

Abstract
Endoplasmic reticulum stress contributes to ischemia-reperfusion (I/R) injury in rodent and cell models. However, the contribution of endoplasmic reticulum stress in the pathogenesis of endothelial I/R injury in humans is unknown. We tested the hypothesis that compared with placebo, inhibition of endoplasmic reticulum stress via ingestion of tauroursodeoxycholic acid would prevent the attenuation of endothelium-dependent vasodilation following I/R injury. Twelve young adults (6 women) were studied following ingestion of a placebo or 1,500 mg tauroursodeoxycholic acid (TUDCA). Endothelium-dependent vasodilation was assessed via brachial artery flow-mediated dilation (duplex ultrasonography) before and after I/R injury, which was induced by 20 min of arm ischemia followed by 20 min of reperfusion. Endothelium-independent vasodilation (glyceryl trinitrate-mediated vasodilation) was also assessed after I/R injury. Compared with placebo, TUDCA ingestion increased circulating plasma concentrations by 145 ± 90 ng/ml and increased concentrations of the taurine unconjugated form, ursodeoxycholic acid, by 560 ± 156 ng/ml (both P < 0.01). Ischemia-reperfusion injury attenuated endothelium-dependent vasodilation, an effect that did not differ between placebo (pre-I/R, 5.0 ± 2.1% vs. post-I/R, 3.5 ± 2.2%) and TUDCA (pre-I/R, 5.6 ± 2.1% vs. post-I/R, 3.9 ± 2.1%; P = 0.8) conditions. Similarly, endothelium-independent vasodilation did not differ between conditions (placebo, 19.6 ± 4.8% vs. TUDCA, 19.7 ± 6.1%; P = 0.9). Taken together, endoplasmic reticulum stress does not appear to contribute to endothelial I/R injury in healthy young adults.
Keywords: endoplasmic reticulum stress, flow-mediated dilation, ischemia-reperfusion, tauroursodeoxycholic acid
INTRODUCTION
From 1995 to 2012, the number of hospitalizations associated with an ischemic event nearly doubled in the United States (4). Importantly, this translates to a reduced quality of life for the survivors of an ischemic event and ultimately increases cardio- and cerebrovascular morbidity and mortality. The tissue and organ damage associated with an ischemic event is mediated, in part, by local hypoxia that occurs secondary to a reduction in blood flow. Paradoxically, the successful restoration of blood flow exacerbates damage of the previously ischemic tissue and is referred to as ischemia-reperfusion (I/R) injury (14, 35, 43). While the pathophysiology of I/R injury is thought to be multifactorial and is yet to be fully elucidated, a local vascular endothelial mechanism has emerged as a strong contributor (16, 43).
Endothelial I/R injury is characterized by an attenuated endothelium-dependent vasodilator function (24) that can lead to a sustained malperfusion of the previously ischemic tissue despite interventions that restore vessel patency, a condition known as the “no-reflow” phenomenon (22). Notably, transient or persistent no-reflow occurs in ∼1 in 20 patients undergoing percutaneous coronary intervention and is associated with adverse in-hospital and 30-day clinical outcomes (11). In the most severe cases, endothelial I/R injury can cause remote organ injury and multiorgan dysfunction and ultimately lead to death (13, 44). Despite these adverse clinical outcomes associated with the no-reflow phenomenon, the mechanisms attenuating endothelium-dependent vasodilator function in humans remain unclear.
The endoplasmic reticulum is a multipurpose organelle found in most human cells, including the endothelium of blood vessels. One of the primary functions of the endoplasmic reticulum is the posttranslational folding of new proteins and the reprocessing of misfolded or damaged proteins (3, 21). Stress conditions, such as hypoxia, can lead to the accumulation of unfolded/misfolded proteins in the endoplasmic reticulum, thus triggering the unfolded protein response, which is a quality control system that maintains endoplasmic reticulum homeostasis (3). However, with prolonged or severe exposure to endoplasmic reticulum stress inducers, the unfolded protein response can augment the formation of reactive oxygen species and inflammatory meditators that can decrease the formation of vasodilator substances such as nitric oxide (3, 7, 21). In addition, endoplasmic reticulum stress can reduce nitric oxide bioavailability independent of the effects on inflammatory mediators and reactive oxygen species by directly suppressing endothelial nitric oxide synthase activity (28).
Despite evidence suggesting that endoplasmic reticulum stress contributes to I/R injury in rodent and cell models (23, 29, 40, 45) and other investigations suggesting that endoplasmic reticulum stress impairs vasodilator function in pathophysiological conditions that share common etiologies (41), the contribution to endothelial I/R injury in humans is unknown. Therefore, the purpose of this study was to determine if endoplasmic reticulum stress contributes to the pathogenesis of endothelial I/R injury. We tested the hypothesis that compared with placebo, inhibition of endoplasmic reticulum stress via ingestion of tauroursodeoxycholic acid (TUDCA) would prevent the attenuation of endothelium-dependent vasodilation following I/R injury.
METHODS
Participants
Before subject enrollment, we performed a power analysis to estimate the minimum number of subjects required to test our hypothesis. Sample size and the power calculations were based on the estimated Δflow-mediated dilation between placebo and TUDCA conditions following I/R injury. Using data from pilot testing and data from a prior study (15), a sample size of n = 12 provides 80% power to detect a 3.3% difference between conditions with an expected standard deviation of 1.8% and using an alpha = 0.05. Therefore, 12 participants (6 women) were recruited to participate in this study.
