

Keywords: AMP-activated protein kinase, apoptosis, Bax, caspase 3, cisplatin, nephrotoxicity, p53
Abstract
Cisplatin, a commonly used anticancer drug, has been shown to induce acute kidney injury, which limits its clinical use in cancer treatment. Emerging evidence has suggested that AMP-activated protein kinase (AMPK), which functions as a cellular energy sensor, is activated by various cellular stresses that deplete cellular ATP. However, the potential role of AMPK in cisplatin-induced apoptosis of renal tubular epithelial cells has not been studied. In this study, we demonstrated that cisplatin activates AMPK (Thr172 phosphorylation) in cultured renal tubular epithelial cells in a time-dependent manner, which was associated with p53 phosphorylation. Compound C, a selective AMPK inhibitor, suppressed cisplatin-induced AMPK activation, p53 phosphorylation, Bax induction, and caspase 3 activation. Furthermore, silencing AMPK expression by siRNA attenuated cisplatin-induced p53 phosphorylation, Bax induction, and caspase 3 activation. In a mouse model of cisplatin-induced kidney injury, compound C inhibited p53 phosphorylation, Bax expression, caspase 3 activation, and apoptosis, protecting the kidney from injury and dysfunction. Taken together, these results suggest that the AMPK-p53-Bax signaling pathway plays a crucial role in cisplatin-induced tubular epithelial cell apoptosis.
INTRODUCTION
Cisplatin is a widely used and highly effective chemotherapeutic agent for the treatment of various solid-organ malignancies such as lung, head and neck, testicular, and ovarian cancer (8). One of the most common dose-limiting factors of cisplatin is nephrotoxicity (10). Cisplatin exerts its therapeutic effect by reacting with DNA, creating intrastrand DNA-cisplatin cross-links that cause cytotoxic lesions (8). Direct DNA damage, inflammatory injury, and oxidative stress have been proposed as the mechanisms of cisplatin-induced renal injury [acute kidney injury (AKI)] (2, 10, 18, 23, 36). Tubular epithelial cells are the most affected cell type during cisplatin-induced nephrotoxicity. The damage to the tubular cells includes necrosis and apoptosis, which depend on the concentration of cisplatin (10). However, the exact molecular mechanism underlying cisplatin-induced nephrotoxicity is not fully understood.
Recent evidence indicates that p53 is involved in the cisplatin-induced apoptosis in kidney tubular cells (11, 13, 16, 26). The cisplatin-induced apoptosis is ameliorated by inhibition of p53, including pharmacological inhibitors, deacetylation, and gene deficiency. Activated p53 exerts its transcriptional function to increase the expression of Bcl-2 family proteins (3, 4). It has been reported that p53 contributes to the cisplatin-induced apoptosis in tubular epithelial cells through transcriptional activation of Bax, which results in cytochrome c release and cell apoptosis (34).
It remains unclear how cisplatin activates p53 in tubular cells during its nephrotoxicity. The activation of p53 was determined by both phosphorylation of its NH2-terminal end to prevent mouse double minute 2 homolog binding and acetylation of its COOH-terminal end, which expose its DNA-binding domain for transcriptional function (3, 9). It has been reported that AMP-activated protein kinase (AMPK) could contribute to the activation of p53 (1, 9, 21, 35). AMPK is a ubiquitous serine/threonine kinase and a heterotrimeric protein, composed of a catalytic α-subunit and two regulatory (β and γ) subunits. Each subunit has multiple isoforms (α1, α2, β1, β2, γ1, γ2, and γ3) with different subcellular and tissue distributions (15). The catalytic α-subunit is composed of three functional domains, including an NH2-terminal serine/threonine protein kinase domain, a central autoinhibitory region, and a COOH-terminal regulatory subunit-binding domain. AMPK functions as an intracellular energy sensor by monitoring cellular energy levels. Under conditions in which intracellular ATP is reduced and AMP level rises, AMP activates AMPK allosterically, which switches off anabolic pathways and turns on catabolic pathways that generate ATP, thereby maintaining energy balance within cells (15). In addition to allosteric activation, AMPK can be activated by phosphorylation of the α-subunit at Thr172 by several upstream kinases including liver kinase B1 (LKB1) (6), Ca2+/calmodulin-dependent protein kinase (7, 8), and transforming growth factor-β1 activated kinase-1 (TAK1) (15). AMPK activation triggers a phosphorylation cascade that regulates the activity of various downstream targets including transcription factors such as p53 (28). Therefore, AMPK may mediate the activation of p53 in cisplatin-induced tubular epithelial cell apoptosis.
