

Keywords: bicarbonate, blood glucose, cholinergic anti-inflammatory pathway, clinical trial, complement, glomerular filtration rate, metabolic acidosis, pH
Abstract
Sodium bicarbonate (NaHCO3) has been recognized as a possible therapy to target chronic kidney disease (CKD) progression. Several small clinical trials have demonstrated that supplementation with NaHCO3 or other alkalizing agents slows renal functional decline in patients with CKD. While the benefits of NaHCO3 treatment have been thought to result from restoring pH homeostasis, a number of studies have now indicated that NaHCO3 or other alkalis may provide benefit regardless of the presence of metabolic acidosis. These data have raised questions as to how NaHCO3 protects the kidneys. To date, the physiological mechanism(s) that mediates the reported protective effect of NaHCO3 in CKD remain unclear. In this review, we first examine the evidence from clinical trials in support of a beneficial effect of NaHCO3 and other alkali in slowing kidney disease progression and their relationship to acid-base status. Then, we discuss the physiological pathways that have been proposed to underlie these renoprotective effects and highlight strengths and weaknesses in the data supporting each pathway. Finally, we discuss how answering key questions regarding the physiological mechanism(s) mediating the beneficial actions of NaHCO3 therapy in CKD is likely to be important in the design of future clinical trials. We conclude that basic research in animal models is likely to be critical in identifying the physiological mechanisms underlying the benefits of NaHCO3 treatment in CKD. Gaining an understanding of these pathways may lead to the improved implementation of NaHCO3 as a therapy in CKD and perhaps other disease states.
INTRODUCTION
Clinical trials in patients with chronic kidney disease (CKD) have raised questions regarding the potential therapeutic use of sodium bicarbonate (NaHCO3). Over the past two decades, there has been interest in the use of NaHCO3 to prevent the negative effects of acidosis in patients with CKD. The kidneys are responsible for balancing the daily acid load. In patients with CKD, where the kidney can often no longer efficiently perform this task, the development of metabolic acidosis is common (7, 70). Ingestion of NaHCO3 or other alkali limits the need for the kidneys to excrete an acid load (46), which acts to improve pH homeostasis. While restoring pH homeostasis was predicted to prevent the negative effects of metabolic acidosis, including bone and muscle wasting, a surprising result of this research has been observations that NaHCO3 or other alkali may also protect the kidney from injury. A number of small clinical trials have now supported the use of NaHCO3 to slow kidney functional decline, despite the absence of a clear pathological mechanism linking metabolic acidosis with kidney injury (Table 1). Additional data from clinical studies indicate that NaHCO3 may also improve glucose homeostasis in patients with CKD (6). Even more surprising is evidence that this protective effect may be observed in patients without preexisting metabolic acidosis. These findings raise important questions regarding the mechanism of action of NaHCO3 and its potential use in other conditions.
Table 1.
Clinical trials evaluating NaHCO3 in CKD progression
| Reference | Year | Study Design and Duration | Intervention | CKD Stage | Acid/Base Status | Participant Number | Outcomes |
|---|---|---|---|---|---|---|---|
|
Benefit | |||||||
| de Brito-Ashurst et al. (28) | 2009 | Single-center, randomized, open label; 2 yr | NaHCO3 vs. SC | 4 and 5 | Serum (mmol/L); Pre: NaHCO3 (19.8) vs. SC (19.9), Post: NaHCO3 (∼24) vs. SC (∼20) | NaHCO3 (67) vs. SC (67) | Nutritional status, GFR, ESKD, change in Cr clearance |
| Mahajan et al. (83) | 2010 | Prospective, randomized, placebo-controlled, blinded; 5 yr | NaHCO3 vs. NaCl vs. placebo | 2 and 3 | Venous total CO2 (mM); Pre: NaHCO3 (26.2) vs. NaCl (26.4) vs. placebo (26.0), Post: NaHCO3 (26.4) vs. NaCl (26.3) vs. placebo (26.1) | NaHCO3 (40) vs. NaCl (40) vs. placebo (40) | Rate of estimated GFR decline, plasma Cr, urinary markers of kidney injury |
| Goraya et al. (43) | 2014 | Single-center, randomized, open label; 3 yr | NaHCO3 vs. F+V vs.SC | 3 | Venous Pco2; Pre: NaHCO3 (41.6) vs. F+V (41.5) vs. SC (41.4), Post: NaHCO3 (41.9) vs. F+V (41.9) vs. SC (41.3) | NaHCO3 (36) vs. F+V (36) vs. SC (36) | Change in estimated GFR, urinary markers of kidney injury |
| Jeong et al. (55) | 2014 | Single-center, randomized, paralleled; 1 yr | NaHCO3 vs. SC | 4 and 5 | Total CO2 (meq/L); Pre: NaHCO3 (18.5) vs. SC (18.9), Post: NaHCO3 (19.8) vs. SC (16.5) | NaHCO3 (40) vs. SC (40) | Change in estimated GFR, renal replacement therapy, nutritional status |
| Dubey et al. (35) | 2018 | Single-center, randomized, open label; 6 mo | NaHCO3 vs. SC | 3 and 4 | Serum (meq/L); Pre: NaHCO3 (18.1) vs. SC (18.1), Post: NaHCO3 (23.5) vs. SC (17.8) | NaHCO3 (94) vs. SC (94) | Change in estimated GFR, lean body mass, mid-arm muscle circumference |
| Di Iorio et al. (32) | 2019 | Single-center, randomized, open-label; 3 yr | NaHCO3 vs. SC | 3–5 | Serum (meq/L); Pre: NaHCO3 (21.5) vs. SC (21.4), Post: NaHCO3 (26.1) vs. SC (21.9) | NaHCO3 (376) vs. SC (364) | Cr doubling, all-cause mortality, time to renal replacement therapy |
| Goraya et al. (42) | 2019 | Single-center, open-label; 5 yr | NaHCO3 vs. F+V vs. SC | 3 | NA | NaHCO3 (36) vs. F+V (36) vs. SC (36) | Change in estimated GFR, systolic blood pressure, cardiovascular events, serum low-density lipoprotein, serum |
| Alva et al. (2) | 2020 | Single-center, randomized, paralleled; 9 mo | NaHCO3 vs. SC | 4 | Serum (meq/L); Pre: NaHCO3 (16.6) vs. SC (16.8), Post: NaHCO3 (19.8) vs. SC (16.3) | NaHCO3 (33) vs. SC (34) | Estimated GFR, serum , muscle mass, serum albumin |
|
No benefit | |||||||
| BiCARB Study Group (8) | 2020 | Multicenter, randomized, double-blind, placebo controlled; 2 yr | NaHCO3 vs. placebo | 4 and 5 | Serum (mmol/L); Pre: NaHCO3 (20.6) vs. placebo (20.1), Post: NaHCO3 (∼22.6) vs. placebo (∼22.1) | NaHCO3 (116) vs. placebo (104) | Short physical performance battery, blood pressure, renal function, quality of life measure, anthropometry, bone and vascular health markers, incremental cost |
| Melamed et al. (88) | 2020 | Multicenter, randomized, double-blind, placebo controlled; 2 yr | NaHCO3 vs. placebo | 3 and 4 | Serum (meq/L); Pre: NaHCO3 (24.0) vs. placebo (24.1), Post: NaHCO3 (24.4) vs. placebo (23.6) | NaHCO3 (74) vs. placebo (75) | Sit-to-stand time, handgrip strength, bone mineral density, muscle biopsy |
| Raphael et al. (111) | 2020 | Single-center, randomized, double-blind, placebo controlled; 6 mo | NaHCO3 vs. placebo | 3–5 | Serum total CO2 (meq/L); Pre: NaHCO3 (24.0) vs. placebo (24.0), Post: NaHCO3 (25.0) vs. placebo (23.8) | NaHCO3 (35) vs. placebo (39) | Urinary transforming growth factor-β1/Cr, urinary kidney injury markers |
CKD, chronic kidney disease; Cr, creatinine; ESKD, end-stage kidney disease; GFR, glomerular filtration rate; F+V, fruits + vegetables; NA, not applicable; SC, standard care.
Despite a number of clinical trials reporting data for the affirmative, the therapeutic benefit of NaHCO3 to slow glomerular filtration rate (GFR) decline remains to be proven. Most studies supporting the use of NaHCO3 to slow GFR decline in patients with CKD are small, single-center trials, and not all studies have reported a benefit (Table 1). Additional large multicenter, double-blind, placebo-controlled, clinical trials are required before any beneficial action of NaHCO3 to slow GFR decline can be established. In the absence of a clear physiological mechanism mediating the benefit of NaHCO3 on GFR decline, though, how should these studies be designed? The progression of CKD itself is poorly understood. Which population should be targeted so that any potential benefit of NaHCO3 treatment is not missed? What dose of NaHCO3 should be used and how should it be administered? What level of plasma bicarbonate ()/pH, if any, should be the goal of treatment? While to date this field has been primarily driven by clinical observations, the answers to many of these questions may be most efficiently found through animal studies.
This goal of this review is to outline what is known regarding the therapeutic use of alkali in CKD and to identify key gaps in our knowledge that must be answered to drive the field forward. We will attempt to critically examine the mechanisms so far proposed to underlie the benefits of alkali in CKD. Furthermore, we will highlight how answers obtained from animal studies may impact the design of clinical trials.
CLINICAL EVIDENCE FOR A BENEFIT OF ALKALI SUPPLEMENTATION ON GFR DECLINE IN CKD
Acidosis in CKD and Its Relationship to GFR Decline
CKD represents an enormous health burden (18a, 96), and new treatments to slow the progression of CKD are needed. CKD is defined as abnormalities of kidney structure or function and/or a prolonged decrease in GFR of >3 mo in duration (53). Diabetes and hypertension are the major risk factors for CKD and the primary diagnosis in the majority of patients (96); however, only a fraction of patients with these diseases develop CKD (57, 74, 150). As such, in most cases, the pathophysiology of CKD remains poorly understood. CKD is progressive (18a), with both the risk of morbidity and mortality (96) for patients and the cost of treatment (18b) increasing as CKD worsens.
Associations between metabolic acidosis and the rate of GFR decline in patients with CKD suggest metabolic acidosis may drive CKD progression. Metabolic acidosis is defined by a serum of < 22 meq/L and a Pco2 of <40 mmHg (16, 71). Data implicating low-serum in the progression of CKD came from the Chronic Renal Insufficiency Cohort Study (34). It was reported that among 3,939 participants with CKD stages 2–4, the risk of developing a renal end point [defined as a 50% reduction in estimated GFR (eGFR) or the initiation of dialysis or kidney transplantation for end-stage renal disease] was 3% lower per 1.0 meq/L increase in serum level. As increased dietary acid load may be associated with low serum , a number of studies have investigated the relationship between dietary acid load and CKD progression (4, 75, 104, 114). Data from the National Health and Nutrition Examination Survey demonstrated that among 12,293 participants for whom dietary acid was estimated, higher levels of net acid excretion were associated with greater odds of albuminuria and a trend toward low eGFR that persisted after adjustment for confounders (4). Together, these data suggest that low-serum or high dietary acid loads may drive GFR decline.
Evidence That Alkali Supplementation May Slow GFR Decline in Patients With Metabolic Acidosis
Several small clinical trials have now indicated that NaHCO3 supplementation is of benefit to slow GFR decline in patients with metabolic acidosis (Table 1). de Brito-Ashurst et al. (28) found that among 134 patients with stage 4 and 5 CKD and metabolic acidosis, GFR decline over 2 yr was slower with NaHCO3 supplementation (1.88 mL/min/1.73 m2) compared with standard care (5.93 mL/min/1.73 m2, P < 0.0001). Furthermore, patients supplemented with NaHCO3 were less likely to experience rapid progression of CKD (9% vs. 45%, P < 0.0001), and fewer developed end-stage kidney disease (ESKD; 6.5% vs. 33%). Serum levels increased significantly from an average of ∼19 mmol/L to ∼24 mmol/L following 2 yr of treatment with NaHCO3 (Table 1). More recently, Goraya et al. (43) found similar protective effects on GFR decline with either NaHCO3 treatment or a diet high in alkali-producing fruits and vegetables. In a 3-yr trial, including 108 patients with metabolic acidosis and stage 3 CKD due to hypertensive nephropathy, eGFR decline remained lower in patients receiving NaHCO3 or fruits and vegetables compared with controls (Table 1). Jeong et al. (55) also reported a beneficial effect of NaHCO3 treatment to slow eGFR decline in patients with metabolic acidosis. Over 12 mo, in 40 patients with stage 4 CKD and a total CO2 of <22 meq/L, eGFR was found to decrease by −2.30 ± 4.49 mL/min/1.73 m2 in patients given NaHCO3 versus −6.58 ± 6.32 mL/min/1.73 m2 (P < 0.05) in the control group. In 2019, Di Iorio et al. (32) published the results of a randomized, open-label, controlled trial to determine the effect of NaHCO3 supplementation versus standard care in patients with stage 3–5 CKD and metabolic acidosis over 3 yr. Serum levels were similar between the two groups at the start of the study but increased significantly in the NaHCO3-treated group following 1 yr of treatment to 25.0 ± 2.4 mmol/L versus 22.3 ± 1.9 mmol/L in controls, and this difference was sustained for the duration of the study. 17.0% of patients receiving standard care had a doubling of serum creatinine compared with 6.6% of patients supplemented with NaHCO3. Similarly, there were significant differences in the time to renal replacement therapy between the two groups, as 12.3% of participants receiving standard care versus 6.9% receiving NaHCO3 started dialysis. Importantly, all-cause mortality was significantly reduced. 6.8% of participants receiving standard care died compared with 3.1% supplemented with NaHCO3 (Table 1). Dubey et al. (35) also recently reported a clinical benefit of NaHCO3 supplementation in patients with metabolic acidosis. Patients with stage 3 and 4 CKD were randomized to receive standard care or standard care plus NaHCO3 supplementation to maintain serum within normal ranges for 1 yr. GFR in the NaHCO3-treated group was higher at the end of the study following NaHCO3 supplementation (33 vs. 28 mL/min/1.73 m2, P ≤ 0.001). Furthermore, a rapid decline in GFR was documented in 41.5% of patients in the standard care group and only 20.2% of patients in the NaHCO3-supplemented group (P = 0.001). Serum increased in NaHCO3-supplemented patients compared with controls (23.45 vs. 17.8 mmol/L, P < 0.001). Most recently, Alva et al. (2) reported the results from a single-center, parallel, randomized, controlled trial, including 67 patients with CKD and metabolic acidosis. In this study of 9-mo duration, NaHCO3 treatment raised serum levels and prevented eGFR decline compared with controls. In all, eight clinical trials have now been completed that reported a significant beneficial effect of NaHCO3 on CKD progression (Table 1).