Written informed consent was obtained from all participants following a verbal and written briefing of all experimental procedures. This study was approved by the North Texas Regional Institutional Review Board (Project No. 1590377-2) and was performed in accordance with the principles outlined in the Declaration of Helsinki, except for registration in a database. Participants were deemed free from cardiometabolic disease following the completion of an in-depth medical history questionnaire and a resting 12-lead electrocardiogram. Participants were required to abstain from caffeine, supplements, alcohol, and exercise for 24 h before the study. Participants were also required to abstain from over the counter or prescription medications at the time of the study. Women were studied during the early follicular phase of the menstrual cycle (days 1–5) or during the placebo phase if using an oral contraceptive (n = 1). Body mass was measured via dual energy X-ray absorptiometry (Lunar Prodigy, GE Healthcare, Chicago, IL). Participant physical characteristics are shown in Table 1.
Table 1.
Participant characteristics
| Male/Female | 6/6 |
| Age, yr | 26 ± 5 |
| Height, cm | 170 ± 11 |
| Weight, kg | 71 ± 20 |
| Lean mass, kg | 46 ± 12 |
| Fat mass, kg | 21 ± 9 |
| Body mass index, kg/m2 | 23.6 ± 4.6 |
Data are presented as means ± SD.
Experimental Approach
We utilized a single-blind, placebo-controlled experimental design. Experiments were randomized, counterbalanced, and separated by at least 7 days. Participants were studied ∼8 h after ingesting a placebo (Tackett Compounding Pharmacy, Hudson Oaks, TX) or 1,500 mg TUDCA (Olympus Laboratories, Las Vegas, NV). Tauroursodeoxycholic acid is a chemical chaperone that alleviates endoplasmic reticulum stress by augmenting protein stability, preventing protein aggregation, restoring trafficking of misfolded proteins, and upregulating the function of endogenous molecular chaperones (18). This pharmacological approach has been utilized in humans to increase circulating concentrations of TUDCA and the taurine unconjugated form, ursodeoxycholic acid, which also alleviates endoplasmic reticulum stress (39, 41). Plasma concentrations of TUDCA peak ∼8 h after ingestion and return to baseline at 24 h (41). In addition, TUDCA does not appear to have any direct cardiovascular effects in healthy young adults under resting conditions (41).
For each experiment, participants reported to the laboratory at ∼8:30 AM after an overnight fast. Participants were provided a standardized breakfast before experimentation. Following instrumentation and a ∼20-min rest period, endothelium-dependent vasodilation was assessed in the right arm (pre-I/R). Approximately 20 min thereafter, I/R injury was induced in the right arm by placing a pneumatic cuff (SC10, Hokanson, Bellevue, WA) immediately distal to the axilla and rapidly inflating it to 250 mmHg (E20 Rapid Cuff Inflator, Hokanson, Bellevue, WA). The cuff was always placed proximal to the ultrasonography site, and complete circulatory arrest was confirmed by absence of a palpable radial pulse. After 20 min of ischemia, the cuff was rapidly deflated, and reperfusion was allowed for 20 min. Endothelium-dependent vasodilation was reassessed immediately upon completion of reperfusion (post-I/R). Endothelium-independent vasodilation was then assessed 15–20 min thereafter at the same imaging site.
Measurements
Participants were in the supine position throughout experimentation and were asked to remain quiet and relaxed during measurements. The arm of each participant was abducted to 90° and supported at heart level during assessments of vasodilator function. Laboratory temperature was maintained at ∼21°C.
Hemodynamics.
Arterial blood pressure was measured using an automated sphygmomanometer (Tango M2, SunTech Medical, Morrisville, NC) placed on the left arm. Heart rate was monitored continuously via electrocardiogram (Solar 8000M, GE Healthcare, Chicago, IL). Brachial artery diameter and blood velocity were measured in the right arm via duplex ultrasonography (11 MHz, Phillips iE33, Andover, MA) using a linear-array transducer and an insonation angle of 60°. The ultrasound was interfaced with a computer running custom software to capture blood velocity (DUC2). An outline of the ultrasound transducer was marked on the skin to ensure consistent placement throughout each experiment. In addition, care was taken to match ultrasound settings (e.g., sample volume size and depth) between experiments to ensure consistent probe placement.
Vasodilator function.
Endothelium-dependent vasodilation was assessed via flow-mediated dilation of the brachial artery in accordance with recent guidelines (20, 38). Briefly, a pneumatic cuff (SC5D, Hokanson, Bellevue, WA) was placed on the forearm, immediately distal to the antecubital fossa. Arterial inflow to the forearm was occluded by rapidly inflating the cuff to 220 mmHg for 5 min (E20 Rapid Cuff Inflator, Hokanson, Bellevue, WA). Before cuff inflation, brachial artery diameter and blood velocity were recorded during a 1-min baseline period and resumed 20 s before cuff deflation and continued for 3 min thereafter.
Endothelium-independent vasodilation was assessed via glyceryl trinitrate-mediated dilation of the brachial artery. Brachial artery diameter and blood velocity were recorded during a 1-min baseline period and continued for 10 min after lingual administration of 400 µg glyceryl trinitrate (Nitroglycerin Lingual Spray, Perrigo Pharmaceuticals, Allegan MI). Endothelium-independent vasodilation was not assessed at the pre-I/R time point to avoid possible carry-over effects of glyceryl trinitrate administration.
Blood sampling and analysis.
