

Keywords: calcineurin inhibitor, magnesium, NaCl cotransporter, transient receptor potential subfamily M member 6, uromodulin
Abstract
The genetic disease Gitelman syndrome, knockout mice, and pharmacological blockade with thiazide diuretics have revealed that reduced activity of the NaCl cotransporter (NCC) promotes renal Mg2+ wasting. NCC is expressed along the distal convoluted tubule (DCT), and its activity determines Mg2+ entry into DCT cells through transient receptor potential channel subfamily M member 6 (TRPM6). Several other genetic forms of hypomagnesemia lower the drive for Mg2+ entry by inhibiting activity of basolateral Na+-K+-ATPase, and reduced NCC activity may do the same. Lower intracellular Mg2+ may promote further Mg2+ loss by directly decreasing activity of Na+-K+-ATPase. Lower intracellular Mg2+ may also lower Na+-K+-ATPase indirectly by downregulating NCC. Lower NCC activity also induces atrophy of DCT cells, decreasing the available number of TRPM6 channels. Conversely, a mouse model with increased NCC activity was recently shown to display normal Mg2+ handling. Moreover, recent studies have identified calcineurin and uromodulin (UMOD) as regulators of both NCC and Mg2+ handling by the DCT. Calcineurin inhibitors paradoxically cause hypomagnesemia in a state of NCC activation, but this may be related to direct effects on TRPM6 gene expression. In Umod−/− mice, the cause of hypomagnesemia may be partly due to both decreased NCC expression and lower TRPM6 expression on the cell surface. This mini-review discusses these new findings and the possible role of altered Na+ flux through NCC and ultimately Na+-K+-ATPase in Mg2+ reabsorption by the DCT.
INTRODUCTION
Mg2+ is the second most abundant intracellular divalent cation and plays an essential role in many fundamental cellular processes (15). It is a cofactor for Na+-K+-ATPase and many kinases, regulates stabilization of nucleotides, and is critical for mitochondrial function, ion channels, and neuromuscular excitability (15, 29). Therefore, Mg2+ deficiency is associated with systemic defects but often presents with symptoms of neuromuscular hyperexcitability such as tremor, muscle cramps, tetany, and generalized seizures (15). Hypomagnesemia is frequently associated with hypokalemia and/or hypocalcemia, which can result in cardiac arrhythmia and sudden death (15). Mg2+ deficiency is also linked to various diseases including type 2 diabetes mellitus, hypertension, atherosclerosis, dyslipidemia, myocardial infarction, and psychiatric disorders (15).
Plasma Mg2+ concentration is maintained by the interplay of renal reabsorption, intestinal absorption, and bone exchange (29). In the kidney, ∼70% of total plasma Mg2+ is filtered by the glomeruli, of which ∼96% is reabsorbed along the nephron. Most reabsorption occurs along the thick ascending limb of the loop of Henle (TAL; 50–70%), with less along the proximal convoluted tubule (PCT; 10–25%) (54). Only 3–7% of the filtered Mg2+ is reabsorbed in the distal convoluted tubule (DCT), but this segment determines urinary excretion since it is the major site of regulated reabsorption (54). The critical role of the TAL in Mg2+ handling is demonstrated by the effects of claudin disruption of Mg2+ handling by this segment. However, the DCT plays a critical role in fine tuning Mg2+ homeostasis, as demonstrated by the presentation of hypomagnesemia as a central feature of Gitelman syndrome, caused by inactivating mutations in the DCT-specific NaCl cotransporter (NCC) (74), and it can also occur as a side effect of thiazide diuretics, which inhibit NCC. Moreover, several other congenital syndromes that lead to DCT dysfunction can present with renal Mg2+ wasting, hypomagnesemia, and hypocalciuria, resembling Gitelman syndrome (84), and thus confirming the critical role of the DCT in Mg2+ homeostasis.
Epidermal growth factor (EGF) is highly expressed along the DCT, and proEGF is cleaved to generate active EGF that binds to EGF receptor (EGFR) at the basolateral surface (31). EGF signaling has been shown to play an important role in Mg2+ homeostasis. Recently, uromodulin (UMOD; also known as Tamm-Horsfall protein) and calcineurin have been identified as novel factors modulating Mg2+ handling by the DCT, providing new insights into the relationship between NCC activity and Mg2+ homeostasis. Other recent studies have explored the link between Mg2+ and NCC activity more directly, using dietary Mg2+ restriction and a genetic mouse model that results in hyperactive NCC. We also briefly discuss the possible role of changes in intracellular Cl− concentration in Mg2+ handling. This mini-review discusses these new findings, focusing on how altered Na+ flux through NCC and ultimately Na+-K+-ATPase may be a central regulator of Mg2+ reabsorption.
Mg2+ REABSORPTION ALONG THE DCT
Along the DCT, Mg2+ is transported via active transcellular pathways, which has been functionally divided into early (DCT1) and late (DCT2) subsegments (50, 54). Apical Mg2+ entry along DCT1 and DCT2 is mediated by transient receptor potential channel subfamily M member 6 (TRPM6) (69, 86), likely in complex with transient receptor potential channel subfamily M member 7, which is required for maximal expression of TRPM6 (68). Mg2+ entry through the channel is driven by the electrical potential across the apical membrane (−70 mV), which may be established by the voltage-gated K+ channel (Kv1.1) (Fig. 1) (24, 52). TRPM6 mutations result in hypomagnesemia and renal Mg2+ wasting (86), whereas patients with Kv1.1 mutations fail to lower fractional renal Mg2+ excretion in the presence of hypomagnesemia (24). Intestine-specific Trpm6−/− mice suffer from hypomagnesemia, whereas kidney-specific Trpm6−/− mice show normal Mg2+ homeostasis (11). It remains unclear what causes this discrepancy between mice and humans; however, it is well established that inhibition of EGFR that increases TRPM6 transcription and activation (37, 78) induces hypomagnesemia with renal Mg2+ wasting in both mice and humans (16, 65). In addition, mutations in the gene encoding EGF result in isolated, recessive renal hypomagnesemia (31).
Fig. 1.

Apical and basolateral membrane proteins that participate in Mg2+ handling in the early distal convoluted tubule (DCT1). NaCl cotransporter (NCC), voltage-gated K+ channel Kv1.1, and transient receptor potential channel subfamily M member 6 (TRPM6) are expressed in the apical membrane, whereas Na+-K+-ATPase, Kir4.1/Kir5.1, voltage-gated Cl− channel ClC-K2, SLC41A1, and cyclin and CBS domain divalent metal cation transport mediator-2 (CNNM2) are expressed in the basolateral membrane. Mg2+ entry through TRPM6 is driven by the electrical potential across the apical membrane (−70 mV), which may be established by Kv1.1. Parvalbumin (PV) has been proposed to act as the intracellular Mg2+ shuttle. SLC41A1 and CNNM2 may play a key role in basolateral Mg2+ extrusion. Yellow circles represent Mg2+. FXYD2, Na+-K+-ATPase γ-subunit.
The intracellular protein that binds and transports Mg2+ to maintain normal intracellular Mg2+ levels during transcellular reabsorption has not been identified (54), but the cytoplasmic Ca2+/Mg2+-binding proteins parvalbumin (PV) and calbindin D28K (CaBP28) are relatively highly expressed along the DCT and may serve this role. PV is specifically expressed in DCT1 (7, 50), whereas CaBP28 is expressed along with apical Ca2+ transporter and basolateral Ca2+-extruding proteins in DCT2 and the connecting segment. This different distribution suggests that PV may act as the intracellular Mg2+ shuttle and that the major site for transcellular Mg2+ reabsorption is the DCT1 (Fig. 1). In support of a dominant role for PV, Pv−/− mice may display mild urinary Mg2+ wasting (63), whereas Cabp28−/− mice do not (47). However, the question remains of whether there is even the need for intracellular proteins to buffer intracellular Mg2+. The presence of CaBP28 keeps intracellular Ca2+ very low (0.12 µmol/L) despite ongoing transepithelial flux (54), whereas the intracellular concentration of Mg2+ is much higher (∼800 µmol/L) and is similar to tubular and plasma Mg2+ concentrations (52, 54).
The basolateral Mg2+ extrusion mechanism remains unclear, but SLC41A1, which acts as a Na+/Mg2+ exchanger, has been implicated (Fig. 1) (43). A SLC41A1 mutation that reduces its expression in patients with nephronophthisis-related disorders by an in-frame deletion of a transmembrane helix shows a defect in Mg2+ transport activity in human embryonic kidney (HEK)-293 cells (35). Another Na+/Mg2+ exchanger, SLC41A3, is also highly expressed in the DCT (14), but its role is less clear. Slc41a3−/− mice showed hypomagnesemia but no change in urinary Mg2+ excretion (14). Furthermore, it has been previously reported that SLC41A3 is a mitochondrial Mg2+ transporter rather than a plasma membrane protein (51). Cyclin and CBS domain divalent metal cation transport mediator-2 (CNNM2) might also play a key role in the Mg2+ reabsorptive mechanism (Fig. 1), although its true function has not yet been clarified (23). CNNM2 localizes at the basolateral membrane of the DCT in the human kidney, and mutations in patients cause renal Mg2+ wasting and hypomagnesemia (75). Recently, Cnnm2+/− mice and kidney-specific Cnnm2−/− mice have been reported to display lower Mg2+ levels in serum but no increase in urinary Mg2+ excretion, suggesting no defect in renal Mg2+ reabsorption (21). Of the three, SLC41A3 and CNNM2 deficiency do not cause renal Mg2+ wasting, so SLC41A1 is considered to be the prime candidate to facilitate Mg2+ extrusion; generation of DCT-specific Slc41a1−/− mice might resolve the issue of basolateral Mg2+ extrusion.
