

Abstract
Bronchopulmonary dysplasia (BPD)-associated pulmonary hypertension (PH) is a chronic infantile lung disease that lacks curative therapies. Infants with BPD-associated PH are often exposed to hyperoxia and additional insults such as sepsis that contribute to disease pathogenesis. Animal models that simulate these scenarios are necessary to develop effective therapies; therefore, we investigated whether lipopolysaccharide (LPS) and hyperoxia exposure during saccular lung development cooperatively induce experimental BPD-PH in mice. C57BL/6J mice were exposed to normoxia or 70% O2 (hyperoxia) during postnatal days (PNDs) 1–5 and intraperitoneally injected with varying LPS doses or a vehicle on PNDs 3–5. On PND 14, we performed morphometry, echocardiography, and gene and protein expression studies to determine the effects of hyperoxia and LPS on lung development, vascular remodeling and function, inflammation, oxidative stress, cell proliferation, and apoptosis. LPS and hyperoxia independently and cooperatively affected lung development, inflammation, and apoptosis. Growth rate and antioxidant enzyme expression were predominantly affected by LPS and hyperoxia, respectively, while cell proliferation and vascular remodeling and function were mainly affected by combined exposure to LPS and hyperoxia. Mice treated with lower LPS doses developed adaptive responses and hyperoxia exposure did not worsen their BPD phenotype, whereas those mice treated with higher LPS doses displayed the most severe BPD phenotype when exposed to hyperoxia and were the only group that developed PH. Collectively, our data suggest that an additional insult such as LPS may be necessary for models utilizing short-term exposure to moderate hyperoxia to recapitulate human BPD-PH.
Keywords: bronchopulmonary dysplasia, hyperoxia, inflammation, lipopolysaccharide, pulmonary hypertension
INTRODUCTION
Bronchopulmonary dysplasia (BPD) is a developmental lung disorder of preterm infants that is primarily caused by immature host defense mechanisms, which prevent tissue injury and facilitate repair (62). Although its incidence has not changed over the past few decades (89), BPD increases economic burden (64) and long-term morbidity (60, 95, 121, 123); for instance, the hospitalization cost incurred by an infant with BPD in the first year of life is $377,871, more than twice that of infants without BPD (79). Pulmonary hypertension (PH) is a common comorbidity of BPD, with a pooled prevalence of 6, 12, and 39%, in mild, moderate, and severe BPD, respectively (7). Importantly, the presence of PH increases both short- and long-term morbidities and mortality in BPD infants (7, 14, 29, 98, 124). Further, there are no curative therapies for this disease complex.
Supplemental oxygen and mechanical ventilation are life-sustaining therapies administered to infants with respiratory failure; however, these therapies are associated with oxidative stress and inflammation, which disrupt normal lung development and lead to BPD and PH in both infants and mice (63, 95, 126). Additional postnatal risk factors that contribute to BPD pathogenesis include sepsis, patent ductus arteriosus, and respiratory microbial dysbiosis (9, 54, 67, 78, 105, 118, 132). The majority of infants who develop BPD-PH are exposed to multiple insults; however, preclinical studies primarily use a single insult to phenotype human BPD-PH. Thus, animal models that incorporate more than one insult may be necessary to determine the clinically relevant molecular mechanisms of BPD-PH and discover novel effective therapies for this multifactorial disease complex.
At birth, murine lungs are equivalent to the developmental lung stage of a 25–26-wk-old infant (151); therefore, mice are the most common laboratory animal used to model human BPD. Several preclinical studies, including ours, have shown that exposing neonatal mice to hyperoxia leads to a phenotype that models BPD and PH in infants (8, 52, 110). Although mechanical ventilation is another common factor that causes BPD, it is technically challenging to expose neonatal mice to a mechanical ventilator long enough to develop the BPD-PH phenotype. Among the remaining risk factors, postnatal sepsis has been increasingly recognized as a major risk factor that leads to BPD and its associated morbidities (46, 66, 74). Lipopolysaccharide (LPS) is widely used to model sepsis in experimental animals (43, 86), as it is the major biologically active component and primary recognition structure in gram-negative bacteria (53). Recently, we demonstrated that the lung phenotype of mice exposed to LPS during the saccular phase of lung development is similar to human BPD (122); however, the experimental BPD-PH phenotype of mice exposed to both neonatal hyperoxia and postnatal sepsis has not been well characterized.
In this study, we exposed neonatal mice to hyperoxia and LPS to model the clinical scenario of infants with BPD-PH and tested the hypothesis that chronic LPS and hyperoxia exposure during the saccular phase of lung development cooperatively disrupt lung development and recapitulate experimental BPD-PH in neonatal mice. We demonstrated that short-term exposure to LPS or hyperoxia modestly recapitulates human BPD without PH, while short-term exposure to both LPS and hyperoxia is necessary to recapitulate human BPD with PH.
MATERIALS AND METHODS
Animals.
Institutional Animal Care and Use Committee of Baylor College of Medicine approved this study and we adhered to the federal guidelines for the humane care and use of laboratory animals. C57BL/6J wild-type mice were obtained from The Jackson Laboratory (Bar Harbor, ME). Timed-pregnant mice were raised in our facility for our studies. The dams were fed standard mice food and water ad libitum, and all study groups were maintained under 12:12-h day-night cycles.
Hyperoxia exposure.
Male and female pups from 16 litters were pooled, equally redistributed among the dams, and exposed to 21% oxygen (normoxia) or 70% oxygen (hyperoxia) during postnatal days (PNDs) 1–5. Hyperoxia exposure was achieved by continuously delivering oxygen into plexiglass chambers using an oxygen blender to yield a constant 70% oxygen level. To prevent oxygen toxicity in the dams, they were rotated between normoxia- and hyperoxia-exposed litters every day during the exposure period (110).
LPS treatment.
To induce a systemic inflammatory response, mice exposed to normoxia and hyperoxia were injected intraperitoneally once daily on PNDs 3–5 with Escherichia coli O55:B5 LPS (Sigma-Aldrich, St. Louis, MO; L2880) or an equivalent volume of the vehicle (PBS). Varying LPS doses were used to determine the effects of, and interactions between, LPS and hyperoxia on survival rate (3, 6, or 10 mg·kg−1·day−1 of LPS), experimental BPD (3 or 6 mg·kg−1·day−1 of LPS), and PH (6 mg·kg−1·day−1 of LPS).
Tissue preparation for lung morphometry.
Vehicle- and LPS-treated animals exposed to normoxia or hyperoxia were euthanized on PND 14 (n = 4–6/group), and their lungs were inflated at a pressure of 25 cm H2O and fixed with 10% formalin via the trachea, as described previously (120). The lung morphometric measurements were obtained by observers who were blinded to the study groups.
Alveolarization and lung vascularization analyses.
To quantify alveolarization, the radial alveolar count (RAC) was measured, as described by Cooney and Thurlbeck (28), and the mean linear intercept (MLI) was assessed, as described previously (136), using images taken from at least 10 nonoverlapping lung fields (original magnification, ×100) per animal. Lung blood vessel density was estimated by quantifying the number of von Willebrand factor (vWF)-stained vessels in images from at least 10 nonoverlapping lung fields (original magnification, ×200) per animal, as described previously (122).
Real-time quantitative-PCR assays.
Total RNA extracted from lung tissues (n = 3–7/group) was reverse transcribed to cDNA as described before (149). RT-qPCR analysis was then performed using the TaqMan gene expression master mix and the following gene-specific primers (Applied Biosystems, Grand Island, NY): BCL2-associated X protein (BAX; Mm00432051_m1), chemokine (C-C motif) ligand 2 (CCL2; Mm00441242_m1), heme oxygenase 1 (HO-1; Mm00516005_m1), intercellular adhesion molecule 1 (ICAM-1; Mm00516023_m1), interleukin (IL)-1β (IL-1β; Mm00434228_m1), NAD(P)H quinone dehydrogenase 1 (NQO1; Mm01253561_m1), tumor necrosis factor-α (TNF-α; Mm00443258_m1), tumor necrosis factor receptor superfamily, member 10b (TNFRSF10B; Mm00457866_m1), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Mm99999915_g1). GAPDH was detected as the reference gene.
Immunoblot assays.
Protein lysates were prepared from the experimental animals (n = 3–7/group) and subjected to immunoblotting, as described previously (122), using the antibodies against Akt (Cell Signaling Technology, Danvers, MA; 4691, dilution 1:1,000), phospho(p)-Akt (Cell Signaling Technology; 4060, dilution 1:1,000), β-actin (Santa Cruz Biotechnology, Santa Cruz, CA; sc-47778, dilution 1: 5,000), endothelial nitric oxide synthase (eNOS; BD Transduction Laboratories, San Jose, CA; 610296, dilution 1:1,000), phosphor(p)-eNOS [eNOS(Ser-1177); BD Transduction Laboratories; 612392, dilution 1:1000], HO1 (Enzo Life Sciences, Farmingdale, NY; ADI-SPA-896, dilution 1:1,000), ICAM-1 (Abcam, Cambridge, MA; ab179707, dilution 1:1,000), NQO1 (Cell Signaling Technology; 62262, dilution 1:1,000), proliferating cell nuclear antigen (PCNA; Thermo Fisher Scientific, Waltham, MA; MA5-11358, dilution 1:1,000), signal transducer and activator of transcription 1 (STAT1; Cell Signaling Technology; 9172, dilution 1: 1,000), phospho(p)-STAT1 (STAT1[Tyr701]; Cell Signaling Technology; 7649, dilution 1: 1,000), STAT3 (Cell Signaling Technology; 12640, dilution 1: 1,000), p-STAT3 (STAT3[Tyr705]; Cell Signaling Technology; 9145, dilution 1: 1,000), STAT6 (Cell Signaling Technology; 9362, dilution 1: 1,000), p-STAT6 [STAT6(Tyr-641); Cell Signaling Technology; 56554, dilution 1: 1,000], SLUG (Cell Signaling Technology; 9585, dilution 1: 1,000), and vascular endothelial growth factor 2 (VEGFR2; Cell Signaling Technology; 2479, dilution 1:1,000). The primary antibodies were detected by incubation with appropriate horseradish peroxidase-conjugated secondary antibodies. The immunoreactive bands were detected by chemiluminescence methods, and the band densities were quantified using Image Laboratory software (Chemidoc touch imaging system, Bio-Rad Laboratories, Hercules, CA).
Enzyme-linked immunosorbent assay.
The IL-1β protein levels in the lung homogenates were measured by the ELISA technique. Mouse IL-1 β/IL-1F2 Quantikine (R&D Systems, Minneapolis, MN; MLB00C) ELISA kit was used to quantify IL-1β protein, according to the manufacturer’s recommendations.
Transthoracic echocardiography.
On PND 14, right ventricular (RV) systolic time intervals were measured by functional echocardiography to detect PH, as described previously (110). Pulsed-wave Doppler (PWD) recordings of pulmonary blood flow were obtained at the aortic valve level in parasternal short axis view (134) using a VisualSonics Vevo 2100 and a 40-MHz linear transducer to measure pulmonary acceleration time (PAT) and RV ejection time (ET). RV systolic pressure (RVSP) was calculated using the following regression formula: RVSP = 64.5 – (83.5 × PAT/ET) (134). The measurements were obtained by investigators blinded to the experimental groups.
Right ventricle left ventricle free wall thickness.
Serial 5-µm sections of the paraffin-embedded heart (n = 4–6/group) were stained with hematoxylin and eosin to analyze right ventricle/left ventricle (RV/LV) free wall thickness, as described previously (6, 34, 35, 37). Briefly, RV and LV free wall thickness were assessed in a transverse section free of papillary muscle projections at ×125 magnification, and their ratio (RV:LV) was used to determine ventricular free wall thickness. Morphometric measurements were obtained by observers blinded to slide identity.
Pulmonary vascular remodeling.
Pulmonary vascular remodeling, a marker and a cause of significant PH, was estimated by quantifying the medial thickness index of resistance pulmonary blood vessels (20- to 150- µm external diameter). The paraffin-embedded tissues were deparaffinized and subjected to staining with α-smooth muscle actin (α-SMA) antibody (Sigma-Aldrich, St. Louis, MO; A5228), according to the manufacturer’s recommendations. The medial thickness index was calculated using the equation: [(areaext − areaint)/areaext] × 100, where areaext and areaint are the areas within the external and internal boundaries of the α-SMA layer, respectively (8).
Statistical analyses.
Data were analyzed using GraphPad Prism 5 software and were expressed as means ± SD. Interactions between hyperoxia and LPS and their effects on survival were determined using Log-rank (Mantel-Cox) tests, while their interactions and effects on experimental BPD-PH were determined by ANOVA. Post hoc Bonferroni’s multiple-comparison tests were performed if variables or interactions were found to be statistically significant (P < 0.05) by ANOVA.
RESULTS
Survival and growth rates of mice exposed to early LPS and hyperoxia.
