

Abstract
Nano luciferase (NLuc), the smallest luciferase known, was used as the fusion partner with a nanobody against Aflatoxin B1 to develop a bioluminescent enzyme-linked immunosorbent assay (BLEIA) for detection of the aflatoxin B1 in cereal. Nanobody (clone G8) against Aflatoxin B1 was fused with nano-luciferase and cloned into a pET22b expression vector, then transformed into E. coli. The nanobody fusion gene contained a hexahistidine tag for purification by immobilized metal affinity chromatography, yielding a biologically active fusion protein. The fusion protein G8-Nluc retained binding properties of the original nanobody. Concentration of the coelenterazine substrate and buffer composition were also optimized to provide high intensity and long half-life of the luminescent signal. The G8–Nluc was used as a detection antibody to establish a competitive bioluminescent ELISA for the detection of Aflatoxin B1 in cereals successfully. Compared with classical ELISA, this novel assay showed more than 20-fold improvement in detection sensitivity, with an IC50 value of 0.41 ng/ml and linear range from 0.10 ng/mL to 1.64 ng/ml. In addition, the entire BLEIA detection procedure can be completed in one step within two hours, from sample preparation to data analysis. These results suggest that nanobody fragments fused with nano-luciferase might serve as useful and highly sensitive dual functional reagents for the development of rapid and highly sensitive immunoanalytical methods.
Keywords: Aflatoxin B1, Nano luciferase, One–step ultra-sensitive bioluminescent immunoassay
Graphical Abstract
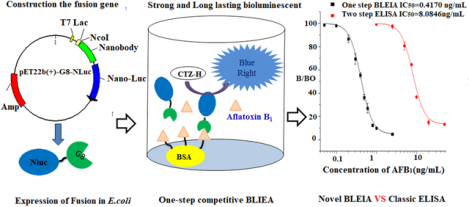
Introduction
Mycotoxins are a problematic toxic group of small organic molecules that are produced as secondary metabolites by several fungal species that colonize crops1. They lead to contamination at both the field and postharvest stages of food production with a range of cereals. Aflatoxins are a group of naturally occurring mycotoxins, produced mainly by A. flavus and Aspergillus parasiticus. More than 20 types of aflatoxins and some metabolites have been identified, among which four aflatoxins (B1, B2, G1 and G2) occur naturally (Figure 1). Aflatoxin B1 is the most toxic and one of the most potent carcinogens in nature2, classified as a class 1 carcinogen to humans3. Due to the high toxicity and carcinogenicity of aflatoxins, many countries have set up strict maximum limits for AFB1 or total aflatoxins permissible in food and agri-products with the range from 2 to 20 μg/kg4. Numerous analytical methods have been developed for aflatoxin determination, including chromatography based instrumental analytical methods5 and immunoassay based rapid methods (e.g. ELISA, lateral flow strips and sensors)6–8. In past decades, numerous antibodies against aflatoxins have been developed by polyclonal and monoclonal techniques and applied in various immunoassay-based rapid methods for the detection of aflatoxins. With the advent of molecular engineering and phage display technology, recombinant antibodies produced in host bacteria or other cell lines have become more useful reagents compared with conventional animal derived antibodies. Aflatoxin-specific recombinant antibodies such as Fab and ScFv have been utilized in ELISAs and biosensors9–10. Recently, a novel kind of recombinant antibody derived from camelids or related species, called nanobodies, have attracted much more attention. Lacking the light chain seen in monoclonal antibodies, these recombinant heavy chain only antibodies retain the antigen binding capacity of antibodies, but are much smaller in size, averaging approximately 15 kDa. Nanobodies are promising reagents for the next generation of immunoassays, due to their high thermostability and high expression yields compared to the conventional recombinant antibody fragments. Currently, nanobody technology has been widely used in the clinical diagnostic field targeting protein biomarkers and an increasing number of nanobodies have been isolated against small molecules for detection in rapid immunoassays11. An aflatoxin-specific nanobody was successfully obtained from an immune alpaca phage-display VHH library constructed by our laboratory. In our previous study, it has been applied in a colorimetric immunoassay12 and immuno-affinity column preparation13, proving to be a powerful analytical reagent to recognize Aflatoxin B1 in various foods.
Figure 1.
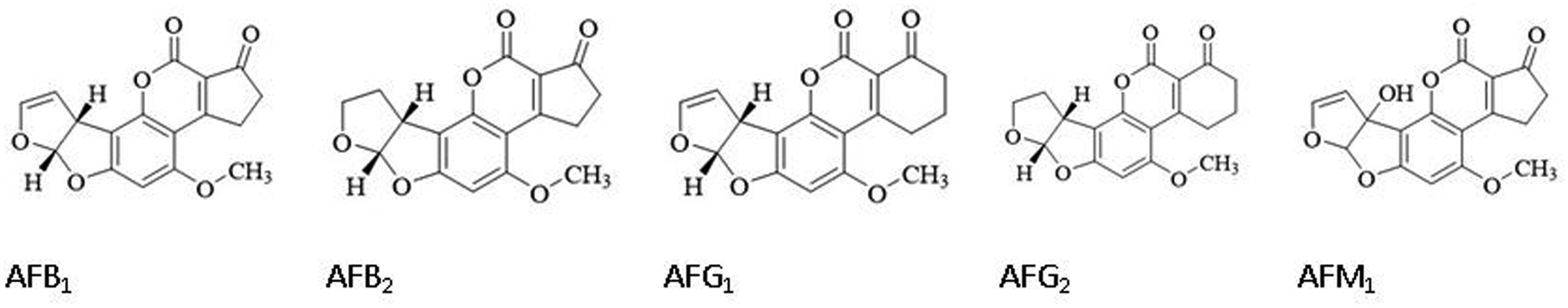
The structure of aflatoxin analogs and metabolite.
Recently, construction of antibody-reporter (enzyme/photoprotein) fusions based on recombinant DNA technology has shown a significant advantage in immunoassays. Fusions directly linking recombinant antibody fragments with the reporter enzyme like alkaline phosphatase are excellent probes for immunoassays and have been shown to be promising reagents for detection of protein antigens and small chemicals, like Bacillus anthracis14, dermonercrotoxin15, and O,O-diethyl organophosphorus pesticides16. AP -Nanobody fusions have also been utilized for the detection of small molecules, such as ochratoxin A17, tetrabromobisphenol A18, and parathion19. Another group of reporters are fluorescent proteins (i.e. GFP and RFP), which have been used for construction of fusion proteins with antibody fragments20–22. The one-step immunoassay based on antibody–reporter fusions can reduce long assay times, which is a drawback for traditional immunoassays requiring secondary or tertiary antibody enzyme conjugates. Another important group of photoproteins are luciferases. These enzymes from a variety of organisms catalyze a light-emitting reaction and are widely utilized as reporter proteins. The first-generation protein species is firefly luciferase (FLuc)23 , which requires ATP for the oxidation of luciferin. Other popular luciferases are derived from the sea pansy Renilla reniformis (RLuc)24 and the marine copepod Gaussia princeps (GLuc)25. They can oxidize their substrate coelenterazine without ATP or Coenzyme A, which simplifies their use in a number of reporter applications. Genetic fusions between Coelenterazine based luciferases and antibody fragments have been applied in clinical diagnostics and utilized as analytical tools26–27. Nano-luciferase (NLuc) derived from the deep-sea shrimp Oplophorus gracilirostris, is the newest commercially available luciferase enzyme. It is only 19.1 kDa in size and produces high intensity, glow-type luminescence. Nluc is one of the smallest luciferases in the world28, exhibiting several excellent physical properties superior to traditional luciferases; it is a thermo-stable enzyme which retains activity after 30 min incubation at 55 °C and is active over a broad pH range (fully active between pH 7–9, and also retains significant degree of activity at pH 5–7). Furthermore, its monomeric nature facilitates its use as transcriptional reporter or fusion partner. NLuc also contains no post-translational modifications, including disulfide bonds, making it a powerful luminescent probe, which can be used as a fusion partner with antibodies or other binding proteins/peptides for developing immunoassays29–31.
Although the aforementioned luciferase enzymes have been available to researchers for over a decade, their application as reporters in immunoassays have been limited due to some inherent caveats such as substrate stability and the bioluminescence signal half-life. Various substrates and buffer systems have been developed to alleviate some of these problems. However, many of these reagents are expensive. Recently, new coelenterazine substrates have been developed and are highly sensitive for the detection of luciferase, as low as the attomole level. However, the combination of different coelenterazine substrates with this latest luciferase Nluc has not yet been well studied. A few studies have reported on the application of Nanobody-Nluc fusions for the one-step detection, yet their full potential has yet to be revealed. In our study, we developed a novel fusion protein as a powerful dual functional reagent and its matching substrate system. Then, an ultra-sensitive one-step competitive bioluminescent enzyme-linked immunosorbent assay (BL-ELISA) based on Nb-Nluc fusion was developed and used for quantification of aflatoxin B1 in cereals.
Material and methods
Chemicals and reagent
Restriction enzymes NotI , SALI, NcoI and Sfi I, Phusion High-Fidelity DNA Polymerase and T4 DNA ligase were obtained from New England Biolabs (Beverly, MA, USA); Invitrogen blot TM 4–12% bis-tris plus, Disposable 5mL Polypropylene columns, 96 Flat Bottom White Polystyrol (442404); Bovine serum albumin (BSA), Halt protease inhibitor cocktail(EDTA-FREE, 100X), B-PER™ bacterial protein extraction and HisPur™ Ni-NTA resin were purchased from Thermo Fisher Scientific (Rockford, IL, USA); Skim milk powder was purchased from Magermilchpulver (Billerica MA,USA); Black 96 well (E18073CV) and white 96 well (E180538G) microplates were purchased from Greiner bio-one; coelenterazine substrates (CTZ naive ,CTZ 400a and CTZ-H) were purchased from NanoLight Technology, Anti-6X His tag antibody (HRP) was purchased from Abcam; Fumonisin B1 (FB1), aflatoxin B1 (AFB1), ochratoxin A (OTA), deoxynivalenol (DON), zearalenone (ZEN) were purchased from Fermentek (Jerusalem, Israel); pNPP was purchased from American Sigma. Primers used for polymerase chain reaction (PCR) amplification were synthesized and purified by Invitrogen.
