

Abstract
Marburg virus (MARV) causes severe disease in humans and is known to activate Nrf2, the major transcription factor of the antioxidant response. Canonical activation of Nrf2 involves oxidative or electrophilic stress that prevents Keap1 targeted degradation of Nrf2, leading to Nrf2 stabilization and activation of the antioxidant response. MARV activation of Nrf2 is non-canonical, with the MARV VP24 protein (mVP24) interacting with Keap1, freeing Nrf2 from degradation. A high throughput screening (HTS) assay was developed to identify inhibitors of mVP24-induced Nrf2 activity and used to screen more than 55,000 compounds. Hit compounds were further screened against secondary HTS assays for inhibition of antioxidant activity induced by additional canonical and non-canonical mechanisms. This pipeline identified 14 compounds that suppress the response, dependent on the inducer, with 50% inhibitory concentrations below 5 μM and selectivity index values greater than 10. Notably, several of the identified compounds specifically inhibit mVP24-induced Nrf2 activity.
Keywords: Nrf2, antioxidant response, Marburg virus, high-throughput screen
The Nuclear factor erythroid 2-related factor 2 (Nrf2) transcription factor is a major regulator of the cellular antioxidant response, inducing expression of genes involved in the establishment of a cytoprotective state1. In addition to cellular detoxification, the Nrf2 pathway also has important roles in disease, with either activation or inhibition contributing to cancer, inflammatory diseases, metabolic reprogramming and virus pathogenicity and replication2–12. Therefore, regulators of Nrf2 activity are of interest.
Under homeostatic conditions, Nrf2 is targeted for ubiquitination and proteasomal degradation by Kelch-like ECH-associated protein 1 (Keap1), a cellular substrate adaptor protein of the Cul3/Rbx1 ubiquitin E3 ligase complex (reviewed in10). During canonical activation, reactive oxygen species lead to conformational changes in Keap1, preventing the targeted degradation of Nrf2. Newly synthesized Nrf2 accumulates, translocates to the nucleus and leads to the upregulation of genes with promoters containing antioxidant response elements (ARE). ARE genes include phase II detoxifying enzymes and regulators of redox reactions, such as NADPH quinone oxidoreductase 1 (NQO1) and heme-oxygenase 1 (HO-1). Additional, non-canonical, mechanisms of Nrf2 activation have been described. These include somatic mutations of Nrf2; mutations of Keap1 in cancer cells that cause constitutive activation of the pathway; and the interaction of Keap1 with other proteins, such as the autophagy adaptor protein p62, that compete with Nrf2 for Keap1 binding thereby releasing Nrf2 from degradation13–15.
Several viruses have been demonstrated to activate the Nrf2 antioxidant pathway indirectly, via the induction of oxidative stress or other signaling pathways, with both pro- and antiviral effects demonstrated3, 4, 6–8, 11. Notably, Marburg virus (MARV) directly activates the antioxidant response through the interaction of MARV VP24 protein (mVP24) with Keap1, which competes for Nrf2 binding5, 9. Although the impact of Nrf2 activation on MARV-infected cells has yet to be clarified, Nrf2-deficient mice are better able to control MARV infection as compared to wildtype mice9. While wildtype mice succumbed to infection by day 8, mice lacking Nrf2 demonstrated 50% survival as well as lower viral titers on day 4 post infection and complete clearance of virus in surviving mice by day 9. This suggests a role by which Nrf2 contributes to MARV pathogenesis, although the full extent of the contribution is not yet understood9.
Given the variety of disease etiologies linked to Nrf2 activation, inhibitors of the antioxidant pathway are of significant interest. In this study, we initially screened compounds for inhibition of ARE activity induced non-canonically by mVP24 expression. Inhibition by the resulting hits was then further assessed with counterscreening assays, where ARE responses were induced by tert-butyl hydroquinone (tBHQ) treatment, p62 over-expression or Nrf2 overexpression. The data identify compounds that inhibit antioxidant gene expression in an inducer-dependent manner, including several compounds that specifically suppress mVP24-induced Nrf2 activity.
