

Abstract
Background
Cardiac fibroblasts (CFs) have the ability to sense stiffness changes and respond to biochemical cues to modulate their states as either quiescent or activated myofibroblasts. Given the potential for secretion of bioactive molecules to modulate the cardiac microenvironment, we sought to determine how the CF secretome changes with matrix stiffness and biochemical cues and how this affects cardiac myocytes via paracrine signaling.
Methods and Results
Myofibroblast activation was modulated in vitro by combining stiffness cues with TGFβ1 (transforming growth factor β 1) treatment using engineered poly (ethylene glycol) hydrogels, and in vivo with isoproterenol treatment. Stiffness, TGFβ1, and isoproterenol treatment increased AKT (protein kinase B) phosphorylation, indicating that this pathway may be central to myofibroblast activation regardless of the treatment. Although activation of AKT was shared, different activating cues had distinct effects on downstream cytokine secretion, indicating that not all activated myofibroblasts share the same secretome. To test the effect of cytokines present in the CF secretome on paracrine signaling, neonatal rat ventricular cardiomyocytes were treated with CF conditioned media. Conditioned media from myofibroblasts cultured on stiff substrates and activated by TGFβ1 caused hypertrophy, and one of the cytokines in that media was insulin growth factor 1, which is a known mediator of cardiac myocyte hypertrophy.
Conclusions
Culturing CFs on stiff substrates, treating with TGFβ1, and in vivo treatment with isoproterenol all caused myofibroblast activation. Each cue had distinct effects on the secretome or genes encoding the secretome, but only the secretome of activated myofibroblasts on stiff substrates treated with TGFβ1 caused myocyte hypertrophy, most likely through insulin growth factor 1.
Keywords: fibrosis, hydrogels, mechanotransduction, secretome
Subject Categories: Remodeling, Basic Science Research, Fibrosis, Growth Factors/Cytokines
Nonstandard Abbreviations and Acronyms
- AKT
protein kinase B
- CF
cardiac fibroblast
- CM
conditioned medium
- ECM
extracellular matrix
- NRVM
neonatal rat ventricular myocyte
- PEG
poly (ethylene glycol)
- RT‐qPCR
quantitative reverse transcription–polymerase chain reaction
- αSMA
α‐smooth muscle actin
- SMAD
small mothers against decapentaplegic
- TGFβ
transforming growth factor β
Clinical Perspective
What Is New?
This work identifies novel secreted factors that are upregulated with myofibroblast activation and may contribute to heart disease progression through paracrine signaling.
What Are the Clinical Implications?
Identification of these secreted factors can inform therapeutics to treat or prevent cardiac fibrosis, or can serve as markers for diagnosis of cardiac fibrosis.
Better diagnoses and treatments for fibrosis can lead to earlier intervention and better outcomes for patients with cardiovascular disease.
Cardiac fibrosis, characterized by increased cardiac stiffness, accompanies many forms of heart disease and exacerbates preexisting heart conditions by changing the cardiac microenvironment. Currently, there are limited therapies to treat cardiac fibrosis, highlighting the need to better understand signaling in the fibrotic heart. 1 Identifying secreted proteins associated with cardiac fibrosis serves the dual purpose of identifying novel diagnostic markers and targeting secreted proteins to treat cardiac fibrosis.
Cardiac fibroblasts (CFs) are the main source of proteins that cause cardiac fibrosis. 2 , 3 CFs play a vital role in the injured heart by secreting proteins that prevent cardiac rupture. 4 , 5 CFs deposit extracellular matrix (ECM) that forms a proteinaceous network around cells, reinforcing the cardiac architecture. CFs also secrete signaling molecules to surrounding immune cells and cardiomyocytes that affect the inflammatory response, contraction, and hypertrophy. 3 , 6 , 7 Conditioned medium (CM), which contains a plethora of different proteins, is generally used to determine the effects of the CF secretome on other cell types, but there is a need to identify specific factors that CFs secrete that elicit pathological paracrine signaling. RNA sequencing data on CFs after myocardial infarction have revealed changes in gene expression of cytokines, which change from proinflammatory and leukocyte recruiting 1 day after myocardial infarction to profibrotic and proangiogenic 3 days after myocardial infarction. 8 Although initial activation of this reparative response is critical for healing, persistent activation can result in buildup of ECM and long‐term activation of pathological signaling.
When CFs respond to injury, they transition from quiescent fibroblasts to activated myofibroblasts. 2 Quiescent fibroblasts are characterized by the expression of transcription factor 21, whereas activated myofibroblasts express α‐smooth muscle actin (αSMA) and periostin. 2 Myofibroblast activation is initiated by changes in the physical and biochemical cues in the microenvironment, which include increases in ECM and in TGFβ (transforming growth factor β ) levels. TGFβ is the most well‐characterized profibrotic cue that triggers activation to the myofibroblast phenotype both in vitro and in vivo. 9 In animal models, TGFβ receptor knockouts have been shown to have reduced cardiac fibrosis, 9 and TGFβ neutralizing antibodies can reduce fibrosis by suppressing ECM deposition in many organs, including the liver, kidney, lung, and heart. 10 , 11 TGFβ exerts its effects through activation of small mothers against decapentaplegic (SMAD) transcription factors, which increase expression of αSMA, collagen, and connective tissue growth factor. 12 , 13 TGFβ signaling is difficult to target for treatment of cardiac fibrosis, specifically because of its pleotropic effects in other tissues 14 ; however, targeting cytokine secretion downstream of TGFβ signaling may provide a means to treat fibrotic signaling. 15
Although TGFβ plays an important role in myofibroblast activation and consequent fibrosis, recent research has revealed an equally important role of physical cues underneath and surrounding cells on cell behavior, including stem cell differentiation, migration, and myofibroblast activation. 16 , 17 , 18 In fibrotic diseases, excessive ECM deposition causes tissue stiffening. 19 The stiff network of cross‐linked collagen that forms with cardiac fibrosis is thought to contribute to a positive feedback loop that leads to persistent myofibroblast activation with disease progression. The effects of stiffness have been studied in vitro using synthetic hydrogels that can be tuned to match the stiffness observed in healthy or injured tissue. 18 , 20 , 21 On stiff hydrogels, fibroblasts activate to myofibroblasts as they do in a fibrotic heart. 22 The cues that initiate activation have been characterized; however, their downstream effects on cytokine secretion have not been well studied. In addition, it is not understood if TGFβ or stiffness‐induced activation results in the same myofibroblast phenotype or if the 2 cues may synergize to influence the CF secretome.
In vivo, increased ECM stiffness, TGFβ, and changes in a variety of other factors act together to promote myofibroblast activation. Herein, isoproterenol treatment in vivo was used as a model for cardiac injury to determine the relevance of certain cytokines in an in vivo model of cardiac fibrosis. 23 Isoproterenol treatment in rats causes increased expression of both ECM and TGFβ in the heart. 24 Although physical and biochemical cues are intimately tied to each other in vivo, where growth factors are sequestered in and bound to ECM, 25 the 2 cues can be decoupled in vitro to determine how TGFβ and stiffness individually or synergistically influence cytokine secretion and signaling to cardiomyocytes. Using both an in vitro and in vivo model of fibrosis allowed us to determine whether changes in the secretome in vitro were consistent with changes observed in vivo with isoproterenol. This combination of treatments allowed us to identify unique protein signatures that change with each cue. Understanding how the fibrotic microenvironment affects CF cytokine secretion and paracrine signaling is critical to define the mechanisms involved in cardiac fibrosis and to prevent the downward spiral of events that can lead to heart failure. The information gained through these experiments can be used to inform diagnostics and treatments for fibrosis.
Methods
Transparency and Openness
The data that support the findings of this study are available from the corresponding author on reasonable request. The authors declare that all supporting data are available within the article and its supplementary files.
