

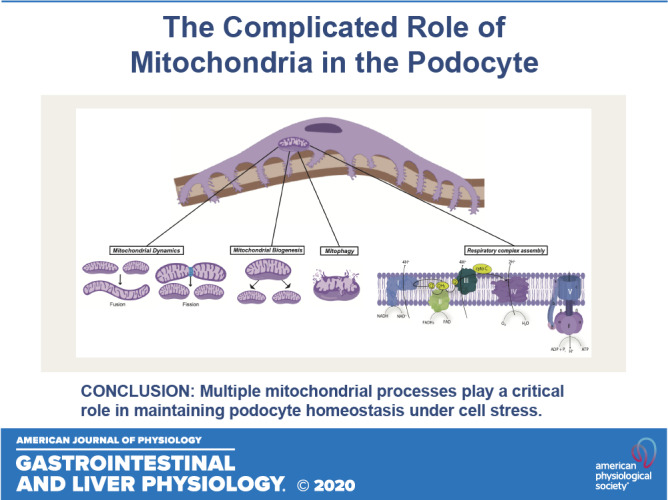
Keywords: glomerulosclerosis, kidney disease, mitochondria, podocytes, proteinuria
Abstract
Mitochondria play a complex role in maintaining cellular function including ATP generation, generation of biosynthetic precursors for macromolecules, maintenance of redox homeostasis, and metabolic waste management. Although the contribution of mitochondrial function in various kidney diseases has been studied, there are still avenues that need to be explored under healthy and diseased conditions. Mitochondrial damage and dysfunction have been implicated in experimental models of podocytopathy as well as in humans with glomerular diseases resulting from podocyte dysfunction. Specifically, in the podocyte, metabolism is largely driven by oxidative phosphorylation or glycolysis depending on the metabolic needs. These metabolic needs may change drastically in the presence of podocyte injury in glomerular diseases such as diabetic kidney disease or focal segmental glomerulosclerosis. Here, we review the role of mitochondria in the podocyte and the factors regulating its function at baseline and in a variety of podocytopathies to identify potential targets for therapy.
INTRODUCTION
Human kidneys filter ∼180 liters of blood daily and are the second highest energy-demanding organs in the human body (24). Mitochondria are essential organelles that fulfill the cell’s energy requirement through generation of ATP (62). The kidney has the second highest density of mitochondria, which helps meet the energy requirement to carry out essential functions including filtration of blood to remove waste, reabsorption of nutrients, maintenance of electrolyte and fluid homeostasis, hormone secretion, and regulation of blood pressure (6). Although there is a disproportionate distribution of mitochondria density in proximal tubular cells because of their high energy demand for active electrolyte, metabolite, and nutrient exchange, recent studies have demonstrated a critical role for mitochondria in podocytes, the visceral epithelial cells that maintain the glomerular filtration barrier (17).
Podocytes are highly specialized and terminally differentiated visceral epithelial cells essential for the maintenance of the glomerular filtration barrier. These cells are highly dynamic and require a high energy demand to maintain the organization of cytoskeletal and extracellular matrix proteins, motility, and remodeling of their foot processes in the glomerulus (26, 62, 89). Since these cells are terminally differentiated epithelial cells with minimal capacity to regenerate, they are highly sensitive to cell stress. In fact, podocyte injury and loss are central to the development of albuminuria and eventual progression of several glomerular diseases such as diabetic kidney disease (DKD) and focal segmental glomerulosclerosis (FSGS). Regardless of the inciting factor contributing to podocyte injury in these glomerular diseases, the inevitable consequence is destabilization of the actin cytoskeleton, loss of terminal differential markers, apical redistribution or loss of slit diaphragm proteins, and loss of structural integrity, leading to eventual podocyte detachment (85). Nonetheless, mechanisms by which these inciting factors contribute to podocyte injury is an area of active investigation because of the scarcity of therapies to treat these disorders.
Several laboratories have underscored the critical role of mitochondria in the maintenance of podocyte homeostasis as well as in the development and progression of podocytopathy (4, 5, 7, 11–13, 16, 34, 40, 60). Mitochondrial respiration accounts for 77% of cellular respiration, and 75% of this is coupled to ATP synthesis in mouse podocytes (2). In addition, mitochondria also participate in various other cellular functions, including calcium homeostasis and induction of cell apoptosis (32). Mitochondria are highly dynamic and heterogeneous organelles that constantly change shape, size, and numbers to maintain cellular homeostasis (17). Imbalances in mitochondrial homeostasis, the molecular control of mitochondrial formation (biogenesis), morphology (fusion/fission), and degradation (mitophagy) have been implicated in development and progression of various glomerular diseases (71). Specifically, acute and chronic insults might evoke mtDNA damage, reduce matrix density, and impair mitochondrial membrane integrity in podocytes (21). Interestingly, a majority of mtDNA mutations and deletions have been reported to cause glomerular injury associated with diseases such as FSGS or global glomerulosclerosis (67). These disturbances in mitochondrial dynamics lead to reduced ATP generation and calcium signaling, increased oxidative stress, and eventually apoptosis (21). In addition, mitochondria are major sites of intracellular reactive oxygen species (ROS) generation; electrons leaking from the electron transport chain combine with oxygen molecules to form superoxide and other mediators of ROS. ROS can increase the release of intermembrane space proteins such as cytochrome c to the cytosol and cause initiation of cell apoptosis. ROS triggers opening of the mitochondrial permeability transition pore (MPTP), which can lead to mitochondrial dysfunction, organelle swelling, and eventual cell death (93). Furthermore, mtDNA is directly exposed to ROS because of the lack of histone protection and incomplete repair mechanisms, thereby making it highly susceptible to damage and mutations (85). Here, we highlight the key aspects of mitochondrial dynamics in the maintenance of podocyte health as well as provide potential areas in need of future investigation.
MITOCHONDRIAL BIOGENESIS IN THE PODOCYTE
Peroxisome proliferator-activated receptor (PPAR)-γ coactivator-1α (PGC-1α) is an inducible upstream transcriptional coactivator and a key regulator of mitochondrial biogenesis and function (36). Induction of PGC-1α expression has been observed in conditions requiring increased energy demands such as in the setting of podocyte stress (36). PGC-1α regulates the activity of a large number of transcription factors including PPAR-γ and nuclear respiratory factor 1 (NRF1), which enhance fatty acid oxidation and regulate the expression of assembly factors of the electron transport chain for oxidative phosphorylation, respectively (50). PGC-1α plays a complex role in regulating the expression of ROS-scavenging factors such as superoxide dismutase (SOD2). Although PGC-1α is abundantly expressed in the kidney, the role and regulation of PGC-1α in the podocyte remain complex. For instance, several regulators of PGC-1α expression and function have been reported with cell culture and murine models of DKD, which include AMP-activated protein kinase (AMPK), sirtuin 1 (Sirt1), and lncRNA taurine-upregulated gene 1 (Tug1) (9, 19, 57, 90).
The mechanism by which PGC-1α responds to energy requirements is determined by whether it has been phosphorylated by AMPK or deacetylated by Sirt1 (59). The complex signaling pathways of PGC-1α and its downstream targets have been studied in various kidney diseases, including disorders involving podocytopathy (59). Dugan et al. (19) reported reduced gene expression and protein level of PGC-1α and concurrent decrease in AMPK activity in diabetic mouse glomeruli as well as in human diabetic nephropathy. AMPK activation by 5-aminoimidazole-4-carboxamide-1-β-d-ribofuranoside (AICAR), an AMPK activator, restored PGC-1α levels in glomeruli of diabetic mice and improved diabetic kidney disease. Expression of SIRT1, an upstream regulator of PGC-1α, is reduced in podocytes and glomerular cells of human diabetic kidneys as well as in kidneys in db/db mice (39). Podocyte-specific knockout of Sirt1 accelerated DKD in streptozotocin (STZ)-treated mice as well as glomerular injury in an adriamycin (ADR)-induced podocytopathy murine model (14, 90). Conversely, Hong et al. (39) showed that podocyte-specific overexpression of Sirt1 induced deacetylation of PGC-1α and activation of PPARγ-targeted gene expression in cultured podocytes as well as reducing oxidative stress in glomeruli in diabetic OVE26 mice, thereby attenuating podocyte injury and improving albuminuria.
A combination of recent studies has demonstrated that tight regulation of PGC-1α levels is required to maintain podocyte homeostasis. Decreased PGC-1α expression and mitochondrial transcripts in microdissected glomeruli from human kidney biopsies with diabetic and nondiabetic chronic kidney disease (CKD) have been reported (50). Furthermore, STZ-treated mice as well as ADR-treated mice exhibit reduced expression of PGC-1α in glomeruli (49, 92). Not surprisingly, PGC-1α silencing leads to induction of podocyte injury by disrupting mitochondrial function in cultured podocytes (Table 1). Although the loss of PGC-1α in the podocyte exacerbates podocyte injury, the renoprotective role of PGC-1α overexpression, interestingly, remains debatable. Long et al. (57) identified that a lncRNA, Tug1, regulates PGC-1α transcription and mitochondrial biogenesis in podocytes in DKD. Podocyte-specific overexpression of Tug1 in diabetic mice improved mitochondrial bioenergetics by increasing PGC-1α expression in murine models of DKD (51, 57). Overexpression of PGC-1α in ADR-treated cultured mouse podocytes also improved mitochondrial function, restored mature podocyte markers, and reduced apoptosis (92). However, a recent study by Li et al. (49) demonstrated that induction of podocyte-specific PGC-1α expression in mice contributes to a loss of podocyte differentiation markers with subsequent glomerular epithelial cell proliferation, leading to collapsing glomerulopathy (Table 2). Collectively, these studies suggest a disease context-specific role for PGC-1α as well as a potential need to maintain tight regulation of PGC-1α expression to preserve podocyte homeostasis.