NCC AND Na+-K+-ATPASE IN DCT Mg2+ TRANSPORT
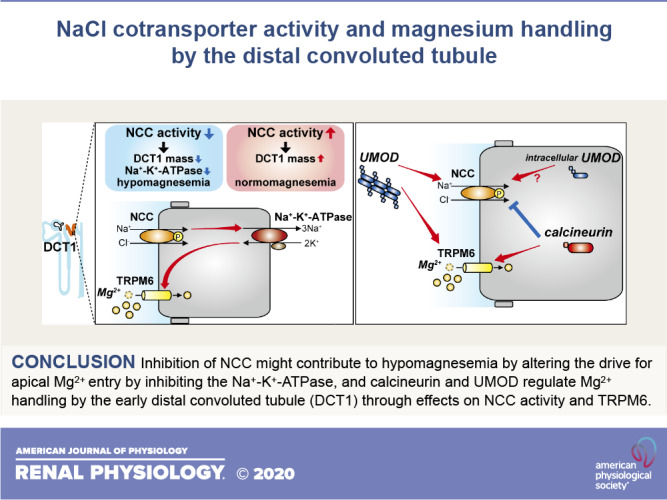
DCT Mg2+ transport depends upon NCC function. Loss-of-function mutations in SLC12A3, which encodes NCC, cause Gitelman syndrome, characterized by hypokalemia, hypocalciuria, metabolic alkalosis, and renal Mg2+ wasting and hypomagnesemia (74). Ncc−/− mice and mice treated with thiazide diuretics, which specifically inhibit NCC, phenotypically resemble Gitelman syndrome and display marked atrophy of DCT1 (50, 60). Although hydrochlorothiazide treatment also inhibits the Na+-driven Cl–/ exchanger and pendrin in β-intercalated cells of the collecting duct (48), this is probably not relevant to DCT Mg2+ handling. Furthermore, mice lacking STE20/SPS1-related proline-alanine-rich protein kinase (SPAK), a kinase that phosphorylates NCC, display almost complete ablation of NCC activity and decreased DCT1 mass (28). These findings strongly suggest that NCC activity influences DCT1 mass (Fig. 2, A and B). Trpm6 mRNA and protein expression are significantly lower in Ncc−/− mice (60). DCT1 atrophy might therefore simply result in lower TRPM6 expression as a consequence of lower DCT1 mass (Fig. 2A). However, TRPM6 expression levels were reduced in mice with 6 days of thiazide treatment despite strong NCC expression and no evidence of DCT atrophy (60). More recently, experiments in 1-day-old Ncc−/− mice showed lower TRPM6 protein expression before impaired DCT1 outgrowth was observed (70). Thus, NCC may indirectly influence Trpm6 mRNA and protein expression before DCT1 atrophy is manifested, through unidentified pathways.
Fig. 2.

Schematic model of the effect of NaCl cotransporter (NCC) activity on distal convoluted tubule (DCT) Mg2+ handling. A: NCC inactivation reduces early DCT (DCT1) mass and Na+-K+-ATPase activity, leading to hypomagnesemia. This is caused by decreased Mg2+ entry through transient receptor potential channel subfamily M member 6 (TRPM6). B: NCC activation increases DCT mass, but TRPM6 per unit DCT1 mass is downregulated, resulting in a minimal effect on Mg2+ entry. C: Na+-K+-ATPase activity is a central determinant of DCT Mg2+ reabsorption because it provides the driving force for Mg2+ entry via TRPM6. Hepatocyte nuclear factor-1β (HNF1B) plays a key role in Mg2+ handling, since it promotes transcription of Na+-K+-ATPase subunit-γ (FXYD2) and Kir5.1, which enhance Na+-K+-ATPase activity. Kir4.1/Kir5.1 activity is closely related to NCC activity. D: Mg2+ deficiency may promote NCC degradation via ubiquitin ligase neuronal precursor cell developmentally downregulated 4-2 (NEDD4-2). Increased Na+ delivery to downstream segments can promote K+ secretion. Mg2+ deficiency upregulates TRPM6 expression, which enhances Mg2+ entry, but NCC downregulation can cause DCT1 atrophy. Combined with reduced levels of the intracellular Mg2+ concentration ([Mg2+]i) for Na+-K+-ATPase activity, this would be expected to reduce Mg2+ entry via TRPM6 channels.
Although loss of NCC activity causes hypomagnesemia, increased NCC activity does not cause hypermagnesemia. NCC is activated by phosphorylation of several serine and threonine residues at its NH2 terminal. The pathways involved in this process are complex (72), but in simple terms, with no lysine kinase 4 (WNK4) phosphorylates and activates SPAK, which then directly phosphorylates NCC. This NCC-activating pathway is regulated by inhibition of WNK4, which can occur by two mechanisms. First, direct binding of Cl− prevents WNK4 autophosphorylation (5). Second, abundance of WNK4 is decreased by proteasomal degradation following ubiquitination by an E3 ubiquitin ligase consisting of the scaffold cullin 3, the substrate adaptor Kelch-like 3 (KLHL3), and the ubiquitin conjugating ligase RING (12). The disease familial hyperkalemic hypertension (FHHt) is caused by mutations in the genes encoding WNK4, cullin 3, and KLHL3 as well as WNK1 (which results in its ectopic expression) (53). The net effect of these mutations is to inappropriately activate NCC, as reflected by the fact that all features of FHHt (which also include metabolic acidosis and hypercalciuria) are reversed by thiazides. FHHt is the mirror image of Gitelman syndrome, with the notable exception that it is not associated with Mg2+ imbalance (53). Consistent with this, in mice with DCT1-specific expression of constitutively active mutant SPAK (CA-SPAK), which display NCC hyperactivation, serum Mg2+, urinary Mg2+ excretion, and Trpm6 mRNA levels were similar to those of control mice (83). Since CA-SPAK mice display increased DCT1 mass (27), this suggests compensatory downregulation of TRPM6 (lower TRPM6 per unit DCT1 mass) to maintain Mg2+ homeostasis. One caveat is that mRNA abundances may not be directly match protein levels, so TRPM6 protein abundances and activities need to be evaluated. Another recent study confirmed that NCC activation is not associated with altered Mg2+ homeostasis (Fig. 2B). Low dietary K+ intake strongly activates NCC via WNK/SPAK-mediated phosphorylation via activation of the Kir4.1/Kir5.1 heterotetramer in the basolateral membrane of the DCT (88). van der Wijst et al. (82) demonstrated that although low-K+ diet activated NCC, it did not alter Trpm6 mRNA expression, serum Mg2+ concentration, or Mg2+ excretion. In both studies, TRPM6 currents were not measured, and compensatory mechanisms in other segments may also contribute to maintenance of Mg2+ homeostasis. Another explanation proposed for the different effects of NCC inactivation and activation is that membrane potential does not change much with NCC activation (52). However, the actual membrane potential of the DCT has not been measured while NCC activity was manipulated. Although activation or inactivation of NCC activity would affect Kir4.1/Kir5.1 activity through Na+-K+-ATPase coupling, this may not change the basolateral membrane potential because Cl− efflux through voltage-gated Cl− channel ClC-K2 should also change in the same direction (see potential role of intracellular cl− in mg2+ handling by the dct for more details). Another explanation is that the effect of NCC activation on DCT1 mass may be smaller than that of NCC inactivation (Fig. 2, A and B). Indeed, the DCT1 mass of Ncc−/− mice is reduced to <0.1-fold (50), whereas that of CA-SPAK mice is increased to only ∼1.4-fold (27), and TRPM6 mRNA abundance is decreased in Ncc−/− mice but not in CA-SPAK mice. Thus, reduced TRPM6 due to DCT atrophy may be central in causing Mg2+ wasting with NCC inactivation, but with NCC activation DCT mass and TRPM6 are disconnected.
Genetic disorders that influence Na+-K+-ATPase function and cause urinary Mg2+ wasting confirm the importance of ATPase activity for DCT Mg2+ reabsorption (84). These include loss-of-function mutations of other genes highly expressed in DCT, including Na+-K+-ATPase subunit-γ (FXYD2) (55), inward rectifying K+ channel subfamily J member 10 (KCNJ10) (66), hepatocyte nuclear factor-1β (HNF1B) (1), and pterin-4-α-carbinolamine dehydratase 1 (PCBD1) (18). FXYD2 interacts with Na+-K+-ATPase, modulating its activity (39, 52). Fxyd2−/− mice show destabilized and depressed Na+-K+-ATPase activity (39) and a nonsignificant trend to increased urinary Mg2+ excretion (39, 52). In addition, Na+-K+-ATPase activity depends on K+ recycling via Kir4.1/Kir5.1 (encoded by KCNJ10/KCNJ16) expressed in the basolateral membrane of the DCT (Fig. 2C). Kir4.1/Kir5.1 activity is closely related to NCC activity (Fig. 2C), as demonstrated by dramatically lower reduced NCC expression and phosphorylation in Kir4.1 knockout mice (13); thus, kidney-specific deletion of Kir4.1 in mice causes DCT atrophy (76), which may also contribute to hypomagnesemia. HNF1β binds the FXYD2 promoter and activates its transcription (Fig. 2C), whereas PCBD1 regulates FXYD2 transcription as a dimerization cofactor for HNF1B (18). A recent study has demonstrated that HNF1β also activates the Kcnj16 promoter, and kidney-specific Hnf1b disruption downregulated mRNA expression of not only Kir5.1 but also Kir4.1 (44). Thus, HFN1B promotes Mg2+ reabsorption by regulating Na+-K+-ATPase activity both directly and indirectly (Fig. 2C).