In this study, we discovered that AMPK plays an important role in cisplatin-induced tubular epithelial cell apoptosis both in vitro and in vivo. Cisplatin activates AMPK. Activation of AMPK results in phosphorylation of p53, which promotes Bax transcription and subsequent caspase 3 activation and tubular epithelial cell apoptosis. Inhibition of AMPK suppresses p53 activation, Bax induction, caspase 3 activation, and tubular epithelial cell apoptosis and protects the kidney from cisplatin-induced kidney dysfunction.
MATERIALS AND METHODS
Chemicals and reagents.
cis-Diammineplatinum (II) dichloride (cisplatin) and 6-[4-(2-piperidin-1-ylethoxy) phenyl]-3-pyridin-4-ylpyrazolo-[1,5-a]-pyrimidine (compound C; AMPK inhibitor) were purchased from Sigma-Aldrich (St. Louis, MO). The antibodies used in this study included AMPK-α1/2, phosphorylated AMPK1/2, p53, phosphorylated p53, and cleaved caspase 3 from Cell Signaling Technology (Danvers, MA); monoclonal anti-Bax from BioGenex (San Ramon, CA); and monoclonal anti-GAPDH from Sigma-Aldrich. All antibodies have been validated in our prior publications (12, 33, 36). The In Situ Cell Death Detection Kit was purchased from Roche Applied Science (Millipore). The QuantiChrom Creatinine Assay Kit (DICT 500) was purchased from BioAssay Systems (Hayward, CA). All siRNA reagents including AMPK-α1/2 siRNA (sc-45313), control siRNA (sc-37007), siRNA transfection reagent (sc-29528), and siRNA transfection medium (sc-36868) were purchased from Santa Cruz Biotechnology (Dallas, TX).
Cell culture.
A mouse kidney epithelial cell line (TCMK-1, CCL-139) was purchased from the American Type Culture Collection (Manassas, VA) and cultured in DMEM supplemented with 10% FBS and 1% antibiotics in a 37°C incubator with an atmosphere of 5% CO2. TCMK-1 cells were retrieved using 0.25% trypsin and then seeded on 6-cm dishes at 30,000 cells/well. After overnight culture, cells at 80% confluence were used for experiments.
Cisplatin treatment of TCMK-1 cells.
Cells were starved for 12 h in DMEM containing 1% FBS before cisplatin treatment. Cisplatin was freshly prepared by dissolving in DMSO at 100 mM, and an appropriate volume of cisplatin was then added directly to the culture medium with 1% FBS to get the desired concentration. An equal volume of DMSO was used as a control. After incubation for 18–20 h, cells were monitored morphologically and harvested for further biochemical analyses. For cell lysis, both floating and adherent cells were collected.
Western blot analysis.
Total proteins were extracted from cultured cells using Pierce RIPA Buffer (ThermoFisher Scientific, Rockford, IL) containing Halt Protease and Phosphatase Inhibitor Cocktail and 5 mM EDTA solution (ThermoFisher Scientific) (19). Protein concentrations were determined using the Bio-Rad Protein Assay (Bio-Rad Laboratories). Equal amounts of proteins were separated on SDS-polyacrylamide gels and transferred onto nitrocellulose membrane. After being blocked, membranes were incubated with primary antibodies. The appropriate secondary antibodies conjugated with fluorescent molecules were applied on the membranes to visualize the corresponding proteins using an Odyssey Imaging System (LI-COR Biosciences, Lincoln, NE).
siRNA transfection.
TCMK-1 cells were seeded on six-well plates with 2 × 105 cells/well in 2 mL of media containing 10% FBS without antibiotics and incubated for 24 h. Cells were transfected with AMPK-α1/2 siRNA or control siRNA according to the manufacturer’s protocol. Briefly, cells were transfected by incubating with a mixture of 25 nM siRNA, 5 μL of transfection reagents, and 100 μL of transfection medium. After incubation at 37°C in a 5% CO2 incubator for 6 h, normal medium was added and incubated for additional 24 h. Cells were treated with cisplatin as indicated.
Animals.
Animal experiments were conducted according to the “Guidelines of Laboratory Animal Care” and were approved by the Institutional Animal Care and Use Committee of Baylor College of Medicine. Wild-type C57BL/6 male mice were purchased from the Jackson Laboratory (Bar Harbor, ME). Cisplatin was resolved in 0.9% sodium chloride at 1 mg/mL. Compound C was suspended in sesame oil through vigorous stirring. Mice (aged 8–10 wk) were orally administrated compound C (20 mg/kg) in sesame oil. Sesame oil without compound C was administrated orally by equitable volume as a vehicle control. One hour after the administration of compound C or vehicle, cisplatin was injected intraperitoneally at 20 mg/kg. The same volume of saline was injected intraperitoneally as a control. Compound C was administered daily for 3 days. Mice were euthanized 72 h after cisplatin treatment with free access to water and food.