Initially, we investigated the dose-dependent effects of LPS and hyperoxia on the survival of neonatal mice and found that LPS decreased the survival rate in a dose-dependent manner (Fig. 1A). Under normoxic conditions, the survival rate did not significantly differ in animals treated with the vehicle or 3 mg/kg of LPS, but decreased by 25% and 50% in animals treated with 6 and 10 mg/kg of LPS, respectively. Although the survival rates of animals treated with 3 or 6 mg/kg of LPS did not significantly differ under normoxic and hyperoxic conditions, the effect of LPS on survival was potentiated in the 10 mg/kg of LPS-treated group exposed to hyperoxia, suggesting that hyperoxia and high LPS doses exert an interactive effect on survival. The survival rates of the 10 mg/kg of LPS-treated group over PNDs 1–5 were 50% and 0% under normoxic and hyperoxic conditions, respectively (Fig. 1A); therefore, we excluded this group from subsequent experiments.
Fig. 1.
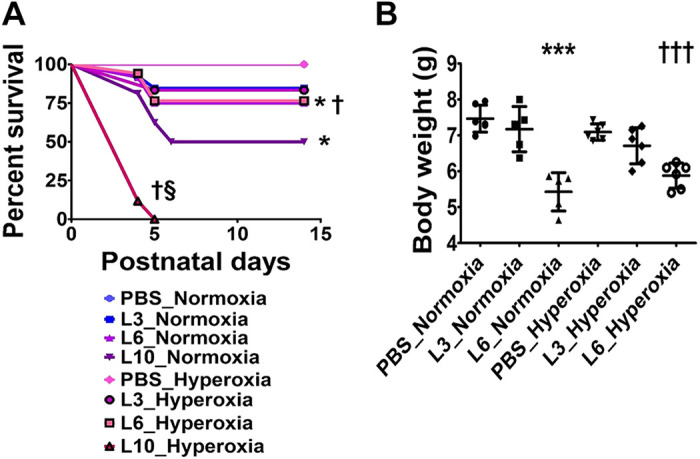
Effects of early LPS and hyperoxia exposure on the survival rate (A) and body weight (B) of C57BL/6J wild-type mice. Newborn C57BL/6J wild-type mice were treated intraperitoneally with 3 (L3), 6 (L6), or 10 (L10) mg/kg of LPS or vehicle (PBS) on postnatal days (PNDs) 3–5 and exposed to 21% (normoxia) or 70% (hyperoxia) during PNDs 1–5. Their survival and weight gain were monitored until PND 14. A: percentage survival of PBS- and LPS-treated mice exposed to normoxia or hyperoxia (n = 9–17/group). Significant differences between PBS- and LPS-treated mice are indicated by *P < 0.05 under normoxic conditions and by †P < 0.05 under hyperoxic conditions. Significant differences between treatment-matched mice under normoxic and hyperoxic conditions are indicated by §P < 0.05 (Log-rank [Mantel-Cox] test). B: body weight (g) of PBS- and LPS-treated mice exposed to normoxia or hyperoxia. Values are expressed as means ± SD (n = 5 or 6/group). Significant differences between PBS- and LPS-treated mice are indicated by ***P < 0.001 under normoxic conditions and by †††P < 0.001 under hyperoxic conditions.
LPS exerted similar effects on the growth rates of mice under normoxic and hyperoxic conditions. At PND 3, the body weight of all our experimental animals was comparable. However, at PND 14, the body weight of animals treated with 6 mg/kg of LPS was lower than those treated with the vehicle (Fig. 1B). Hyperoxia did not independently affect the growth rate in the experimental animals (Fig. 1B).
Effects of early LPS and hyperoxia exposure on lung inflammation.
We evaluated the extent of lung inflammation by quantifying the generation of proinflammatory cytokines (CCL2, ICAM-1, IL-1β, and TNF-α) in lung tissues by real-time RT-PCR, immunoblotting, and ELISA. LPS doses of 3 or 6 mg/(kg/day) only affected IL-1β mRNA expression (Fig. 2C; increased by ≥2-fold) in normoxic mice at PND 14, whereas hyperoxia did not affect IL-1β (Fig. 2C) but did increase CCL2 (Fig. 2A) and ICAM-1 (Fig. 2B) mRNA expression by ≥1.5-fold at PND 14. We also observed that LPS and hyperoxia had interactive effects on the expression of these proinflammatory cytokines; mice exposed to 3 mg/kg of LPS and hyperoxia displayed lower cytokine generation of ICAM-1 (Fig. 2B) and IL-1β (Fig. 2C) mRNA levels than vehicle-treated mice exposed to hyperoxia, indicating that low LPS doses may mitigate or delay the hyperoxia-induced lung inflammatory response. By contrast, IL-1β mRNA levels remained significantly elevated in mice treated with 6 mg/kg of LPS and exposed to hyperoxia (Fig. 2C). Next, we estimated the ICAM-1 protein levels by immunoblotting. Hyperoxia increased ICAM-1 protein expression in mice treated with 6 mg/kg of LPS compared with vehicle-treated mice (Fig. 2, E and F). By contrast, ICAM-1 protein levels were significantly decreased in mice treated with 3 mg/kg of LPS compared with vehicle-treated mice under hyperoxic conditions, a finding that was consistent with our gene expression studies. We also estimated IL-1β protein levels by ELISA. Congruent with our IL-1β mRNA finding, mice treated with 6 mg/kg of LPS had elevated IL-1β protein levels in both normoxic and hyperoxic conditions compared with other treatment groups (Fig. 2G).
Fig. 2.

Lung inflammation indices in C57BL/6J wild-type mice exposed to LPS and hyperoxia during the saccular phase of lung development. C57BL/6J wild-type mice were exposed to 21% O2 (normoxia) or 70% O2 (hyperoxia) during postnatal days (PNDs) 1–5 and injected intraperitoneally with 3 (L3) or 6 (L6) mg/kg of LPS or the vehicle (PBS) on PNDs 3–5. Whole-lung tissues were harvested on PND 14 for real-time RT-PCR, ELISA, and immunoblot analyses. A–D: real-time RT-PCR analysis-based determination of CCL2 (A), ICAM-1 (B), IL-1β (C), and TNF-α (D) mRNA expression. E: immunoblot determination of ICAM-1 and β-actin protein levels. F: ICAM-1 band intensities were quantified and normalized to β-actin. G: ELISA-based determination of IL-1β protein expression. H: immunoblot determination of p-STAT3, STAT3, p-STAT1, STAT1, p-STAT6, STAT6, and β-actin protein levels. I–J: p-STAT3 band intensity was quantified and normalized to STAT3 (I) and p-STAT6 to STAT6 (J). Values represent the means ± SD (n = 3–7 mice/group). Significant differences between PBS- and LPS-treated mice are indicated by *P < 0.05, **P < 0.01, and ***P < 0.001 under normoxic conditions and by †P < 0.05, ††P < 0.01, and †††P < 0.001 under hyperoxic conditions. Significant differences between treatment-matched mice under normoxic and hyperoxic conditions are indicated by §P < 0.05, §§P < 0.01, and §§§P < 0.001.
Next, we examined the activation of transcription factors that regulate inflammation. LPS or hyperoxia exposure during the saccular phase of lung development failed to activate STAT1, as evidenced by a lack of p-STAT1 expression in our experimental animals (Fig. 2H). However, early LPS or hyperoxia exposure independently activated STAT3, with the p-STAT3/total STAT3 ratio 1.5- and 1.9-fold higher in normoxic animals treated with 3 mg/(kg/day) or 6 mg/(kg/day) LPS, respectively, than in the vehicle-treated animals (Fig. 2, H and I). Likewise, the p-STAT3/total STAT3 ratio was 1.5-fold higher in vehicle-treated animals exposed to hyperoxia than normoxia (Fig. 2, H and I). Consistent with their effects on proinflammatory cytokine expression, LPS and hyperoxia had interactive effects on STAT3 activation. STAT3 activation decreased when mice treated with 3 mg/(kg/day) or 6 mg/(kg/day) LPS were exposed to hyperoxia, with the p-STAT3/total STAT3 ratio decreasing by 1.5-fold in both of these groups compared with normoxia-exposed mice (Fig. 2, H and I). We also determined the extent of STAT6 activation in our experimental animals, observing that 3 mg/kg of LPS had an independent effect, while 6 mg/kg of LPS and hyperoxia did not affect STAT6 activation (Fig. 2, H and J). Treatment with 3 mg/kg of LPS decreased the p-STAT6/total STAT6 ratio by 5.7- and 7.8-fold under normoxic and hyperoxic conditions, respectively, compared with corresponding vehicle-treated mice (Fig. 2, H and J).
Alveolarization of mice exposed to early LPS and hyperoxia.
Alveolarization was quantified by measuring RAC and MLI on PND 14. LPS and hyperoxia exposure independently disrupted alveolar development (alveolar simplification). In animals breathing room air (normoxia), exposure to 3 or 6 mg/(kg/day) of LPS induced alveolar simplification compared with exposure to vehicle. LPS exposure significantly decreased RACs (Fig. 3, A–C, G), indicating that there were fewer alveoli, compared with vehicle-treated animals. Additionally, the MLIs of these mice significantly increased (Fig. 3, A–C, H), indicating that their alveoli were also larger than those of vehicle-treated mice. Hyperoxia also induced alveolar simplification, as evidenced by the significantly lower RAC and higher MLI in vehicle-treated mice under hyperoxic conditions than under normoxic conditions (Fig. 3, A, D, G, H). The extent of alveolar simplification in mice treated with 3 mg/kg of LPS was not increased by hyperoxia exposure (Fig. 3, B, E, G, H) and was similar to that observed in vehicle-treated mice exposed to hyperoxia (Fig. 3, D, G, H). Conversely, the extent of alveolar simplification increased in mice treated with 6 mg/kg of LPS upon hyperoxia exposure (Fig. 3, C, F, G, H) and was significantly greater than that observed in vehicle-treated mice exposed to hyperoxia (Fig. 3, D, G, H). Taken together, these observations suggest that hyperoxia and LPS exert an interactive effect on alveolarization. Moreover, while hyperoxia did not affect alveolarization in mice exposed to low LPS doses, it did augment alveolar simplification in those exposed to higher LPS doses.
Fig. 3.

Alveolarization deficits in 2-wk-old C57BL/6J wild-type mice exposed to LPS and hyperoxia during the saccular phase of lung development. C57BL/6J wild-type mice were exposed to either 21% O2 (normoxia) or 70% O2 (hyperoxia) during postnatal days (PNDs) 1–5 and injected intraperitoneally with 3 (L3) or 6 (L6) mg/kg of LPS or the vehicle (PBS) on PNDs 3–5. The mice were euthanized on PND 14 for lung morphometry. A–F: representative hematoxylin-and-eosin-stained lung sections from mice treated with PBS (A and D), L3 (B and E), or L6 (C and F) and exposed to normoxia (A–C) or hyperoxia (D–F). Scale bar = 100 µm. G and H: alveolarization was quantified by determining RAC (G) and MLI (H). Values are expressed as means ± SD (n = 4–6 mice/group). Significant differences between PBS- and LPS-treated mice are indicated by **P < 0.01 and ***P < 0.001 under normoxic conditions and by ††P < 0.01 and †††P < 0.001 under hyperoxic conditions. Significant differences between treatment-matched mice under normoxic and hyperoxic conditions are indicated by §P < 0.05, §§P < 0.01, and §§§P < 0.001.
Lung vascularization of mice exposed to early LPS and hyperoxia.
Next, we examined the effects of early LPS and hyperoxia exposure on pulmonary vascularization by quantifying the number of vWF-stained lung blood vessels (pulmonary vascular simplification). LPS and hyperoxia exposure independently induced pulmonary vascular simplification at PND 14. Pulmonary vascular simplification was significantly greater in animals treated with 6 mg/kg of LPS than in those treated with 3 mg/kg of LPS under normoxic conditions (Fig. 4, A–C, G). Hyperoxia exposure also induced pulmonary vascular simplification, as evidenced by significantly fewer vWF-stained lung blood vessels in vehicle-treated mice under hyperoxic conditions than under normoxic conditions (Fig. 4, A, D, G). Consistent with effects of LPS and hyperoxia on alveolarization, treatment with 3 mg/kg of LPS did not increase the extent of pulmonary vascular simplification in mice exposed to hyperoxia (Fig. 4, B, E, G) and was similar to vehicle-treated mice exposed to hyperoxia (Fig. 4, D and G). Conversely, the extent of pulmonary vascular simplification in mice treated with 6 mg/kg of LPS increased upon exposure to hyperoxia (Fig. 4, C, F, G) and was significantly higher than in vehicle-treated mice exposed to hyperoxia (Fig. 4, D and G). These observations suggest that LPS and hyperoxia interactively affect pulmonary vascularization. While hyperoxia did not decrease lung vascularization in mice exposed to low LPS doses, it did potentiate the pulmonary vascular phenotype of mice exposed to high LPS doses.
Fig. 4.