CF Isolation and Isoproterenol Treatment
CFs from adult rat left ventricles (LVs) of male rats were isolated using an established protocol. 26 Briefly, adult, male Sprague Dawley rats (200–250 g at arrival) were acclimated to the facility for 1 week before isolation. Rats were injected intraperitoneally with 1 mL, 390 mg/mL pentobarbital (Fatal Plus), left for 15 minutes, checked for responsiveness with foot pinch, before the heart was removed. The LV was isolated, minced, and digested with collagenase II (1 mg/mL) for 3 hours. The resulting cell mixture was centrifuged, and cell pellets were resuspended and plated for 2 hours before plates were washed vigorously to remove nonadherent cells. The cell population was negative for vascular endothelial‐cadherin (endothelial cells) and positive for vimentin (fibroblasts). For all experiments, CFs were cultured for no more than 4 days on tissue culture plastic in 10% fetal bovine serum DMEM/F12 (Gibco) before seeding on hydrogels. Animal procedures were done in accordance with Institutional Animal Care and Use Committee approved protocol No. 2351. For the isoproterenol treatment studies, adult, male Sprague Dawley rats (200–250 g at arrival) were implanted randomly with either ascorbic acid (vehicle) or isoproterenol pumps (4 mg/kg per day) for 3 days, rats were euthanized, and fibroblasts were isolated using the above method. Isoproterenol was dissolved in 1 μmol/L ascorbic acid. For administration of isoproterenol, subcutaneous administration was achieved by osmotic mini‐pump implantation. For mini‐pump implantation, rats were anaesthetized with isoflurane (5%) via spontaneous inhalation and then placed on a 37°C recirculated heating pad. For the remainder of the procedure, animals were maintained on 2% isoflurane via spontaneous inhalation. All surgical procedures were performed under aseptic conditions with sterile surgical instruments and on a sterile field. Fur was removed from the surgical site by shaving. The operating field was disinfected with Nolvasan surgical scrub and 70% ethanol twice in alternate. Rats received a single injection of sustained‐release buprenorphine (1.0 mg/kg, subcutaneous) before beginning the procedure. An incision was made slightly caudal to the scapulae with scissors. The skin and the muscle layers were separated with curved hemostats. The sterile mini‐pumps containing either isoproterenol or vehicle (1 μmol/L ascorbic acid) were placed under the skin. The incision was closed with a surgical stapler. Animals were observed for any signs of distress and returned to the vivarium where they were observed daily for the duration of the experiment. Any animals showing visible signs of distress were removed from the study and euthanized. The procedures followed were in accordance with institutional guidelines.
Hydrogel Synthesis
Synthesis of poly (ethylene glycol) (PEG) functionalized with norbornene end groups was done following an established protocol. 27 PEG thiol was purchased from JenKem. Hydrogels were polymerized using a thiol‐ene photo‐click reaction and to promote cell adhesion a cysteine functionalized linear RGD (2 mmol/L) (Custom order Bachem) was included in all formulations (Table 1). The photo‐click reaction was initiated using 2 mmol/L lithium phenyl‐2,4,6‐trimethylbenzoylphosphinate and 365 nm light for 3 minutes at 5 mW/cm2.
Table 1.
PEG Hydrogel Formulations
| PEG‐Norbornene Arms | PEG‐Norbornene MW, kDa | Norbornene, mmol/L | PEG‐Thiol Arms | PEG‐Thiol MW, kDa | Thiol, mmol/L | Young Modulus, kPa |
|---|---|---|---|---|---|---|
| 8 | 47 | 8 | 2 | 5 | 2.96 | 6 |
| 8 | 27 | 40 | 8 | 13 | 2.25 | 59 |
MW indicates molecular weight; and PEG, poly (ethylene glycol).
Rheology
In situ rheology was used to measure the modulus of each gel formulation. A Discovery HR‐3 Hybrid Rheometer was used to measure the shear storage modulus (G') of each replicate of each formulation. UV light (365 nm at 5 mW/cm2) was applied through a quartz plate to polymerize the hydrogel (180 seconds), while the rheometer applied stress at an angular frequency of 1 rad/s and 1% strain. The resulting storage modulus for each gel was converted to a Young modulus for comparisons to other literature studies and characterization of heart muscle. 19 Assuming relative incompressibility of the PEG gels 28 and a Poisson ratio (ν) of 0.5, the E (Young modulus) was calculated as: E=2G'(1+ν). The in situ modulus was assumed to closely approximate the final modulus used for cell studies.
Cell Seeding on Hydrogels, Treatment, and CM Collection
For immunostaining and quantitative reverse transcription–polymerase chain reaction (RT‐qPCR) experiments, CFs were seeded onto hydrogels (5000 cells/cm2) and cultured for 5 days before fixation, lysis for RNA isolation, or CM collection. For TGFβ1 treatment, CFs were grown with 10 ng/mL human TGFβ1 (Fisher, Pepro Tech NC0427897) or untreated cells were treated with vehicle (citric acid). For CM collection, CFs were grown for 5 days on selected hydrogel formulations (Table 1). At day 2 of culture, medium was refreshed and collected 5 days after seeding. CM was centrifuged to remove particulates. For all experiments on hydrogels, CFs were cultured in 1% fetal bovine serum DMEM/F12 (Gibco).
RNA Isolation and RT‐qPCR
RNA was isolated using RNAeasy Micro Kit (Qiagen catalog No. 74004) or Trizol extraction. cDNA synthesis was performed using BioRad iScript Reverse Transcriptase cDNA synthesis kit (1708840). RT‐qPCR was performed using IQ BioRad SYBR Green Supermix (170‐8880). Expression of the gene of interest was normalized to GAPDH expression for hydrogel experiments and TBP (TATA‐binding protein) for isoproterenol experiments. Primer sequences are listed in Table 2.
Table 2.
Primer Sequences
| Primer | Forward (5′‐3′) | Reverse (5′‐3′) |
|---|---|---|
| GAPDH | AGGTCGGTGTGAACGGATTTG | TGTAGACCATGTAGTTGAGGTCA |
| TBP | TGCACAGGAGCCAAGAGTGAA | CACATCACAGCTCCCCACCA |
| TCF21 | GGCCAACGACAAGTACGAGAA | CACACCTCCAAGGTCAGGATG |
| Periostin | CAGGCTCGCCTTCAATG | TCGTGGAACCAAAAATTAAAGTC |
| Osteopontin | GAAGCCAGCCAAGGTAAGC | CACTGCCAGTCTCATGGTTG |
| Osteoprotegerin | GAGGTTTCCAGAGGACCACA | TGTCCATTCAATGATGTCCAA |
| IGFBP‐3 | AGCCTGAGTGCCTACCTCC | GCATTGTGGTCCTCCTCAGA |
| IGFBP‐5 | TACGGCGAGCAAACCAAGAT | ACCTTCGGGGAGTAGGTCTC |
| VEGF | CGGGCCTCTGAAACCATGAA | GCTTTCTGCTCCCCTTCTGT |
| WISP‐1 | GACATCCGACCACACATCAA | GAAGTTCGTGGCCTCCTCT |
Immunostaining, Imaging, and Quantification
CFs cultured on hydrogels were fixed in 4% paraformaldehyde for 15 minutes. Samples were permeabilized with 0.1% Triton for 1 hour, then blocked with 5% BSA overnight. Primary antibodies were added for 1 hour and washed, and secondary antibodies were applied for 1 hour. The primary antibodies were αSMA (Abcam Ab7817) at 1:1000 and SMAD 2/3 (Santa Cruz SC‐133098) at 1:200. The secondary antibodies were goat anti‐mouse (AbCam ab150117) at 1:400 and cellmask orange (Thermo Fisher H32713) at 1:5000, and nuclei were stained with 4′,6‐diamidino‐2‐phenylindole (Lifetech D1306) at 1:1000. Imaging was performed using the Perkin Elmer high‐throughput imaging microscope (Operetta). Activation was quantified using Harmony software: the cytoplasm of the cells was identified using cellmask staining, and the intensity of α‐SMA, SMAD2/3, or cellmask was quantified.