Measurement of kidney function.
Serum creatinine was measured using a creatinine assay kit (BioAssay Systems) as previously reported (12). Blood urea nitrogen was determined fluorometrically as previously described (12).
Kidney morphology.
Kidney tissues were fixed in 10% buffered formalin, embedded in paraffin, and cut into 4-mm-thick sections. After deparaffinization and rehydration, sections were stained with hematoxylin and eosin. Tissue damage was examined in a blinded manner and scored according to the percentage of damaged tubules: 0, no damage; 1, 1–25% damage; 2, 25–50% damage; 3, 50–75% damage; and 4, 75–100% damage, as previously reported (12).
Immunohistochemistry.
Immunohistochemistry staining was performed on paraffin sections (5, 32). Antigen retrieval was performed with antigen unmasking solution (Vector Laboratories, Burlingame, CA). Endogenous peroxidase activity was quenched with 3% hydrogen peroxide. After being blocked with 5% normal serum, slides were incubated with primary antibodies in a humidified chamber overnight. After being washed, slides were incubated with the appropriate secondary antibodies and ABC solution sequentially according to the ABC kit protocol (Vector Laboratories). Slides were then visualized by incubation in diaminobenzidine solution. Nuclear staining was performed with hematoxylin. Slides were dehydrated, cleared, and mounted. Images from these slides were obtained and analyzed by NIS Element software (Nikon Instruments, Melville, NY) with a Nikon microscope imaging system (Nikon Instruments).
Apoptosis detection in the kidney.
A TUNEL assay was performed to evaluate apoptosis using an ApopTag plus Peroxidase In Situ Apoptosis Detection Kit (Millipore, Billerica, MA) according to the manufacturer’s instruction (12). Numbers of TUNEL-positive cells per high-power field were counted and analyzed in a blinded manner.
Statistical analysis.
Qualitative data obtained in this study are expressed as means ± SE. One-way ANOVA followed by Tukey’s post hoc test was used to compare multiple treatment groups. P < 0.05 was considered a significant difference.
RESULTS
Cisplatin activates AMPK in kidney tubular epithelial cells.
To determine whether cisplatin can activate AMPK in kidney tubular epithelial cells, TCMK-1 cells were treated with cisplatin at 50 μM for different periods of time. Western blot analysis showed that cisplatin treatment resulted in AMPK activation identified as increased AMPK-α phosphorylation in a time-dependent manner, which occurred as early as 30 min and peaked at 2 h (Fig. 1, A and B).
Fig. 1.

Cisplatin activates AMP-activated protein kinase-α (AMPK-α) and p53 in tubular epithelial cells. A: representative Western blots showing phosphorylated (p-)AMPK-α, AMPK-α, p-p53, p53, and GAPDH 9 loading control in tubular epithelial cells. B: quantitative analysis of p-AMPK-α/AMPK-α in tubular epithelial cells. **P < 0.01. C: quantitative analysis of p-p53/p53 in tubular epithelial cells. **P < 0.01.
Since p53 is critically involved in cisplatin-induced tubular epithelial cell apoptosis, we examined whether there is a temporal relationship between AMPK and p53. Western blot analysis revealed that the activation of p53, as indicated by p53 phosphorylation, followed the pattern of AMPK activation (Fig. 1, A and C).
Inhibition of AMPK with compound C blocks p53 phosphorylation, Bax induction, and caspase 3 activation induced by cisplatin in tubular epithelial cells.
To determine the role of AMPK in cisplatin-induced p53 activation, TCMK-1 cells were pretreated with compound C (10 μM), a selective AMPK inhibitor, for 30 min and then treated with cisplatin at 50 μM for 2 h. The results showed that compound C significantly attenuated cisplatin-induced p53 phosphorylation in tubular epithelial cells (Fig. 2, A and B).
Fig. 2.

Compound C blocks p53 phosphorylation, Bax induction, and caspase 3 activation induced by cisplatin in tubular epithelial cells. A: representative Western blots showing phosphorylated (p-)p53, p53, Bax, cleaved caspase 3, and GAPDH in tubular epithelial cells. B: quantitative analysis of p-p53/p53 in tubular epithelial cells. **P < 0.01. C: quantitative analysis of Bax expression in tubular epithelial cells. **P < 0.01. D: quantitative analysis of cleaved caspase 3 in tubular epithelial cells. **P < 0.01.