Altered NCC activity might also be expected to affect Mg2+ homeostasis through effects on Na+-K+-ATPase activity. NCC activation and the following elevation of intracellular Na+ concentration might be expected to increase Na+-K+-ATPase activity, but, as discussed above, NCC activation (e.g., in FHHt) is not associated with altered Mg2+ handling in part due to effects on TRPM6 expression (Fig. 2B). Whether NCC activation significantly alters Na+-K+-ATPase activity has not been determined. In contrast, NCC inactivation and the resulting decrease in NaCl entry may have a significant inhibitory effect on Na+-K+-ATPase, thus reducing the driving force for Mg2+ entry via TRPM6 (Fig. 2A) (52). Indeed, Na+-K+-ATPase activity of rat DCT segments is ∼30–50% lower after thiazide treatment (22, 56). Although the link between NCC activity and basolateral Mg2+ extrusion is unclear, Mg2+ extrusion via SLC41A1 is considered to be driven by the inward Na+ electrochemical gradient generated by Na+-K+-ATPase (52). Thus, NCC inactivation may also affect the driving forces for basolateral Mg2+ extrusion.
The DCT is clearly important for Mg2+ reabsorption, so Mg2+ depletion exerts significant effects on DCT function. Dietary Mg2+ depletion stimulates expression of Trpm6 and PV (30). The highest Na+-K+-ATPase activity along the renal tubule is found in the DCT (40). Mg2+ can directly influence Na+-K+-ATPase activity (2) and is required for mitochondrial production of ATP to power Na+-K+-ATPase (29). Thus, there is reason to suspect that Mg2+ depletion may impair DCT Na+ transport by reducing Na+-K+-ATPase function (Fig. 2D), and this segment may be particularly susceptible. Mg2+ depletion may also impair DCT Na+ transport by reducing NCC activity. We recently found that dietary Mg2+ restriction as short as 3 days decreased abundances of total and phosphorylated NCC (17). NCC is ubiquitinated and targeted for degradation by neural precursor cell expressed, developmentally downregulated 4-2 (NEDD4-2) (Fig. 2D) (3). In mice lacking tubular NEDD4-2, Mg2+ restriction no longer reduced total or phosphorylated NCC, suggesting that Mg2+ depletion increases NEDD4-2 activity (17). Although the mechanism by which Mg2+ depletion activates NEDD4-2 has not been determined, one possibility is that the decrease in intracellular Mg2+ induced by dietary Mg2+ restriction elevates the intracellular free ionized Ca2+ concentration (90). NEDD4-2 can be activated by either directly binding Ca2+ via its Calb/C2-binding domain, which releases its autoinhibition, or after binding to substrate (87). Reduced Na+ entry would also lower Na+ available to drive Na+-K+-ATPase, exacerbating reductions in Na+ and Mg2+ entry; DCT1 atrophy would have an additional detrimental effect. However, Mg2+ restriction upregulates protein abundance of the Na+-K+-ATPase activator HNF1B (81). To resolve the relative contributions of these opposing forces on Na+-K+-ATPase activity, further studies are needed to evaluate DCT1 mass and Na+-K+-ATPase activity during Mg2+ restriction. For an extensive recent discussion of the mechanisms linking Na+ and Mg2+ reabsorption along the DCT, the reader is referred to Franken et al. (19).
Mg2+ depletion has long been known to promote urinary K+ excretion, sustaining hypokalemia (34). Classically, this phenomenon is explained by Mg2+-mediated suppression of outward K+ currents through the renal outer medullary K+ channel (ROMK) in the connecting segment and collecting duct, such that Mg2+ depletion allows unbridled K+ secretion through ROMK (34). We recently found in mice that the effect of dietary Mg2+ restriction to downregulate NCC overrode the effect of K+ restriction to activate it (17). This observation provides a plausible mechanism for increased Na+ delivery and flow in the setting of combined hypomagnesemia and hypokalemia.
POTENTIAL ROLE OF INTRACELLULAR Cl− IN Mg2+ HANDLING BY THE DCT
Changes in intracellular Cl− concentration play an important role in regulating NCC. Cl− prevents WNK4 autophosphorylation and activation by directly binding to its catalytic domain (5), so decreased intracellular Cl− concentration phosphorylates and activates the WNK4-SPAK-NCC pathway (Fig. 3). ClC-K2 is mainly expressed in the TAL and DCT (32) and is known to be the main pathway for basolateral Cl− exit in DCT cells (32, 42). The driving force for Cl− extrusion via ClC-K2 is provided by the inside negativity of the membrane potential, which is determined by Kir4.1/Kir5.1 activity (32, 88). Recently, Su et al. (77) generated a transgenic mouse model expressing an optogenetic kidney-specific Cl− sensor and found that chemical inhibition of Kir4.1/Kir5.1 significantly increased intracellular Cl− concentration. Thus, intracellular Cl− levels are in a large part determined by Kir4.1/Kir5.1 activity and regulate NCC activation (Fig. 3). Cl− efflux through ClC-K2 may also be modulated independently of Kir4.1/Kir5.1, but this has not yet been demonstrated.
Fig. 3.

Schematic model of the effect of intracellular Cl− on distal convoluted tubule (DCT) Mg2+ handling. Left: decreased intracellular Cl− levels ([Cl−]i) activate the with no lysine kinase 4 (WNK4)-STE20/SPS1-related proline/alanine-rich kinase (SPAK)-NaCl cotransporter (NCC) pathway. The driving force for Cl− extrusion via the voltage-gated chloride channel ClC-K2 is provided by the inside negativity of the membrane potential, which is determined by Kir4.1/Kir5.1 activity. Serum K+ levels regulate Kir4.1/5.1 activity. Right: ClC-K2 deficiency is predicted to increased [Cl−]i, leading to inhibition of the WNK4-SPAK-NCC pathway. This induces DCT1 atrophy, decreasing transient receptor potential channel subfamily M member 6 (TRPM6) expression and activity, with the consequence of Mg2+ wasting. FXYD2, Na+-K+-ATPase subunit-γ.
Loss-of-function mutations in the CLCNK2 gene that encodes ClC-K2 cause Bartter syndrome type 3, but some patients display renal Mg2+ wasting and phenotypes similar to Gitelman syndrome (20, 61). Therefore, CLC-K2 may affect not only Mg2+ reabsorption along the TAL but also along the DCT1 by modulating the WNK4-SPAK-NCC cascade. Recently, Hennings et al. (32) generated Clcnk2−/− mice that displayed a phenotype resembling this syndrome and a profound decrease in total and phosphorylated NCC. Consistent with this, these mice exhibited no response to furosemide but also a severely blunted response to thiazide (32). Interestingly, Clcnk2−/− mice showed a marked atrophy of the DCT1 and renal Mg2+ wasting (32). These effects of Clcnk2−/− mice are similar to those of renal epithelium-specific kcnj10−/− mice (13, 76, 91) that phenocopy the human disease epilepsy, ataxia, sensorineural deafness, and tubulopathy (EAST) syndrome (also called SeSAME) caused by mutations in KCNJ10 (8, 71). ClC-K2 deficiency would be predicted to increased intracellular Cl− concentration, leading to inhibition of WNK4. This would lower NCC activity and hence induce DCT1 atrophy, decreasing TRPM6 expression and activity, with the consequence of Mg2+ wasting (Fig. 3).
DCT Mg2+ HANDLING AND CALCINEURIN INHIBITION
Calcineurin inhibitors (CNIs) such as tacrolimus (previously referred to as FK506) and cyclosporin A are immunosuppressants widely used to prevent rejection after solid-organ transplantation and to treat autoimmune disease. CNI use is frequently accompanied by hypomagnesemia and hypertension through effects on the kidney (57). Calcineurin is a Ca2+/calmodulin-dependent serine/threonine phosphatase that influences expression of downstream genes by dephosphorylating transcription factors, such as nuclear factor of activated T cells, but can also act through nongenomic mechanisms (49). The DCT expresses both the calcineurin-α and -β isoforms (45), suggesting that effects on this segment may contribute to CNI side effects; studies in rodents support this.