Not all studies have identified a benefit of alkali supplementation to slow GFR decline in CKD. Data from the BiCARB Study Group (8), a multicenter, double-blind, placebo-controlled trial (Table 1), revealed that there was no significant treatment effect of NaHCO3 on renal outcomes over 2 yr. None of eGFR (0.6 mL/min/1.73 m2, P = 0.39), the risk of commencing renal replacement therapy (P = 0.43), or time to either a doubling of serum creatinine, a 40% reduction in eGFR, or initiation of renal replacement therapy (P = 0.53) were different with NaHCO3 treatment. Additionally, both Raphael et al. (111) and Melamed et al. (88) also failed to observe any significant differences between NaHCO3-supplemented groups and placebo on eGFR decline at the completion of their respective studies. Raphael et al. (111) observed a small but not statistically significant increase in eGFR in the NaHCO3-supplemented group following 6 mo of treatment (4.0 mL/min/1.73 m2). Melamed et al. (88) observed no significant differences in eGFR following 24 mo of treatment with either placebo (37.1 ± 12.2 vs. 36.3 ± 12.9 mL/min/1.73 m2) or NaHCO3 (39.2 ± 14.5 vs. 38.5 ± 17.7 mL/min/1.73 m2).
There are several reasons that may explain the contrasting results of clinical trials in regard to the benefits of NaHCO3 treatment on GFR decline. All of the three studies discussed that failed to observe a significant beneficial effect of NaHCO3 were multicenter, double-blind, placebo-controlled clinical trials. As discussed by Hu et al. (49), failure to appropriately blind and lack of a placebo control in clinical studies showing a benefit of NaHCO3 on CKD progression could have introduced bias that may have affected the results. Improved experimental design in the three negative studies, then, could potentially explain the absence of an observed effect. There are, however, other important differences between these studies that could also explain the contrasting results. For both the BiCARB Study Group (8) and Raphael et al. (111) studies, the mean age of study participants was around 72.5 yr of age. As denoted by Hu et al. (49), in studies showing a benefit of NaHCO3, the average age of the participants with CKD ranged from 41 to 65 yr old, with the majority of trials having recruited patients with a mean age <56 yr. Unrecognized differences in the etiology of CKD between populations may explain why some individuals are not responsive to NaHCO3. GFR typically declines with age (26). It is possible that NaHCO3 may not be as effective in older patients with CKD as it is in younger patients with CKD, where GFR decline may have been more rapid due to additional pathologies. A potential reflection of these differences is the greater rate of GFR decline observed in generally younger control patients in studies in which NaHCO3 slowed GFR decline. These issues highlight the necessity to better understand the mechanism(s) through which NaHCO3 may impart benefit in CKD.
Evidence of the Beneficial Effects of Alkali Supplementation on GFR Decline May Be Independent of Serum
Data from both clinical and preclinical studies indicate that the beneficial effects of NaHCO3 to slow GFR decline may be observed independently of metabolic acidosis. To date, most clinical trials of NaHCO3 have been performed in patients with metabolic acidosis (Table 1). In addition to renal protection, these studies have also demonstrated that NaHCO3 supplementation increased serum levels (2, 28, 32, 35). These data are consistent with the hypothesis that metabolic acidosis drives more rapid loss of kidney function. An alternative interpretation of these data, however, may be that reduced serum simply indicates already lower underlying renal function in patients more susceptible to rapid decline, despite similar eGFR. In this scenario, increases in serum associated with alkali treatment may not drive the beneficial response. In support of this, Mahajan et al. (83) demonstrated that NaHCO3 supplementation provided protection from GFR decline in patients with stage 2 CKD without metabolic acidosis. In their study, Mahajan et al. (83) found that the rate of GFR decline over 5 yr in subjects treated with NaHCO3 (−1.47 ± 0.19 mL/min/yr) was significantly less than that observed in the placebo group (−2.13 ± 0.19 mL/min/yr, P = 0.014) or the NaCl-treated control group (−2.05 ± 0.19 mL/min/yr, P = 0.029). While these differences are mild, GFR decline in both the control and NaCl-treated groups was less than that observed in most clinical studies that have shown a benefit of NaHCO3 treatment (28, 32, 35, 55). Additionally, the slowing of GFR decline by ∼0.6 mL/min/yr represents almost a 30% reduction in the rate of GFR decline. Entry and 5-yr serum total CO2 were not different among the three groups. It is important to note, however, that while early clinical work by Mahajan et al. (83) revealed a beneficial effect of NaHCO3 treatment in nonacidotic CKD, the findings of the recent study by Melamed et al. (88) stand in contrast to this. In patients with CKD stages 3 and 4 with average baseline serum levels of 24.0 ± 2.2 meq/L, Melamed et al. (88) failed to observe any significant differences in eGFR between patients given NaHCO3 or placebo.
Evidence from rodent models also indicates that the benefits of alkali treatment to slow CKD progression may be independent of serum acid-base status. The two-thirds nephrectomy rat model is a model of early-stage CKD that does not develop metabolic acidosis (142). Wesson and Simoni (142) found that GFR declined in control rats over a 24-wk period but that dietary supplementation with calcium bicarbonate [Ca()2] prevented any GFR decline (Table 2). Our own data in the Dahl salt-sensitive rat model also indicate that NaHCO3 supplementation limits renal injury independent of metabolic acidosis (113). The Dahl salt-sensitive rat model is a model of salt-sensitive hypertension that develops high blood pressure and progressive renal injury when fed a high-salt diet (108). In our study, we found that arterial blood was elevated above normal following high salt (8% salt) feeding in both vehicle (0.1 M NaCl drinking) and 0.1 M NaHCO3 drinking animals (likely secondary to mild hypokalemia) and was not significantly different between the two groups (P. M. O’Connor, unpublished observations). Despite the absence of a difference in plasma concentration and a tendency toward systemic alkalosis, NaHCO3 supplementation in the drinking water greatly attenuated tubular hyaline cast formation (Fig. 1) compared with NaCl treatment. This effect was associated with lower levels of tubular interstitial fibrosis and was independent of any difference in blood pressure (113). Furthermore, two studies have found that acidosis does not drive CKD progression in rodents (54, 128). Together, these data indicate that alkali supplementation may be renoprotective, even in the absence of metabolic acidosis.
Table 2.
Alkali treatment in rodent models of renal injury progression
| Reference | Year | Model | Intervention | Acid/Base Status | Outcomes |
|---|---|---|---|---|---|
|
Benefit | |||||
| Nath et al. (95) | 1985 | Remnant kidney model (infarction); 1¾ nephrectomy | Dietary NaHCO3 (2.5 meq of per 10 g of food intake) vs. equimolar dietary NaCl | Plasma (mM); NaHCO3 (31) vs. NaCl (20.4) | Total urinary protein excretion, urinary low-molecular-weight protein excretion, GFR, transport maximum for p-aminohippurate |
| Tolins et al. (129) | 1987 | Chronic K+ deficiency | 150 mM NaHCO3 or equimolar NaCl in drinking water | Arterial concentration (meq/L); NaHCO3 (34.7) vs. NaCl (21.7) |
Urine output, urinary total protein excretion, urinary low-molecular-weight protein, GFR, renal plasma flow, renal NH3 metabolism, tubulointerstitial injury |
| Torres et al. (130) | 1994 | Autosomal dominant polycystic kidney disease | 300 mM NH4Cl or 300 mM KHCO3 or 200 mM KHCO3 or 200 mM KCl or 200 mM NaHCO3 or 200 mM NaCl | Plasma (meq/L); male: control (21.3) vs. 300 KHCO3 (24.5) vs. 200 KHCO3 (23.7) vs. KCl (21.4) vs. NaHCO3 (25.5) vs. NaCl (19.4), female: control (23.5) vs. NH4Cl (17.7) vs. 200 KHCO3 (23.8) vs. KCl (20.9) vs. NaHCO3 (27.9) vs. NaCl (20.4) | Plasma creatinine, urinary creatinine, urinary ammonium, renal cystic changes, immune cell infiltration |
| Tanner (125) | 1998 | Autosomal dominant polycystic kidney disease | Potassium citrate/citric acid (55 mM tripotassium citrate/67 mM citric acid) or tap water | NA | Terminal blood pressure, GFR, plasma potassium, urinary potassium, kidney fibrosis, widening of internephron space, inflammatory cells, individual cyst/lumen area, cortical cyst number |
| Gadola et al. (37) | 2004 | Remnant kidney model (incision); 5/6 nephrectomy | Captopril (500 mg/L) in drinking water or calcium citrate (1.45 g/100 g food) in food or captopril and calcium citrate or nontreated | Arterial (meq/L); captopril (20.5) vs. calcium citrate (20.7) vs. captopril + calcium citrate (20.3) vs. nontreated (16.3) vs. sham (20.5) | Proteinuria, blood pressure, inulin clearance, plasma calcium, renal fibrosis markers |
| Wesson and Simoni (142) | 2010 | Remnant kidney model (incision); 2/3 nephrectomy | Dietary Ca(HCO3)2 (125 μmol/g, 17 g/day) and/or darusentan (20 mg/kg/day) and/or eplerenone (100 mg/kg/day) | Arterial total CO2 (mmol/L); arterial partial CO2 (mmol/L) |
GFR, plasma ET-1, plasma aldosterone, urinary net acid excretion |
| Kim et al. (62) | 2014 | Remnant kidney model (incision); 5/6 nephrectomy | Dietary NaHCO3 (174 μM/g) or dietary NaCl (174 μM/g) | Serum total CO2 (mmol/L); NaHCO3 (23.5) vs. NaCl (16.1) |
Sodium balance, glomerulosclerosis, tubulointerstitial injury, renal sodium and acid-base transporter expression, kidney tissue ET-1 levels |
| Kim et al. (64) | 2014 | Remnant kidney model (incision); 5/6 nephrectomy | Standard diet or dietary sodium citrate (174 μM/g) or dietary NaCl (174 μM/g) | Serum total CO2 (mmol/L); remnant: standard diet (24.8) vs. dietary sodium citrate (24.5) vs. dietary NaCl (21.5), sham: standard diet (25.5) vs. dietary sodium citrate (25.5) vs. dietary NaCl (25.5) |
Blood pressure, urine sodium excretion, glomerulosclerosis, tubulointerstitial injury, renal sodium and acid-base transporter expression, kidney tissue ET-1 levels |
| Ray et al. (113) | 2018 | Dahl salt-sensitive | High-salt diet (8%) and 0.1 M NaHCO3 or equimolar NaCl in drinking water | Arterial (mM); NaHCO3 (26.1) vs. NaCl (25.5) |
Blood pressure, renal arterial blood velocity, glomerulosclerosis, fibrosis, protein casts, proteinuria |
|
No benefit | |||||
| Throssell et al. (128) | 1995 | Remnant kidney model (infarction); 5/6 nephrectomy | Dietary NaHCO3 (1.9 g/100 g food) or NaCl (1.3 g/100 g food) in Na+-deficient diet | Arterial pH, NaHCO3 (7.27) vs. NaCl (6.95) | Blood pressure, GFR, proteinuria, glomerulosclerosis, tubular dilatation, cast formation, interstitial fibrosis, total kidney collagen content, time to development of terminal uremia |
ET-1, endothelin-1; GFR, glomerular filtration rate; NA, not applicable.
Fig. 1.
