

Abstract
Background
Cardiovascular disease is currently the leading cause of death in patients with human immunodeficiency virus on combination antiretroviral therapy. Although the use of the protease inhibitor ritonavir has been associated with increased prevalence of cardiovascular disease, the underlying mechanisms remain ill‐defined. Herein, we tested the hypothesis that ritonavir‐mediated lipoatrophy causes endothelial dysfunction via reducing endothelial leptin signaling.
Methods and Results
Long‐term (4 weeks) but not short‐term (3 days) treatment with ritonavir reduced body weight, fat mass, and leptin levels and induced endothelial dysfunction in mice. Moreover, ritonavir increased vascular NADPH oxidase 1, aortic H2O2 levels as well as interleukin‐1β, GATA3 (GATA binding protein 3), the macrophage marker (F4/80), and C‐C chemokine receptor type 5 (CCR5) expression. Reactive oxygen species scavenging with tempol restored endothelial function, and both NADPH oxidase 1 and CCR5 deletion in mice protected from ritonavir‐mediated endothelial dysfunction and vascular inflammation. Remarkably, leptin infusion markedly improved endothelial function and significantly reduced vascular NADPH oxidase 1, interleukin‐1β, GATA3, F4/80, and CCR5 levels in ritonavir‐treated animals. Selective deficiency in endothelial leptin receptor abolished the protective effects of leptin infusion on endothelial function. Conversely, selective increases in endothelial leptin signaling with protein tyrosine phosphatase deletion blunted ritonavir‐induced endothelial dysfunction.
Conclusions
All together, these data indicate that ritonavir‐associated endothelial dysfunction is a direct consequence of a reduction in adiposity and leptin secretion, which decreases endothelial leptin signaling and leads to a NADPH oxidase 1–induced, CCR5‐mediated reduction in NO bioavailability. These latter data also introduce leptin deficiency as an additional contributor to cardiovascular disease and leptin as a negative regulator of CCR5 expression, which may provide beneficial avenues for limiting human immunodeficiency virus infection.
Keywords: CCR5, HIV, leptin, lipoatrophy, NADPH oxidase, ritonavir
Subject Categories: Basic Science Research, Endothelium/Vascular Type/Nitric Oxide, Inflammation, Translational Studies, Vascular Biology
Nonstandard Abbreviations and Acronyms
- cART
combination antiretroviral therapy
- CCL5
C‐C motif chemokine ligand 5
- CCR5
C‐C chemokine receptor type 5
- D:A:D
Data Collection on Adverse Events of Anti‐HIV Drugs
- EC
endothelial cells
- GATA3
GATA binding protein 3
- IL‐1β
interleukin 1β
- LepR
leptin receptor
- l‐NAME
l
‐NG-nitro arginine methyl ester
- Nox1
NADPH oxidase 1
- Ptp1b
protein tyrosine phosphatase 1B
- PWH
patients with HIV
Clinical Perspective
What Is New?
Ritonavir causes endothelial dysfunction via lipoatrophy‐induced reduction in circulating leptin levels.
Reduction in endothelial leptin signaling impairs endothelial function via NADPH oxidase 1–induced, C‐C chemokine receptor type 5–mediated decreases in NO bioavailability; leptin negatively regulates vascular NADPH oxidase 1 and C‐C chemokine receptor type 5 expression.
Endothelial leptin signaling contributes to vascular homeostasis regulation.
What Are the Clinical Implications?
Alterations in adipose tissue endocrine function are a cause of endothelial dysfunction.
Leptin treatment restores endothelial function and reduces vascular inflammation in situations associated with leptin deficiency.
Leptin likely protects from cardiovascular disease and HIV viral infection via reducing C‐C chemokine receptor type 5 expression.
Over the past few decades, the introduction of combination antiretroviral therapy (cART) has led to a profound suppression of human immunodeficiency virus‐1 (HIV‐1) replication and increased the longevity of patients with HIV, which now approaches that of the general population. 1 , 2 As a consequence, the spectrum of diseases related to HIV has shifted from opportunistic AIDS‐related diseases towards long‐term age‐related complications. Notably, atherosclerosis‐associated cardiovascular disease (CVD) has markedly increased in patients with HIV (PWH) in the post‐cART era, and CVD is currently the leading cause of death in PWH on cART. 3 , 4 , 5 , 6 , 7 A current limitation to the prevention and treatment of accelerated atherosclerosis in PWH is our limited knowledge of the underlying mechanisms, which have been almost exclusively obtained through clinical observations. 4
Despite our limited knowledge regarding the cause of CVD in PWH, certain classes of antiretroviral drugs such as the HIV protease inhibitors have been strongly implicated in the process, and the contribution of the protease inhibitor ritonavir was pointed out by several studies. Indeed, ritonavir has been shown to have a small but statistically significant effect on the progression of carotid‐wall thickness, 8 to be associated with increased risk of myocardial infarction, 9 and to increase cardiovascular mortality of heart failure in PWH. 10 More recently, the D:A:D (Data Collection on Adverse Events of Anti‐HIV Drugs) study, a 7‐year surveillance study involving a cohort of patients from all over the world, further highlighted the potential contribution of ritonavir by indicating that ritonavir‐boosted darunavir use was associated with increased risk of CVD, with an incidence rate ratio of 1.59. 11 Ritonavir is currently a major component of second‐line cART regimens 12 , 13 , 14 but also a compound considered for the treatment of several forms of cancer, 15 including multiple myeloma, 16 for the treatment of hepatitis C, 17 and more recently for the treatment of adults hospitalized with severe coronavirus 2019. 18 Therefore, it remains critical to understand the mechanisms whereby ritonavir could promote CVD.
Previous clinical and basic science reports have associated ritonavir with lipoatrophy and dyslipidemia 9 , 19 , 20 , 21 and showed that acute in vitro exposure of vessels or endothelial cells to ritonavir impairs endothelial function via increasing oxidative stress and reducing NO bioavailability. 22 , 23 Despite numerous studies, the underlying mechanisms whereby ritonavir impairs endothelial function, a major precursor and contributor to CVD, remain poorly defined in an in vivo setting. Herein, we treated C57bl/6 mice with ritonavir for 4 weeks to test the hypothesis that ritonavir‐mediated lipoatrophy impairs endothelial function via reducing leptin‐mediated decrease in endothelial oxidative stress and inflammation.
Methods
The data that support the findings of this study are available from the corresponding author on reasonable request.
Experimental Animals
Male and female C57BL/6 mice (33 male and 10 female mice), males deficient in NADPH oxidase 1 (Nox1−/−, gift from Dr. DW. Stepp, Vascular Biology Center, Augusta University) (N=8), C‐C chemokine receptor type 5 (CCR5−/− mice, B6.129P2‐Ccr5tm1Kuz/J Jackson Laboratory) (N=8), and males with selective deletion of either Ptp1b (protein tyrosine phosphatase 1B) (N=7) or leptin receptor (LepR) (N=7), in endothelial cells (EC) have been used at 8 to 14 weeks of age. Inducible endothelial Ptp1b‐ and LepR‐deficient mice have been generated by crossing Cdh5‐CreERT2 mice (from R. Adams, Max‐Planck‐Institute) with Ptp1bflox/flox and LepRflox/flox, respectively (Jackson Laboratory). Endothelial‐specific deletion of Ptp1b or LepR was induced via activation of the endothelial‐specific Cre recombinase in 6‐ to 8‐week‐old mice by 5 for Ptp1b or 14 for LepR consecutive daily intraperitoneal injections of tamoxifen (0.1 mL of a 20 mg/mL solution in corn oil). 24 , 25 Animals receiving tamoxifen injections (−/−) were compared with vehicle (corn oil)‐treated animals (+/+).
All animals were fed standard mouse chow, and tap water was provided ad libitum. Mice were housed in an American Association of Laboratory Animal Care–approved animal care facility at Augusta University. Augusta University Institutional Animal Care and Use Committee approved all protocols (IACUC protocol #2011‐0108).
