

Keywords: furosemide, loop diuretics, pressure myography, renal lymphatics
Abstract
Similar to other organs, renal lymphatics remove excess fluid, solutes, and macromolecules from the renal interstitium. Given the kidney’s unique role in maintaining body fluid homeostasis, renal lymphatics may be critical in this process. However, little is known regarding the pathways involved in renal lymphatic vessel function, and there are no studies on the effects of drugs targeting impaired interstitial clearance, such as diuretics. Using pressure myography, we showed that renal lymphatic collecting vessels are sensitive to changes in transmural pressure and have an optimal range of effective pumping. In addition, they are responsive to vasoactive factors known to regulate tone in other lymphatic vessels including prostaglandin E2 and nitric oxide, and their spontaneous contractility requires Ca2+ and Cl−. We also demonstrated that Na+-K+-2Cl− cotransporter Nkcc1, but not Nkcc2, is expressed in extrarenal lymphatic vessels. Furosemide, a loop diuretic that inhibits Na+-K+-2Cl− cotransporters, induced a dose-dependent dilation in lymphatic vessels and decreased the magnitude and frequency of spontaneous contractions, thereby reducing the ability of these vessels to propel lymph. Ethacrynic acid, another loop diuretic, had no effect on vessel tone. These data represent a significant step forward in our understanding of the mechanisms underlying renal lymphatic vessel function and highlight potential off-target effects of furosemide that may exacerbate fluid accumulation in edema-forming conditions.
INTRODUCTION
Recent identification of markers that differentiate lymphatic vessels from blood vessels has led to the recognition that the lymphatic vascular network has an important role in health and disease (11). Aside from immunosurveillance, the principal function of the lymphatics is clearance of fluids, solutes, macromolecules, and cells from the interstitial space and returning these elements to the circulation. Inadequate lymphatic transport has been associated with disease in the heart, lungs, gut, and joints including atherosclerosis (30), cardiac fibrosis (51), inflammatory bowel disease (1), and rheumatoid arthritis (7).
Like blood vessels, lymphatic vessel contractility is regulated by endothelium-derived factors including nitric oxide (NO) (10, 54) and prostaglandins (36, 44, 49). Recent reports have also documented the contribution of biomechanical factors [e.g., pressure and shear stress (41)], neuronal factors [e.g., α-adrenoreceptors (35)], and ion channels [e.g., Cl− channels (61) and ATP-gated K+ (KATP) channels (12)] to lymphatic vessel contractility and pumping. Interestingly, diseases such as diabetes, rheumatoid arthritis, and hypertension alter lymphatic vessel contractions through mechanisms that affect NO bioavailability, increased sympathetic tone, and modulation of lymphatic vessel ion channels and transporters (33, 47, 56).
Among various lymphatic vessels, accumulating evidence suggests a high degree of structural and molecular heterogeneity of mechanisms underlying fluid absorption and pumping (17, 50). For instance, lymphatic capillaries have discontinuous button-like endothelial junctions to allow for fluid influx, but as these capillaries transition to larger collecting vessels, the endothelial junctions become zipper like, making them less permeable (3). Moreover, collecting vessels that transport lymph acquire layers of smooth muscle cells and valves to move lymph along the lymphatic network. Some vessels, particularly those embedded in skeletal and heart muscle, rely on extrinsic tissue forces to compress and expand the vessels to move lymph. In contrast, mesenteric lymphatic vessels among others rely on an intrinsic lymphatic pumping mechanism to generate contractions that drive lymph movement (60).
The kidneys are central in fluid homeostasis. The kidneys filter the entire blood volume >35 times each day, reclaiming >99% of the filtrate that then enters the renal interstitium. The interstitium is a critical site for accumulation of bioactive particles, cells, and fluid that can initiate and propagate tissue remodeling and fibrosis. Although the renal venous network transports much of the reabsorbed fluid and solutes, lymphatics also contribute to clearance of the kidney interstitium. Renal lymphatic vessels begin as blind-ended capillaries originating in the cortex. These capillaries, lacking a basement membrane and smooth muscle, form a dense network of intrarenal vessels that run in close proximity to the renal tubules but do not enter the renal corpuscle. Renal lymph drainage begins with lymphatic vessels in the cortical interstitium, which drain into arcades at the corticomedullary junction and then parallel interlobular blood vessels into the renal pelvis, finally emptying into the hilar lymphatic vessels (21, 58). As intrarenal lymphatics transition to extrarenal collecting vessels, they acquire layers of smooth muscle, a continuous basement membrane, and valves (45, 50). Various renal injuries and diseases cause a lymphangiogenic response (6, 58, 59). Thus, animal models including unilateral ureteral obstruction, acute and chronic kidney injury, diabetes, hypertension, and cystic disease are accompanied by robust lymphangiogenesis (22, 27, 31, 40). Moreover, renal biopsy patients with diabetic kidney disease, IgA nephropathy, lupus, tubulointerstitial nephritis, and transplants show increased cross-sectional areas of lymphatics (22, 25). However, currently very little is known about extrarenal lymphatic vessel contractility or the mechanisms that drive renal lymphatic pumping. This is particularly relevant since the treatment of interstitial edema specifically targets ion channels that may also be functional in lymphatic vessels. Furosemide, an inhibitor of Na+-K+-2Cl− cotransporters (NKCCs), is the cornerstone in treating fluid retention and has been shown to modulate blood vessel tone (14). The aim of the present study was to elucidate the physiology of extrarenal lymphatic collecting vessels and determine the impact of biologically relevant vasoactive factors.
METHODS
Rats.
Adult male Sprague-Dawley rats (175–250 g, Harlan Laboratories) were housed and maintained in a temperature-controlled room with a 12:12-h light-dark cycle, with free access to standard rat chow and water. Rats were euthanized using an overdose of isoflurane anesthesia followed by exsanguination immediately prior to tissue collection. All experimental protocols were approved by the Vanderbilt University Institutional Animal Care and Use Committee.
Pressure myography.