Lung vascularization deficits in 2-wk-old C57BL/6J wild-type mice exposed to LPS and hyperoxia during the saccular phase of lung development. C57BL/6J wild-type mice were exposed to either 21% O2 (normoxia) or 70% O2 (hyperoxia) during postnatal days (PNDs) 1–5 and injected intraperitoneally with 3 (L3) or 6 (L6) mg/kg of LPS or the vehicle (PBS) on PNDs 3–5. The mice were euthanized on PND 14 for lung morphometry and protein expression studies. A–F: Representative von Willebrand factor (vWF)-immunostained lung sections from mice treated with PBS (A and D), L3 (B and E), or L6 (C and F) and exposed to normoxia (A–C) or hyperoxia (D–F). Scale bar = 100 µm. Pulmonary vascularization was quantified by counting the number of vWF-stained lung blood vessels (G). VEGFR2, p-eNOS, eNOS, and β-actin protein levels were determined by immunoblotting (H). (I and J) VEGFR2 band intensities were quantification and normalized against β-actin (I) and p-eNOS against eNOS (J). Values represent the mean ± SD (n = 3–7 mice/group). Significant differences between PBS- and LPS-treated mice are indicated by *P < 0.05, **P < 0.01, and ***P < 0.001 under normoxic conditions and by †P < 0.05, ††P < 0.01, and †††P < 0.001 under hyperoxic conditions. Significant differences between treatment-matched mice under normoxic and hyperoxic conditions are indicated by §P < 0.05.
The major pathways required for lung vascular homeostasis are the VEFGR2 and eNOS signaling pathways; therefore, we examined their expression and activation in our experimental model. While 6 mg/kg of LPS independently decreased VEGFR2 protein expression, hyperoxia had a differential effect on VEGFR2 expression (Fig. 4, H and I). Hyperoxia exposure significantly increased VEGFR2 protein expression only in mice treated with 3 mg/kg of LPS. Hyperoxia also had a differential effect on eNOS activation in our experimental conditions. While hyperoxia significantly increased phosphorylated eNOS protein levels in mice treated with 3 mg/kg or 6 mg/kg of LPS, it did not affect the expression of phosphorylated eNOS protein levels in vehicle-treated mice (Fig. 4, H and J). These findings indicate that hyperoxia and LPS exert an interactive effect on VEGFR2 expression and eNOS activation.
Effects of early LPS and hyperoxia exposure on pulmonary antioxidant enzymes, NQO1 and HO1.
At PND 14, we also quantified the mRNA and protein expression of well-known antioxidant enzymes (NQO1 and HO1) because they are shown to modulate hyperoxia-induced neonatal lung injury. Real-time RT-PCR and immunoblot analyses indicated that early LPS exposure had no effect on the mRNA or protein expression of these enzymes, while hyperoxia increased NQO1 and HO1 mRNA expression in mice treated with the vehicle or 6 mg/kg of LPS (Fig. 5, A and B). However, when we analyzed protein expression, which is more functionally relevant, we found that neither LPS nor hyperoxia had an independent effect on NQO1 expression (Fig. 5, C and D). Likewise, LPS did not affect HO1 protein expression. However, hyperoxia increased HO1 expression by 1.5-fold only in vehicle-treated animals, indicating that hyperoxia exerts an independent effect on HO1 protein expression (Fig. 5, C and E).
Fig. 5.

Pulmonary antioxidant enzyme levels in C57BL/6J wild-type mice exposed to LPS and hyperoxia during the saccular phase of lung development. C57BL/6J wild-type mice were exposed to 21% O2 (normoxia) or 70% O2 (hyperoxia) during postnatal days (PNDs) 1–5 and injected intraperitoneally with 3 (L3) or 6 (L6) mg/kg of LPS or the vehicle (PBS) on PNDs 3–5. Whole-lung tissues were harvested on PND 14 for real-time RT-PCR and immunoblot analyses. A and B: real-time RT-PCR analysis-based determination of NQO1 (A) and HO1 (B) mRNA expression. C: immunoblot determination of NQO1, HO1, and β-actin protein levels. densitometric analysis wherein NQO1 (D) and HO1 (E) band intensity was quantified and normalized to β-actin. Values are expressed as means ± SD (n = 3–7 mice/group). Significant differences between PBS- and LPS-treated mice are indicated by †P < 0.05 and †††P < 0.001 under hyperoxic conditions. Significant differences between treatment-matched mice under normoxic and hyperoxic conditions are indicated by §P < 0.05, §§P < 0.01 and §§§P < 0.001.
Effects of early LPS and hyperoxia exposure on lung cell apoptosis and proliferation.
To determine the mechanisms of alveolar and pulmonary vascular simplification in our model, we examined the effects of LPS and hyperoxia on lung cell apoptosis and proliferation. LPS predominantly increased the expression of the extrinsic pathway proapoptotic gene TNFRSF10B by over 2.5-fold (Fig. 6B), whereas hyperoxia predominantly increased that of the intrinsic pathway gene, BAX1, by 1.8-fold (Fig. 6A). BAX1 mRNA levels were comparable between the vehicle- and 6 mg/kg of LPS-treated mice upon hyperoxia exposure; however, there was no corresponding increase in its expression when mice treated with 3 mg/kg of LPS were exposed to hyperoxia (Fig. 6A). These findings indicate that mice exposed to low LPS doses display decreased intrinsic pathway-mediated apoptosis under hyperoxic conditions.
Fig. 6.

Apoptosis and cell proliferation indices in the lungs of C57BL/6J wild-type mice exposed to LPS and hyperoxia during the saccular phase of lung development. C57BL/6J wild-type mice were exposed to 21% O2 (normoxia) or 70% O2 (hyperoxia) during postnatal days (PNDs) 1–5 and injected intraperitoneally with 3 (L3) or 6 (L6) mg/kg of LPS or the vehicle (PBS) on PNDs 3–5. Whole-lung tissues were harvested on PND 14 for real-time RT-PCR and immunoblot analyses. Real-time RT-PCR analysis-based determination of BAX (A) and TNFRSF10B (B) mRNA expression. C: immunoblot determination of p-Akt, Akt, PCNA, SLUG, and β-actin protein levels. Quantification and normalization of p-Akt (D) band intensity to Akt, and PCNA (E) and SLUG (F) band intensities to β-actin. Values are expressed as means ± SD (n = 3–7 mice/group). Significant differences between PBS- and LPS-treated mice are indicated by ***P < 0.001 under normoxic conditions and by †P < 0.05, ††P < 0.01, and †††P < 0.001 under hyperoxic conditions. Significant differences between treatment-matched mice under normoxic and hyperoxic conditions are indicated by §§P < 0.01 and §§§P < 0.001.
We also analyzed the expression and activation of proteins that modulate apoptosis and cell proliferation. Mice treated with 3 mg/kg of LPS and exposed to hyperoxia displayed increased Akt activation, with the pAKT/total AKT ratio increasing by 3.4-fold compared with corresponding normoxia-exposed mice (Fig. 6, C and D). Similarly, PCNA expression was 1.5-fold higher in hyperoxia-exposed mice treated with 3 mg/kg of LPS compared with corresponding normoxia-exposed mice (Fig. 6, C and E). These findings suggest that hyperoxia interacts with LPS to affect AKT activation and PCNA expression. In contrast, neither LPS nor hyperoxia independently affected SLUG protein expression (Fig. 6, C and F). Collectively, hyperoxia did not independently affect the expression of these apoptosis- and cell proliferation-modulating proteins in our experimental model.
Effects of early LPS and hyperoxia exposure on pulmonary arterial pressure.
We also determined the extent of PH in our experimental model. We excluded mice treated with 3 mg/kg of LPS for our echocardiographic studies due to the following reasons. In mice exposed to hyperoxia, the lung development was comparable between the vehicle- and 3 mg/kg of LPS-treated groups. Further, other lung injury markers were significantly lower in mice treated with 3 mg/kg of LPS and exposed to hyperoxia. Neither LPS nor hyperoxia exposure independently affected PH indices (PAT or PAT/ET ratio) in our model (Fig. 7); however, mice treated with 6 mg/kg of LPS and exposed to hyperoxia displayed a modest decrease in PAT/ET ratio (Fig. 7, D and F) and a modest increase in estimated RVSP (Fig. 7G) than the vehicle-treated animals exposed to normoxia. There were no differences in PAT (Fig. 7E) between the experimental groups, and the heart rate was comparable in all experimental groups (Fig. 7H).
Fig. 7.

Pulmonary hypertension (PH) indices at PND14 in C57BL/6J wild-type mice exposed to LPS and hyperoxia during the saccular phase of lung development. High-resolution echocardiography was performed on postnatal day (PND) 14 in C57BL/6J wild-type mice exposed to 21% O2 (normoxia) or 70% O2 (hyperoxia) during PNDs 1–5 and injected intraperitoneally with 6 mg/kg of LPS (L6) or the vehicle (PBS) on PNDs 3–5. Representative PWD echocardiography recordings of pulmonary artery blood flow obtained from mice treated with PBS (A and C) or L6 (B and D) and exposed to normoxia (A and B) or hyperoxia (C and D). PAT (E), PAT/ET ratio (F), RVSP (G), and heart rate (H) were estimated from the PWD Echo pulmonary artery blood flow recordings. Values are expressed as means ± SD (n = 5 or 6 mice/group). Significant differences between PBS- and LPS-treated mice under hyperoxic conditions are indicated by †P < 0.05. Significant differences between treatment-matched mice under normoxic and hyperoxic conditions are indicated by §P < 0.05.
Effects of early LPS and hyperoxia exposure on RV free wall thickness.
Severe and/or chronic PH causes RV hypertrophy. Estimation of RV hypertrophy by Fulton’s index in neonatal mice may be inaccurate due to the technical challenges associated with dissection of the small heart, which is required to estimate the index. Alternatively, RV/LV free wall thickness ratio obtained from hematoxylin-and-eosin-stained heart sections is a reliable marker of RV hypertrophy in neonatal mice. Therefore, we used this measurement in our studies. RV/LV free wall thickness ratio was comparable in all our experimental groups (Fig. 8), indicating that hyperoxia and LPS did not have any independent or interactive effects on RV hypertrophy in our mouse model.
Fig. 8.

Right ventricle/left ventricle (RV/LV) free wall thickness ratio at PND14 in C57BL/6J wild-type mice exposed to LPS and hyperoxia during the saccular phase of lung development. C57BL/6J wild-type mice were exposed to either 21% O2 (normoxia) or 70% O2 (hyperoxia) during postnatal days (PNDs) 1–5 and injected intraperitoneally with 6 (L6) mg/kg of LPS or the vehicle (PBS) on PNDs 3–5. The mice were euthanized on PND 14 for heart morphometry. The RV/LV free wall thickness ratio was estimated from hematoxylin-and-eosin-stained heart sections. Values are expressed as means ± SD (n = 4–6 mice/group).
Effects of early LPS and hyperoxia exposure on pulmonary vascular remodeling.
Finally, we quantified pulmonary vascular remodeling in our experimental model because it is one of the major determinants of pulmonary vascular disease, including PH across all age groups. The extent of muscularization of resistance pulmonary blood vessels was measured by immunostaining with the α-SMA antibody to determine the independent and interactive effects of hyperoxia and LPS exposure on pulmonary vascular remodeling. Neither LPS nor hyperoxia independently affected the extent of muscularization of resistance pulmonary blood vessels, as determined by quantifying the medial thickness index of α-SMA-immunostained vessels (Fig. 9, A–C, E). However, consistent with our echocardiographic studies, mice treated with 6 mg/kg of LPS and exposed to hyperoxia exhibited a significant increase in the muscularization of pulmonary blood vessels (Fig. 9, D and E), indicating an interactive effect of LPS and hyperoxia on pulmonary vascular remodeling in our experimental model.
Fig. 9.

Pulmonary vascular remodeling at postnatal day (PND) 14 in C57BL/6J wild-type mice exposed to LPS and hyperoxia during the saccular phase of lung development. C57BL/6J wild-type mice were exposed to either 21% O2 (normoxia) or 70% O2 (hyperoxia) during postnatal days (PNDs) 1–5 and injected intraperitoneally with 6 (L6) mg/kg of LPS or the vehicle (PBS) on PNDs 3–5. The mice were euthanized on PND 14 for quantifying pulmonary vascular remodeling. Representative α-SMA-stained pulmonary blood vessels (arrows) from mice treated with PBS (A and C) or L6 (B and D) and exposed to normoxia (A and B) or hyperoxia (C and D). Scale bar = 100 µm. Quantitative analysis of pulmonary vascular remodeling by medial thickness index (E). Values are expressed as means ± SD (n = 4–6 mice/group). Significant differences between PBS- and LPS-treated mice are indicated by †††P < 0.001 under hyperoxic conditions. Significant differences between treatment-matched mice under normoxic and hyperoxic conditions are indicated by §§P < 0.01.
DISCUSSION
In this study, we elucidated the effects of hyperoxia and systemic LPS exposure on saccular murine lungs and demonstrated that the disruptive effects of early LPS and hyperoxia exposure on lung development persist at PND 14. We also analyzed gene and protein expression to determine the fundamental molecular mechanisms via which LPS and hyperoxia independently and cooperatively disrupt lung development and demonstrated the interactive effects between LPS and hyperoxia exposure on pulmonary vascular function using high-resolution trans-thoracic echocardiography.
