Hindricks G, Jais P, Akoum N, Marchlinski F, Kholmovski E, Burgon N, Hu N, Mont L, et al. Association of atrial tissue fibrosis identified by delayed enhancement MRI and atrial fibrillation catheter ablation: the DECAAF study. JAMA. 2014;311:498–506. [DOI] [PubMed] [Google Scholar]
- 40. Vidmar D, Narayan SM, Rappel WJ. Phase synchrony reveals organization in human atrial fibrillation. Am J Physiol Heart Circ Physiol. 2015;309:H2118–H2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Knecht S, Wright M, Matsuo S, Nault I, Lellouche N, Sacher F, Kim SJ, Morgan D, Afonso V, Shinzuke M, et al. Impact of pharmacological autonomic blockade on complex fractionated atrial electrograms. J Cardiovasc Electrophysiol. 2010;21:766–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Baykaner T, Rogers AJ, Meckler GL, Zaman J, Navara R, Rodrigo M, Alhusseini M, Kowalewski CAB, Viswanathan MN, Narayan SM, et al. Clinical implications of ablation of drivers for atrial fibrillation: a systematic review and meta‐analysis. Circ Arrhythm Electrophysiol. 2018;11:e006119 DOI: 10.1161/CIRCEP.117.006119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sanders P, Berenfeld O, Hocini M, Jais P, Vaidyanathan R, Hsu LF, Garrigue S, Takahashi Y, Rotter M, Sacher F, et al. Spectral analysis identifies sites of high‐frequency activity maintaining atrial fibrillation in humans. Circulation. 2005;112:789–797. [DOI] [PubMed] [Google Scholar]
- 44. Schuessler RB, Kay MW, Melby SJ, Branham BH, Boineau JP, Damiano RJ Jr. Spatial and temporal stability of the dominant frequency of activation in human atrial fibrillation. J Electrocardiol. 2006;39:S7–S12. [DOI] [PubMed] [Google Scholar]
- 45. Platonov PG, Mitrofanova LB, Orshanskaya V, Ho SY. Structural abnormalities in atrial walls are associated with presence and persistency of atrial fibrillation but not with age. J Am Coll Cardiol. 2011;58:2225–2232. [DOI] [PubMed] [Google Scholar]
- 46. Heijman J, Guichard JB, Dobrev D, Nattel S. Translational challenges in atrial fibrillation. Circ Res. 2018;122:752–773. [DOI] [PubMed] [Google Scholar]
- 47. Hansen BJ, Csepe TA, Zhao J, Ignozzi AJ, Hummel JD, Fedorov VV. Maintenance of atrial fibrillation: are reentrant drivers with spatial stability the key? Circ Arrhythm Electrophysiol. 2016;9:e004398 DOI: 10.1161/CIRCEP.116.004398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Maffit SK, Sellitto AD, Al‐Dadah AS, Schuessler RB, Damiano RJ Jr, Lawton JS. Diazoxide maintains human myocyte volume homeostasis during stress. J Am Heart Assoc. 2012;1:jah3 ‐e000778 DOI: 10.1161/JAHA.112.000778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhao J, Butters TD, Zhang H, LeGrice IJ, Sands GB, Smaill BH. Image‐based model of atrial anatomy and electrical activation: a computational platform for investigating atrial arrhythmia. IEEE Trans Med Imaging. 2013;32:18–27. [DOI] [PubMed] [Google Scholar]
- 50. Seemann G, Hoper C, Sachse FB, Dossel O, Holden AV, Zhang H. Heterogeneous three‐dimensional anatomical and electrophysiological model of human atria. Philos Trans A Math Phys Eng Sci. 2006;364:1465–1481. [DOI] [PubMed] [Google Scholar]
- 51. Wilhelms M, Hettmann H, Maleckar MM, Koivumaki JT, Dossel O, Seemann G. Benchmarking electrophysiological models of human atrial myocytes. Front Physiol. 2012;3:487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zhao J, Butters TD, Zhang H, Pullan AJ, LeGrice IJ, Sands GB, Smaill BH. An image‐based model of atrial muscular architecture: effects of structural anisotropy on electrical activation. Circ Arrhythm Electrophysiol. 2012;5:361–370. [DOI] [PubMed] [Google Scholar]
- 53. Gonzales MJ, Vincent KP, Rappel WJ, Narayan SM, McCulloch AD. Structural contributions to fibrillatory rotors in a patient‐derived computational model of the atria. Europace. 2014;16(suppl 4):iv3–iv10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Smaill BH, Zhao J, Trew ML. Three‐dimensional impulse propagation in myocardium: arrhythmogenic mechanisms at the tissue level. Circ Res. 2013;112:834–848. [DOI] [PubMed] [Google Scholar]
- 55. Kazbanov IV, ten Tusscher KH, Panfilov AV. Effects of heterogeneous diffuse fibrosis on arrhythmia dynamics and mechanism. Sci Rep. 2016;6:20835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zhao J, Hansen BJ, Csepe TA, Lim P, Wang Y, Williams M, Mohler PJ, Janssen PM, Weiss R, Hummel JD, et al. Integration of high‐resolution optical mapping and 3‐dimensional micro‐computed tomographic imaging to resolve the structural basis of atrial conduction in the human heart. Circ Arrhythm Electrophysiol. 2015;8:1514–1517. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Videos S1
Videos S2
Videos S3
Videos S4
Videos S5
Videos S6
Videos S7
Videos S8
Videos S9
Videos S10
Videos S11