Western Blot
CFs were grown on hydrogels for 5 days and lysed in radioimmunoprecipitation assay buffer. Protein was quantified using microBCA assay. Protein was loaded into BioRad 50 μL (5%–20% gradient gels), transferred to a polyvinylidene difluoride membrane, stained for total protein (LI‐COR 926‐11011), imaged using Typhoon, blocked with 5% BSA, cut and probed for phosphorylated AKT (protein kinase B) (Cell Signaling 9275S), stripped and probed for AKT (Cell Signaling C73H10), and αSMA was probed separately (Abcam Ab7817) at 1:1000. Secondary antibodies were purchased from Jackson Immunoresearch (115‐035‐003), and chemiluminescent substrate was purchased (Thermo Fisher Scientific 34075). Signal for αSMA was normalized to total protein (entire lane of protein was used for normalization). The same protocol was used to probe phosphorylated AKT (Cell Signaling 9275S) and AKT (Cell Signaling C73H10) in LV tissue.
Histology, Imaging, and Atomic Force Microscopy
LV tissue samples from vehicle (saline treated) or isoproterenol treated rats were collected and embedded in optimal cutting temperature (OCT) compound, flash frozen, and cryosectioned. Sections were fixed in 4% paraformaldehyde, stained for vimentin (Thermo Fisher PA1‐10003) at 1:5000, SMAD 2/3 (Santa Cruz SC‐133098) at 1:200, and nuclei were stained with 4′,6‐diamidino‐2‐phenylindole (Lifetech D1306) at 1:1000. The secondary antibody used was goat anti‐mouse 488 (AbCam ab150117) at 1:400 and goat anti‐chicken 650 (AbCam ab96954). Sections were imaged using a Nikon Spinning Disc confocal microscope with a ×40 objective. Imaris software was used to quantify the nuclear/cytoplasmic ratio of SMAD 2/3 in vimentin‐positive cells. Vimentin was used to identify the cytoplasm, and 4′,6‐diamidino‐2‐phenylindole was used to identify the nuclei.
Mechanical characterization was performed on an Asylum Research MFP‐3D (Oxford Instruments) atomic force microscope. Atomic force microscopy was performed on LV tissue sections, where N=3 male rats treated for 7 days with either vehicle or isoproterenol. Tissue sections were submerged in PBS and allowed to thermally equilibrate to room temperature for at least 30 minutes before measurements. A spherical probe was created by attaching a borosilicate glass sphere (10 μm diameter, Duke Standards) to a tipless cantilever (Budget Sensors All‐in‐One TL) using a UV curable resin (Loon Outdoors UV Clear). The spring constant of the cantilever was determined using the thermal method 29 to be 0.21 N m−1, in agreement with the manufacturer's specification (0.2 N m−1). Force‐volume mapping was performed in PBS in a 32×32 grid over a 90×90 μm area, with a ramp speed of 4 μm s−1 and a trigger point of 5 nN. The Young modulus was determined at each pixel using an Oliver‐Pharr indentation model, assuming incompressibility for the heart tissue (Poisson ratio=0.5).
Cytokine Array and ELISAs
A Proteome Profiler XL cytokine array (ARY030) was used to characterize changes in the CF secretome. For each cytokine array, N=3, with the exception of the 6 kPa TGFβ (N=2). One array was incubated with media to subtract out secreted protein from protein in the media. Each N represents a different isolation of CFs. For each isolation, and incubation on hydrogels, CFs from 4 male rats were pooled together. The cytokine array was analyzed using ImageQuant TL (Array Analysis). Signal was normalized to positive control on the array and total protein. Supplemental: osteopontin ELISA (R&D MOST00), lysyl oxidase activity assay (ab112139), and lysyl oxidase activity were normalized to total protein in the media.
Neonatal Rat Ventricular Myocyte Isolation and Treatment
Neonatal rat ventricular myocytes (NRVMs) were isolated from Sprague Dawley rats (50–60 rats pooled, male and female, 1 day old), kept on ice before isolation, and isolated using previously identified method. 30 NRVMs were cultured for 24 hours in growth media, rinsed, and cultured 24 hours in serum‐free media (minimum essential media (MEM), HEPES, B12, Bromodeoxyuridine (BrdU), BSA, insulin, transferrin) to prevent growth of fibroblasts. CM from fibroblasts was used to treat NRVMs for 48 hours before measuring cell size. SD208 (Sigma Aldrich S7071) was used in combination with CF CM treatment of NRVMs at 3μmol/L. The same protocol was used for insulin growth factor 1 (IGF‐1) and osteopontin treatments. IGF‐1 (R&D Systems 4326‐RG‐050) and osteopontin (R&D Systems 6359‐OP‐050) were added NRVMs in serum‐free media and cultured for 48 hours before NRVMs were trypsinized and cell size was measured by Coulter Counter.
Graphics
All schematics in figures were created with BioRender.com. Heat map for cytokines was created using Clustervis using unit variance scaling.
Statistical Analysis
GraphPad Prism 7.0e was used for data analysis. When 1‐way ANOVAs were used, Tukey multiple comparisons test was performed. When 2‐way ANOVAs were used, Tukey multiple comparisons test was performed. For T tests, a 2‐tailed, unpaired, test was used. For all statistics, 95% confidence level was used to determine statistical significance, where P≤0.05 was considered statistically significant.
Results
Stiffness and TGFβ1 Treatment Induce Myofibroblast Activation
We probed the role of stiffness versus TGFβ in initiating myofibroblast activation, using a hydrogel platform to culture CFs from the LV of male rats. A thiol‐ene photo‐click reaction was used to synthesize PEG hydrogels for the culture of adult rat CFs. This method provides a tunable and cytocompatible platform to measure mechanosensitive responses of CFs. Hydrogels of different stiffnesses were generated by adjusting the cross‐linking density of PEGs functionalized with either norbornene or thiol arms. The Young modulus was calculated from rheological measurements of the shear storage modulus and targeted to match that of cardiac tissue. 19 The soft hydrogel formulation had a Young modulus of ≈6 kPa, whereas the stiff hydrogel formulation had a Young modulus of ≈59 kPa (Figure S1). In addition, TGFβ1 (10 ng/mL) was added exogenously to the culture media to observe the combined effect of stiffness and TGFβ1 treatment on CF myofibroblast activation (Figure 1A). Culturing CFs on soft hydrogels preserved the quiescent CF phenotype, allowing us to observe changes in myofibroblast activation with treatment (Figure 1A).
Figure 1. Cardiac fibroblast (CF) activation is modulated by culturing on different stiffness hydrogels and by adding TGFβ1 (transforming growth factor β 1) .