Results
Development of high-throughput screen to identify inhibitors of mVP24-induced ARE activity
We developed and optimized a high-throughput screening (HTS) assay in 384-well format based on the capacity of mVP24 expression to stimulate antioxidant response element (ARE) activity (Figure 1A)5. A loss of mVP24-induced ARE activity is expected to result from compounds that disrupt the mVP24:Keap1 interaction, or compounds that inhibit the Nrf2 antioxidant pathway downstream of mVP24. HEK293T cells stably expressing an ARE firefly luciferase reporter and either GFP alone (ARE/GFP), as a negative control, or GFP and mVP24 (ARE/mVP24) were generated. Stable expression of mVP24 was demonstrated over at least eight passages of the reporter cell line (Figure S1). The expression of mVP24 results in constitutive activation of the Nrf2-dependent ARE pathway, allowing for luciferase activity to be assessed at any time point (data not shown). For use as an HTS assay, we chose to assess luciferase activity at 24h post-compound addition. The assay is robust, with a Z-factor consistently greater than 0.5, a signal to background ratio (S/B) of ~80 and a coefficient of variation (CV) less than 6%, with no effect of DMSO concentrations at 0.25% (Figure 1B).
Figure 1. Development of a high-throughput screen (HTS) for identifying inhibitors of mVP24-induced antioxidant response.
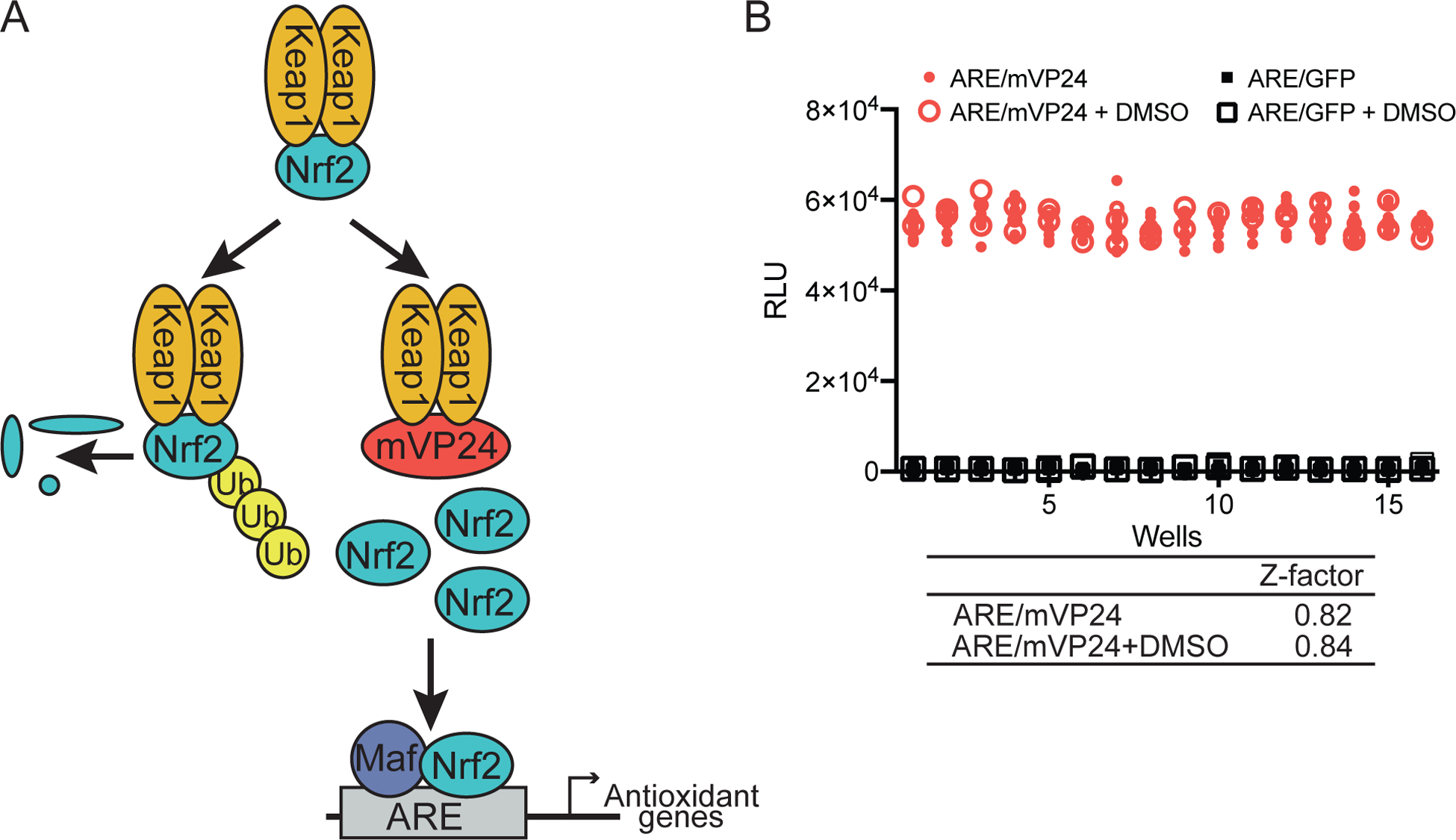
(A) Under homeostatic conditions, Keap1 targets Nrf2 for poly-ubiquitination and proteasomal degradation. mVP24 competes with Nrf2 for interaction with Keap1, allowing Nrf2 to accumulate, translocate to the nucleus and upregulate antioxidant target genes. (B) A quality control plate was used to assess the robustness of the HTS assay, comparing ARE/mVP24 cells to ARE/GFP cells with or without DMSO. Z-factor values were calculated using the formula Z-factor = 1 − [(3σc+ + 3σc−)/(|μc+ − μc−|)]. ARE/mVP24, red circles, 144 wells; ARE/mVP24 + 0.25% DMSO, red open circles, 48 wells; ARE/GFP; black squares, 144 wells; ARE/GFP + 0.25% DMSO, black open squares, 48 wells.
Development of secondary screening assays to assess compound specificity