Afferent extrarenal lymphatic collecting vessels were isolated by microdissection, and lymphangions were mounted on glass pipettes in vessel perfusion chambers (Fig. 1A) (19, 43). Briefly, chambers were placed on inverted microscopes equipped with a digital image capture system (IonOptix) to record prevalve intraluminal diameters. Vessels were warmed to 37°C, pressurized to 0.5 mmHg using a column of Krebs buffer, and allowed to equilibrate (20–60 min). Vessels that failed to contract spontaneously were excluded from further study. For preload experiments, the inflow pressure was increased in a stepwise manner (0.5–4.0 mmHg), whereas the outflow pressure was held constant. For drug-response experiments, intraluminal pressure was increased in a stepwise manner to 2.5 mmHg and held constant. Vessels were then challenged with the following factors: N-nitro-l-arginine methyl ester (l-NAME; 10−3M, Cayman Chemical), PGE2 (10−9 M, Cayman Chemical), pinacidil (10−4 M, Tocris), niflumic acid (10−7–10−3 M, Sigma-Aldrich), and nifedipine (10−9–10−8 M, Sigma-Aldrich). Some vessels also received furosemide (10−8–10−3M, Hospira) or ethacrynic acid (10−8–10−3M, Sigma-Aldrich) with and without indomethacin (10−6 M, Tocris). After each factor was added, lumen diameters were allowed to plateau (20–40 min). After each drug challenge, fresh Krebs buffer was circulated to facilitate washout and return lumen diameters to baseline conditions (20–40 min). Single vessels were exposed to multiple compounds over the course of each experiment. The order in which each compound was added varied regularly to test for changes in vessel response or viability. We found no difference in response or viability based on the order of compound administration.
Fig. 1.

Extrarenal afferent lymphatic vessels were isolated from adult male rats. Lymphangions were mounted in microvessel perfusion chambers for assessment in pressure myography assays (A). Lymphatic vessel identity was confirmed using immunostaining for podoplanin (brown staining, arrows) and α-smooth muscle actin (purple staining, arrowheads). Nuclei were stained with methyl green (B). A digital image capture system was used to measure the frequency of spontaneous contractions, end-diastolic lumen diameter (C), and end-systolic lumen diameter (D). EDD, end-diastolic diameter; ESD, end-systolic diameter.
Buffers.
Standard Krebs buffer contained the following: 109 mM NaCl, 4.7 mM KCl, 2.5 mM CaCl2, 0.9 mM MgSO4, 1 mM KH2PO4, 11.1 mM glucose, and 34 mM NaHCO3. Bicarbonate-free buffer was made by replacing NaHCO3 with 25 mM HEPES. Cl−-free buffer was made by replacing NaCl and KCl with 10 mM HEPES and replacing CaCl2 with 1 mM EDTA. Ca2+-free buffer was made by replacing CaCl2 with 1 mM EDTA. Control Krebs + HEPES buffer was made by adding 10 mM HEPES to standard Krebs buffer. All buffers were titrated to pH 7.3.
Contractility calculations.
As previously reported (48), the amplitude of contraction was measured as the difference between end-diastolic diameter (EDD) and end-systolic diameter (ESD) (Fig. 1, C and D). Ejection fraction was calculated as follows: (EDD2 − ESD2)/EDD2.
Conventional PCR.
Total RNA was isolated from rat extrarenal lymphatic vessels and kidney cortex lysates (positive control) by the RNase Mini Kit (Qiagen). Reverse transcription was performed using the high-capacity cDNA Reverse Transcription Kit (Applied Biosystems). PCR was performed in a total reaction volume of 25 µL using 12.5 µL of Universal Master Mix II, 1.25 µL of forward and reverse primers [Nkcc1 (SLC12A2), Nkcc2 (SLC12A1), α-smooth muscle actin (α-SMA, ACTA2), podoplanin, lymphatic vessel endothelial receptor 1 (LYVE1), and β-actin (ACTB) (ThermoFisher)], and 11.25 µL cDNA (10 ng/µL). cDNA synthesis was carried out using the CFX96TM Real-Time PCR Detection System (Bio-Rad Laboratories) with the following cycling parameters: polymerase activation for 10 min at 95°C and amplification for 40 cycles of 15 s at 95°C and 60 s at 60°C. Amplified cDNA was run on a 1% agarose gel and photographed with a gel imaging system (Bio-Rad).
Immunostaining.
Extrarenal lymphatic collecting vessels from rats were fixed in 4% paraformaldehyde/PBS, paraffin embedded, and sectioned for immunostaining. Double staining for podoplanin and α-SMA used citrate buffer for antigen retrieval followed by primary antibody podoplanin (1:400, Novus). ImmPRESS reagents (Vector Laboratories) were used as secondary antibodies. The signal was visualized by diaminobenzidine. Sections were incubated with rabbit anti-rat α-SMA antibody followed by anti-rabbit horseradish peroxidase, and the signal was visualized by the Impact VIP peroxidase substrate kit (Vector Laboratories). Methyl green was used to counterstain the nucleus.
Statistical analysis.
Data are expressed as means, with error bars representing SEs. Experimental results were compared with baseline results using one-way ANOVA with Dunnett’s multiple-comparison tests. P values of <0.05 were considered statistically significant. n values represent the number of individual vessels used in each experiment.
RESULTS
Preload regulates extrarenal lymphatic vessel pumping.
Although physiological and pathophysiological conditions alter the pressure within the kidney interstitium (4), there is little information regarding how this may affect renal collecting lymphatic vessels. Transmural pressure is a key determinant of contractile activity in mesenteric, thoracic, and femoral collecting vessels (17). Therefore, we used pressure myography assays to define the contractile activity of isolated extrarenal lymphatics in response to increasing preload pressure. Renal collecting lymphangions were isolated from rats and mounted in microvessel perfusion chambers (Fig. 1A). Lymphatic vessel identity was confirmed using immunostaining for the lymphatic marker podoplanin and the smooth muscle marker α-SMA (Fig. 1B). These vessels exhibited spontaneous contractions (Supplemental Movie S1, available online at https://doi.org/10.6084/m9.figshare.12573707.v1). To better understand the role of preload pressure, or the transmural stretching of the vessel prior to contraction, vessels were subjected to stepwise increases in inflow pressure (0.5–4.0 mmHg). This range is reflective of in vivo rat cortical interstitial pressure (0.5 ± 0.3 to 2.7 ± 0.4 mmHg) (29). As pressure increased, end-diastolic and end-systolic intraluminal diameters increased (Fig. 2A). In contrast, the amplitude of contraction increased from 0.5 to 2.5 mmHg and then declined at higher pressures (Fig. 2B). Similarly, ejection fraction, a calculated value that indicates the pumping capacity of a lymphatic vessel, increased and then declined at higher pressures (Fig. 2D). The frequency of contractions increased from 0.5 to 3.5 mmHg and then declined (Fig. 2C). These data demonstrate that renal vessels have an optimum preload pressure range under which they pump most effectively.