CNI-induced hypertension is mediated in part by increased NCC activity due to reduced NCC dephosphorylation (Fig. 4) (33). To inhibit calcineurin and exert immunosuppressive effects, tacrolimus must bind to the 12-kDa FK506-binding protein (FKBP12), but it can also have calcineurin-independent effects. To address whether the effect of tacrolimus is calcineurin dependent, Lazelle et al. (45) generated inducible renal epithelium-specific FKBP12 knockout (KS-Fkbp12−/−) mice and found that tacrolimus-induced NCC activation and hypertension were absent in KS-Fkbp12−/− mice. These results strongly suggest that tacrolimus influences the calcineurin pathway (Fig. 4).
Fig. 4.

Role of 12-kDa FK506-binding protein (FKBP12) in distal convoluted tubule (DCT) Mg2+ handling. NaCl cotransporter (NCC) activity and transient receptor potential channel subfamily M member 6 (TRPM6) expression under normal conditions (left) and after tacrolimus treatment (right) are shown. TRPM6 is regulated by epidermal growth factor (EGF) that is generated by cleavage from proEGF and stimulates EGF receptor (EGFR). Stimulation of EGFR promotes TRPM6 transcription via the EGF-ERK1/2- activator protein-1 (AP-1) pathway and increases cell surface protein abundance and activation. The tacrolimus-FKBP12 complex inhibits calcineurin, leading to hypertension and hypomagnesemia as a result of NCC activation and TRPM6 downregulation. Tacrolimus treatment can decrease Egf mRNA expression and transcriptional activity of AP-1, leading to TRPM6 downregulation.
Dietary K+ alters NCC phosphorylation via modulation of the WNK-SPAK signaling pathway (88). High-K+ diet may also activate calcineurin by inducing cell depolarization, which opens voltage-dependent Ca2+ channels, providing another possible pathway for NCC regulation (73). Furthermore, CNIs increase abundance of WNK kinases and SPAK (33). Recent findings demonstrate that tacrolimus increases WNK4 abundance by preventing calcineurin-mediated dephosphorylation of the ubiquitin-ligase complex component KLHL3 (38). Phosphorylation of KLHL3 impairs degradation of WNK4. Thus, tacrolimus prevents WNK4 degradation, resulting in increased SPAK activity, with more NCC phosphorylation, increasing DCT Na+ reabsorption and blood pressure (Fig. 4).
The effects of tacrolimus raise a paradox since tacrolimus causes NCC activation associated with lower Mg2+ reabsorption and hypomagnesemia. A recent study by Gratreak et al. (26) exploring Mg2+ homeostasis in KS-Fkbp12−/− mice may provide a possible explanation. Administration of tacrolimus to rats profoundly downregulates Trpm6 mRNA and protein levels, leading to renal Mg2+ wasting (Fig. 4) (59). In KS-Fkbp12−/− mice, tacrolimus-induced hypomagnesemia and reduction of Trpm6 mRNA were completely abolished (26), suggesting that calcineurin downregulates TRPM6 expression through a pathway that differs from its NCC regulatory pathway. CNI-treated rats have shown downregulation of Egf mRNA paralleling effects on Trpm6 (Fig. 4) (46) that may hint at mechanism. In HEK-293 cells, EGFR activation upregulated TRPM6 transcription via the ERK1/2 activator protein-1 (AP-1) pathway and increased TRPM6 cell surface protein abundance and activation (Fig. 4) (37). In rats, EGF administration did not rescue CNI-induced TRPM6 downregulation, despite the fact that its administration to control rats upregulated TRPM6 expression (46). In addition, in renal tubular epithelial cell lines, CNI inhibited the ERK1/2-AP-1 pathway that upregulates Trpm6 mRNA (36). Therefore, calcineurin may affect Mg2+ transport by regulating TRPM6 expression via a EGF-ERK1/2-AP-1 pathway (Fig. 4). However, conflicting data from Kiely et al. (41) showed that CNI activated ERK1/2 in Madin-Darby canine kidney cells. Whether this difference is a consequence of the cell type examined or other technical differences remains unclear. Decreased EGF-EGFR-ERK1/2 pathway activation may also affect NCC activity. A recent study by Cheng et al. (10) reported that EGFR tyrosine kinase inhibition resulted in lower ERK1/2 phosphorylation, lower NCC activity, and increased Na+ excretion, suggesting a direct effect of EGFR on NCC. This NCC inactivation should contribute to decreased Mg2+ entry via TRPM6 by decreased Na+-K+-ATPase activity. However, the effect of EGF-EGFR-ERK1/2 pathway by CNI on NCC is likely to be smaller than that of WNK4-SPAK pathway because CNI activates NCC as discussed above.
Several questions remain. How is it that CNIs stimulate NCC activity but suppress TRPM6 expression? Is this because they influence DCT1 and DCT2 differently? Do they alter cell morphology or number in the DCT? Since calcineurin can act independently of nuclear factor of activated T cells (49), another possibility is differential interactions with other signaling molecules. Further studies will be required to evaluate these hypotheses.
UMOD AND DCT Mg2+ HANDLING
UMOD is a glycosylphosphatidyl-inositol-anchored protein that is targeted to the apical membrane of tubular cells, where it is subsequently cleaved and released into the lumen by the serine protease hepsin (9). UMOD is highly expressed in the TAL and after cleavage is excreted in the urine, where it is the most abundant protein in healthy subjects (89). Urinary UMOD protects against urinary tract infections and renal stone formation and stimulates renal salt reabsorption (89).
Umod−/− mice exhibit renal Mg2+ wasting with compensatory upregulation of mRNA expression of Trpm6, Hnf1b, Egf, Fxyd2, and Pv (58). Since UMOD enhances the surface expression of the Na+-K+-2Cl− cotransporter (NKCC2) and ROMK (89), its deletion is likely to indirectly affect paracellular Mg2+ along the TAL. However, although Umod−/− mice display increased urinary Mg2+ wasting, administration of a loop diuretic to these mice increases urinary Mg2+ to a degree similar to that seen in control mice (58). This suggests effects of UMOD deletion on additional segments. Indeed, in the DCT, despite increasing Trpm6 mRNA, UMOD deletion decreased apical TRPM6 protein abundance (58). Cotransfection experiments in HEK-293 cells showed that UMOD enhanced TRPM6 cell surface abundance and current density. Whole cell patch-clamp recording of HEK-293 cells treated with purified UMOD enhanced TRPM6 cell surface abundance and current density by impairing TRPM6 endocytosis (58). UMOD could exert similar effects on TRMP6 in native DCT1 cells to influence Mg2+ handling by this segment (Fig. 5A).
Fig. 5.

Schematic model of the effect of uromodulin (UMOD) on distal convoluted tubule (DCT) Mg2+ handling. A: UMOD levels and transient receptor potential channel subfamily M member 6 (TRPM6) cell surface abundance differ between normal conditions (left) and low-Mg2+ conditions (right). UMOD can enhance TRPM6 cell surface abundance and current density by impairing TRPM6 endocytosis. Furthermore, luminal UMOD levels increase under low-Mg2+ conditions. Thus, luminal UMOD may play a role in Mg2+ reabsorption along the DCT by regulating cell surface expression of TRPM6 and by altering NaCl cotransporter (NCC) activity. B: effects of intracellular UMOD on NCC. Left, UMOD can facilitate NCC phosphorylation and activation. Right, UMOD deficiency is associated with decreased phosphorylated NCC in the early DCT (DCT1), leading to increased NCC phosphorylation in the late DCT (DCT2). Decreased NCC activity in DCT1 may cause inhibition of Na+-K+-ATPase activity, further contributing to decreased Mg2+ entry via TRPM6.
Although UMOD is classically thought of as a TAL marker, Tokonami et al. (80) found significant UMOD expression in mouse and human DCT1, at ∼10% of TAL expression levels. The Ca2+-sensing receptor (CaSR) plays a facilitating role, since its activation decreased UMOD excretion by reducing intracellular levels of cAMP, which is involved in the trafficking of many proteins in tubular cells (79). Another study showed that genetic and pharmacological inhibition of ROMK lowered urinary UMOD excretion, suggesting that a membrane potential-dependent trafficking mechanism might influence UMOD excretion (67). Since hepsin, CaSR, and ROMK are all expressed in the DCT (9, 25, 85), they could regulate UMOD trafficking and release not only in the TAL but also along the DCT. Therefore, UMOD synthesized in the DCT1 itself may also modulate Mg2+ reabsorption by this segment via direct effects on TRPM6 as discussed above.
Global disruption of UMOD was associated with decreased phosphorylated NCC in DCT1, consistent with a coexpression study of UMOD and NCC in HEK-293 cells, suggesting a facilitating role for UMOD in NCC activation (Fig. 5B) (80). UMOD excretion was significantly increased in Mg2+-restricted wild-type mice (58). Therefore, UMOD might be expected to stimulate Mg2+ reabsorption along the DCT by activating NCC, but we found lower phosphorylated NCC in Mg2+-restricted mice (17), suggesting that other pathways (e.g., NEDD4-2 activation) override the ability of UMOD to stimulate NCC during Mg2+ deficiency. However, there is evidence that UMOD may contribute to Mg2+ handling by the DCT. Tokonami et al. (80) reported that NCC activity was lower in DCT1 but higher in DCT2 in Umod−/− mice at baseline. Chronic furosemide treatment increased NCC phosphorylation along DCT2 but not along DCT1 in Umod−/− mice, despite increased NCC phosphorylation along DCT1 in control mice (80). Importantly, acute addition of hydrochlorothiazide to chronic furosemide treatment decreased Mg2+ excretion in wild-type mice but not in Umod−/− mice. NCC inactivation along DCT1 in Umod−/− mice should inhibit Na+-K+-ATPase activity, further contributing to decreased Mg2+ entry via TRPM6 (Fig. 5B). Thus, regulation of NCC activity by UMOD along DCT1 might also contribute to normal Mg2+ handling.