To allow the specificity of compound inhibitory activities to be assessed, we developed several secondary screens. First, to ensure the detected activity was not due to inhibition of firefly luciferase, we generated an assay in 384-well format using cells transfected with either a plasmid encoding constitutively expressed firefly luciferase (ce-FF) or, as a negative control, empty vector (pCAGGS) (Figure 2A). Activity was robust, with DMSO at 0.25% having a limited effect. Secondly, to assess the breadth of inhibition of the antioxidant response, we generated three assays where Nrf2-dependent responses were stimulated at different levels of the pathway (Figure 2B–E). tBHQ is a chemical activator of Nrf2 that acts through modification of cysteine residues on Keap1, leading to the stabilization of Nrf216. Treatment of the stable ARE/GFP cell line with 10 μM tBHQ was sufficient to induce ARE luciferase activity over the untreated ARE/GFP cells, with a S/B of 19 (Figure 2C). Addition of DMSO at 0.25% resulted in a slight decrease of tBHQ-induced ARE firefly luciferase activity, however, the S/B remained greater than 15. Host protein p62, an autophagy adapter protein, interacts with a site on Keap1 that overlaps with that of the mVP24 interaction with Keap19, 14, 17. p62 is a known non-canonical activator of the Nrf2 antioxidant response that acts in an analogous manner to mVP24, competing with the interaction of Nrf2 for Keap1 leading to the stabilization of Nrf214. As expected, expression of p62 activates the ARE luciferase reporter, with minimal effects of DMSO at 0.25% and a S/B greater than 10 (Figure 2D). Finally, over-expression of Nrf2 itself bypasses the negative regulation by endogenous Keap1, activating the ARE reporter without the need for additional inducers and resulting in a S/B greater than 250 in the presence or absence of DMSO (Figure 2E). Each of these secondary assays was robust, with Z-factor values greater than 0.5 and S/B greater than 10, indicating their capacity to be used for primary high-throughput screening as well as in a confirmatory manner (Figure 2A–E).
Figure 2. Development of secondary screening assays.