Because p53 phosphorylation induces Bax induction and caspase 3 activation in cisplatin-induced tubular epithelial cell apoptosis, we then assessed whether inhibition of AMPK with compound C affects Bax expression and caspase 3 activation. TCMK-1 cells were pretreated with compound C (10 μM) or vehicle for 30 min and then treated with cisplatin (50 μM) for 24 h. Western blot analysis demonstrated that inhibition of AMPK with compound C markedly suppressed cisplatin-induced Bax expression and caspase 3 activation in tubular epithelial cells (Fig. 2, A, C, and D).
Knockdown of AMPK-α with siRNA abolishes p53 phosphorylation, Bax induction, and caspase 3 activation in tubular epithelial cells.
To further evaluate the role of AMPK-α in cisplatin-induced p53 phosphorylation, Bax induction, and caspase 3 activation, TCMK-1 cells were transfected with siRNA to knock down AMPK-α expression. Cells transfected with scrambled siRNA were used as a control. Twenty-four hours after transfection, the AMPK-α protein expression level was significantly reduced in cells transfected with siRNA against AMPK-α compared with scrambled siRNA (Fig. 3, A and B). These cells were then treated with 50 μM cisplatin or vehicle. Western blot analysis showed that knockdown of AMPK-α considerably inhibited p53 phosphorylation, Bax expression, and caspase 3 activation (Fig. 3, A and C–E).
Fig. 3.

Knockdown of AMP-activated protein kinase-α (AMPK-α) inhibits p53 phosphorylation, Bax induction, and caspase 3 activation induced by cisplatin in tubular epithelial cells. A: representative Western blots showing AMPK-α, phosphorylated (p-)p53, p53, Bax, cleaved caspase 3 (C-caspase 3), and GAPDH in tubular epithelial cells. B: quantitative analysis of AMPK-α expression levels in tubular epithelial cells transfected with scrambled control siRNA or AMPK-α siRNA. **P < 0.01. C: quantitative analysis of p-p53 in tubular epithelial cells. **P < 0.01. D: quantitative analysis of Bax expression in tubular epithelial cells. **P < 0.01. E: quantitative analysis of cleaved caspase 3 in tubular epithelial cells. **P < 0.01.
Compound C inhibits p53 activation and Bax expression in the kidney during cisplatin-induced AKI.
To investigate whether AMPK has a role in p53 activation in vivo, wild-type mice on a C57/BL6J background were treated with compound C or vehicle daily for 3 days in a well-characterized model of cisplatin-induced AKI (36). Immunohistochemical analysis with an antibody against phosphorylated p53 showed that cisplatin treatment resulted in a marked increase in p53 phosphorylation in the kidney, which was significantly inhibited by compound C (Fig. 4, A and B). These results indicate that AMPK mediates p53 activation in the kidney in vivo.
Fig. 4.

Compound C inhibits p53 activation and Bax expression in the kidney during cisplatin-induced acute kidney injury. A: representative photomicrographs of kidney sections stained for phosphorylated (p-)p53 (brown) and counterstained with hematoxylin (blue). Scale bar = 50 μm. B: quantitative analysis of positive p-p53 staining in kidney sections. **P < 0.01. C: representative photomicrographs of kidney sections stained for Bax (brown) and counterstained with hematoxylin (blue). Scale bar = 50 μm. D: quantitative analysis of positive Bax staining in the kidney sections. **P < 0.01. HPF, high-powered field.
We next performed immunohistochemical staining to examine the expression level of Bax, a downstream target of p53, in the kidney in cisplatin-induced AKI. The results showed that Bax protein levels in the kidney increased considerably in cisplatin-induced AKI, whereas compound C administration significantly reduced Bax protein induction in the kidney with cisplatin-induced AKI (Fig. 4, C and D).
Compound C suppresses caspase 3 activation and apoptosis in the kidney with cisplatin-induced AKI.
Induction of Bax results in caspase 3 activation. We conducted immunohistochemical staining to determine the effect of compound C on caspase 3 activation in cisplatin-induced AKI. The results revealed that cisplatin treatment resulted in a marked increase in cleaved caspase, a marker for caspase 3 activation (Fig. 5, A and B). Administration of compound C considerably reduced cisplatin-induced caspase 3 activation, as reflected in the reduction of cleaved caspase 3 levels in the kidney.
Fig. 5.