Table 1.
Mitochondrial regulatory factors studied in cultured podocytes
| Gene | In Vitro Model | Effects | Mitochondrial Health | Reference |
|---|---|---|---|---|
| PGC-1α | Human podocytes | PGC-1α expression increased with differentiation | ↔ | (41) |
| PGC-1α | Human podocytes treated with HG | Reduced expression of PGC-1α and TFAM | ↓ | (41) |
| PGC-1α | Overexpression of PGC-1α in mouse podocytes | Increase in mitochondrial biogenesis and function; induction of podocyte proliferation and dedifferentiation | ↑ | (49) |
| PGC-1α | Mouse podocytes treated with ADR | Podocyte injury and mitochondrial dysfunction; decreased PGC-1α expression | ↓ | (92) |
| PGC-1α | Overexpression of PGC-1α in mouse podocytes treated with ADR | Protective against podocyte injury and mitochondrial dysfunction | ↑ | (92) |
| PGC-1α | Knockdown of PGC-1α in human podocytes | Reduced mitochondrial function and improved glycolysis | ↓ | (8) |
| SIRT1 | Knockdown of Sirt1 in human podocytes with HG | Reduced expression of PGC-1α, NRF-1, and Tfam; decreased mtDNA | ↓ | (39) |
| TUG1 | Knockout of Tug1 in mouse podocytes treated with HG | Increased mitochondrial ROS and apoptosis; reduced complex I and III activity; reduced ATP | ↓ | (57) |
| TUG1 | Overexpression of Tug1 in mouse podocytes treated with HG | Restored complex I and III activity and ATP level | ↑ | (57) |
| TUG1 | Primary mouse podocytes from db/db Tug1PodTg mice | Increased mitochondrial content and ATP level compared with db/db mice | ↑ | (57) |
| KLF6 | Knockdown and overexpression of KLF6 in ADR-treated mouse and human podocytes | Knockdown of KLF6 in podocytes increased mitochondrial fragmentation and apoptosis; overexpression of KLF6 was able to rescue these changes | ↓ ↑ |
(60) |
| KLF6 | HIV-1-infected human podocytes | Reduced KLF6 expression | ↓ | (60) |
| SCO2 | Knockdown of SCO2 in human podocytes | Increased caspase-9 and release of cytochrome c from mitochondria | ↓ | (60) |
| KLF6 | Knockout and overexpression of KLF6 in human podocytes under HG conditions | Knockout of KLF6 caused mitochondrial injury and apoptosis that was rescued by overexpression of KLF6 | ↓ | (40) |
| TFAM | Knockout of TFAM in human podocytes | Reduced mitochondrial function and increased glycolysis | ↓ | (8) |
| DRP1 | Knockout of DRP1 in human podocytes | No significant change in mitochondrial or glycolytic function | ↔ | (8) |
| DRP1 | Primary podocytes from db/db;Drp1fl/fl mice | Increase in basal and maximal OCR; increased ATP levels | ↑ | (4) |
| DRP1 | Cultured mouse podocyte with Drp1 deletion under HG conditions | Reversed the effect on HG; increased OCR; increased ATP content; reduced ROS production | ↑ | (4) |
| DRP1 | Cultured mouse podocyte with Drp1 knockdown treated with aldosterone | Suppressed mitochondrial fission; attenuated mitochondrial dysfunction and podocyte injury | ↑ | (86) |
| ROCK1 | Knockout of ROCK1 in mouse podocytes treated with HG | Maintained elongated mitochondria; reduced Drp1 recruitment | ↑ | (80) |
ADR, adriamycin; DRP1, dynamin-related protein-1; HG, high glucose; HIV-1, human immunodeficiency virus-1; KLF6, Krüppel-like factor 6; NRF-1, nuclear respiratory factor 1; OCR, oxygen consumption rate; PGC-1α, peroxisome proliferator-activated receptor-γ coactivator-1α; ROCK1, Rho-associated coiled coil-containing protein kinase 1; ROS, reactive oxygen species; SIRT1, sirtuin 1; TFAM, mitochondrial transcription factor A; TUG1, taurine-upregulated gene 1.
Table 2.
Mitochondrial regulator factors studied in murine models of kidney disease
| Gene | Murine Model | Additional Stress | Effects | Mitochondrial Function | Reference |
|---|---|---|---|---|---|
| PGC-1α | Inducible podocyte-specific overexpression of PGC-1α | Induced collapsing FSGS | ↓ | (49) | |
| PGC-1α | Inducible podocyte-specific overexpression of PGC-1α | Diabetes (STZ) | Mice not protected from diabetes-induced albuminuria | ↓ | (49) |
| PGC-1α | Male Sprague-Dawley rats | ADR-induced FSGS | Podocyte mitochondrial dysfunction; increase in ROS and cell apoptosis and decrease in PGC-1α expression | ↓ | (92) |
| PGC-1α | Podocyte-specific knockout of PGC-1α | Reduced mitochondrial biogenesis but no impact on mitochondrial substrate preferences | ↓ | (8) | |
| SIRT1 | Global inducible and reversible Sirt1 knockdown | No significant effect in glomerular function under basal conditions | ↔ | (14) | |
| SIRT1 | Global inducible and reversible Sirt1 knockdown | Diabetes (STZ) | Promotes mitochondrial injury in glomerular cells | ↓ | (14) |
| SIRT1 | Inducible podocyte-specific Sirt1 knockdown | ADR-induced FSGS | Enhanced susceptibility to ADR-induced FSGS; mtDNA damage | ↓ | (14) |
| SIRT1 | Podocyte-specific Sirt1 knockdown | Diabetes (db/db mice) | Increased susceptibility to diabetic nephropathy | ↓ | (55) |
| SIRT1 | Inducible podocyte-specific Sirt1 knockdown | Aging | Aggravated glomerular and podocyte injury; decrease in PGC-1α | ↓ | (13) |
| SIRT1 | Inducible podocyte-specific SIRT1 overexpression | Diabetes (OVE26 mice) | Attenuated DKD progression and podocyte loss | ↑ | (39) |
| TUG1 | Podocyte-specific overexpression of Tug1 | Diabetes (db/db mice) | Attenuated DKD progression; regulates PGC-1α | ↑ | (57) |
| KLF6 | Podocyte-specific knockdown of Klf6 | ADR-induced FSGS | Induced mitochondrial dysfunction; reduced mitochondrial transcripts; reduced ATP levels and OCR | ↓ | (60) |
| KLF6 | Podocyte-specific knockdown of Klf6 | Diabetes (STZ) | Increased susceptibility to diabetic nephropathy; reduced complex IV activity and ATP levels | ↓ | (40) |
| ROCK1 | Global knockout of Rock1 | Diabetes (STZ; db/db mice) | Decrease in albuminuria | ↑ | (80) |
| ROCK1 | Podocyte-specific inducible knockin of constitutively active ROCK1 | Promoted podocyte loss; albuminuria; increase in mtROS and apoptosis | ↓ | (80) | |
| DRP1 | Inducible podocyte-specific Drp1 knockout | Diabetes (db/db mice) | Ameliorates key diabetic nephropathy features and podocytes have elongated mitochondria | ↑ | (4) |
| ROCK1- mediated phosphorylated DRP1 | Global Drp1S600A knockin | Diabetes (db/db mice) | Protective against diabetic nephropathy by preventing recruitment on Drp1 to mitochondria | ↑ | (27) |
| TFAM | Podocyte-specific Tfam knockout | No change in overall podocyte health | ↔ | (8) |
ADR, adriamycin; DKD, diabetic kidney disease; DRP1, dynamin-related protein-1; Drp1S600A, Drp1 serine 600 mutated to alanine; FSGS, focal segmental glomerulosclerosis; KLF6, Krüppel-like factor 6; OCR, oxygen consumption rate; PGC-1α, peroxisome proliferator-activated receptor-γ coactivator-1α; ROCK1, Rho-associated coiled coil-containing protein kinase 1; ROS, reactive oxygen species; SIRT1, sirtuin 1; STZ, streptozotocin; TFAM, mitochondrial transcription factor A; TUG1, taurine-upregulated gene 1.
Given the need for tight regulation of PGC-1α in the podocyte, it is not surprising that mechanisms mediating mitochondria turnover in podocytes is an area of active investigation. Rinschen et al. (73) used stable isotope labeling by amino acids in cell culture (SILAC)-labeled glomerular lysates to determine the rate of synthesis of mitochondrial proteins in podocyte and nonpodocyte cell fractions. Overall protein incorporation was slower in podocytes compared with nonpodocytes, whereas the rate of incorporation of mitochondrial proteins was significantly higher in podocytes compared with nonpodocytes (73). Furthermore, the number of amino acids incorporated into the mitochondrial proteins did not differ between the two cell fractions, suggesting that the podocytes may have a lower total mitochondrial mass compared with the nonpodocytes despite the high turnover of mitochondria (73). However, further studies are required to determine whether the rates of mitochondrial protein turnover are altered in the setting of podocyte stress.