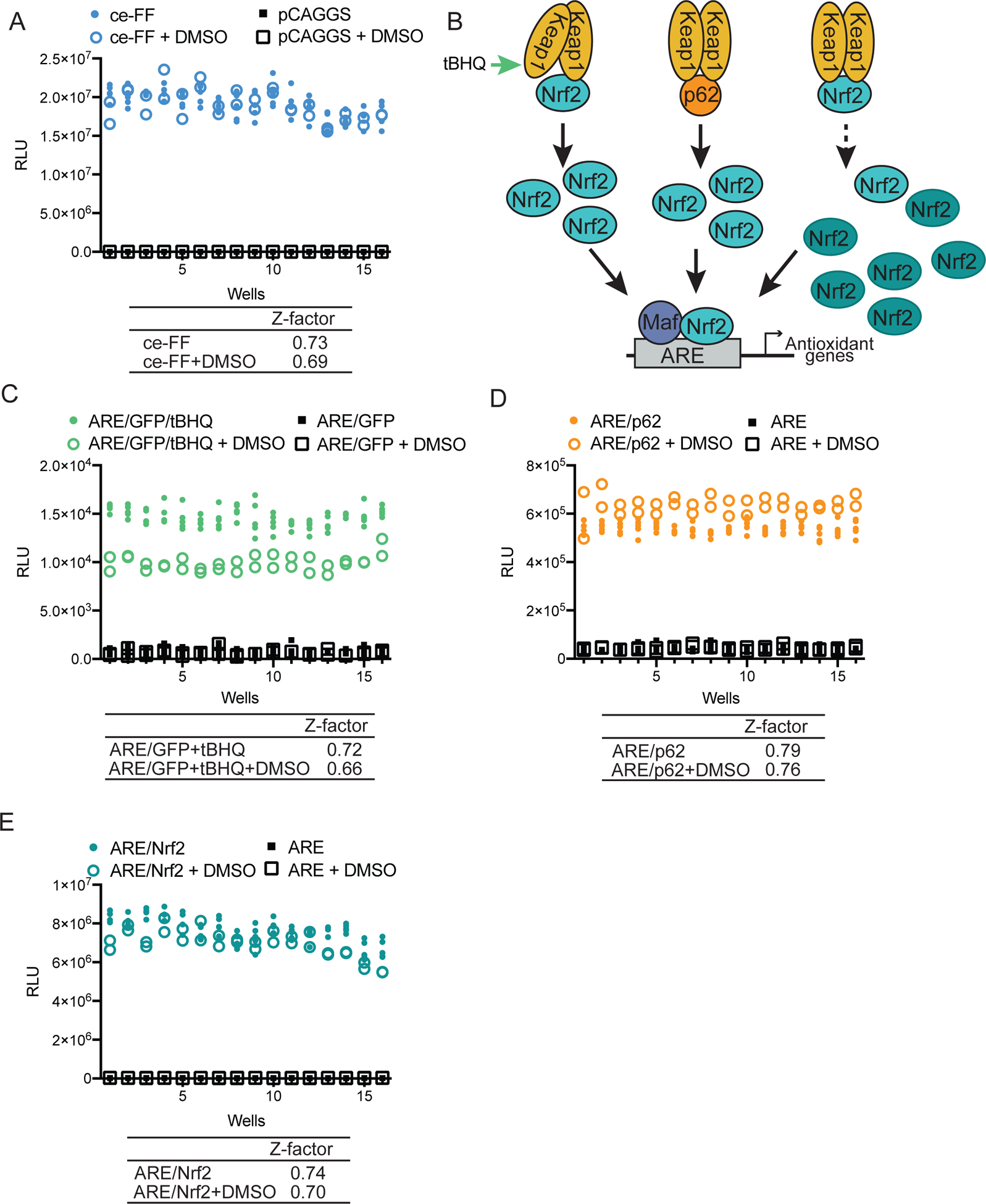
(A) HEK293T cells transfected with a constitutively expressing firefly reporter, pCAGGS firefly (ce-FF), or empty vector (pCAGGS) were plated in a 384-well plate and were treated with 0.25% DMSO as indicated. Twenty-four hours post treatment, firefly luciferase activity was assessed. ce-FF, blue circles, 80 wells; ce-FF + DMSO, blue open circles, 32 wells; pCAGGS, black squares, 80 wells; pCAGGS + DMSO, black open squares, 32 wells. (B) Schematic diagram illustrating different mechanisms by which the Nrf2 antioxidant pathway can be activated: tBHQ treatment (green arrow), p62 transfection, or Nrf2 overexpression (dark teal, Nrf2 labeled ovals). (C) ARE/GFP HEK293T cells were plated in a 384-well plate and were treated with10 μM tert-butylhydroquinone (tBHQ) and 0.25% DMSO as indicated. Twenty-four hours post-treatment, firefly luciferase activity was assessed. ARE/GFP/tBHQ, green circles, 80 wells; ARE/GFP/tBHQ + DMSO, green open circles, 32 wells; ARE/GFP, black squares, 80 wells; ARE/GFP + DMSO, black open squares, 32 wells. (D) HEK293T cells transfected with a plasmid encoding an ARE firefly reporter and pCAGGS encoding HA-tagged p62 as indicated, were plated in a 384-well plate and were treated with 0.25% DMSO as indicated. Twenty-four hours post treatment, firefly luciferase activity was assessed. ARE/p62, orange circles, 80 wells; ARE/p62 + DMSO, orange open circles, 32 wells; ARE, black squares, 80 wells; ARE + DMSO, black open squares, 32 wells. (E) HEK293T cells transfected with a plasmid encoding an ARE firefly reporter and pCAGGS encoding Flag-tagged Nrf2 as indicated, were plated in a 384-well plate and were treated with 0.25% DMSO as indicated. Twenty-four hours post treatment, firefly luciferase activity was assessed. ARE/Nrf2, teal circles, 80 wells; ARE/Nrf2 + DMSO, teal open circles, 32 wells; ARE, black squares, 80 wells; ARE + DMSO, black open squares, 32 wells. (A, C–E) Z-factor values were calculated using the formula Z-factor = 1 − [(3σc+ + 3σc−)/(|μc+ − μc−|)] comparing in each subpanel the indicated cell line in colored circles to that in black squares, with or without DMSO.