Ritonavir and Leptin Treatments
Mice were submitted to daily intraperitoneal injections of either ritonavir (5 mg/kg for 4 weeks) or vehicle as previously described. 21 , 26 After 3 weeks of ritonavir treatment, mice were separated into 2 groups and treated with either leptin (0.3 mg/kg per day, ProSpec, Israel) or vehicle via subcutaneous osmotic mini‐pumps (ALZET, Cupertino, CA; model 1007D, 0.5 μL/h) for 7 more days, as previously described by our group. 27 , 28 , 29 A short‐term ritonavir treatment (5 mg/kg for 3 days) was also used to test the potential direct effects of ritonavir on endothelial function.
Statistical Analysis
All data are presented as mean±SEM. P<0.05 were considered significant. Differences in means between 2 groups for nonrepeated variables were compared by the Student's t test. Differences in means among groups and treatments were compared by 2‐way ANOVA with repeated measures, when appropriate. Tukey test was used as the post hoc test (GraphPad).
Detailed description of the methods used is available in Data S1. The sequence of the primers is included in Table S1.
Results
Ritonavir Induces Endothelial Dysfunction Via Reducing Leptin Biosynthesis
Following 4 weeks of ritonavir treatment, male mice exhibited a lipoatrophic phenotype characterized by a significant reduction in body weight (Figure 1A), fat mass (Figure 1B), and leptin levels (Figure 1F). As reported in Figure 1B through 1E, fat mass reduction affected gonadal, subcutaneous, and perivascular adipose depots. Lean mass, glycemia, and plasma lipids levels remained intact in ritonavir‐treated mice (Table S2). While investigating the effects of ritonavir treatment in endothelial function, we reported that ritonavir markedly reduced acetylcholine‐ but not sodium nitroprusside–induced relaxation of the aortic rings (Figure 1G and 1H), which supports a dysfunction at the levels of the endothelium. In female mice, 4 weeks of ritonavir treatment reduced body weight, fat mass, and impaired endothelial function to a similar extent as in males (Figure S1A through S1C), suggesting that ritonavir‐mediated metabolic and vascular alterations are not sex‐specific. Treatment of male animals with ritonavir for 3 days did not reduce body weight, fat mass or leptin levels, nor impair endothelial function (Figure S1D through S1G). These latter data rule out direct effects of ritonavir on endothelial function and support the contribution of ritonavir‐induced lipoatrophy to endothelial dysfunction. To further test the contribution of fat mass reduction to endothelial dysfunction, we investigated whether restoration of the levels of the adipokine leptin improved endothelial function. As reported in Figure 1G, leptin treatment markedly improved endothelial function despite further reducing body weight (Figure 1A through 1F), suggesting that decreases in leptin levels mediates endothelial dysfunction in ritonavir‐treated animals.
Figure 1. Ritonavir induces endothelial dysfunction via reducing leptin secretion.
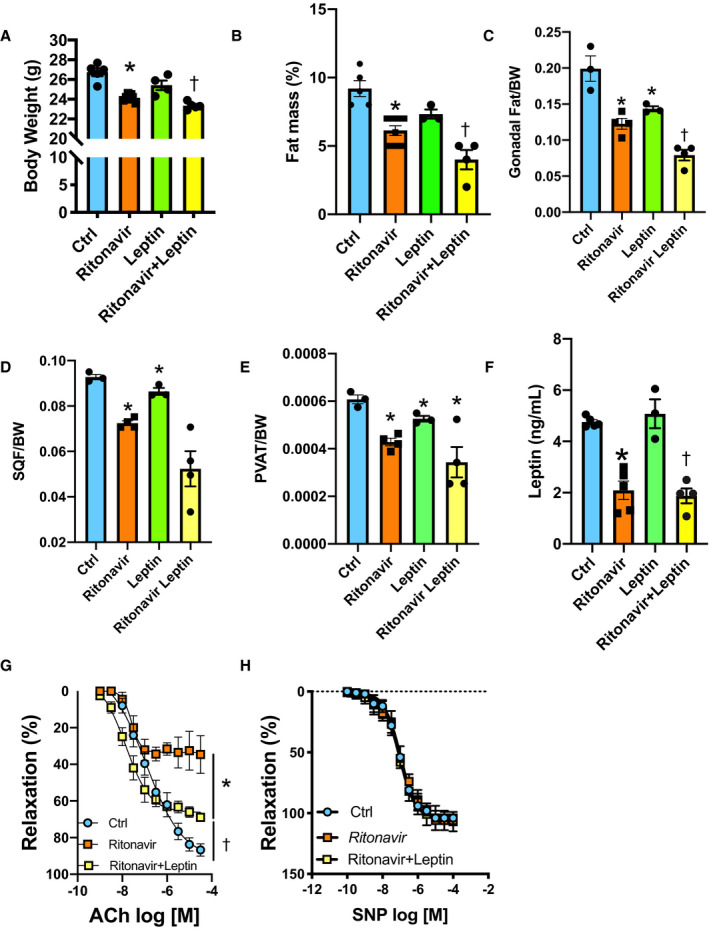
Body weight (A), percentage of fat mass (B), gonadal fat depot (C), subcutaneous fat depot (D), PVAT, (E) leptin plasma levels (F), and concentration response curves to ACh (G) and SNP (H) in aortic rings from control (Ctrl, vehicle‐treated) and ritonavir‐treated mice (ritonavir, 5 mg/kg per day for 4 weeks, ip) in the presence or absence of leptin treatment (0.3 mg/kg per day for 1 week, via osmotic mini‐pump). Data are presented as mean±SEM. N=5 to 8; *P<0.05 vs Ctrl; † P<0.05 vs Ctrl and ritonavir. ACh indicates acetylcholine; BW, body weight; Ctrl, control; PVAT, perivascular adipose tissue; and SQF, subcutaneous fat.
Ritonavir‐Induced Endothelial Dysfunction and Inflammation are Nox1‐Dependent
While investigating the mechanisms whereby ritonavir impairs endothelial function, we revealed a reduced NO bioavailability reflected by a complete abolition of ACh‐mediated relaxation in l‐NG‐nitro arginine methyl ester–treated aortic rings in both control and ritonavir‐treated animals (Figure 2A). Reactive oxygen species are a major cause of reduced NO bioavailability. Therefore, we repeated the relaxation response curve to ACh in the presence of the reactive oxygen species scavenger tempol, which fully restored endothelial function in ritonavir‐treated mice and revealed that ritonavir‐mediated endothelial dysfunction involves oxidative stress (Figure 2B). Concomitantly, ritonavir increased Nox1, NoxA1 transcript expression without altering NoxO1, Nox2, and Nox4 expression (Figure 2C). Ritonavir also increased aortic H2O2 levels (Figure 2D) and induced vascular inflammation as reflected by marked increases in aorta interleukin 1β (IL‐1β), GATA3 (GATA binding protein 3), F4/80, and CCR5 expression as well as in CCR5 ligand, C‐C motif chemokine ligand 5 (CCL5) (Figure 2E). The crucial role of Nox1 in ritonavir‐mediated endothelial dysfunction and vascular inflammation was demonstrated by reporting that Nox1 deficiency protected mice from ritonavir‐mediated endothelial dysfunction (Figure 2F) and increases in vascular IL‐1β, GATA3, F4/80, CCR5, and CCL5 (Figure 2E) without blunting ritonavir‐mediated decreases in body weight and fat mass (Figure S2A and S2B). The potential contribution of endothelial Nox1 to vascular inflammation was investigated via overexpression of Nox1, NoxA1, and NoxO1 in EC. This approach revealed that high Nox1 expression in EC is associated with a 5‐fold increase in CCR5 expression (Figure 2G).
Figure 2. Ritonavir‐induced endothelial dysfunction and inflammation are Nox1‐dependent.