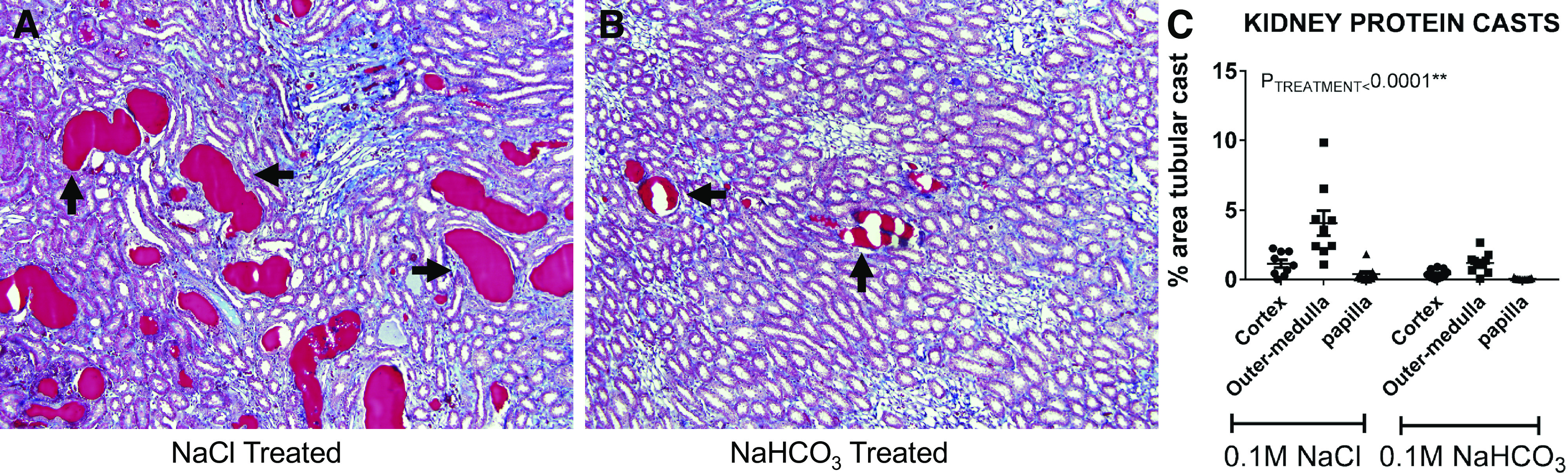
Tubular casts in the kidney of Dahl salt-sensitive rats. Dahl salt-sensitive rats develop waxy proteinaceous casts (arrows) in renal tubules following high salt feeding. Two weeks of treatment with 0.1 M NaHCO3 solution limits the formation of these tubular casts in the renal medulla (B) compared with 0.1 M NaCl solution (A). Images are shown at ×10 original magnification. Average cast areas in the renal cortex, outer medulla, and papilla in NaCl-treated (n = 9) and NaHCO3-treated (n = 11) Dahl salt-sensitive rats fed a high-salt diet (8%) are shown in C. Data are expressed as means ± SE. Symbols represent individual animals. PTREATMENT is the result of two-way ANOVA comparing kidney region and treatment. [The figure modified from Ray et al. (113).]
How could alkali supplementation protect the kidney without altering systemic acid-base status? One possibility is that the compensatory response of the kidney to alkali supplementation is beneficial. Changes in dietary acid/alkali loads are not generally reflected as large changes in systemic pH/serum status in healthy subjects or in subjects with moderate renal impairment. These stimuli do, however, alter the function of the kidney, which is reflected in changes in the composition of urine such as net acid excretion. It is possible that changes in renal function in response to alkali loading mediate the protective effects of NaHCO3 independent of systemic pH status. Another, related possibility to explain the apparent disconnect between serum and evidence that acidosis drives renal functional decline is the proposed phenomenon of “subclinical acidosis” (139). Subclinical acidosis, or “eubicarbonatemic metabolic acidosis,” describes a condition that has been reported to occur early in CKD, wherein patients are unable to completely excrete their daily acid load and thus retain acid; however, this is insufficient to decrease serum levels (139). Increased dietary acid has been demonstrated to increase kidney cortical acid content and urinary net acid excretion in the absence of any significant differences in arterial pH in rodents with normal kidney function or two-thirds subtotal nephrectomy (143). Consistent with this concept, studies from humans with stage 2 CKD revealed that these individuals excrete less urinary acid in the face of an acute NaHCO3 bolus compared to patients with a normal eGFR, suggesting that patients with stage 2 CKD have greater acid retention basally (144). It is also possible that proton shifts between the intracellular and extracellular compartments may prevent changes in cellular acid-base status from being reflected in changes in serum . Another possibility, as discussed later, is that extrarenal mechanisms that respond to alkali supplementation are responsible for protecting the kidney.
POTENTIAL MECHANISMS OF PROTECTION
The physiological mechanism(s) underlying the reported beneficial actions of NaHCO3 supplementation on GFR decline remains unclear, and identifying this mechanism(s) is likely to advance the use of alkali as a therapeutic. In the absence of a clear physiological mechanism, investigators can only speculate at the answers to many key questions, which are required to design ongoing clinical trials in this field. Ultimately, while the efficacy of any therapeutic must be assessed in a clinical trial, animal studies may provide insight into the physiological mechanism(s) mediating the observed benefit. That being said, identifying the mechanism providing physiological benefit from NaHCO3 supplementation in CKD is likely to be challenging, as the pathogenesis of CKD itself is not well understood. Furthermore, animal models do not replicate all of the features of CKD in the human population, and not all animal studies have identified a benefit of NaHCO3 treatment. A list of animal studies in which alkali supplementation has been investigated in regard to its effects on renal injury, which is categorized by outcome (benefit/no benefit), is shown in Table 2. Five studies have shown alkali treatment slows GFR decline (37, 62, 125, 130, 142). Conversely, Throssell et al. (128) found that NaHCO3 did not slow CKD progression in rats with acidosis, and Jara et al. (54) have reported that metabolic acidosis may even halt the progression of kidney disease in rats fed a high-phosphate diet. Possible explanations for differences in the outcome of these rodent studies include differences in the severity of the models used and whether Na+ was given as the cation with . For example, in the remnant kidney model, Wesson and Simoni (142), who found alkali treatment slowed GFR decline, used a more moderate surgical incision in the two-thirds nephrectomy, model followed rats for the longest period (24 wk), and used a Na+-free alkali [Ca()2]. Similarly, Gadola et al. (37) used the surgical incision remnant kidney model and Na+-free alkali (calcium citrate) to prevent the development of severe hypertension. In contrast, Throssell et al. (128) used the more severe infarction five-sixth nephrectomy model and administered Na+ as the cation with treatment. These are important differences as the method of renal mass reduction is critically important in determining the development of subsequent hypertension and glomerular injury, with both being more severe in the infarction model (44). It is possible that alkali treatment is insufficient to slow GFR decline in the more rapidly developing renal infarction model of CKD or in animals that develop significant hypertension from Na+ loading. Nevertheless, studies in animal models have already revealed several candidate pathways that could underlie the benefits of alkali supplementation in CKD.
In the following section, we will outline the physiological mechanisms proposed so far to potentially underlie the benefit conferred by alkali supplementation in CKD. We will then attempt to highlight the strengths and weaknesses of the data supporting each pathway. Finally, we will comment on how potential confirmation of each given pathway could inform the design of future clinical trials.
DOES THE RENAL COMPENSATORY RESPONSE TO AN ACID LOAD DRIVE GFR DECLINE?
Activation of Intrarenal Complement by High Interstitial Ammonium Concentration
It has been suggested that increased single-nephron ammoniagenesis activates complement within the kidney, contributing to GFR decline (20, 95). The complement pathway is a critical component of the innate immune system. In some cases, however, aberrant activation of the complement pathway may promote unwanted damage. The renal tubules normally act to excrete an acid load through a combination of tubular H+ secretion and production of ammonium ions (). As nephron mass is reduced, individual nephrons must produce relatively more (47, 82, 139). Even if the kidneys are able to maintain serum acid-base homeostasis, this increased single-nephron production may lead to increases in local concentrations within the renal interstitium. It has been proposed that this increased local concentration can disrupt the internal thioester bond of complement protein C3, leading to activation of the alternative complement pathway. This then may result in tissue injury and tubulointerstitial fibrosis, driving GFR decline (22, 95, 109). As alkali treatment reduces the requirement of the kidneys to excrete an acid load and the tubules to produce , this may limit complement activation and GFR decline (95, 129).
Nath et al. (95) demonstrated that in the five-sixth nephrectomy rat remnant kidney model of CKD, administration of oral NaHCO3 resulted in less severe tubulointerstitial injury and reduced peritubular C3 and C5b-9 deposition. Systemic acid-base status was assessed in this study, and NaHCO3-supplemented animals had a significantly higher plasma concentration (31.0 ± 0.7 vs. 20.4 ± 1.6 meq/L) and a significantly lower mean renal vein ammonia concentration (145 ± 20 vs. 240 ± 20 µM) compared with NaCl-supplemented controls. Tolins et al. (129) also implicated ammoniagenesis toward driving renal pathology in a K+ depletion model of renal injury. Furthermore, treatment of metabolic acidosis in humans reduces urine, but not serum, levels of complement activation proteins (91). Together, these findings are consistent with the hypothesis that intrarenal complement activation is ameliorated by reducing tubular ammoniagenesis (95, 129).
The hypothesis that increased concentration drives GFR decline is consistent with the association between net acid excretion (which reflects dietary acid load) and CKD progression. With larger dietary acid loads, there is a greater need for production. Such a relationship would also explain the absence of metabolic acidosis as a requirement to drive GFR decline, as single-nephron production may be upregulated even when acid-base status is maintained. Moreover, the relationship to reduced nephron mass may explain why acid loading does not promote renal injury under normal conditions, despite similarly elevated net acid excretion. In the absence of prior nephron loss, local renal interstitial concentrations may not rise to the same level and, therefore, would not activate complement.
Although the concept that increased single-nephron ammoniagenesis drives GFR decline is consistent with a number of observations, important caveats remain. Studies have revealed a complex relationship between and CKD progression. Nath et al. (95) found that the activation of complement proteins by was biphasic, with complement activity being maximally increased at NH4Cl concentrations around 0.25 mM, whereas NH4Cl concentrations above 0.5 mM significantly reduced complement activity. Furthermore, in more than 1,000 patients with CKD, it was found that lower, rather than higher, urinary was a risk factor for the development of ESKD (135). A study of participants in the African American Study of Kidney Disease and Hypertension (AASK; n = 1,044) study identified a similar association between low excretion and increased risk of death or ESKD (110). Specifically, this study found that patients who were part of the lowest tertile of urinary excretion had a 46% higher risk of death or developing ESKD after adjusting for confounders. Scialla et al. (120) also reported that both the total net acid excretion and the percentage of urinary acid that was excreted as were lower in patients with low eGFRs. These data suggest that factors, such as the degree of prior nephron loss, make the relationship between renal production and CKD progression more complex. It also remains unclear whether complement activation in response to changes in interstitial concentration is sufficient to mediate progression of renal injury. Studies in complement-deficient animals with CKD in which acid-base status is altered experimentally would provide insight into whether biological changes in production drive CKD progression specifically via complement activation.
Another potential caveat of the study by Nath et al. (95) is that blood pressure may have been different between the treatment and control groups. Salt sensitivity is common in rats with a remnant kidney, and NaCl may be more prohypertensive than NaHCO3 or Na+ coupled to other anions (12, 13, 64, 68, 145). This may have led to greater elevations in blood pressure in NaCl-fed rats, which acted as the control. Even small elevations in blood pressure have been reported to drive CKD progression in the remnant kidney model (10). While the authors did show that there was no difference in the acute blood pressure response to NaHCO3 or NaCl, blood pressure was not measured across the entire 6 wk of this study. This leaves the possibility that differences in renal injury were secondary to blood pressure elevation in the control group. Elevated proteinuria in the NaCl-treated control group would be consistent with the elevated blood pressure, which would result in greater glomerular stretch (148). Potential differences in the prohypertensive effects of NaCl and NaHCO3 loading are an important caveat in all preclinical studies, where NaCl is used as a control and highlight the need for careful monitoring of blood pressure in such studies.
If the hypothesis that NaHCO3 slows CKD progression via limiting complement activation can be confirmed, this may impact the design of future clinical trials. Urinary production or, if one can be identified, a more specific indicator of renal complement activity, would likely be the preferred marker(s) over serum acid-base status. As nephron loss is common to all forms of CKD, this would suggest that minimizing single-nephron production with alkali would likely benefit most patients with CKD.
Acid Stimulation of Renal Endothelin-1/Angiotensin II
Increased local production of endothelin (ET)-1 and angiotensin II (ANG II) within the kidney is thought to occur to help excrete an acid load and may also promote renal injury. ET is produced at a number of sites along the nephron, including the proximal tubule and collecting duct (67, 101). While the mechanisms through which the kidney senses and responds to an acid load are not fully elucidated, renal ET activity has been reported to increase in response to dietary acid loading (58). Decreased interstitial and intracellular pH has also been shown to promote ET production (139). ET is thought to promote tubular acid secretion by stimulating Na+/H+ exchange (72) and promoting aldosterone release by the adrenal cortex, which increases distal tubular H+-ATPase activity (59, 87, 101). Renal ET levels have also been demonstrated to be increased in the kidney of five-sixth nephrectomy rats (62, 104). Both systemic and intrarenal ANG II levels have also been reported to be increased in rats with remnant kidneys (41, 81, 98). Like ET, ANG II can be produced within the kidney (89, 97), and local renal ANG II signaling is thought to be important in regulation of renal acid excretion (94). ANG II appears to contribute to renal acid excretion through a number of mechanisms, including stimulation of Na+/H+ exchanger 3 (NHE3) and H+-ATPase (39, 77, 78, 137) as well as stimulation of tubular ammoniagenesis (92). Acid loading also increases ANG II type 1 receptor expression in renal tubular cells, sensitizing the nephron to ANG II (93).
Wesson et al. (139) proposed that decreased renal interstitial and intracellular pH drive tubular injury and fibrosis in CKD via upregulation of intrarenal ET-1 and the local renin-angiotensin-aldosterone system (RAAS). In support of this, treatment of rats with an ET type A receptor antagonist and a mineralocorticoid receptor antagonist or the alkali Ca(HCO3)2 significantly slowed GFR decline in two-third nephrectomy animals over 24 wk of treatment (142). Furthermore, when animals were supplemented with Ca(HCO3)2 in combination with both an ET type A receptor antagonist as well as a mineralocorticoid receptor antagonist, there was no additive protection. These data were interpreted to indicate that alkali loading slows renal injury progression by downregulating the production of these hormones (142). Gadola et al. also found that both the alkali calcium citrate and the angiotensin-converting enzyme inhibitor captopril independently slowed GFR decline in the remnant kidney model (37). Although the average GFR decline was less with combined alkali and captopril therapy, this effect was not significantly greater than either treatment alone (37). These data are also consistent with a role of reduced ANG II in mediating the protective effects of alkali on GFR decline. This hypothesis is consistent with evidence indicating that ET-1, aldosterone, and ANG II can directly promote renal fibrosis and tissue injury (25, 66, 132, 136).