The systemic LPS dose used to model sepsis in this study was similar to that used in other rodent studies (19, 40, 90, 150), and our use of multiple LPS doses is consistent with the notion that repetitive doses are necessary to model a chronic inflammatory disorder, such as BPD (122, 140). Likewise, the concentration of oxygen used in this study was comparable to that used in previous studies (51, 52, 81, 138). Two-hit rodent models have used hyperoxia and postnatal LPS insults to phenotype experimental BPD (70, 77, 127); however, our study differs from these by limiting exposure to the saccular phase of lung development and elucidating the individual and combined effects of these exposures on pulmonary vascular function by echocardiography.
Initially, we determined the survival rates of mice exposed to LPS and hyperoxia. Consistent with our previous study, we observed a lower survival rate in mice treated with 6 or 10 mg/kg of LPS and exposed to normoxia (122), and we demonstrated that this effect was substantially greater in mice treated with 10 mg/kg of LPS and exposed to hyperoxia. Although we did not investigate the mechanisms of decreased survival, high LPS doses have been shown to increase mortality by damaging organs that are vital for cardiorespiratory function and survival, such as the brain, heart, and lungs (144). It is possible that the effects of high-dose LPS are exacerbated when concurrently exposed to hyperoxia, since the latter is also known to damage these vital organs (21, 94, 107, 115), or that high-dose LPS primes these organs for further damage by a second insult such as hyperoxia (141). Since decreased growth rates are a morbidity of sepsis and BPD in infants (2, 108), we quantified the growth rate of our experimental animals. LPS independently decreased growth rate, consistent with previous studies (19, 41, 122). Hyperoxia has been shown to decrease growth rate via various mechanisms in neonatal mouse models of experimental BPD (23, 45, 48, 65, 111); however, we may not have observed this effect due to the limited exposure duration in our study.
Inflammation is a necessary biological process via which the host eliminates pathogens, damaged cells, or irritants; however, it is important that immune homeostasis is restored once these harmful agents have been eliminated, which requires the balanced and coordinated signaling of the innate and adaptive immune systems. Failure to achieve homeostasis leads to several inflammation-mediated disorders (56), including BPD (11, 68, 114). Inflammatory stimuli such as infection, mechanical ventilation, and hyperoxia have been shown to disrupt growth factor signaling, extracellular matrix assembly, and cell proliferation in developing lungs and contribute to BPD pathogenesis (8, 24, 125, 131). In this study, we found that LPS and hyperoxia had different effects on the expression of inflammatory cytokines and chemokines. While LPS predominantly affected IL-1β mRNA expression, hyperoxia affected CCL2 and ICAM1 expression. Notably, IL-1β expression was lower in the low-dose LPS group and higher in the high-dose LPS group upon exposure to hyperoxia. IL-1 is a cytokine implicated in the pathogenesis of many acute and chronic inflammatory diseases, with elevated levels known to increase the risk of BPD in infants (5, 16, 20). Moreover, IL-1 has been directly implicated in the pathogenesis of experimental BPD (17, 83, 102, 112, 139). Bui et al. (18) recently demonstrated that the IL-1 receptor antagonist (IL-1Ra) reduces both the short- and long-term detrimental effects of neonatal hyperoxia on murine alveoli and lung vasculature, indicating that elevated IL-1β levels may be partly responsible for the BPD-PH phenotype observed in the high-dose LPS-treated mice exposed to hyperoxia in this study. Transcription factors such as STAT3 regulate cell proliferation during the development, injury, and repair of organs (39, 128, 130), as well as inflammation (59, 142), the biological processes that play a major role in the pathogenesis of BPD. For instance, STAT3 activation increases pulmonary vascularization (106), which is critical for healthy lung development. Therefore, we demonstrated the effects of LPS and hyperoxia on the expression and activation of STAT3, finding that early LPS and hyperoxia exposure activated STAT3 in the lungs, which was consistent with previous studies (73, 122, 143). We also observed that STAT3 activation is lower in LPS-treated mice under hyperoxic conditions than under normoxic conditions. The molecular mechanisms responsible for these differences are currently unclear and warrant further investigation in future studies, alongside the mechanistic role of STAT3 activation in LPS- and hyperoxia-induced neonatal lung injury. STAT6 activation is primarily known to protect murine lungs against several insults. For instance, Nepal et al. (100) elegantly demonstrated that STAT6 activation is necessary to clear inflammatory cells and resolve lung injury in an LPS-induced model of acute respiratory distress syndrome, while several other studies have also shown that STAT6 activation exerts anti-inflammatory effects and mediates the resolution of lung injury (30, 38, 119). Low LPS doses decreased STAT6 activation, while high LPS doses and hyperoxia did not affect STAT6 activation. LPS treatment may downregulate the Th2 response necessary for STAT6 activation (36), while low-dose LPS pretreatment has been shown to modulate lung immune homeostasis and prevent worsening lung injury when exposed to a second insult (116). Regardless, the extent of interrupted lung development was similar in both hyperoxia and low-dose LPS groups, indicating that low-dose LPS exposure impedes healthy lung development. Additionally, mice treated with high-dose LPS and exposed to hyperoxia have increased levels of IL-1β, an interleukin that is associated with an increased risk for BPD-PH.
Preterm infants are born with immature lungs and require therapies such as positive-pressure ventilatory support and supplemental oxygen to support their lung function; however, these life-saving therapies, along with postnatal sepsis, can disrupt lung maturation and cause BPD (10, 85). Sepsis is associated with an increased risk of BPD development in preterm infants (46, 66, 69, 74, 76, 88), while microbial products, such as LPS, have been shown to disrupt lung development in experimental animals (26, 27, 57, 90, 92, 122). Our findings are consistent with this notion. Moderate hyperoxia exposure during the saccular lung developmental phase also disrupts alveolarization in rodents (81, 148). In agreement with these studies, short-term hyperoxia exposure decreased alveolarization in our study. However, in contrast to these studies, we also observed that short-term hyperoxia reduces lung vascularization. This discrepancy may be due to the duration of hyperoxia exposure, which was 5 days in our study but only 4 days in the two previous studies. The discrepancy may also be due to the differences in the method used to quantify pulmonary vascularization. Leary et al. (81) determined the extent of vascularization by quantifying the intensity of vWF staining, whereas we analyzed vascularization by counting the actual number of vWF-positive blood vessels.
Combined insults are known to interact and potentiate the extent of interrupted lung development compared with single insults in experimental animals (70, 77, 127, 137). Similarly, we observed that our higher LPS dose (6 mg·kg−1·day−1) interacted with and augmented the experimental BPD phenotype induced by hyperoxia; however, the BPD phenotype did not worsen in mice treated with the lower LPS dose (3 mg·kg−1·day−1) upon hyperoxia exposure. There is evidence that strongly supports the vascular hypothesis of lung development, that is, that lung vascular and alveolar development are interdependent and that interrupted lung angiogenesis leads to alveolar simplification (1). Therefore, the synergistic effect of a high LPS dose and hyperoxia on lung vascular development may partly explain the severe BPD phenotype observed in our two-hit model. Many studies have investigated the VEGF and nitric oxide (NO) signaling pathways and demonstrated that they are necessary for lung development in health and disease in neonatal animals (49, 50, 61, 80, 125, 133, 145). VEGF has also been shown to restore alveolar and pulmonary vascular structure and function via the eNOS pathway in experimental BPD and PH (72, 84, 129). Endothelial nitric oxide synthase (eNOS) is the most important vasculoprotective enzyme among the three isoforms of nitric oxide synthases. Nitric oxide (NO) generated by eNOS mediates vasodilation (58), prevents vascular smooth muscle proliferation (47, 55, 97), and inhibits vascular inflammation by regulating the expression of proinflammatory adhesion molecules and cytokines (32, 33, 71). Further, disrupted eNOS signaling contributes to interrupted lung development and PH, suggesting that endogenous NO generated by eNOS is crucial to promote angiogenesis and alveolarization and regulate vascular tone and function (12, 13, 44, 82, 96, 117). Therefore, we examined the expression and activation of this pathway to understand the major mechanisms via which LPS and hyperoxia collectively regulate lung development. Consistent with a previous study (57), we found that high-dose LPS decreases VEGFR2 signaling under normoxic conditions. However, hyperoxia increased VEGFR2 and eNOS signaling in LPS-treated mice, particularly the low-dose LPS group, indicating that low-dose LPS exposure is associated with an adaptive response (e.g., increased VEGFR2 and eNOS signaling) that prevents further disruption to lung development upon exposure to hyperoxia. In contrast, the severe BPD phenotype observed in the high-dose LPS group exposed to hyperoxia suggests that this adaptive response may be insufficient to protect the lungs or that a different angiogenic pathway may be affected under these conditions. Further investigations are required to elucidate the molecular mechanisms underlying these findings.
Increased generation of reactive oxygen species (ROS) contributes to BPD pathogenesis by altering signal transduction pathways and modifying the structure and function of protein, lipids, and DNA (15, 113). NQO1 protects cells and tissues against oxidant injury induced by toxins (103) and hyperoxia (31), by conjugating and scavenging the reactive electrophiles and lipid peroxidation products generated by oxidant stress (25). Similarly, HO-1 exerts antioxidant effects by modulating the expression of the prooxidant molecule, heme (104), and is necessary and sufficient to protect against experimental BPD in rodents (42, 147). Therefore, the increased HO-1 protein expression observed in the vehicle-treated mice exposed to hyperoxia in this study may be an adaptive response toward decreasing ROS. The reason for the lack of a similar response in LPS-treated mice exposed to hyperoxia remains unclear and warrants further study. It is possible that exposure to LPS is associated with either decreased production or augmented elimination of ROS or both in the context of hyperoxic lung injury. It is also important to note that quantifying the antioxidant enzymes at 14 d may not truly reflect an acute response to either LPS and/or hyperoxia.
To determine the molecular mechanisms that disrupt lung development, we demonstrated the effects of early LPS and hyperoxia exposure on apoptosis and cell proliferation. Hyperoxia primarily modulates apoptosis via the intrinsic or Bcl-2-mediated pathway (87, 99, 146), whereas LPS regulates cell death via the extrinsic or receptor-mediated pathway (22). Our findings are consistent with these studies and show that extrinsic pathway activation is attenuated when LPS-treated animals are exposed to hyperoxia, with animals treated with low LPS doses protected against hyperoxia-induced apoptosis. To understand this phenomenon further, we determined the expression and activation of Akt, a serine/threonine kinase, which plays a crucial role in regulating cellular apoptosis and proliferation (101). Activation of Akt is necessary for healthy lung development and is sufficient to decrease apoptosis of alveolar epithelial cells and mitigate alveolar simplification in hyperoxia-induced developmental lung injury (4). Akt activation also mitigates hyperoxia-induced cell death via mitochondrial mechanisms in human pulmonary microvascular endothelial cells, which are necessary for healthy lung development (3). We observed that Akt activation might be partly responsible for the attenuated apoptosis observed in the low-dose LPS group exposed to hyperoxia. The low-dose LPS group under hyperoxic conditions also displayed other adaptive responses such as an increase in the protein expression of PCNA, a protein that facilitates cell proliferation and lung development. The molecular mechanisms leading to these adaptive responses are poorly understood and warrants further investigation. Finally, we determined the effects of LPS and hyperoxia on lung vascular function and vascular remodeling. Cardiac catheterization is the gold standard for PH diagnosis; however, echocardiography is being increasingly used to phenotype PH in mice due to the technical difficulty of obtaining accurate pulmonary arterial (PA) pressure by cardiac catheterization in neonatal mice. There are several advantages to using echocardiography. For instance, it is a noninvasive, cost effective, and rapid technique. It is also ideal for follow-up studies in mice. Moreover, the RV systolic time intervals, such as PAT and PAT/ET ratio, estimated using high-frequency echocardiography closely correlate with PA pressures measured by cardiac catheterization and, thus, can be used as alternative PA pressure indices in rodents (110, 134, 135). Since increased PA pressures cause the pulmonary valve to close prematurely and decrease the PAT and PAT/ET ratio, our findings suggest that the PH phenotype is only induced when exposed to a combination of LPS and hyperoxia. Similarly, pulmonary vascular remodeling was induced only in the group exposed to both LPS and hyperoxia. However, the PAT was not decreased and the RV/LV free wall thickness ratio was not increased in any of our experimental groups, indicating that PH was not severe in our model. In addition, moderate hyperoxia for a short duration failed to induce PH. Although several previous studies, including ours, have demonstrated that hyperoxia independently induces PH in rodents (37, 51, 75, 81, 93, 110), the oxygen concentration was higher or the duration of oxygen exposure was longer in these studies. Importantly, there are certain limitations to echocardiography studies. They are operator-dependent, and it is challenging to obtain an optimal acoustic window in small animals. Additionally, echocardiography studies rely on geometric assumptions that can be altered in diseased states (109). Therefore, it is possible the echocardiography studies may have underestimated the true incidence of PH in our experimental mice. Cardiac magnetic resonance imaging (cMRI) studies, which are less operator dependent and do not rely on geometric assumptions, can address some of the limitations of echocardiography and provide a more accurate incidence of PH. However, cMRI studies are expensive and time-consuming.