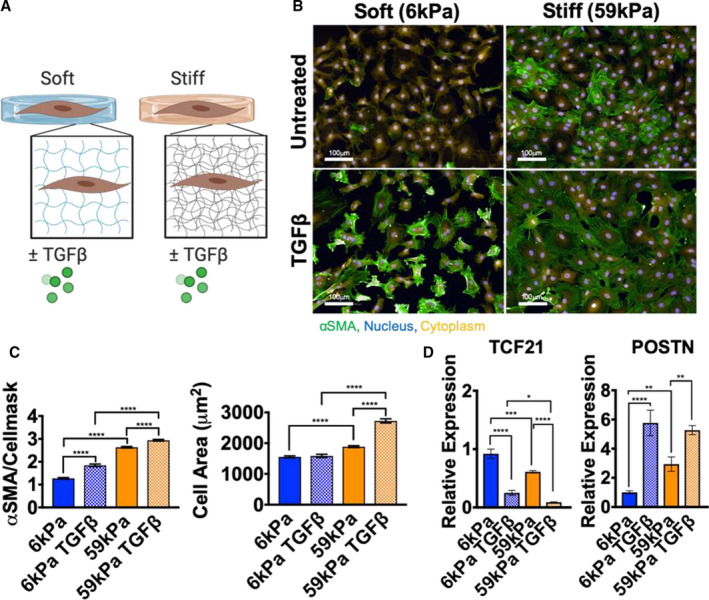
A, Schematic representation of experimental design to probe changes in myofibroblast activation in CFs. B, Representative images of immunofluorescence staining of CFs cultured for 5 days on hydrogels ±10 ng/mL TGFβ1 (green, α‐smooth muscle actin [αSMA]; yellow, cellmask; blue, nuclei; bar=100 μm). C, Changes in αSMA/cellmask intensity and changes in cell area quantified (N=individual cells from 2 experiments, error bars show 95% CI, 1‐way ANOVA, ****P≤0.0001). D, Quantitative reverse transcription–polymerase chain reaction measurement of periostin (POSTN) and transcription factor 21 (TCF21) expression in CFs on hydrogels±TGFβ1 (N=3, error bars show mean and SEM, 1‐way ANOVA, *P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001).
CFs were cultured for 5 days on soft (E=≈6 kPa) or stiff (E=≈59 kPa) hydrogels, with or without TGFβ1, and measured for myofibroblast activation using αSMA as a marker of activation. CFs cultured on stiff hydrogels exhibited higher expression of αSMA protein compared with CFs cultured on soft hydrogels (Figure 1B and 1C). Regardless of whether CFs were cultured on stiff or soft hydrogels, TGFβ1 was able to further increase expression of αSMA and stress fiber formation on both hydrogel stiffnesses. Measurements of cell area showed that cell spreading was increased on stiff hydrogels and with TGFβ1 treatment, which may be related to αSMA stress fiber formation (Figure 1C). As additional markers of myofibroblast quiescence and activation, transcription factor 21 and periostin expression were measured by RT‐qPCR. Periostin increased with stiffness and TGFβ1 treatment, whereas transcription factor 21 expression decreased with both activation cues (Figure 1D). These results indicated that this platform could be used to measure changes in the quiescent versus activated myofibroblast secretome because increased stiffness and TGFβ1 treatment caused CF activation to the myofibroblast phenotype.
Stiffness and TGFβ1 Treatment Act Through Different Pathways to Induce Myofibroblast Activation
To determine whether stiffness changes and TGFβ1 treatment activate CFs through the same pathway, SMAD and AKT signaling were measured. SMAD2 and SMAD3 are transcription factors that localize to the nucleus when TGFβ receptor signaling is active. 9 As expected, nuclear localization of SMAD 2/3 was increased with TGFβ1 treatment. However, SMAD localization was not affected by stiffness changes (Figure 2A and 2B). This result indicated that the increase in αSMA levels with stiffness was not mediated by changes in SMAD signaling.
Figure 2. Small mothers against decapentaplegic (SMAD) signaling was activated with TGFβ1 (transforming growth factor β 1) treatment, but not stiffness, and AKT (protein kinase B) phosphorylation increased with both cues.
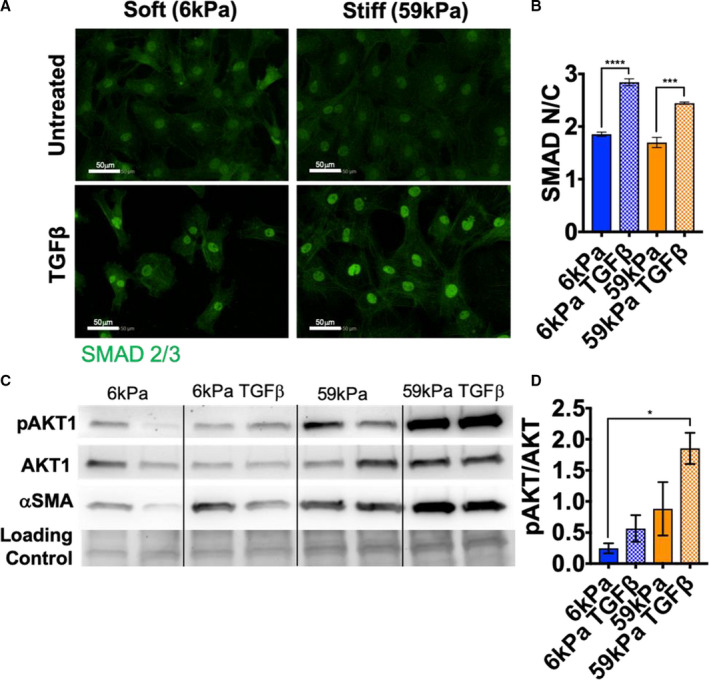
A, Representative images of immunofluorescence staining of cardiac fibroblasts (CFs) cultured for 5 days on hydrogels ±10ng/mL TGFβ1 (green, SMAD 2/3; bar=50 μm). B, Quantification of changes in SMAD 2/3 nuclear/cytoplasm (N/C) intensity (N=3, error bars show 95% CI, 1‐way ANOVA, ***P≤0.001, ****P≤0.0001). C, Western blot images of phosphorylated AKT 1 (pAKT1), total AKT1, α‐smooth muscle actin (αSMA), and loading control (total protein stain). D, Quantification of pAKT1/AKT1 (N=2, error bars show mean and SEM, 1‐way ANOVA, *P≤0.05).
To examine pathways involved in mechanosensing, changes in AKT phosphorylation were examined with stiffness and TGFβ1 treatment. AKT has been shown to modulate mechanosensing in valvular interstitial cells and regulates a multitude of processes in cells, including protein synthesis, proliferation, and migration. 18 Although SMAD localization was unchanged with stiffness, AKT phosphorylation was increased with both stiffness and TGFβ1 treatment (Figure 2C and 2D). Unlike SMAD localization, the change in AKT phosphorylation corresponded to changes in αSMA protein levels, indicating that this pathway is likely involved in myofibroblast activation with both stiffness and TGFβ1 treatment. In addition, activation of SMAD and AKT was examined in an in vivo fibrosis model (isoproterenol treatment). SMAD localization did not change in vivo with isoproterenol (Figure S2A and S2B); however, AKT phosphorylation increased in LV tissue with isoproterenol (Figure S2C and S2D). This result indicated that the myofibroblast activation caused by isoproterenol treatment in vivo corresponded to increased AKT signaling, which was increased in vitro with both TGFβ and stiffness cues. To determine whether LV stiffness changes with isoproterenol treatment, atomic force microscopy was used to measure the modulus of tissue sections. The Young modulus of the LV was not significantly different with isoproterenol treatment compared with vehicle treatment, indicating that stiffness changes in the LV were not activating AKT signaling (Figure S3A through S3C).