Whether increases in ET and ANG II that occur in the acidotic kidney mediate renal injury through hemodynamic versus direct mechanisms remains unknown; however, experimental evidence indicates that hemodynamic pathways are likely to dominate. The concept of direct renal injury from various vasoactive hormones is well documented. Much of the data supporting this, however, comes from studies where pharmacological levels of these hormones were administered, resulting in severe hypertension, or from in vitro studies that may not be directly applicable to the in vivo condition. Most of the injurious effects of these hormones appear to be mediated by changes in hemodynamics and renal barotrauma. For example, there is little evidence of renal injury under physiological conditions in which ANG II and aldosterone are upregulated absent hypertension, such as during a low-salt diet (21, 38). An additional example in which intrarenal and systemic ANG II levels are markedly elevated but renal injury is limited is the clipped kidney in the two-kidney, one-clip hypertensive model. In this model, invasion of inflammatory cells, morphological changes, glomerular sclerosis, and renal tubular interstitial injury are almost exclusively observed in the contralateral unclipped kidney exposed to elevated arterial pressure. This is despite greater elevations in intrarenal ANG II in the normotensive clipped kidney (36, 107). Injury progression in the remnant kidney model used by Wesson et al. is also highly blood pressure dependent, and the protective effects of renin-angiotensin system blockade in this model have been shown to be attributable primarily to their effect in lowering blood pressure (9). Even subtle changes in blood pressure that may not be easily detected may promote more rapid injury progression over long periods because of impairments in renal autoregulation and enhanced glomerular transmission of the blood pressure in this model (45).
One caveat of the study by Wesson and Simoni (142) is that there was no decline in GFR in the Ca(HCO3)2 alone-treated group (142). This would likely have prevented any further benefit of antagonism of both the ET and mineralocorticoid receptors being detected even if present, as GFR decline could not be further reduced. An alternative interpretation of these data then may be that either Ca (HCO3)2 or ET/mineralocorticoid receptor antagonism limits GFR decline in this model. These effects, however, may not be through a common mechanism.
Although further studies are needed to define the role of acid-induced ET and ANG II in the progression of CKD, if confirmed this result would likely impact the design of clinical trials. Confirmation of the role of ANG II in mediating the beneficial effects of alkali would suggest that patients treated with high-dose RAAS inhibitors may be less responsive to alkali treatment, as both alkali and RAAS inhibitors may work through a common mechanism.
Prevention of Tubular Cast Formation
We have previously reported that luminal alkalization following NaHCO3 acts to limit the formation of tubular casts and associated renal damage in Dahl salt-sensitive rats (113). Nath et al. (95) also reported lower levels of tubular cast formation in NaHCO3-treated remnant kidney rats. The Dahl salt-sensitive rat strain develops hypertension and progressive renal injury when fed a high-salt diet and mimics many of the traits observed in salt-sensitive patients (11, 17, 24, 60, 69, 124). Waxy proteinaceous casts are associated with CKD severity and are a prominent histological finding in Dahl salt-sensitive rats following high salt feeding (Fig. 1) (1, 90). The formation of proteinaceous casts may block fluid flow through the nephron, resulting in nephron failure, cell loss, and fibrosis (102). Proteinaceous casts may form in CKD secondary to loss of the glomerular filtration barrier, resulting in increased filtration of blood proteins. Filtered acidic blood proteins have been reported to precipitate out of solution, which may be facilitated by low luminal fluid pH (149). By lowering the rate of H+ secretion by the tubules, alkali loading may increase the pH of the luminal fluid, limiting the precipitation of acid proteins and proteinaceous cast formation. We found that Dahl salt-sensitive rats given either 0.1 M NaHCO3 or equimolar NaCl in the drinking water and placed on a high-salt (8%) diet for 2 wk did not exhibit any differences in blood pressure. Furthermore, glomerular injury and urinary protein excretion were similar between the two groups (113). We found that animals supplemented with NaHCO3, however, exhibited a marked reduction in tubular cast formation and tissue fibrosis compared with NaCl-treated controls (Fig. 1) (113). A similar phenotype was observed in Dahl salt-sensitive rats lacking the voltage-gated proton channel Hv1, which is expressed on the apical membrane of medullary thick ascending limb segments and mediates outward proton movement (29, 56, 99, 113). Together, these data suggest that NaHCO3 supplementation may limit renal injury by reducing tubular H+ secretion and slowing tubular cast formation.
NaHCO3 supplementation slowed cast formation in Dahl salt-sensitive rats in the absence of any detectable change in serum acid-base parameters. We found that high salt feeding alone elevated blood levels, likely secondary to the development of mild hypokalemia (P. M. O’Connor, unpublished observations). Interestingly, NaHCO3 supplementation had no additional effect to increase blood pH or plasma levels but did reduce urinary net acid excretion (113). These data are consistent with the concept that the beneficial actions of alkali loading can dissociate from serum acid-base status and may be better reflected by urinary net acid excretion.
The hypothesis that alkali loading may protect the kidney by limiting tubular H+ secretion and subsequent protein cast formation is consistent with data linking tubular cast formation with more rapid progression of CKD (1). Furthermore, such a mechanism may explain the reported association with net acid excretion and faster GFR decline (119). Hyaline cast formation is more prominent in individuals carrying apolipoprotein L1 (APOL1) gene risk variants (73). As significant levels of acidic proteins may be filtered by the remaining functional nephrons of patients with CKD (148), the pathogenesis of cast formation may also explain why acid loads alone do not damage the healthy kidney. These data may also explain, in part, the effect of dietary protein source on GFR decline, with diets high in low-molecular-weight acidic proteins being more likely to promote cast formation (117).
As with other mechanisms related to the renal compensatory response to an acid load, a direct link between prevention of this pathology and GFR decline is lacking. We did not measure GFR decline in our study, as when placed on very high-salt diets (8%), Dahl salt-sensitive rats typically die from stroke or heart failure from malignant hypertension before major reductions in GFR are observed. Furthermore, it remains unclear whether proteinaceous casts are simply a result of nephron failure (i.e., slow-moving fluid) or whether they also contribute to CKD progression by tubular blockage or through other mechanisms.
If a causative relationship between prevention of tubular cast formation by alkali and slowing of GFR decline can be established, this would indicate that luminal H+ secretion drives CKD progression. To this end, urinary pH or titratable acids would likely be the preferred indicator used for alkali dosing in the clinical setting. Treatments that slow cast formation may also be most effective in patient populations where tubular cast formation is more prominent, such as in patients with APOL1 risk variants (73).
POTENTIAL EXTRARENAL MECHANISMS OF PROTECTION FROM GFR DECLINE
Activation of the Cholinergic Anti-Inflammatory Pathway by Oral Ingestion of Alkali
Evidence from preclinical models points toward a role of chronic inflammatory processes in the progression of CKD (18, 115). Recent data from human CKD populations also support a role of inflammation in GFR decline. The Chronic Renal Insufficiency Study found that elevated inflammatory markers were associated with rapid loss of kidney function in patients with CKD (3). Furthermore, treatment with TNF-α antagonists has been associated with an attenuation in renal functional decline in patients with rheumatoid arthritis and CKD (61).
Activation of the cholinergic anti-inflammatory pathway (CAIP) is one of the most promising approaches for the treatment of inflammatory diseases (15, 19, 48). The CAIP is an innate anti-inflammatory pathway thought to act via vagal efferent signaling to limit activation of the innate immune system. Activation of the CAIP by vagal nerve stimulation has been reported to attenuate renal injury in rodent models of ischemic (40, 116, 147) and nephrotoxic (52, 133) acute kidney injury, two-kidney one-clip hypertension (76), in a mouse model of systemic lupus erythematosus (103), and in the unilateral ureter obstruction model (121) of CKD. Given the reported association between inflammation and CKD progression as well as mounting preclinical evidence of a renoprotective effect of activation of the CAIP, we speculate that activation of the CAIP may be protective in patients with CKD.
We have previously reported that oral ingestion of NaHCO3 promotes an anti-inflammatory response in both rats and healthy human subjects and that this is likely mediated by activation of the CAIP (112). We found that rats that drank 0.1 M NaHCO3 solution for 3 days demonstrated a robust anti-inflammatory immune cell phenotype within the kidney compared with NaCl-treated controls (112). Most prominently, classically activated proinflammatory M1 macrophages were decreased and alternatively activated anti-inflammatory M2 macrophages were increased in the kidney following NaHCO3 treatment. The CAIP is thought to be dependent on splenic macrophages and is characterized by vagal activation of α7-nicotinic acetylcholine receptors (15). Consistent with activation of the CAIP, pharmacological blockade of α7-nicotinic acetylcholine receptors abolished the anti-inflammatory action of NaHCO3 in rats. Furthermore, the ability of oral NaHCO3 to promote M2 macrophage polarization in the kidney of rats was dependent on the presence of the spleen (112). These data support activation of the CAIP in the anti-inflammatory effect of NaHCO3. We demonstrated a similar anti-inflammatory effect in the blood of healthy human subjects following ingestion of 2 g NaHCO3 in 250 mL water. In 12 healthy human subjects, ingestion of a solution of NaHCO3 resulted in a decrease in M1 macrophages and an increase in M2 macrophages in the blood within 3 h. This effect was significantly different to control subjects who ingested an equimolar solution of NaCl (112). Although the physiological pathways that promote an anti-inflammatory response following ingestion of NaHCO3 solution have yet to be fully elucidated, our data indicate that stimulation of these pathway(s) likely requires gastric acid secretion, as the anti-inflammatory response can be inhibited by a proton pump inhibitor (112). We and others have argued that a physiological role of the CAIP is to sense ingestion of a meal and dampen the inflammatory response to antigens that are absorbed during digestion (14, 85, 131). By increasing the pH of the gastric fluid, ingestion of NaHCO3 mimics meal consumption, which activates the CAIP. In support of this, the hunger peptide ghrelin, which induces similar physiological actions (acid secretion/vagal activation) and high fat feeding, have also been reported to activate the CAIP (79, 86, 131, 146). Activation of the CAIP by ingestion of NaHCO3 may then slow GFR decline by limiting kidney inflammation.
In addition to a beneficial effect to slow GFR decline, a systemic anti-inflammatory response may explain a number of the purported benefits of alkali ingestion. A gastrointestinal-immune mechanism would also explain the independence of any beneficial effect from increases in serum . Further studies are required however, to confirm that ingestion of NaHCO3 activates the cholinergic anti-inflammatory pathway and that this slows GFR decline. There is currently no evidence that activation of the CAIP can slow GFR decline in patients with CKD. Whether inflammatory processes act as a common pathway to drive CKD progression or are simply associated with renal injury also remains unclear.
If activation of the CAIP was identified as the primary mechanism limiting GFR decline in response to oral ingestion of NaHCO3, this finding would have major repercussions. As the benefit of alkali in CKD has been largely attributed to systemic alkalizing effects, metabolic alkali such as diets high in fruits and vegetables have been used as substitutes to oral ingestion of buffers like NaHCO3. Goraya et al. (42) have reported fruits and vegetables were similarly effective in reducing GFR decline as NaHCO3. One way to interpret this result is that all of the benefits of a diet high in fruits and vegetables in preventing GFR decline are mediated by systemic alkalization. It is likely, however, that diets high in fruits and vegetables provide additional protective benefit against GFR decline over NaHCO3 through factors, such as increased K+ load, weight loss, or reduced blood pressure (42, 51). Given this, metabolic alkali may be expected to out-perform NaHCO3 unless additional protective mechanisms are activated with NaHCO3 treatment. While metabolic alkali-like citrate salts would not stimulate significant gastric acid secretion, these treatments have been demonstrated to slow GFR decline in rodent models (37, 125). To the authors’ knowledge, no direct comparison between alkali that would buffer stomach acid and metabolic alkali has been performed. Citrate itself may also provide renal protective benefit outside its alkalizing actions. For example, filtered citrate acts to chelate luminal Ca2+, which may slow CKD progression (123, 134). Therefore, it remains unknown whether stimulation of gastric acid secretion by buffers like NaHCO3 may provide additional benefit. If activation of the CAIP by stimulation of gastric acid secretion mediates some of the protective effects of NaHCO3, dietary intervention with NaHCO3 or newer Na+-free alkali such as alkali like veverimer (140, 141) may provide additive benefit to metabolic alkali. Further, it is possible that concurrent therapies, such as proton pump inhibitors, may limit the efficacy of oral NaHCO3 in CKD. Additionally, if only gastric, but not systemic, alkalization is required to activate the anti-inflammatory response, the dose of alkali administered may be reduced, given the much smaller volume of the stomach compartment compared with systemic volume. Smaller doses that are given more frequently may be more effective, as they may result in more frequent activation of the CAIP. Identification of the CAIP as the pathway mediating the beneficial effects of NaHCO3 in CKD would also provide an impetus for investigation of this approach to treat other inflammatory diseases.