Construction of recombinant plasmid, expression and purification
All the nucleotide sequences of primers used are listed in table s1. The Nano-luciferase encoding gene was first amplified by PCR with primers 1,2. After restriction with NotI and SalI and purification with a PCR cleanup kit, the resulting nano-luciferase gene was subcloned into a modified pET22b vector digested by NotI and SalI. The resulting positive vector was confirmed by DNA sequencing. The gene for the nanobody containing the upper hinge at the C-terminal (G8) was amplified with primers 3,4 and sub-cloned into the pET22b vector carrying the nano-luciferase gene using NcoI and SfiI as described above. The constructed vector carrying G8–Nanoluc was confirmed by DNA sequencing, and the full length of the protein sequence of the fusion is shown in Table s1. The vector of Nano-luciferase and the vector of G8-Nanoluc fusion protein were transformed into E. coli BL21(DE3) competent cells. The transformed cells were grown on LB agar plates containing ampicillin and single colonies were used to inoculate 5 mL LB broth containing ampicillin and incubated at 37 °C, 220 rpm overnight. The culture was used to inoculate 200 mL LB broth containing ampicillin and incubated at 37 °C, 220 rpm until the OD600 reached approximately 0.5 (more than three hours). To induce expression of the target proteins, IPTG was added to the culture at a final concentration of 0.1 mM. After incubation for 12 h at 20 °C and 180 rpm, cells were harvested by centrifugation at 5000 × g for 10 min. The periplasmic extract was obtained by using Bacterial Protein Extraction Reagent (B-PER): 4 mL of B-PER and 2 ul of lysozyme were added for each gram of cells. After the cells were lysed thoroughly for 30 min at room temperature, the supernatant was collected by centrifugation at 15000 × g for 5 min. Nanoluciferase and G8-Nlu fusion each contain a Hexahistidine tag at their C-terminals and were purified by using HisPur Ni-NTA resin according to the manufacturer’s protocol. The eluted fractions were analyzed by SDS-PAGE and stained according to a standard protocol. The concentration of the nano-luciferase and G8-Nlu fusion were determined by absorbance at 280 nm using a Nanodrop.
Catalytic activity of the fusion protein
10 ul of G8-Nanoluciferase solution in 10 mM PBS was added per well (white 96-well plate) and mixed with 100 ul of solutions of different coelentrazine substrate analogues (CTZ Naive, CTZ 400a and CTZ-H). After mixing, the bioluminescent signal was measured by a Tecan1000 plate reader in the luminescence mode.
Horseradish peroxidase (HRP) was 2-fold serially diluted with 10 mM PBS (3.97×106 fmol) to 1.2×104 fmol). 10 ul of HRP serial dilutions were mixed separately with100 ul TMB substrate solution, at 37 °C for 10 min. The reactions were stopped with 50 ul of 2 M H2SO4 and the absorbance was read at 450 nm. Similarly, 10 ul of 2-fold serial dilutions of AP enzyme (2.9×105 fmol) to 2.8×102 fmol, in 1×PBS) were separately mixed with 100 ul of BBTP fluorescent substrate in corresponding buffer, incubated at 37 °C for 30 min, and the fluorescent intensities were measured at 550 nm after excitation at 450 nm. The nanoluciferase and G8-nanoluc fusion proteins were 2-fold serially diluted in 10 mM PBS, from 1.5×104 fmol to 14.7 fmol for nanoluciferase and 1.06×104 fmol to 10.35 fmol for G8-Nluc, respectively. 10 ul of nano-luciferase or G8-Nluc fusion serial dilutions were separately mixed with 100 ul of CTZ-H chemiluminescent substrate in white 96-well plates and the bioluminescence intensity was measured with Tecan 1000 within 60 s after mixing.
Formulation of substrate buffer for coelenterazine based bioluminescent detection
Coelenterazine-H is a suitable substrate for Nanoluciferase. The 100X stock solution (0.5 mg/mL) was prepared by dissolving coelenterazine-H in 75% methanol, 15% glycerin, and 100 μM ascorbic acid. In biological luminescence measurement systems, the maximum luminescent intensity and the dynamic stability of the luminescence signal are critical for high-throughput detection. A series of luminescence assay buffers were formulated and their luminescence performance with nano-luciferase and the coelenterazine-H substrate was evaluated. All luminescence assay buffers contained BSA, EDTA.Na2, Triton X-100, and Tergitol NP-10. 10 mM PBS (pH 8) buffers containing different quantities of the following additives were prepared: BSA (0.06 mg/mL, 0.12 mg/mL, 0.25 mg/mL, 0.5 mg/mL,1.0 mg/mL), EDTA.Na2 (1.1 mM, 2.20 mM, 4.4 mM, 8.8 mM, 17.6 mM), Tergitol NP-10 (0.25%, 0.5%, 1%, 2%, 4%,8%). 100 ul of 1x coelenterazine-h substrate solutions in different luminescence assay buffers were loaded onto the white plate. The luminescence reactions were started by the addition of 10 ul of the G8-Nluc (70 ng/mL) working solution and the luminescence intensity was recorded in 60 s intervals for 1200 s by using a Tecan1000 instrument. The maximum intensity of luminescence (Imax) was shown as a relative light unit (RLU). RLU and signal half-life were measured for reactions of CTZ-H with G8-Nluc fusion protein in a variety of luminescence assay buffers. After determining the optimal luminescence assay buffer, its characteristics were compared with two commercial luminescence assay buffers (passive™ lysis buffer and Glo™ lysis buffer from Promega).
Competitive ELISA based on G8 nanobody and G8-Nluc fusion
Optimal concentrations of G8 nanobody, G8-Nluc fusion and coating antigen AFB1-BSA were determined by checkerboard titration. For this assay, a 96-well microtiter plate was coated with AFB1-BSA (2 μg/mL in 10 mM PBS) at 100 ul/well overnight at 4 °C; the liquid was removed, the plate was washed three times with 0.01% PBST, and the plate was blocked with 300 ul/well skim milk at 37 °C for 1 h. After removal of the blocking solution the plate was washed three times with PBST; 50 ul/well of G8 nanobody(2.75 μg/mL) or G8-Nlu fusion protein (3.1 μg/mL) and an equal volume of serial concentrations of AFB1 standard were added, the plate was mixed and incubated at 37 °C for 30 min after the plate was washed 3 times with PBST. For the two-step ELISA, 100 ul of HRP-labeled mouse anti-His Tag secondary antibody (1:1000) was added, and incubated at 37°C for 30 min. After washing 3 times with PBST, TMB substrate solution was added, and the plate was incubated at 37 °C for 7 min. The reaction was stopped by adding 50 ul of 3M H2SO4, and the absorbance was measured at 450 nm. The binding rate (%) of each standard concentration was calcululated as B/B0×100% (B0 is the OD450 in the presence of AFB1, and B is in absence of AFB1). Taking the logarithm of the AFB1 standard concentration as the abscissa and the binding rate as the ordinate, an inhibition standard curve was obtained by plotting the binding rate against the AFB1 concentration, which was analyzed using the program Origin 7.5 with the four parameters logistic formula listed below.
Competitive BLEIA based on G8-Nluc fusion
The coating and blocking steps for BLEIA were the same as those used in the ELISA. In one-step BLEIA, the concentration of AFB1-BSA and the G8-Nluc were optimized by checkerboard titration. A 96-well high-binding white microtiter plate was coated with AFB1-BSA (1 μg/mL in 10 mm PBS) overnight at 4 °C, followed by blocking with 5 % skim milk. 50 ul/well of G8-Nlu fusion protein (0.5 μg/mL) and an equal volume of serial concentrations of AFB1 standard were added, the plate was mixed and incubated at 37 °C for 30 min. After washing, 100 ul/well of 5 μg/mL of CZT-H substrate solution was added and the bioluminescent signal was recorded immediately. The standard curve was obtained by using the same formula as above for the ELISA. The BLEIA assay buffer has three critical parameters, including pH value, ionic strength, and methanol concentration, all of which were assessed in this study. The following BLEIA assay buffers were evaluated with varying methanol concentrations (0%, 5%, 10%, 20%, 40%, 80%), ionic strength (0 mM, 10 mM, 20 mM, 50 mM), and pH (5, 6, 7, 8, 9). BLEIA under different conditions was carried out using the same procedure, and their RLUmax and IC50 values were compared.
For cross-reactivity analysis, various concentrations of AFB1, FB1, OTA, ZEN, and DON standard solutions were prepared using optimized assay buffer for BLEIA, and a competitive inhibition standard curve was drawn to calculate IC50 and the cross-reactivities (CR) value using the following formula:
Sample extraction and Analysis by BLEIA and LC-MS/MS
For the spiking and recovery study, 5 g of crushed grain samples (corn, wheat), shown to be AFB1 free by LC-MS/MS, were placed in three 50 mL centrifuge tubes and fortified with 5, 10, and 20 micrograms per kilogram of AFB1 standard, respectively. For the extraction, 15 mL of 80% methanol-PBS was added to the tube. After sonication for 15 min, the tube was centrifuged at 8000 rpm for 15 min and the supernatant was collected and filtered through a 0.22 μm filter. The resulting extract was diluted with assay buffer and then subjected to BLEIA. Each sample was measured in triplicate. Experimental cereal samples purchased from the supermarket were treated in the same manner.