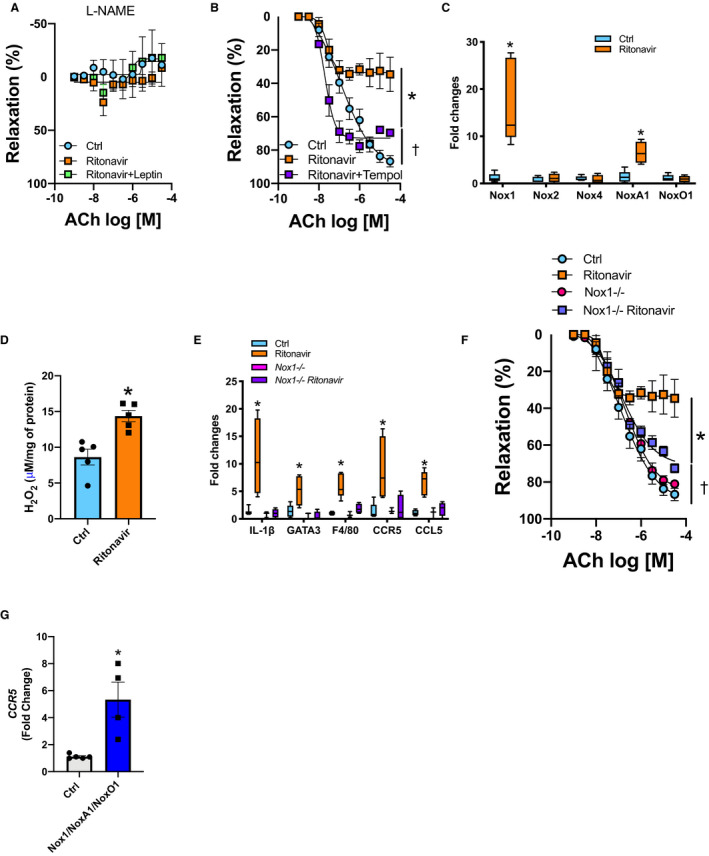
CRC to ACh in aortic rings in the presence of l‐NAME (100 μmol/L) (A) or tempol (100 μmol/L) (B). Real‐time PCR quantification of aortic NADPH oxidases subunits (C). Aortic H2O2 levels measured by Amplex Red (D) from control (Ctrl, vehicle‐treated) and ritonavir‐treated mice (ritonavir, 5 mg/kg per day for 4 weeks, ip) in the presence or absence of leptin treatment (0.3 mg/kg per day for 1 week, via osmotic mini‐pump). Real‐time PCR quantification of inflammatory markers (E) and CRC to ACh (F) in aortic segments from Nox1‐deficient mice (Nox1−/−) treated or not with ritonavir. G, CCR5 gene expression in human umbilical vein endothelial cells transduced with Nox1/NoxA1/NoxO1. Data are presented as mean±SEM. Gene expression data are presented as Min. to Max. N=3 to 8; *P<0.05 vs Ctrl; † P<0.05 vs Ctrl and ritonavir. ACh indicates acetylcholine; Ctrl, control; CCL5, C‐C motif chemokine ligand 5; CCR5, C‐C chemokine receptor 5; CRC, concentration response curves; IL1‐β, interleukin 1‐β; F4/80, the macrophage marker F4/80; GATA3, GATA binding protein 3; IL1‐β, interleukin 1‐β; l‐NAME, l‐NG‐nitro arginine methyl ester; NADPH, reduced nicotinamide adenine dinucleotide phosphate; Nox1, NADPH oxidase 1; and PCR, polymerase chain reaction.
Ritonavir‐Induced Endothelial Dysfunction and Vascular Inflammation Are CCR5‐Dependent
As ritonavir‐mediated increases in Nox1 led to CCR5 overexpression, we investigated CCR5 contribution to endothelial dysfunction and vascular inflammation. CCR5 deficiency did not protect mice from ritonavir‐mediated reductions in body weight and fat mass (Figure S3A and S3B) but protected mice from endothelial dysfunction (Figure 3A) to a similar extent as Nox1 deletion (Figure S4) and increases in IL‐1β, GATA3, and F4/80 (Figure 3B). As reported in Figure 3C, CCR5 deficiency also protected from ritonavir‐mediated increases in Nox1 and NoxA1.
Figure 3. Ritonavir‐induced endothelial dysfunction and vascular inflammation are CCR5 dependent.
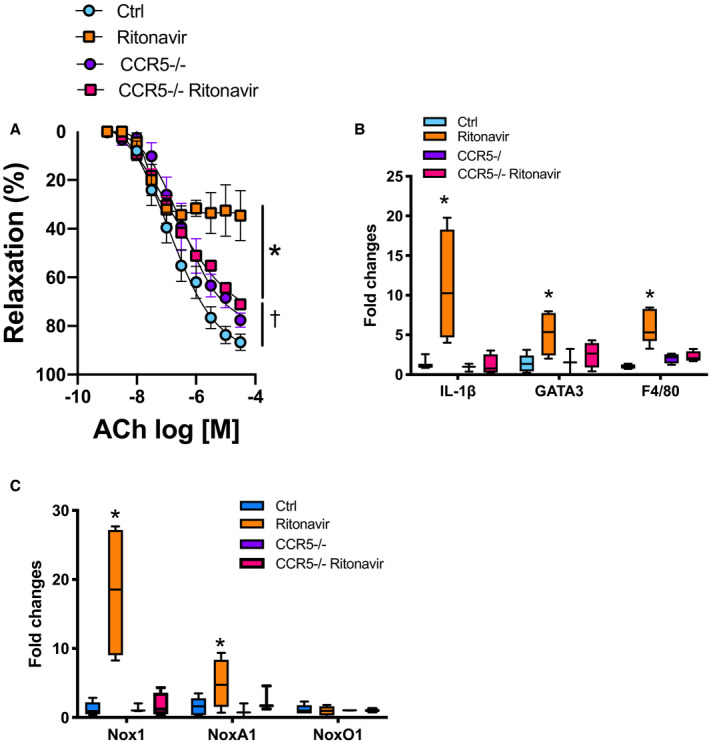
Concentration response curves to ACh in aortic rings (A), real‐time polymerase chain reaction quantification of aortic inflammatory gene markers (B), and aortic reduced nicotinamide adenine dinucleotide phosphate oxidases subunits (C) from control (Ctrl, vehicle treatment) and CCR5‐deficient mice (CCR5−/−) in the presence or absence of ritonavir treatment (ritonavir, 5 mg/kg per day for 4 weeks, ip). Vascular reactivity data are presented as mean±SEM. Gene expression data are presented as Min. to Max. N=3 to 8; *P<0.05 vs Ctrl; † P<0.05 vs Ctrl and ritonavir. ACh indicates acetylcholine; CCL5, C‐C motif chemokine ligand 5; CCR5, C‐C chemokine receptor 5; F4/80, the macrophage marker F4/80; GATA3, GATA binding protein 3; IL1‐β, interleukin 1‐β; and Nox1, NADPH oxidase 1.
Leptin Treatment Decreases Nox1 Expression and Vascular Inflammation
As leptin treatment restored endothelial function, we investigated whether leptin would also reduce Nox1 and CCR5 expression as well as vascular inflammation. We also assessed whether Nox1 and CCR5 deletion, as well as ritonavir treatment affect vascular leptin signaling. As reported in Figure 4A through 4C, leptin treatment blunted ritonavir‐mediated increases in aortic Nox1 and NoxA1, in aortic H2O2 production, as well as in IL‐1β, GATA3, F4/80, CCR5, and CCL5 transcript expression. We also showed that vascular leptin receptor expression remained intact with Nox1 and CCR5 deletion as well as ritonavir treatment (Figure 4D).
Figure 4. Leptin treatment decreases Nox1 expression and vascular inflammation.