A 22-gauge intravenous catheter was placed in an antecubital vein of the participant’s right arm. Venous blood samples obtained before the first assessment of endothelial function were collected into a Vacutainer and centrifuged at 1,300 g within 5 min. Blood plasma was then aliquoted into cryogenic vials, immediately frozen in liquid nitrogen, and stored at −80°C until analysis. Plasma concentrations of TUDCA and ursodeoxycholic acid were quantified via high-performance liquid chromatography in the Sterol Analysis Laboratory at Oregon Health & Science University.
Data and Statistical Analyses
Blood velocity measurements were determined from Doppler ultrasound audio recordings using an intensity-weighted algorithm (custom software), subsequent to demodulation of forward and reverse Doppler frequencies (9, 32–34). Blood velocities were then thin-beam corrected using an average correction factor of 0.975 ± 0.033 (9, 26). Correction factors were based on a measured ultrasound probe beam-width of 3.67 mm and an insonation depth of 1.76 ± 0.35 cm. Vessel diameter was determined using custom edge-detection and wall-tracking software (6). Peak diameter measured during flow-mediated dilation and glyceryl trinitrate-mediated dilation was determined using an algorithm previously described by Black and colleagues (6). Blood flow was calculated by multiplying the cross-sectional area of the brachial artery by mean blood velocity (reported in ml/min). Shear stress (i.e., the frictional drag of red blood cells along the lumen the blood vessel) was estimated using shear rate, which was calculated by multiplying 8 by the quotient of blood velocity and vessel diameter (expressed as s−1).
Hemodynamic indexes were analyzed using a two-way (condition × time) mixed model ANOVA with repeated measures (JMP 14; SAS Institute, Cary, NC). Follow-up tests were performed using Tukey’s post hoc procedure. Flow-mediated dilation was assessed using the allometric modeling solution proposed by Atkinson et al. (1, 2), subsequent to verification of the presence of inadequate scaling by examining the slope of the relation between logarithmically transformed baseline and peak diameter. Shear rate area under the curve summed through peak diameter was also entered into the model as a covariate to account for changes in shear stimulus. Glyceryl trinitrate-mediated dilation conditions was analyzed using a paired t test. Plasma concentrations of TUDCA and ursodeoxycholic acid were analyzed using a paired t test. Data are reported as means ± SD.
RESULTS
Plasma Analysis
Absolute plasma concentrations of TUDCA and ursodeoxycholic acid are shown in Fig. 1. Relative to the placebo condition, TUDCA ingestion increased plasma concentrations by Δ145 ± 90 ng/ml and increased ursodeoxycholic acid concentrations by Δ560 ± 156 ng/ml (both P < 0.01 vs. 0 change).
Fig. 1.

Plasma ursodeoxycholic acid (UDCA; A) and tauroursodeoxycholic acid (TUDCA; B) concentrations are shown for placebo and TUDCA conditions. Men, n = 6; women, n = 6. Data were analyzed using a paired t test. ‡P < 0.01 vs. placebo.
Baseline Hemodynamics
Baseline hemodynamics are shown in Table 2. Heart rate did not differ from pre- to post-I/R for placebo (P = 0.4) and TUDCA (P = 0.07) conditions. Likewise, mean arterial pressure did not differ from pre- to post-I/R for placebo (P = 0.6) and TUDCA (P = 0.5) conditions. Brachial artery blood flow did not differ between conditions or across time (P = 0.8)
Table 2.
Baseline hemodynamics
| Placebo |
TUDCA |
|||
|---|---|---|---|---|
| Pre-I/R | Post-I/R | Pre-I/R | Post-I/R | |
| Heart rate, beats/min | 72 ± 8 | 69 ± 9 | 70 ± 9 | 68 ± 12 |
| Mean arterial pressure, mmHg | 85 ± 6 | 87 ± 6 | 86 ± 6 | 83 ± 9 |
| Brachial artery blood flow, ml/min | 45 ± 29 | 46 ± 33 | 47 ± 35 | 47 ± 27 |
Values are means ± SD. TUDCA, tauroursodeoxycholic acid; I/R, ischemia-reperfusion.
Vasodilator Function
Endothelium-dependent vasodilation assessed via brachial artery flow-mediated dilation is shown in Fig. 2. Additional measures relevant to the assessment of endothelium-dependent vasodilation are presented in Table 3. We were unable to collect data for one subject in the placebo condition due to the coronavirus-related closure of the laboratory. Endothelium-dependent vasodilation did not differ between conditions before I/R injury (P = 0.3). Ischemia-reperfusion injury attenuated endothelium-dependent vasodilation (P < 0.01), an effect that did not differ between placebo and TUDCA conditions (P = 0.8).
Fig. 2.