Further complexity is added when UMOD regulatory proteins expressed along both the TAL and DCT are considered. CaSR inhibits UMOD but activates NCC. CaSR also binds and responds to Mg2+, and DCT cells modulate intracellular signals in response to changes in extracellular Mg2+ as well as in extracellular Ca2+ (4). Since CaSR is expressed in the apical membrane of DCT (25), reduced luminal Mg2+ might lower CaSR activation. Lower CaSR activity would be predicted to increase UMOD release from the DCT1 by lowering CaSR-mediated cAMP inhibition of UMOD trafficking, stimulating TRPM6 to prevent Mg2+ loss. In contrast, CaSR stimulation activates NCC through the KLHL3-WNK4-SPAK pathway in vitro and in vivo (6). Therefore, lower CaSR activation during Mg2+ deficiency might result in NCC inhibition.
Hepsin, which cleaves and releases UMOD, is expressed at similar levels in the DCT and TAL (9). However, Olinger et al. (62) showed that in mice carrying a hepsin missense mutation cellular UMOD accumulation affected NKCC2 activity but not total and phosphorylated NCC. This raises the possibility that UMOD is differentially regulated in the two segments. However, given that UMOD deletion differentially influences NCC activation in DCT1 and DCT2, and since NCC was only evaluated in whole kidney, the two segments should be evaluated separately. On the other hand, DCT1-made UMOD might be still released because another protease, prostasin, which directly cleaves UMOD in vitro (9), is strongly expressed in the DCT (64). Although its deletion did not change urinary UMOD levels (9), the effect of prostasin on UMOD release from DCT1 remains unknown. Whether hepsin activity changes in response to changing dietary Mg2+ intake is unknown. Finally, how ROMK stimulation of urinary UMOD excretion contributes to Mg2+ handling has not been explored.
There are other unanswered questions regarding the contribution of UMOD synthesized by the DCT1 to Mg2+ handling. Can it increase TRPM6 membrane abundance before its hepsin-mediated cleavage? Although urinary UMOD excretion increases with Mg2+ restriction, is this additional UMOD derived from both the TAL and DCT1? What are the relative contributions of UMOD effects along the TAL and DCT in renal Mg2+ handling? To fully understand the roles of UMOD and its regulators in Mg2+ handling by the DCT, TAL- and DCT-specific knockout models will be needed.
SUMMARY
Recent studies have identified calcineurin and UMOD as regulators of renal Mg2+ handling through effects on the DCT. Several lines of evidence suggest that inhibition of NCC might contribute to hypomagnesemia by altering the drive for apical Mg2+ entry by inhibiting Na+-K+-ATPase. This is supported by the etiology of other monogenic diseases that resemble Gitelman syndrome. Mg2+ deficiency may itself promote Mg2+ loss by directly lowering Na+-K+-ATPase activity since it serves as a cofactor, and downregulation of NCC by decreased intracellular Mg2+ may exacerbate this. In contrast, inappropriate activation of NCC, as seen in FHHt or CA-SPAK mice, does not lead to altered Mg2+ handling. This may be due to the fact that Na+-K+-ATPase is high along the DCT compared with other segments, rendering it more susceptible to conditions that lower Na+-K+-ATPase activity. CNIs paradoxically cause hypomagnesemia in a state of NCC activation, but this may be related to direct effects on TRPM6 gene expression or selective effects on DCT2. Further studies are needed to work out precisely how these regulating factors interact with dietary Mg2+ and the mechanisms underlying altered DCT Mg2+ entry.
GRANTS
Y.M. received a postdoctoral fellowship from Uehara Memorial Foundation, and J.A.M. is funded by National Institute of Diabetes and Digestive and Kidney Diseases Grant DK098141.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
Y.M. drafted manuscript and prepared figures; J.A.M. edited and revised manuscript; Y.M. and J.A.M. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Evan Ray (University of Pittsburgh) for helpful discussion and critical feedback on the manuscript.
REFERENCES
- 1.Adalat S, Woolf AS, Johnstone KA, Wirsing A, Harries LW, Long DA, Hennekam RC, Ledermann SE, Rees L, van’t Hoff W, Marks SD, Trompeter RS, Tullus K, Winyard PJ, Cansick J, Mushtaq I, Dhillon HK, Bingham C, Edghill EL, Shroff R, Stanescu H, Ryffel GU, Ellard S, Bockenhauer D. HNF1B mutations associate with hypomagnesemia and renal magnesium wasting. J Am Soc Nephrol 20: 1123–1131, 2009. doi: 10.1681/ASN.2008060633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Apell HJ, Hitzler T, Schreiber G. Modulation of the Na,K-ATPase by magnesium ions. Biochemistry 56: 1005–1016, 2017. doi: 10.1021/acs.biochem.6b01243. [DOI] [PubMed] [Google Scholar]
- 3.Arroyo JP, Lagnaz D, Ronzaud C, Vázquez N, Ko BS, Moddes L, Ruffieux-Daidié D, Hausel P, Koesters R, Yang B, Stokes JB, Hoover RS, Gamba G, Staub O. Nedd4-2 modulates renal Na+-Cl− cotransporter via the aldosterone-SGK1-Nedd4-2 pathway. J Am Soc Nephrol 22: 1707–1719, 2011. doi: 10.1681/ASN.2011020132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bapty BW, Dai LJ, Ritchie G, Jirik F, Canaff L, Hendy GN, Quamme GA. Extracellular Mg2+- and Ca2+-sensing in mouse distal convoluted tubule cells. Kidney Int 53: 583–592, 1998. doi: 10.1046/j.1523-1755.1998.00790.x. [DOI] [PubMed] [Google Scholar]
- 5.Bazúa-Valenti S, Chávez-Canales M, Rojas-Vega L, González-Rodríguez X, Vázquez N, Rodríguez-Gama A, Argaiz ER, Melo Z, Plata C, Ellison DH, García-Valdés J, Hadchouel J, Gamba G. The effect of WNK4 on the Na+-Cl− cotransporter is modulated by intracellular chloride. J Am Soc Nephrol 26: 1781–1786, 2015. doi: 10.1681/ASN.2014050470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bazúa-Valenti S, Rojas-Vega L, Castañeda-Bueno M, Barrera-Chimal J, Bautista R, Cervantes-Pérez LG, Vázquez N, Plata C, Murillo-de-Ozores AR, González-Mariscal L, Ellison DH, Riccardi D, Bobadilla NA, Gamba G. The calcium-sensing receptor increases activity of the renal NCC through the WNK4-SPAK pathway. J Am Soc Nephrol 29: 1838–1848, 2018. doi: 10.1681/ASN.2017111155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Belge H, Gailly P, Schwaller B, Loffing J, Debaix H, Riveira-Munoz E, Beauwens R, Devogelaer JP, Hoenderop JG, Bindels RJ, Devuyst O. Renal expression of parvalbumin is critical for NaCl handling and response to diuretics. Proc Natl Acad Sci USA 104: 14849–14854, 2007. doi: 10.1073/pnas.0702810104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bockenhauer D, Feather S, Stanescu HC, Bandulik S, Zdebik AA, Reichold M, et al. Epilepsy, ataxia, sensorineural deafness, tubulopathy, and KCNJ10 mutations. N Engl J Med 360: 1960–1970, 2009. doi: 10.1056/NEJMoa0810276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brunati M, Perucca S, Han L, Cattaneo A, Consolato F, Andolfo A, Schaeffer C, Olinger E, Peng J, Santambrogio S, Perrier R, Li S, Bokhove M, Bachi A, Hummler E, Devuyst O, Wu Q, Jovine L, Rampoldi L. The serine protease hepsin mediates urinary secretion and polymerisation of Zona Pellucida domain protein uromodulin. eLife 4: e08887, 2015. doi: 10.7554/eLife.08887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cheng L, Poulsen SB, Wu Q, Esteva-Font C, Olesen ET, Peng L, Olde B, Leeb-Lundberg LM, Pisitkun T, Rieg T, Dimke H, Fenton RA. Rapid aldosterone-mediated signaling in the DCT increases activity of the thiazide-sensitive NaCl cotransporter. J Am Soc Nephrol 30: 1454–1470, 2019. doi: 10.1681/ASN.2018101025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chubanov V, Ferioli S, Wisnowsky A, Simmons DG, Leitzinger C, Einer C, Jonas W, Shymkiv Y, Bartsch H, Braun A, Akdogan B, Mittermeier L, Sytik L, Torben F, Jurinovic V, van der Vorst EP, Weber