Stiffness and TGFβ1‐Induced Myofibroblast Activation Result in Different Secretomes
Although myofibroblast activation was observed with both cues, the fact that different pathways appear to be used by each cue to activated myofibroblasts pointed to possible changes in downstream responses, including changes in cytokine secretion. To examine changes in cytokine secretion with stiffness changes or TGFβ treatment, CFs were cultured under conditions (Figure 3A) to produce populations of quiescent fibroblasts (soft/TGFβ−) and activated myofibroblasts (soft/TGFβ+; stiff/TGFβ−; stiff/TGFβ+). Medium was collected from cultured CFs, defined as CM, and used to measure relative changes in 79 different cytokines. Changes in cytokine secretion of CFs grown under 4 different conditions (soft/TGFβ−, soft/TGFβ+, stiff/TGFβ−, and stiff/TGFβ+) are shown in Figure 3B. To examine whether there were secretory markers that matched myofibroblast activation markers, αSMA and periostin, correlation analyses were performed. Vascular endothelial growth factor secretion correlated with the activation marker αSMA (Figure S4A), whereas IGF‐1 correlated with periostin expression (Figure S4B). However, cytokine secretion did not always follow linear trends that correlated with activation markers. The cytokines that changed significantly with stiffness or TGFβ1 treatment are shown in Figure 4A through 4E, where different subsets of cytokines changed levels depending on whether cells were cultured on soft or stiff hydrogels. Chemokine (C‐C motif) ligand 20 (CCL20), granulocyte‐macrophage colony‐stimulating factor, CCL3/4, and prolactin levels changed in the media of CFs grown on soft versus stiff hydrogels (Figure 4A and 4E). WISP‐1 (Wnt1 inducible signaling pathway protein 1), fetuin‐A, IGF‐1, and granulocyte‐macrophage colony‐stimulating factor were increased in the CM of CFs treated with TGFβ1 on soft and stiff hydrogels (Figure 4B, 4D, and 4E), whereas 11 cytokines were decreased in the CM of CFs treated with TGFβ1 (Figure 4B, 4D, and 4E). Endostatin, hepassocin, and granulocyte‐macrophage colony‐stimulating factor were significantly different between the soft/TGFβ+ and stiff/TGFβ+ conditions (Figure 4C). In addition, osteopontin, which has been shown to regulate collagen cross‐linking via lysyl oxidase activity, 31 was significantly increased with TGFβ1 and stiffness (Figure S5A). Lysyl oxidase activity in CM did not correlate with osteopontin secretion, suggesting that osteopontin was not regulating collagen cross‐linking in fibroblasts cultured in vitro (Figure S5B).
Figure 3. Cytokine arrays were used to determine the changes in relative levels of 79 different cytokines secreted with myofibroblast activation by stiffness, TGFβ1 (transforming growth factor β 1), or both, in conditioned medium (CM).
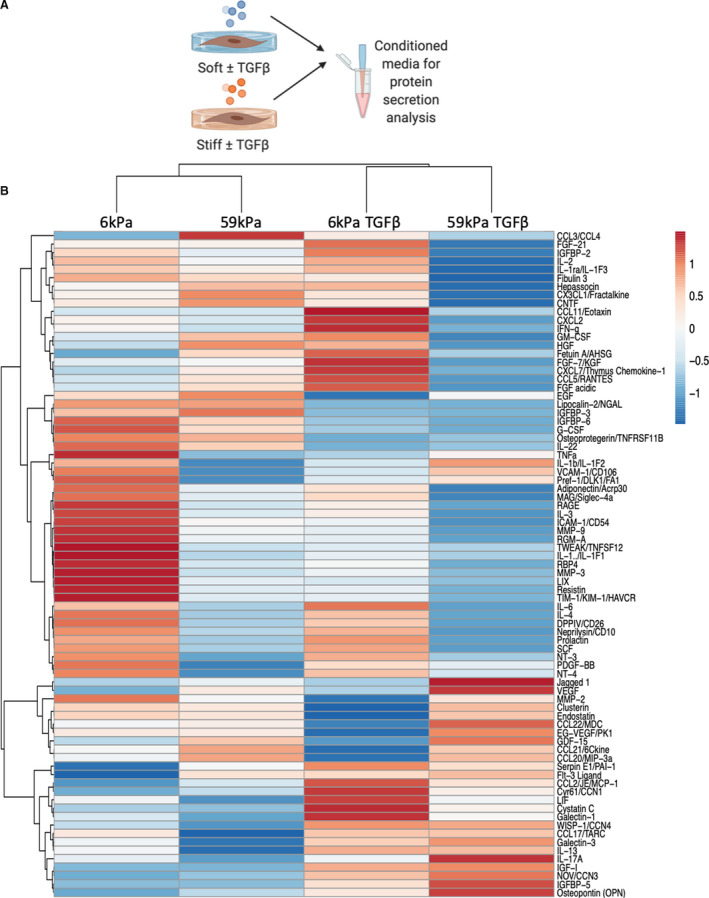
A, Schematic representation of CM collection. B, Heat map showing changes in relative levels of 79 different cytokines using unit variance scaling (created using Clustervis, average values N=2 isolations [4 rats pooled for each isolation]).
Figure 4. Cytokines that significantly increased or decreased in the conditioned media of cardiac fibroblasts (CFs) cultured on soft vs stiff hydrogels ±10 ng/mL TGFβ1 (transforming growth factor β 1).
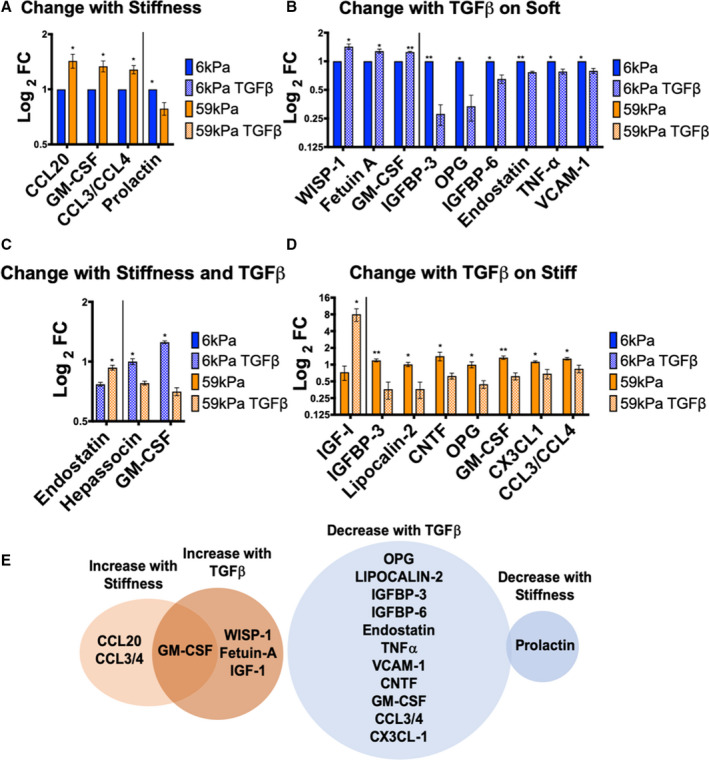
A, Cytokines that change by culturing on different stiffnesses, *P≤0.05. B, Cytokines that change with TGFβ1 treatment on soft hydrogels, *P≤0.05, **P≤0.01. C, Cytokines that change between CFs grown on soft hydrogels with TGFβ1 vs CFs grown on stiff hydrogels with TGFβ1, *P≤0.05. D, Cytokines that change with TGFβ1 treatment on stiff hydrogels, *P≤0.05, **P≤0.01. E, Cytokines that significantly increased with myofibroblast activation (P≤0.05) are shown in orange and cytokines that decreased with activation are shown in blue. Overlap between treatment with stiffness and TGFβ1 is shown in the Venn diagrams. Cytokines that change with TGFβ1 treatment on either soft or stiff hydrogels are displayed in the Venn diagrams. All data are normalized to 6 kPa condition for each isolation. (N=3 isolations for 6 kPa, 59 kPa, 59 kPa TGFβ, N=2 for 6 kPa TGFβ, error bars show mean and SEM, 2‐tailed unpaired T test). FC indicates fold change; GM‐CSF, granulocyte‐macrophage colony‐stimulating factor; IGFBP, insulin growth factor binding protein; OPG, osteoprotegerin; TNF‐α, tumor necrosis factor‐α; VCAM‐1, vascular cell adhesion molecule 1; WISP‐1, Wnt1 inducible signaling pathway protein 1; CCL, Chemokine (C‐C motif) ligand; and CNTF, Ciliary neurotrophic factor.