Compound C suppresses caspase 3 activation and apoptosis in the kidney with cisplatin-induced acute kidney injury. A: representative photomicrographs of kidney sections stained for cleaved caspase 3 (brown) and counterstained with hematoxylin (blue). Scale bar = 50 μm. B: quantitative analysis of positive cleaved caspase 3 staining in kidneys. **P < 0.01. C: representative photomicrographs of kidney sections stained for apoptotic cells (brown) and counterstained with methyl green (green). Scale bar = 50 μm. D: quantitative analysis of apoptotic cells in kidneys. **P < 0.01. HPF, high-powered field.
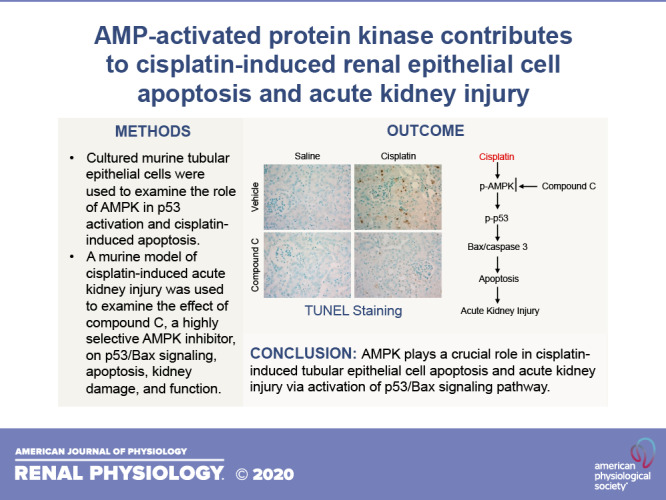
Activation of caspase 3 causes tubular epithelial cell apoptosis, contributing to cisplatin-induced AKI. We next performed a TUNEL assay to examine the effect of compound C on cisplatin-induced apoptotic cell death in the kidney. The results showed that cisplatin caused severe apoptotic cell death in the kidney, and the numbers of apoptotic cells in the kidney were significantly suppressed by compound C (Fig. 5, C and D).
Compound C protects against cisplatin-induced AKI.
To determine the role of AMPK in cisplatin-induced AKI in vivo, hematoxylin and eosin staining was performed. The results demonstrated that cisplatin induced severe tubular damage, displaying tubular dilation, cast formation, sloughing of tubular epithelial cells, and loss of the brush border in the kidney (Fig. 6, A and B). In comparison, administration of compound C significantly attenuated the severity of tubular damage in the kidney of cisplatin-treated mice. Furthermore, compared with mice treated with saline, cisplatin-treated mice displayed significant kidney dysfunction, as reflected by elevated serum creatinine and blood urea nitrogen (Fig. 6, C and D). In contrast, mice treated with compound C exhibited less severe kidney dysfunction with lower serum creatinine and blood urea nitrogen in cisplatin-induced AKI.
Fig. 6.

Compound C protects the kidney from cisplatin-induced acute kidney injury. A: representative photomicrographs of kidney sections stained with hematoxylin and eosin. Scale bar = 50 μm. B: quantitative assessment of tubular damage induced by cisplatin treatment. **P < 0.01. C: effect of compound C on serum creatinine. **P < 0.01. D: effect of compound C on blood urea nitrogen. **P < 0.01.
DISCUSSION
Cisplatin is a widely used antineoplasm drug, which has a considerable side effect on the kidney. At present, the molecular mechanism of cisplatin-induced nephrotoxicity is not well understood. In this study, we demonstrated that AMPK plays an important role in cisplatin-induced apoptosis in tubular epithelial cells in vitro and in vivo. Inhibition of AMPK with compound C or knockdown of AMPK with siRNA attenuates cisplatin-induced p53 phosphorylation, Bax induction, and caspase 3 activation in tubular epithelial cells in vitro. Administration of compound C inhibits p53 phosphorylation, Bax induction, caspase 3 activation, and apoptosis, protecting the kidney from cisplatin-induced AKI. These findings suggest that AMPK is involved in the cisplatin-induced nephrotoxicity through activation of p53 signaling pathway.
It has been previously reported that cisplatin produced the nephrotoxicity in kidney tubular cells through activation of the proapoptosis signaling pathway of p53 (20, 31). However, it is not known how p53 is activated in cisplatin-induced apoptosis. In this study, we demonstrated that cisplatin activated AMPK in a time-dependent manner, which was followed by the activation of p53 in tubular epithelial cells. These findings suggest that AMPK may be involved in cisplatin-induced p53 activation in tubular epithelial cells. Furthermore, pharmacological inhibition of AMPK-α with compound C or molecular knockdown of AMPK-α abolishes cisplatin-induced p53 phosphorylation, Bax induction, and caspase 3 activation in tubular epithelial cells. These results strongly support that AMPK is critically involved in cisplatin-induced apoptosis through activation of p53.