MITOCHONDRIAL STRUCTURAL DYNAMICS IN THE PODOCYTE
The mitochondrion undergoes fission and fusion to maintain its physiological function, and this dynamic nature is critical to initiate rapid repair of damaged mitochondria (Fig. 1). Specifically, mitochondrial fission is a critical mediator of increased ROS production, which causes damage to mtDNA and respiratory chain complexes (66). Dynamin-related protein-1 (Drp1) is a large GTPase of the dynamin superfamily that plays an essential role in mitochondrial fission in mammalian cells (10, 87). During the process of mitochondrial fission, Drp1 is recruited from the cytosol to the outer mitochondrial membrane, where it self-assembles to form oligomers that constrict to sever both the mitochondrial membranes (22). Drp1 activity is also regulated by various posttranslational modifications (10, 22, 61). Increased Drp1 activity and mitochondrial fission, along with release of cytochrome c and generation of ROS, have been reported as the primary initiating event that leads to cell apoptosis under diabetic conditions (15). Conditional podocyte-specific knockout of Drp1 in diabetic mice as well as Drp1−/− cultured podocytes under high glucose conditions improved mitochondrial fitness of podocytes and ameliorated podocyte loss and the progression of DKD (Table 1) (4). Pharmacological inhibition of Drp1 by mitochondrial division inhibitor-1 (mdivi-1) in an aldosterone-induced injury model leads to inhibition of mitochondrial fission and attenuates aldosterone-induced podocyte injury. Aldosterone was found to induce p53 expression in a dose-dependent manner, and knockout of p53, a transcription regulator of Drp1 in cultured podocytes, leads to downregulation of Drp1 expression and attenuates aldosterone-induced mitochondrial fragmentation, podocyte injury, and mitochondrial dysfunction. Cultured mouse podocytes with Drp1 knockdown showed inhibition of MPTP opening and restoration of mitochondrial membrane potential (86). Glycogen synthase kinase (GSK)-3β interacts with mitochondrial membrane proteins such as cyclophilin F (Cyp-F), leading to opening of the MPTP, which results in eradication of the mitochondrial membrane potential and leak in mitochondrial inner membrane followed by cell death. Disruption of the interaction of cyclophilin F with GSK-3β by either pharmacological inhibition of GSK-3β or podocyte-specific knockout of GSK-3β in a mouse model protects against ADR nephropathy by preventing mitochondrial fragmentation, destruction of the mitochondrial inner membrane, and mitochondrial cristae disruption; similar results have been observed in GSK-3β knockout podocytes (48, 81).
Fig. 1.
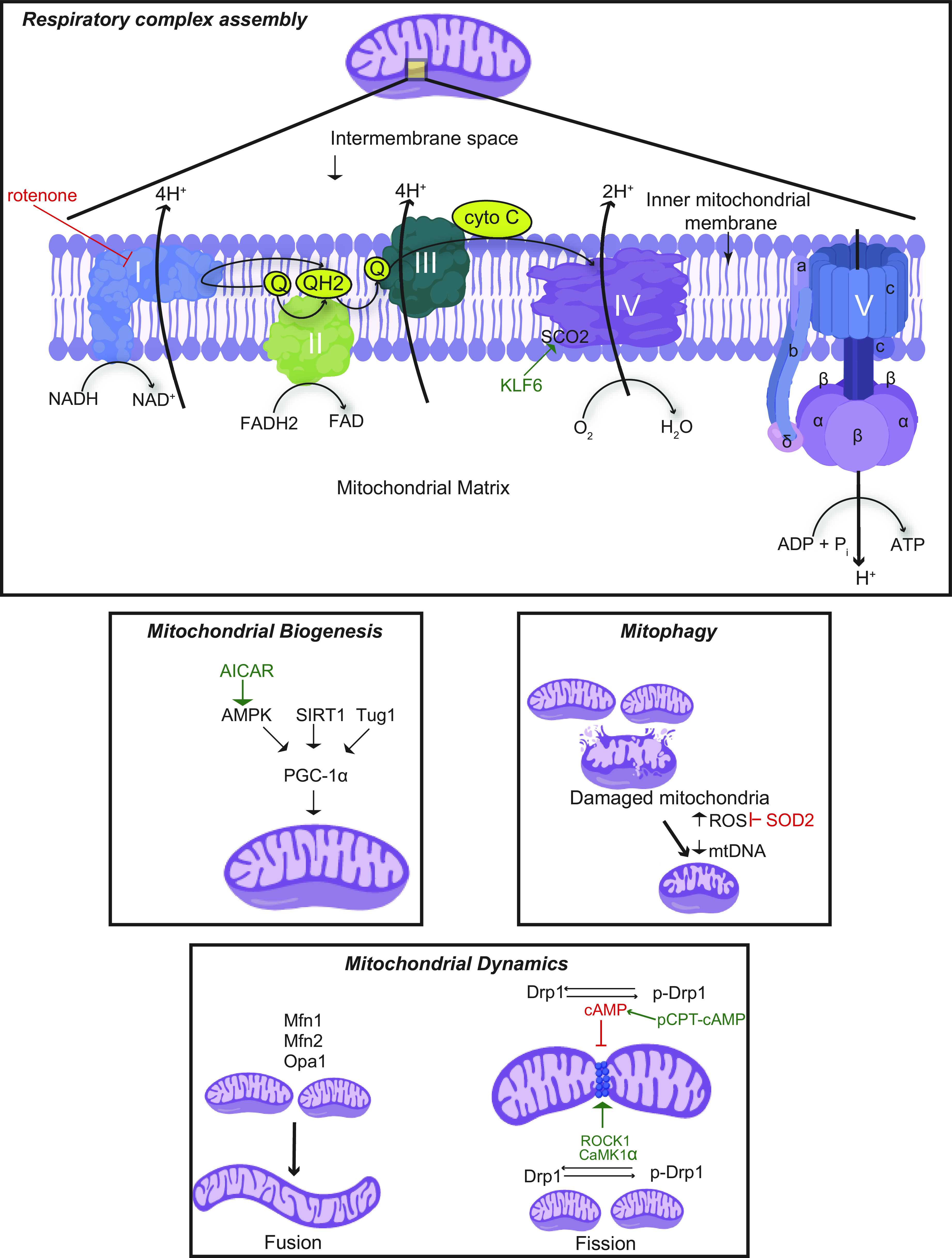
Mitochondrial dynamics and respiratory complex. Mitochondrial biogenesis is mainly regulated by peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α), and mitochondrial fission and fusion are regulated by dynamin-related protein-1 (Drp1) and mitofusin-1 (Mfn1), mitofusin-2 (Mfn2), and optic atrophy 1 (Opa1) respectively. The damaged mitochondria result in excessive reactive oxygen species (ROS) generation that can be inhibited by mitochondrial enzymes such as superoxide dismutase 2 (SOD2). Damaged mitochondria with decreased mtDNA undergo mitophagy. The electron transport chain (ETC) in the inner mitochondrial membrane consists of five respiratory complexes (complexes I–V). The flow of electrons through these complexes along with proton gradient leads to the generation of ATP. A wide array of genes involved in mitochondrial homeostasis and ETC have been identified to play a critical role in maintaining podocyte health and function and therefore have been associated with various podocytopathies. 5-Aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR) has been reported to increase mitochondrial biogenesis through AMP-activated protein kinase (AMPK)-mediated phosphorylation of PGC-1α. Other regulators of PGC-1α, such as sirtuin 1 (SIRT1) and taurine-upregulated gene 1 (Tug1), have been also studied in podocytopathies. Drp1 phosphorylation by Rho-associated coiled coil-containing protein kinase 1 (ROCK1) and Ca2+/calmodulin-dependent kinase-1α (CaMK1α) promotes mitochondrial fission, whereas phosphorylation by cAMP and its activator, 8-(4-chlorophenylthio)adenosine cAMP (pCPT-cAMP), leads to inhibition of mitochondrial fission. Inhibition of respiratory complex II by rotenone has been found to attenuate diabetic kidney disease (DKD); transcriptional factor Krüppel-like factor 6 (KLF6) and its downstream target, mitochondrial cytochrome c oxidase assembly gene (SCO2), a complex IV assembly factor, have been reported to play a role in glomerulopathies.
Drp1 phosphorylation is mediated by Rho-associated coiled coil-containing protein kinase 1 (ROCK1) or Ca2+/calmodulin-dependent protein kinase-1α (CaMK1α), which leads to its translocation to mitochondria and increased mitochondrial fission (22, 35, 80). Recent studies have demonstrated that introduction of site-specific mutation at the Drp1 phosphorylation site attenuates ROS production, albuminuria, mesangial matrix expansion, and podocyte effacement (27). Conversely, podocyte-specific ROCK1 activation promotes mitochondrial fission, ROS production, and cellular apoptosis through Drp1 translocation to mitochondria in mice (80). However, a recent study has demonstrated that podocyte-specific conditional knockout of Drp1 under physiological conditions showed no sign of podocyte foot process effacement (8), raising the need to investigate potential compensatory mechanisms.