Enhanced Blood Glucose Control and Improved Metabolic Status
Alkali treatment may also slow GFR decline by improving glycemic control or lipid profiles. Tight glucose control may slow GFR decline in patients with diabetic kidney disease (80), and improving metabolic status may also slow GFR decline in nondiabetic patients. In a 2019 meta-analysis of over 51,000 patients with stage 3 and 4 CKD, including 35 randomized controlled trials, Taylor et al. (126) reported that patients taking glycemic control or lipid-modifying drugs may have slower progression of CKD. Although diabetes is one of the leading causes of CKD, nondiabetic patients with CKD also exhibit insulin resistance and impaired glucose tolerance (50). In ESKD, insulin resistance has been demonstrated to be due to downstream impairments in insulin signaling in insulin-sensitive tissues (30). Most recently, results from the Study of Glucose and Insulin in Renal Disease (or SUGAR) demonstrated that nondiabetic patients with CKD have lower insulin sensitivity and impaired insulin clearance compared with healthy controls (27). In this study, an eGFR of <60 mL/min/1.73m2 was moderately positively associated with reduced insulin sensitivity and clearance (r = 0.72, P < 0.001), although no relationships between eGFR and insulin release or glucose tolerance were observed. Suggestive of a direct effect of CKD on insulin resistance, this study found that adjusting for differences in lifestyle as well as body fat mass only partially explained the impairments in insulin sensitivity observed in this cohort of patients with CKD. Rodent models of nondiabetic CKD also demonstrate insulin resistance (33). Together, these data suggest that treatment strategies that improve glucose control could also improve outcomes in patients with CKD, even in the absence of diabetes.
Some evidence associates insulin resistance with the degree of acidosis (122, 127). The first evidence to link metabolic acidosis to insulin resistance came from DeFronzo and Beckles (31). When ammonium chloride (NH4Cl) is metabolized in the liver, it consumes and promotes metabolic acidosis. In 1979, DeFronzo and Beckles (31) demonstrated administration of NH4Cl resulted in a decreased tissue sensitivity to insulin in 16 healthy volunteers. More recently, Kobayashi et al. (65) demonstrated that insulin resistance correlated linearly with decline in renal function in 29 patients with CKD without diabetes. This study also found that there was a close correlation between insulin-dependent glucose metabolism and plasma , demonstrating a high predictive value of the degree of acidosis for the presence of insulin resistance in patients with CKD. Becker et al. (5) found reduced insulin sensitivity in 277 nondiabetic patients with CKD compared with 76 healthy subjects, as indicated by worsening homeostatic model assessment of insulin resistance (HOMA-IR) scores. These data suggest that metabolic acidosis, rather than CKD, mediates worsening insulin resistance, as HOMA-IR score in this population was not significantly associated with kidney function.
Treatment with NaHCO3 may improve glucose homeostasis in patients with kidney disease. Evidence that NaHCO3 supplementation may improve glucose homeostasis in uremia comes from Mak (84). In this study of eight patients with CKD and metabolic acidosis, who were receiving maintenance hemodialysis, the investigator found that 2 wk of oral NaHCO3 treatment increased both insulin sensitivity and insulin secretion. More substantial evidence that NaHCO3 supplementation may improve glucose homeostasis in patients with CKD with metabolic acidosis comes from Bellasi et al. (6). In a total of 145 subjects with CKD and type 2 diabetes treated with oral antidiabetic drugs, Bellasi et al. (6) reported that subjects treated with NaHCO3 demonstrated lower insulin levels (13.4 ± 5.2 vs. 19.9 ± 6.3 μIU, P < 0.001), HOMA-IR scores (5.9 vs. 6.3, P = 0.01), and need for oral antidiabetic drugs compared with controls. Additionally, NaHCO3 treatment prevented a worsening of hemoglobin A1c over time in patients given NaHCO3 [7.7 ± 3.7 (control) vs. 6.70 ± 0.9 (NaHCO3), P = 0.0028]. Importantly, the authors found that there was a nonlinear relationship (i.e., a U-shaped curve) between serum levels and HOMA-IR scores, with the greatest reduction in HOMA-IR scores occurring at normal serum levels of 24–28 mmol/L. Taken together, these data suggest a relationship between CKD, acid-base status, and glucose homeostasis, such that NaHCO3 supplementation may improve glucose homeostasis in patients with CKD with metabolic acidosis.
Experimental data indicate mechanisms unrelated to pH homeostasis may improve metabolic parameters following alkali ingestion. Dahl salt-sensitive rats develop insulin resistance when fed a high-salt diet (100). Data from our laboratory indicates NaHCO3 supplementation may improve blood glucose control in Dahl salt-sensitive rats absent metabolic acidosis. Following 7 or 14 days of a high-salt diet, spot glucose levels were lower in NaHCO3-treated versus NaCl-treated controls (113). Furthermore, experimental data support a link between activation of the CAIP and improved metabolic status. In a randomized trial of 60 patients with metabolic syndrome, activation of vagal signaling by administration of the central cholinergic activator galantamine alleviated inflammation and insulin resistance (23). In this study, galantamine resulted in lower plasma levels of proinflammatory molecules and higher levels of anti-inflammatory molecules. Galantamine treatment also produced significant reductions in plasma insulin and HOMA-IR values as well as vagal alteration of heart rate variability (23). Similar effects of galantamine attributed to the CAIP have been reported in mice, including improved glucose tolerance and metabolic function (118, 138). It is possible, then, that the protective effect of NaHCO3 is not limited to its action on acid-base status, and improved outcomes in CKD can be attributed to stimulation of extrarenal pathways.
As insulin resistance may be present in both patients with diabetic kidney disease and nondiabetic patients with CKD, improved metabolic control may provide a common mechanism for the benefit of alkali supplementation in CKD. However, a number of caveats remain. Whether improvements in insulin resistance in patients with CKD may slow GFR decline remains uncertain. Furthermore, to the authors’ knowledge, there is currently no evidence in animal models of CKD that alkali slow GFR decline via improvements in metabolic control.
If improved metabolic status by alkali supplementation is identified as a mechanism of renal protection, this would suggest that patients with CKD and type 2 diabetes or insulin resistance may benefit the most from alkali treatment. Most evidence indicates a direct link between metabolic acidosis and insulin resistance. This suggests that titration of serum acid-base status, so that parameters fell within the normal range by alkali loading, would be the most efficacious approach. If, however, activation of the CAIP by gastrointestinal stimuli was found to mediate improvements in metabolic status in these patients, caveats related to activation of these pathways independent of serum acid-base status would apply.
CONCLUSIONS AND FUTURE DIRECTIONS
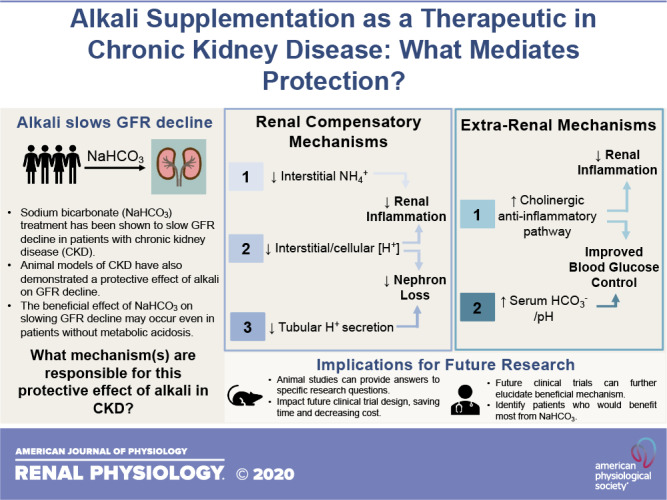
Both clinical and basic science research has demonstrated beneficial effects of NaHCO3 on slowing kidney functional decline. A number of potential pathways through which alkali loading may protect the kidney, including both renal compensatory responses and extrarenal mechanisms, have been identified (Fig. 2). The physiological mechanisms through which alkali supplementation protects the kidney, however, remain uncertain. To identify the populations that may benefit most from alkali loading, future research should incorporate more targeted questions to move beyond established associations. Several mechanisms through which NaHCO3 may slow CKD progression have been identified, but none have been shown to be the dominant mechanism in slowing GFR decline. Other currently unknown pathways of protection may exist. The use of animal models in biomedical research provides the opportunity for more targeted interventional studies to identify mechanisms that may mediate kidney protection by alkali. It is hoped that clinical studies that build on these observations will be better able to identify the populations that would receive the most benefit from alkali supplementation and identify the most efficacious therapeutic approaches. Current evidence suggests that alkali loading may have a beneficial impact on the long-term outcomes of patients with CKD and could have implications for other disease states as well.
Fig. 2.

Proposed renoprotective mechanisms of alkali in chronic kidney disease (CKD). Several potential mechanisms have been proposed that underlie the beneficial effect of alkali supplementation in slowing the decline in glomerular filtration rate (GFR) and ultimately the progression of CKD. Solid lines represent established phenomena; dotted lines represent areas of ongoing research. Several of these mechanisms have been proposed to be mediated through renal compensatory responses to alkali, including 1) decreasing interstitial ammonium levels that lead to decreases in complement activation; 2) decreasing interstitial acidosis, which decreases local production of endothelin-1 (ET-1) and angiotensin II (ANG II); or 3) decreasing tubular H+ secretion, which can limit tubular cast formation. It is possible that other, extrarenal mechanisms also mediate a protective effect, either through 1) activation of the cholinergic anti-inflammatory pathway, which can decrease renal inflammation, or 2) correction of acidosis that can lead to enhanced blood glucose control.
GRANTS
This work was supported by National Institutes of Health Grants P01HL134604 (to P.M.O.) and R21AI150723 (to P.M.O.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the author(s).
AUTHOR CONTRIBUTIONS
E.C.M. prepared figures; E.C.M. and P.M.O. drafted manuscript; E.C.M. and P.M.O. edited and revised manuscript; E.C.M. and P.M.O. approved final version of manuscript.
REFERENCES
- 1.Adachi M, Hoshi M, Ushimaru S, Hayashi A, Nakamoto K, Kanbe A, Furuta N, Inagaki I, Ito H, Seishima M.. [Clinical significance of hyaline casts in the new CKD risk classification (KDIGO 2009)]. Rinsho Byori 61: 104−111, 2013. [PubMed] [Google Scholar]
- 2.Alva S, Divyashree M, Kamath J, Prakash PS, Prakash KS. A study on effect of bicarbonate supplementation on the progression of chronic kidney disease. Indian J Nephrol 30: 91–97, 2020. doi: 10.4103/ijn.IJN_93_19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Amdur RL, Feldman HI, Gupta J, Yang W, Kanetsky P, Shlipak M, Rahman M, Lash JP, Townsend RR, Ojo A, Roy-Chaudhury A, Go AS, Joffe M, He J, Balakrishnan VS, Kimmel PL, Kusek JW, Raj DS; CRIC Study Investigators . Inflammation and progression of CKD: the CRIC study. Clin J Am Soc Nephrol 11: 1546–1556, 2016. doi: 10.2215/CJN.13121215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Banerjee T, Crews DC, Wesson DE, Tilea A, Saran R, Rios Burrows N, Williams DE, Powe NR; Centers for Disease Control and Prevention Chronic Kidney Disease Surveillance Team . Dietary acid load and chronic kidney disease among adults in the United States. BMC Nephrol 15: 137–137, 2014. doi: 10.1186/1471-2369-15-137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Becker B, Kronenberg F, Kielstein JT, Haller H, Morath C, Ritz E, Fliser D; MMKD Study Group . Renal insulin resistance syndrome, adiponectin and cardiovascular events in patients with kidney disease: the mild and moderate kidney disease study. J Am Soc Nephrol 16: 1091–1098, 2005. doi: 10.1681/ASN.2004090742. [DOI] [PubMed] [Google Scholar]
- 6.Bellasi A, Di Micco L, Santoro D, Marzocco S, De Simone E, Cozzolino M, Di Lullo L, Guastaferro P, Di Iorio B; UBI study investigators . Correction of metabolic acidosis improves insulin resistance in chronic kidney disease. BMC Nephrol 17: 158, 2016. doi: 10.1186/s12882-016-0372-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bello AK, Alrukhaimi M, Ashuntantang GE, Basnet S, Rotter RC, Douthat WG, Kazancioglu R, Köttgen A, Nangaku M, Powe NR, White SL, Wheeler DC, Moe O. Complications of chronic kidney disease: current state, knowledge gaps, and strategy for action. Kidney Int Suppl 7: 122−129, 2017. doi: 10.1016/j.kisu.2017.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.BiCARB study group Clinical and cost-effectiveness of oral sodium bicarbonate therapy for older patients with chronic kidney disease and low-grade acidosis (BiCARB): a pragmatic randomised, double-blind, placebo-controlled trial. BMC Med 18: 91, 2020. doi: 10.1186/s12916-020-01542-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bidani AK, Griffin KA, Bakris G, Picken MM. Lack of evidence of blood pressure-independent protection by renin-angiotensin system blockade after renal ablation. Kidney Int 57: 1651–1661, 2000. doi: 10.1046/j.1523-1755.2000.00009.x. [DOI] [PubMed] [Google Scholar]
- 10.Bidani AK, Polichnowski AJ, Licea-Vargas H, Long J, Kliethermes S, Williamson GA, Griffin KA. BP fluctuations and the real-time dynamics of renal blood flow responses in conscious rats. J Am Soc Nephrol 31: 324–336, 2020. doi: 10.1681/ASN.2019070718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bloch MJ, Basile J. African American patients with hypertensive chronic kidney disease receive no benefit on kidney disease progression from the currently recommended blood pressure goal of <130/80 mmHg unless there is significant proteinuria at baseline: long-term follow-up of the AASK study. J Clin Hypertens (Greenwich) 13: 214–216, 2011. doi: 10.1111/j.1751-7176.2010.00409.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boegehold MA, Kotchen TA. Importance of dietary chloride for salt sensitivity of blood pressure. Hypertension 17, Suppl: I158–I161, 1991. doi: 10.1161/01.HYP.17.1_Suppl.I158. [DOI] [PubMed] [Google Scholar]
- 13.Boegehold MA, Kotchen TA. Relative contributions of dietary Na+ and Cl− to salt-sensitive hypertension. Hypertension 14: 579–583, 1989. doi: 10.1161/01.HYP.14.6.579. [DOI] [PubMed] [Google Scholar]
- 14.Bonaz B, Bazin T, Pellissier S. The vagus nerve at the interface of the microbiota-gut-brain axis. Front Neurosci 12: 49, 2018. doi: 10.3389/fnins.2018.00049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature 405: 458–462, 2000. doi: 10.1038/35013070. [DOI] [PubMed] [Google Scholar]
- 16.Burger M, Schaller DJ. Metabolic acidosis In: StatPearls. Treasure Island, FL: StatPearls Publishing, 2020. [PubMed] [Google Scholar]
- 17.Campese VM. Salt sensitivity in hypertension. Renal and cardiovascular implications. Hypertension 23: 531–550, 1994. doi: 10.1161/01.HYP.23.4.531. [DOI] [PubMed] [Google Scholar]
- 18.Cao Q, Harris DC, Wang Y. Macrophages in kidney injury, inflammation, and fibrosis. Physiology (Bethesda) 30: 183–194, 2015. doi: 10.1152/physiol.00046.2014. [DOI] [PubMed] [Google Scholar]
- 18a.Centers for Disease Control and Prevention Chronic Kidney Disease Basics. https://www.cdc.gov/kidneydisease/basics.html.