C, Yildirim OA, Sotlar K, Schürmann A, Zierler S, Zischka H, Ryazanov AG, Gudermann T. Epithelial magnesium transport by TRPM6 is essential for prenatal development and adult survival. eLife 5: e20914, 2016. doi: 10.7554/eLife.20914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cornelius RJ, Ferdaus MZ, Nelson JW, McCormick JA. Cullin-Ring ubiquitin ligases in kidney health and disease. Curr Opin Nephrol Hypertens 28: 490–497, 2019. doi: 10.1097/MNH.0000000000000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cuevas CA, Su XT, Wang MX, Terker AS, Lin DH, McCormick JA, Yang CL, Ellison DH, Wang WH. Potassium sensing by renal distal tubules requires Kir4.1. J Am Soc Nephrol 28: 1814–1825, 2017. doi: 10.1681/ASN.2016090935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.de Baaij JH, Arjona FJ, van den Brand M, Lavrijsen M, Lameris AL, Bindels RJ, Hoenderop JG. Identification of SLC41A3 as a novel player in magnesium homeostasis. Sci Rep 6: 28565, 2016. doi: 10.1038/srep28565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.de Baaij JH, Hoenderop JG, Bindels RJ. Magnesium in man: implications for health and disease. Physiol Rev 95: 1–46, 2015. doi: 10.1152/physrev.00012.2014. [DOI] [PubMed] [Google Scholar]
- 16.Dimke H, van der Wijst J, Alexander TR, Meijer IM, Mulder GM, van Goor H, Tejpar S, Hoenderop JG, Bindels RJ. Effects of the EGFR inhibitor erlotinib on magnesium handling. J Am Soc Nephrol 21: 1309–1316, 2010. doi: 10.1681/ASN.2009111153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ferdaus MZ, Mukherjee A, Nelson JW, Blatt PJ, Miller LN, Terker AS, Staub O, Lin DH, McCormick JA. Mg2+ restriction downregulates NCC through NEDD4-2 and prevents its activation by hypokalemia. Am J Physiol Renal Physiol 317: F825–F838, 2019. doi: 10.1152/ajprenal.00216.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferrè S, de Baaij JH, Ferreira P, Germann R, de Klerk JB, Lavrijsen M, van Zeeland F, Venselaar H, Kluijtmans LA, Hoenderop JG, Bindels RJ. Mutations in PCBD1 cause hypomagnesemia and renal magnesium wasting. J Am Soc Nephrol 25: 574–586, 2014. doi: 10.1681/ASN.2013040337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Franken GA, Adella A, Bindels RJ, de Baaij JH. Mechanisms coupling sodium and magnesium reabsorption in the distal convoluted tubule of the kidney. Acta Physiol (Oxf) 2020: e13528, 2020. doi: 10.1111/apha.13528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fukuyama S, Okudaira S, Yamazato S, Yamazato M, Ohta T. Analysis of renal tubular electrolyte transporter genes in seven patients with hypokalemic metabolic alkalosis. Kidney Int 64: 808–816, 2003. doi: 10.1046/j.1523-1755.2003.00163.x. [DOI] [PubMed] [Google Scholar]
- 21.Funato Y, Yamazaki D, Miki H. Renal function of cyclin M2 Mg2+ transporter maintains blood pressure. J Hypertens 35: 585–592, 2017. doi: 10.1097/HJH.0000000000001211. [DOI] [PubMed] [Google Scholar]
- 22.Garg LC, Narang N. Effects of hydrochlorothiazide on Na-K-ATPase activity along the rat nephron. Kidney Int 31: 918–922, 1987. doi: 10.1038/ki.1987.86. [DOI] [PubMed] [Google Scholar]
- 23.Giménez-Mascarell P, Schirrmacher CE, Martínez-Cruz LA, Müller D. Novel aspects of renal magnesium homeostasis. Front Pediatr 6: 77, 2018. doi: 10.3389/fped.2018.00077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Glaudemans B, van der Wijst J, Scola RH, Lorenzoni PJ, Heister A, van der Kemp AW, Knoers NV, Hoenderop JG, Bindels RJ. A missense mutation in the Kv1.1 voltage-gated potassium channel-encoding gene KCNA1 is linked to human autosomal dominant hypomagnesemia. J Clin Invest 119: 936–942, 2009. doi: 10.1172/JCI36948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Graca JA, Schepelmann M, Brennan SC, Reens J, Chang W, Yan P, Toka H, Riccardi D, Price SA. Comparative expression of the extracellular calcium-sensing receptor in the mouse, rat, and human kidney. Am J Physiol Renal Physiol 310: F518–F533, 2016. doi: 10.1152/ajprenal.00208.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gratreak BD, Swanson EA, Lazelle RA, Jelen SK, Hoenderop J, Bindels RJ, Yang CL, Ellison DH. Tacrolimus-induced hypomagnesemia and hypercalciuria requires FKBP12 suggesting a role for calcineurin. Physiol Rep 8: e14316, 2020. doi: 10.14814/phy2.14316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grimm PR, Coleman R, Delpire E, Welling PA. Constitutively active SPAK causes hyperkalemia by activating NCC and remodeling distal tubules. J Am Soc Nephrol 28: 2597–2606, 2017. doi: 10.1681/ASN.2016090948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Grimm PR, Taneja TK, Liu J, Coleman R, Chen YY, Delpire E, Wade JB, Welling PA. SPAK isoforms and OSR1 regulate sodium-chloride co-transporters in a nephron-specific manner. J Biol Chem 287: 37673–37690, 2012. doi: 10.1074/jbc.M112.402800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gröber U, Schmidt J, Kisters K. Magnesium in prevention and therapy. Nutrients 7: 8199–8226, 2015. doi: 10.3390/nu7095388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Groenestege WM, Hoenderop JG, van den Heuvel L, Knoers N, Bindels RJ. The epithelial Mg2+ channel transient receptor potential melastatin 6 is regulated by dietary Mg2+ content and estrogens. J Am Soc Nephrol 17: 1035–1043, 2006. doi: 10.1681/ASN.2005070700. [DOI] [PubMed] [Google Scholar]
- 31.Groenestege WM, Thébault S, van der Wijst J, van den Berg D, Janssen R, Tejpar S, van den Heuvel LP, van Cutsem E, Hoenderop JG, Knoers NV, Bindels RJ. Impaired basolateral sorting of pro-EGF causes isolated recessive renal hypomagnesemia. J Clin Invest 117: 2260–2267, 2007. doi: 10.1172/JCI31680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hennings JC, Andrini O, Picard N, Paulais M, Huebner AK, Cayuqueo IK, Bignon Y, Keck M, Cornière N, Böhm D, Jentsch TJ, Chambrey R, Teulon J, Hübner CA, Eladari D. The ClC-K2 chloride channel is critical for salt handling in the distal nephron. J Am Soc Nephrol 28: 209–217, 2017. doi: 10.1681/ASN.2016010085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hoorn EJ, Walsh SB, McCormick JA, Fürstenberg A, Yang CL, Roeschel T, Paliege A, Howie AJ, Conley J, Bachmann S, Unwin RJ, Ellison DH. The calcineurin inhibitor tacrolimus activates the renal sodium chloride cotransporter to cause hypertension. Nat Med 17: 1304–1309, 2011. doi: 10.1038/nm.2497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huang CL, Kuo E. Mechanism of hypokalemia in magnesium deficiency. J Am Soc Nephrol 18: 2649–2652, 2007. doi: 10.1681/ASN.2007070792. [DOI] [PubMed] [Google Scholar]
- 35.Hurd TW, Otto EA, Mishima E, Gee HY, Inoue H, Inazu M, Yamada H, Halbritter J, Seki G, Konishi M, Zhou W, Yamane T, Murakami S, Caridi G, Ghiggeri G, Abe T, Hildebrandt F. Mutation of the Mg2+ transporter SLC41A1 results in a nephronophthisis-like phenotype. J Am Soc Nephrol 24: 967–977, 2013. doi: 10.1681/ASN.2012101034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ikari A, Okude C, Sawada H, Takahashi T, Sugatani J, Miwa M. Down-regulation of TRPM6-mediated magnesium influx by cyclosporin A. Naunyn Schmiedebergs Arch Pharmacol 377: 333–343, 2008. doi: 10.1007/s00210-007-0212-4. [DOI] [PubMed] [Google Scholar]
- 37.Ikari A, Sanada A, Okude C, Sawada H, Yamazaki Y, Sugatani J, Miwa M. Up-regulation of TRPM6 transcriptional activity by AP-1 in renal epithelial cells. J Cell Physiol 222: 481–487, 2010. [DOI] [PubMed] [Google Scholar]
- 38.Ishizawa K, Wang Q, Li J, Yamazaki O, Tamura Y, Fujigaki Y, Uchida S, Lifton RP, Shibata S. Calcineurin dephosphorylates Kelch-like 3, reversing phosphorylation by angiotensin II and regulating renal electrolyte handling. Proc Natl Acad Sci USA 116: 3155–3160, 2019. doi: 10.1073/pnas.1817281116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jones DH, Li TY, Arystarkhova E, Barr KJ, Wetzel RK, Peng J, Markham K, Sweadner KJ, Fong GH, Kidder GM. Na,K-ATPase from mice lacking the gamma subunit (FXYD2) exhibits altered Na+ affinity and decreased thermal stability. J Biol Chem 280: 19003–19011, 2005. doi: 10.1074/jbc.M500697200. [DOI] [PubMed] [Google Scholar]