The main strength of our study is that we examined the isolated and combined effects of repetitive LPS (chronic inflammatory state) and moderate hyperoxia during the saccular phase of lung development to model the clinical scenario of preterm infants. Furthermore, we used high-resolution echocardiography to assess the functional effect of LPS and hyperoxia on pulmonary vascular function. However, our study has several limitations, which we plan to address in future studies. For instance, our study is descriptive rather than mechanistic in nature, showing an association rather than a direct cause and effect between the combined hyperoxia and LPS exposure and the severity of experimental BPD-PH. Furthermore, we did not examine sex- or cell-specific effects of LPS and hyperoxia in the developing lungs. Additionally, we did not measure lung function in our experimental animals. Finally, we did not determine the long-term interactive effects of neonatal hyperoxia and LPS exposure on the lungs and hearts.
In conclusion, this study elucidates the effects of LPS and hyperoxia exposure on saccular lungs and identifies potential mechanisms via which these exposures disrupt lung development and induce a BPD-PH phenotype. Specifically, we demonstrated that in murine models utilizing short-term exposure to moderate hyperoxia, an additional insult such as LPS might be necessary to recapitulate human BPD-PH. Furthermore, we showed that pretreatment with low-dose LPS induces several adaptive mechanisms and prevents worsening lung injury upon exposure to hyperoxia. Finally, we identified molecular and biological processes such as increased IL-1β and pulmonary vascular remodeling, respectively, that are exclusive to combined LPS and hyperoxia exposure.
GRANTS
This work was supported by NIH Grants (HL139594 to B.S., U54 HG006348 to the Mouse Phenotyping Core at Baylor College of Medicine (BCM), and P30DK056338 to the Digestive Disease Center Core at the BCM), and Texas Children’s Hospital Pediatric Pilot Award (to B.S.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.K.S., R.T.M., R.B., C.R. and B.S. conceived and designed research; A.K.S., R.T.M., C.R. and B.S. performed experiments; A.K.S., R.T.M., A.E.S., R.B., C.R. and B.S. analyzed data; A.K.S., R.T.M., A.E.S., R.B., C.R. and B.S. interpreted results of experiments; A.K.S., R.T.M., A.E.S., C.R. and B.S. prepared figures; A.K.S., R.T.M. and B.S. drafted manuscript; B.S. edited and revised manuscript; A.K.S., R.T.M., A.E.S., R.B., C.R. and B.S. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Pamela Parsons for timely processing of histopathology slides.
REFERENCES
- 1.Abman SH. Bronchopulmonary dysplasia: “a vascular hypothesis”. Am J Respir Crit Care Med 164: 1755–1756, 2001. doi: 10.1164/ajrccm.164.10.2109111c. [DOI] [PubMed] [Google Scholar]
- 2.Abman SH, Collaco JM, Shepherd EG, Keszler M, Cuevas-Guaman M, Welty SE, Truog WE, McGrath-Morrow SA, Moore PE, Rhein LM, Kirpalani H, Zhang H, Gratny LL, Lynch SK, Curtiss J, Stonestreet BS, McKinney RL, Dysart KC, Gien J, Baker CD, Donohue PK, Austin E, Fike C, Nelin LD; Bronchopulmonary Dysplasia Collaborative . Interdisciplinary care of children with severe bronchopulmonary dysplasia. J Pediatr 181: 12–28.e1, 2017. doi: 10.1016/j.jpeds.2016.10.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ahmad A, Ahmad S, Chang LY, Schaack J, White CW. Endothelial Akt activation by hyperoxia: role in cell survival. Free Radic Biol Med 40: 1108–1118, 2006. doi: 10.1016/j.freeradbiomed.2005.10.045. [DOI] [PubMed] [Google Scholar]
- 4.Alphonse RS, Vadivel A, Coltan L, Eaton F, Barr AJ, Dyck JR, Thébaud B. Activation of Akt protects alveoli from neonatal oxygen-induced lung injury. Am J Respir Cell Mol Biol 44: 146–154, 2011. doi: 10.1165/rcmb.2009-0182OC. [DOI] [PubMed] [Google Scholar]
- 5.Ambalavanan N, Carlo WA, D’Angio CT, McDonald SA, Das A, Schendel D, Thorsen P, Higgins RD; Eunice Kennedy Shriver National Institute of Child Health and Human Development Neonatal Research Network . Cytokines associated with bronchopulmonary dysplasia or death in extremely low birth weight infants. Pediatrics 123: 1132–1141, 2009. doi: 10.1542/peds.2008-0526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ambalavanan N, Nicola T, Hagood J, Bulger A, Serra R, Murphy-Ullrich J, Oparil S, Chen YF. Transforming growth factor-β signaling mediates hypoxia-induced pulmonary arterial remodeling and inhibition of alveolar development in newborn mouse lung. Am J Physiol Lung Cell Mol Physiol 295: L86–L95, 2008. doi: 10.1152/ajplung.00534.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arjaans S, Zwart EAH, Ploegstra MJ, Bos AF, Kooi EMW, Hillege HL, Berger RMF. Identification of gaps in the current knowledge on pulmonary hypertension in extremely preterm infants: A systematic review and meta-analysis. Paediatr Perinat Epidemiol 32: 258–267, 2018. doi: 10.1111/ppe.12444. [DOI] [PubMed] [Google Scholar]
- 8.Aslam M, Baveja R, Liang OD, Fernandez-Gonzalez A, Lee C, Mitsialis SA, Kourembanas S. Bone marrow stromal cells attenuate lung injury in a murine model of neonatal chronic lung disease. Am J Respir Crit Care Med 180: 1122–1130, 2009. doi: 10.1164/rccm.200902-0242OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baker CD, Abman SH, Mourani PM. Pulmonary hypertension in preterm infants with bronchopulmonary dysplasia. Pediatr Allergy Immunol Pulmonol 27: 8–16, 2014. doi: 10.1089/ped.2013.0323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baker CD, Alvira CM. Disrupted lung development and bronchopulmonary dysplasia: opportunities for lung repair and regeneration. Curr Opin Pediatr 26: 306–314, 2014. doi: 10.1097/MOP.0000000000000095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balany J, Bhandari V. Understanding the impact of infection, inflammation, and their persistence in the pathogenesis of bronchopulmonary dysplasia. Front Med (Lausanne) 2: 90, 2015. doi: 10.3389/fmed.2015.00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Balasubramaniam V, Maxey A, Abman S. Inhaled nitric oxide reverses hypoxia induced lung hypoplasia in endothelial nitric oxide synthase-deficient mice. Chest 128, Suppl: 613S–614S, 2005. doi: 10.1378/chest.128.6_suppl.613S. [DOI] [PubMed] [Google Scholar]
- 13.Balasubramaniam V, Maxey AM, Morgan DB, Markham NE, Abman SH. Inhaled NO restores lung structure in eNOS-deficient mice recovering from neonatal hypoxia. Am J Physiol Lung Cell Mol Physiol 291: L119–L127, 2006. doi: 10.1152/ajplung.00395.2005. [DOI] [PubMed] [Google Scholar]
- 14.Berkelhamer SK, Mestan KK, Steinhorn R. An update on the diagnosis and management of bronchopulmonary dysplasia (BPD)-associated pulmonary hypertension. Semin Perinatol 42: 432–443, 2018. doi: 10.1053/j.semperi.2018.09.005. [DOI] [PubMed] [Google Scholar]
- 15.Bhandari V. Hyperoxia-derived lung damage in preterm infants. Semin Fetal Neonatal Med 15: 223–229, 2010. doi: 10.1016/j.siny.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bose CL, Dammann CE, Laughon MM. Bronchopulmonary dysplasia and inflammatory biomarkers in the premature neonate. Arch Dis Child Fetal Neonatal Ed 93: F455–F461, 2008. doi: 10.1136/adc.2007.121327. [DOI] [PubMed] [Google Scholar]
- 17.Bry K, Whitsett JA, Lappalainen U. IL-1β disrupts postnatal lung morphogenesis in the mouse. Am J Respir Cell Mol Biol 36: 32–42, 2007. doi: 10.1165/rcmb.2006-0116OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bui CB, Kolodziej M, Lamanna E, Elgass K, Sehgal A, Rudloff I, Schwenke DO, Tsuchimochi H, Kroon MAGM, Cho SX, Maksimenko A, Cholewa M, Berger PJ, Young MJ, Bourke JE, Pearson JT, Nold MF, Nold-Petry CA. Interleukin-1 receptor antagonist protects newborn mice against pulmonary hypertension. Front Immunol 10: 1480, 2019. doi: 10.3389/fimmu.2019.01480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cardoso FL, Herz J, Fernandes A, Rocha J, Sepodes B, Brito MA, McGavern DB, Brites D. Systemic inflammation in early neonatal mice induces transient and lasting neurodegenerative effects. J Neuroinflammation 12: 82, 2015. doi: 10.1186/s12974-015-0299-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cayabyab RG, Jones CA, Kwong KY, Hendershott C, Lecart C, Minoo P, Ramanathan R. Interleukin-1beta in the bronchoalveolar lavage fluid of premature neonates: a marker for maternal chorioamnionitis and predictor of adverse neonatal outcome. J Matern Fetal Neonatal Med 14: 205–211, 2003. doi: 10.1080/jmf.14.3.205.211. [DOI] [PubMed] [Google Scholar]
- 21.Chang JL, Bashir M, Santiago C, Farrow K, Fung C, Brown AS, Dettman RW, Dizon MLV. Intrauterine growth restriction and hyperoxia as a cause of white matter injury. Dev Neurosci 40: 344–357, 2018. doi: 10.1159/000494273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chen G, Goeddel DV. TNF-R1 signaling: a beautiful pathway. Science 296: 1634–1635, 2002. doi: 10.1126/science.1071924. [DOI] [PubMed] [Google Scholar]
- 23.Chen HJ, Chiang BL. Effect of hyperoxia on retinoid metabolism and retinoid receptor expression in the lungs of newborn mice. PLoS One 10: e0140343, 2015. doi: 10.1371/journal.pone.0140343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen S, Rong M, Platteau A, Hehre D, Smith H, Ruiz P, Whitsett J, Bancalari E, Wu S. CTGF disrupts alveolarization and induces pulmonary hypertension in neonatal mice: implication in the pathogenesis of severe bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 300: L330–L340, 2011. doi: 10.1152/ajplung.00270.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cho HY, Jedlicka AE, Reddy SP, Kensler TW, Yamamoto M, Zhang LY, Kleeberger SR. Role of NRF2 in protection against hyperoxic lung injury in mice. Am J Respir Cell Mol Biol 26: 175–182, 2002. doi: 10.1165/ajrcmb.26.2.4501. [DOI] [PubMed] [Google Scholar]
- 26.Choi CW, Lee J, Oh JY, Lee SH, Lee HJ, Kim BI. Protective effect of chorioamnionitis on the development of bronchopulmonary dysplasia triggered by postnatal systemic inflammation in neonatal rats. Pediatr Res 79: 287–294, 2016. doi: 10.1038/pr.2015.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Collins JJ, Kuypers E, Nitsos I, Jane Pillow J, Polglase GR, Kemp MW, Newnham JP, Cleutjens JP, Frints SG, Kallapur SG, Jobe AH, Kramer BW. LPS-induced chorioamnionitis and antenatal corticosteroids modulate Shh signaling in the ovine fetal lung. Am J Physiol Lung Cell Mol Physiol 303: L778–L787, 2012. doi: 10.1152/ajplung.00280.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cooney TP, Thurlbeck WM. The radial alveolar count method of Emery and Mithal: a reappraisal 1—postnatal lung growth. Thorax 37: 572–579, 1982. doi: 10.1136/thx.37.8.572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Critser PJ, Higano NS, Tkach JA, Olson ES, Spielberg DR, Kingma PS, Fleck RJ, Lang SM, Moore RA, Taylor MD, Woods JC. Cardiac magnetic resonance imaging evaluation of neonatal bronchopulmonary dysplasia–associated pulmonary hypertension. Am J Respir Crit Care Med 201: 73–82, 2020. doi: 10.1164/rccm.201904-0826OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.D’Alessio FR, Craig JM, Singer BD, Files DC, Mock JR, Garibaldi BT, Fallica J, Tripathi A, Mandke P, Gans JH, Limjunyawong N, Sidhaye VK, Heller NM, Mitzner W, King LS, Aggarwal NR. Enhanced resolution of experimental ARDS through IL-4-mediated lung macrophage reprogramming. Am J Physiol Lung Cell Mol Physiol 310: L733–L746, 2016. doi: 10.1152/ajplung.00419.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Das A, Kole L, Wang L, Barrios R, Moorthy B, Jaiswal AK. BALT development and augmentation of hyperoxic lung injury in mice deficient in NQO1 and NQO2. Free Radic Biol Med 40: 1843–1856, 2006. doi: 10.1016/j.freeradbiomed.2006.01.025. [DOI] [PubMed] [Google Scholar]