- 18b.Centers for Disease Control and Prevention Chronic Kidney Disease Surveillance System−United States. https://nccd.cdc.gov/CKD/detail.aspx?Qnum=Q673#refreshPosition.
- 19.Chavan SS, Pavlov VA, Tracey KJ. Mechanisms and therapeutic relevance of neuro-immune communication. Immunity 46: 927–942, 2017. doi: 10.1016/j.immuni.2017.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen W, Abramowitz MK. Metabolic acidosis and the progression of chronic kidney disease. BMC Nephrol 15: 55, 2014. doi: 10.1186/1471-2369-15-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cianciaruso B, Bellizzi V, Minutolo R, Tavera A, Capuano A, Conte G, De Nicola L. Salt intake and renal outcome in patients with progressive renal disease. Miner Electrolyte Metab 24: 296–301, 1998. doi: 10.1159/000057385. [DOI] [PubMed] [Google Scholar]
- 22.Clark EC, Nath KA, Hostetter MK, Hostetter TH. Role of ammonia in tubulointerstitial injury. Miner Electrolyte Metab 16: 315–321, 1990. [PubMed] [Google Scholar]
- 23.Consolim-Colombo FM, Sangaleti CT, Costa FO, Morais TL, Lopes HF, Motta JM, Irigoyen MC, Bortoloto LA, Rochitte CE, Harris YT, Satapathy SK, Olofsson PS, Akerman M, Chavan SS, MacKay M, Barnaby DP, Lesser ML, Roth J, Tracey KJ, Pavlov VA. Galantamine alleviates inflammation and insulin resistance in patients with metabolic syndrome in a randomized trial. JCI Insight 2: e93340, 2017. doi: 10.1172/jci.insight.93340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cowley AW., Jr Renal medullary oxidative stress, pressure-natriuresis, and hypertension. Hypertension 52: 777–786, 2008. doi: 10.1161/HYPERTENSIONAHA.107.092858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Crowley SD, Rudemiller NP. Immunologic effects of the renin-angiotensin system. J Am Soc Nephrol 28: 1350–1361, 2017. doi: 10.1681/ASN.2016101066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Davies DF, Shock NW. Age changes in glomerular filtration rate, effective renal plasma flow, and tubular excretory capacity in adult males. J Clin Invest 29: 496–507, 1950. doi: 10.1172/JCI102286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.de Boer IH, Zelnick L, Afkarian M, Ayers E, Curtin L, Himmelfarb J, Ikizler TA, Kahn SE, Kestenbaum B, Utzschneider K. Impaired glucose and insulin homeostasis in moderate-severe CKD. J Am Soc Nephrol 27: 2861–2871, 2016. doi: 10.1681/ASN.2015070756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.de Brito-Ashurst I, Varagunam M, Raftery MJ, Yaqoob MM. Bicarbonate supplementation slows progression of CKD and improves nutritional status. J Am Soc Nephrol 20: 2075–2084, 2009. doi: 10.1681/ASN.2008111205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Decoursey TE. Voltage-gated proton channels. Compr Physiol 2: 1355–1385, 2012. doi: 10.1002/cphy.c100071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeFronzo RA, Alvestrand A, Smith D, Hendler R, Hendler E, Wahren J. Insulin resistance in uremia. J Clin Invest 67: 563–568, 1981. doi: 10.1172/JCI110067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.DeFronzo RA, Beckles AD. Glucose intolerance following chronic metabolic acidosis in man. Am J Physiol Endocrinol Metab 236: E328–E334, 1979. doi: 10.1152/ajpendo.1979.236.4.E328. [DOI] [PubMed] [Google Scholar]
- 32.Di Iorio BR, Bellasi A, Raphael KL, Santoro D, Aucella F, Garofano L, Ceccarelli M, Di Lullo L, Capolongo G, Di Iorio M, Guastaferro P, Capasso G; UBI Study Group . Treatment of metabolic acidosis with sodium bicarbonate delays progression of chronic kidney disease: the UBI Study. J Nephrol 32: 989–1001, 2019. doi: 10.1007/s40620-019-00656-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dion F, Dumayne C, Henley N, Beauchemin S, Arias EB, Leblond FA, Lesage S, Lefrançois S, Cartee GD, Pichette V. Mechanism of insulin resistance in a rat model of kidney disease and the risk of developing type 2 diabetes. PLoS One 12: e0176650, 2017. doi: 10.1371/journal.pone.0176650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dobre M, Yang W, Chen J, Drawz P, Hamm LL, Horwitz E, Hostetter T, Jaar B, Lora CM, Nessel L, Ojo A, Scialla J, Steigerwalt S, Teal V, Wolf M, Rahman M; CRIC Investigators . Association of serum bicarbonate with risk of renal and cardiovascular outcomes in CKD: a report from the Chronic Renal Insufficiency Cohort (CRIC) study. Am J Kidney Dis 62: 670–678, 2013. doi: 10.1053/j.ajkd.2013.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dubey AK, Sahoo J, Vairappan B, Haridasan S, Parameswaran S, Priyamvada PS. Correction of metabolic acidosis improves muscle mass and renal function in chronic kidney disease stages 3 and 4: a randomized controlled trial. Nephrol Dial Transplant 35: 121–129, 2020. doi: 10.1093/ndt/gfy214. [DOI] [PubMed] [Google Scholar]
- 36.Eng E, Veniant M, Floege J, Fingerle J, Alpers CE, Menard J, Clozel JP, Johnson RJ. Renal proliferative and phenotypic changes in rats with two-kidney, one-clip Goldblatt hypertension. Am J Hypertens 7: 177–185, 1994. doi: 10.1093/ajh/7.2.177. [DOI] [PubMed] [Google Scholar]
- 37.Gadola L, Noboa O, Márquez MN, Rodriguez MJ, Nin N, Boggia J, Ferreiro A, García S, Ortega V, Musto ML, Ponte P, Sesser P, Pizarrosa C, Ravaglio S, Vallega A. Calcium citrate ameliorates the progression of chronic renal injury. Kidney Int 65: 1224–1230, 2004. doi: 10.1111/j.1523-1755.2004.00496.x. [DOI] [PubMed] [Google Scholar]
- 38.Garofalo C, Borrelli S, Provenzano M, De Stefano T, Vita C, Chiodini P, Minutolo R, De Nicola L, Conte G. Dietary salt restriction in chronic kidney disease: a meta-analysis of randomized clinical trials. Nutrients 10: 732, 2018. doi: 10.3390/nu10060732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Geibel J, Giebisch G, Boron WF. Angiotensin II stimulates both Na+-H+ exchange and Na+/ cotransport in the rabbit proximal tubule. Proc Natl Acad Sci USA 87: 7917–7920, 1990. doi: 10.1073/pnas.87.20.7917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gigliotti JC, Huang L, Ye H, Bajwa A, Chattrabhuti K, Lee S, Klibanov AL, Kalantari K, Rosin DL, Okusa MD. Ultrasound prevents renal ischemia-reperfusion injury by stimulating the splenic cholinergic anti-inflammatory pathway. J Am Soc Nephrol 24: 1451–1460, 2013. doi: 10.1681/ASN.2013010084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gilbert RE, Wu LL, Kelly DJ, Cox A, Wilkinson-Berka JL, Johnston CI, Cooper ME. Pathological expression of renin and angiotensin II in the renal tubule after subtotal nephrectomy. Implications for the pathogenesis of tubulointerstitial fibrosis. Am J Pathol 155: 429–440, 1999. doi: 10.1016/S0002-9440(10)65139-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Goraya N, Munoz-Maldonado Y, Simoni J, Wesson DE. Fruit and vegetable treatment of chronic kidney disease-related metabolic acidosis reduces cardiovascular risk better than sodium bicarbonate. Am J Nephrol 49: 438–448, 2019. doi: 10.1159/000500042. [DOI] [PubMed] [Google Scholar]
- 43.Goraya N, Simoni J, Jo CH, Wesson DE. Treatment of metabolic acidosis in patients with stage 3 chronic kidney disease with fruits and vegetables or oral bicarbonate reduces urine angiotensinogen and preserves glomerular filtration rate. Kidney Int 86: 1031–1038, 2014. doi: 10.1038/ki.2014.83. [DOI] [PubMed] [Google Scholar]
- 44.Griffin KA, Picken M, Bidani AK. Method of renal mass reduction is a critical modulator of subsequent hypertension and glomerular injury. J Am Soc Nephrol 4: 2023–2031, 1994. [DOI] [PubMed] [Google Scholar]
- 45.Griffin KA, Picken MM, Bidani AK. Blood pressure lability and glomerulosclerosis after normotensive 5/6 renal mass reduction in the rat. Kidney Int 65: 209–218, 2004. doi: 10.1111/j.1523-1755.2004.00356.x. [DOI] [PubMed] [Google Scholar]
- 46.Hamm LL, Nakhoul N, Hering-Smith KS. Acid-base homeostasis. Clin J Am Soc Nephrol 10: 2232–2242, 2015. doi: 10.2215/CJN.07400715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hayslett JP. Functional adaptation to reduction in renal mass. Physiol Rev 59: 137–164, 1979. doi: 10.1152/physrev.1979.59.1.137. [DOI] [PubMed] [Google Scholar]
- 48.Hoover DB. Cholinergic modulation of the immune system presents new approaches for treating inflammation. Pharmacol Ther 179: 1–16, 2017. doi: 10.1016/j.pharmthera.2017.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hu MK, Witham MD, Soiza RL. Oral bicarbonate therapy in non-haemodialysis dependent chronic kidney disease patients: a systematic review and meta-analysis of randomised controlled trials. J Clin Med 8: 208, 2019. doi: 10.3390/jcm8020208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hutchings RH, Hegstrom RM, Scribner BH. Glucose intolerance in patients on long-term intermittent dialysis. Ann Intern Med 65: 275–285, 1966. doi: 10.7326/0003-4819-65-2-275. [DOI] [Google Scholar]
- 51.Ibrahim HN, Hostetter TH. Role of dietary potassium in the hyperaldosteronism and hypertension of the remnant kidney model. J Am Soc Nephrol 11: 625–631, 2000. [DOI] [PubMed] [Google Scholar]
- 52.Ibrahim SM, Al-Shorbagy MY, Abdallah DM, El-Abhar HS. Activation of α7 nicotinic acetylcholine receptor ameliorates zymosan-induced acute kidney injury in BALB/c Mice. Sci Rep 8: 16814, 2018. doi: 10.1038/s41598-018-35254-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Inker LA, Astor BC, Fox CH, Isakova T, Lash JP, Peralta CA, Kurella Tamura M, Feldman HI. KDOQI US commentary on the 2012 KDIGO clinical practice guideline for the evaluation and management of CKD. Am J Kidney Dis 63: 713–735, 2014. doi: 10.1053/j.ajkd.2014.01.416. [DOI] [PubMed] [Google Scholar]
- 54.Jara A, Felsenfeld AJ, Bover J, Kleeman CR. Chronic metabolic acidosis in azotemic rats on a high-phosphate diet halts the progression of renal disease. Kidney Int 58: 1023–1032, 2000. doi: 10.1046/j.1523-1755.2000.00260.x. [DOI] [PubMed] [Google Scholar]