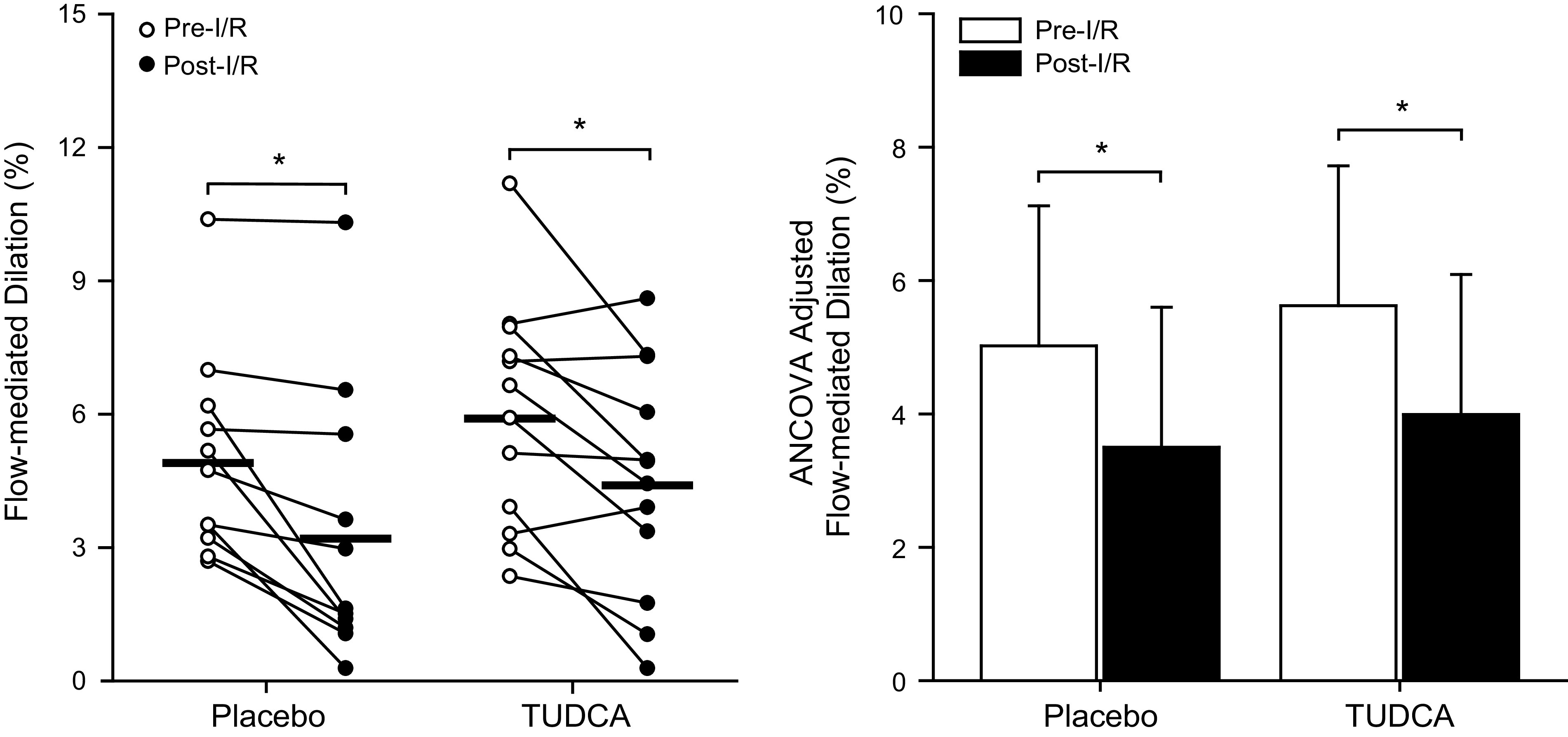
Individual (left) and ANCOVA-adjusted (right) flow-mediated vasodilatory responses are shown at preischemia-reperfusion (pre-I/R) and post-I/R time points for placebo and tauroursodeoxycholic acid (TUDCA) conditions. Individual data were analyzed using a two-way (condition × time) mixed model analysis of variance with repeated measures. All data were then adjusted by entering logarithmically transformed baseline diameter and shear rate area under the curve summed through peak diameter into the model as covariates to account for their influence on the flow-mediated vasodilatory response. Placebo: men, n = 6; women, n = 5; TUDCA: men, n = 6; women, n = 6. *P < 0.01 vs. pre-I/R.
Table 3.
Brachial artery hemodynamics during assessment of endothelium-dependent vasodilation
| Placebo |
TUDCA |
|||
|---|---|---|---|---|
| Pre-I/R | Post-I/R | Pre-I/R | Post-I/R | |
| Baseline diameter, cm | 0.353 ± 0.062 | 0.362 ± 0.064 | 0.347 ± 0.055 | 0.350 ± 0.053 |
| Peak diameter, cm | 0.370 ± 0.062 | 0.373 ± 0.060 | 0.367 ± 0.054 | 0.365 ± 0.053 |
| ΔDiameter, cm | 0.017 ± 0.007 | 0.011 ± 0.009* | 0.020 ± 0.008 | 0.015 ± 0.008* |
| Time to peak diameter, s | 49 ± 34 | 48 ± 29 | 54 ± 40 | 62 ± 55 |
| Shear rate AUC | 42,267 ± 13,302 | 42,380 ± 14,990 | 48,773 ± 23,773 | 48,912 ± 21,547 |
Values are means ± SD. Shear rate AUC, shear rate area under the curve through peak diameter; TUDCA, tauroursodeoxycholic acid; I/R, ischemia-reperfusion.
P < 0.01 vs. pre-I/R.
Endothelium-independent vasodilation assessed via glyceryl trinitrate-mediated dilation of the brachial artery is shown in Fig. 3. Glyceryl trinitrate was not administered to 1 subject due to a contraindication of low resting blood pressure. Glyceryl trinitrate-mediated dilation did not differ between placebo and TUDCA conditions after I/R injury (P = 0.9).
Fig. 3.