Fig. 2.

Renal lymphatic vessel contractility is regulated by transmural pressure. Isolated vessels were subjected to increasing preload pressure. End-diastolic and end-systolic lumen diameters (A), amplitude of contraction (B), frequency of contractions (C), and calculated ejection fraction (D) were measured at each pressure. Data are expressed as means ± SE. Experimental values were compared with values acquired at 0.5 mmHg to determine statistical significance. n = 6 vessels. EDD, end-diastolic diameter; ESD, end-systolic diameter.
Vasoactive factors regulate extrarenal lymphatic contractility.
Similar to the vascular smooth muscle of blood vessels, biochemical factors that affect lymphatic muscle cell contractility also play a critical role in collecting lymphatic vessel dynamics (37, 44, 54). The ability of vasoactive factors to regulate renal collecting vessels has not been studied. Therefore, we disrupted NO and prostaglandin signaling pathways in isolated extrarenal vessels and evaluated spontaneous contractile activity. Inhibition of NO signaling via l-NAME resulted in a decrease in end-diastolic and end-systolic vessel diameter compared with baseline values (Fig. 3, A and B). l-NAME reduced the amplitude of contraction (Fig. 3D), which contributed to a decrease in the overall pumping efficiency as measured by the calculated ejection fraction (Fig. 3E). In contrast, activation of prostaglandin signaling via PGE2 resulted in vasodilation evidenced by increases in both EDD and ESD (Fig. 3, A and B). However, the amplitude of contraction fell, as did ejection fraction (Fig. 3E). Both l-NAME and PGE2 increased the frequency of contraction (Fig. 3C), which may be a compensatory response to reduced contraction amplitude and ejection fraction.
Fig. 3.

Renal lymphatic vessels respond to factors known to regulate lymphatic vasoactivity. Vessels were challenged with the nitric oxide synthase inhibitor N-nitro-l-arginine methyl ester (l-NAME; 10−3M), PGE2 (10−9 M), or the ATP-gated K+ (KATP) channel activator pinacidil (10−4 M). The following contractile parameters were measured: end-diastolic diameter (EDD; A), end-systolic diameter (ESD; B), frequency of contraction (C), amplitude of contraction (D), and ejection fraction (E). Baseline values were measured immediately prior to drug addiction. Data are expressed as means ± SE. Experimental values were compared with baseline values to determine statistical significance. PGE2 values were also compared with pinacidil values to determine statistically significant differences. n ≥ 7 vessels. EDD, end-diastolic diameter; ESD, end-systolic diameter.
KATP channels have been shown to regulate the frequency of spontaneous contractions in mesenteric lymphatics (34). Furthermore, NO-induced KATP channel activation contributes to mesenteric lymphatic dysfunction in the setting of inflammatory bowel disease (33). We challenged extrarenal lymphatic vessels with pinacidil, a KATP channel activator. Pinacidil dramatically increased end-systolic and end-diastolic vessel diameter even more than PGE2 (Fig. 3, A and B). Pinacidil also caused a striking reduction in the contraction amplitude that virtually abolished ejection fraction (Fig. 3, D and E). In contrast to PGE2, the KATP channel activator impeded lymphatic vessel contractility (Fig. 3C).
Ca2+ and Cl− are required for extrarenal lymphatic contractility.
Lymphatic smooth muscle contractility is tightly coupled to membrane potential. Electrophysiological studies have demonstrated that contraction is primarily regulated by Ca2+ influx via voltage-dependent Ca2+ channels (VDCCs), although the resting membrane potential of lymphatic smooth muscle cells is dependent on K+ and Cl− channels (48). It is well known that Cl− channels play a role in vascular smooth muscle contractility (32). Cl− efflux causes membrane depolarization, activation of voltage-sensitive Ca2+ channels, and smooth muscle contraction. Intracellular Cl− accumulates via several transport mechanisms including NKCCs, Ca2+-activated Cl− channels (CaCCs), and Cl−-bicarbonate anion exchangers (AEs). Recently, CaCCs have been implicated in mesenteric and inguinal collecting vessel contractility (35, 61). We sought to determine the extent to which Cl− regulates extrarenal lymphatic vessel tone. Isolated vessels were maintained in control Krebs buffer containing bicarbonate, Cl−, and Ca2+. Vessels were then exposed to various exclusion buffers to determine the effects of removing bicarbonate, Cl−, or Ca2+ (Supplemental Fig. S1, available online at https://doi.org/10.6084/m9.figshare.12573662.v1). In each case, absence of bicarbonate, Cl−, or Ca2+ resulted in the cessation of spontaneous contractions (Fig. 4, A and B), demonstrating their requirement for maintaining renal lymphatic pumping. In addition, vessels were treated with increasing concentrations of nifedipine, a Ca2+ channel blocker, or niflumic acid, a CaCC inhibitor. Both nifedipine and niflumic acid inhibited contractions in a dose-dependent manner (Fig. 4, D and F; Supplemental Fig. S1). Interestingly, both caused significant increases in ESD, whereas EDD remained relatively unchanged (Fig. 4, C and E). Taken together, these data highlight the requirement of Ca2+ and Cl− for maintaining spontaneous extrarenal lymphatic vessel contractions.
Fig. 4.

Cl− and Ca2+ are required for renal lymphatic contractility. Vessels were exposed to standard Krebs, Krebs + HEPES, NaHCO3-free Krebs, Cl−-free Krebs, or Ca2+-free Krebs buffer. The frequency of contractions and ejection fraction under each condition were quantified (A and B). Vessels were also challenged with increasing doses of nifedipine, a Ca2+ channel blocker, or niflumic acid, a Ca2+-activated Cl− channel inhibitor. End-diastolic diameter (EDD) and end-systolic diameter (ESD; C and E) as well as ejection fraction (D and F) were quantified and expressed as a percentage of baseline values. Data are expressed as means ± SE. In A and B, experimental values were compared with baseline values (control Krebs buffer) to determine statistical significance. In C–F, experimental values were compared with baseline values to determine statistical significance. n ≥ 6 vessels.
Furosemide dilates and decreases renal lymphatic vessel contractility.