- 32.Davenpeck KL, Gauthier TW, Lefer AM. Inhibition of endothelial-derived nitric oxide promotes P-selectin expression and actions in the rat microcirculation. Gastroenterology 107: 1050–1058, 1994. doi: 10.1016/0016-5085(94)90229-1. [DOI] [PubMed] [Google Scholar]
- 33.De Caterina R, Libby P, Peng HB, Thannickal VJ, Rajavashisth TB, Gimbrone MA Jr, Shin WS, Liao JK. Nitric oxide decreases cytokine-induced endothelial activation. Nitric oxide selectively reduces endothelial expression of adhesion molecules and proinflammatory cytokines. J Clin Invest 96: 60–68, 1995. doi: 10.1172/JCI118074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.de Visser YP, Walther FJ, Laghmani H, Boersma H, van der Laarse A, Wagenaar GT. Sildenafil attenuates pulmonary inflammation and fibrin deposition, mortality and right ventricular hypertrophy in neonatal hyperoxic lung injury. Respir Res 10: 30, 2009. doi: 10.1186/1465-9921-10-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.de Visser YP, Walther FJ, Laghmani H, Steendijk P, Middeldorp M, van der Laarse A, Wagenaar GT. Phosphodiesterase 4 inhibition attenuates persistent heart and lung injury by neonatal hyperoxia in rats. Am J Physiol Lung Cell Mol Physiol 302: L56–L67, 2012. doi: 10.1152/ajplung.00041.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ding F, Liu B, Zou W, Tian D, Li Q, Dai J, Luo Z, Fu Z. LPS exposure in early life protects against mucus hypersecretion in ovalbumin-induced asthma by down-regulation of the IL-13 and JAK-STAT6 pathways. Cell Physiol Biochem 46: 1263–1274, 2018. doi: 10.1159/000489109. [DOI] [PubMed] [Google Scholar]
- 37.Dolma K, Freeman AE, Rezonzew G, Payne GA, Xu X, Jilling T, Blalock JE, Gaggar A, Ambalavanan N, Lal CV. Effects of hyperoxia on alveolar and pulmonary vascular development in germ-free mice. Am J Physiol Lung Cell Mol Physiol 318: L421–L428, 2020. doi: 10.1152/ajplung.00316.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duan JX, Zhou Y, Zhou AY, Guan XX, Liu T, Yang HH, Xie H, Chen P. Calcitonin gene-related peptide exerts anti-inflammatory property through regulating murine macrophages polarization in vitro. Mol Immunol 91: 105–113, 2017. doi: 10.1016/j.molimm.2017.08.020. [DOI] [PubMed] [Google Scholar]
- 39.Dutzmann J, Daniel JM, Bauersachs J, Hilfiker-Kleiner D, Sedding DG. Emerging translational approaches to target STAT3 signalling and its impact on vascular disease. Cardiovasc Res 106: 365–374, 2015. doi: 10.1093/cvr/cvv103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Erickson MA, Hartvigson PE, Morofuji Y, Owen JB, Butterfield DA, Banks WA. Lipopolysaccharide impairs amyloid β efflux from brain: altered vascular sequestration, cerebrospinal fluid reabsorption, peripheral clearance and transporter function at the blood-brain barrier. J Neuroinflammation 9: 150, 2012. doi: 10.1186/1742-2094-9-150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fan LW, Kaizaki A, Tien LT, Pang Y, Tanaka S, Numazawa S, Bhatt AJ, Cai Z. Celecoxib attenuates systemic lipopolysaccharide-induced brain inflammation and white matter injury in the neonatal rats. Neuroscience 240: 27–38, 2013. doi: 10.1016/j.neuroscience.2013.02.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fernandez-Gonzalez A, Alex Mitsialis S, Liu X, Kourembanas S. Vasculoprotective effects of heme oxygenase-1 in a murine model of hyperoxia-induced bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 302: L775–L784, 2012. doi: 10.1152/ajplung.00196.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fink MP. Animal models of sepsis. Virulence 5: 143–153, 2014. doi: 10.4161/viru.26083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Förstermann U, Münzel T. Endothelial nitric oxide synthase in vascular disease: from marvel to menace. Circulation 113: 1708–1714, 2006. doi: 10.1161/CIRCULATIONAHA.105.602532. [DOI] [PubMed] [Google Scholar]
- 45.Fritzell JA Jr, Mao Q, Gundavarapu S, Pasquariello T, Aliotta JM, Ayala A, Padbury JF, De Paepe ME. Fate and effects of adult bone marrow cells in lungs of normoxic and hyperoxic newborn mice. Am J Respir Cell Mol Biol 40: 575–587, 2009. doi: 10.1165/rcmb.2008-0176OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garcia-Munoz Rodrigo F, Urquia Marti L, Galan Henriquez G, Rivero Rodriguez S, Figueras-Aloy J, Vento M. Intercenter variability and factors associated with survival without bronchopulmonary dysplasia in extremely preterm newborns. J Matern Fetal Neonatal Med 33: 1–8, 2020. doi: 10.1080/14767058.2019.1585423. [DOI] [PubMed] [Google Scholar]
- 47.Garg UC, Hassid A. Nitric oxide-generating vasodilators and 8-bromo-cyclic guanosine monophosphate inhibit mitogenesis and proliferation of cultured rat vascular smooth muscle cells. J Clin Invest 83: 1774–1777, 1989. doi: 10.1172/JCI114081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gortner L, Monz D, Mildau C, Shen J, Kasoha M, Laschke MW, Roolfs T, Schmiedl A, Meier C, Tutdibi E. Bronchopulmonary dysplasia in a double-hit mouse model induced by intrauterine hypoxia and postnatal hyperoxia: closer to clinical features? Ann Anat 195: 351–358, 2013. doi: 10.1016/j.aanat.2013.02.010. [DOI] [PubMed] [Google Scholar]
- 49.Han RN, Babaei S, Robb M, Lee T, Ridsdale R, Ackerley C, Post M, Stewart DJ. Defective lung vascular development and fatal respiratory distress in endothelial NO synthase-deficient mice: a model of alveolar capillary dysplasia? Circ Res 94: 1115–1123, 2004. doi: 10.1161/01.RES.0000125624.85852.1E. [DOI] [PubMed] [Google Scholar]
- 50.Han RN, Stewart DJ. Defective lung vascular development in endothelial nitric oxide synthase-deficient mice. Trends Cardiovasc Med 16: 29–34, 2006. doi: 10.1016/j.tcm.2005.11.004. [DOI] [PubMed] [Google Scholar]
- 51.Hansmann G, Fernandez-Gonzalez A, Aslam M, Vitali SH, Martin T, Mitsialis SA, Kourembanas S. Mesenchymal stem cell-mediated reversal of bronchopulmonary dysplasia and associated pulmonary hypertension. Pulm Circ 2: 170–181, 2012. doi: 10.4103/2045-8932.97603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Heilman RP, Lagoski MB, Lee KJ, Taylor JM, Kim GA, Berkelhamer SK, Steinhorn RH, Farrow KN. Right ventricular cyclic nucleotide signaling is decreased in hyperoxia-induced pulmonary hypertension in neonatal mice. Am J Physiol Heart Circ Physiol 308: H1575–H1582, 2015. doi: 10.1152/ajpheart.00569.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Heine H, Rietschel ET, Ulmer AJ. The biology of endotoxin. Mol Biotechnol 19: 279–296, 2001. doi: 10.1385/MB:19:3:279. [DOI] [PubMed] [Google Scholar]
- 54.Higgins RD, Jobe AH, Koso-Thomas M, Bancalari E, Viscardi RM, Hartert TV, Ryan RM, Kallapur SG, Steinhorn RH, Konduri GG, Davis SD, Thebaud B, Clyman RI, Collaco JM, Martin CR, Woods JC, Finer NN, Raju TNK. Bronchopulmonary dysplasia: executive summary of a workshop. J Pediatr 197: 300–308, 2018. doi: 10.1016/j.jpeds.2018.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hogan M, Cerami A, Bucala R. Advanced glycosylation endproducts block the antiproliferative effect of nitric oxide. Role in the vascular and renal complications of diabetes mellitus. J Clin Invest 90: 1110–1115, 1992. doi: 10.1172/JCI115928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hotchkiss RS, Monneret G, Payen D. Sepsis-induced immunosuppression: from cellular dysfunctions to immunotherapy. Nat Rev Immunol 13: 862–874, 2013. doi: 10.1038/nri3552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hou Y, Liu M, Husted C, Chen C, Thiagarajan K, Johns JL, Rao SP, Alvira CM. Activation of the nuclear factor-κB pathway during postnatal lung inflammation preserves alveolarization by suppressing macrophage inflammatory protein-2. Am J Physiol Lung Cell Mol Physiol 309: L593–L604, 2015. doi: 10.1152/ajplung.00029.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huang PL, Huang Z, Mashimo H, Bloch KD, Moskowitz MA, Bevan JA, Fishman MC. Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 377: 239–242, 1995. doi: 10.1038/377239a0. [DOI] [PubMed] [Google Scholar]
- 59.Ikegami M, Whitsett JA, Martis PC, Weaver TE. Reversibility of lung inflammation caused by SP-B deficiency. Am J Physiol Lung Cell Mol Physiol 289: L962–L970, 2005. doi: 10.1152/ajplung.00214.2005. [DOI] [PubMed] [Google Scholar]
- 60.Islam JY, Keller RL, Aschner JL, Hartert TV, Moore PE. Understanding the short- and long-term respiratory outcomes of prematurity and bronchopulmonary dysplasia. Am J Respir Crit Care Med 192: 134–156, 2015. doi: 10.1164/rccm.201412-2142PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jakkula M, Le Cras TD, Gebb S, Hirth KP, Tuder RM, Voelkel NF, Abman SH. Inhibition of angiogenesis decreases alveolarization in the developing rat lung. Am J Physiol Lung Cell Mol Physiol 279: L600–L607, 2000. doi: 10.1152/ajplung.2000.279.3.L600. [DOI] [PubMed] [Google Scholar]
- 62.Jobe AH. Animal models, learning lessons to prevent and treat neonatal chronic lung disease. Front Med (Lausanne) 2: 49, 2015. doi: 10.3389/fmed.2015.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jobe AJ. The new BPD: an arrest of lung development. Pediatr Res 46: 641–643, 1999. doi: 10.1203/00006450-199912000-00007. [DOI] [PubMed] [Google Scholar]
- 64.Johnson TJ, Patel AL, Jegier BJ, Engstrom JL, Meier PP. Cost of morbidities in very low birth weight infants. J Pediatr 162: 243–49.e1, 2013. doi: 10.1016/j.jpeds.2012.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jones CV, Alikhan MA, O’Reilly M, Sozo F, Williams TM, Harding R, Jenkin G, Ricardo SD. The effect of CSF-1 administration on lung maturation in a mouse model of neonatal hyperoxia exposure. Respir Res 15: 110, 2014. doi: 10.1186/s12931-014-0110-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jung E, Lee BS. Late-onset sepsis as a risk factor for bronchopulmonary dysplasia in extremely low birth weight infants: a nationwide cohort study. Sci Rep 9: 15448, 2019. doi: 10.1038/s41598-019-51617-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kalikkot Thekkeveedu R, Guaman MC, Shivanna B. Bronchopulmonary dysplasia: A review of pathogenesis and pathophysiology. Respir Med 132: 170–177, 2017. doi: 10.1016/j.rmed.2017.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kallapur SG, Jobe AH. Contribution of inflammation to lung injury and development. Arch Dis Child Fetal Neonatal Ed 91: F132–F135, 2006. doi: 10.1136/adc.2004.068544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Klinger G, Levy I, Sirota L, Boyko V, Lerner-Geva L, Reichman B; Israel Neonatal Network . Outcome of early-onset sepsis in a national cohort of very low birth weight infants. Pediatrics 125: e736–e740, 2010. doi: 10.1542/peds.2009-2017. [DOI] [PubMed] [Google Scholar]
- 70.Kroon AA, Wang J, Huang Z, Cao L, Kuliszewski M, Post M. Inflammatory response to oxygen and endotoxin in newborn rat lung ventilated with low tidal volume. Pediatr Res 68: 63–69, 2010. doi: 10.1203/PDR.0b013e3181e17caa. [DOI] [PubMed] [Google Scholar]
- 71.Kubes P, Suzuki M, Granger DN. Nitric oxide: an endogenous modulator of leukocyte adhesion. Proc Natl Acad Sci USA 88: 4651–4655, 1991. doi: 10.1073/pnas.88.11.4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Kunig AM, Balasubramaniam V, Markham NE, Seedorf G, Gien J, Abman SH. Recombinant human VEGF treatment transiently increases lung edema but enhances lung structure after neonatal hyperoxia. Am J Physiol Lung Cell Mol Physiol 291: L1068–L1078, 2006. doi: 10.1152/ajplung.00093.2006. [DOI] [PubMed] [Google Scholar]