LC-MS/MS analysis of aflatoxin B1 was performed on an Agilent SL liquid chromatograph connected to a 4000 Qtrap mass spectrometer. The separation was performed on a Kinetex C18 column (30 mm × 4.6 mm, 2.6 μm). The column temperature was set at 50°C. Water (solution A) and acetonitrile containing 0.1% (v:v) acetic acid (solution B) were used as the mobile phases with a flow rate of 0.6 mL/min. The sample injection volume was 5 ul, and each sample had a running time of 3 min. The gradient is shown in Table S-2a. The mass spectrometer was operated in negative ESI mode and multiple reaction monitoring mode. The optimized ion source parameters and MRM method are shown in Table S-2b and 2c, respectively. 12-(3-cyclohexyl-ureido)-dodecanoic acid with a final concentration of 200nmol/L was mixed with analytes as an internal standard to account for ionization suppression.
Results and discussion
To evaluate the utility of nano-Luciferase as a fusion partner for nanobodies, we selected a common competitive assay system for detection of aflatoxin B1, the major mycotoxin in cereals and the most harmful to humans as a model antigen. In our study, we focused on the following three scientific questions: (1) whether nano-Luciferase fusion protein has the same binding and catalytic ability as its parents; (2) whether the bioluminescent immunoassay based on the fusion protein achieves higher sensitivity, with specific substrate and buffer conditions and (3) whether the novel one-step BLEIA is superior to the classical ELISA, for the detection of Aflatoxin B1 in cereals.
Expression, purification and identification of the NB-Nanoluc fusion protein
The first step toward the creation of the fusion protein is construction of the recombinant plasmid. First, an oligo of the sequence of Nano-luciferase was sub-cloned in the modified pET22b vector, and the nanoluciferase was expressed with high yield in E. coli as in the previous study. Though the gene of Nano-luciferase is derived from deep sea shrimp, it was considered as the potential fusion partner because of its small size (19 kD), and lack of glycosylation and disulfide-bonds. The anti-AFB1 specific nanobody G8 was chosen as the partner of Nano-luciferase. Its gene was amplified from the original plasmid and sub-cloned into the modified pET22b vector containing the nano-luciferase gene. Using the special primer (Table S-1), G8 VHH was amplified with the upper hinge at the C-terminus, and then fused with the N-terminus of nano-luciferase resulting in a fusion protein with fusion partners being separated with the upper hinge as the spacer. The full sequence of the fusion gene is shown in Table S1. After transformation, the resulting recombinant plasmid was confirmed by DNA sequencing and then transformed into competent cells for expressing the soluble fusion protein. The crude fusion protein extracted by the B-PER reagent was purified by a Ni-NTA affinity column, and SDS-PAGE was used to determine the size and purity of the Nb-AP fusion protein (Figure 2). The gel shown has a band at approximately 35 kDa, as expected for the Nb-Nluc fusion protein. Though the yield of the fusion protein is lower than the parent nanobody G8 and the parent nano-luciferase, we obtained almost 5 mg purified fusion protein from 1 L of culture.
Figure 2.
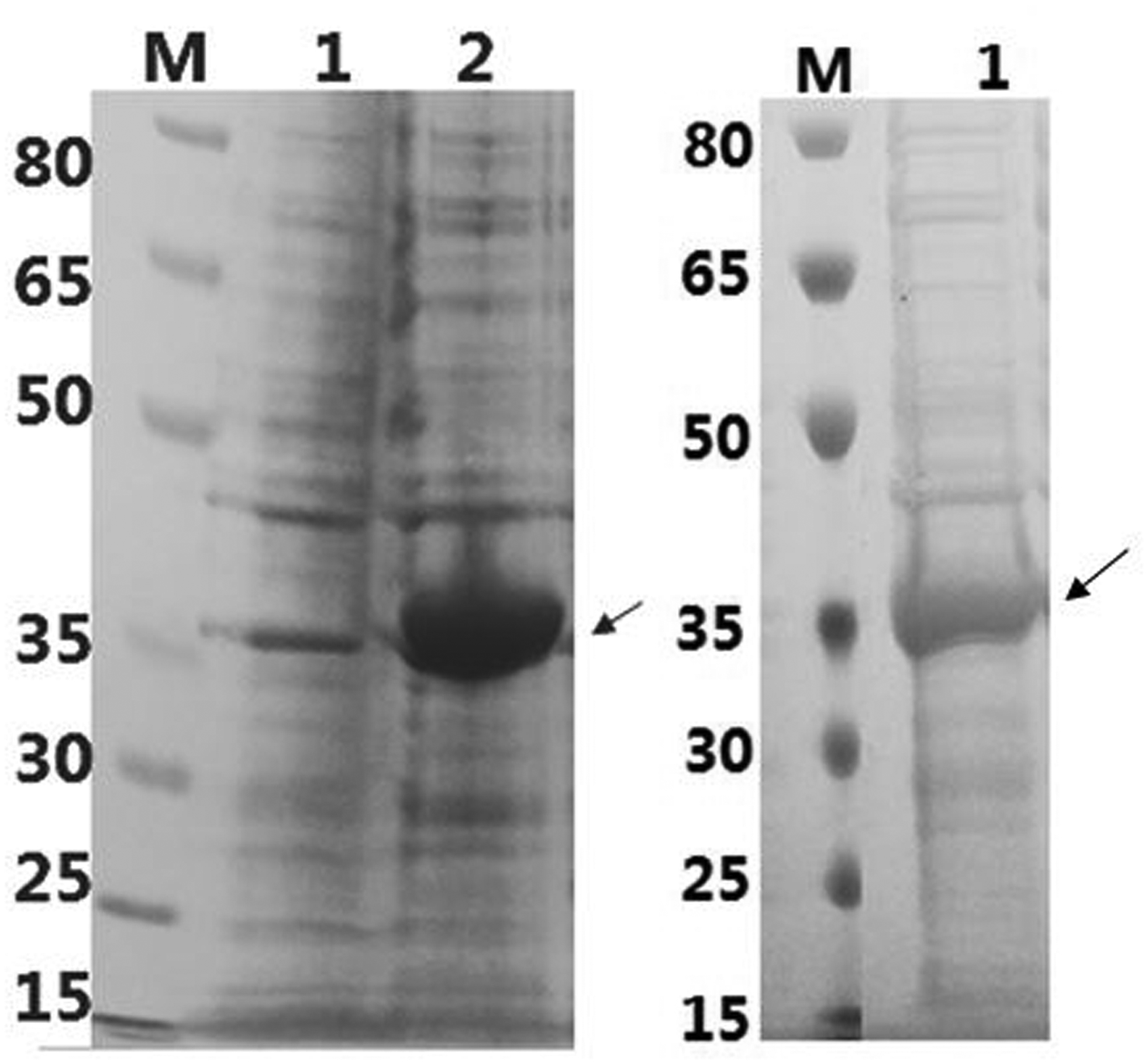
SDS–PAGE analysis of G8-nanlucc expression (SB medium; IPTG: 0.1mM; 25°, 12h) Left: Lane 1for the whole cell extract before induced; Lane 2 for the whole cell extract after induced
Right: lane 1for purified G8-Nanoluc fusion protein eluted by 50 Mm imidazole.
Determination of luminescence activity of fusion protein with different CTZ substrates
Coelenterazine (CTZ) is the natural substrate for luciferase, and over a dozen of CTZ analogs have been synthesized and are now available commercially. These CTZ analogs can function as substrates for Renilla and Guassia luciferase and have different bioluminescence properties32. CTZ-h (2-deoxy derivative of native Coelenterazine), has a 2-fold higher initial light-output than native CTZ from Renilla sp. CTZ-400a is a CTZ derivative that serves as a substrate for luciferase from Renilla sp and generates an emission peak centered around 400 nm. Only few studies reported to use the CTZ and its analogues with the latest Nano-luciferase enzyme. In this study, we intended to seek CTZ analogues compatible with nano-luciferase. Three CTZ anlogues, including CTZ- Native, CTZ- 400a and CTZ-H, were chosen as substrates for nanoluciferase to generate a luminescence signal. The order of the initial luminescence intensity (RLUmax) for the purified nano-luciferases was CTZ-H (2.3×106) > CTZ-400a (1.5×106) > CTZ-native (2.5×105) (Figure 3). The colenterazine-H showed the strongest signal compared with two other analogues. These results indicated that 2-deoxy substituent on the phenyl group of coelenterazine significantly affects the catalytic function of nanoluciferase, since there was a 10-fold improvement in signal intensity observed compared with CTZ-naitve. A similar order obtained with colenternzine-1 for reactions with G8-Nluc showed that CTZ-H is a good substrate for both the nano-luciferase and its fused protein. In addition, the cost of CTZ-H is almost the same as the common CTZ-Native, and significantly cheaper than the patented CTZ analogue fuzimazine from Promega.
Figure3.