Loop diuretics act by inhibiting NKCCs in tubular epithelial cells of the kidney. Although NKCCs are now recognized to affect vascular tone, their effect on the renal lymphatics is unknown. Conventional PCR was used to identify the presence of specific NKCC channel isoforms in isolated renal lymphatic tissue. The lymphatic vessel identity of the extrarenal lymphatic vessel samples (lane 1) was confirmed using markers for lymphatic vessels (podoplanin and LYVE1) and smooth muscle (α2-smooth muscle) (Fig. 5A). Nkcc1, but not Nkcc2, was expressed in extrarenal lymphatic vessels (lane 1), whereas both isoforms were expressed in whole kidney samples (lane 2; Fig. 5A). To evaluate the extent to which loop diuretics affect lymphatic vessel tone, isolated vessels were challenged with increasing concentrations of furosemide and another loop diuretic, ethacrynic acid. Furosemide dilated renal collecting vessels and decreased the frequency of contractions in a dose-dependent manner (Fig. 5, A−C). In addition, furosemide reduced the magnitude of contractions and ejection fraction (Fig. 5, E and F). Some have hypothesized that the vasoactive effects of furosemide are dependent on stimulating prostaglandin production (14). Therefore, we treated vessels with indomethacin, a potent prostaglandin inhibitor, in the presence of furosemide. The effects of furosemide on extrarenal lymphatic tone and contractility were unaltered by indomethacin, suggesting that furosemide acts directly on Nkcc1 cotransporters present on renal lymphatic vessels. In contrast, ethacrynic acid had relatively little effect on lymphatic vasoactivity. These data suggest that furosemide, but not all loop diuretics, can alter extrarenal lymphatic vessel tone and pumping efficiency via inhibition of Nkcc1.
Fig. 5.

Furosemide regulates renal lymphatic contractility via Na+-K+-2Cl− cotransporter NKCC1. Images of conventional PCR gels show amplification products for Na+-K+-2Cl− cotransporters Nkcc1 and Nkcc2. Lane 1 shows extrarenal lymphatic vessel samples, lane 2 shows whole kidney samples, and lane 3 shows the water-negative control. α2-Smooth muscle actin (Acta2), podoplanin (Pdpn), and lymphatic vessel endothelial receptor 1 (Lyve1) were used to confirm the lymphatic vessel identity of samples in lane 1. The housekeeping gene β-actin was used as a loading control (A). Renal lymphatic vessels were exposed to increasing concentrations of the loop diuretics furosemide or ethacrynic acid. The following contractile parameters were quantified: frequency of contraction (B), end-diastolic diameter (EDD; C), end-systolic diameter (ESD; D) amplitude of contraction (E), and ejection fraction (F). Baseline values were measured immediately prior to drug addiction. Vessels that received the highest dose of furosemide were then challenged with indomethacin. Data points represent means ± SE. Experimental values were compared with baseline values to determine statistical significance. n ≥ 6 vessels.
DISCUSSION
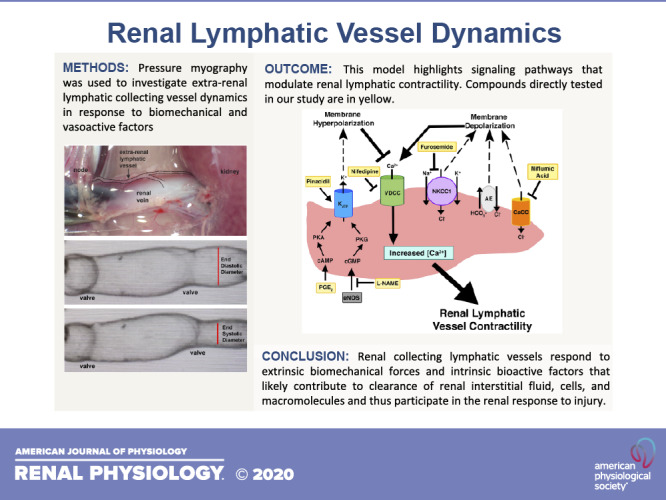
In the past several decades, significant progress has been made in identifying lymphatic vessel markers, understanding lymphangiogenesis, and developing techniques to isolate lymphatic endothelial cells. Our study is the first physiological analysis of renal lymphatic collecting vessel dynamics. The central role of lymphatics in modulating interstitial fluid homeostasis may be particularly important in the kidneys, which reclaim almost all filtered fluid returning it to the circulation and preventing edema formation. As in other vascular beds, Starling forces regulate interstitial fluid exchange, with interstitial and venous volume and hydrostatic/oncotic pressures driving lymph formation in the kidney (8, 45). Lymph drainage is achieved by coordinated contractions of adjacent lymphangions linked by unidirectional valves. This contractile activity is intrinsic to some lymphatic smooth muscle cells and is triggered by activation of VDCCs (Fig. 6). We show that extrarenal lymphatic vessel contractility is dependent on VDCCs as exposure to nifedipine or Ca2+-depleted buffer blocks contractions (Fig. 4).
Fig. 6.

Biochemical regulation of renal lymphatic vessels. This model highlights the signaling pathways that modulate renal lymphatic contractility. Compounds that were directly tested in our study are in yellow. CaCC, Ca2+-activated Cl− channels; NKCC1, Na+-K+-2Cl− cotransporter; AE, anion exchanger; eNOS, endothelial nitric oxide synthase.
Lymphatic contractility is also modulated by several other factors, including fluid load. Previous studies have shown that mesenteric lymphatic vessels can adjust their pumping output to mitigate increases in preload or the stretch on the lymphatic wall during the diastolic filling phase of the contraction cycle (48, 62). We found that renal collecting vessels increased the frequency and magnitude of contractions in response to a relatively narrow pressure range (0.5−2.5 mmHg). However, the contractile machinery began to fail and pumping became less efficient at pressures above 2.5–3.5 mmHg (Fig. 2). This phenomenon may correspond to lymphatic dysfunction observed when interstitial pressure is elevated, lymph production is increased, and edema forms (10, 15, 46). This may be especially germane in the kidneys, which are encapsulated organs that can become susceptible to the adverse consequences of increased interstitial pressure in various pathophysiological conditions. Interestingly, acute volume expansion in rats caused a more than fivefold increase in hydrostatic pressure within the renal interstitial compartment (29). Thus, it is possible that renal disease increases pressure in the renal interstitium leading to diminished renal lymphatic contractility, which, in turn, reduces interstitial clearance. Another possibility is that other factors such as pulsations of adjacent arteries not captured by myographic assessments contribute to in vivo lymphatic contractility and interstitial clearance.