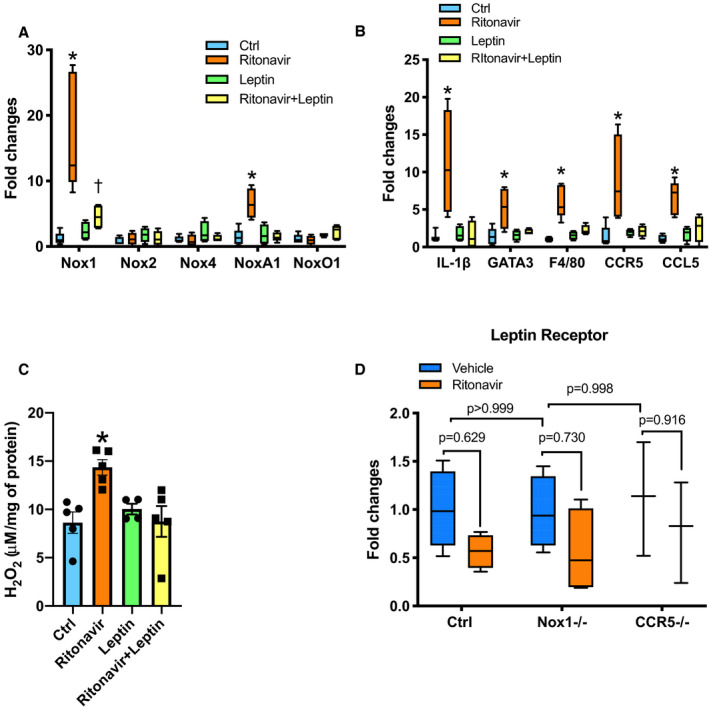
Real‐time PCR quantification of aortic NADPH oxidases subunits (A), aortic inflammatory gene markers (B). Aortic H2O2 levels measured by Amplex Red (C) from Ctrl and ritonavir‐treated mice (ritonavir, 5 mg/kg per day for 4 weeks, ip) in the presence or absence of leptin treatment (0.3 mg/kg per day for 1 week, via osmotic mini‐pump). Real‐time PCR quantification of aortic leptin receptor (D) from control (Ctrl), Nox1‐deficient mice (Nox1−/−), CCR5‐deficient mice (CCR5−/−), and ritonavir‐treated mice (ritonavir, 5 mg/kg per day for 4 weeks, ip). H2O2 content data are presented as mean±SEM. Gene expression data are presented as Min. to Max. N=4 to 8; *P<0.05 vs Ctrl; † P<0.05 vs leptin and ritonavir. CCL5 indicates C‐C motif chemokine ligand 5; CCR5, C‐C chemokine receptor 5; Ctrl, control; F4/80, the macrophage marker F4/80; IL‐1β, interleukin‐1β; GATA3, GATA binding protein 3; Nox1, NADPH oxidase 1; PCR, polymerase chain reaction.
Protective Effects of Leptin Require Endothelial Leptin Signaling Activation
To investigate the origin of the protective effects of leptin, we repeated the ritonavir experiments in inducible endothelial leptin receptor–deficient mice as well as in inducible endothelial Ptp1b–deficient mice that present increased endothelial leptin signaling. 30 As reported in Figure 5A and 5B, endothelial leptin deficiency blunted the protective effects of leptin treatment on endothelial function as well as on Nox1 and NoxA1. However, increases in endothelial leptin signaling with Ptp1b deletion blunted ritonavir‐mediated endothelial dysfunction and increases in Nox1 and NoxA1 expression. Neither endothelial leptin receptor nor Ptp1b deletion affected ritonavir‐mediated decrease in body weight and fat mass (Figure S5).
Figure 5. Leptin‐mediated vascular protection involves endothelial‐leptin signaling.
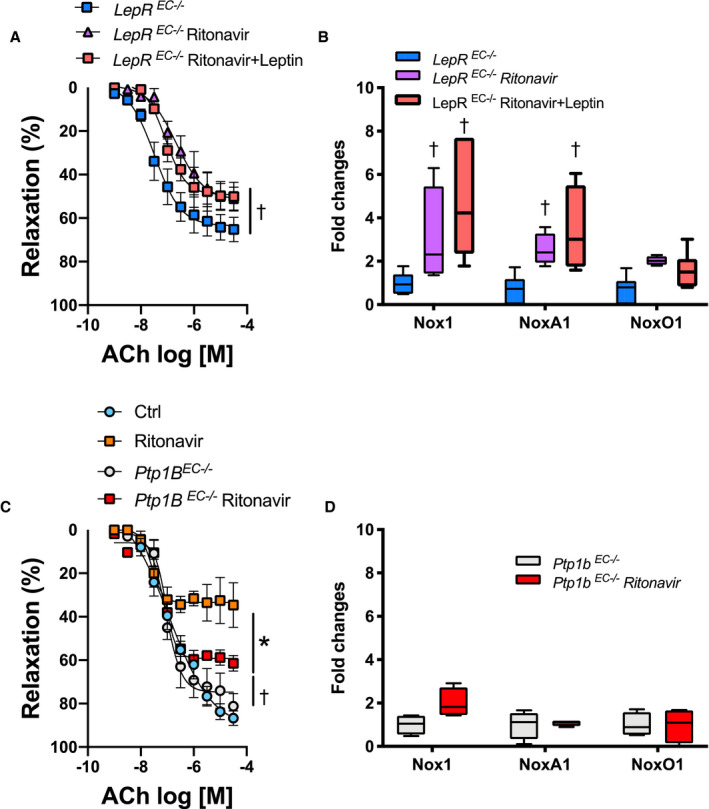
Concentration response curves to ACh in aortic rings (A and C) and real‐time polymerase chain reaction quantification of aortic NADPH oxidases subunits (B and D) in wild type, endothelial LepR–deficient (LepR EC−/−, A and B) or endothelial Ptp1B–deficient mice (Ptp1B EC−/−, C and D) treated with vehicle (Ctrl) or ritonavir (5 mg/kg per day for 4 weeks, ip) in the presence or absence of leptin treatment (0.3 mg/kg per day for 1 week, via osmotic mini‐pump). Vascular reactivity data are presented as mean±SEM. Gene expression data are presented as Min. to Max. N=3 to 8; *P<0.05 vs Ctrl; † P<0.05 vs LepR EC−/− or Ptp1b EC−/−. ACh indicates acetylcholine; Ctrl, control; LepR, leptin receptor; Nox1, NADPH oxidase 1; and Ptp1B, protein tyrosine phosphatase 1B.
Ritonavir Increases Vascular Adrenergic Contractility Via Nox1‐Dependent Mechanisms
Lastly, we investigated whether ritonavir‐mediated endothelial dysfunction could result in increased vascular adrenergic contractility. As reported in Figure 6A, ritonavir increased vascular contractility to phenylephrine. Both leptin treatment (Figure 6A) and Nox1 deficiency (Figure 6B) restored vascular contractility. In addition, l‐NG‐nitro arginine methyl ester abolished the protective effects of leptin treatment and Nox1 deletion (Figure 6C and 6D), indicating that leptin treatment and Nox1 deletion reduced contractility via restoration of NO bioavailability. Remarkably, ritonavir treatment, leptin infusion, and Nox1 deletion did not affect general vascular contractility because none of these treatments altered vascular constriction in response to vascular smooth muscle cell depolarization (KCl‐mediated constriction, Figure 6E).
Figure 6. Leptin treatment reduces ritonavir‐induced adrenergic hypercontractility via Nox1‐dependent mechanisms.