High-throughput screen to identify inhibitors
We screened a total of 55,108 compounds from libraries of known bioactive compounds and other discrete small molecule compounds to identify inhibitors of mVP24-induced ARE activity (Figure 3). 428 compounds were identified as initial hits (Figure 3). Hit compounds identified from the Microbiotix Library were screened to exclude pan-assay interference compounds (PAINS) and historical data was used to discard promiscuous hits active in more than two unrelated screens. Remaining hit compounds were then subjected to confirmatory dose response assays and candidates were removed when inhibitory activity mirrored cytotoxicity, leaving 46 compounds.
Figure 3. Schematic of screening campaign.
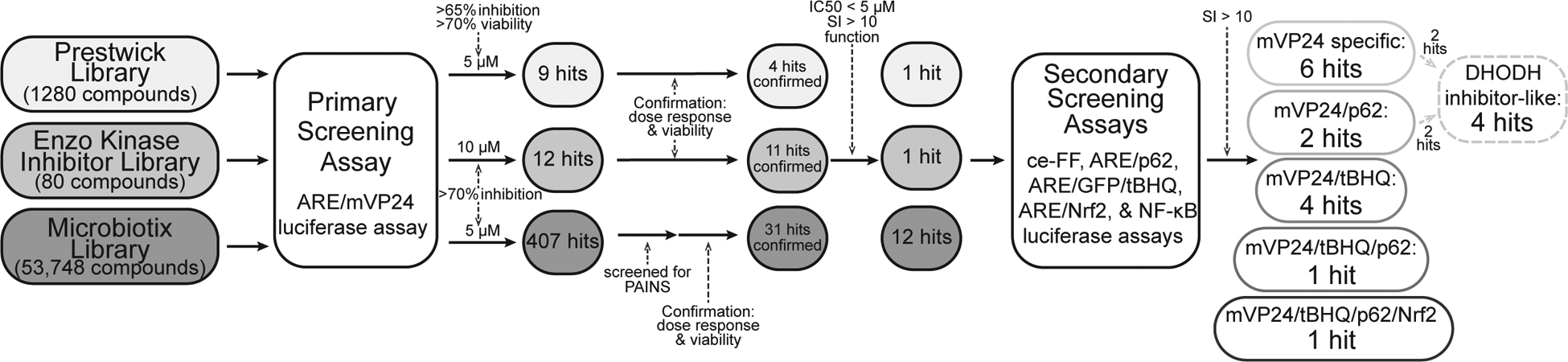
55,108 compounds were screened against the ARE/mVP24 luciferase assay. Hit compounds were screened for pan-assay interference (PAINS) and confirmed through dose response and viability assays. Compounds with a 50% inhibitory concentration (IC50) < 5 μM and a selectivity index (SI) > 10 were moved forward and activity was assessed against the secondary assays; ce-FF (constitutively expressed firefly), ARE/p62, ARE/GFP/tBHQ, ARE/Nrf2 and NF-κB luciferase assays. Compounds were considered to demonstrate specific inhibitory activity with an SI > 10.