- 55.Jeong J, Kwon SK, Kim H-Y. Effect of bicarbonate supplementation on renal function and nutritional indices in predialysis advanced chronic kidney disease. Electrolyte Blood Press 12: 80–87, 2014. doi: 10.5049/EBP.2014.12.2.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jin C, Sun J, Stilphen CA, Smith SM, Ocasio H, Bermingham B, Darji S, Guha A, Patel R, Geurts AM, Jacob HJ, Lambert NA, O’Connor PM. HV1 acts as a sodium sensor and promotes superoxide production in medullary thick ascending limb of Dahl salt-sensitive rats. Hypertension 64: 541–550, 2014. doi: 10.1161/HYPERTENSIONAHA.114.03549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Judd E, Calhoun DA. Management of hypertension in CKD: beyond the guidelines. Adv Chronic Kidney Dis 22: 116–122, 2015. doi: 10.1053/j.ackd.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Khanna A, Simoni J, Hacker C, Duran M-J, Wesson DE. Increased endothelin activity mediates augmented distal nephron acidification induced by dietary protein. J Am Soc Nephrol 15: 2266–2275, 2004. doi: 10.1097/01.ASN.0000138233.78329.4E. [DOI] [PubMed] [Google Scholar]
- 59.Khanna A, Simoni J, Wesson DE. Endothelin-induced increased aldosterone activity mediates augmented distal nephron acidification as a result of dietary protein. J Am Soc Nephrol 16: 1929–1935, 2005. doi: 10.1681/ASN.2004121054. [DOI] [PubMed] [Google Scholar]
- 60.Kidambi S, Kotchen JM, Krishnaswami S, Grim CE, Kotchen TA. Cardiovascular correlates of insulin resistance in normotensive and hypertensive African Americans. Metabolism 60: 835–842, 2011. doi: 10.1016/j.metabol.2010.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Kim HW, Lee CK, Cha HS, Choe JY, Park EJ, Kim J. Effect of anti-tumor necrosis factor alpha treatment of rheumatoid arthritis and chronic kidney disease. Rheumatol Int 35: 727–734, 2015. doi: 10.1007/s00296-014-3146-4. [DOI] [PubMed] [Google Scholar]
- 62.Kim S, Lee J, Heo NJ, Lee JW, Han JS. Alkali therapy attenuates the progression of kidney injury via Na/H exchanger inhibition in 5/6 nephrectomized rats. J Korean Med Sci 29: 691–698, 2014. doi: 10.3346/jkms.2014.29.5.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim S, Yang JY, Jung ES, Lee J, Heo NJ, Lee JW, Na KY, Han JS. Effects of sodium citrate on salt sensitivity and kidney injury in chronic renal failure. J Korean Med Sci 29: 1658–1664, 2014. doi: 10.3346/jkms.2014.29.12.1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kobayashi S, Maesato K, Moriya H, Ohtake T, Ikeda T. Insulin resistance in patients with chronic kidney disease. Am J Kidney Dis 45: 275–280, 2005. doi: 10.1053/j.ajkd.2004.09.034. [DOI] [PubMed] [Google Scholar]
- 66.Kohan DE, Barton M. Endothelin and endothelin antagonists in chronic kidney disease. Kidney Int 86: 896–904, 2014. doi: 10.1038/ki.2014.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kohan DE, Rossi NF, Inscho EW, Pollock DM. Regulation of blood pressure and salt homeostasis by endothelin. Physiol Rev 91: 1–77, 2011. doi: 10.1152/physrev.00060.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kotchen TA, Luke RG, Ott CE, Galla JH, Whitescarver S. Effect of chloride on renin and blood pressure responses to sodium chloride. Ann Intern Med 98: 817–822, 1983. doi: 10.7326/0003-4819-98-5-817. [DOI] [PubMed] [Google Scholar]
- 69.Kotchen TA, Zhang HY, Covelli M, Blehschmidt N. Insulin resistance and blood pressure in Dahl rats and in one-kidney, one-clip hypertensive rats. Am J Physiol Endocrinol Metab 261: E692–E697, 1991. doi: 10.1152/ajpendo.1991.261.6.E692. [DOI] [PubMed] [Google Scholar]
- 70.Kraut JA, Kurtz I. Metabolic acidosis of CKD: diagnosis, clinical characteristics, and treatment. Am J Kidney Dis 45: 978–993, 2005. doi: 10.1053/j.ajkd.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 71.Kraut JA, Madias NE. Metabolic acidosis of CKD: an update. Am J Kidney Dis 67: 307–317, 2016. doi: 10.1053/j.ajkd.2015.08.028. [DOI] [PubMed] [Google Scholar]
- 72.Laghmani K, Preisig PA, Moe OW, Yanagisawa M, Alpern RJ. Endothelin-1/endothelin-B receptor-mediated increases in NHE3 activity in chronic metabolic acidosis. J Clin Invest 107: 1563–1569, 2001. doi: 10.1172/JCI11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Larsen CP, Beggs ML, Saeed M, Ambruzs JM, Cossey LN, Messias NC, Walker PD, Freedman BI. Histopathologic findings associated with APOL1 risk variants in chronic kidney disease. Modern Pathol 28: 95–102, 2015. doi: 10.1038/modpathol.2014.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lash JP, Go AS, Appel LJ, He J, Ojo A, Rahman M, Townsend RR, Xie D, Cifelli D, Cohan J, Fink JC, Fischer MJ, Gadegbeku C, Hamm LL, Kusek JW, Landis JR, Narva A, Robinson N, Teal V, Feldman HI; Chronic Renal Insufficiency Cohort (CRIC) Study Group . Chronic Renal Insufficiency Cohort (CRIC) study: baseline characteristics and associations with kidney function. Clin J Am Soc Nephrol 4: 1302–1311, 2009. doi: 10.2215/CJN.00070109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Levey AS, Greene T, Sarnak MJ, Wang X, Beck GJ, Kusek JW, Collins AJ, Kopple JD. Effect of dietary protein restriction on the progression of kidney disease: long-term follow-up of the Modification of Diet in Renal Disease (MDRD) Study. Am J Kidney Dis 48: 879–888, 2006. doi: 10.1053/j.ajkd.2006.08.023. [DOI] [PubMed] [Google Scholar]
- 76.Li DJ, Evans RG, Yang ZW, Song SW, Wang P, Ma XJ, Liu C, Xi T, Su DF, Shen FM. Dysfunction of the cholinergic anti-inflammatory pathway mediates organ damage in hypertension. Hypertension 57: 298–307, 2011. doi: 10.1161/HYPERTENSIONAHA.110.160077. [DOI] [PubMed] [Google Scholar]
- 77.Liu FY, Cogan MG. Angiotensin II stimulation of hydrogen ion secretion in the rat early proximal tubule. Modes of action, mechanism, and kinetics. J Clin Invest 82: 601–607, 1988. doi: 10.1172/JCI113638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu FY, Cogan MG. Angiotensin II: a potent regulator of acidification in the rat early proximal convoluted tubule. J Clin Invest 80: 272–275, 1987. doi: 10.1172/JCI113059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Luyer MD, Greve JW, Hadfoune M, Jacobs JA, Dejong CH, Buurman WA. Nutritional stimulation of cholecystokinin receptors inhibits inflammation via the vagus nerve. J Exp Med 202: 1023–1029, 2005. doi: 10.1084/jem.20042397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.MacIsaac RJ, Jerums G, Ekinci EI. Effects of glycaemic management on diabetic kidney disease. World J Diabetes 8: 172–186, 2017. doi: 10.4239/wjd.v8.i5.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mackie FE, Meyer TW, Campbell DJ. Effects of antihypertensive therapy on intrarenal angiotensin and bradykinin levels in experimental renal insufficiency. Kidney Int 61: 555–563, 2002. doi: 10.1046/j.1523-1755.2002.00141.x. [DOI] [PubMed] [Google Scholar]
- 82.Maclean AJ, Hayslett JP. Adaptive change in ammonia excretion in renal insufficiency. Kidney Int 17: 595–606, 1980. doi: 10.1038/ki.1980.70. [DOI] [PubMed] [Google Scholar]
- 83.Mahajan A, Simoni J, Sheather SJ, Broglio KR, Rajab MH, Wesson DE. Daily oral sodium bicarbonate preserves glomerular filtration rate by slowing its decline in early hypertensive nephropathy. Kidney Int 78: 303–309, 2010. doi: 10.1038/ki.2010.129. [DOI] [PubMed] [Google Scholar]
- 84.Mak RH. Effect of metabolic acidosis on insulin action and secretion in uremia. Kidney Int 54: 603–607, 1998. doi: 10.1046/j.1523-1755.1998.00023.x. [DOI] [PubMed] [Google Scholar]
- 85.Mannon EC, Sun J, Wilson K, Brands M, Martinez-Quinones P, Baban B, O’Connor PM. A basic solution to activate the cholinergic anti-inflammatory pathway via the mesothelium? Pharmacol Res 141: 236–248, 2019. doi: 10.1016/j.phrs.2019.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Mao Y, Tokudome T, Kishimoto I, Otani K, Nishimura H, Yamaguchi O, Otsu K, Miyazato M, Kangawa K. Endogenous ghrelin attenuates pressure overload-induced cardiac hypertrophy via a cholinergic anti-inflammatory pathway. Hypertension 65: 1238–1244, 2015. doi: 10.1161/HYPERTENSIONAHA.114.04864. [DOI] [PubMed] [Google Scholar]
- 87.Mazzocchi G, Rebuffat P, Meneghelli V, Malendowicz LK, Kasprzak A, Nussdorfer GG. Effects of prolonged infusion with endothelin-1 on the function and morphology of rat adrenal cortex. Peptides 11: 767–772, 1990. doi: 10.1016/0196-9781(90)90193-9. [DOI] [PubMed] [Google Scholar]
- 88.Melamed ML, Horwitz EJ, Dobre MA, Abramowitz MK, Zhang L, Lo Y, Mitch WE, Hostetter TH. Effects of sodium dicarbonate in CKD stages 3 and 4: a randomized, placebo-controlled, multicenter clinical trial. Am J Kidney Dis 75: 225–234, 2020. doi: 10.1053/j.ajkd.2019.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mendelsohn FA. Angiotensin II: evidence for its role as an intrarenal hormone. Kidney Int Suppl 12: S78–S81, 1982. [PubMed] [Google Scholar]
- 90.Mori T, Polichnowski A, Glocka P, Kaldunski M, Ohsaki Y, Liang M, Cowley AW Jr. High perfusion pressure accelerates renal injury in salt-sensitive hypertension. J Am Soc Nephrol 19: 1472–1482, 2008. doi: 10.1681/ASN.2007121271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Morita Y, Ikeguchi H, Nakamura J, Hotta N, Yuzawa Y, Matsuo S. Complement activation products in the urine from proteinuric patients. J Am Soc Nephrol 11: 700–707, 2000. [DOI] [PubMed] [Google Scholar]
- 92.Nagami GT. Ammonia production and secretion by S3 proximal tubule segments from acidotic mice: role of ANG II. Am J Physiol Renal Physiol 287: F707–F712, 2004. doi: 10.1152/ajprenal.00189.2003. [DOI] [PubMed] [Google Scholar]
- 93.Nagami GT. Role of angiotensin II in the enhancement of ammonia production and secretion by the proximal tubule in metabolic acidosis. Am J Physiol Renal Physiol 294: F874–F880, 2008. doi: 10.1152/ajprenal.00286.2007. [DOI] [PubMed] [Google Scholar]
- 94.Nagami GT, Kraut JA. Acid-base regulation of angiotensin receptors in the kidney. Curr Opin Nephrol Hypertens 19: 91–97, 2010. doi: 10.1097/MNH.0b013e32833289fd. [DOI] [PubMed] [Google Scholar]
- 95.Nath KA, Hostetter MK, Hostetter TH. Pathophysiology of chronic tubulo-interstitial disease in rats. Interactions of dietary acid load, ammonia, and complement component C3. J Clin Invest 76: 667–675, 1985. doi: 10.1172/JCI112020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health Kidney Disease Statistics for the United States, 2016. https://www.niddk.nih.gov/health-information/health-statistics/kidney-disease.