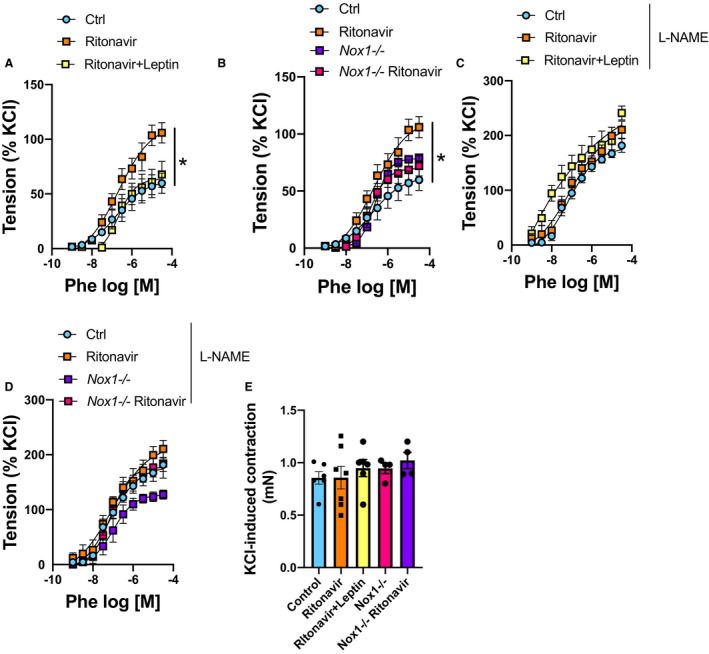
Concentration response curves to Phe (A through D) in presence or absence of l‐NAME (C and D) in aortic rings from wild type or Nox1‐deficient mice (Nox1−/−) (B and D) treated or not with ritonavir (5 mg/kg per day for 4 weeks, ip) in the presence or absence of leptin treatment (0.3 mg/kg per day for 1 week, via osmotic mini‐pump) (A and C). E, KCl induced vascular contraction. Data are presented as mean±SEM. N=3 to 7; *P<0.05 vs Ctrl. Ctrl indicates control; l‐NAME, l‐NG‐nitro arginine methyl ester; Nox1, NADPH oxidase 1; and Phe, phenylephrine.
Discussion
In the present study, we investigated the mechanisms whereby ritonavir, a protease inhibitor associated with CVD in numerous clinical studies, 8 , 9 , 10 , 11 impairs endothelial function, in an in vivo setting. We identified for the first time that ritonavir‐mediated reduction in adipose mass causes endothelial dysfunction via reducing the secretion of the adipokine leptin and lessening the activation of endothelial leptin signaling. We extended our findings by providing the first demonstration that reduction in endothelial leptin signaling induces an increase in Nox1 expression, which elevates vascular CCR5 levels, promotes vascular inflammation, reduces NO bioavailability, and consequently leads to impaired endothelium‐dependent relaxation (mechanisms summarized in Figure 7). Relevant to these observations are the roles of fat mass, endothelial leptin receptor, Nox1, and CCR5 in ritonavir‐associated endothelial dysfunction.
Figure 7. Schematic representation of the mechanisms whereby endothelial leptin signaling maintains endothelial cell integrity and promotes vascular relaxation.
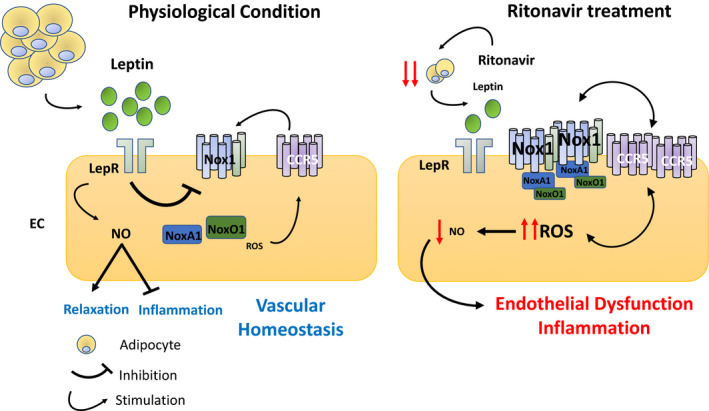
EC indicates endothelial cells; CCR5, C‐C chemokine receptor type 5; Nox1, NADPH oxidase 1; LepR, leptin receptor; and ROS, reactive oxygen species.
Reduction in fat mass and adipokines secretion have been among the first side effects reported in PWH on protease inhibitors 19 , 31 ; however, no study had investigated the cardiovascular consequences of alterations in adipose tissue endocrine function. Herein, in agreement with previous clinical studies, 19 , 32 , 33 , 34 , 35 , 36 we report that ritonavir reduced adipose mass and the secretion of the adipokine leptin. We demonstrated that reduction in leptin levels and its consequent decrease in endothelial leptin signaling are the causes of endothelial dysfunction. Indeed, we showed that leptin supplementation markedly improved endothelial function, and while selective endothelial leptin receptor deficiency abolished the protective effects of leptin supplementation, selective increases in endothelial leptin signaling protected against ritonavir‐mediated endothelial dysfunction. In contradiction to our results, previous in vitro studies in EC in culture 37 and ex vivo experiments in isolated arteries 23 , 38 support direct toxic effects of ritonavir on EC. However, our current in vivo study, added to the work by others, minimizes the contribution of the latter mechanism and supports ritonavir‐mediated decreases in adipose mass as a primary cause of endothelial dysfunction. Indeed, while toxic effects of ritonavir would have been expected to induce endothelial dysfunction after only a few days of exposure, endothelial function remained intact after 3 days of ritonavir treatment, which did not reduce adipose mass. In addition, ritonavir treatment did not induce endothelial dysfunction in healthy volunteers, in the absence of changes in adipose mass. 39 , 40 While these results provide strong arguments to support the contribution of ritonavir‐associated fat mass reduction to endothelial dysfunction, these results also raised the question of the fat depot responsible for the decreases in leptin levels. The subcutaneous adipose tissue is the adipose depot preferentially affected by protease inhibitor. Indeed, PWH on protease inhibitors do commonly present with reduced limb and face subcutaneous fat mass but with intact or increased visceral fat mass. 19 , 41 , 42 In the present mouse study, ritonavir similarly reduced subcutaneous, visceral, as well as perivascular adipose tissue. Thus, based on the reports that the subcutaneous adipose tissue is the main source of leptin, 43 we could speculate that the reduction in subcutaneous fat mass causes endothelial dysfunction. However, our data do not rule out the potential contribution of a localized decrease in leptin mediated by a reduction in perivascular adipose tissue. Therefore, further investigations are required to identify the primary fat depot responsible for the maintenance of EC homeostasis.
Dyslipidemia has been presented as another key contributor to ritonavir‐mediated endothelial dysfunction. 20 However, the present study as well as work by others do also challenge this concept. Endothelial dysfunction was not associated with significant increases in cholesterol and plasma triglycerides in our ritonavir‐treated mice, and reduction in plasma cholesterol and lipid levels did not restore endothelial function in PWH on ritonavir treatment. 44 Moreover, despite raising triglyceride levels, liponavir/ritonavir treatment did not induce endothelial dysfunction in healthy volunteers exhibiting no alteration in body mass index. 40
Further evidence to support the contribution of adipose mass reduction and reduced leptin levels in endothelial dysfunction are provided by recent observations in obese PWH. Indeed, recent clinical evidence reports no negative impact of increased adiposity on carotid intima‐media thickness and flow‐mediated dilation in PWH on cART, 45 , 46 , 47 and large epidemiology studies suggest no increased risk for myocardial infarction among higher body mass index patients on cART. 9 , 48 , 49 Remarkably, higher plasma levels of the adipocyte‐derived hormone leptin have been associated with healthier arterial structure and function in PWH on cART. 50 Together with studies reporting that leptin treatment improves metabolic function in patients and animals on cART, 51 , 52 , 53 these data indicate that preserving adequate leptin levels may represent an avenue to limit metabolic and cardiovascular disorders in patients on cART.