Many studies have shown that tubular epithelial cell apoptosis is a major cause of cisplatin-induced nephrotoxicity (10, 18, 22, 36). Activation of p53 and subsequent Bax induction and caspase 3 activation have been shown to mediate cisplatin-induced AKI (31). In the present study, we have shown that compound C, a selective AMPK inhibitor, suppresses p53 activation, Bax induction, caspase 3 activation, and apoptosis. Furthermore, inhibition of AMPK with compound C protects the kidney from cisplatin-induced AKI and preserves kidney function in vivo. These results indicate that compound C could be a novel therapeutic agent for the treatment of cisplatin-induced nephrotoxicity.
AMPK is a multifunctional kinase that regulates energy metabolism, cell proliferation, apoptosis, and autophagy. We have recently shown that AMPK stimulates fibroblast activation and development of kidney fibrosis through regulation of myocardin-related transcription factor A signaling (30). Nevertheless, the role of AMPK in kidney injury is not clearly defined. A previous study has shown that activation of AMPK with metformin can inhibit apoptosis and promote autophagy (17). Another study reported that 5-aminoimidazole-4-carboxamide-1-β-D-ribofuranoside (AICAR) activates AMPK to ameliorate cisplatin-induced tubular epithelial cell apoptosis through the JAK/STAT/suppressor of cytokine signaling pathway (29). However, metformin and AICAR are known to have AMPK-independent effects (14, 24). Indeed, it has been previously reported that the renal protective effect of metformin is independent of AMPK in the kidney (7). Furthermore, it is unclear whether autophagy has a protective or a detrimental role in the kidney and other organs (6, 25, 27). The role of autophagy in cisplatin-induced AKI remains to be defined. Recently, Taub and Cutuli (28) have shown that activation of AMPK contributes to the increased apoptosis of renal proximal tubules in cystinosis. In the present study, we have demonstrated that inhibition of AMPK with compound C attenuates cisplatin-induced tubular epithelial cell apoptosis and protects the kidney from cisplatin-induced AKI.
In summary, our study identifies a critical role of AMPK in cisplatin-induced tubular epithelial cell apoptosis and nephrotoxicity. In response to cisplatin administration, activated AMPK phosphorylates p53, resulting in Bax induction, caspase 3 activation, tubular epithelial cell apoptosis, and kidney injury and dysfunction. These findings suggest that AMPK could serve as a novel therapeutic target for cisplatin-induced nephrotoxicity.
GRANTS
This work was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grant R01DK95835 and US Department of Veterans Affairs Grant I01BX02650 (to Y.W.).
DISCLAIMERS
The contents of the article do not represent the views of the United States Department of Veterans Affairs or the United States Government.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
X.J. and Y.W. conceived and designed research; X.J. performed experiments; X.J. analyzed data; X.J., C.A., B.J., R.L.S., and Y.W. interpreted results of experiments; X.J. and Y.W. prepared figures; X.J. drafted manuscript; C.A., B.J., R.L.S., and Y.W. edited and revised manuscript; X.J., C.A., B.J., R.L.S., and Y.W. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Christopher Bonin and Dr. Geneva Hargis for manuscript editing.