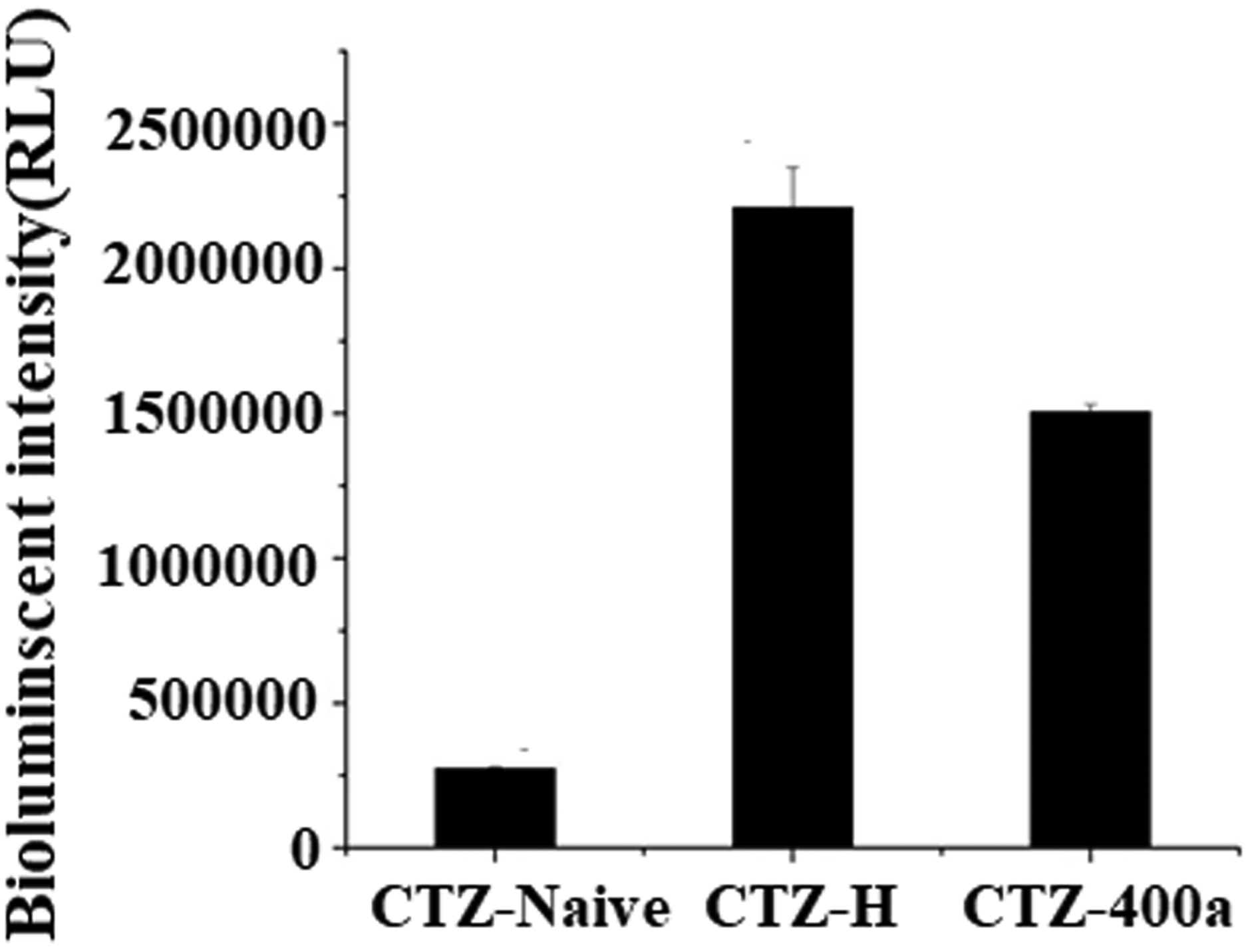
Identification of luminescence activity of nano-luciferase with different CZE substrates
Next, the luminescent signals for the serial dilutions of G8-NLuc with CTZ-H substrate were measured to determine the sensitivity of G8-NLuc detection. The limit of detection (LOD) for the catalytic enzyme was defined as the amount (per assay) that provided 2-fold higher signals than the signals at zero concentration. As shown in Figure 4, the detection limit for G8-Nluc was as low as 50 fmol, at the same level as for the nano-luciferase. This result indicated that G8-Nluc has similar catalytic activity to the original Nluc. For comparison, detection sensitivities for alkaline phosphosphase (AP) and horseradish peroxidase (HRP) were measured in fluorescent and colorimetric formats, respectively. The sensitivity of G8-Nluc detection in bioluminescent format was significantly higher compared to that for AP and HRP, the LOD which were 1000 fmol and 10000 fmol, respectively. Thus, by using well matched CTZ-H substrate, G8-Nluc could be detected at femtomole level, which is 20- and 200-fold more sensitive than detection of AP with fluorescence readout and HRP with colorimetric readout, respectively. This result shows that Nluc is a very promising fusion partner for the development of Nluc-Ab fusion proteins that could be considered as ultra-sensitive enzyme reagents for an immunoassay.
Figure 4.
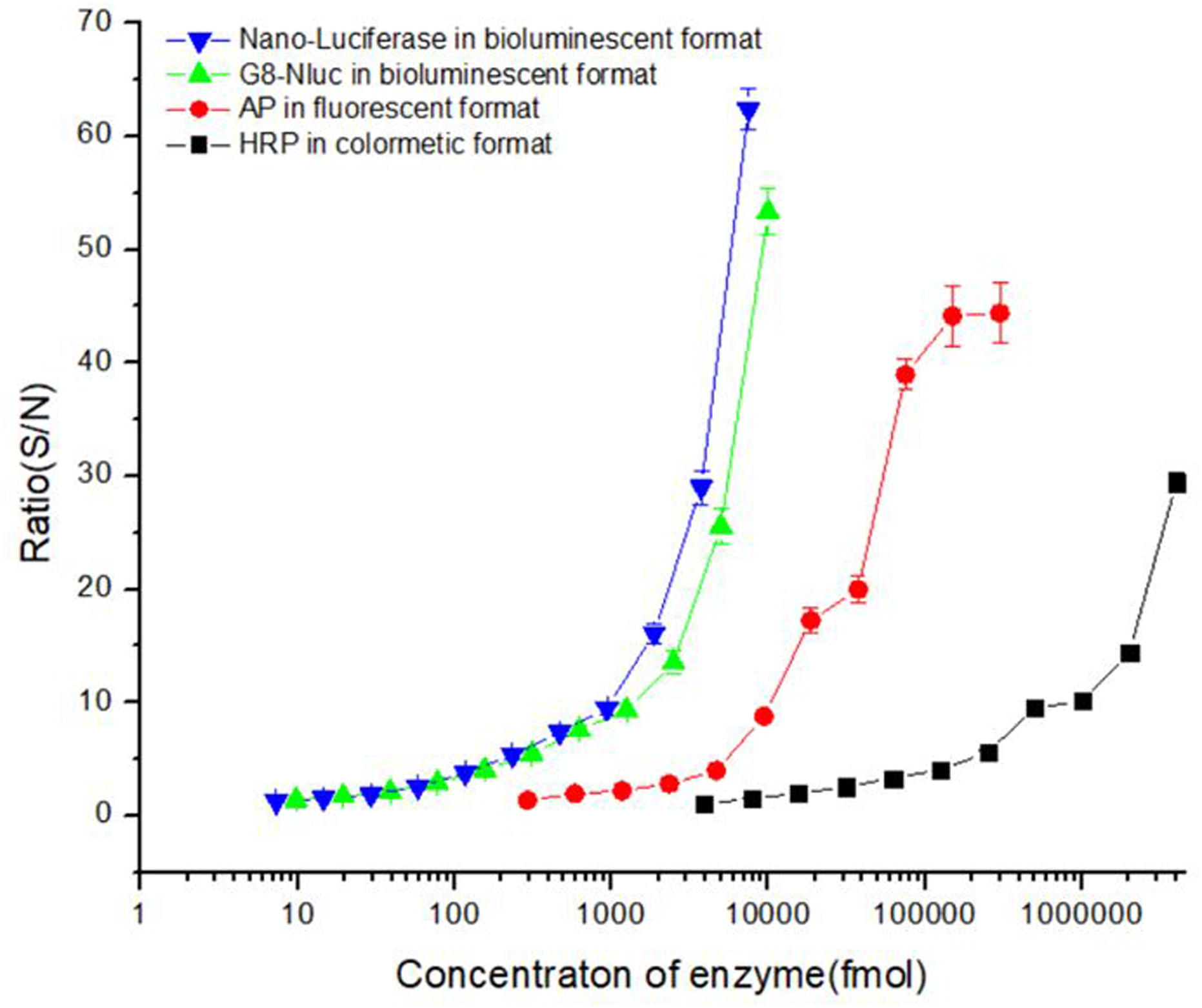
The detection limit of fusion protein in bioluminescent format
Optimization of the luminescence assay buffer
Luciferase is able to generate a glow-type luminescent signal by catalyzing the oxidation of specific substrates like luciferins and CTZ, with the concurrent emission of a photon. However, the signal half-life can decrease significantly, likely due to rapid depletion of substrate. Meanwhile, a substrate like CTZ is unstable in a weakly basic aqueous solution, which is the optimal pH value for many biological experiments. Thus, variation of the luminescence signal between the first sample and the last sample may become apparent when a large number of samples is processed sequentially. Many strategies were applied to address this problem, such as measurement of many samples simultaneously by using an injection based reader to reduce the variation of luminescence.33 Alternatively, some luciferase variants have been constructed whose luminescence half-life is stable over several minutes, although the specific activity of these variants is significantly lower34. In contrast to those complex solutions, a simple approach to achieve stable luminescent signal consists of adding detergent to lysis buffer when extracting the Gaussia Luciferase from the cell34. Many commercial lysis buffers containing detergents have been developed for the detection of luminescence directly in mammalian cells, but the high cost of these buffers prohibits their use for immunoassay where large volumes of luminescence assay buffer is needed. In addition, suitability of these commercial luminescence assay buffers for nano-luciferase and CTZ-H combination is unknown. Therefore, we decided to develop an efficient luminescence assay buffer that enhances the half-life of the bioluminescence signal. Therefore, we prepared 10 mM PBS buffers containing various amounts of Triton X-100, Tergitol NP-10, BSA and EDTA.Na2 as additives. Next, a luminescence signal from reactions of G8-Nluc with CTZ-H substrate in each of these substrate buffers was recorded in 60 s intervals for more than 1200 s. When compared to blank PBS buffer (Figure 5), the surfactant additives like Triton X-100 and Tergitol NP-10 depressed the luminescence intensity slightly, but they both dramatically retarded the decay of luminescence. Surprisingly, BSA and EDTA.Na2 increased the maximum intensity of the luminescence signal by 2- and 4-fold, respectively, while having no effect on the signal half-life. To improve the signal intensity and the half-life of luminescence, the concentrations of Triton X-100, BSA, and EDTA.Na2, were optimized for the final buffer formulations. This luminescence assay buffer (pH 8.0, 10 mM PBS containing 1% Tergitol NP-10, 0.25 mg/mL BSA, and 8.8 mM EDTA.Na2) has great compatibility with the combination of nano-luciferase and CTX-H substrate and performs significantly better than commercial Glo-lysis and Passive lysis buffers, purchased from Promega (Figure 6). The present formulation of luminescence assay buffer shows great promise with comparable high luminescence intensity and more than 20 min half-life, which permits its application in high-throughput immunoassays based on nano-luciferase and its fusions.