Several studies have correlated high leptin levels with increased prevalence of CVD 54 and associated excess leptin with NO synthase inhibition, oxidative stress, vascular inflammation, and increases in release of endothelin‐1. 55 , 56 , 57 However, very few studies have investigated the potential cardiovascular consequences of reduced leptin levels. Consistent with previous work by our group in a mouse model of congenital lipodystrophy, 28 we reported that reduced endothelial leptin signaling activation elevates Nox1 expression and impairs endothelial dysfunction via reactive oxygen species–dependent mechanisms. Interestingly, we made the new observation that elevated vascular Nox1 levels result in the increased expression of a major regulator of inflammation, CCR5, its ligand, CCL5 as well as GATA3, F4/80, and IL‐1β, which are respective markers of T cells, macrophages, and inflammation in general. The role of Nox1 as a driver of inflammation and vascular dysfunction was demonstrated by reporting that Nox1 deletion prevented ritonavir‐mediated increases in CCR5, GATA3, and F4/80 and abolished ritonavir‐induced endothelial dysfunction and increased vascular adrenergic contractility. Further arguments in support of Nox1 were provided by showing that increases in Nox1 expression in EC elevated CCR5 expression. Remarkably, similarly to Nox1 deletion, CCR5 deletion protected from ritonavir‐mediated endothelial dysfunction and vascular inflammation, without altering adipose mass. CCR5, which is expressed in immune and vascular cells, including EC, 58 plays a crucial role in atherogenesis through the regulation of chemoattraction of immune cells and infiltration into the inflammatory site. 59 The role of CCR5 in the control of endothelium‐dependent relaxation is, however, unknown. Although our study did not enable us to delineate the mechanisms whereby CCR5 deletion restores NO bioavailability, it presents, for the first time, CCR5 as a regulator of endothelium‐dependent relaxation. Based on the reduced expression of Nox1, GATA3, F4/80, and IL‐1β with CCR5 deletion, we can speculate that CCR5 deficiency increases NO bioavailability via reducing macrophage‐ and T cell–derived reactive oxygen species production. However, we cannot rule out a potential direct role of CCR5 in the control of endothelial nitric oxide synthase function. Therefore, additional experiments are required to investigate the contribution of CCR5 signaling to EC homeostasis.
CCR5 function is not limited to chemoattraction and the progression of atherosclerosis. CCR5 is also a main co‐receptor for HIV involved in the entry of the virus into the cell as well as in the spread of the virus. 60 Therefore, the novel observation that leptin downregulates CCR5 expression via Nox1‐dependent mechanisms could potentially have important implications beyond the regulation of endothelial function. Leptin‐mediated decreases in CCR5 expression can potentially limit the spread of the viral infection and may contribute to explaining the slower HIV disease progression in PWH with high body mass index, 61 as well as the better immune reconstitution over time in obese PWH. 62 Although our study demonstrates that leptin‐mediated CCR5 regulation is Nox1 dependent, additional studies will be required to delineate the molecular mechanisms whereby leptin downregulates Nox1 expression and whereby Nox1 controls CCR5 expression.
A limitation of the current study is the lack of measurement of blood pressure. Hypertension, which affects 35% of PWH on cART, is both a consequence of and a contributor to endothelial dysfunction. 63 In addition, lipodystrophy has been identified as a contributor to hypertension in PWH on cART. Therefore, one can speculate that ritonavir‐associated endothelial dysfunction could induce hypertension or that ritonavir‐associated lipodystrophy induces hypertension, which causes endothelial dysfunction. However, our results do suggest that ritonavir‐mediated alterations in endothelial function are independent of blood pressure. Indeed, while leptin has been associated with vascular inflammation, increases in sympathetic activity, and blood pressure, 64 , 65 leptin treatment improved endothelial function and reduced vascular adrenergic contractility as well as vascular inflammation, which do not appear compatible with elevations in blood pressure. However, additional studies are required to confirm this hypothesis.
In conclusion, the present study provides the first evidence that endothelial dysfunction associated with the use of the protease inhibitor ritonavir is a direct consequence of a decrease in adipose mass and leptin secretion, which reduces endothelial leptin signaling activation and triggers an increase in CCR5 expression and inflammation via Nox1‐dependent mechanisms. Taken together, these findings highlight the crucial role played by the adipose tissue in the control of cardiovascular function and indicate that excessive reduction in fat mass associated with protease inhibitor, chemotherapy, and anorexia likely primes the cardiovascular system for more severe alterations.
Sources of Funding
This work was supported by a Postdoctoral Fellowship (17POST33410363), a K99 (1K99HL140139‐01A1), and a R00 (4R00HL14013903) from the NHLBI to Bruder‐Nascimento. This study was also supported by an Established Investigator Award (19EIA34760167) from the American Heart Association, R01s (1R01HL130301‐01 and 1R01HL147639‐01A1) from the NHLBI, and Start‐up funds from Augusta University to Belin de Chantemèle.
Disclosures
None.
Supporting information
Acknowledgments
Author contributions: TBN and EBC participated in the conception, design of the work, acquisition of the data, analysis and interpretation of the data, and redaction of the manuscript. TCK participated in the acquisition of the data and SK generated the mouse models.
(J Am Heart Assoc. 2020;9:e018074 DOI: 10.1161/JAHA.120.018074.)
Supplementary Materials for this article are available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.120.018074
For Sources of Funding and Disclosures, see page 12.
References
- 1. Michaels SH, Clark R, Kissinger P. Declining morbidity and mortality among patients with advanced human immunodeficiency virus infection. N Engl J Med. 1998;405–406. [DOI] [PubMed] [Google Scholar]
- 2. Bozzette SA, Ake CF, Tam HK, Phippard A, Cohen D, Scharfstein DO, Louis TA. Long‐term survival and serious cardiovascular events in HIV‐infected patients treated with highly active antiretroviral therapy. J Acquir Immune Defic Syndr. 2008;338–341. [DOI] [PubMed] [Google Scholar]
- 3. Smith CJ, Ryom L, Weber R, Morlat P, Pradier C, Reiss P, Kowalska JD, de Wit S, Law M, el Sadr W, et al. Trends in underlying causes of death in people with HIV from 1999 to 2011 (D:A:D): a multicohort collaboration. Lancet. 2014;241–248. [DOI] [PubMed] [Google Scholar]
- 4. Kearns A, Gordon J, Burdo TH, Qin X. HIV‐1-associated atherosclerosis: unraveling the missing link. J Am Coll Cardiol. 2017;3084–3098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Currier JS, Lundgren JD, Carr A, Klein D, Sabin CA, Sax PE, Schouten JT, Smieja M, Working G. Epidemiological evidence for cardiovascular disease in HIV‐infected patients and relationship to highly active antiretroviral therapy. Circulation. 2008;e29–e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Boccara F, Lang S, Meuleman C, Ederhy S, Mary‐Krause M, Costagliola D, Capeau J, Cohen A. HIV and coronary heart disease: time for a better understanding. J Am Coll Cardiol. 2013;511–523. [DOI] [PubMed] [Google Scholar]
- 7. Vachiat A, McCutcheon K, Tsabedze N, Zachariah D, Manga P. HIV and ischemic heart disease. J Am Coll Cardiol. 2017;73–82. [DOI] [PubMed] [Google Scholar]
- 8. Currier JS, Kendall MA, Zackin R, Henry WK, Alston‐Smith B, Torriani FJ, Schouten J, Mickelberg K, Li Y, Hodis HN, et al. Carotid artery intima‐media thickness and HIV infection: traditional risk factors overshadow impact of protease inhibitor exposure. AIDS. 2005;927–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Group DADS , Friis‐Moller N, Reiss P, Sabin CA, Weber R, Monforte A, El‐Sadr W, Thiebaut R, De Wit S, Kirk O, Fontas E, et al. Class of antiretroviral drugs and the risk of myocardial infarction. N Engl J Med. 2007;1723–1735. [DOI] [PubMed] [Google Scholar]
- 10. Alvi RM, Neilan AM, Tariq N, Awadalla M, Afshar M, Banerji D, Rokicki A, Mulligan C, Triant VA, Zanni MV, et al. Protease inhibitors and cardiovascular outcomes in patients with HIV and heart failure. J Am Coll Cardiol. 2018;518–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ryom L, Lundgren JD, El‐Sadr W, Reiss P, Kirk O, Law M, Phillips A, Weber R, Fontas E, d' Arminio Monforte A, et al. Cardiovascular disease and use of contemporary protease inhibitors: the D:A:D international prospective multicohort study. Lancet HIV. 2018;e291–e300. [DOI] [PubMed] [Google Scholar]
- 12. (WHO) WHO . Update of recommendations on first‐ and second‐line antiretroviral regimens. 2019:16.