Glyceryl trinitrate (GTN)-mediated vasodilation is shown for placebo and tauroursodeoxycholic acid (TUDCA) conditions following ischemia-reperfusion (I/R) injury. Individual values are reported as open circles where bars indicate the mean. Data were analyzed using a paired t test. Placebo: men, n = 6; women, n = 5; TUDCA: men, n = 6; women, n = 5.
DISCUSSION
The purpose of this study was to determine if endoplasmic reticulum stress contributes to the pathogenesis of endothelial I/R injury. Contrary to our hypothesis, inhibition of endoplasmic reticulum stress via ingestion of TUDCA did not prevent the attenuation of endothelium-dependent vasodilation following I/R injury. Thus endoplasmic reticulum stress does not appear to contribute to endothelial I/R injury in humans
Endoplasmic Reticulum Stress and Endothelial I/R Injury
Investigations utilizing rodent models have demonstrated that endoplasmic reticulum stress contributes to cardiac, renal, and hepatic I/R injury (23, 29, 40, 45). Endoplasmic reticulum stress also contributes to hypoxia-reoxygenation injury (i.e., an in vitro model of I/R) in cultured rat and human endothelial cells (5, 42). Despite this evidence, the contribution of endoplasmic reticulum stress to the pathogenesis of endothelial I/R injury was unknown. Notably, endoplasmic reticulum stress has been shown to impair vasodilator function in conditions that share some common etiologies with I/R injury. For example, Tampakakis et al. (37) demonstrated that the intravenous infusion of lipids induced endoplasmic reticulum stress in venous endothelial cells and peripheral blood mononuclear cells and reduced microvascular dilator function in healthy young adults. More recently, Walsh and colleagues (41) demonstrated that inhibition of endoplasmic reticulum stress, via ingestion of TUDCA, prevented hyperglycemia-induced endothelial dysfunction of the brachial artery. Thus a clear relationship exists whereby various endoplasmic reticulum stress inducers can impair vasodilator function in humans.
Our data suggest that endoplasmic reticulum stress does not contribute to endothelial I/R injury in healthy young adults. The model of endothelial I/R injury utilized herein acutely and reversibly impairs endothelium-dependent vasodilation in humans (e.g., Refs. 8, 16, 23, 30, 34, 36, 37). While this experimental model can safely function as a human surrogate for I/R injury, the vasodeleterious consequences and associated mechanisms may vary depending on factors that influence the severity of ischemia and/or reperfusion. For example, it is possible that a more prolonged ischemic insult or a severe hypo/hyperreperfusion could sufficiently induce endoplasmic reticulum stress. In addition, we included only healthy young adults in this study who can more effectively buffer endoplasmic reticulum stress without the untoward vascular consequences associated with induction of the unfolded protein response (19). Thus it remains possible that ingestion of TUDCA could prevent endothelial I/R injury in older adults who have an attenuated capacity to buffer endoplasmic reticulum stress (19). Future studies should explore the relation between endoplasmic reticulum stress and the age-related susceptibility to endothelial I/R injury (12).
Emerging Mechanisms of Human Endothelial I/R Injury
Surprisingly, to date, a limited number of studies have explored the mechanisms contributing to the pathogenesis of endothelial I/R injury in humans. While I/R injury augments sympathetic nervous system outflow (25), the intravenous infusion of trimetaphan does not reverse the attenuation of vasodilator function (27) suggesting that endothelial I/R injury is not autonomic in origin and is likely mediated locally at the vascular level. To that end, Pleiner and colleagues (31) demonstrated that the intra-arterial infusion of ascorbic acid prevented the attenuation of microvascular dilator function (assessed via acetylcholine-mediated hyperemia) following I/R injury, which suggests a mechanistic role for oxidative stress. However, these findings do not appear to extend to the macrovasculature given that antioxidant ingestion fails to reverse the I/R-induced attenuation of brachial artery flow-mediated dilation (10). In addition, cyclooxygenase inhibition does not alter vasodilator function following I/R injury, suggesting that prostanoid signaling is not an obligatory mechanism (10). Finally, we recently demonstrated that concentrations of the potent vasoconstrictor endothelin-1 are not elevated in venous blood draining from the arm exposed endothelial I/R injury (16). Taken together, the local mechanism(s) mediating human endothelial I/R injury remain elusive. This remains an exciting area for future investigation.
Experimental Considerations
Several experimental considerations warrant discussion. First, we did not directly measure endoplasmic reticulum stress. Plasma measures of endoplasmic reticulum stress are currently unavailable; thus the invasive collection of endothelial and/or smooth muscle cells is required to accurately quantify endoplasmic reticulum stress and/or activation of the unfolded protein response, both of which are beyond the exploratory scope of this study. Second, it is possible that the dose and/or duration of TUDCA ingestion utilized in this study was unable to fully inhibit endoplasmic reticulum stress. However, it should be noted that TUDCA has been shown to be a potent inhibitor of endoplasmic reticulum stress in numerous in vitro and in vivo studies. In addition, an identical dose of TUDCA was successfully utilized by Walsh et al. (41) to prevent hyperglycemia-induced endothelial dysfunction. Thus increasing the dose of TUDCA may not be more efficacious within the context of human endothelial I/R injury. Finally, endothelium-independent vasodilation was not assessed at the pre-I/R time point to avoid possible carry-over effects of glyceryl trinitrate administration.