- 97.Navar LG, Imig JD, Zou L, Wang CT. Intrarenal production of angiotensin II. Semin Nephrol 17: 412–422, 1997. [PubMed] [Google Scholar]
- 98.Navar LG, Kobori H, Prieto-Carrasquero M. Intrarenal angiotensin II and hypertension. Curr Hypertens Rep 5: 135–143, 2003. doi: 10.1007/s11906-003-0070-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.O’Connor PM, Guha A, Stilphen CA, Sun J, Jin C. Proton channels and renal hypertensive injury: a key piece of the Dahl salt-sensitive rat puzzle? Am J Physiol Regul Integr Comp Physiol 310: R679–R690, 2016. doi: 10.1152/ajpregu.00115.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ogihara T, Asano T, Ando K, Sakoda H, Anai M, Shojima N, Ono H, Onishi Y, Fujishiro M, Abe M, Fukushima Y, Kikuchi M, Fujita T. High-salt diet enhances insulin signaling and induces insulin resistance in Dahl salt-sensitive rats. Hypertension 40: 83–89, 2002. doi: 10.1161/01.HYP.0000022880.45113.C9. [DOI] [PubMed] [Google Scholar]
- 101.Pallini A, Hulter HN, Muser J, Krapf R. Role of endothelin-1 in renal regulation of acid-base equilibrium in acidotic humans. Am J Physiol Renal Physiol 303: F991–F999, 2012. doi: 10.1152/ajprenal.00309.2012. [DOI] [PubMed] [Google Scholar]
- 102.Patel R, McKenzie JK, McQueen EG. Tamm-Horsfall urinary mucoprotein and tubular obstruction by casts in acute renal failure. Lancet 1: 457–461, 1964. doi: 10.1016/S0140-6736(64)90794-9. [DOI] [PubMed] [Google Scholar]
- 103.Pham GS, Wang LA, Mathis KW. Pharmacological potentiation of the efferent vagus nerve attenuates blood pressure and renal injury in a murine model of systemic lupus erythematosus. Am J Physiol Regul Integr Comp Physiol 315: R1261–R1271, 2018. doi: 10.1152/ajpregu.00362.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Phisitkul S, Hacker C, Simoni J, Tran RM, Wesson DE. Dietary protein causes a decline in the glomerular filtration rate of the remnant kidney mediated by metabolic acidosis and endothelin receptors. Kidney Int 73: 192–199, 2008. doi: 10.1038/sj.ki.5002647. [DOI] [PubMed] [Google Scholar]
- 107.Prieto MC, González-Villalobos RA, Botros FT, Martin VL, Pagán J, Satou R, Lara LS, Feng Y, Fernandes FB, Kobori H, Casarini DE, Navar LG. Reciprocal changes in renal ACE/ANG II and ACE2/ANG 1-7 are associated with enhanced collecting duct renin in Goldblatt hypertensive rats. Am J Physiol Renal Physiol 300: F749–F755, 2011. doi: 10.1152/ajprenal.00383.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Raij L, Azar S, Keane W. Mesangial immune injury, hypertension, and progressive glomerular damage in Dahl rats. Kidney Int 26: 137–143, 1984. doi: 10.1038/ki.1984.147. [DOI] [PubMed] [Google Scholar]
- 109.Raphael KL. Metabolic acidosis and subclinical metabolic acidosis in CKD. J Am Soc Nephrol 29: 376–382, 2018. doi: 10.1681/ASN.2017040422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Raphael KL, Carroll DJ, Murray J, Greene T, Beddhu S. Urine ammonium predicts clinical outcomes in hypertensive kidney disease. J Am Soc Nephrol 28: 2483–2490, 2017. doi: 10.1681/ASN.2016101151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Raphael KL, Greene T, Wei G, Bullshoe T, Tuttle K, Cheung AK, Beddhu S. Sodium bicarbonate supplementation and urinary TGF-β1 in nonacidotic diabetic kidney disease: a randomized, controlled trial. Clin J Am Soc Nephrol 15: 200–208, 2020. doi: 10.2215/CJN.06600619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ray SC, Baban B, Tucker MA, Seaton AJ, Chang KC, Mannon EC, Sun J, Patel B, Wilson K, Musall JB, Ocasio H, Irsik D, Filosa JA, Sullivan JC, Marshall B, Harris RA, O’Connor PM. Oral NaHCO3 activates a splenic anti-inflammatory pathway: evidence that cholinergic signals are transmitted via mesothelial cells. J Immunol 200: 3568–3586, 2018. doi: 10.4049/jimmunol.1701605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Ray SC, Patel B, Irsik DL, Sun J, Ocasio H, Crislip GR, Jin CH, Chen J, Baban B, Polichnowski AJ, O’Connor PM. Sodium bicarbonate loading limits tubular cast formation independent of glomerular injury and proteinuria in Dahl salt-sensitive rats. Clin Sci (Lond) 132: 1179–1197, 2018. doi: 10.1042/CS20171630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rebholz CM, Coresh J, Grams ME, Steffen LM, Anderson CAM, Appel LJ, Crews DC. Dietary acid load and incident chronic kidney disease: results from the ARIC study. Am J Nephrol 42: 427–435, 2015. doi: 10.1159/000443746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Rudemiller NP, Crowley SD. The role of chemokines in hypertension and consequent target organ damage. Pharmacol Res 119: 404–411, 2017. doi: 10.1016/j.phrs.2017.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sadis C, Teske G, Stokman G, Kubjak C, Claessen N, Moore F, Loi P, Diallo B, Barvais L, Goldman M, Florquin S, Le Moine A. Nicotine protects kidney from renal ischemia/reperfusion injury through the cholinergic anti-inflammatory pathway. PLoS One 2: e469, 2007. doi: 10.1371/journal.pone.0000469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sanders PW, Booker BB, Bishop JB, Cheung HC. Mechanisms of intranephronal proteinaceous cast formation by low molecular weight proteins. J Clin Invest 85: 570–576, 1990. doi: 10.1172/JCI114474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Satapathy SK, Ochani M, Dancho M, Hudson LK, Rosas-Ballina M, Valdes-Ferrer SI, Olofsson PS, Harris YT, Roth J, Chavan S, Tracey KJ, Pavlov VA. Galantamine alleviates inflammation and other obesity-associated complications in high-fat diet-fed mice. Mol Med 17: 599–606, 2011. doi: 10.2119/molmed.2011.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Scialla JJ, Appel LJ, Astor BC, Miller ER III, Beddhu S, Woodward M, Parekh RS, Anderson CA; African American Study of Kidney Disease and Hypertension Study Group . Net endogenous acid production is associated with a faster decline in GFR in African Americans. Kidney Int 82: 106–112, 2012. doi: 10.1038/ki.2012.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Scialla JJ, Asplin J, Dobre M, Chang AR, Lash J, Hsu CY, Kallem RR, Hamm LL, Feldman HI, Chen J, Appel LJ, Anderson CA, Wolf M, Appel LJ, Feldman HI, Go AS, He J, Kusek JW, Lash JP, Ojo A, Rahman M, Townsend RR; Chronic Renal Insufficiency Cohort Study Investigators . Higher net acid excretion is associated with a lower risk of kidney disease progression in patients with diabetes. Kidney Int 91: 204–215, 2017. doi: 10.1016/j.kint.2016.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Silva RC, Terra FF, Guise YF, Prado MA, Prado VF, Hiyane MI, Costa Malheiros DM, Prado CM, Camara NO, Braga TT. Reduced expression of VAChT increases renal fibrosis. Pathophysiology 23: 229−236, 2016. doi: 10.1016/j.pathophys.2016.07.002. [DOI] [PubMed] [Google Scholar]
- 122.Souto G, Donapetry C, Calviño J, Adeva MM. Metabolic acidosis-induced insulin resistance and cardiovascular risk. Metab Syndr Relat Disord 9: 247–253, 2011. doi: 10.1089/met.2010.0108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Soygür T, Akbay A, Küpeli S. Effect of potassium citrate therapy on stone recurrence and residual fragments after shockwave lithotripsy in lower caliceal calcium oxalate urolithiasis: a randomized controlled trial. J Endourol 16: 149–152, 2002. doi: 10.1089/089277902753716098. [DOI] [PubMed] [Google Scholar]
- 124.Sullivan JM. Salt sensitivity. Definition, conception, methodology, and long-term issues. Hypertension 17, Suppl: I61–I68, 1991. doi: 10.1161/01.HYP.17.1_Suppl.I61. [DOI] [PubMed] [Google Scholar]
- 125.Tanner GA. Potassium citrate/citric acid intake improves renal function in rats with polycystic kidney disease. J Am Soc Nephrol 9: 1242–1248, 1998. [DOI] [PubMed] [Google Scholar]
- 126.Taylor KS, Mclellan J, Verbakel JY, Aronson JK, Lasserson DS, Pidduck N, Roberts N, Fleming S, O’Callaghan CA, Bankhead CR, Banerjee A, Hobbs FR, Perera R. Effects of antihypertensives, lipid-modifying drugs, glycaemic control drugs and sodium bicarbonate on the progression of stages 3 and 4 chronic kidney disease in adults: a systematic review and meta-analysis. BMJ Open 9: e030596, 2019. doi: 10.1136/bmjopen-2019-030596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Teta D. Insulin resistance as a therapeutic target for chronic kidney disease. J Ren Nutr 25: 226–229, 2015. doi: 10.1053/j.jrn.2014.10.019. [DOI] [PubMed] [Google Scholar]
- 128.Throssell D, Brown J, Harris KP, Walls J. Metabolic acidosis does not contribute to chronic renal injury in the rat. Clin Sci (Lond) 89: 643–650, 1995. doi: 10.1042/cs0890643. [DOI] [PubMed] [Google Scholar]
- 129.Tolins JP, Hostetter MK, Hostetter TH. Hypokalemic nephropathy in the rat. Role of ammonia in chronic tubular injury. J Clin Invest 79: 1447–1458, 1987. doi: 10.1172/JCI112973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Torres VE, Mujwid DK, Wilson DM, Holley KH. Renal cystic disease and ammoniagenesis in Han:SPRD rats. J Am Soc Nephrol 5: 1193–1200, 1994. [DOI] [PubMed] [Google Scholar]
- 131.Tracey KJ. Fat meets the cholinergic antiinflammatory pathway. J Exp Med 202: 1017–1021, 2005. doi: 10.1084/jem.20051760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Turner JM, Bauer C, Abramowitz MK, Melamed ML, Hostetter TH. Treatment of chronic kidney disease. Kidney Int 81: 351–362, 2012. doi: 10.1038/ki.2011.380. [DOI] [PubMed] [Google Scholar]
- 133.Uni R, Inoue T, Nakamura Y, Fukaya D, Hasegawa S, Wu CH, Fujii R, Surattichaiyakul B, Peerapanyasut W, Ozeki A, Akimitsu N, Wada Y, Nangaku M, Inagi R. Vagus nerve stimulation even after injury ameliorates cisplatin-induced nephropathy via reducing macrophage infiltration. Sci Rep 10: 9472, 2020. doi: 10.1038/s41598-020-66295-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Uribarri J. Chronic kidney disease and kidney stones. Curr Opin Nephrol Hypertens 29: 237–242, 2020. doi: 10.1097/MNH.0000000000000582. [DOI] [PubMed] [Google Scholar]
- 135.Vallet M, Metzger M, Haymann JP, Flamant M, Gauci C, Thervet E, Boffa JJ, Vrtovsnik F, Froissart M, Stengel B, Houillier P; NephroTest Cohort Study group . Urinary ammonia and long-term outcomes in chronic kidney disease. Kidney Int 88: 137–145, 2015. doi: 10.1038/ki.2015.52. [DOI] [PubMed] [Google Scholar]
- 136.Vernerová Z, Kujal P, Kramer HJ, Bäcker A, Cervenka L, Vanecková I. End-organ damage in hypertensive transgenic Ren-2 rats: influence of early and late endothelin receptor blockade. Physiol Res 58, Suppl 2: S69–S78, 2009. [DOI] [PubMed] [Google Scholar]
- 137.Wagner CA, Giebisch G, Lang F, Geibel JP. Angiotensin II stimulates vesicular H+-ATPase in rat proximal tubular cells. Proc Natl Acad Sci USA 95: 9665–9668, 1998. doi: 10.1073/pnas.95.16.9665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Wang L, Opland D, Tsai S, Luk CT, Schroer SA, Allison MB, Elia AJ, Furlonger C, Suzuki A, Paige CJ, Mak TW, Winer DA, Myers MG Jr, Woo M. Pten deletion in RIP-Cre neurons protects against type 2 diabetes by activating the anti-inflammatory reflex. Nat Med 20: 484–492, 2014. doi: 10.1038/nm.3527. [DOI] [PubMed] [Google Scholar]
- 139.Wesson DE, Buysse JM, Bushinsky DA. Mechanisms of metabolic acidosis-induced kidney injury in chronic kidney disease. J Am Soc Nephrol 31: 469–482, 2020. doi: 10.1681/ASN.2019070677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Wesson DE, Mathur V, Tangri N, Stasiv Y, Parsell D, Li E, Klaerner G, Bushinsky DA. Long-term safety and efficacy of veverimer in patients with metabolic acidosis in chronic kidney disease: a multicentre, randomised, blinded, placebo-controlled, 40-week extension. Lancet 394: 396–406, 2019. doi: 10.1016/S0140-6736(19)31388-1. [DOI] [PubMed] [Google Scholar]
- 141.Wesson DE, Mathur V, Tangri N, Stasiv Y, Parsell D, Li E, Klaerner G, Bushinsky DA. Veverimer versus placebo in patients with metabolic acidosis associated with chronic kidney disease: a multicentre, randomised, double-blind, controlled, phase 3 trial. Lancet 393: 1417–1427, 2019. doi: 10.1016/S0140-6736(18)32562-5. [DOI] [PubMed] [Google Scholar]
- 142.Wesson DE, Simoni J. Acid retention during kidney failure induces endothelin and aldosterone production which lead to progressive GFR decline, a situation ameliorated by alkali diet. Kidney Int 78: 1128–1135, 2010. doi: 10.1038/ki.2010.348. [DOI] [PubMed] [Google Scholar]
- 143.Wesson DE, Simoni J. Increased tissue acid mediates a progressive decline in the glomerular filtration rate of animals with reduced nephron mass. Kidney Int 75: 929–935, 2009. doi: 10.1038/ki.2009.6. [DOI] [PubMed] [Google Scholar]
- 144.Wesson DE, Simoni J, Broglio K, Sheather S. Acid retention accompanies reduced GFR in humans and increases plasma levels of endothelin and aldosterone. Am J Physiol Renal Physiol 300: F830–F837, 2011. doi: 10.1152/ajprenal.00587.2010. [DOI] [PubMed] [Google Scholar]
- 145.Whitescarver SA, Ott CE, Jackson BA, Guthrie GP Jr, Kotchen TA. Salt-sensitive hypertension: contribution of chloride. Science 223: 1430–1432, 1984. doi: 10.1126/science.6322303. [DOI] [PubMed] [Google Scholar]
- 146.Wu R, Dong W, Ji Y, Zhou M, Marini CP, Ravikumar TS, Wang P. Orexigenic hormone ghrelin attenuates local and remote organ injury after intestinal ischemia-reperfusion. PLoS One 3: e2026, 2008. doi: 10.1371/journal.pone.0002026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Yeboah MM, Xue X, Duan B, Ochani M, Tracey KJ, Susin M, Metz CN. Cholinergic agonists attenuate renal ischemia-reperfusion injury in rats. Kidney Int 74: 62–69, 2008. doi: 10.1038/ki.2008.94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Yoshioka T, Shiraga H, Yoshida Y, Fogo A, Glick AD, Deen WM, Hoyer JR, Ichikawa I. “Intact nephrons” as the primary origin of proteinuria in chronic renal disease. Study in the rat model of subtotal nephrectomy. J Clin Invest 82: 1614–1623, 1988. doi: 10.1172/JCI113773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zager RA. Precipitable tissue proteins can cause experimental acute renal failure. Lab Invest 59: 798–808, 1988. [PubMed] [Google Scholar]
- 150.Zelnick LR, Weiss NS, Kestenbaum BR, Robinson-Cohen C, Heagerty PJ, Tuttle K, Hall YN, Hirsch IB, de Boer IH. Diabetes and CKD in the United States population, 2009−2014. Clin J Am Soc Nephrol 12: 1984–1990, 2017. doi: 10.2215/CJN.03700417. [DOI] [PMC free article] [PubMed] [Google Scholar]