- 13. (WHO) WHO . Consolidated Guidelines on the USE of Antiretroviral Drugs for Treating and Preventing HIV Infection Recommendations for a Public Health Approach. 2nd ed WHO Library Cataloguing‐in-Publication Data; 2016:480 Available at: https://apps.who.int/iris/bitstream/handle/10665/208825/9789241549684_eng.pdf?sequence=1. Accessed September 15, 2020. [Google Scholar]
- 14. Moorhouse M, Maartens G, Venter WDF, Moosa MY, Steegen K, Jamaloodien K, Fox MP, Conradie F. Third‐line antiretroviral therapy program in the south african public sector: cohort description and virological outcomes. J Acquir Immune Defic Syndr. 2019;73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eatemadi A, Aiyelabegan HT, Negahdari B, Mazlomi MA, Daraee H, Daraee N, Eatemadi R, Sadroddiny E. Role of protease and protease inhibitors in cancer pathogenesis and treatment. Biomed Pharmacother. 2017;221–231. [DOI] [PubMed] [Google Scholar]
- 16. Mendez‐Lopez M, Sutter T, Driessen C, Besse L. HIV protease inhibitors for the treatment of multiple myeloma. Clin Adv Hematol Oncol. 2019;615–623. [PubMed] [Google Scholar]
- 17. Klibanov OM, Gale SE, Santevecchi B. Ombitasvir/paritaprevir/ritonavir and dasabuvir tablets for hepatitis C virus genotype 1 infection. Ann Pharmacother. 2015;566–581. [DOI] [PubMed] [Google Scholar]
- 18. Cao B, Wang Y, Wen D, Liu W, Wang J, Fan G, Ruan L, Song B, Cai Y, Wei M, et al. A trial of lopinavir‐ritonavir in adults hospitalized with severe covid‐19. N Engl J Med. 2020;1787–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Carr A, Samaras K, Burton S, Law M, Freund J, Chisholm DJ, Cooper DA. A syndrome of peripheral lipodystrophy, hyperlipidaemia and insulin resistance in patients receiving HIV protease inhibitors. AIDS. 1998;F51–F58. [DOI] [PubMed] [Google Scholar]
- 20. Stein JH, Klein MA, Bellehumeur JL, McBride PE, Wiebe DA, Otvos JD, Sosman JM. Use of human immunodeficiency virus‐1 protease inhibitors is associated with atherogenic lipoprotein changes and endothelial dysfunction. Circulation. 2001;257–262. [DOI] [PubMed] [Google Scholar]
- 21. Mencarelli A, Francisci D, Renga B, D'Amore C, Cipriani S, Basile F, Schiaroli E, Baldelli F, Fiorucci S. Ritonavir‐induced lipoatrophy and dyslipidaemia is reversed by the anti‐inflammatory drug leflunomide in a PPAR‐gamma‐dependent manner. Antivir Ther. 2012;669–678. [DOI] [PubMed] [Google Scholar]
- 22. Reyskens KM, Essop MF. HIV protease inhibitors and onset of cardiovascular diseases: a central role for oxidative stress and dysregulation of the ubiquitin‐proteasome system. Biochim Biophys Acta. 2014;256–268. [DOI] [PubMed] [Google Scholar]
- 23. Wang X, Chai H, Lin PH, Yao Q, Chen C. Roles and mechanisms of human immunodeficiency virus protease inhibitor ritonavir and other anti‐human immunodeficiency virus drugs in endothelial dysfunction of porcine pulmonary arteries and human pulmonary artery endothelial cells. Am J Pathol. 2009;771–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. DeFalco J, Tomishima M, Liu H, Zhao C, Cai X, Marth JD, Enquist L, Friedman JM. Virus‐assisted mapping of neural inputs to a feeding center in the hypothalamus. Science. 2001;2608–2613. [DOI] [PubMed] [Google Scholar]
- 25. Lanahan AA, Lech D, Dubrac A, Zhang J, Zhuang ZW, Eichmann A, Simons M. PTP1b is a physiologic regulator of vascular endothelial growth factor signaling in endothelial cells. Circulation. 2014;902–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Cipriani S, Francisci D, Mencarelli A, Renga B, Schiaroli E, D'Amore C, Baldelli F, Fiorucci S. Efficacy of the CCR5 antagonist maraviroc in reducing early, ritonavir‐induced atherogenesis and advanced plaque progression in mice. Circulation. 2013;2114–2124. [DOI] [PubMed] [Google Scholar]
- 27. Bruder‐Nascimento T, Butler BR, Herren DJ, Brands MW, Bence KK, Belin de Chantemele EJ. Deletion of protein tyrosine phosphatase 1b in proopiomelanocortin neurons reduces neurogenic control of blood pressure and protects mice from leptin‐ and sympatho‐mediated hypertension. Pharmacol Res. 2015;235–244. [DOI] [PubMed] [Google Scholar]
- 28. Bruder‐Nascimento T, Faulkner JL, Haigh S, Kennard S, Antonova G, Patel VS, Fulton DJR, Chen W, Belin de Chantemele EJ. Leptin restores endothelial function via endothelial PPARgamma‐Nox1-mediated mechanisms in a mouse model of congenital generalized lipodystrophy. Hypertension. 2019;1399–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huby AC, Antonova G, Groenendyk J, Gomez‐Sanchez CE, Bollag WB, Filosa JA, Belin de Chantemele EJ. Adipocyte‐derived hormone leptin is a direct regulator of aldosterone secretion, which promotes endothelial dysfunction and cardiac fibrosis. Circulation. 2015;2134–2145. [DOI] [PubMed] [Google Scholar]
- 30. Belin de Chantemele EJ, Muta K, Mintz J, Tremblay ML, Marrero MB, Fulton DJ, Stepp DW. Protein tyrosine phosphatase 1B, a major regulator of leptin‐mediated control of cardiovascular function. Circulation. 2009;753–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Addy CL, Gavrila A, Tsiodras S, Brodovicz K, Karchmer AW, Mantzoros CS. Hypoadiponectinemia is associated with insulin resistance, hypertriglyceridemia, and fat redistribution in human immunodeficiency virus‐infected patients treated with highly active antiretroviral therapy. J Clin Endocrinol Metab. 2003;627–636. [DOI] [PubMed] [Google Scholar]
- 32. Estrada V, Serrano‐Rios M, Martinez Larrad MT, Villar NG, Gonzalez Lopez A, Tellez MJ, Fernandez C. Leptin and adipose tissue maldistribution in HIV‐infected male patients with predominant fat loss treated with antiretroviral therapy. J Acquir Immune Defic Syndr. 2002;32–40. [DOI] [PubMed] [Google Scholar]
- 33. Gleason RL Jr, Caulk AW, Seifu D, Parker I, Vidakovic B, Getenet H, Assefa G, Amogne W. Current Efavirenz (EFV) or ritonavir‐boosted lopinavir (LPV/r) use correlates with elevate markers of atherosclerosis in HIV‐infected subjects in Addis Ababa, Ethiopia. PLoS One. 2015;9:e0117125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nagy GS, Tsiodras S, Martin LD, Avihingsanon A, Gavrila A, Hsu WC, Karchmer AW, Mantzoros CS. Human immunodeficiency virus type 1‐related lipoatrophy and lipohypertrophy are associated with serum concentrations of leptin. Clin Infect Dis. 2003;795–802. [DOI] [PubMed] [Google Scholar]
- 35. Tiliscan C, Arama V, Mihailescu R, Munteanu DI, Streinu‐Cercel A, Ion DA, Radulescu MA, Popescu C, Lobodan AE, Negru AR, et al. Leptin expression in HIV‐infected patients during antiretroviral therapy. Germs. 2015;92–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Paganelli R, Mezzaroma I, Mazzone AM, Pinter E, Aiuti F. Leptin levels in HIV‐positive patients treated with HAART. AIDS. 1999;2479. [DOI] [PubMed] [Google Scholar]
- 37. Zhong DS, Lu XH, Conklin BS, Lin PH, Lumsden AB, Yao Q, Chen C. HIV protease inhibitor ritonavir induces cytotoxicity of human endothelial cells. Arterioscler Thromb Vasc Biol. 2002;1560–1566. [DOI] [PubMed] [Google Scholar]
- 38. Conklin BS, Fu W, Lin PH, Lumsden AB, Yao Q, Chen C. HIV protease inhibitor ritonavir decreases endothelium‐dependent vasorelaxation and increases superoxide in porcine arteries. Cardiovasc Res. 2004;168–175. [DOI] [PubMed] [Google Scholar]