To further focus the list of hit compounds, we advanced candidates that had 50% inhibitory concentration (IC50) values less than 5 μM and a selectivity index (SI; 50% cytotoxicity concentration (CC50)/IC50) greater than 10 in the primary mVP24 screen (Figure 3, Table 1). Among these, we identified three compounds with a shared function, podophyllotoxin, colchicine and nocodazole, which all destabilize microtubules through interaction with tubulin18–20. We therefore chose to proceed with one representative microtubule destabilizer, podophyllotoxin.
Table 1.
Activity of hit compounds across primary and secondary screens
| Compound | CC50 (μM) | ARE/mVP24 | ARE/GFP/tBHQ | ARE/p62 | ARE/Nrf2 | Ce-FF | NF-κB | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IC50 (μM) | SI | IC50 (μM) | SI | IC50 (μM) | SI | IC50 (μM) | SI | IC50 (μM) | SI | IC50 (μM) | SI | ||
| 5-lodotubercidin | >50 | 0.44 | >114 | 0.20 | >250 | 0.058 | >862 | 1.2 | >41.7 | 15 | >3.33 | 6.5 | >7.69 |
| Podophyllotoxin | >50 | 0.050 | >1000 | 0.026 | >1923 | 3.1 | >16.1 | >50 | >50 | >50 | |||
| F0015–0327 | >50 | 0.61 | >82.0 | 0.33 | >152 | 12 | >4.17 | >50 | >50 | >50 | |||
| F2336–0041 | >50 | 1.5 | >33.3 | 0.71 | >70.4 | >50 | >50 | >50 | >50 | ||||
| F5001–2941 | >50 | 2.5 | >20.0 | 2.7 | >18.5 | >50 | >50 | >50 | >50 | ||||
| F5540–0057 | >50 | 2.5 | >20.0 | 1.4 | >35.7 | >50 | >50 | >50 | 18 | >2.78 | |||
| J107–0140 | 19 | 1.0 | 19 | 7.3 | 2.60 | 1.9 | 10.0 | >50 | >50 | >50 | |||
| J107–0307 | >50 | 0.35 | >143 | 16 | >3.13 | 0.97 | >51.5 | >50 | >50 | >50 | |||
| J107–0137 | >50 | 2.1 | >23.8 | >50 | >50 | >50 | >50 | >50 | |||||
| J107–0181 | 24 | 2.2 | 10.9 | 18 | 1.33 | 5.6 | 4.29 | >50 | >50 | >50 | |||
| C798–0517 | 24 | 2.0 | 12.0 | 49 | 0.490 | 48 | 0.500 | >50 | >50 | >50 | |||
| E975–1448 | >50 | 1.6 | >31.3 | 17 | >2.94 | >50 | >50 | >50 | >50 | ||||
| F2468–0184 | >50 | 3.8 | >13.2 | >50 | >50 | >50 | >50 | >50 | |||||
| F2468–0196 | >50 | 2.4 | >20.8 | >50 | >50 | >50 | >50 | >50 | |||||
The fourteen compounds remaining after this selection process were subjected to secondary screening assays to assess specificity (Figure 3 and Table 1). We also tested the compounds against a NF-κB-dependent firefly luciferase assay activated by TNFα as an additional control for non-specific inhibition of an inducible luciferase reporter (Table 1). Through this set of secondary assays, we identified compounds that demonstrated inhibition against various combinations of inducers of Nrf2-dependent antioxidant responses.
Compounds that inhibit ARE gene expression induced by multiple activators
5-iodotubercidin, an adenosine kinase inhibitor, was identified in our screens as an inhibitor of the four tested inducers of ARE reporter activity (Table 1). Inhibition of mVP24-induced ARE activity occurred with an IC50 of 0.44 μM, while inhibition of activity induced by tBHQ (IC50 0.20 μM), over-expression of p62 (IC50 0.058 μM) and Nrf2 (IC50 1.2 μM) was also demonstrated (Table 1 and Figure 4A). Additionally, 5-iodotubercidin demonstrated inhibition of the NF-κB reporter assay, with an IC50 of 6.5 μM, which is approximately 15-fold higher than the IC50 value for mVP24-induced ARE activity. Therefore, although 5-iodotubercidin inhibition is not limited to the Nrf2-dependent ARE pathway, greater specificity is demonstrated in these assays for the ARE pathway than that of NF-κB (Table 1).