Figure5.
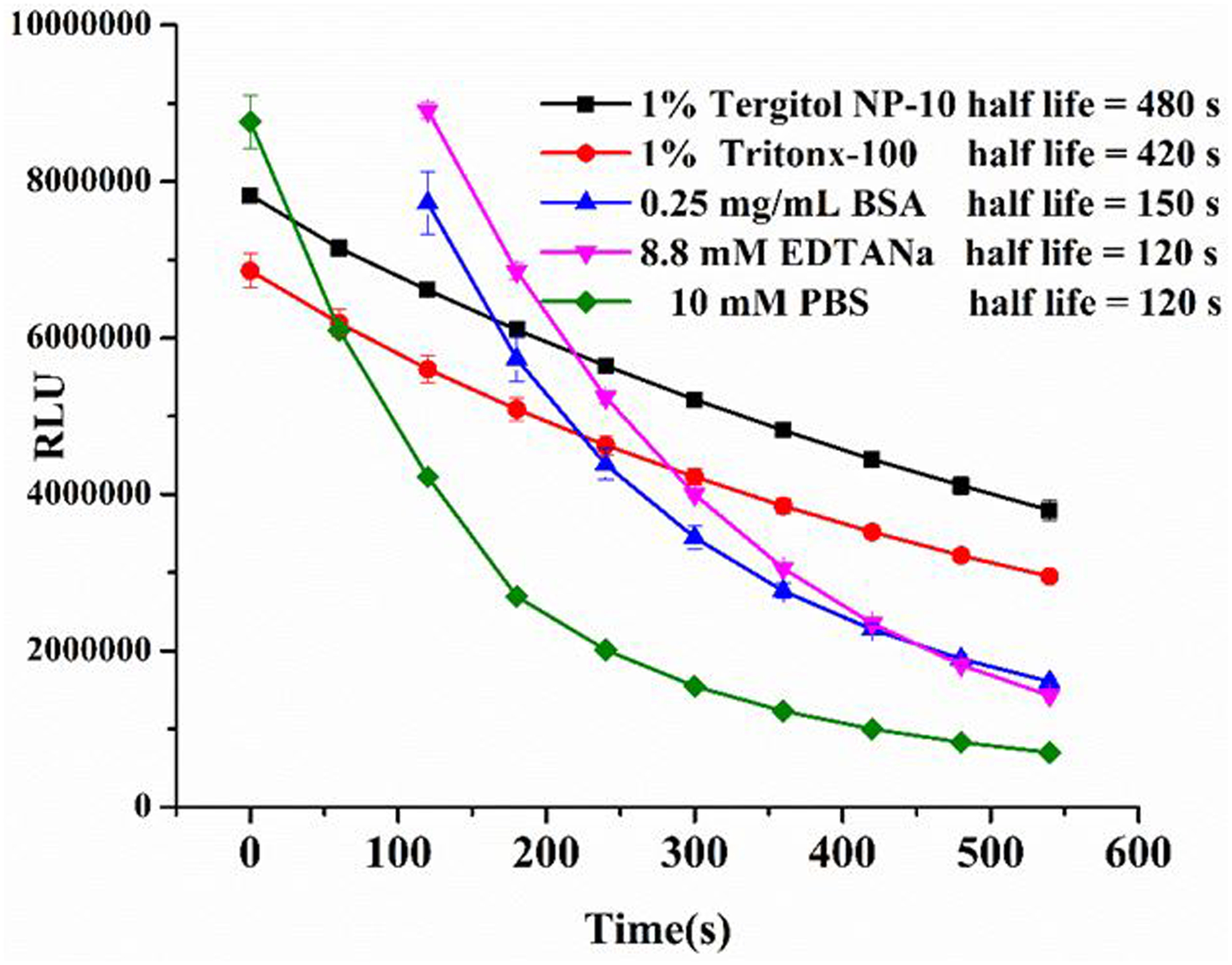
Kinetic analysis of substrate buffer mixed with different components
Figure6.
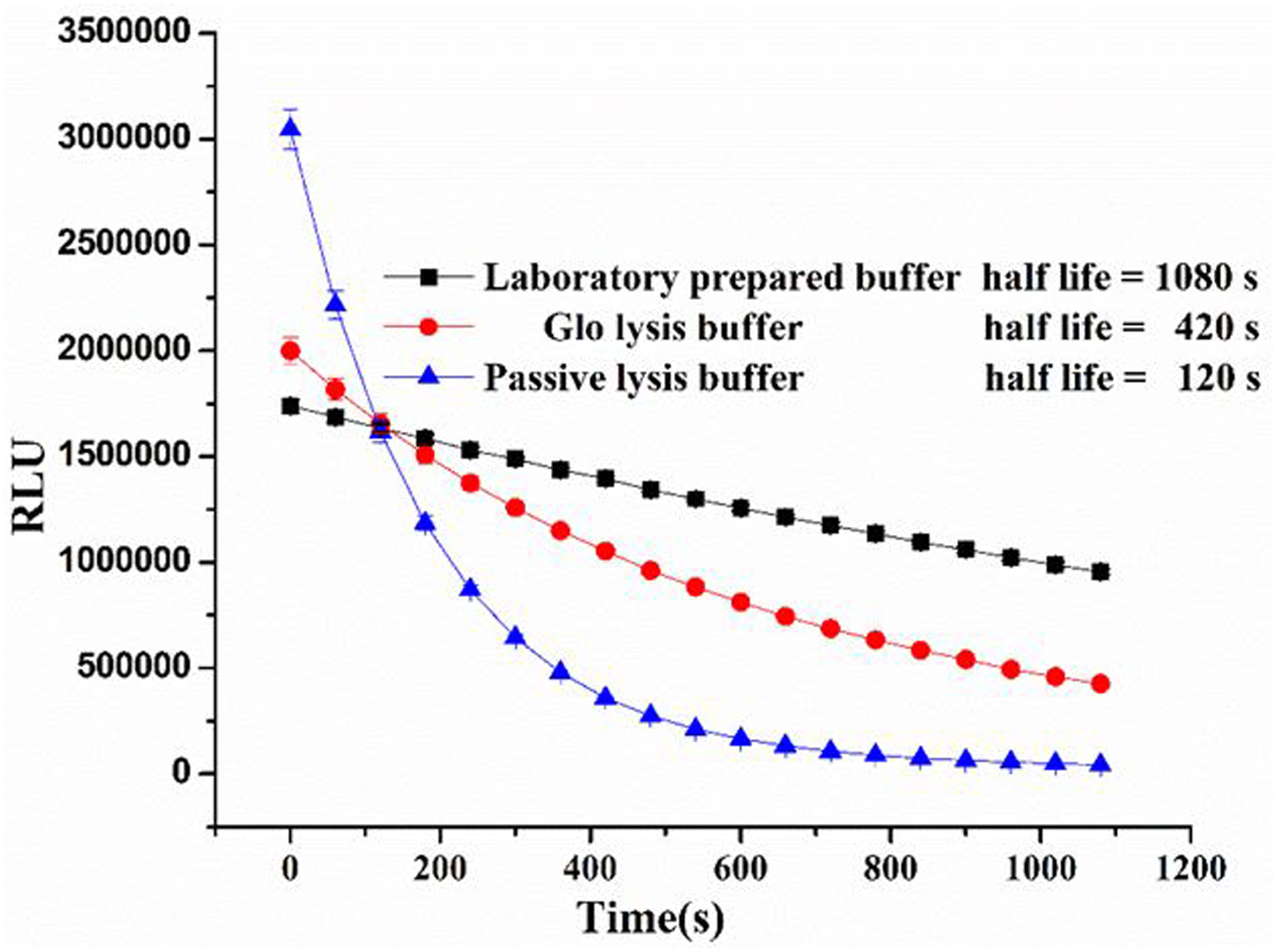
Comparison the half-life of lab prepared substrate buffer with commercial substrate buffers
The binding capacity of fusion protein compared with the parent nanobody
The G8-Nlu fusion protein could serve as a dual functional reagent in an immunoassay, not only for antigen binding, but also the reporter reagent for signal amplification. Thus, the binding properties of the fusion protein is a key feature to guarantee the sensitivity of a bioluminescent ELISA. To evaluate the sensitivity of the G8-Nluc and the parent G8 nanobody, classic two-step ELISAs were performed. Anti-6xHisTag HRP-conjugated antibody was used as a secondary antibody to recognize the His-tag at the C-terminus of both recombinant proteins. As shown in Figure 7, the binding between G8 or G8-Nluc with the coating antigen could be inhibited by AFB1. The IC50 for both ELISAs are comparable, with 8.14 ng/ml for G8 and 10.43 ng/ml for G8-Nluc, respectively. This result indicated that the developed G8-Nluc fusion protein maintains the binding properties of the parent G8 nanobody. In this study, the spacer between the nanobody and nano luciferase was the flexible linker from the upper hinge of the nanobody’s C terminus (Table S1). This result indicated that the developed G8-Nluc fusion was designed properly, where the two parts of the fusion protein were separated by a spacer and their natural protein conformations were maintained.