Lymphatic vessel contractility is also regulated by vasoactive factors. Similar to blood vessels, lymphatic smooth muscle cells are sensitive to endothelial NO synthase or inducible NO synthase derived from lymphatic endothelial cells or activated immune cells, respectively (48). Activation of NO signaling triggers the cGMP/PKG pathway, which activates KATP channels, leading to membrane hyperpolarization, decreased intracellular Ca2+ accumulation, and subsequent vasodilation (Fig. 6) (52, 54). In a guinea pig model of inflammatory bowel disease, NO activation of KATP channels resulted in severely dilated mesenteric collecting vessels and dramatically decreased contractility (57). We found that inhibition of NO signaling in renal collecting vessels using l-NAME-induced vasoconstriction reduced contraction amplitude and ejection fraction and increased contraction frequency (Fig. 3). NO and cGMP levels are decreased in chronic kidney disease (13, 42). Our findings suggest the novel concept that decreased NO signaling also impairs extrarenal lymphatic contractility. Interestingly, Scallan et al. (47) showed that the metabolic disruption induced by diabetes mellitus causes lymphatic vessel dysfunction linked to reduced NO bioavailability. Since defective lymphatic transport can promote disease, it is possible that reduced/impaired NO prevailing in chronic kidney disease contributes to its complications, e.g., hypertension, progressive kidney damage, and cardiovascular diseases, through mechanisms that include defective lymphatic function.
Like NO, prostaglandin metabolites are inflammatory mediators capable of regulating lymphatic contractility. To date, much of the prostaglandin work has been done in mesenteric vessels to identify the metabolites involved in inflammatory-mediated lymphatic dysfunction. Depending on the specific metabolite, prostaglandins can have excitatory or inhibitory effects on contractility. Several studies have demonstrated that PGE2 and prostacyclin inhibit mesenteric vessel contractility, whereas PGF2α and thromboxane A2 stimulate contraction (16, 23, 44, 49). Although circulating prostaglandins likely play a role, prostaglandin metabolites are also synthesized by lymphatic endothelial cells and can regulate contractility in a paracrine fashion (24, 28, 36, 53). Similar to other lymphatic vessels, our results revealed that PGE2 induced vasodilation and decreased the magnitude of contraction but increased the frequency of contractions in renal collecting vessels (Fig. 3). Indeed, Rehal et al. (44) also observed an increase in contraction frequency of mesenteric vessels when exposed to a 1 μM dose of PGE2 but a decrease in frequency after administration of a 10 μM dose. In any case, it is likely that PGE2 asserts its effects on extrarenal vessels via the cAMP/PKA signaling cascade (44, 55), with contribution from PGE2-mediated activation of KATP channels (52) as have both been shown for mesenteric lymphatic vessels (Fig. 6). In the current study, we demonstrated that activation of KATP channels with pinacidil profoundly dilates renal vessels to the point of precluding contractions (Fig. 3).
In addition to the classic vasodilators, NO and PGE2, recent studies support a critical role for Cl− in regulating lymphatic vessel contractility. In vascular smooth muscle, the intracellular concentration of Cl− is high and maintained by Cl− channels, AEs, and cotransporters. Activation of these channels/transporters causes Cl− efflux and cell membrane depolarization, which, in turn, activates VDCCs, leading to muscle cell contraction (9). A similar process occurs in lymphatic smooth muscle cells (Fig. 6). Studies using human thoracic duct and mesenteric lymphatics have shown that Cl− is essential to lymphatic vessel contraction (35). Similarly, Zawieja et al. (61) determined that the CaCC Ano1 mediates pressure-induced contractility in mouse inguinal axillary lymphatic vessels. We found that Cl− also plays an integral role in renal lymphatic contractility. Pharmacological inhibition of CaCCs or exposure to Cl−-depleted buffer caused profound loss of vessel contractility (Figs. 4 and 5). Importantly, vessels exposed to the NKCC inhibitor furosemide dilated and had diminished ability to contract. These findings are consistent with a report showing that another NKCC inhibitor, bumetanide, decreased contractility in mesenteric lymphatics (20). This is notable because furosemide is the cornerstone and first-line drug for managing abnormal fluid homeostasis, i.e., edema. Acting on the thick ascending limb of the loop of Henle, furosemide inhibits NKCC2, leading to increased renal excretion of water and solutes from the body. Our data indicate that furosemide can act on NKCC1 transporters in renal lymphatic collecting vessels causing vasodilation and decreased contractility. It is notable that furosemide-mediated dilation of blood vessels has been suggested as a contributing factor in the therapeutic reduction of elevated blood pressure (14). Indeed, inhibition of NKCC is rapidly developing as a novel antihypertensive strategy (38). The effects of NKCC transporters in the lymphatic vascular network are currently unclear. If renal disease increases expression/activity of NKCC transporters and lymphatic constriction, inhibition may increase lymphatic flow and interstitial clearance. In contrast, inhibition of NKCC in distended or otherwise compromised lymphatic vessels may cause further dilation and reduced contractility, which predicts impaired drainage of the renal interstitium. In this context, Pedersen et al. (39) found that furosemide increased overall water content in renal tissue. Of note, ethacrynic acid, another loop diuretic, had minimal effect on extrarenal vessels. Others have noted differential effects of these drugs (2, 5, 26); however, the underlying mechanism is unresolved. Of note, Greenwood et al. (18) demonstrated that furosemide, but not ethacrynic acid, could regulate Ca2+ release from the sarcoplasmic reticulum of vascular smooth muscle cells. This difference in Ca2+ handling may account for the differential effects we observed in extrarenal vessel contractility.
In summary, renal collecting lymphatic vessels are dynamic and can respond to both extrinsic biomechanical forces and intrinsic bioactive factors. This study represents a significant step forward in our understanding of the mechanisms underlying renal lymphatic vessel function and highlights potential off-target effects of loop diuretics that may exacerbate fluid accumulation in edema-forming conditions. The contribution of these vessels to renal, cardiovascular, and autoimmune disease progression should not be overlooked as they may provide an opportunity for novel clinical intervention strategies.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant 1P01HL116263 (to V.K.) and an American Physiological Society, Vanderbilt Center for Kidney Disease PILOT grant (to H.C.Y.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
E.L.S., H-C.Y., and V.K. conceived and designed research; E.L.S. and J.Z. performed experiments; E.L.S., H-C.Y., and V.K. analyzed data; E.L.S., H-C.Y., J.Z., and V.K. interpreted results of experiments; E.L.S. prepared figures; E.L.S. and V.K. drafted manuscript; E.L.S., H-C.Y., J.Z., M.M.S., and V.K. edited and revised manuscript; E.L.S., H-C.Y., M.M.S., and V.K. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. Fred Lamb for conceptual contributions to this work.