Figure 4. Inhibitors of multiple levels of ARE promoter activity.
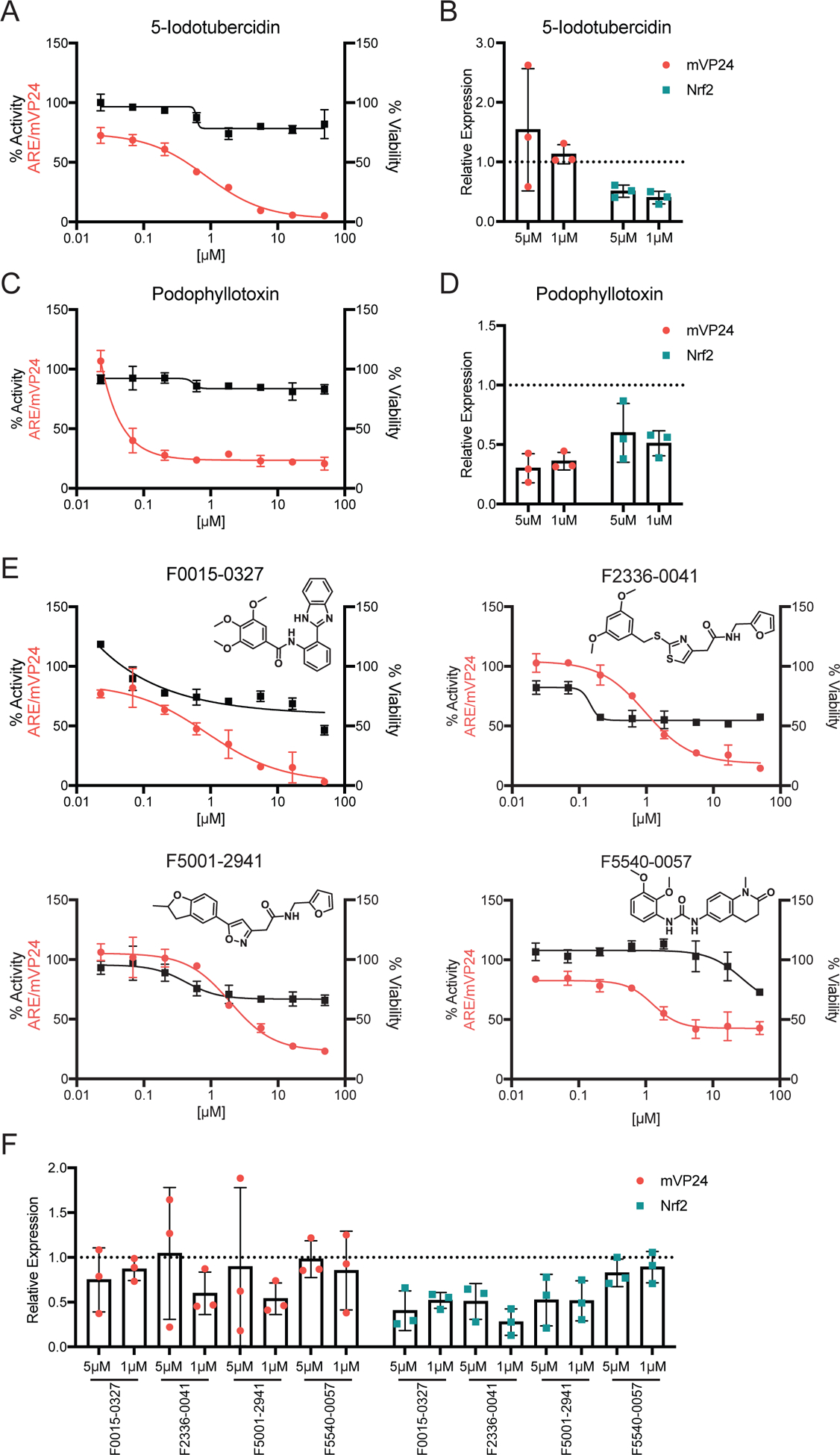
(A) ARE/mVP24 HEK293T cells were distributed in a 384-well plate and treated with increasing concentrations of 5-iodotubercidin in triplicate. Twenty-four hours post-treatment firefly luciferase activity was assessed (left axis, red circles). In parallel, HEK293T cells were plated in a 384-well plate and treated in triplicate with increasing concentrations of compounds. Twenty-four hours post-treatment, ATP content was assessed to determine viability (right axis, black squares). Error bars represent the standard deviation. (B) Expression of Flag-tagged mVP24 (red circles) and endogenous Nrf2 (teal squares) expression determined relative to DMSO control for ARE/mVP24 HEK293T cells treated with 5-iodotubercidin at 5 and 1 μM for 24h. The dotted line represents the DMSO control, individual values for each of the triplicates are indicated and the error bars represent the standard deviation. (C) ARE/mVP24 HEK293T cells were treated with podophyllotoxin and analyzed as in (A). (D) Relative expression of Flag-tagged mVP24 and Nrf2 determined relative to the DMSO control. (E) ARE/mVP24 HEK293T cells were treated with the indicated compounds and analyzed, as in (A). Structures of the compounds are indicated. (F) ARE/mVP24 HEK293T cells were treated with the indicated compounds and analyzed as in (B).