Mitochondrial fusion involves tethering of two adjacent mitochondria, which facilitates the exchange of materials and aids repair of defective mitochondria (74, 86). Mitofusins (Mfn1 and Mfn2) regulate the fusion of the outer mitochondrial membrane, and optic atrophy 1 (Opa1) mediates the fusion of the inner mitochondrial membrane. The three GTPases (Mfn1, Mfn2, and Opa1) work in a synchronized fashion to fuse both mitochondrial membranes. Knockout of Mfn1, Mfn2, and Opa1 causes embryonic lethality in mice, suggesting their critical role in embryonic development (63). In a podocyte-specific PGC-1α overexpression mouse model that exhibited collapsing glomerulopathy, expression of these mitochondrial fusion genes (Mfn1, Mfn2, and Opa1) was significantly increased, whereas no changes were observed in mitochondrial fission genes Drp1 and fission 1 (Table 2) (49). Puromycin and ADR induce mitochondrial fission in podocytes by repressing Mfn1, which can be reversed with 8-(4-chlorophenylthio)adenosine cAMP (pCPT-cAMP), a cAMP/PKA activator that induces Mfn1 expression and Drp1 phosphorylation (53). Drp1 phosphorylation by cAMP/PKA results in the release of Drp1 from the mitochondria to the cytosol, leading to inhibition of mitochondrial fission and subsequent cell apoptosis (Table 1) (22). Elamipretide, a mitochondria-targeted peptide that selectively targets cardiolipin on the inner mitochondrial membrane and thereby protects the cristae curvature, stabilizes mitochondrial structure, minimizes ROS, and facilitates electron transport chain, has been reported to protect against mitochondrial damage in the podocyte caused during the early stages of metabolic syndrome in swine (88). Further studies are required to determine the mechanisms mediating the balance between mitochondrial fission and fusion in podocytes at baseline as well as in the setting of cell stress.
REGULATION OF RESPIRATORY COMPLEX ASSEMBLY IN THE PODOCYTE
The terminal component of cellular respiration is oxidative phosphorylation (OXPHOS), which takes place in the mitochondria and leads to the generation of ATP. Mitochondrial complexes embedded in the inner mitochondrial membrane form the electron transport chain and orchestrate the generation of ATP via a series of electron and proton transfers. Complexes I and II receive electrons in the form of NADH and FADH2, respectively. These electrons get shuttled from complexes I and II through complex III until complex IV uses the electrons to reduce oxygen to form water (Fig. 1). Coenzyme Q10 shuttles electrons from complexes I and II to complex III (72), and cytochrome c serves as an electron shuttle between complex III and complex IV (28). Simultaneously, complexes I, III, and IV all provide a proton gradient necessary for complex V (ATP synthase) to produce ATP. Although OXPHOS is efficient at making ATP, the process also leads to the generation of reactive oxygen and nitrogen species at basal levels, which is exacerbated in the setting of mitochondrial dysfunction (20, 77). Regardless of the cell type, it is almost universal that decreased respiration, decreased ATP, increased mitochondrial ROS generation, and oxidative damage precede mitochondrial loss (69). However, there is still controversy as to whether dysregulated OXPHOS is a driver or merely a consequence of podocyte injury. If mitochondria were not such an integral part of podocyte metabolism, it is hard to explain why cultured human podocytes spend so much energy increasing their mitochondrial network with differentiation. Furthermore, there is a stepwise increase in OXPHOS in human podocytes with an increase in PGC-1α, mitochondrial transcription factor A (Tfam), mtDNA, and the mitochondrial network during differentiation (41).
Furthermore, primary mouse podocytes have a significantly greater oxygen consumption rate (OCR) capacity compared with immortalized mouse podocytes, reflecting their increased demand for OXPHOS (2), Rinschen et al. (73) showed that there is a lower abundance of phospholipid metabolism and tight junction-related proteins in an immortalized podocyte cell line compared with native podocytes. Podocytes under cell stress undergo significant changes in OXPHOS compared with basal conditions. For instance, Abe et al. (1) showed that in the presence of transforming growth factor-β1 (TGF-β1), there is an increase in OCR and thereby increase in ROS production in primary mouse podocytes, which may be partially related to the mammalian target of rapamycin complex 1 pathway. In cultured mouse podocytes, long-term exposure to high glucose (30 mM glucose) showed an increase in basal respiration rate, protein leak rate, and maximum respiratory capacity under permissive (differentiated) conditions compared with nonpermissive (undifferentiated, proliferating) conditions (76). This increase was further augmented with the use of TGF-β1 (76). However, in cultured human podocytes, introduction of high glucose states (20 mM) leads to a significant decrease in OXPHOS with a glycolytic switch leading to increased lactate concentrations and decreases in mtDNA biogenesis accompanied by decreased TFAM, PGC-1α, AMPK, and serine threonine liver kinase B1 (41). Since various studies on mouse or human podocytes cultured in the presence of high glucose have reported conflicting results, there is a critical need for more studies to clearly understand the effects of high glucose on mitochondrial function and the overall ATP production in podocytes.
Recent studies have demonstrated that there is a nonlinear increase in respiratory chain complex proteins with differentiation by label-free quantification proteomics, with subunits of complex I increasing fastest from day 0 to day 7 of differentiation followed by increases in complexes IV and V from day 3 to day 15 of differentiation (41). Synthesis of the α- and β-subunits of the F1 catalytic domain of the F1F0-ATP synthase complex (complex V) was further increased at the final stages of podocyte differentiation (41). Likewise, label-free quantification proteomics showed that most glycolytic enzymes were decreased with podocyte differentiation while proteins involved with fueling OXPHOS with fatty acids, amino acids, or ketones were upregulated (41).
Several lines of investigation reveal that mitochondrial function is compromised because of genetic or environmental factors leading to glomerular disease. Mitochondria isolated from mice with diffuse glomerular damage after aldosterone administration showed a decrease in mitochondrial membrane potential and ATP production (91). This was accompanied by a reduction in complex I, complex III, and complex IV activity by 40–50% (91). A reduction in OCR has also been observed in human glomerulopathies. For instance, earlier case reports such as a Romanian 18-mo-old boy with collapsing glomerulopathy demonstrated a reduction in complex II and III activity in the kidney cortex due to a coenzyme Q2 deficiency (5). ADR-treated immortalized mouse podocytes also exhibit a decrease in ATP-linked OCR along with a decrease in ATP production, whereas treatment with pCPT-cAMP leads to increased baseline OCR and ATP-linked OCR with a significant increase in ATP production, suggesting that mitochondrial dysfunction might be a key driver in ADR-induced podocyte injury (83).
Dysregulation of respiratory complexes has been widely seen in both murine and human diabetic samples. In microdissected glomerular biopsies from a Pima Indian DKD cohort, there was significant upregulation of genes encoding complex I, IV, and V subunits (18). Meanwhile, there was significant downregulation of genes encoding complex II subunit in the same cohort (18). Although these changes are not necessarily consistent with those observed in cell culture and murine models, it is important to note that these kidney biopsies were from individuals with an average glomerular filtration rate of 157–192 mL/min and an albumin-to-creatinine ratio of 24–57 mg/g, suggesting early diabetic nephropathy (18).
Assembly of each respiratory complex is a very elaborate process that requires a host of assembly factors to work in cohesion (75). Given the complexity of the assembly process, there might still be many assembly factors that have yet to be identified or fully explored. Assembly factors play a critical role in the structure and function of respiratory complexes, and mutations in these factors have been associated with various diseases such as Leigh syndrome as well as proximal tubulopathies and congenital nephrotic syndrome (30). For instance, mutations in mitochondrial chaperone BCS1 (BCSL1) and cytochrome c oxidase subunit X (COX10) are associated with neonatal proximal tubulopathy and encephalopathy with proximal tubulopathy (30, 67). Mutations in genes such as prenyl (decaprenyl) diphosphate synthase subunit 2 (PDSS2), coenzyme Q2 (COQ2), and coenzyme Q6 (COQ6) that are essential for the biosynthesis of coenzyme Q10 have also been associated with nephrotic syndrome (16, 29, 38, 58, 67, 70). The mechanism(s) mediating critical components of respiratory complex assembly in the podocyte at baseline as well as in injury is in need of further exploration.
Understanding the factors that regulate respiratory complex assembly in podocytes under basal conditions and in stress might provide potential targets for therapy. Krüppel-like factor 6 (KLF6), a zinc finger transcription factor, is an early inducible injury response gene that plays a critical role in regulating complex IV assembly in proteinuric murine models of FSGS and DKD (ADR- and STZ-induced diabetic murine models) (40, 60). We observed that podocyte-specific expression of KLF6 in human kidney biopsies is significantly reduced in late-stage DKD compared with early-stage DKD. Podocyte-specific knockdown of Klf6 exacerbates podocyte injury and FSGS after ADR in the resistant C57BL/6 mouse strain. Loss of KLF6 in cultured human podocytes exacerbated mitochondrial dysfunction, whereas induction of KLF6 attenuated mitochondrial dysfunction and apoptosis in the setting of ADR treatment. Interestingly, KLF6 binding sites occupy the promoter of the mitochondrial cytochrome c oxidase assembly gene (SCO2), which plays a critical role in complex IV assembly. Podocyte-specific knockdown of Klf6 also worsened glomerular injury in a murine model of DKD (STZ treatment) and in human podocytes under high glucose conditions, with reduced SCO2 expression leading to mitochondrial injury and eventually podocyte loss (40).
To assess whether inhibition of respiratory complex activity exacerbates podocyte injury under diabetic conditions, Wu et al. (82) assessed the effects of rotenone, a potent complex I inhibitor, in a murine model of DKD (uninephrectomy + STZ). Although rotenone is certainly not specific to podocytes, interestingly, there was a significant increase in podocin and nephrin expression with a decrease in glomerular volume and urine albumin excretion in diabetic mice (82). Treatment with rotenone in these diabetic mice led to a significant decrease in the overactivity of OXPHOS and other metabolic pathways along with a decrease in mitochondria-derived ROS and restoration of redox homeostasis. Although this study needs to be reproduced, they suggest the complexity of targeting respiratory complex activity as a potential therapeutic target in DKD.