REFERENCES
- 1.Alexander JS, Chaitanya GV, Grisham MB, Boktor M. Emerging roles of lymphatics in inflammatory bowel disease. Ann N Y Acad Sci 1207, Suppl 1: E75–E85, 2010. doi: 10.1111/j.1749-6632.2010.05757.x. [DOI] [PubMed] [Google Scholar]
- 2.Alguire PC, Bailie MD, Weaver WJ, Taylor DG, Hook JB. Differential effects of furosemide and ethacrynic acid on electrolyte excretion in anesthetized dogs. J Pharmacol Exp Ther 190: 515–522, 1974. [PubMed] [Google Scholar]
- 3.Baluk P, Fuxe J, Hashizume H, Romano T, Lashnits E, Butz S, Vestweber D, Corada M, Molendini C, Dejana E, McDonald DM. Functionally specialized junctions between endothelial cells of lymphatic vessels. J Exp Med 204: 2349–2362, 2007. doi: 10.1084/jem.20062596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Basile DP, Anderson MD, Sutton TA. Pathophysiology of acute kidney injury. Compr Physiol 2: 1303–1353, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beutler JJ, Boer WH, Koomans HA, Dorhout Mees EJ. Comparative study of the effects of furosemide, ethacrynic acid and bumetanide on the lithium clearance and diluting segment reabsorption in humans. J Pharmacol Exp Ther 260: 768–772, 1992. [PubMed] [Google Scholar]
- 6.Bierer S, Herrmann E, Köpke T, Neumann J, Eltze E, Hertle L, Wülfing C. Lymphangiogenesis in kidney cancer: expression of VEGF-C, VEGF-D and VEGFR-3 in clear cell and papillary renal cell carcinoma. Oncol Rep 20: 721–725, 2008. [PubMed] [Google Scholar]
- 7.Bouta EM, Bell RD, Rahimi H, Xing L, Wood RW, Bingham CO III, Ritchlin CT, Schwarz EM. Targeting lymphatic function as a novel therapeutic intervention for rheumatoid arthritis. Nat Rev Rheumatol 14: 94–106, 2018. doi: 10.1038/nrrheum.2017.205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Breslin JW, Yang Y, Scallan JP, Sweat RS, Adderley SP, Murfee WL. Lymphatic vessel network structure and physiology. Compr Physiol 9: 207–299, 2018. doi: 10.1002/cphy.c180015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bulley S, Jaggar JH. Cl− channels in smooth muscle cells. Pflugers Arch 466: 861–872, 2014. doi: 10.1007/s00424-013-1357-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chakraborty S, Davis MJ, Muthuchamy M. Emerging trends in the pathophysiology of lymphatic contractile function. Semin Cell Dev Biol 38: 55–66, 2015. doi: 10.1016/j.semcdb.2015.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cueni LN, Detmar M. The lymphatic system in health and disease. Lymphat Res Biol 6: 109–122, 2008. doi: 10.1089/lrb.2008.1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Davis MJ, Kim HJ, Zawieja SD, Castorena-Gonzalez JA, Gui P, Li M, Saunders BT, Zinselmeyer BH, Randolph GJ, Remedi MS, Nichols CG. Kir6.1-dependent KATP channels in lymphatic smooth muscle and vessel dysfunction in mice with Kir6.1 gain-of-function. J Physiol 598: 3107–3127, 2020. doi: 10.1113/JP279612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Di Pietro N, Giardinelli A, Sirolli V, Riganti C, Di Tomo P, Gazzano E, Di Silvestre S, Panknin C, Cortese-Krott MM, Csonka C, Kelm M, Ferdinandy P, Bonomini M, Pandolfi A. Nitric oxide synthetic pathway and cGMP levels are altered in red blood cells from end-stage renal disease patients. Mol Cell Biochem 417: 155–167, 2016. doi: 10.1007/s11010-016-2723-0. [DOI] [PubMed] [Google Scholar]
- 14.Dormans TP, Pickkers P, Russel FG, Smits P. Vascular effects of loop diuretics. Cardiovasc Res 32: 988–997, 1996. doi: 10.1016/S0008-6363(96)00134-4. [DOI] [PubMed] [Google Scholar]
- 15.Ebah LM, Wiig H, Dawidowska I, O’Toole C, Summers A, Nikam M, Jayanti A, Coupes B, Brenchley P, Mitra S. Subcutaneous interstitial pressure and volume characteristics in renal impairment associated with edema. Kidney Int 84: 980–988, 2013. doi: 10.1038/ki.2013.208. [DOI] [PubMed] [Google Scholar]
- 16.Elias RM, Johnston MG. Modulation of fluid pumping in isolated bovine mesenteric lymphatics by a thromboxane/endoperoxide analogue. Prostaglandins 36: 97–106, 1988. doi: 10.1016/0090-6980(88)90105-0. [DOI] [PubMed] [Google Scholar]
- 17.Gashev AA, Davis MJ, Delp MD, Zawieja DC. Regional variations of contractile activity in isolated rat lymphatics. Microcirculation 11: 477–492, 2004. doi: 10.1080/10739680490476033. [DOI] [PubMed] [Google Scholar]
- 18.Greenwood IA, Hogg RC, Large WA. Effect of frusemide, ethacrynic acid and indanyloxyacetic acid on spontaneous Ca-activated currents in rabbit portal vein smooth muscle cells. Br J Pharmacol 115: 733–738, 1995. doi: 10.1111/j.1476-5381.1995.tb14994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hooper CW, Delaney C, Streeter T, Yarboro MT, Poole S, Brown N, Slaughter JC, Cotton RB, Reese J, Shelton EL. Selective serotonin reuptake inhibitor exposure constricts the mouse ductus arteriosus in utero. Am J Physiol Heart Circ Physiol 311: H572–H581, 2016. doi: 10.1152/ajpheart.00822.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hugues GA. The effect of Na+-K+-2Cl− cotransport inhibition and chloride channel blockers on membrane potential and contractility in rat lymphatic smooth muscle in vitro. J Physiol 518: 127P, 1999. [Google Scholar]