Perspectives and Significance
The mechanisms contributing to the pathogenesis of endothelial I/R injury in humans remain unclear. Our findings suggest that endoplasmic reticulum stress does not appear to contribute to endothelial I/R injury in healthy young adults. However, it remains unclear if this finding extends to other populations that are at an increased risk for an ischemic event (e.g., older adults) and who have an attenuated ability to buffer endoplasmic reticulum stress. Importantly, elucidating the mechanisms by which I/R injury impairs vasodilator function will aid in the development of therapeutic interventions that ultimately improve quality of life and decrease cardio- and cerebrovascular morbidity and mortality.
GRANTS
Funding was provided by the National Institute on Aging Grants R01-AG-059314 and T32-AG-020494 and laboratory startup funds from the University of North Texas Health Science Center.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
H.W.H. and S.A.R. conceived and designed research; H.W.H., A.M.M., A.H.O., and S.A.R. performed experiments; H.W.H. and S.A.R. analyzed data; H.W.H., A.M.M., and S.A.R. interpreted results of experiments; H.W.H. and S.A.R. prepared figures; H.W.H. and S.A.R. drafted manuscript; H.W.H., A.M.M., A.H.O., and S.A.R. edited and revised manuscript; A.M.M., A.H.O., and S.A.R. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank the participants who cheerfully participated in this research study. We also thank Dr. John R. Halliwill for the development and use of the Doppler ultrasound capture and calculate (DUC2) program.
REFERENCES
- 1.Atkinson G, Batterham AM, Thijssen DH, Green DJ. A new approach to improve the specificity of flow-mediated dilation for indicating endothelial function in cardiovascular research. J Hypertens 31: 287–291, 2013. doi: 10.1097/HJH.0b013e32835b8164. [DOI] [PubMed] [Google Scholar]
- 2.Atkinson G, Batterham AM. The percentage flow-mediated dilation index: a large-sample investigation of its appropriateness, potential for bias and causal nexus in vascular medicine. Vasc Med 18: 354–365, 2013. doi: 10.1177/1358863X13508446. [DOI] [PubMed] [Google Scholar]
- 3.Battson ML, Lee DM, Gentile CL. Endoplasmic reticulum stress and the development of endothelial dysfunction. Am J Physiol Heart Circ Physiol 312: H355–H367, 2017. doi: 10.1152/ajpheart.00437.2016. [DOI] [PubMed] [Google Scholar]
- 4.Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Das SR, Delling FN, Djousse L, Elkind MS, Ferguson JF, Fornage M, Jordan LC, Khan SS, Kissela BM, Knutson KL, Kwan TW, Lackland DT, Lewis TT, Lichtman JH, Longenecker CT, Loop MS, Lutsey PL, Martin SS, Matsushita K, Moran AE, Mussolino ME, O’Flaherty M, Pandey A, Perak AM, Rosamond WD, Roth GA, Sampson UK, Satou GM, Schroeder EB, Shah SH, Spartano NL, Stokes A, Tirschwell DL, Tsao CW, Turakhia MP, VanWagner LB, Wilkins JT, Wong SS, Virani SS; American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee . Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation 139: e56–e528, 2019. doi: 10.1161/CIR.0000000000000659. [DOI] [PubMed] [Google Scholar]
- 5.Bi X, He X, Xu M, Zhao M, Yu X, Lu X, Zang W. Acetylcholine ameliorates endoplasmic reticulum stress in endothelial cells after hypoxia/reoxygenation via M3 AChR-AMPK signaling. Cell Cycle 14: 2461–2472, 2015. doi: 10.1080/15384101.2015.1060383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Black MA, Cable NT, Thijssen DH, Green DJ. Importance of measuring the time course of flow-mediated dilatation in humans. Hypertension 51: 203–210, 2008. doi: 10.1161/HYPERTENSIONAHA.107.101014. [DOI] [PubMed] [Google Scholar]
- 7.Brown DI, Griendling KK. Regulation of signal transduction by reactive oxygen species in the cardiovascular system. Circ Res 116: 531–549, 2015. doi: 10.1161/CIRCRESAHA.116.303584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brunt VE, Jeckell AT, Ely BR, Howard MJ, Thijssen DH, Minson CT. Acute hot water immersion is protective against impaired vascular function following forearm ischemia-reperfusion in young healthy humans. Am J Physiol Regul Integr Comp Physiol 311: R1060–R1067, 2016. doi: 10.1152/ajpregu.00301.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buck TM, Sieck DC, Halliwill JR. Thin-beam ultrasound overestimation of blood flow: how wide is your beam? J Appl Physiol (1985) 116: 1096–1104, 2014. doi: 10.1152/japplphysiol.00027.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carter SE, Faulkner A, Rakobowchuk M. The role of prostaglandin and antioxidant availability in recovery from forearm ischemia-reperfusion injury in humans. J Hypertens 32: 339–351, 2014. doi: 10.1097/HJH.0000000000000033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chan W, Stub D, Clark DJ, Ajani AE, Andrianopoulos N, Brennan AL, New G, Black A, Shaw JA, Reid CM, Dart AM, Duffy SJ; Melbourne Interventional Group Investigators . Usefulness of transient and persistent no reflow to predict adverse clinical outcomes following percutaneous coronary intervention. Am J Cardiol 109: 478–485, 2012. doi: 10.1016/j.amjcard.2011.09.037. [DOI] [PubMed] [Google Scholar]