REFERENCES
- 1.Adamovich Y, Adler J, Meltser V, Reuven N, Shaul Y. AMPK couples p73 with p53 in cell fate decision. Cell Death Differ 21: 1451–1459, 2014. doi: 10.1038/cdd.2014.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arany I, Safirstein RL. Cisplatin nephrotoxicity. Semin Nephrol 23: 460–464, 2003. doi: 10.1016/S0270-9295(03)00089-5. [DOI] [PubMed] [Google Scholar]
- 3.Aubrey BJ, Kelly GL, Janic A, Herold MJ, Strasser A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ 25: 104–113, 2018. doi: 10.1038/cdd.2017.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brázda V, Fojta M. The rich world of p53 DNA binding targets: the role of DNA structure. Int J Mol Sci 20: 5605, 2019. doi: 10.3390/ijms20225605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen G, Lin SC, Chen J, He L, Dong F, Xu J, Han S, Du J, Entman ML, Wang Y. CXCL16 recruits bone marrow-derived fibroblast precursors in renal fibrosis. J Am Soc Nephrol 22: 1876–1886, 2011. doi: 10.1681/ASN.2010080881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen W, Sun Y, Liu K, Sun X. Autophagy: a double-edged sword for neuronal survival after cerebral ischemia. Neural Regen Res 9: 1210–1216, 2014. doi: 10.4103/1673-5374.135329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Christensen M, Jensen JB, Jakobsen S, Jessen N, Frøkiær J, Kemp BE, Marciszyn AL, Li H, Pastor-Soler NM, Hallows KR, Nørregaard R. Renoprotective effects of metformin are independent of organic cation transporters 1 & 2 and AMP-activated protein kinase in the kidney. Sci Rep 6: 35952, 2016. doi: 10.1038/srep35952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dasari S, Tchounwou PB. Cisplatin in cancer therapy: molecular mechanisms of action. Eur J Pharmacol 740: 364–378, 2014. doi: 10.1016/j.ejphar.2014.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He G, Zhang YW, Lee JH, Zeng SX, Wang YV, Luo Z, Dong XC, Viollet B, Wahl GM, Lu H. AMP-activated protein kinase induces p53 by phosphorylating MDMX and inhibiting its activity. Mol Cell Biol 34: 148–157, 2014. doi: 10.1128/MCB.00670-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holditch SJ, Brown CN, Lombardi AM, Nguyen KN, Edelstein CL. Recent advances in models, mechanisms, biomarkers, and interventions in cisplatin-induced acute kidney injury. Int J Mol Sci 20: 3011, 2019. doi: 10.3390/ijms20123011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Huang D, Wang C, Duan Y, Meng Q, Liu Z, Huo X, Sun H, Ma X, Liu K. Targeting Oct2 and P53: formononetin prevents cisplatin-induced acute kidney injury. Toxicol Appl Pharmacol 326: 15–24, 2017. doi: 10.1016/j.taap.2017.04.013. [DOI] [PubMed] [Google Scholar]
- 12.Jin X, Chen J, Hu Z, Chan L, Wang Y. Genetic deficiency of adiponectin protects against acute kidney injury. Kidney Int 83: 604–614, 2013. doi: 10.1038/ki.2012.408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim N, Min WK, Park MH, Lee JK, Jin HK, Bae JS. Neuropeptide Y protects kidney against cisplatin-induced nephrotoxicity by regulating p53-dependent apoptosis pathway. BMB Rep 49: 288–292, 2016. doi: 10.5483/BMBRep.2016.49.5.231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kirchner J, Brüne B, Namgaladze D. AICAR inhibits NFκB DNA binding independently of AMPK to attenuate LPS-triggered inflammatory responses in human macrophages. Sci Rep 8: 7801, 2018. doi: 10.1038/s41598-018-26102-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kurumbail RG, Calabrese MF. Structure and regulation of AMPK. Exp Suppl 107: 3–22, 2016. doi: 10.1007/978-3-319-43589-3_1. [DOI] [PubMed] [Google Scholar]
- 16.Lee CG, Kang YJ, Kim HS, Moon A, Kim SG. Phlda3, a urine-detectable protein, causes p53 accumulation in renal tubular cells injured by cisplatin. Cell Biol Toxicol 31: 121–130, 2015. doi: 10.1007/s10565-015-9299-4. [DOI] [PubMed] [Google Scholar]