- 21.Ishikawa Y, Akasaka Y, Kiguchi H, Akishima-Fukasawa Y, Hasegawa T, Ito K, Kimura-Matsumoto M, Ishiguro S, Morita H, Sato S, Soh S, Ishii T. The human renal lymphatics under normal and pathological conditions. Histopathology 49: 265–273, 2006. doi: 10.1111/j.1365-2559.2006.02478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jafree DJ, Long DA. Beyond a passive conduit: implications of lymphatic biology for kidney diseases. J Am Soc Nephrol 31: 1178–1190, 2020. doi: 10.1681/ASN.2019121320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Johnston MG, Gordon JL. Regulation of lymphatic contractility by arachidonate metabolites. Nature 293: 294–297, 1981. doi: 10.1038/293294a0. [DOI] [PubMed] [Google Scholar]
- 24.Karnezis T, Shayan R, Caesar C, Roufail S, Harris NC, Ardipradja K, Zhang YF, Williams SP, Farnsworth RH, Chai MG, Rupasinghe TW, Tull DL, Baldwin ME, Sloan EK, Fox SB, Achen MG, Stacker SA. VEGF-D promotes tumor metastasis by regulating prostaglandins produced by the collecting lymphatic endothelium. Cancer Cell 21: 181–195, 2012. doi: 10.1016/j.ccr.2011.12.026. [DOI] [PubMed] [Google Scholar]
- 25.Kerjaschki D, Regele HM, Moosberger I, Nagy-Bojarski K, Watschinger B, Soleiman A, Birner P, Krieger S, Hovorka A, Silberhumer G, Laakkonen P, Petrova T, Langer B, Raab I. Lymphatic neoangiogenesis in human kidney transplants is associated with immunologically active lymphocytic infiltrates. J Am Soc Nephrol 15: 603–612, 2004. doi: 10.1097/01.ASN.0000113316.52371.2E. [DOI] [PubMed] [Google Scholar]
- 26.Kirkendall WM, Stein JH. Clinical pharmacology of furosemide and ethacrynic acid. Am J Cardiol 22: 162–167, 1968. doi: 10.1016/0002-9149(68)90221-X. [DOI] [PubMed] [Google Scholar]
- 27.Kneedler SC, Phillips LE, Hudson KR, Beckman KM, Lopez Gelston CA, Rutkowski JM, Parrish AR, Doris PA, Mitchell BM. Renal inflammation and injury are associated with lymphangiogenesis in hypertension. Am J Physiol Renal Physiol 312: F861–F869, 2017. doi: 10.1152/ajprenal.00679.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koller A, Mizuno R, Kaley G. Flow reduces the amplitude and increases the frequency of lymphatic vasomotion: role of endothelial prostanoids. Am J Physiol 277: R1683–R1689, 1999. doi: 10.1152/ajpregu.1999.277.6.R1683. [DOI] [PubMed] [Google Scholar]
- 29.Kon V, Hughes ML, Ichikawa I. Blood flow dependence of postglomerular fluid transfer and glomerulotubular balance. J Clin Invest 72: 1716–1728, 1983. doi: 10.1172/JCI111131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kutkut I, Meens MJ, McKee TA, Bochaton-Piallat ML, Kwak BR. Lymphatic vessels: an emerging actor in atherosclerotic plaque development. Eur J Clin Invest 45: 100–108, 2015. doi: 10.1111/eci.12372. [DOI] [PubMed] [Google Scholar]
- 31.Lee AS, Lee JE, Jung YJ, Kim DH, Kang KP, Lee S, Park SK, Lee SY, Kang MJ, Moon WS, Kim HJ, Jeong YB, Sung MJ, Kim W. Vascular endothelial growth factor-C and -D are involved in lymphangiogenesis in mouse unilateral ureteral obstruction. Kidney Int 83: 50–62, 2013. doi: 10.1038/ki.2012.312. [DOI] [PubMed] [Google Scholar]
- 32.Matchkov VV, Secher Dam V, Bødtkjer DM, Aalkjær C. Transport and function of chloride in vascular smooth muscles. J Vasc Res 50: 69–87, 2013. doi: 10.1159/000345242. [DOI] [PubMed] [Google Scholar]
- 33.Mathias R, von der Weid PY. Involvement of the NO-cGMP-KATP channel pathway in the mesenteric lymphatic pump dysfunction observed in the guinea pig model of TNBS-induced ileitis. Am J Physiol Gastrointest Liver Physiol 304: G623–G634, 2013. doi: 10.1152/ajpgi.00392.2012. [DOI] [PubMed] [Google Scholar]
- 34.Mizuno R, Ono N, Ohhashi T. Involvement of ATP-sensitive K+ channels in spontaneous activity of isolated lymph microvessels in rats. Am J Physiol Heart Circ Physiol 277: H1453–H1456, 1999. doi: 10.1152/ajpheart.1999.277.4.H1453. [DOI] [PubMed] [Google Scholar]
- 35.Mohanakumar S, Majgaard J, Telinius N, Katballe N, Pahle E, Hjortdal V, Boedtkjer D. Spontaneous and α-adrenoceptor-induced contractility in human collecting lymphatic vessels require chloride. Am J Physiol Heart Circ Physiol 315: H389–H401, 2018. doi: 10.1152/ajpheart.00551.2017. [DOI] [PubMed] [Google Scholar]
- 36.Oguogho A, Aghajanian AA, Sinzinger H. Prostaglandin synthesis in human lymphatics from precursor fatty acids. Lymphology 33: 62–66, 2000. [PubMed] [Google Scholar]
- 37.Ohhashi T, Kawai Y, Azuma T. The response of lymphatic smooth muscles to vasoactive substances. Pflugers Arch 375: 183–188, 1978. doi: 10.1007/BF00584242. [DOI] [PubMed] [Google Scholar]
- 38.Orlov SN, Tremblay J, Hamet P. NKCC1 and hypertension: a novel therapeutic target involved in the regulation of vascular tone and renal function. Curr Opin Nephrol Hypertens 19: 163–168, 2010. doi: 10.1097/MNH.0b013e3283360a46. [DOI] [PubMed] [Google Scholar]
- 39.Pedersen M, Vajda Z, Stødkilde-Jørgensen H, Nielsen S, Frøkiaer J. Furosemide increases water content in renal tissue. Am J Physiol Renal Physiol 292: F1645–F1651, 2007. doi: 10.1152/ajprenal.00060.2006. [DOI] [PubMed] [Google Scholar]
- 40.Perretta-Tejedor N, Jafree DJ, Long DA. Endothelial-epithelial communication in polycystic kidney disease: Role of vascular endothelial growth factor signalling. Cell Signal 72: 109624, 2020. doi: 10.1016/j.cellsig.2020.109624. [DOI] [PubMed] [Google Scholar]