Despite the observations of mitochondrial dysfunction in many podocytopathies, recent studies have raised the question of whether the mitochondria are the main source of energy under basal conditions for the podocyte. Ozawa et al. (68) initially ascertained the level of contribution to energy status from OXPHOS in cultured mouse podocytes. Podocytes were exposed to oligomycin, rotenone, and antimycin (inhibitors of complexes V, I, and III, respectively) in a dose-dependent manner. Each reagent caused ATP levels to decrease by 50%, which suggests that OXPHOS contributes to more than 50% of ATP production. However, despite the observed contribution of OXPHOS to ATP production, inhibition of OXPHOS with antimycin failed to induce apoptosis or exhibit a significant change in lamellipodia formation in the cultured podocytes. Conversely, the use of 2-deoxy-d-glucose, which blocks glycolysis, with pyruvate supplementation prevented the formation of lamellipodia. Additionally, blockade of glycolysis significantly reduced podocyte migratory ability, induced cleaved caspase-3, and increased TUNEL-positive cells (68). Nonetheless, OXPHOS and glycolysis were both essential for synaptopodin expression, thereby suggesting a context-dependent role for the source of ATP production in podocytes.
Brinkkoetter et al. (8) identified anaerobic glycolysis as the primary metabolic pathway in podocytes under physiological conditions. Mice with podocyte-specific knockout of Pgc-1α, Drp1, and Tfam were generated to determine whether functional changes in OXPHOS lead to podocytopathy. Primary podocytes with Tfam and Pgc-1α knockdown led to significant decreases in basal respiration, ATP-linked respiration, and maximum respiration, whereas Drp1 knockdown did not show significant changes in basal or ATP-linked respiration but instead showed an increase in maximum respiration. Protein expression of NADH:ubiquinone oxidoreductase subunit B8 (NDUFB8), succinate dehydrogenase complex iron sulfur subunit B (SDHB), and mitochondria-encoded genes mitochondrially encoded cytochrome c oxidase I (mtCO1) and ATP5A were all significantly decreased with knockdown of Tfam, leading to the decreased activity of respiratory complexes I, IV, and V, respectively. Despite the functional changes in OXPHOS, there were no changes in body weight, urinary albumin excretion, or glomerular morphological changes in histology for either Pgc-1α, Tfam, or Drp1 knockout mice. Although Brinkkoetter et al. suggested that this might be due to reliance on anaerobic glycolysis versus OXPHOS for energy by the podocyte under basal conditions, it is unclear whether these changes will be reflected in the setting of cell stress. Not surprisingly, ultrastructural changes under transmission electron microscopy showed elongated and swollen mitochondria in podocytes with Drp1 knockdown; however, there were no significant changes in mitochondria morphology observed in podocytes with Tfam and Pgc-1α knockdown (8), which could be attributed to the residual protein expression from partial knockdown of the genes using siRNA. Collectively, these data suggest a need for future studies to focus on the mechanisms mediating the change in energy demand under podocyte stress compared with basal conditions.
REGULATION OF MITOPHAGY IN THE PODOCYTE
Macroautophagy is an intracellular system that allows for the catabolism of cytoplasmic components including damaged organelles. It is characterized by the formation of autophagosomes, double-membrane organelles responsible for sequestering the cytosolic contents (79). Mitochondria accumulate damage with age, which leads to mitochondrial dysfunction and has been linked to many degenerative diseases, aging, and podocytopathies (64). Mitochondria are eliminated through a process called mitophagy when they cannot be restored to a healthier state with fission and fusion (Fig. 1) (25). When there is a loss in mitochondrial membrane potential, mitophagy is mediated by the PTEN-induced putative kinase protein 1 (PINK1) and Parkin (PARK2) system (15, 25, 45, 46, 84). They produce ubiquitin and nonubiquitin tags that allow for recognition by sequestosome 1 (SQSTM1), optineurin (OPTN), calcium binding and coiled‐coil domain 2 (CALCOCO2), and light chain 3 (LC3) by the autophagosome (25). The autophagosome can now find the damaged mitochondria and form an isolation membrane, a double membrane that sequesters the cytosolic contents, which then fuses with a lysosome allowing hydrolytic degradation (84). The multiple steps of autophagy, including the expansion of the isolation membrane and autophagosome production, are controlled by autophagy-related (Atg) proteins (84). In yeast, Atg32 is the primary target for macroautophagic signals, and it is thought that BCL2-like 13 (BCL2L13) is the analogous receptor in mouse and human cells (25).
Previous studies have demonstrated that disruption of mitophagy allows dysfunctional mitochondria to persist, which contributes to altered cell respiration. Early studies to test this hypothesis were conducted by knocking down Pink1 and Park2 in Drosophila. In Pink1−/− Drosophila, mitochondria had decreased complex I and IV activity, which was restored with the expression of Drp1 (56). Although this leads to the idea that mitophagy and fission are linked, the mechanism by which these two processes work in concert is not known. In comparison, Parkin knockdown Drosophila had dysmorphic mitochondria on electron microscopy, decreased oxidative phosphorylation, and increased ROS that preceded the eventual cell injury (33).
Dysregulated mitophagy has been previously shown to exacerbate podocyte injury in proteinuric murine models. Inability to remove dysfunctional mitochondria can cause significant damage to the cells. Under high glucose conditions, there is a decrease in mitophagy in cultured mouse podocytes and in the glomeruli of STZ-induced diabetic mice that causes an imbalance in mitochondrial homeostasis. Conversely, forkhead box O1 (FoxO1) overexpression promotes mitophagy via interaction with the mitophagy-related downstream targets such as PINK1, thereby protecting the high glucose-treated podocytes and diabetic mice against mitochondrial dysfunction (52). Upregulation and activation of PINK1/Parkin pathway in podocytes in a model of palmitic acid-induced mitophagy plays a protective role in apoptosis of podocytes by removing damage mitochondria, and Parkin knockout results in mitochondrial dysfunction, ROS production, and increased apoptosis of podocytes, due to accumulation of damaged mitochondria (43, 65). Podocyte-specific Atg5-deficient mice had exacerbated glomerulosclerosis in aging, type 1 diabetes mellitus and type 2 diabetes mellitus murine models (37, 47, 78). In addition, knockout of Atg5 or Atg7 in mice disrupts normal autophagic pathways, resulting in FSGS in 4-mo-old mice with segmental lesions of scarring, loss of capillary loops with increased matrix and hyaline deposition, adhesions to Bowman’s capsule, and epithelial cell hyperplasia (44). Transmission electron microscopy demonstrated ultrastructural changes in podocytes, enlarged mitochondria with loss of cristae, and increased ROS, leading to kidney failure by 6 mo of age (44). In cultured podocytes, hyperglycemia initially induces autophagy and persistent exposure to hyperglycemia downregulates autophagy through persistent activation of the mammalian target of rapamycin and β-arrestin pathways (3, 23, 31, 42, 54). Mitophagy plays an essential role in maintaining podocytes in a healthy state by removal of damaged mitochondria; however, there is a need to further explore the mechanism mediating dysregulated mitophagy in podocytes in the setting of cell stress.
CONCLUSIONS
For a healthy kidney, it is essential to support the metabolic needs of podocytes. Various mitochondrial processes such as biogenesis, dynamics, electron transport chain assembly, and mitophagy have been identified to play an essential role in maintaining podocyte health under physiological as well as disease states, as discussed in this review. A balance in the biogenesis of mitochondria promoted by PGC-1α is critical for maintaining podocyte health; however, increased mitochondrial fission by Drp1 has a detrimental effect on podocytes under cell stress. Transcriptional regulation of mitochondrial electron transport chain complexes is essential to meet the changing metabolic demand of podocytes. Although our understanding of the role of mitochondria in podocytes has dramatically improved in recent years, many questions remain unanswered. Podocytes are reported to undergo a metabolic switch depending on the extrinsic environment and disease state. It is therefore important to understand the factors that regulate this metabolic switch to identify therapeutic targets for various podocytopathies such as DKD and FSGS. In vitro podocyte cultures do not fully replicate the complex cell-cell interactions in glomeruli, and there is a dire need for better in vivo and ex vivo models to decipher the complex role of mitochondria in the podocytes.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants DK-102519 and DK-112984 and by Dialysis Clinic Incorporated (to S.K.M.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
N.A.G., J.M.V., and S.K.M. conceived and designed this review; N.A.G., J.M.V., and S.K.M. prepared figures; N.A.G., J.M.V., and S.K.M. drafted manuscript; N.A.G., J.M.V., D.F.B., and S.K.M. edited and revised manuscript; N.A.G., J.M.V., D.F.B., and S.K.M. approved final version of manuscript.