- 39. Grubb JR, Dejam A, Voell J, Blackwelder WC, Sklar PA, Kovacs JA, Cannon RO, Masur H, Gladwin MT. Lopinavir‐ritonavir: effects on endothelial cell function in healthy subjects. J Infect Dis. 2006;1516–1519. [DOI] [PubMed] [Google Scholar]
- 40. Dube MP, Shen C, Greenwald M, Mather KJ. No impairment of endothelial function or insulin sensitivity with 4 weeks of the HIV protease inhibitors atazanavir or lopinavir‐ritonavir in healthy subjects without HIV infection: a placebo‐controlled trial. Clin Infect Dis. 2008;567–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Carr A, Samaras K, Thorisdottir A, Kaufmann GR, Chisholm DJ, Cooper DA. Diagnosis, prediction, and natural course of HIV‐1 protease‐inhibitor‐associated lipodystrophy, hyperlipidaemia, and diabetes mellitus: a cohort study. Lancet. 1999;2093–2099. [DOI] [PubMed] [Google Scholar]
- 42. Domingo P, Gutierrez Mdel M, Gallego‐Escuredo JM, Torres F, Mateo MG, Villarroya J, Lamarca K, Domingo JC, Vidal F, Villarroya F, et al. A 48‐week study of fat molecular alterations in HIV naive patients starting tenofovir/emtricitabine with lopinavir/ritonavir or efavirenz. J Acquir Immune Defic Syndr. 2014;457–465. [DOI] [PubMed] [Google Scholar]
- 43. Schoof E, Stuppy A, Harig F, Carbon R, Horbach T, Stohr W, Rascher W, Dotsch J. Comparison of leptin gene expression in different adipose tissues in children and adults. Eur J Endocrinol. 2004;579–584. [DOI] [PubMed] [Google Scholar]
- 44. Murphy RL, Berzins B, Zala C, Fichtenbaum C, Dube MP, Guaraldi G, Torriani F, Belsey E, Mitchell C, Stein JH, et al. Change to atazanavir/ritonavir treatment improves lipids but not endothelial function in patients on stable antiretroviral therapy. AIDS. 2010;885–890. [DOI] [PubMed] [Google Scholar]
- 45. Koethe JR, Grome H, Jenkins CA, Kalams SA, Sterling TR. The metabolic and cardiovascular consequences of obesity in persons with HIV on long‐term antiretroviral therapy. AIDS. 2016;83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mangili A, Polak JF, Skinner SC, Gerrior J, Sheehan H, Harrington A, Wanke CA. HIV infection and progression of carotid and coronary atherosclerosis: the CARE study. J Acquir Immune Defic Syndr. 2011;148–153. [DOI] [PubMed] [Google Scholar]
- 47. Hsue PY, Ordovas K, Lee T, Reddy G, Gotway M, Schnell A, Ho JE, Selby V, Madden E, Martin JN, et al. Carotid intima‐media thickness among human immunodeficiency virus‐infected patients without coronary calcium. Am J Cardiol. 2012;742–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Freiberg MS, Chang CC, Kuller LH, Skanderson M, Lowy E, Kraemer KL, Butt AA, Bidwell Goetz M, Leaf D, Oursler KA, et al. HIV infection and the risk of acute myocardial infarction. JAMA Intern Med. 2013;614–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Womack JA, Chang CC, So‐Armah KA, Alcorn C, Baker JV, Brown ST, Budoff M, Butt AA, Gibert C, Goetz MB, et al. HIV infection and cardiovascular disease in women. J Am Heart Assoc. 2014;9:e001035 DOI: 10.1161/JAHA.114.001035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Longenecker CT, Dunn W, Jiang Y, Debanne SM, McComsey GA. Adipokines and vascular health in treated HIV infection: an obesity paradox? AIDS. 2013;1353–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lee JH, Chan JL, Sourlas E, Raptopoulos V, Mantzoros CS. Recombinant methionyl human leptin therapy in replacement doses improves insulin resistance and metabolic profile in patients with lipoatrophy and metabolic syndrome induced by the highly active antiretroviral therapy. J Clin Endocrinol Metab. 2006;2605–2611. [DOI] [PubMed] [Google Scholar]
- 52. Tsiodras S, Mantzoros C. Leptin and adiponectin in the HIV associated metabolic syndrome: physiologic and therapeutic implications. Am J Infect Dis. 2006;141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Riddle TM, Fichtenbaum CJ, Hui DY. Leptin replacement therapy but not dietary polyunsaturated fatty acid alleviates HIV protease inhibitor‐induced dyslipidemia and lipodystrophy in mice. J Acquir Immune Defic Syndr. 2003;564–570. [DOI] [PubMed] [Google Scholar]
- 54. Koh KK, Park SM, Quon MJ. Leptin and cardiovascular disease: response to therapeutic interventions. Circulation. 2008;3238–3249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Bouloumie A, Marumo T, Lafontan M, Busse R. Leptin induces oxidative stress in human endothelial cells. FASEB J. 1999;1231–1238. [PubMed] [Google Scholar]
- 56. Yamagishi S‐I, Edelstein D, Du X‐L, Kaneda Y, Guzmán M, Brownlee M. Leptin induces mitochondrial superoxide production and monocyte chemoattractant protein‐1 expression in aortic endothelial cells by increasing fatty acid oxidation via protein kinase A. J Biol Chem. 2001;25096–25100. [DOI] [PubMed] [Google Scholar]
- 57. Quehenberger P, Exner M, Sunder‐Plassmann R, Ruzicka K, Bieglmayer C, Endler G, Muellner C, Speiser W, Wagner O. Leptin induces endothelin‐1 in endothelial cells in vitro. Circ Res. 2002;711–718. [DOI] [PubMed] [Google Scholar]
- 58. Berger O, Gan X, Gujuluva C, Burns AR, Sulur G, Stins M, Way D, Witte M, Weinand M, Said J, et al. CXC and CC chemokine receptors on coronary and brain endothelia. Mol Med. 1999;795–805. [PMC free article] [PubMed] [Google Scholar]
- 59. Jones KL, Maguire JJ, Davenport AP. Chemokine receptor CCR5: from AIDS to atherosclerosis. Br J Pharmacol. 2011;1453–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, Di Marzio P, Marmon S, Sutton RE, Hill CM, et al. Identification of a major co‐receptor for primary isolates of HIV‐1. Nature. 1996;661–666. [DOI] [PubMed] [Google Scholar]
- 61. Koethe JR, Hulgan T, Niswender K. Adipose tissue and immune function: a review of evidence relevant to HIV infection. J Infect Dis. 2013;1194–1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Li X, Ding H, Geng W, Liu J, Jiang Y, Xu J, Zhang Z, Shang H. Predictive effects of body mass index on immune reconstitution among HIV‐infected HAART users in China. BMC Infect Dis. 2019;373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Brandes RP. Endothelial dysfunction and hypertension. Hypertension. 2014;924–928. [DOI] [PubMed] [Google Scholar]
- 64. Faulkner JL, Belin de Chantemele EJ. Sex differences in mechanisms of hypertension associated with obesity. Hypertension. 2018;15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Belin de Chantemele EJ. Sex differences in leptin control of cardiovascular function in health and metabolic diseases. Adv Exp Med Biol. 2017;87–111. [DOI] [PubMed] [Google Scholar]
- 66. da Costa RM, Fais RS, Dechandt CRP, Louzada‐Junior P, Alberici LC, Lobato NS, Tostes RC. Increased mitochondrial ROS generation mediates the loss of the anti‐contractile effects of perivascular adipose tissue in high‐fat diet obese mice. Br J Pharmacol. 2017;3527–3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Chen F, Barman S, Yu Y, Haigh S, Wang Y, Black SM, Rafikov R, Dou H, Bagi Z, Han W, et al. Caveolin‐1 is a negative regulator of NADPH oxidase‐derived reactive oxygen species. Free Radic Biol Med. 2014;201–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.