- 41.Planas-Paz L, Lammert E. Mechanical forces in lymphatic vascular development and disease. Cell Mol Life Sci 70: 4341–4354, 2013. doi: 10.1007/s00018-013-1358-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Reddy YS, Kiranmayi VS, Bitla AR, Krishna GS, Rao PV, Sivakumar V. Nitric oxide status in patients with chronic kidney disease. Indian J Nephrol 25: 287–291, 2015. doi: 10.4103/0971-4065.147376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Reese J, O’Mara PW, Poole SD, Brown N, Tolentino C, Eckman DM, Aschner JL. Regulation of the fetal mouse ductus arteriosus is dependent on interaction of nitric oxide and COX enzymes in the ductal wall. Prostaglandins Other Lipid Mediat 88: 89–96, 2009. doi: 10.1016/j.prostaglandins.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rehal S, Blanckaert P, Roizes S, von der Weid PY. Characterization of biosynthesis and modes of action of prostaglandin E2 and prostacyclin in guinea pig mesenteric lymphatic vessels. Br J Pharmacol 158: 1961–1970, 2009. doi: 10.1111/j.1476-5381.2009.00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Russell PS, Hong J, Windsor JA, Itkin M, Phillips ARJ. Renal Lymphatics: Anatomy, Physiology, and Clinical Implications. Front Physiol 10: 251, 2019. doi: 10.3389/fphys.2019.00251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Scallan J, Huxley VH, Korthuis RJ. Capillary Fluid Exchange: Regulation, Functions, and Pathology. Colloquium Series on Integrated Systems Physiology: From Molecule to Function. San Rafael, CA: Morgan & Claypool Life Sciences, 2010, vol 2. [PubMed] [Google Scholar]
- 47.Scallan JP, Hill MA, Davis MJ. Lymphatic vascular integrity is disrupted in type 2 diabetes due to impaired nitric oxide signalling. Cardiovasc Res 107: 89–97, 2015. doi: 10.1093/cvr/cvv117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Scallan JP, Zawieja SD, Castorena-Gonzalez JA, Davis MJ. Lymphatic pumping: mechanics, mechanisms and malfunction. J Physiol 594: 5749–5768, 2016. doi: 10.1113/JP272088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sinzinger H, Kaliman J, Mannheimer E. Regulation of human lymph contractility by prostaglandins and thromboxane. Lymphology 17: 43–45, 1984. [PubMed] [Google Scholar]
- 50.Ulvmar MH, Mäkinen T. Heterogeneity in the lymphatic vascular system and its origin. Cardiovasc Res 111: 310–321, 2016. doi: 10.1093/cvr/cvw175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vieira JM, Norman S, Villa Del Campo C, Cahill TJ, Barnette DN, Gunadasa-Rohling M, Johnson LA, Greaves DR, Carr CA, Jackson DG, Riley PR. The cardiac lymphatic system stimulates resolution of inflammation following myocardial infarction. J Clin Invest 128: 3402–3412, 2018. doi: 10.1172/JCI97192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.von der Weid PY. ATP-sensitive K+ channels in smooth muscle cells of guinea-pig mesenteric lymphatics: role in nitric oxide and beta-adrenoceptor agonist-induced hyperpolarizations. Br J Pharmacol 125: 17–22, 1998. doi: 10.1038/sj.bjp.0702026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.von der Weid PY. Review article: lymphatic vessel pumping and inflammation--the role of spontaneous constrictions and underlying electrical pacemaker potentials. Aliment Pharmacol Ther 15: 1115–1129, 2001. doi: 10.1046/j.1365-2036.2001.01037.x. [DOI] [PubMed] [Google Scholar]
- 54.von der Weid PY, Zhao J, Van Helden DF. Nitric oxide decreases pacemaker activity in lymphatic vessels of guinea pig mesentery. Am J Physiol Heart Circ Physiol 280: H2707–H2716, 2001. doi: 10.1152/ajpheart.2001.280.6.H2707. [DOI] [PubMed] [Google Scholar]
- 55.von der Weid PY, Rehal S, Dyrda P, Lee S, Mathias R, Rahman M, Roizes S, Imtiaz MS. Mechanisms of VIP-induced inhibition of the lymphatic vessel pump. J Physiol 590: 2677–2691, 2012. doi: 10.1113/jphysiol.2012.230599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang Y, Oliver G. Current views on the function of the lymphatic vasculature in health and disease. Genes Dev 24: 2115–2126, 2010. doi: 10.1101/gad.1955910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu TF, Carati CJ, Macnaughton WK, von der Weid PY. Contractile activity of lymphatic vessels is altered in the TNBS model of guinea pig ileitis. Am J Physiol Gastrointest Liver Physiol 291: G566–G574, 2006. doi: 10.1152/ajpgi.00058.2006. [DOI] [PubMed] [Google Scholar]
- 58.Yazdani S, Navis G, Hillebrands JL, van Goor H, van den Born J. Lymphangiogenesis in renal diseases: passive bystander or active participant? Expert Rev Mol Med 16: e15, 2014. doi: 10.1017/erm.2014.18. [DOI] [PubMed] [Google Scholar]
- 59.Zarjou A, Black LM, Bolisetty S, Traylor AM, Bowhay SA, Zhang MZ, Harris RC, Agarwal A. Dynamic signature of lymphangiogenesis during acute kidney injury and chronic kidney disease. Lab Invest 99: 1376–1388, 2019. doi: 10.1038/s41374-019-0259-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zawieja DC. Contractile physiology of lymphatics. Lymphat Res Biol 7: 87–96, 2009. doi: 10.1089/lrb.2009.0007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zawieja SD, Castorena JA, Gui P, Li M, Bulley SA, Jaggar JH, Rock JR, Davis MJ. Ano1 mediates pressure-sensitive contraction frequency changes in mouse lymphatic collecting vessels. J Gen Physiol 151: 532–554, 2019. doi: 10.1085/jgp.201812294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Zhang R, Gashev AA, Zawieja DC, Lane MM, Davis MJ. Length-dependence of lymphatic phasic contractile activity under isometric and isobaric conditions. Microcirculation 14: 613–625, 2007. doi: 10.1080/10739680701436160. [DOI] [PubMed] [Google Scholar]