REFERENCES
- 1.Abe Y, Sakairi T, Beeson C, Kopp JB. TGF-β1 stimulates mitochondrial oxidative phosphorylation and generation of reactive oxygen species in cultured mouse podocytes, mediated in part by the mTOR pathway. Am J Physiol Renal Physiol 305: F1477–F1490, 2013. doi: 10.1152/ajprenal.00182.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abe Y, Sakairi T, Kajiyama H, Shrivastav S, Beeson C, Kopp JB. Bioenergetic characterization of mouse podocytes. Am J Physiol Cell Physiol 299: C464–C476, 2010. doi: 10.1152/ajpcell.00563.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Audzeyenka I, Rogacka D, Piwkowska A, Angielski S, Jankowski M. Viability of primary cultured podocytes is associated with extracellular high glucose-dependent autophagy downregulation. Mol Cell Biochem 430: 11–19, 2017. doi: 10.1007/s11010-017-2949-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ayanga BA, Badal SS, Wang Y, Galvan DL, Chang BH, Schumacker PT, Danesh FR. Dynamin-related protein 1 deficiency improves mitochondrial fitness and protects against progression of diabetic nephropathy. J Am Soc Nephrol 27: 2733–2747, 2016. doi: 10.1681/ASN.2015101096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barisoni L, Diomedi-Camassei F, Santorelli FM, Caridi G, Thomas DB, Emma F, Piemonte F, Ghiggeri GM. Collapsing glomerulopathy associated with inherited mitochondrial injury. Kidney Int 74: 237–243, 2008. doi: 10.1038/sj.ki.5002767. [DOI] [PubMed] [Google Scholar]
- 6.Bhargava P, Schnellmann RG. Mitochondrial energetics in the kidney. Nat Rev Nephrol 13: 629–646, 2017. doi: 10.1038/nrneph.2017.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boerries M, Grahammer F, Eiselein S, Buck M, Meyer C, Goedel M, Bechtel W, Zschiedrich S, Pfeifer D, Laloë D, Arrondel C, Gonçalves S, Krüger M, Harvey SJ, Busch H, Dengjel J, Huber TB. Molecular fingerprinting of the podocyte reveals novel gene and protein regulatory networks. Kidney Int 83: 1052–1064, 2013. doi: 10.1038/ki.2012.487. [DOI] [PubMed] [Google Scholar]
- 8.Brinkkoetter PT, Bork T, Salou S, Liang W, Mizi A, Özel C, Koehler S, Hagmann HH, Ising C, Kuczkowski A, Schnyder S, Abed A, Schermer B, Benzing T, Kretz O, Puelles VG, Lagies S, Schlimpert M, Kammerer B, Handschin C, Schell C, Huber TB. Anaerobic glycolysis maintains the glomerular filtration barrier independent of mitochondrial metabolism and dynamics. Cell Rep 27: 1551−1566.e5, 2019. doi: 10.1016/j.celrep.2019.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cantó C, Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr Opin Lipidol 20: 98–105, 2009. doi: 10.1097/MOL.0b013e328328d0a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang CR, Blackstone C. Dynamic regulation of mitochondrial fission through modification of the dynamin-related protein Drp1. Ann NY Acad Sci 1201: 34–39, 2010. doi: 10.1111/j.1749-6632.2010.05629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Che R, Yuan Y, Huang S, Zhang A. Mitochondrial dysfunction in the pathophysiology of renal diseases. Am J Physiol Renal Physiol 306: F367–F378, 2014. doi: 10.1152/ajprenal.00571.2013. [DOI] [PubMed] [Google Scholar]
- 12.Cheong HI, Chae JH, Kim JS, Park HW, Ha IS, Hwang YS, Lee HS, Choi Y. Hereditary glomerulopathy associated with a mitochondrial tRNA(Leu) gene mutation. Pediatr Nephrol 13: 477–480, 1999. doi: 10.1007/s004670050641. [DOI] [PubMed] [Google Scholar]
- 13.Chuang PY, Cai W, Li X, Fang L, Xu J, Yacoub R, He JC, Lee K. Reduction in podocyte SIRT1 accelerates kidney injury in aging mice. Am J Physiol Renal Physiol 313: F621–F628, 2017. doi: 10.1152/ajprenal.00255.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chuang PY, Xu J, Dai Y, Jia F, Mallipattu SK, Yacoub R, Gu L, Premsrirut PK, He JC. In vivo RNA interference models of inducible and reversible Sirt1 knockdown in kidney cells. Am J Pathol 184: 1940–1956, 2014. doi: 10.1016/j.ajpath.2014.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441: 1162–1166, 2006. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- 16.Diomedi-Camassei F, Di Giandomenico S, Santorelli FM, Caridi G, Piemonte F, Montini G, Ghiggeri GM, Murer L, Barisoni L, Pastore A, Muda AO, Valente ML, Bertini E, Emma F. COQ2 nephropathy: a newly described inherited mitochondriopathy with primary renal involvement. J Am Soc Nephrol 18: 2773–2780, 2007. doi: 10.1681/ASN.2006080833. [DOI] [PubMed] [Google Scholar]
- 17.Duann P, Lin PH. Mitochondria damage and kidney disease In: Mitochondrial Dynamics in Cardiovascular Medicine, edited by Santulli G. Cham, Switzerland: Springer, 2017, p. 529–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ducasa GM, Mitrofanova A, Mallela SK, Liu X, Molina J, Sloan A, Pedigo CE, Ge M, Santos JV, Hernandez Y, Kim JJ, Maugeais C, Mendez AJ, Nair V, Kretzler M, Burke GW, Nelson RG, Ishimoto Y, Inagi R, Banerjee S, Liu S, Szeto HH, Merscher S, Fontanesi F, Fornoni A. ATP-binding cassette A1 deficiency causes cardiolipin-driven mitochondrial dysfunction in podocytes. J Clin Invest 129: 3387–3400, 2019. doi: 10.1172/JCI125316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dugan LL, You YH, Ali SS, Diamond-Stanic M, Miyamoto S, DeCleves AE, Andreyev A, Quach T, Ly S, Shekhtman G, Nguyen W, Chepetan A, Le TP, Wang L, Xu M, Paik KP, Fogo A, Viollet B, Murphy A, Brosius F, Naviaux RK, Sharma K. AMPK dysregulation promotes diabetes-related reduction of superoxide and mitochondrial function. J Clin Invest 123: 4888–4899, 2013. doi: 10.1172/JCI66218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Eid AA, Gorin Y, Fagg BM, Maalouf R, Barnes JL, Block K, Abboud HE. Mechanisms of podocyte injury in diabetes: role of cytochrome P450 and NADPH oxidases. Diabetes 58: 1201–1211, 2009. doi: 10.2337/db08-1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Eirin A, Lerman A, Lerman LO. The emerging role of mitochondrial targeting in kidney disease. Handb Exp Pharmacol 240: 229–250, 2017. doi: 10.1007/164_2016_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Elgass K, Pakay J, Ryan MT, Palmer CS. Recent advances into the understanding of mitochondrial fission. Biochim Biophys Acta 1833: 150–161, 2013. doi: 10.1016/j.bbamcr.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 23.Fang L, Zhou Y, Cao H, Wen P, Jiang L, He W, Dai C, Yang J. Autophagy attenuates diabetic glomerular damage through protection of hyperglycemia-induced podocyte injury. PLoS One 8: e60546, 2013. doi: 10.1371/journal.pone.0060546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Feher J. (editor) 7.2—Functional anatomy of the kidneys and overview of kidney function In: Quantitative Human Physiology. Boston, MA: Academic, 2012, p. 626–632. [Google Scholar]
- 25.Galluzzi L, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cecconi F et al. Molecular definitions of autophagy and related processes. EMBO J 36: 1811–1836, 2017. doi: 10.15252/embj.201796697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Galvan DL, Green NH, Danesh FR. The hallmarks of mitochondrial dysfunction in chronic kidney disease. Kidney Int 92: 1051–1057, 2017. doi: 10.1016/j.kint.2017.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Galvan DL, Long J, Green N, Chang BH, Lin JS, Schumacker P, Truong LD, Overbeek P, Danesh FR. Drp1S600 phosphorylation regulates mitochondrial fission and progression of nephropathy in diabetic mice. J Clin Invest 129: 2807–2823, 2019. doi: 10.1172/JCI127277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Garrido C, Galluzzi L, Brunet M, Puig PE, Didelot C, Kroemer G. Mechanisms of cytochrome c release from mitochondria. Cell Death Differ 13: 1423–1433, 2006. doi: 10.1038/sj.cdd.4401950. [DOI] [PubMed] [Google Scholar]
- 29.Gasser DL, Winkler CA, Peng M, An P, McKenzie LM, Kirk GD, Shi Y, Xie LX, Marbois BN, Clarke CF, Kopp JB. Focal segmental glomerulosclerosis is associated with a PDSS2 haplotype and, independently, with a decreased content of coenzyme Q10. Am J Physiol Renal Physiol 305: F1228–F1238, 2013. doi: 10.1152/ajprenal.00143.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ghezzi D, Zeviani M. Human diseases associated with defects in assembly of OXPHOS complexes. Essays Biochem 62: 271–286, 2018. doi: 10.1042/EBC20170099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gödel M, Hartleben B, Herbach N, Liu S, Zschiedrich S, Lu S, Debreczeni-Mór A, Lindenmeyer MT, Rastaldi MP, Hartleben G, Wiech T, Fornoni A, Nelson RG, Kretzler M, Wanke R, Pavenstädt H, Kerjaschki D, Cohen CD, Hall MN, Rüegg MA, Inoki K, Walz G, Huber TB. Role of mTOR in podocyte function and diabetic nephropathy in humans and mice. J Clin Invest 121: 2197–2209, 2011. doi: 10.1172/JCI44774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Granata S, Dalla Gassa A, Tomei P, Lupo A, Zaza G. Mitochondria: a new therapeutic target in chronic kidney disease. Nutr Metab (Lond) 12: 49–49, 2015. doi: 10.1186/s12986-015-0044-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci USA 100: 4078–4083, 2003. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hagiwara M, Yamagata K, Capaldi RA, Koyama A. Mitochondrial dysfunction in focal segmental glomerulosclerosis of puromycin aminonucleoside nephrosis. Kidney Int 69: 1146–1152, 2006. doi: 10.1038/sj.ki.5000207. [DOI] [PubMed] [Google Scholar]