- 12.DeVan AE, Umpierre D, Harrison ML, Lin HF, Tarumi T, Renzi CP, Dhindsa M, Hunter SD, Tanaka H. Endothelial ischemia-reperfusion injury in humans: association with age and habitual exercise. Am J Physiol Heart Circ Physiol 300: H813–H819, 2011. doi: 10.1152/ajpheart.00845.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eltzschig HK, Collard CD. Vascular ischaemia and reperfusion injury. Br Med Bull 70: 71–86, 2004. doi: 10.1093/bmb/ldh025. [DOI] [PubMed] [Google Scholar]
- 14.Eltzschig HK, Eckle T. Ischemia and reperfusion--from mechanism to translation. Nat Med 17: 1391–1401, 2011. doi: 10.1038/nm.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Engelland RE, Hemingway HW, Tomasco OG, Olivencia-Yurvati AH, Romero SA. Acute lower leg hot water immersion protects macrovascular dilator function following ischaemia-reperfusion injury in humans. Exp Physiol 105: 302–311, 2020. doi: 10.1113/EP088154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Engelland RE, Hemingway HW, Tomasco OG, Olivencia-Yurvati AH, Romero SA. Neural control of blood pressure is altered following isolated leg heating in aged humans. Am J Physiol Heart Circ Physiol 318: H976–H984, 2020. doi: 10.1152/ajpheart.00019.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Engin F, Hotamisligil GS. Restoring endoplasmic reticulum function by chemical chaperones: an emerging therapeutic approach for metabolic diseases. Diabetes Obes Metab 12, Suppl 2: 108–115, 2010. doi: 10.1111/j.1463-1326.2010.01282.x. [DOI] [PubMed] [Google Scholar]
- 19.Estébanez B, de Paz JA, Cuevas MJ, González-Gallego J. Endoplasmic reticulum unfolded protein response, aging and exercise: An update. Front Physiol 9: 1744, 2018. doi: 10.3389/fphys.2018.01744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harris RA, Nishiyama SK, Wray DW, Richardson RS. Ultrasound assessment of flow-mediated dilation. Hypertension 55: 1075–1085, 2010. doi: 10.1161/HYPERTENSIONAHA.110.150821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hotamisligil GS. Endoplasmic reticulum stress and the inflammatory basis of metabolic disease. Cell 140: 900–917, 2010. doi: 10.1016/j.cell.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jerome SN, Akimitsu T, Korthuis RJ. Leukocyte adhesion, edema, and development of postischemic capillary no-reflow. Am J Physiol 267: H1329–H1336, 1994. doi: 10.1152/ajpheart.1994.267.4.H1329. [DOI] [PubMed] [Google Scholar]
- 23.Jian L, Lu Y, Lu S, Lu C. Chemical chaperone 4-phenylbutyric acid reduces cardiac ischemia/reperfusion injury by alleviating endoplasmic reticulum stress and oxidative stress. Med Sci Monit 22: 5218–5227, 2016. doi: 10.12659/MSM.898623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kharbanda R, Kattenhorn M, Deanfield J, Klein N, Walton B, Vallance P, Peters M, Mullen M, Macallister R. Ischemic preconditioning prevents endothelial injury and immune cell activation during ischemia-reperfusion in humans in vivo. Circulation 102: 119–119, 2000. [DOI] [PubMed] [Google Scholar]
- 25.Lambert EA, Thomas CJ, Hemmes R, Eikelis N, Pathak A, Schlaich MP, Lambert GW. Sympathetic nervous response to ischemia-reperfusion injury in humans is altered with remote ischemic preconditioning. Am J Physiol Heart Circ Physiol 311: H364–H370, 2016. doi: 10.1152/ajpheart.00369.2016. [DOI] [PubMed] [Google Scholar]
- 26.Limberg JK, Casey DP, Trinity JD, Nicholson WT, Wray DW, Tschakovsky ME, Green DJ, Hellsten Y, Fadel PJ, Joyner MJ, Padilla J. Assessment of resistance vessel function in human skeletal muscle: guidelines for experimental design, Doppler ultrasound, and pharmacology. Am J Physiol Heart Circ Physiol 318: H301–H325, 2020. doi: 10.1152/ajpheart.00649.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Loukogeorgakis SP, Panagiotidou AT, Broadhead MW, Donald A, Deanfield JE, MacAllister RJ. Remote ischemic preconditioning provides early and late protection against endothelial ischemia-reperfusion injury in humans: role of the autonomic nervous system. J Am Coll Cardiol 46: 450–456, 2005. doi: 10.1016/j.jacc.2005.04.044. [DOI] [PubMed] [Google Scholar]
- 28.Lu Y, Cheng J, Chen L, Li C, Chen G, Gui L, Shen B, Zhang Q. Endoplasmic reticulum stress involved in high-fat diet and palmitic acid-induced vascular damages and fenofibrate intervention. Biochem Biophys Res Commun 458: 1–7, 2015. doi: 10.1016/j.bbrc.2014.12.123. [DOI] [PubMed] [Google Scholar]
- 29.Montie HL, Kayali F, Haezebrouck AJ, Rossi NF, Degracia DJ. Renal ischemia and reperfusion activates the eIF 2 alpha kinase PERK. Biochim Biophys Acta 1741: 314–324, 2005. doi: 10.1016/j.bbadis.2005.04.007. [DOI] [PubMed] [Google Scholar]