- 40.Katz AI, Doucet A, Morel F. Na-K-ATPase activity along the rabbit, rat, and mouse nephron. Am J Physiol Renal Physiol 237: F114–F120, 1979. doi: 10.1152/ajprenal.1979.237.2.F114. [DOI] [PubMed] [Google Scholar]
- 41.Kiely B, Feldman G, Ryan MP. Modulation of renal epithelial barrier function by mitogen-activated protein kinases (MAPKs): mechanism of cyclosporine A-induced increase in transepithelial resistance. Kidney Int 63: 908–916, 2003. doi: 10.1046/j.1523-1755.2003.00804.x. [DOI] [PubMed] [Google Scholar]
- 42.Kobayashi K, Uchida S, Mizutani S, Sasaki S, Marumo F. Intrarenal and cellular localization of CLC-K2 protein in the mouse kidney. J Am Soc Nephrol 12: 1327–1334, 2001. [DOI] [PubMed] [Google Scholar]
- 43.Kolisek M, Nestler A, Vormann J, Schweigel-Röntgen M. Human gene SLC41A1 encodes for the Na+/Mg2+ exchanger. Am J Physiol Cell Physiol 302: C318–C326, 2012. doi: 10.1152/ajpcell.00289.2011. [DOI] [PubMed] [Google Scholar]
- 44.Kompatscher A, de Baaij JH, Aboudehen K, Hoefnagels AP, Igarashi P, Bindels RJ, Veenstra GJ, Hoenderop JG. Loss of transcriptional activation of the potassium channel Kir5.1 by HNF1β drives autosomal dominant tubulointerstitial kidney disease. Kidney Int 92: 1145–1156, 2017. doi: 10.1016/j.kint.2017.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lazelle RA, McCully BH, Terker AS, Himmerkus N, Blankenstein KI, Mutig K, Bleich M, Bachmann S, Yang CL, Ellison DH. Renal deletion of 12 kDa FK506-binding protein attenuates tacrolimus-induced hypertension. J Am Soc Nephrol 27: 1456–1464, 2016. doi: 10.1681/ASN.2015040466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ledeganck KJ, Boulet GA, Horvath CA, Vinckx M, Bogers JJ, Van Den Bossche R, Verpooten GA, De Winter BY. Expression of renal distal tubule transporters TRPM6 and NCC in a rat model of cyclosporine nephrotoxicity and effect of EGF treatment. Am J Physiol Renal Physiol 301: F486–F493, 2011. doi: 10.1152/ajprenal.00116.2011. [DOI] [PubMed] [Google Scholar]
- 47.Lee CT, Ng HY, Lee YT, Lai LW, Lien YH. The role of calbindin-D28k on renal calcium and magnesium handling during treatment with loop and thiazide diuretics. Am J Physiol Renal Physiol 310: F230–F236, 2016. doi: 10.1152/ajprenal.00057.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leviel F, Hübner CA, Houillier P, Morla L, El Moghrabi S, Brideau G, Hassan H, Parker MD, Kurth I, Kougioumtzes A, Sinning A, Pech V, Riemondy KA, Miller RL, Hummler E, Shull GE, Aronson PS, Doucet A, Wall SM, Chambrey R, Eladari D. The Na+-dependent chloride-bicarbonate exchanger SLC4A8 mediates an electroneutral Na+ reabsorption process in the renal cortical collecting ducts of mice. J Clin Invest 120: 1627–1635, 2010. doi: 10.1172/JCI40145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li H, Rao A, Hogan PG. Interaction of calcineurin with substrates and targeting proteins. Trends Cell Biol 21: 91–103, 2011. doi: 10.1016/j.tcb.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Loffing J, Vallon V, Loffing-Cueni D, Aregger F, Richter K, Pietri L, Bloch-Faure M, Hoenderop JG, Shull GE, Meneton P, Kaissling B. Altered renal distal tubule structure and renal Na+ and Ca2+ handling in a mouse model for Gitelman’s syndrome. J Am Soc Nephrol 15: 2276–2288, 2004. doi: 10.1097/01.ASN.0000138234.18569.63. [DOI] [PubMed] [Google Scholar]
- 51.Mastrototaro L, Smorodchenko A, Aschenbach JR, Kolisek M, Sponder G. Solute carrier 41A3 encodes for a mitochondrial Mg2+ efflux system. Sci Rep 6: 27999, 2016. doi: 10.1038/srep27999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mayan H, Farfel Z, Karlish SJ. Renal Mg handling, FXYD2 and the central role of the Na,K-ATPase. Physiol Rep 6: e13843, 2018. doi: 10.14814/phy2.13843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mayan H, Vered I, Mouallem M, Tzadok-Witkon M, Pauzner R, Farfel Z. Pseudohypoaldosteronism type II: marked sensitivity to thiazides, hypercalciuria, normomagnesemia, and low bone mineral density. J Clin Endocrinol Metab 87: 3248–3254, 2002. doi: 10.1210/jcem.87.7.8449. [DOI] [PubMed] [Google Scholar]
- 54.McCormick JA, Ellison DH. Distal convoluted tubule. Compr Physiol 5: 45–98, 2015. doi: 10.1002/cphy.c140002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meij IC, Koenderink JB, van Bokhoven H, Assink KF, Groenestege WT, de Pont JJ, Bindels RJ, Monnens LA, van den Heuvel LP, Knoers NV. Dominant isolated renal magnesium loss is caused by misrouting of the Na+,K+-ATPase gamma-subunit. Nat Genet 26: 265–266, 2000. doi: 10.1038/81543. [DOI] [PubMed] [Google Scholar]
- 56.Morsing P, Velázquez H, Wright FS, Ellison DH. Adaptation of distal convoluted tubule of rats. II. Effects of chronic thiazide infusion. Am J Physiol Renal Physiol 261: F137–F143, 1991. doi: 10.1152/ajprenal.1991.261.1.F137. [DOI] [PubMed] [Google Scholar]
- 57.Naesens M, Kuypers DR, Sarwal M. Calcineurin inhibitor nephrotoxicity. Clin J Am Soc Nephrol 4: 481–508, 2009. doi: 10.2215/CJN.04800908. [DOI] [PubMed] [Google Scholar]
- 58.Nie M, Bal MS, Liu J, Yang Z, Rivera C, Wu XR, Hoenderop JG, Bindels RJ, Marciano DK, Wolf MT. Uromodulin regulates renal magnesium homeostasis through the ion channel transient receptor potential melastatin 6 (TRPM6). J Biol Chem 293: 16488–16502, 2018. doi: 10.1074/jbc.RA118.003950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Nijenhuis T, Hoenderop JG, Bindels RJ. Downregulation of Ca2+ and Mg2+ transport proteins in the kidney explains tacrolimus (FK506)-induced hypercalciuria and hypomagnesemia. J Am Soc Nephrol 15: 549–557, 2004. doi: 10.1097/01.ASN.0000113318.56023.B6. [DOI] [PubMed] [Google Scholar]
- 60.Nijenhuis T, Vallon V, van der Kemp AW, Loffing J, Hoenderop JG, Bindels RJ. Enhanced passive Ca2+ reabsorption and reduced Mg2+ channel abundance explains thiazide-induced hypocalciuria and hypomagnesemia. J Clin Invest 115: 1651–1658, 2005. doi: 10.1172/JCI24134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nozu K, Iijima K, Kanda K, Nakanishi K, Yoshikawa N, Satomura K, Kaito H, Hashimura Y, Ninchoji T, Komatsu H, Kamei K, Miyashita R, Kugo M, Ohashi H, Yamazaki H, Mabe H, Otsubo A, Igarashi T, Matsuo M. The pharmacological characteristics of molecular-based inherited salt-losing tubulopathies. J Clin Endocrinol Metab 95: E511–E518, 2010. doi: 10.1210/jc.2010-0392. [DOI] [PubMed] [Google Scholar]
- 62.Olinger E, Lake J, Sheehan S, Schiano G, Takata T, Tokonami N, Debaix H, Consolato F, Rampoldi L, Korstanje R, Devuyst O. Hepsin-mediated processing of uromodulin is crucial for salt-sensitivity and thick ascending limb homeostasis. Sci Rep 9: 12287, 2019. doi: 10.1038/s41598-019-48300-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Olinger E, Schwaller B, Loffing J, Gailly P, Devuyst O. Parvalbumin: calcium and magnesium buffering in the distal nephron. Nephrol Dial Transplant 27: 3988–3994, 2012. doi: 10.1093/ndt/gfs457. [DOI] [PubMed] [Google Scholar]
- 64.Peters DE, Szabo R, Friis S, Shylo NA, Uzzun Sales K, Holmbeck K, Bugge TH. The membrane-anchored serine protease prostasin (CAP1/PRSS8) supports epidermal development and postnatal homeostasis independent of its enzymatic activity. J Biol Chem 289: 14740–14749, 2014. doi: 10.1074/jbc.M113.541318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Petrelli F, Borgonovo K, Cabiddu M, Ghilardi M, Barni S. Risk of anti-EGFR monoclonal antibody-related hypomagnesemia: systematic review and pooled analysis of randomized studies. Expert Opin Drug Saf 11, Suppl 1: S9–S19, 2012. doi: 10.1517/14740338.2011.606213. [DOI] [PubMed] [Google Scholar]