Taken together, these results indicate that CFs activated by stiffness and TGFβ1 treatment have different secretomes, but there were also synergies between the stiffness and TGFβ1 treatment that likely contribute to changes in cytokine expression. For example, TGFβ1 treatment on stiff hydrogels had significantly different effects than TGFβ1 treatment on soft hydrogels (Figure 4C), suggesting that physical cues influence cellular responses to biochemical cues. These changes in the secretome are likely mediated by different signaling mechanisms, SMAD (for TGFβ1 treatment) and AKT (for stiffness and TGFβ1 treatment).
Measurements of Cytokine mRNA Expression in Vivo Agree With in Vitro Measurements
To validate the physiological relevance of changes in cytokine levels observed with in vitro myofibroblast activation, results were compared with changes measured in CFs isolated from isoproterenol treated animals (Figure 5C). Specifically, cytokine gene expression was measured in cultured CFs in vitro and from CFs isolated from in vivo experiments. First, to determine whether the changes in protein observed from the cytokine array were seen at the mRNA level, RT‐qPCR was used to quantify in vitro samples (Figure 5A). For the 6 cytokines measured, changes in gene expression correlated well with changes in protein secretion, including osteopontin, osteoprotegerin, vascular endothelial growth factor, WISP‐1, and IGFBP (insulin growth factor binding protein) 3 and 5 (Figure 5B and 5D). RT‐qPCR was then used to determine whether cytokine mRNA expression levels changed in the CFs of rats treated with isoproterenol (Figure 5C). Osteopontin, IGFBP‐5, and IGFBP‐3 mRNA expression levels were significantly different between vehicle and isoproterenol treated groups (Figure 5E). The changes in RNA expression observed in vivo with isoproterenol treatment matched well with activation changes in vitro with stiffness or TGFβ1 treatment, with the exception of IGFBP‐3, which decreased with activation in vitro and increased with activation in vivo.
Figure 5. Changes in protein secretion in conditioned media match changes in gene expression in both in vitro and in vivo models of fibrosis.
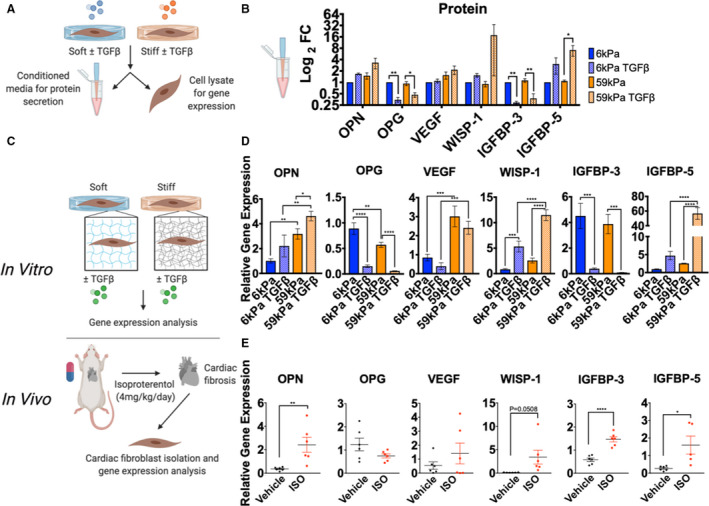
A, Schematic of experimental layout for measuring changes in protein secretion in conditioned media and gene expression in cardiac fibroblasts (CFs) cultured on hydrogels. B, Protein changes in select cytokines from cytokine array (normalized to 6 kPa condition, N=3, error bars show mean and SEM, 1‐way ANOVA, *P≤0.05, **P≤0.01). C, Schematic representation of experimental design to compare changes in cytokine gene expression in vitro with changes in vivo with isoproterenol (ISO). D, Changes in gene expression of cytokines from CFs cultured on hydrogels±TGFβ1 (transforming growth factor β 1) for 5 days (N=3, error bars show mean and SEM, 1‐way ANOVA, *P≤0.05, **P≤0.01, ***P≤0.001, ****P≤0.0001). E, Changes in gene expression of cytokines from CFs isolated from rats treated with vehicle or ISO (4 mg/kg per day for 3 days, N=6 for each group, error bars show mean and SEM, 2‐tailed unpaired T test, *P≤0.05, **P≤0.01, ****P≤0.0001). FC indicates fold change; IGFBP, insulin growth factor binding protein; OPG, osteoprotegerin; OPN, osteopontin; VEGF, vascular endothelial growth factor; and WISP‐1, Wnt1 inducible signaling pathway protein 1.
Cytokines From TGFβ‐Activated Myofibroblasts Cause Cardiomyocyte Hypertrophy
In the heart, fibroblasts are located adjacent to cardiomyocytes, where cross talk between the 2 cell types can lead to myocyte hypertrophy. 32 To test whether myocytes would respond to cytokines secreted by activated myofibroblasts, NRVMs were treated with CM from the quiescent (soft/TGFβ−) and activated myofibroblasts (soft/TGFβ+, stiff/TGFβ−, and stiff/TGFβ+), and NRVM cell volume was measured (Figure 6A). Myocytes showed a hypertrophic response to the stiff/TGFβ+ CM, even in the presence of SD208 (a TGFβ receptor inhibitor), indicating that secreted factors in the media were inducing cardiomyocyte hypertrophy independent of signaling through the TGFβ receptor (Figure 6B). Because there was an increase in hypertrophy with stiff/TGFβ+ CM compared with stiff/TGFβ− CM, cytokines that increased with TGFβ1 treatment on stiff hydrogels were examined, which included IGF‐1 and osteopontin. It is well established that IGF‐1 induces cardiomyocyte hypertrophy, 33 and we recapitulated that observation herein. To test whether osteopontin could cause hypertrophy, NRVMs were treated with different concentrations of osteopontin (and IGF‐1) and cell size was measured. The myocytes showed a dose‐dependent increase in size with IGF‐1, but not osteopontin (Figure 6C), indicating that IGF‐1 secreted by activated myofibroblasts can signal to myocytes to induce a hypertrophic response (Figure 6D).
Figure 6. The cardiac fibroblast (CF) secretome increases neonatal rat ventricular myocyte (NRVM) size.
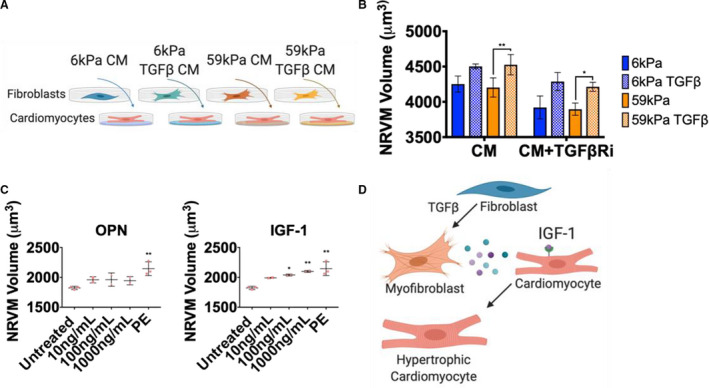
A, Schematic of conditioned medium (CM) treatment of NRVMs (48 hours treatment with conditioned media from CFs). B, Coulter counter measurements of NRVMs treated for 48 hours with CM from CFs with or without 3 μmol/L SD208 to inhibit effects caused by TGFβ1 (transforming growth factor β 1) (N=CM from 3 CF isolations, 4 rats pooled per isolation, error bars show mean and SEM, 1‐way ANOVA, *P≤0.05, **P≤0.01). C, Coulter counter measurements of NRVM volume when treated with different concentrations of osteopontin (OPN), insulin growth factor 1 (IGF‐1), or phenylephrine (PE) (10 μmol/L), (N=2 isolations of NRVMs, N=3 for controls [same controls used for OPN and IGF‐1 treatment], error bars show SD, 1‐way ANOVA, significance compared with untreated, *P≤0.05, **P≤0.01). D, Schematic of CF signaling to NRVMs to induce hypertrophy through IGF‐1 signaling.