Figure7.
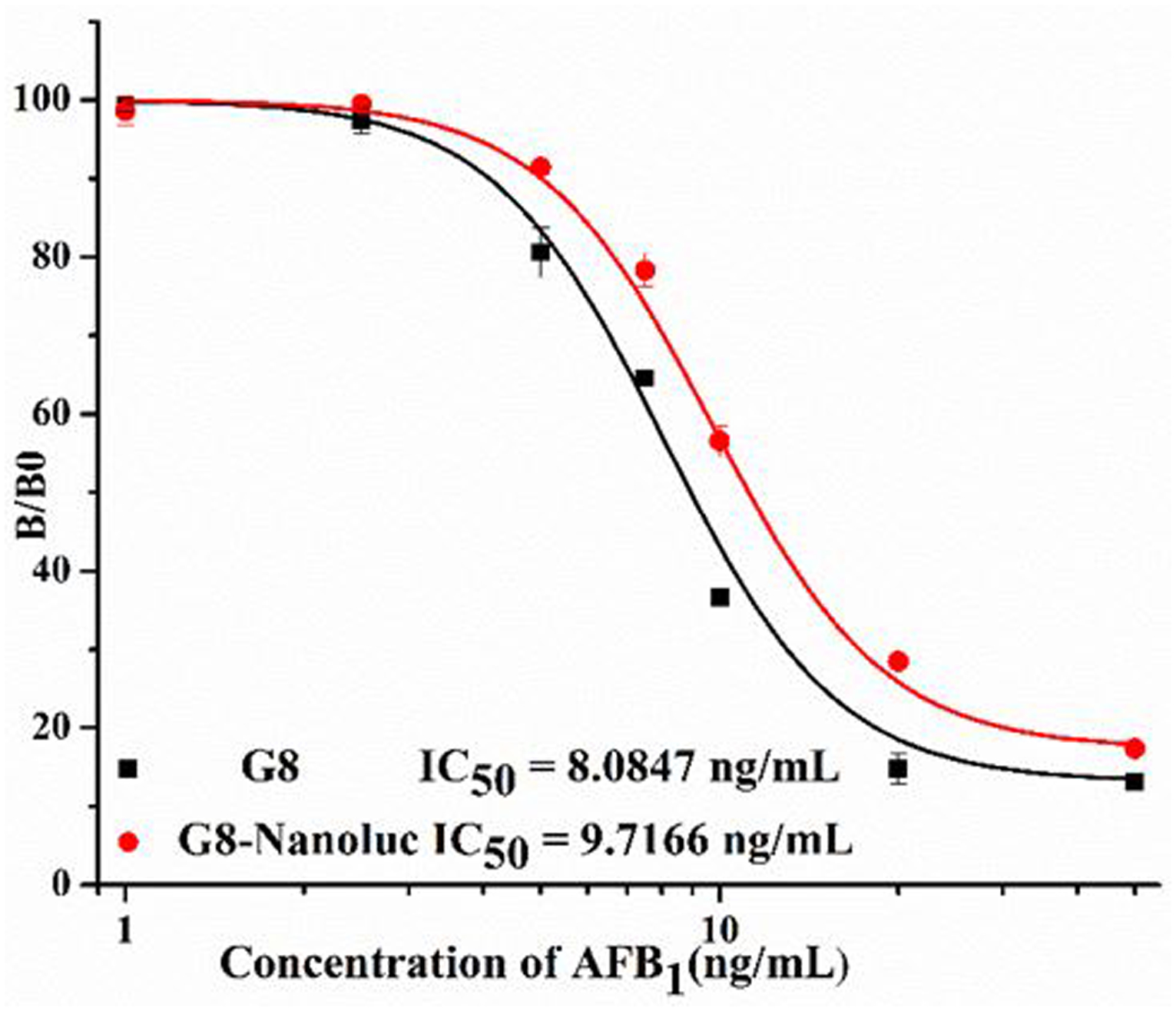
Standard curve of Fusion Protein and parent Nanobody in two step ELISA
Performance of BLEIA using the fusion protein in one-step format
As shown above, the G8-Nluc is a dual functional reagent, which contains a recognition domain that can bind the antigen and a reporter domain which generates the luminescent signal. We next evaluated the usefulness of this dual functional reagent for the detection of AFB1. Therefore, a one-step direct-competitive bioluminescent enzyme-linked immunoassay (BLEIA) was developed. Optimal concentrations of G8-Nluc and the coating antigen were determined by using checkerboard titration. Due to the high intensity of our luminescent system (the combination of G8-Nluc with CTZ-H in the luminescent assay buffer generated in house), the concentration of immuno-reagents could be dramatically reduced compared with a classical two-step ELISA. G8-Nluc at 0.5 μg/mL and AFB1-BSA at 1 μg/mL were optimal based on the two performance parameters: the IC50 and the maximum relative bioluminescence intensity (RLUmax). Since other assay parameters such as pH, ionic strength and organic solvent could also influence immunoreactions, these parameters were optimized to increase the sensitivity of the assay. The BLEIA for detection AFB1 was carried out in different conditions, including 5 different pH values of PBS, 4 different ionic strengths, 6 different concentrations of methanol (Figure S4). The lowest IC50 and the highest RLUmax were obtained at 50mM PBS, pH 6.0 and 10% methanol (Table 1). Under the optimized conditions, a standard curve was established for one-step BLEIA (Figure 8). The standard curve had a good correlation coefficient of 0.997 and a limit of detection of 0.05 ng/mL. The assay had an IC50 of 0.4170 ng/mL with a linear range of 0.10–1.64 ng/mL. The IC50 of the BLEIA based on the G8-Nluc fusion protein was almost 20 times lower than that of the classical two step ELISA based on the parent nanobody (IC50 = 8.14 ng/mL). The specificity of the G8-Nluc fusion protein was studied with 5 mycotoxin analogues (Table 1). The cross-reactivity was calculated using the following formula: [(IC50 of AFB1) / (IC50 of tested mycotoxin)] ×100%. As shown in Table 2, no cross reactivity was observed for any of the mycotoxins tested, consistent with the cross-reactivity pattern for the parent Nanobody12. This result indicated that the G8-Nluc fusion preserved the recognition features of the parent nanobody. Negligible cross reactivity with other mycotoxins demonstrated that the BLEIA based on the novel fusion protein had good selectivity and could effectively and reliably detect AFB1.
Table 1.
The assay condition for best performance in BLEIA. The vertical bars indicate the standard deviation (n = 3).
| Assay buffer factor | The best condition | Performance of assay(IC50) | RLU |
|---|---|---|---|
| PH value | 6.0 | 0.42 ng/mL | 6233088 |
| PBS ionic strength | 0.05M | 0.53ng/mL | 2607437 |
| Menthol concentrion | 10 % | 0.63 ng/mL | 2540141 |
Figure8.
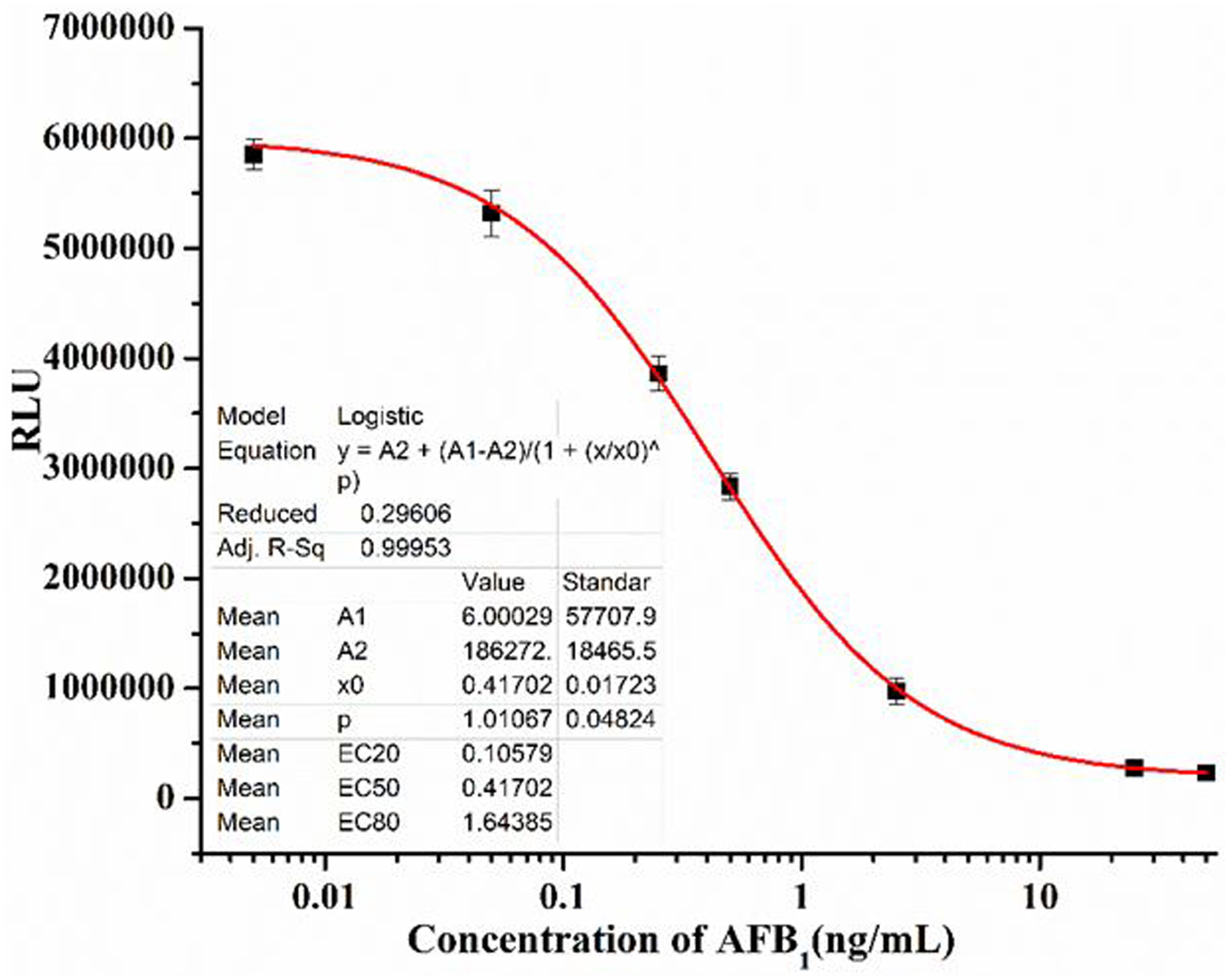
Standard curve of one-step BLEIA for detection of AflatoxinB1
Table2.