- 66.Reichold M, Zdebik AA, Lieberer E, Rapedius M, Schmidt K, Bandulik S, Sterner C, Tegtmeier I, Penton D, Baukrowitz T, Hulton SA, Witzgall R, Ben-Zeev B, Howie AJ, Kleta R, Bockenhauer D, Warth R. KCNJ10 gene mutations causing EAST syndrome (epilepsy, ataxia, sensorineural deafness, and tubulopathy) disrupt channel function. Proc Natl Acad Sci USA 107: 14490–14495, 2010. doi: 10.1073/pnas.1003072107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Schiano G, Glaudemans B, Olinger E, Goelz N, Müller M, Loffing-Cueni D, Deschenes G, Loffing J, Devuyst O. The urinary excretion of uromodulin is regulated by the potassium channel ROMK. Sci Rep 9: 19517, 2019. doi: 10.1038/s41598-019-55771-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schlingmann KP, Waldegger S, Konrad M, Chubanov V, Gudermann T. TRPM6 and TRPM7−gatekeepers of human magnesium metabolism. Biochim Biophys Acta 1772: 813–821, 2007. doi: 10.1016/j.bbadis.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 69.Schlingmann KP, Weber S, Peters M, Niemann Nejsum L, Vitzthum H, Klingel K, Kratz M, Haddad E, Ristoff E, Dinour D, Syrrou M, Nielsen S, Sassen M, Waldegger S, Seyberth HW, Konrad M. Hypomagnesemia with secondary hypocalcemia is caused by mutations in TRPM6, a new member of the TRPM gene family. Nat Genet 31: 166–170, 2002. doi: 10.1038/ng889. [DOI] [PubMed] [Google Scholar]
- 70.Schnoz C, Carrel M, Loffing J. Loss of sodium chloride co-transporter impairs the outgrowth of the renal distal convoluted tubule during renal development. Nephrol Dial Transplant 35: 411–432, 2020. doi: 10.1093/ndt/gfz172. [DOI] [PubMed] [Google Scholar]
- 71.Scholl UI, Choi M, Liu T, Ramaekers VT, Häusler MG, Grimmer J, Tobe SW, Farhi A, Nelson-Williams C, Lifton RP. Seizures, sensorineural deafness, ataxia, mental retardation, and electrolyte imbalance (SeSAME syndrome) caused by mutations in KCNJ10. Proc Natl Acad Sci USA 106: 5842–5847, 2009. doi: 10.1073/pnas.0901749106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Shekarabi M, Zhang J, Khanna AR, Ellison DH, Delpire E, Kahle KT. WNK kinase signaling in ion homeostasis and human disease. Cell Metab 25: 285–299, 2017. doi: 10.1016/j.cmet.2017.01.007. [DOI] [PubMed] [Google Scholar]
- 73.Shoda W, Nomura N, Ando F, Mori Y, Mori T, Sohara E, Rai T, Uchida S. Calcineurin inhibitors block sodium-chloride cotransporter dephosphorylation in response to high potassium intake. Kidney Int 91: 402–411, 2017. doi: 10.1016/j.kint.2016.09.001. [DOI] [PubMed] [Google Scholar]
- 74.Simon DB, Nelson-Williams C, Bia MJ, Ellison D, Karet FE, Molina AM, Vaara I, Iwata F, Cushner HM, Koolen M, Gainza FJ, Gitleman HJ, Lifton RP. Gitelman’s variant of Bartter’s syndrome, inherited hypokalaemic alkalosis, is caused by mutations in the thiazide-sensitive Na-Cl cotransporter. Nat Genet 12: 24–30, 1996. doi: 10.1038/ng0196-24. [DOI] [PubMed] [Google Scholar]
- 75.Stuiver M, Lainez S, Will C, Terryn S, Günzel D, Debaix H, Sommer K, Kopplin K, Thumfart J, Kampik NB, Querfeld U, Willnow TE, Němec V, Wagner CA, Hoenderop JG, Devuyst O, Knoers NV, Bindels RJ, Meij IC, Müller D. CNNM2, encoding a basolateral protein required for renal Mg2+ handling, is mutated in dominant hypomagnesemia. Am J Hum Genet 88: 333–343, 2011. doi: 10.1016/j.ajhg.2011.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Su XT, Ellison DH, Wang WH. Kir4.1/Kir5.1 in the DCT plays a role in the regulation of renal K+ excretion. Am J Physiol Renal Physiol 316: F582–F586, 2019. doi: 10.1152/ajprenal.00412.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Su XT, Klett NJ, Sharma A, Allen CN, Wang WH, Yang CL, Ellison DH. Distal convoluted tubule Cl− concentration is modulated via K+ channels and transporters. Am J Physiol Renal Physiol 319: F534–F540, 2020. doi: 10.1152/ajprenal.00284.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Thebault S, Alexander RT, Tiel Groenestege WM, Hoenderop JG, Bindels RJ. EGF increases TRPM6 activity and surface expression. J Am Soc Nephrol 20: 78–85, 2009. doi: 10.1681/ASN.2008030327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tokonami N, Olinger E, Debaix H, Houillier P, Devuyst O. The excretion of uromodulin is modulated by the calcium-sensing receptor. Kidney Int 94: 882–886, 2018. doi: 10.1016/j.kint.2018.07.022. [DOI] [PubMed] [Google Scholar]
- 80.Tokonami N, Takata T, Beyeler J, Ehrbar I, Yoshifuji A, Christensen EI, Loffing J, Devuyst O, Olinger EG. Uromodulin is expressed in the distal convoluted tubule, where it is critical for regulation of the sodium chloride cotransporter NCC. Kidney Int 94: 701–715, 2018. doi: 10.1016/j.kint.2018.04.021. [DOI] [PubMed] [Google Scholar]
- 81.van Angelen AA, San-Cristobal P, Pulskens WP, Hoenderop JG, Bindels RJ. The impact of dietary magnesium restriction on magnesiotropic and calciotropic genes. Nephrol Dial Transplant 28: 2983–2993, 2013. doi: 10.1093/ndt/gft358. [DOI] [PubMed] [Google Scholar]
- 82.van der Wijst J, Tutakhel OA, Bos C, Danser AH, Hoorn EJ, Hoenderop JG, Bindels RJ. Effects of a high-sodium/low-potassium diet on renal calcium, magnesium, and phosphate handling. Am J Physiol Renal Physiol 315: F110–F122, 2018. doi: 10.1152/ajprenal.00379.2017. [DOI] [PubMed] [Google Scholar]
- 83.van Megen WH, Grimm PR, Welling PA, van der Wijst J. Renal sodium and magnesium reabsorption are not coupled in a mouse model of Gordon syndrome. Physiol Rep 6: e13728, 2018. doi: 10.14814/phy2.13728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Viering DH, de Baaij JH, Walsh SB, Kleta R, Bockenhauer D. Genetic causes of hypomagnesemia, a clinical overview. Pediatr Nephrol 32: 1123–1135, 2017. doi: 10.1007/s00467-016-3416-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wade JB, Fang L, Coleman RA, Liu J, Grimm PR, Wang T, Welling PA. Differential regulation of ROMK (Kir1.1) in distal nephron segments by dietary potassium. Am J Physiol Renal Physiol 300: F1385–F1393, 2011. doi: 10.1152/ajprenal.00592.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Walder RY, Landau D, Meyer P, Shalev H, Tsolia M, Borochowitz Z, Boettger MB, Beck GE, Englehardt RK, Carmi R, Sheffield VC. Mutation of TRPM6 causes familial hypomagnesemia with secondary hypocalcemia. Nat Genet 31: 171–174, 2002. doi: 10.1038/ng901. [DOI] [PubMed] [Google Scholar]
- 87.Wang J, Peng Q, Lin Q, Childress C, Carey D, Yang W. Calcium activates Nedd4 E3 ubiquitin ligases by releasing the C2 domain-mediated auto-inhibition. J Biol Chem 285: 12279–12288, 2010. doi: 10.1074/jbc.M109.086405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wang MX, Cuevas CA, Su XT, Wu P, Gao ZX, Lin DH, McCormick JA, Yang CL, Wang WH, Ellison DH. Potassium intake modulates the thiazide-sensitive sodium-chloride cotransporter (NCC) activity via the Kir4.1 potassium channel. Kidney Int 93: 893–902, 2018. doi: 10.1016/j.kint.2017.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wolf MT, Zhang J, Nie M. Uromodulin in mineral metabolism. Curr Opin Nephrol Hypertens 28: 481–489, 2019. doi: 10.1097/MNH.0000000000000522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhang A, Cheng TP, Altura BM. Magnesium regulates intracellular free ionized calcium concentration and cell geometry in vascular smooth muscle cells. Biochim Biophys Acta 1134: 25–29, 1992. doi: 10.1016/0167-4889(92)90024-6. [DOI] [PubMed] [Google Scholar]
- 91.Zhang C, Wang L, Zhang J, Su XT, Lin DH, Scholl UI, Giebisch G, Lifton RP, Wang WH. KCNJ10 determines the expression of the apical Na-Cl cotransporter (NCC) in the early distal convoluted tubule (DCT1). Proc Natl Acad Sci USA 111: 11864–11869, 2014. doi: 10.1073/pnas.1411705111. [DOI] [PMC free article] [PubMed] [Google Scholar]