- 73.Kunzmann S, Collins JJ, Yang Y, Uhlig S, Kallapur SG, Speer CP, Jobe AH, Kramer BW. Antenatal inflammation reduces expression of caveolin-1 and influences multiple signaling pathways in preterm fetal lungs. Am J Respir Cell Mol Biol 45: 969–976, 2011. doi: 10.1165/rcmb.2010-0519OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kurata H, Ochiai M, Inoue H, Ichiyama M, Yasuoka K, Fujiyoshi J, Matsushita Y, Honjo S, Sakai Y, Ohga S; Neonatal Research Network of Japan . A nationwide survey on tracheostomy for very-low-birth-weight infants in Japan. Pediatr Pulmonol 54: 53–60, 2019. doi: 10.1002/ppul.24200. [DOI] [PubMed] [Google Scholar]
- 75.Ladha F, Bonnet S, Eaton F, Hashimoto K, Korbutt G, Thébaud B. Sildenafil improves alveolar growth and pulmonary hypertension in hyperoxia-induced lung injury. Am J Respir Crit Care Med 172: 750–756, 2005. doi: 10.1164/rccm.200503-510OC. [DOI] [PubMed] [Google Scholar]
- 76.Lahra MM, Beeby PJ, Jeffery HE. Intrauterine inflammation, neonatal sepsis, and chronic lung disease: a 13-year hospital cohort study. Pediatrics 123: 1314–1319, 2009. doi: 10.1542/peds.2008-0656. [DOI] [PubMed] [Google Scholar]
- 77.Lal CV, Olave N, Travers C, Rezonzew G, Dolma K, Simpson A, Halloran B, Aghai Z, Das P, Sharma N, Xu X, Genschmer K, Russell D, Szul T, Yi N, Blalock JE, Gaggar A, Bhandari V, Ambalavanan N. Exosomal microRNA predicts and protects against severe bronchopulmonary dysplasia in extremely premature infants. JCI Insight 3: e93994, 2018. doi: 10.1172/jci.insight.93994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lal CV, Travers C, Aghai ZH, Eipers P, Jilling T, Halloran B, Carlo WA, Keeley J, Rezonzew G, Kumar R, Morrow C, Bhandari V, Ambalavanan N. The airway microbiome at birth. Sci Rep 6: 31023, 2016. doi: 10.1038/srep31023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lapcharoensap W, Bennett MV, Xu X, Lee HC, Dukhovny D. Hospitalization costs associated with bronchopulmonary dysplasia in the first year of life. J Perinatol 40: 130–137, 2019. doi: 10.1038/s41372-019-0548-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Le Cras TD, Markham NE, Tuder RM, Voelkel NF, Abman SH. Treatment of newborn rats with a VEGF receptor inhibitor causes pulmonary hypertension and abnormal lung structure. Am J Physiol Lung Cell Mol Physiol 283: L555–L562, 2002. doi: 10.1152/ajplung.00408.2001. [DOI] [PubMed] [Google Scholar]
- 81.Leary S, Das P, Ponnalagu D, Singh H, Bhandari V. Genetic strain and sex differences in a hperoxia-induced mouse model of varying severity of bronchopulmonary dysplasia. Am J Pathol 189: 999–1014, 2019. doi: 10.1016/j.ajpath.2019.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Leuwerke SM, Kaza AK, Tribble CG, Kron IL, Laubach VE. Inhibition of compensatory lung growth in endothelial nitric oxide synthase-deficient mice. Am J Physiol Lung Cell Mol Physiol 282: L1272–L1278, 2002. doi: 10.1152/ajplung.00490.2001. [DOI] [PubMed] [Google Scholar]
- 83.Liao J, Kapadia VS, Brown LS, Cheong N, Longoria C, Mija D, Ramgopal M, Mirpuri J, McCurnin DC, Savani RC. The NLRP3 inflammasome is critically involved in the development of bronchopulmonary dysplasia. Nat Commun 6: 8977, 2015. doi: 10.1038/ncomms9977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lin YJ, Markham NE, Balasubramaniam V, Tang JR, Maxey A, Kinsella JP, Abman SH. Inhaled nitric oxide enhances distal lung growth after exposure to hyperoxia in neonatal rats. Pediatr Res 58: 22–29, 2005. doi: 10.1203/01.PDR.0000163378.94837.3E. [DOI] [PubMed] [Google Scholar]
- 85.Madurga A, Mizíková I, Ruiz-Camp J, Morty RE. Recent advances in late lung development and the pathogenesis of bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 305: L893–L905, 2013. doi: 10.1152/ajplung.00267.2013. [DOI] [PubMed] [Google Scholar]
- 86.Männel DN. Advances in sepsis research derived from animal models. Int J Med Microbiol 297: 393–400, 2007. doi: 10.1016/j.ijmm.2007.03.005. [DOI] [PubMed] [Google Scholar]
- 87.Mao Q, Gundavarapu S, Patel C, Tsai A, Luks FI, De Paepe ME. The Fas system confers protection against alveolar disruption in hyperoxia-exposed newborn mice. Am J Respir Cell Mol Biol 39: 717–729, 2008. doi: 10.1165/rcmb.2008-0052OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Marshall DD, Kotelchuck M, Young TE, Bose CL, Kruyer L, O’Shea TM; North Carolina Neonatologists Association . Risk factors for chronic lung disease in the surfactant era: a North Carolina population-based study of very low birth weight infants. Pediatrics 104: 1345–1350, 1999. doi: 10.1542/peds.104.6.1345. [DOI] [PubMed] [Google Scholar]
- 89.McEvoy CT, Jain L, Schmidt B, Abman S, Bancalari E, Aschner JL. Bronchopulmonary dysplasia: NHLBI Workshop on the Primary Prevention of Chronic Lung Diseases. Ann Am Thorac Soc 11, Suppl 3: S146–S153, 2014. doi: 10.1513/AnnalsATS.201312-424LD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Menden H, Xia S, Mabry SM, Noel-MacDonnell J, Rajasingh J, Ye SQ, Sampath V. Histone deacetylase 6 regulates endothelial MyD88-dependent canonical TLR signaling, lung inflammation, and alveolar remodeling in the developing lung. Am J Physiol Lung Cell Mol Physiol 317: L332–L346, 2019. doi: 10.1152/ajplung.00247.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Menden HL, Xia S, Mabry SM, Navarro A, Nyp MF, Sampath V. Nicotinamide adenine dinucleotide phosphate oxidase 2 regulates LPS-induced inflammation and alveolar remodeling in the developing lung. Am J Respir Cell Mol Biol 55: 767–778, 2016. doi: 10.1165/rcmb.2016-0006OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Menon RT, Shrestha AK, Reynolds CL, Barrios R, Shivanna B. Long-term pulmonary and cardiovascular morbidities of neonatal hyperoxia exposure in mice. Int J Biochem Cell Biol 94: 119–124, 2018. doi: 10.1016/j.biocel.2017.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mian MOR, He Y, Bertagnolli M, Mai-Vo TA, Fernandes RO, Boudreau F, Cloutier A, Luu TM, Nuyt AM. TLR (Toll-like receptor) 4 antagonism prevents left ventricular hypertrophy and dysfunction caused by neonatal hyperoxia exposure in rats. Hypertension 74: 843–853, 2019. doi: 10.1161/HYPERTENSIONAHA.119.13022. [DOI] [PubMed] [Google Scholar]
- 95.Mourani PM, Abman SH. Pulmonary hypertension and vascular abnormalities in bronchopulmonary dysplasia. Clin Perinatol 42: 839–855, 2015. doi: 10.1016/j.clp.2015.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Murohara T, Asahara T, Silver M, Bauters C, Masuda H, Kalka C, Kearney M, Chen D, Symes JF, Fishman MC, Huang PL, Isner JM. Nitric oxide synthase modulates angiogenesis in response to tissue ischemia. J Clin Invest 101: 2567–2578, 1998. doi: 10.1172/JCI1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nakaki T, Nakayama M, Kato R. Inhibition by nitric oxide and nitric oxide-producing vasodilators of DNA synthesis in vascular smooth muscle cells. Eur J Pharmacol 189: 347–353, 1990. doi: 10.1016/0922-4106(90)90031-R. [DOI] [PubMed] [Google Scholar]
- 98.Nakanishi H, Uchiyama A, Kusuda S. Impact of pulmonary hypertension on neurodevelopmental outcome in preterm infants with bronchopulmonary dysplasia: a cohort study. J Perinatol 36: 890–896, 2016. doi: 10.1038/jp.2016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Natarajan V, Ha AW, Dong Y, Reddy NM, Ebenezer DL, Kanteti P, Reddy SP, Usha Raj J, Lei Z, Maienschein-Cline M, Arbieva Z, Harijith A. Expression profiling of genes regulated by sphingosine kinase1 signaling in a murine model of hyperoxia induced neonatal bronchopulmonary dysplasia. BMC Genomics 18: 664, 2017. doi: 10.1186/s12864-017-4048-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nepal S, Tiruppathi C, Tsukasaki Y, Farahany J, Mittal M, Rehman J, Prockop DJ, Malik AB. STAT6 induces expression of Gas6 in macrophages to clear apoptotic neutrophils and resolve inflammation. Proc Natl Acad Sci USA 116: 16513–16518, 2019. doi: 10.1073/pnas.1821601116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nicholson KM, Anderson NG. The protein kinase B/Akt signalling pathway in human malignancy. Cell Signal 14: 381–395, 2002. doi: 10.1016/S0898-6568(01)00271-6. [DOI] [PubMed] [Google Scholar]
- 102.Nold MF, Mangan NE, Rudloff I, Cho SX, Shariatian N, Samarasinghe TD, Skuza EM, Pedersen J, Veldman A, Berger PJ, Nold-Petry CA. Interleukin-1 receptor antagonist prevents murine bronchopulmonary dysplasia induced by perinatal inflammation and hyperoxia. Proc Natl Acad Sci USA 110: 14384–14389, 2013. doi: 10.1073/pnas.1306859110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.O’Brien PJ. Molecular mechanisms of quinone cytotoxicity. Chem Biol Interact 80: 1–41, 1991. doi: 10.1016/0009-2797(91)90029-7. [DOI] [PubMed] [Google Scholar]
- 104.Otterbein LE, Soares MP, Yamashita K, Bach FH. Heme oxygenase-1: unleashing the protective properties of heme. Trends Immunol 24: 449–455, 2003. doi: 10.1016/S1471-4906(03)00181-9. [DOI] [PubMed] [Google Scholar]
- 105.Pammi M, Lal CV, Wagner BD, Mourani PM, Lohmann P, Luna RA, Sisson A, Shivanna B, Hollister EB, Abman SH, Versalovic J, Connett GJ, Bhandari V, Ambalavanan N. Airway microbiome and development of bronchopulmonary dysplasia in preterm infants: a systematic review. J Pediatr 204: 126–133.e2, 2019. doi: 10.1016/j.jpeds.2018.08.042. [DOI] [PubMed] [Google Scholar]
- 106.Pradhan A, Dunn A, Ustiyan V, Bolte C, Wang G, Whitsett JA, Zhang Y, Porollo A, Hu YC, Xiao R, Szafranski P, Shi D, Stankiewicz P, Kalin TV, Kalinichenko VV. The S52F FOXF1 mutation inhibits STAT3 signaling and causes alveolar capillary dysplasia. Am J Respir Crit Care Med 200: 1045–1056, 2019. doi: 10.1164/rccm.201810-1897OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Ramani M, van Groen T, Kadish I, Bulger A, Ambalavanan N. Neurodevelopmental impairment following neonatal hyperoxia in the mouse. Neurobiol Dis 50: 69–75, 2013. doi: 10.1016/j.nbd.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Ramel SE, Brown LD, Georgieff MK. The impact of neonatal illness on nutritional requirements-one size does not fit all. Curr Pediatr Rep 2: 248–254, 2014. doi: 10.1007/s40124-014-0059-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Reiter G, Reiter U, Rienmüller R, Gagarina N, Ryabikin A. On the value of geometry-based models for left ventricular volumetry in magnetic resonance imaging and electron beam tomography: a Bland-Altman analysis. Eur J Radiol 52: 110–118, 2004. doi: 10.1016/j.ejrad.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 110.Reynolds CL, Zhang S, Shrestha AK, Barrios R, Shivanna B. Phenotypic assessment of pulmonary hypertension using high-resolution echocardiography is feasible in neonatal mice with experimental bronchopulmonary dysplasia and pulmonary hypertension: a step toward preventing chronic obstructive pulmonary disease. Int J Chron Obstruct Pulmon Dis 11: 1597–1605, 2016. doi: 10.2147/COPD.S109510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Rogers LK, Tipple TE, Nelin LD, Welty SE. Differential responses in the lungs of newborn mouse pups exposed to 85% or >95% oxygen. Pediatr Res 65: 33–38, 2009. doi: 10.1203/PDR.0b013e31818a1d0a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Royce SG, Nold MF, Bui C, Donovan C, Lam M, Lamanna E, Rudloff I, Bourke JE, Nold-Petry CA. Airway remodeling and hyperreactivity in a model of bronchopulmonary dysplasia and their modulation by IL-1 receptor antagonist. Am J Respir Cell Mol Biol 55: 858–868, 2016. doi: 10.1165/rcmb.2016-0031OC. [DOI] [PubMed] [Google Scholar]