- 17.Li J, Gui Y, Ren J, Liu X, Feng Y, Zeng Z, He W, Yang J, Dai C. Metformin protects against cisplatin-induced tubular cell apoptosis and acute kidney injury via AMPKα-regulated autophagy induction. Sci Rep 6: 23975, 2016. doi: 10.1038/srep23975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liang H, Zhang Z, He L, Wang Y. CXCL16 regulates cisplatin-induced acute kidney injury. Oncotarget 7: 31652–31662, 2016. doi: 10.18632/oncotarget.9386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liang H, Zhang Z, Yan J, Wang Y, Hu Z, Mitch WE, Wang Y. The IL-4 receptor α has a critical role in bone marrow-derived fibroblast activation and renal fibrosis. Kidney Int 92: 1433–1443, 2017. doi: 10.1016/j.kint.2017.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mi XJ, Hou JG, Wang Z, Han Y, Ren S, Hu JN, Chen C, Li W. The protective effects of maltol on cisplatin-induced nephrotoxicity through the AMPK-mediated PI3K/Akt and p53 signaling pathways. Sci Rep 8: 15922, 2018. doi: 10.1038/s41598-018-34156-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Okoshi R, Ozaki T, Yamamoto H, Ando K, Koida N, Ono S, Koda T, Kamijo T, Nakagawara A, Kizaki H. Activation of AMP-activated protein kinase induces p53-dependent apoptotic cell death in response to energetic stress. J Biol Chem 283: 3979–3987, 2008. doi: 10.1074/jbc.M705232200. [DOI] [PubMed] [Google Scholar]
- 22.Pabla N, Dong Z. Cisplatin nephrotoxicity: mechanisms and renoprotective strategies. Kidney Int 73: 994–1007, 2008. doi: 10.1038/sj.ki.5002786. [DOI] [PubMed] [Google Scholar]
- 23.Perazella MA. Drug-induced acute kidney injury: diverse mechanisms of tubular injury. Curr Opin Crit Care 25: 550–557, 2019. doi: 10.1097/MCC.0000000000000653. [DOI] [PubMed] [Google Scholar]
- 24.Rena G, Hardie DG, Pearson ER. The mechanisms of action of metformin. Diabetologia 60: 1577–1585, 2017. doi: 10.1007/s00125-017-4342-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sciarretta S, Hariharan N, Monden Y, Zablocki D, Sadoshima J. Is autophagy in response to ischemia and reperfusion protective or detrimental for the heart? Pediatr Cardiol 32: 275–281, 2011. doi: 10.1007/s00246-010-9855-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Suliman FA, Khodeer DM, Ibrahiem A, Mehanna ET, El-Kherbetawy MK, Mohammad HMF, Zaitone SA, Moustafa YM. Renoprotective effect of the isoflavonoid biochanin A against cisplatin induced acute kidney injury in mice: effect on inflammatory burden and p53 apoptosis. Int Immunopharmacol 61: 8–19, 2018. doi: 10.1016/j.intimp.2018.05.010. [DOI] [PubMed] [Google Scholar]
- 27.Tang C, Livingston MJ, Liu Z, Dong Z. Autophagy in kidney homeostasis and disease. Nat Rev Nephrol 16: 489–508, 2020. doi: 10.1038/s41581-020-0309-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Taub M, Cutuli F. Activation of AMP kinase plays a role in the increased apoptosis in the renal proximal tubule in cystinosis. Biochem Biophys Res Commun 426: 516–521, 2012. doi: 10.1016/j.bbrc.2012.08.115. [DOI] [PubMed] [Google Scholar]
- 29.Tsogbadrakh B, Ryu H, Ju KD, Lee J, Yun S, Yu KS, Kim HJ, Ahn C, Oh KH. AICAR, an AMPK activator, protects against cisplatin-induced acute kidney injury through the JAK/STAT/SOCS pathway. Biochem Biophys Res Commun 509: 680–686, 2019. doi: 10.1016/j.bbrc.2018.12.159. [DOI] [PubMed] [Google Scholar]
- 30.Wang Y, Jia L, Hu Z, Entman ML, Mitch WE, Wang Y. AMP-activated protein kinase/myocardin-related transcription factor-A signaling regulates fibroblast activation and renal fibrosis. Kidney Int 93: 81–94, 2018. doi: 10.1016/j.kint.2017.04.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wei Q, Dong G, Yang T, Megyesi J, Price PM, Dong Z. Activation and involvement of p53 in cisplatin-induced nephrotoxicity. Am J Physiol Renal Physiol 293: F1282–F1291, 2007. doi: 10.1152/ajprenal.00230.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yan J, Zhang Z, Yang J, Mitch WE, Wang Y. JAK3/STAT6 stimulates bone marrow-derived fibroblast activation in renal fibrosis. J Am Soc Nephrol 26: 3060–3071, 2015. doi: 10.1681/ASN.2014070717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang J, Lin SC, Chen G, He L, Hu Z, Chan L, Trial J, Entman ML, Wang Y. Adiponectin promotes monocyte-to-fibroblast transition in renal fibrosis. J Am Soc Nephrol 24: 1644–1659, 2013. doi: 10.1681/ASN.2013030217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang D, Liu Y, Wei Q, Huo Y, Liu K, Liu F, Dong Z. Tubular p53 regulates multiple genes to mediate AKI. J Am Soc Nephrol 25: 2278–2289, 2014. doi: 10.1681/ASN.2013080902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang WB, Wang Z, Shu F, Jin YH, Liu HY, Wang QJ, Yang Y. Activation of AMP-activated protein kinase by temozolomide contributes to apoptosis in glioblastoma cells via p53 activation and mTORC1 inhibition. J Biol Chem 285: 40461–40471, 2010. doi: 10.1074/jbc.M110.164046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhou J, An C, Jin X, Hu Z, Safirstein RL, Wang Y. TAK1 deficiency attenuates cisplatin-induced acute kidney injury. Am J Physiol Renal Physiol 318: F209–F215, 2020. doi: 10.1152/ajprenal.00516.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]