- 30.van den Munckhof I, Riksen N, Seeger JP, Schreuder TH, Borm GF, Eijsvogels TM, Hopman MT, Rongen GA, Thijssen DH. Aging attenuates the protective effect of ischemic preconditioning against endothelial ischemia-reperfusion injury in humans. Am J Physiol Heart Circ Physiol 304: H1727–H1732, 2013. doi: 10.1152/ajpheart.00054.2013. [DOI] [PubMed] [Google Scholar]
- 31.Pleiner J, Schaller G, Mittermayer F, Marsik C, MacAllister RJ, Kapiotis S, Ziegler S, Ferlitsch A, Wolzt M. Intra-arterial vitamin C prevents endothelial dysfunction caused by ischemia-reperfusion. Atherosclerosis 197: 383–391, 2008. doi: 10.1016/j.atherosclerosis.2007.06.011. [DOI] [PubMed] [Google Scholar]
- 32.Romero SA, Ely MR, Sieck DC, Luttrell MJ, Buck TM, Kono JM, Branscum AJ, Halliwill JR. Effect of antioxidants on histamine receptor activation and sustained postexercise vasodilatation in humans. Exp Physiol 100: 435–449, 2015. doi: 10.1113/EP085030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Romero SA, Gagnon D, Adams AN, Moralez G, Kouda K, Jaffery MF, Cramer MN, Crandall CG. Folic acid ingestion improves skeletal muscle blood flow during graded handgrip and plantar flexion exercise in aged humans. Am J Physiol Heart Circ Physiol 313: H658–H666, 2017. doi: 10.1152/ajpheart.00234.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Romero SA, Hocker AD, Mangum JE, Luttrell MJ, Turnbull DW, Struck AJ, Ely MR, Sieck DC, Dreyer HC, Halliwill JR. Evidence of a broad histamine footprint on the human exercise transcriptome. J Physiol 594: 5009–5023, 2016. doi: 10.1113/JP272177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seal JB, Gewertz BL. Vascular dysfunction in ischemia-reperfusion injury. Ann Vasc Surg 19: 572–584, 2005. doi: 10.1007/s10016-005-4616-7. [DOI] [PubMed] [Google Scholar]
- 36.Seeger JP, Lenting CJ, Schreuder TH, Landman TR, Cable NT, Hopman MT, Thijssen DH. Interval exercise, but not endurance exercise, prevents endothelial ischemia-reperfusion injury in healthy subjects. Am J Physiol Heart Circ Physiol 308: H351–H357, 2015. doi: 10.1152/ajpheart.00647.2014. [DOI] [PubMed] [Google Scholar]
- 37.Tampakakis E, Tabit CE, Holbrook M, Linder EA, Berk BD, Frame AA, Bretón-Romero R, Fetterman JL, Gokce N, Vita JA, Hamburg NM. Intravenous lipid infusion induces endoplasmic reticulum stress in endothelial cells and blood mononuclear cells of healthy adults. J Am Heart Assoc 5: 1–10, 2016. doi: 10.1161/JAHA.115.002574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thijssen DH, Black MA, Pyke KE, Padilla J, Atkinson G, Harris RA, Parker B, Widlansky ME, Tschakovsky ME, Green DJ. Assessment of flow-mediated dilation in humans: a methodological and physiological guideline. Am J Physiol Heart Circ Physiol 300: H2–H12, 2011. doi: 10.1152/ajpheart.00471.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vang S, Longley K, Steer CJ, Low WC. The unexpected uses of urso- and tauroursodeoxycholic acid in the treatment of non-liver diseases. Glob Adv Health Med 3: 58–69, 2014. doi: 10.7453/gahmj.2014.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vilatoba M, Eckstein C, Bilbao G, Smyth CA, Jenkins S, Thompson JA, Eckhoff DE, Contreras JL. Sodium 4-phenylbutyrate protects against liver ischemia reperfusion injury by inhibition of endoplasmic reticulum-stress mediated apoptosis. Surgery 138: 342–351, 2005. doi: 10.1016/j.surg.2005.04.019. [DOI] [PubMed] [Google Scholar]
- 41.Walsh LK, Restaino RM, Neuringer M, Manrique C, Padilla J. Administration of tauroursodeoxycholic acid prevents endothelial dysfunction caused by an oral glucose load. Clin Sci (Lond) 130: 1881–1888, 2016. doi: 10.1042/CS20160501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu XD, Zhang ZY, Sun S, Li YZ, Wang XR, Zhu XQ, Li WH, Liu XH. Hypoxic preconditioning protects microvascular endothelial cells against hypoxia/reoxygenation injury by attenuating endoplasmic reticulum stress. Apoptosis 18: 85–98, 2013. doi: 10.1007/s10495-012-0766-6. [DOI] [PubMed] [Google Scholar]
- 43.Yang Q, He GW, Underwood MJ, Yu CM. Cellular and molecular mechanisms of endothelial ischemia/reperfusion injury: perspectives and implications for postischemic myocardial protection. Am J Transl Res 8: 765–777, 2016. [PMC free article] [PubMed] [Google Scholar]
- 44.Yassin MM, Harkin DW, Barros D’Sa AA, Halliday MI, Rowlands BJ. Lower limb ischemia-reperfusion injury triggers a systemic inflammatory response and multiple organ dysfunction. World J Surg 26: 115–121, 2002. doi: 10.1007/s00268-001-0169-2. [DOI] [PubMed] [Google Scholar]
- 45.Zhang C, Tang Y, Li Y, Xie L, Zhuang W, Liu J, Gong J. Unfolded protein response plays a critical role in heart damage after myocardial ischemia/reperfusion in rats. PLoS One 12: e0179042, 2017. doi: 10.1371/journal.pone.0179042. [DOI] [PMC free article] [PubMed] [Google Scholar]