- 35.Han XJ, Lu YF, Li SA, Kaitsuka T, Sato Y, Tomizawa K, Nairn AC, Takei K, Matsui H, Matsushita M. CaM kinase I α-induced phosphorylation of Drp1 regulates mitochondrial morphology. J Cell Biol 182: 573–585, 2008. doi: 10.1083/jcb.200802164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Handschin C, Spiegelman BM. Peroxisome proliferator-activated receptor gamma coactivator 1 coactivators, energy homeostasis, and metabolism. Endocr Rev 27: 728–735, 2006. doi: 10.1210/er.2006-0037. [DOI] [PubMed] [Google Scholar]
- 37.Hartleben B, Gödel M, Meyer-Schwesinger C, Liu S, Ulrich T, Köbler S, Wiech T, Grahammer F, Arnold SJ, Lindenmeyer MT, Cohen CD, Pavenstädt H, Kerjaschki D, Mizushima N, Shaw AS, Walz G, Huber TB. Autophagy influences glomerular disease susceptibility and maintains podocyte homeostasis in aging mice. J Clin Invest 120: 1084–1096, 2010. doi: 10.1172/JCI39492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heeringa SF, Chernin G, Chaki M, Zhou W, Sloan AJ, Ji Z et al. COQ6 mutations in human patients produce nephrotic syndrome with sensorineural deafness. J Clin Invest 121: 2013–2024, 2011. doi: 10.1172/JCI45693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hong Q, Zhang L, Das B, Li Z, Liu B, Cai G, Chen X, Chuang PY, He JC, Lee K. Increased podocyte Sirtuin-1 function attenuates diabetic kidney injury. Kidney Int 93: 1330–1343, 2018. doi: 10.1016/j.kint.2017.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Horne SJ, Vasquez JM, Guo Y, Ly V, Piret SE, Leonardo AR, Ling J, Revelo MP, Bogenhagen D, Yang VW, He JC, Mallipattu SK. Podocyte-specific loss of Krüppel-like factor 6 increases mitochondrial injury in diabetic kidney disease. Diabetes 67: 2420–2433, 2018. doi: 10.2337/db17-0958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Imasawa T, Obre E, Bellance N, Lavie J, Imasawa T, Rigothier C, Delmas Y, Combe C, Lacombe D, Benard G, Claverol S, Bonneu M, Rossignol R. High glucose repatterns human podocyte energy metabolism during differentiation and diabetic nephropathy. FASEB J 31: 294−307, 2017. doi: 10.1096/fj.201600293R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Inoki K, Mori H, Wang J, Suzuki T, Hong S, Yoshida S, Blattner SM, Ikenoue T, Rüegg MA, Hall MN, Kwiatkowski DJ, Rastaldi MP, Huber TB, Kretzler M, Holzman LB, Wiggins RC, Guan KL. mTORC1 activation in podocytes is a critical step in the development of diabetic nephropathy in mice. J Clin Invest 121: 2181–2196, 2011. doi: 10.1172/JCI44771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jiang XS, Chen XM, Hua W, He JL, Liu T, Li XJ, Wan JM, Gan H, Du XG. PINK1/Parkin mediated mitophagy ameliorates palmitic acid-induced apoptosis through reducing mitochondrial ROS production in podocytes. Biochem Biophys Res Commun 525: 954–961, 2020. doi: 10.1016/j.bbrc.2020.02.170. [DOI] [PubMed] [Google Scholar]
- 44.Kawakami T, Gomez IG, Ren S, Hudkins K, Roach A, Alpers CE, Shankland SJ, D’Agati VD, Duffield JS. Deficient autophagy results in mitochondrial dysfunction and FSGS. J Am Soc Nephrol 26: 1040–1052, 2015. doi: 10.1681/ASN.2013111202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392: 605–608, 1998. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 46.Lemasters JJ, Nieminen AL, Qian T, Trost LC, Elmore SP, Nishimura Y, Crowe RA, Cascio WE, Bradham CA, Brenner DA, Herman B. The mitochondrial permeability transition in cell death: a common mechanism in necrosis, apoptosis and autophagy. Biochim Biophys Acta 1366: 177–196, 1998. doi: 10.1016/S0005-2728(98)00112-1. [DOI] [PubMed] [Google Scholar]
- 47.Lenoir O, Jasiek M, Hénique C, Guyonnet L, Hartleben B, Bork T, Chipont A, Flosseau K, Bensaada I, Schmitt A, Massé JM, Souyri M, Huber TB, Tharaux PL. Endothelial cell and podocyte autophagy synergistically protect from diabetes-induced glomerulosclerosis. Autophagy 11: 1130–1145, 2015. doi: 10.1080/15548627.2015.1049799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Li C, Ge Y, Dworkin L, Peng A, Gong R. The β isoform of GSK3 mediates podocyte autonomous injury in proteinuric glomerulopathy. J Pathol 239: 23–35, 2016. doi: 10.1002/path.4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li SY, Park J, Qiu C, Han SH, Palmer MB, Arany Z, Susztak K. Increasing the level of peroxisome proliferator-activated receptor γ coactivator-1α in podocytes results in collapsing glomerulopathy. JCI Insight 2: e92930, 2017. doi: 10.1172/jci.insight.92930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li SY, Susztak K. The role of peroxisome proliferator-activated receptor γ coactivator 1α (PGC-1α) in kidney disease. Semin Nephrol 38: 121–126, 2018. doi: 10.1016/j.semnephrol.2018.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li SY, Susztak K. The long noncoding RNA Tug1 connects metabolic changes with kidney disease in podocytes. J Clin Invest 126: 4072–4075, 2016. doi: 10.1172/JCI90828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Li W, Du M, Wang Q, Ma X, Wu L, Guo F, Ji H, Huang F, Qin G. FoxO1 promotes mitophagy in the podocytes of diabetic male mice via the PINK1/Parkin pathway. Endocrinology 158: 2155–2167, 2017. doi: 10.1210/en.2016-1970. [DOI] [PubMed] [Google Scholar]
- 53.Li X, Tao H, Xie K, Ni Z, Yan Y, Wei K, Chuang PY, He JC, Gu L. cAMP signaling prevents podocyte apoptosis via activation of protein kinase A and mitochondrial fusion. PLoS One 9: e92003–e92003, 2014. doi: 10.1371/journal.pone.0092003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu J, Li QX, Wang XJ, Zhang C, Duan YQ, Wang ZY, Zhang Y, Yu X, Li NJ, Sun JP, Yi F. β-Arrestins promote podocyte injury by inhibition of autophagy in diabetic nephropathy. Cell Death Dis 7: e2183–e2183, 2016. doi: 10.1038/cddis.2016.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu R, Zhong Y, Li X, Chen H, Jim B, Zhou MM, Chuang PY, He JC. Role of transcription factor acetylation in diabetic kidney disease. Diabetes 63: 2440–2453, 2014. doi: 10.2337/db13-1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu W, Acín-Peréz R, Geghman KD, Manfredi G, Lu B, Li C. Pink1 regulates the oxidative phosphorylation machinery via mitochondrial fission. Proc Natl Acad Sci USA 108: 12920–12924, 2011. doi: 10.1073/pnas.1107332108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Long J, Badal SS, Ye Z, Wang Y, Ayanga BA, Galvan DL, Green NH, Chang BH, Overbeek PA, Danesh FR. Long noncoding RNA Tug1 regulates mitochondrial bioenergetics in diabetic nephropathy. J Clin Invest 126: 4205–4218, 2016. doi: 10.1172/JCI87927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.López LC, Schuelke M, Quinzii CM, Kanki T, Rodenburg RJ, Naini A, Dimauro S, Hirano M. Leigh syndrome with nephropathy and CoQ10 deficiency due to decaprenyl diphosphate synthase subunit 2 (PDSS2) mutations. Am J Hum Genet 79: 1125–1129, 2006. doi: 10.1086/510023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lynch MR, Tran MT, Parikh SM. PGC1α in the kidney. Am J Physiol Renal Physiol 314: F1–F8, 2018. doi: 10.1152/ajprenal.00263.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Mallipattu SK, Horne SJ, D’Agati V, Narla G, Liu R, Frohman MA, Dickman K, Chen EY, Ma’ayan A, Bialkowska AB, Ghaleb AM, Nandan MO, Jain MK, Daehn I, Chuang PY, Yang VW, He JC. Krüppel-like factor 6 regulates mitochondrial function in the kidney. J Clin Invest 125: 1347–1361, 2015. doi: 10.1172/JCI77084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Mishra P, Chan DC. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol 15: 634–646, 2014. doi: 10.1038/nrm3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Müller-Deile J, Schiffer M. The podocyte power-plant disaster and its contribution to glomerulopathy. Front Endocrinol (Lausanne) 5: 209–209, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Murphy E, Ardehali H, Balaban RS, DiLisa F, Dorn GW II, Kitsis RN, Otsu K, Ping P, Rizzuto R, Sack MN, Wallace D, Youle RJ; American Heart Association Council on Basic Cardiovascular Sciences, Council on Clinical Cardiology, and Council on Functional Genomics and Translational Biology . Mitochondrial function, biology, and role in disease: a scientific statement from the American Heart Association. Circ Res 118: 1960–1991, 2016. doi: 10.1161/RES.0000000000000104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Narendra DP, Youle RJ. Targeting mitochondrial dysfunction: role for PINK1 and Parkin in mitochondrial quality control. Antioxid Redox Signal 14: 1929–1938, 2011. doi: 10.1089/ars.2010.3799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nguyen TN, Padman BS, Lazarou M. Deciphering the molecular signals of PINK1/Parkin mitophagy. Trends Cell Biol 26: 733–744, 2016. doi: 10.1016/j.tcb.2016.05.008. [DOI] [PubMed] [Google Scholar]