Cross reactivity of Nb-Nluc to aflatoxin related mycotoxins in one-step BLEIA
| Analyte | Chemical structure | Cross-reactivity (%) | |
|---|---|---|---|
| Aflatoxin B1 |  |
100 | |
| Deoxynivalenol |  |
0.31096 | |
| Zearalenone |  |
0.38006 | |
| Ochratoxin A |  |
0.33902 | |
| Fumonison B1 |  |
0.36818 |
Matrix effect and Validation
Matrix effect is a crucial factor to be considered in an immunoassay since it can interfere with the reaction between the antigen and antibody by quenching or enhancing readouts. To reduce the matrix effect, the extraction solution and method can be optimized. To apply the one-step BLEIA for sample analysis, the matrix effect was evaluated with wheat and corn in representative cereal samples. Aflatoxin B1 is insoluble in water and is easily soluble in oil, fatty acid esters, and various organic solvents including chloroform, methanol, ethanol, and similar organic reagents. Water is miscible with methanol and is the most common extraction solvent used in aflatoxin analysis. To maximum the recovery rate, we extracted the cereal sample using 80% methanol in water. To reduce the solvent effect against the immune reaction, dilution of the resulting sample solution was necessary before loading the sample in the plate. Additionally, fat and protein are added to the methanol-water extraction solution, which can affect the immune reaction. Dilution of the sample with BLEIA assay buffer is the most common method to reduce or eliminate the matrix effect. To apply the one-step BLEIA for sample analysis, the standard inhibition curves generated in BLEIA assay buffer (10% methanol, 1x PBS, MeOH-PBS) were compared to those using diluted AFB1-free grain (corn and oat) extracts. As the sample extract was serially diluted, the OD max and IC50 of the curve were varied as shown in Figures 5–S6. When the extract of corn and wheat were diluted 8 times, it produced a sufficient standard curve and the matrix effect was minimized. The developed BLEIA had a LOD of 0.84 μg/kg and 2.64 μg/kg in wheat and corn, a linear detection range of 1.68–89.28 μg/kg and 1.93–41.32 μg/kg in wheat and corn for AFB1, respectively. These results meet EU regulatory requirements for AFB1 in cereals (5μg/kg). This result indicated that the sample extract could be analyzed directly by diluting 8-fold without losing sensitivity.
The accuracy and reliability of the developed one-step BLIEA for detecting aflatoxin B1 were evaluated by analysis of spiked wheat and corn samples, confirmed to be free of aflatoxin by LC-MS. Wheat and corn were spiked with three different concentrations of Aflatoxin B1 (5,10, and 20 ng/g). After extraction with methanol and subsequent dilution, the samples were analyzed by one-step BLEIA. As shown in Table 3, the average recovery rate for AFB1 measured by the one-step BLEIA ranged from 88 to 113%. In addition, 5 wheat and 5 corn samples collected from the local supermarket were analyzed with one-step BLEIA and LC-MS/MS. As shown in Tables 4 and 5, 10 samples were positive and the concentration of aflatoxin in these cereal samples ranged from 3 to 8 μg/kg. AFB1 was undetectable in most wheat samples, but one sample was contaminated at a level exceeding MRL (5 μg/kg). However, most of the corn samples contained AFB1, though the contamination in 3 of them were less than the MRL. The results obtained from one-step BLEIA and LC-MS were in good agreement with each other, indicating the accuracy and reliability of the developed one-step BLEIA.
Table 3.
The detection of the cereal spiked with aflatoxin and the real sample. Each assay was performed in triplicate.
| Sample matrix | AFB1 spiked | mean ± SD (μg/kg) | Recovery (%) | CV (%) |
|---|---|---|---|---|
| corn | 5 | 5.70±0.54 | 113 | 9 |
| 10 | 9.61±0.37 | 106 | 4 | |
| 20 | 19.51 ±0.50 | 97.5 | 3 | |
| wheat | 5 | 5.32 ±0.38 | 106 | 7 |
| 10 | 8.73±0.72 | 107 | 8 | |
| 20 | 18.19±0.81 | 90.5 | 4 |
Table 4.
The detection of the cereal purchased from supermarket.
| Samples | No. | One-step BLEIA (n = 3)a(μg/kg) | HPLC-MS/MS (n = 3)a(μg/kg) |
|---|---|---|---|
| C1 | 5.94 ± 0.29 | 6.5 ± 0.34 | |
| C2 | NDb | ND | |
| corn | C3 | 3.11 ± 0.05 | 3.5 ± 0.46 |
| C4 | 3.93 ± 0.19 | 4.6 ± 0.34 | |
| C5 | 3.51 ± 0.94 | 3.8 ± 0.35 | |
| W1 | 6.15 ± 0.53 | 7.17 ± 0.3 | |
| W2 | ND | ND | |
| wheat | W3 | ND | ND |
| W4 | ND | ND | |
| W5 | ND | ND |
Each assay was carried out in three replicates on the same day.
Not detectable.
Conclusion
In this study, a sensitive and reliable dc-BLEIA based on a novel G8-Nluc fusion protein for detecting Aflatoxin B1 in cereal samples was successfully developed. The G8-Nluc fusion had both nanobody binding capacity and luciferase catalytic activity. The detection limit of the fusion protein is as low as 50 fmol when using CTZ-H as substrate. A substrate buffer was formulated in house to enhance the half-life of the bioluminescent signal. This dual-functional reagent was used to develop a one-step ultra-sensitive BLEIA. The IC50 of dc-BLEIA was nearly 20 times lower than that of a classical two-step dc-ELISA. The high sensitivity of dc-BLEIA compensated for the loss of sensitivity during the dilution of samples after extraction. In addition, the one-step BLEIA based on the G8-Nluc fusion protein is faster since no secondary antibodies are required. Therefore, the whole procedure for Aflatoxin B1 detection could be completed in 2 hours from sample dilution to data analysis. These results indicated that the nanobody-Nano-luciferase fusion can be considered as an attractive and powerful reagent for an immunoassay, and the one-step BLEIA can be a simple and rapid analytical tool for quantification of the pollutants in commercial foods.
Supplementary Material
ACKNOWLEDGMENT
This work was financially supported by the National Key R&D Program of China(Grants 2018YFC1602203)National Institute of Environmental Health Science Superfund Research Program, P42 ES04699 and the National Natural Science Foundation of China (No. 31471648,31860260,31301479,81660660, 31671924). Jiangxi Province Science Fund for Distinguished Young Scholar (Grants 20171BCB23023), Natural Science Foundation of Jiangxi Province, China (Grants 20181BAB204019), Open Project Program of State Key Laboratory of Food Science and Technology, Nanchang University (SKLF-KF-201825), Major Program of Natural Science Foundation of Jiangxi, China (20152ACB20005); the Jiangxi Province Key Technology R & D Program(20171BBG70072).