Discussion
Currently, few treatments for cardiac fibrosis have proved effective, 34 highlighting the need to better understand the mechanisms that contribute to fibrosis, and how fibrosis, once established in the heart, can initiate a positive feedback loop that perpetuates pathological signaling. Until now, the cytokines that CFs secrete in an activated, myofibroblast state, have remain relatively uncharacterized. This may be, in part, due to a lack of physiologically relevant cell culture platforms, because culturing fibroblasts on stiff tissue culture plastic results in an altered cell phenotype that is not representative of cell phenotype in vivo. 18 This work used an engineered hydrogel system to maintain the quiescent fibroblast phenotype outside the heart, which allowed us to define differences between the quiescent and activated myofibroblast secretome. We found that different mediators of myofibroblast activation, including TGFβ, stiffness, and isoproterenol treatment, all promote CF activation but have distinct effects on the CF secretome (Figure 7). This finding has implications for diagnosis and treatment of fibrosis because different causes of fibrosis may have a unique CF secretory signature. Identifying protein signatures for each type of cardiac disease would help to identify the signaling pathways involved in the response, including whether the response was initiated by a change in physical or biochemical cues, or both. Although isoproterenol affects both the physical and biochemical properties of the heart, including TGFβ signaling and cardiac stiffening, 24 it also causes changes in heart rate and changes in contraction dynamics, which are physical cues that were not studied herein, but have been shown to affect CF profibrotic signaling. 35 Future research directed toward understanding the effects of stretch on CFs will help to elucidate the effects of combinatorial cues on CF myofibroblast activation and cytokine secretion to better understand fibrosis development in patients. Furthermore, in vivo changes with cardiac injury and disease are highly dynamic, and acute injury may result in a dramatically different CF secretome than with chronic cardiac disease. Monitoring changes in the CF secretome over time with cardiac disease would provide further insight into pathological signaling that contributes to heart failure and time windows for preventative treatment. This research lays the framework for future studies on the effects of various physical and biochemical cues on cytokine levels and paracrine signaling in myocardium. Identification of pathways activated by cytokine signaling in the fibrotic heart will guide appropriate diagnosis and treatment for cardiac fibrosis.
Figure 7. Schematic of TGFβ (transforming growth factor β) signaling and signaling through extracellular matrix (ECM) interactions to influence cytokine secretion.
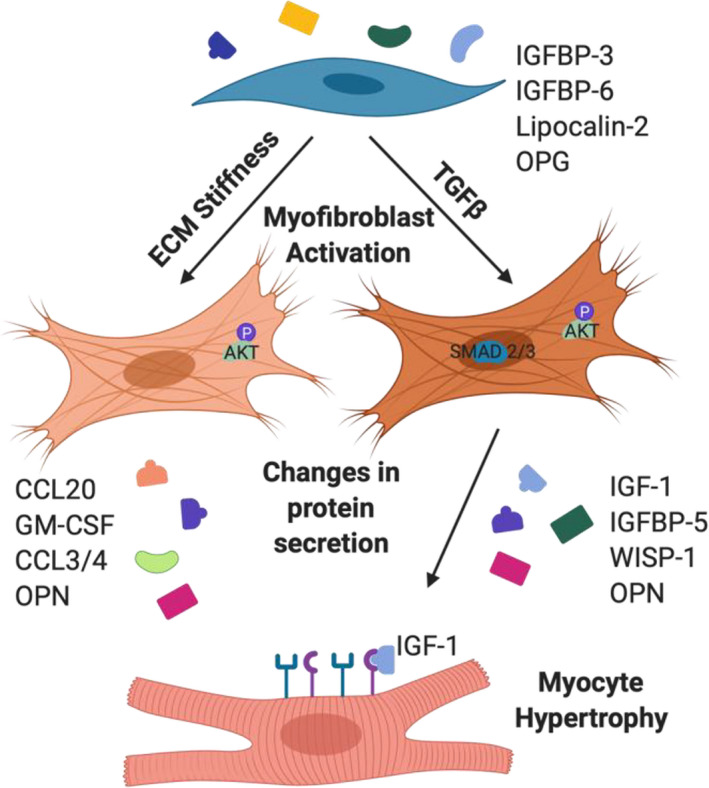
Cytokines that changed with TGFβ signaling vs ECM stiffness sensing are listed, and insulin growth factor 1 (IGF‐1), secreted by TGFβ‐activated myofibroblasts, caused hypertrophy. GM‐CSF indicates granulocyte‐macrophage colony‐stimulating factor; IGFBP, insulin growth factor binding protein; OPN, osteopontin; OPG, osteoprotegerin; WISP‐1, Wnt1 inducible signaling pathway protein 1; and CCL, Chemokine (C‐C motif) ligand.
Sources of Funding
Ceccato was funded by The American Heart Association Predoctoral Fellowship (17PRE33661129) and the National Institutes of Health (NIH) T32 Biophysics Training grant (T32GM065103). Walker was funded by NIH F31 (FHL142223). NIH R01 grants (R01 HL132353 for Anseth and R01 GM029090 for Leinwand) funded this research.
Disclosures
None.
Supporting information
Figures S1–S5
Acknowledgments
The authors thank Ann Robinson for performing the neonatal rat ventricular myocyte isolations, Christa Trexler for performing the Western blots with the isoproterenol tissue, Brian Aguado for his help with Western blots, Angela Peter for her help with the animal surgeries and advice on the manuscript, and the Core Imaging facility at Jennie Smoly Caruthers Biotechnology Building. Specific commercial instruments and materials that are identified in this article are listed to adequately describe the experimental procedure and are not intended to imply endorsement or recommendation by the National Institute of Standards and Technology.
(J Am Heart Assoc. 2020;9:e017025 DOI: 10.1161/JAHA.120.017025.)
For Sources of Funding and Disclosures, see page 13.