- 66.Ni Z, Tao L, Xiaohui X, Zelin Z, Jiangang L, Zhao S, Weikang H, Hongchao X, Qiujing W, Xin L. Polydatin impairs mitochondria fitness and ameliorates podocyte injury by suppressing Drp1 expression. J Cell Physiol 232: 2776–2787, 2017. doi: 10.1002/jcp.25943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.O’Toole JF. Renal manifestations of genetic mitochondrial disease. Int J Nephrol Renovasc Dis 7: 57–67, 2014. doi: 10.2147/IJNRD.S37887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ozawa S, Ueda S, Imamura H, Mori K, Asanuma K, Yanagita M, Nakagawa T. Glycolysis, but not mitochondria, responsible for intracellular ATP distribution in cortical area of podocytes. Sci Rep 5: 18575, 2015. doi: 10.1038/srep18575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Palorini R, De Rasmo D, Gaviraghi M. Oncogenic K-ras expression is associated with derangement of the cAMP/PKA pathway and forskolin-reversible alterations of mitochondrial dynamics and respiration. Oncogene 32: 352−362, 2013. doi: 10.1038/onc.2012.50. [DOI] [PubMed] [Google Scholar]
- 70.Park E, Ahn YH, Kang HG, Yoo KH, Won NH, Lee KB, Moon KC, Seong MW, Gwon TR, Park SS, Cheong HI. COQ6 mutations in children with steroid-resistant focal segmental glomerulosclerosis and sensorineural hearing loss. Am J Kidney Dis 70: 139–144, 2017. doi: 10.1053/j.ajkd.2016.10.040. [DOI] [PubMed] [Google Scholar]
- 71.Qi H, Casalena G, Shi S, Yu L, Ebefors K, Sun Y, Zhang W, D’Agati V, Schlondorff D, Haraldsson B, Böttinger E, Daehn I. Glomerular endothelial mitochondrial dysfunction is essential and characteristic of diabetic kidney disease susceptibility. Diabetes 66: 763–778, 2017. doi: 10.2337/db16-0695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Quinzii CM, Hirano M. Coenzyme Q and mitochondrial disease. Dev Disabil Res Rev 16: 183–188, 2010. doi: 10.1002/ddrr.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rinschen MM, Gödel M, Grahammer F, Zschiedrich S, Helmstädter M, Kretz O, Zarei M, Braun DA, Dittrich S, Pahmeyer C, Schroder P, Teetzen C, Gee H, Daouk G, Pohl M, Kuhn E, Schermer B, Küttner V, Boerries M, Busch H, Schiffer M, Bergmann C, Krüger M, Hildebrandt F, Dengjel J, Benzing T, Huber TB. A multi-layered quantitative in vivo expression atlas of the podocyte unravels kidney disease candidate genes. Cell Rep 23: 2495–2508, 2018. doi: 10.1016/j.celrep.2018.04.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sheridan C, Martin SJ. Mitochondrial fission/fusion dynamics and apoptosis. Mitochondrion 10: 640–648, 2010. doi: 10.1016/j.mito.2010.08.005. [DOI] [PubMed] [Google Scholar]
- 75.Signes A, Fernandez-Vizarra E. Assembly of mammalian oxidative phosphorylation complexes I-V and supercomplexes. Essays Biochem 62: 255–270, 2018. doi: 10.1042/EBC20170098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stieger N, Worthmann K, Teng B, Engeli S, Das AM, Haller H, Schiffer M. Impact of high glucose and transforming growth factor-β on bioenergetic profiles in podocytes. Metabolism 61: 1073–1086, 2012. doi: 10.1016/j.metabol.2011.12.003. [DOI] [PubMed] [Google Scholar]
- 77.Susztak K, Raff AC, Schiffer M, Böttinger EP. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes 55: 225–233, 2006. doi: 10.2337/diabetes.55.01.06.db05-0894. [DOI] [PubMed] [Google Scholar]
- 78.Tagawa A, Yasuda M, Kume S, Yamahara K, Nakazawa J, Chin-Kanasaki M, Araki H, Araki S, Koya D, Asanuma K, Kim EH, Haneda M, Kajiwara N, Hayashi K, Ohashi H, Ugi S, Maegawa H, Uzu T. Impaired podocyte autophagy exacerbates proteinuria in diabetic nephropathy. Diabetes 65: 755–767, 2016. doi: 10.2337/db15-0473. [DOI] [PubMed] [Google Scholar]
- 79.Tooze SA, Yoshimori T. The origin of the autophagosomal membrane. Nat Cell Biol 12: 831–835, 2010. doi: 10.1038/ncb0910-831. [DOI] [PubMed] [Google Scholar]
- 80.Wang W, Wang Y, Long J, Wang J, Haudek SB, Overbeek P, Chang BH, Schumacker PT, Danesh FR. Mitochondrial fission triggered by hyperglycemia is mediated by ROCK1 activation in podocytes and endothelial cells. Cell Metab 15: 186–200, 2012. doi: 10.1016/j.cmet.2012.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang Z, Bao H, Ge Y, Zhuang S, Peng A, Gong R. Pharmacological targeting of GSK3β confers protection against podocytopathy and proteinuria by desensitizing mitochondrial permeability transition. Br J Pharmacol 172: 895–909, 2015. doi: 10.1111/bph.12952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wu M, Li S, Yu X, Chen W, Ma H, Shao C, Zhang Y, Zhang A, Huang S, Jia Z. Mitochondrial activity contributes to impaired renal metabolic homeostasis and renal pathology in STZ-induced diabetic mice. Am J Physiol Renal Physiol 317: F593–F605, 2019. doi: 10.1152/ajprenal.00076.2019. [DOI] [PubMed] [Google Scholar]
- 83.Xie K, Zhu M, Xiang P, Chen X, Kasimumali A, Lu R, Wang Q, Mou S, Ni Z, Gu L, Pang H. Protein kinase A/CREB signaling prevents adriamycin-induced podocyte apoptosis via upregulation of mitochondrial respiratory chain complexes. Mol Cell Biol 38: e00181–e00117, 2017. doi: 10.1128/MCB.00181-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nat Rev Mol Cell Biol 12: 9–14, 2011. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yuan Y, Huang S, Zhang A. Role of mitochondria in podocyte injury. Contrib Nephrol 183: 64–82, 2014. [Google Scholar]
- 86.Yuan Y, Zhang A, Qi J, Wang H, Liu X, Zhao M, Duan S, Huang Z, Zhang C, Wu L, Zhang B, Zhang A, Xing C. p53/Drp1-dependent mitochondrial fission mediates aldosterone-induced podocyte injury and mitochondrial dysfunction. Am J Physiol Renal Physiol 314: F798–F808, 2018. doi: 10.1152/ajprenal.00055.2017. [DOI] [PubMed] [Google Scholar]
- 87.Zhan M, Brooks C, Liu F, Sun L, Dong Z. Mitochondrial dynamics: regulatory mechanisms and emerging role in renal pathophysiology. Kidney Int 83: 568–581, 2013. doi: 10.1038/ki.2012.441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Zhang LH, Zhu XY, Eirin A, Nargesi AA, Woollard JR, Santelli A, Sun IO, Textor SC, Lerman LO. Early podocyte injury and elevated levels of urinary podocyte-derived extracellular vesicles in swine with metabolic syndrome: role of podocyte mitochondria. Am J Physiol Renal Physiol 317: F12–F22, 2019. doi: 10.1152/ajprenal.00399.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zhang T, Chi Y, Ren Y, Du C, Shi Y, Li Y. Resveratrol reduces oxidative stress and apoptosis in podocytes via Sir2-related enzymes, sirtuins1 (SIRT1)/peroxisome proliferator-activated receptor γ co-activator 1α (PGC-1α) axis. Med Sci Monit 25: 1220–1231, 2019. doi: 10.12659/MSM.911714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhong Y, Lee K, He JC. SIRT1 is a potential drug target for treatment of diabetic kidney disease. Front Endocrinol (Lausanne) 9: 624, 2018. doi: 10.3389/fendo.2018.00624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Zhu C, Huang S, Yuan Y, Ding G, Chen R, Liu B, Yang T, Zhang A. Mitochondrial dysfunction mediates aldosterone-induced podocyte damage: a therapeutic target of PPARγ. Am J Pathol 178: 2020–2031, 2011. doi: 10.1016/j.ajpath.2011.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Zhu C, Xuan X, Che R, Ding G, Zhao M, Bai M, Jia Z, Huang S, Zhang A. Dysfunction of the PGC-1α-mitochondria axis confers adriamycin-induced podocyte injury. Am J Physiol Renal Physiol 306: F1410–F1417, 2014. doi: 10.1152/ajprenal.00622.2013. [DOI] [PubMed] [Google Scholar]
- 93.Zorov DB, Juhaszova M, Sollott SJ. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev 94: 909–950, 2014. doi: 10.1152/physrev.00026.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]