Reference
- (1).Shephard GS Current Status of Mycotoxin Analysis: A Critical Review. J AOAC Int .2016, 99, 842–848. [DOI] [PubMed] [Google Scholar]
- (2.Rushing BR; Selim MI Aflatoxin B1: A review on metabolism, toxicity, occurrence in food, occupational exposure, and detoxification methods. Food Chem Toxicol. 2018. [DOI] [PubMed] [Google Scholar]
- (3).Stettler PM; Sengstag C Liver carcinogen aflatoxin B1 as an inducer of mitotic recombination in a human cell line. Mol Carcinog ,2001, 31, 125–38. [DOI] [PubMed] [Google Scholar]
- (4).Van de Perre E; Jacxsens L; Lachat C; El Tahan F; De Meulenaer B Impact of maximum levels in European legislation on exposure of mycotoxins in dried products: case of aflatoxin B1 and ochratoxin A in nuts and dried fruits. Food Chem Toxicol, 2015, 75, 112–7. [DOI] [PubMed] [Google Scholar]
- (5).Zhang Q; Ran CC; Chen D; Li JM; Jiang Y Determination of aflatoxin B1, B2, G1, G2 in Ben Lamge granules by HPLC-FLD after multi-pretreatment clean-up. Zhongguo Zhong Yao Za Zhi ,2015, 40, 3780–5. [PubMed] [Google Scholar]
- (6).Di Nardo F; Alladio E; Baggiani C; Cavalera S; Giovannoli C; Spano G; Anfossi L Colour-encoded lateral flow immunoassay for the simultaneous detection of aflatoxin B1 and type-B fumonisins in a single Test line. Talanta ,2019, 192, 288–294. [DOI] [PubMed] [Google Scholar]
- (7).Pan D; Li G; Hu H; Xue H; Zhang M; Zhu M; Gong X; Zhang Y; Wan Y; Shen Y Direct Immunoassay for Facile and Sensitive Detection of Small Molecule Aflatoxin B1 based on Nanobody. Chemistry, 2018, 24, 9869–9876. [DOI] [PubMed] [Google Scholar]
- (8).Yamasaki T; Miyake S; Sato N; Hirakawa Y; Iwasa S; Narita H; Watanabe T Development of Enzyme-Linked Immunosorbent Assay for Analysis of Total Aflatoxins Based on Monoclonal Antibody Reactive with Aflatoxins B1, B2, G1 and G2. Shokuhin Eiseigaku Zasshi ,2018, 59, 200–205. [DOI] [PubMed] [Google Scholar]
- (9).Edupuganti SR; Edupuganti OP; Hearty S; O’Kennedy R A highly stable, sensitive, regenerable and rapid immunoassay for detecting aflatoxin B1 in corn incorporating covalent AFB1 immobilization and a recombinant Fab antibody. Talanta ,2013, 115, 329–35. [DOI] [PubMed] [Google Scholar]
- (10).Liu A; Ye Y; Chen W; Wang X; Chen F Expression of V(H)-linker-V(L) orientation-dependent single-chain Fv antibody fragment derived from hybridoma 2E6 against aflatoxin B1 in Escherichia coli. J Ind Microbiol Biotechnol ,2015, 42, 255–262. [DOI] [PubMed] [Google Scholar]
- (11).Bever CS; Dong JX; Vasylieva N; Barnych B; Cui Y; Xu ZL; Hammock BD; Gee SJ VHH antibodies: emerging reagents for the analysis of environmental chemicals. Anal Bioanal Chem ,2016, 408, 5985–6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Cao DM; X u Y; T u Z; L I YP; X u L; F JH, One-step enzyme linked immunosorbent assay for detection of aflatoxin B1 using a nanobody alkaline phosphatase fusion protein. Chinese journal of analytical chemistry ,2011, 44. [Google Scholar]
- (13).Xiong Y; Tu Z; Huang X; Xie B; Xiong Y; Xu Y Magnetic beads carrying poly (acrylic acid) brushes as “nanobody containers” for immunoaffinity purification of aflatoxin B1 from corn samples. J RSC Advances .2015, 5, 77380–77387. [Google Scholar]
- (14).Wang SH; Zhang JB; Zhang ZP; Zhou YF; Yang RF; Chen J; Guo YC; You F; Zhang XE Construction of single chain variable fragment (ScFv) and BiscFv-alkaline phosphatase fusion protein for detection of Bacillus anthracis. Analytical Chemistry, 2006, 78, 997–1004. [DOI] [PubMed] [Google Scholar]
- (15).Jiacomini I; Silva SK; Aubrey N; Muzard J; Chavez-Olortegui C; De Moura J; Billiald P; Alvarenga LM Immunodetection of the “brown” spider (Loxosceles intermedia) dermonecrotoxin with an scFv-alkaline phosphatase fusion protein. Immunol Lett ,2016, 173, 1–6. [DOI] [PubMed] [Google Scholar]
- (16).Xu ZL; Dong JX; Wang H; Li ZF; Beier RC; Jiang YM; Lei HT; Shen YD; Yang JY; Sun YM Production and Characterization of a Single-Chain Variable Fragment Linked Alkaline Phosphatase Fusion Protein for Detection of O,O-Diethyl Organophosphorus Pesticides in a One-Step Enzyme-Linked Immunosorbent Assay. J Agr Food Chem, 2012, 60, 5076–5083. [DOI] [PubMed] [Google Scholar]
- (17).Liu X; Xu Y; Wan DB; Xiong YH; He ZY; Wang XX; Gee SJ; Ryu D; Hammock BD Development of a nanobody-alkaline phosphatase fusion protein and its application in a highly sensitive direct competitive fluorescence enzyme immunoassay for detection of ochratoxin A in cereal. Anal Chem 2015, 87, 1387–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Wang J; Majkova Z; Bever CR; Yang J; Gee SJ; Li J; Xu T; Hammock BD One-step immunoassay for tetrabromobisphenol a using a camelid single domain antibody-alkaline phosphatase fusion protein. Anal Chem ,2015, 87, 4741–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Zhang YQ; Xu ZL; Wang F; Cai J; Dong JX; Zhang JR; Si R; Wang CL; Wang Y; Shen YD; Sun Y; Wang H Isolation of Bactrian Camel Single Domain Antibody for Parathion and Development of One-Step dc-FEIA Method Using VHH-Alkaline Phosphatase Fusion Protein. Anal Chem ,2018, 90, 12886–12892. [DOI] [PubMed] [Google Scholar]
- (20).Faulin Tdo E; Guilherme DF; Silva AS; Abdalla DS; Hering VR; Politi MJ; Maranhao AQ GFP-SCFV: expression and possible applications as a tool for experimental investigations of atherosclerosis. Biotechnol Prog, 2014, 30, 1206–13. [DOI] [PubMed] [Google Scholar]
- (21).Rinaldi AS; Freund G; Desplancq D; Sibler AP; Baltzinger M; Rochel N; Mely Y; Didier P; Weiss E The use of fluorescent intrabodies to detect endogenous gankyrin in living cancer cells. Exp Cell Res 2013, 319, 838–49. [DOI] [PubMed] [Google Scholar]
- (22).Wongso D; Dong J; Ueda H; Kitaguchi T Flashbody: A Next Generation Fluobody with Fluorescence Intensity Enhanced by Antigen Binding. Anal Chem, 2017, 89, 6719–6725. [DOI] [PubMed] [Google Scholar]
- (23).Li Y; Li Y; Zhao J; Zheng X; Mao Q; Xia H Development of a Sensitive Luciferase-Based Sandwich ELISA System for the Detection of Human Extracellular Matrix 1 Protein. Monoclon Antib Immunodiagn Immunother ,2016, 35, 273–279. [DOI] [PubMed] [Google Scholar]
- (24).Shifera AS; Hardin JA Factors modulating expression of Renilla luciferase from control plasmids used in luciferase reporter gene assays. Anal Biochem, 2010, 396, 167–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wu N; Rathnayaka T; Kuroda Y Bacterial expression and re-engineering of Gaussia princeps luciferase and its use as a reporter protein. Biochim Biophys Acta,2015, 1854,1392–9. [DOI] [PubMed] [Google Scholar]
- (26).Patel KG; Ng PP; Kuo CC; Levy S; Levy R; Swartz JR Cell-free production of Gaussia princeps luciferase--antibody fragment bioconjugates for ex vivo detection of tumor cells. Biochem Biophys Res Commun ,2009, 390, 971–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Venisnik KM; Olafsen T; Gambhir SS; Wu AM Fusion of Gaussia luciferase to an engineered anti-carcinoembryonic antigen (CEA) antibody for in vivo optical imaging. Mol Imaging Biol ,2007, 9, 267–77. [DOI] [PubMed] [Google Scholar]
- (28).England CG; Ehlerding EB; Cai WB, NanoLuc: A Small Luciferase Is Brightening Up the Field of Bioluminescence. Bioconjugate Chem ,2016, 27, 1175–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Ding Y; Hua X; Chen H; Liu F; Gonzalez-Sapien G; Wang M Recombinant Peptidomimetic-Nano Luciferase Tracers for Sensitive Single-Step Immunodetection of Small Molecules. Anal Chem ,2018, 90, 2230–2237. [DOI] [PubMed] [Google Scholar]
- (30).He SX; Song G; Shi JP; Guo YQ; Guo ZY Nanoluciferase as a novel quantitative protein fusion tag: Application for overexpression and bioluminescent receptor-binding assays of human leukemia inhibitory factor. Biochimie, 2014,106, 140–8. [DOI] [PubMed] [Google Scholar]
- (31).Ling Y; Jiang P; Li N; Yan Q; Wang X A luciferase immunoprecipitation assay for the detection of proinsulin/insulin autoantibodies. Clin Biochem, 2018,54, 51–55. [DOI] [PubMed] [Google Scholar]
- (32).Zhao H; Doyle TC; Wong RJ; Cao Y; Stevenson DK; Piwnica-Worms D; Contag CH Characterization of coelenterazine analogs for measurements of Renilla luciferase activity in live cells and living animals. Mol Imaging, 2004, 3, 43–54. [DOI] [PubMed] [Google Scholar]
- (33).Degeling MH; Bovenberg MS; Lewandrowski GK; de Gooijer MC; Vleggeert-Lankamp CL; Tannous M; Maguire CA; Tannous BA Directed molecular evolution reveals Gaussia luciferase variants with enhanced light output stability. Anal Chem, 2013, 85, 3006–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Maguire CA; Deliolanis NC; Pike L; Niers JM; Tjon-Kon-Fat LA; Sena-Esteves M; Tannous BA Gaussia luciferase variant for high-throughput functional screening applications. Anal Chem, 2009, 81, 7102–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.