- 113.Saugstad OD. Oxygen and oxidative stress in bronchopulmonary dysplasia. J Perinat Med 38: 571–577, 2010. doi: 10.1515/jpm.2010.108. [DOI] [PubMed] [Google Scholar]
- 114.Savani RC. Modulators of inflammation in bronchopulmonary dysplasia. Semin Perinatol 42: 459–470, 2018. doi: 10.1053/j.semperi.2018.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Scheuer T, Sharkovska Y, Tarabykin V, Marggraf K, Brockmöller V, Bührer C, Endesfelder S, Schmitz T. Neonatal hyperoxia perturbs neuronal development in the cerebellum. Mol Neurobiol 55: 3901–3915, 2018. doi: 10.1007/s12035-017-0612-5. [DOI] [PubMed] [Google Scholar]
- 116.Schuijs MJ, Willart MA, Vergote K, Gras D, Deswarte K, Ege MJ, Madeira FB, Beyaert R, van Loo G, Bracher F, von Mutius E, Chanez P, Lambrecht BN, Hammad H. Farm dust and endotoxin protect against allergy through A20 induction in lung epithelial cells. Science 349: 1106–1110, 2015. doi: 10.1126/science.aac6623. [DOI] [PubMed] [Google Scholar]
- 117.Seedorf G, Metoxen AJ, Rock R, Markham N, Ryan S, Vu T, Abman SH. Hepatocyte growth factor as a downstream mediator of vascular endothelial growth factor-dependent preservation of growth in the developing lung. Am J Physiol Lung Cell Mol Physiol 310: L1098–L1110, 2016. doi: 10.1152/ajplung.00423.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sheth S, Goto L, Bhandari V, Abraham B, Mowes A. Factors associated with development of early and late pulmonary hypertension in preterm infants with bronchopulmonary dysplasia. J Perinatol 40: 138–148, 2020. doi: 10.1038/s41372-019-0549-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Shipkowski KA, Taylor AJ, Thompson EA, Glista-Baker EE, Sayers BC, Messenger ZJ, Bauer RN, Jaspers I, Bonner JC. An allergic lung microenvironment suppresses carbon nanotube-induced inflammasome activation via STAT6-dependent inhibition of caspase-1. PLoS One 10: e0128888, 2015. doi: 10.1371/journal.pone.0128888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Shivanna B, Zhang W, Jiang W, Welty SE, Couroucli XI, Wang L, Moorthy B. Functional deficiency of aryl hydrocarbon receptor augments oxygen toxicity-induced alveolar simplification in newborn mice. Toxicol Appl Pharmacol 267: 209–217, 2013. doi: 10.1016/j.taap.2013.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Short EJ, Klein NK, Lewis BA, Fulton S, Eisengart S, Kercsmar C, Baley J, Singer LT. Cognitive and academic consequences of bronchopulmonary dysplasia and very low birth weight: 8-year-old outcomes. Pediatrics 112: e359, 2003. doi: 10.1542/peds.112.5.e359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Shrestha AK, Bettini ML, Menon RT, Gopal VYN, Huang S, Edwards DP, Pammi M, Barrios R, Shivanna B. Consequences of early postnatal lipopolysaccharide exposure on developing lungs in mice. Am J Physiol Lung Cell Mol Physiol 316: L229–L244, 2019. doi: 10.1152/ajplung.00560.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Simpson SJ, Hall GL, Wilson AC. Lung function following very preterm birth in the era of ‘new’ bronchopulmonary dysplasia. Respirology 20: 535–540, 2015. doi: 10.1111/resp.12503. [DOI] [PubMed] [Google Scholar]
- 124.Slaughter JL, Pakrashi T, Jones DE, South AP, Shah TA. Echocardiographic detection of pulmonary hypertension in extremely low birth weight infants with bronchopulmonary dysplasia requiring prolonged positive pressure ventilation. J Perinatol 31: 635–640, 2011. doi: 10.1038/jp.2010.213. [DOI] [PubMed] [Google Scholar]
- 125.Stenmark KR, Abman SH. Lung vascular development: implications for the pathogenesis of bronchopulmonary dysplasia. Annu Rev Physiol 67: 623–661, 2005. doi: 10.1146/annurev.physiol.67.040403.102229. [DOI] [PubMed] [Google Scholar]
- 126.Surate Solaligue DE, Rodríguez-Castillo JA, Ahlbrecht K, Morty RE. Recent advances in our understanding of the mechanisms of late lung development and bronchopulmonary dysplasia. Am J Physiol Lung Cell Mol Physiol 313: L1101–L1153, 2017. doi: 10.1152/ajplung.00343.2017. [DOI] [PubMed] [Google Scholar]
- 127.Syed MA, Bhandari V. Hyperoxia exacerbates postnatal inflammation-induced lung injury in neonatal BRP-39 null mutant mice promoting the M1 macrophage phenotype. Mediators Inflamm 2013: 457189, 2013. doi: 10.1155/2013/457189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Takeda K, Akira S. Multi-functional roles of Stat3 revealed by conditional gene targeting. Arch Immunol Ther Exp (Warsz) 49: 279–283, 2001. [PubMed] [Google Scholar]
- 129.Tang JR, Markham NE, Lin YJ, McMurtry IF, Maxey A, Kinsella JP, Abman SH. Inhaled nitric oxide attenuates pulmonary hypertension and improves lung growth in infant rats after neonatal treatment with a VEGF receptor inhibitor. Am J Physiol Lung Cell Mol Physiol 287: L344–L351, 2004. doi: 10.1152/ajplung.00291.2003. [DOI] [PubMed] [Google Scholar]
- 130.Thangaratnarajah C, Dinger K, Vohlen C, Klaudt C, Nawabi J, Lopez Garcia E, Kwapiszewska G, Dobner J, Nüsken KD, van Koningsbruggen-Rietschel S, von Hörsten S, Dötsch J, Alejandre Alcázar MA. Novel role of NPY in neuroimmune interaction and lung growth after intrauterine growth restriction. Am J Physiol Lung Cell Mol Physiol 313: L491–L506, 2017. doi: 10.1152/ajplung.00432.2016. [DOI] [PubMed] [Google Scholar]
- 131.Thébaud B, Abman SH. Bronchopulmonary dysplasia: where have all the vessels gone? Roles of angiogenic growth factors in chronic lung disease. Am J Respir Crit Care Med 175: 978–985, 2007. doi: 10.1164/rccm.200611-1660PP. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Thébaud B, Goss KN, Laughon M, Whitsett JA, Abman SH, Steinhorn RH, Aschner JL, Davis PG, McGrath-Morrow SA, Soll RF, Jobe AH. Bronchopulmonary dysplasia. Nat Rev Dis Primers 5: 78, 2019. doi: 10.1038/s41572-019-0127-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Thébaud B, Ladha F, Michelakis ED, Sawicka M, Thurston G, Eaton F, Hashimoto K, Harry G, Haromy A, Korbutt G, Archer SL. Vascular endothelial growth factor gene therapy increases survival, promotes lung angiogenesis, and prevents alveolar damage in hyperoxia-induced lung injury: evidence that angiogenesis participates in alveolarization. Circulation 112: 2477–2486, 2005. doi: 10.1161/CIRCULATIONAHA.105.541524. [DOI] [PubMed] [Google Scholar]
- 134.Thibault HB, Kurtz B, Raher MJ, Shaik RS, Waxman A, Derumeaux G, Halpern EF, Bloch KD, Scherrer-Crosbie M. Noninvasive assessment of murine pulmonary arterial pressure: validation and application to models of pulmonary hypertension. Circ Cardiovasc Imaging 3: 157–163, 2010. doi: 10.1161/CIRCIMAGING.109.887109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Urboniene D, Haber I, Fang YH, Thenappan T, Archer SL. Validation of high-resolution echocardiography and magnetic resonance imaging vs. high-fidelity catheterization in experimental pulmonary hypertension. Am J Physiol Lung Cell Mol Physiol 299: L401–L412, 2010. doi: 10.1152/ajplung.00114.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.van Eijl S, Mortaz E, Versluis C, Nijkamp FP, Folkerts G, Bloksma N. A low vitamin A status increases the susceptibility to cigarette smoke-induced lung emphysema in C57BL/6J mice. J Physiol Pharmacol 62: 175–182, 2011. [PubMed] [Google Scholar]
- 137.Velten M, Heyob KM, Rogers LK, Welty SE. Deficits in lung alveolarization and function after systemic maternal inflammation and neonatal hyperoxia exposure. J Appl Physiol (1985) 108: 1347–1356, 2010. doi: 10.1152/japplphysiol.01392.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wang H, Jafri A, Martin RJ, Nnanabu J, Farver C, Prakash YS, MacFarlane PM. Severity of neonatal hyperoxia determines structural and functional changes in developing mouse airway. Am J Physiol Lung Cell Mol Physiol 307: L295–L301, 2014. doi: 10.1152/ajplung.00208.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Wang J, Bao L, Yu B, Liu Z, Han W, Deng C, Guo C. Interleukin-1β promotes epithelial-derived alveolar elastogenesis via αvβ6 integrin-dependent TGF-β activation. Cell Physiol Biochem 36: 2198–2216, 2015. doi: 10.1159/000430185. [DOI] [PubMed] [Google Scholar]
- 140.Wang X, Hellgren G, Löfqvist C, Li W, Hellström A, Hagberg H, Mallard C. White matter damage after chronic subclinical inflammation in newborn mice. J Child Neurol 24: 1171–1178, 2009. doi: 10.1177/0883073809338068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wang X, Stridh L, Li W, Dean J, Elmgren A, Gan L, Eriksson K, Hagberg H, Mallard C. Lipopolysaccharide sensitizes neonatal hypoxic-ischemic brain injury in a MyD88-dependent manner. J Immunol 183: 7471–7477, 2009. doi: 10.4049/jimmunol.0900762. [DOI] [PubMed] [Google Scholar]
- 142.Welte T, Zhang SS, Wang T, Zhang Z, Hesslein DG, Yin Z, Kano A, Iwamoto Y, Li E, Craft JE, Bothwell AL, Fikrig E, Koni PA, Flavell RA, Fu XY. STAT3 deletion during hematopoiesis causes Crohn’s disease-like pathogenesis and lethality: a critical role of STAT3 in innate immunity. Proc Natl Acad Sci USA 100: 1879–1884, 2003. doi: 10.1073/pnas.0237137100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Will JP, Hirani D, Thielen F, Klein F, Vohlen C, Dinger K, Dötsch J, Alejandre Alcázar MA. Strain-dependent effects on lung structure, matrix remodeling, and Stat3/Smad2 signaling in C57BL/6N and C57BL/6J mice after neonatal hyperoxia. Am J Physiol Regul Integr Comp Physiol 317: R169–R181, 2019. doi: 10.1152/ajpregu.00286.2018. [DOI] [PubMed] [Google Scholar]
- 144.Wynn JL, Wong HR. Pathophysiology and treatment of septic shock in neonates. Clin Perinatol 37: 439–479, 2010. doi: 10.1016/j.clp.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Yamamoto H, Yun EJ, Gerber HP, Ferrara N, Whitsett JA, Vu TH. Epithelial-vascular cross talk mediated by VEGF-A and HGF signaling directs primary septae formation during distal lung morphogenesis. Dev Biol 308: 44–53, 2007. doi: 10.1016/j.ydbio.2007.04.042. [DOI] [PubMed] [Google Scholar]
- 146.Yang G, Abate A, George AG, Weng YH, Dennery PA. Maturational differences in lung NF-κB activation and their role in tolerance to hyperoxia. J Clin Invest 114: 669–678, 2004. doi: 10.1172/JCI200419300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yang G, Biswasa C, Lin QS, La P, Namba F, Zhuang T, Muthu M, Dennery PA. Heme oxygenase-1 regulates postnatal lung repair after hyperoxia: role of β-catenin/hnRNPK signaling. Redox Biol 1: 234–243, 2013. doi: 10.1016/j.redox.2013.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Yee M, Chess PR, McGrath-Morrow SA, Wang Z, Gelein R, Zhou R, Dean DA, Notter RH, O’Reilly MA. Neonatal oxygen adversely affects lung function in adult mice without altering surfactant composition or activity. Am J Physiol Lung Cell Mol Physiol 297: L641–L649, 2009. doi: 10.1152/ajplung.00023.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zhang S, Patel A, Chu C, Jiang W, Wang L, Welty SE, Moorthy B, Shivanna B. Aryl hydrocarbon receptor is necessary to protect fetal human pulmonary microvascular endothelial cells against hyperoxic injury: Mechanistic roles of antioxidant enzymes and RelB. Toxicol Appl Pharmacol 286: 92–101, 2015. doi: 10.1016/j.taap.2015.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Zhao J, Kim KD, Yang X, Auh S, Fu YX, Tang H. Hyper innate responses in neonates lead to increased morbidity and mortality after infection. Proc Natl Acad Sci USA 105: 7528–7533, 2008. doi: 10.1073/pnas.0800152105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Zoetis T, Hurtt ME. Species comparison of lung development. Birth Defects Res B Dev Reprod Toxicol 68: 121–124, 2003. doi: 10.1002/bdrb.10014. [DOI] [PubMed] [Google Scholar]