References
- 1. Cojan‐Minzat BO, Zlibut A, Agoston‐Coldea L. Non‐ischemic dilated cardiomyopathy and cardiac fibrosis. Heart Fail Rev. 2020:1‐21 [Published online ahead of print]. DOI: 10.1007/s10741-020-09940-0. [DOI] [PubMed] [Google Scholar]
- 2. Kanisicak O, Khalil H, Ivey MJ, Karch J, Maliken BD, Correll RN, Brody MJ, J Lin S, Aronow BJ, Tallquist MD, et al. Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat Commun. 2016;12260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Takeda N, Manabe I, Uchino Y, Eguchi K, Matsumoto S, Nishimura S, Shindo T, Sano M, Otsu K, Snider P, et al. Cardiac fibroblasts are essential for the adaptive response of the murine heart to pressure overload. J Clin Invest. 2010;254–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Olsen MB, Hildrestrand GA, Scheffler K, Aukrust PL, Bjørå M, Wang J, Neurauter CG, Luna L, Johansen J, Øgaard JDS, et al. NEIL3‐dependent regulation of cardiac fibroblast proliferation prevents myocardial rupture. Cell Rep. 2017;82–92. [DOI] [PubMed] [Google Scholar]
- 5. Kong P, Shinde AV, Su Y, Russo I, Chen B, Saxena A, Conway SJ, Graff JM, Frangogiannis NG. Opposing actions of fibroblast and cardiomyocyte smad3 signaling in the infarcted myocardium. Circulation. 2018;707–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kawaguchi M, Takahashi M, Hata T, Kashima Y, Usui F, Morimoto H, Izawa A, Takahashi Y, Masumoto J, Koyama J, et al. Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation. 2011;594–604. [DOI] [PubMed] [Google Scholar]
- 7. Nagaraju CK, Dries E, Gilbert G, Abdesselem M, Wang N, Amoni M, Driesen RB, Sipido KR. Myofibroblast modulation of cardiac myocyte structure and function. Sci Rep. 2019;1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mouton AJ, Ma Y, Rivera Gonzalez OJ, Daseke MJ, Flynn ER, Freeman TC, Garrett MR, DeLeon‐Pennell KY, Lindsey ML. Fibroblast polarization over the myocardial infarction time continuum shifts roles from inflammation to angiogenesis. Basic Res Cardiol. 2019;1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Khalil H, Kanisicak O, Prasad V, Correll RN, Fu X, Schips T, Vagnozzi RJ, Liu R, Huynh T, Lee SJ, et al. Fibroblast‐specific TGF‐β‐Smad2/3 signaling underlies cardiac fibrosis. J Clin Invest. 2017;3770–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kuwahara F, Kai H, Tokuda K, Kai M, Takeshita A, Egashira K, Imaizumi T. Transforming growth factor‐beta function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure‐overloaded rats. Circulation. 2002;130–135. [DOI] [PubMed] [Google Scholar]
- 11. Walton KL, Johnson KE, Harrison CA. Targeting TGF‐β mediated SMAD signaling for the prevention of fibrosis. Front Pharmacol. 2017;461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen MM, Lam A, Abraham JA, Schreiner GF, Joly AH. CTGF expression is induced by TGF‐beta in cardiac fibroblasts and cardiac myocytes: a potential role in heart fibrosis. J Mol Cell Cardiol. 2000;1805–1819. [DOI] [PubMed] [Google Scholar]
- 13. Lijnen PJ, Petrov VV, Fagard RH. Induction of cardiac fibrosis by transforming growth factor‐β1. Mol Genet Metab. 2000;418–435. [DOI] [PubMed] [Google Scholar]
- 14. Kulkarni AB, Huh CG, Becker D, Geiser A, Lyght M, Flanders KC, Roberts AB, Sporn MB, Ward JM, Karlsson S. Transforming growth factor β1 null mutation in mice causes excessive inflammatory response and early death. Proc Natl Acad Sci USA. 1993;770–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chen S, Liu J, Yang M, Lai W, Ye L, Chen J, Hou X, Ding H, Zhang W, Wu Y, et al. Fn14, a downstream target of the TGF‐β signaling pathway, regulates fibroblast activation. PLoS One. 2015;9:e0143802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Engler AJ, Sen S, Sweeney HL, Discher DE. Matrix elasticity directs stem cell lineage specification. Cell. 2006;677–689. [DOI] [PubMed] [Google Scholar]
- 17. Clark K, Langeslag M, Figdor CG, van Leeuwen FN. Myosin II and mechanotransduction: a balancing act. Trends Cell Biol. 2007;178–186. [DOI] [PubMed] [Google Scholar]
- 18. Wang H, Tibbitt MW, Langer SJ, Leinwand LA, Anseth KS. Hydrogels preserve native phenotypes of valvular fibroblasts through an elasticity‐regulated PI3K/AKT pathway. Proc Natl Acad Sci USA. 2013;19336–19341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Berry MF, Engler AJ, Woo YJ, Pirolli TJ, Bish LT, Jayasankar V, Morine KJ, Gardner TJ, Discher DE, Sweeney HL. Mesenchymal stem cell injection after myocardial infarction improves myocardial compliance. Am J Physiol Heart Circ Physiol. 2006;H2196–H2203. [DOI] [PubMed] [Google Scholar]
- 20. Mabry KM, Lawrence RL, Anseth KS. Dynamic stiffening of poly(ethylene glycol)‐based hydrogels to direct valvular interstitial cell phenotype in a three‐dimensional environment. Biomaterials. 2015;47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Günay KA, Ceccato TL, Silver JS, Bannister KL, Bednarski OJ, Leinwand LA, Anseth KS. PEG‐anthracene hydrogels as an on‐demand stiffening matrix to study mechanobiology. Angew Chem Int Ed Engl. 2019;9912–9916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Herum KM, Choppe J, Kumar A, Engler AJ, McCulloch AD. Mechanical regulation of cardiac fibroblast profibrotic phenotypes. Mol Biol Cell. 2017;1871–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chang SC, Ren S, Rau CD, Wang JJ. Isoproterenol‐induced heart failure mouse model using osmotic pump implantation. Methods Mol Biol. 2018;207–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Omura T, Kim S, Takeuchi K, Iwao H, Takeda T. Transforming growth factor beta 1 and extracellular matrix gene expression in isoprenaline induced cardiac hypertrophy: effects of inhibition of the renin‐angiotensin system. Cardiovasc Res. 1994;1835–1842. [DOI] [PubMed] [Google Scholar]
- 25. Wipff P‐J, Rifkin DB, Meister J‐J, Hinz B. Myofibroblast contraction activates latent TGF‐β1 from the extracellular matrix. J Cell Biol. 2007;1311–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Aguado BA, Schuetze KB, Grim JC, Walker CJ, Cox AC, Ceccato TL, Tan A, Sucharov CC, Leinwand LA, Taylor MRG, et al. Transcatheter aortic valve replacements alter circulating serum factors to mediate myofibroblast deactivation. Sci Transl Med. 2019;11:eaav3233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Fairbanks BD, Schwartz MP, Halevi AE, Nuttelman CR, Bowman CN, Anseth KS. A versatile synthetic extracellular matrix mimic via thiol‐norbornene photopolymerization. Adv Mater. 2009;5005–5010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Joshi R, Tamta K, Chandra B, Kandpal ND. Interactions of poly (ethylene) glycols in aqueous solution at 288.0K: ultrasonic studies. International Journal of Applied Chemistry. 2017;611‐630. [Google Scholar]
- 29. Hutter JL, Bechhoefer J. Calibration of atomic‐force microscope tips. Rev Sci Instrum. 1993;1868–1873. [Google Scholar]
- 30. Maass AH, Buvoli M. Cardiomyocyte preparation, culture, and gene transfer. Methods Mol Biol. 2007;321–330. [DOI] [PubMed] [Google Scholar]
- 31. López B, González A, Lindner D, Westermann D, Ravassa S, Beaumont J, Gallego I, Zudaire A, Brugnolaro C, Querejeta R, et al. Osteopontin‐mediated myocardial fibrosis in heart failure: a role for lysyl oxidase? Cardiovasc Res. 2013;111–120. [DOI] [PubMed] [Google Scholar]
- 32. Cartledge JE, Kane C, Dias P, Tesfom M, Clarke L, Mckee B, Al Ayoubi S, Chester A, Yacoub MH, Camelliti P, et al. Functional crosstalk between cardiac fibroblasts and adult cardiomyocytes by soluble mediators. Cardiovasc Res. 2015;260–270. [DOI] [PubMed] [Google Scholar]
- 33. Troncoso R, Ibarra C, Miguel Vicencio J, Jaimovich E, Lavandero S. New insights into IGF‐1 signaling in the heart. Trends Endocrinol Metab. 2014;128–137. [DOI] [PubMed] [Google Scholar]
- 34. Hinderer S, Schenke‐Layland K. Cardiac fibrosis—a short review of causes and therapeutic strategies. Adv Drug Deliv Rev. 2019;77–82. [DOI] [PubMed] [Google Scholar]
- 35. Lu D, Soleymani S, Madakshire R, Insel PA. ATP released from cardiac fibroblasts via connexin hemichannels activates profibrotic P2Y2 receptors. FASEB J. 2012;2580–2591. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1–S5