A possible mechanism through which inhibition of the antioxidant response can occur is through reduction of mVP24 or Nrf2 expression. To assess if the inhibition is mediated through effects on mVP24 or Nrf2 expression, we treated mVP24/ARE HEK293T cells with the compound at 1 and 5 μM for twenty-four hours, after which expression levels of endogenous Nrf2 and Flag-tagged mVP24 were determined by western blot analysis. Interestingly, the expression of Nrf2 was reduced in the presence of 5-iodotubercidin, relative to the DMSO control, while mVP24 expression was unaffected (Figure 4B and Figure S2). This result suggests that the decrease of Nrf2 protein levels may contribute to the inhibition detected.
Podophyllotoxin, the representative microtubule destabilizer, was a potent inhibitor of mVP24-induced ARE activity, with an IC50 of 0.050 μM, although the inhibitory activity plateaus at approximately 80% inhibition around 1 μM of compound (Figure 4C). Potent activity was also demonstrated against tBHQ-induced ARE activity (IC50 0.026 μM), while inhibition of p62-induced ARE activity was less robust (IC50 3.1 μM) (Table 1). Podophyllotoxin did not inhibit ARE activity following over-expression of Nrf2, suggesting it does not block the function of free Nrf2 or that the inhibitory effect can be overwhelmed by exogenous Nrf2 (Table 1). Notably, expression of both mVP24 and endogenous Nrf2 was affected by treatment at 5 and 1 μM, relative to the DMSO control, suggesting that the decrease in levels of these two proteins may contribute to the inhibition of ARE activity by podophyllotoxin (Figure 4D and Figure S2).
Four additional compounds, F0015–0327, F2336–0041, F5001–2941 and F5540–0057, demonstrated inhibition against both mVP24 and tBHQ-induced ARE activity, with IC50 values between 0.33 and 2.7 μM (Figure 4E and Table 1). Assessment of mVP24 expression following treatment of the mVP24/ARE HEK293T cells with the compounds at 1 and 5 μM for twenty-four hours demonstrated minimal effects on mVP24 expression levels by F0015–0327 and F5540–0057, relative to the DMSO control (Figure 4F and Figure S2). Interestingly, F2336–0041 and F5001–2941 decreased mVP24 expression following treatment at 1 μM, but not 5 μM. Nrf2 expression was decreased with treatment by F0015–0327, F2336–0041 and F5001–2941, while F5540–0057 had no effect (Figure 4F and Figure S2). Together, these data suggest that the inhibition of the antioxidant response by F2336–0041 and F5001–2941 may involve the regulation of mVP24 and Nrf2 expression, F0015–0327 through the regulation of Nrf2 expression and F5540–0057 through a mechanism independent of mVP24 and Nrf2 expression.
DHODH-like inhibitors of mVP24-induced ARE activity
Four of the hit compounds, J107–0140, J107–0307, J107–0137 and J107–0181, shared a similar chemical scaffold (Figure 5A). Two of these compounds, J107–0140 and J107–0307, inhibited both mVP24 and p62-induced ARE reporter activity with IC50 values below 2.0 μM (Table 1). The remaining two compounds inhibited only mVP24-induced ARE activity; J107–0137 (IC50 2.1 μM) and J108–0181 (IC50 2.2 μM). Notably, the similarity in structures was also shared with GSK983, a compound known to inhibit the enzyme dihydroorotate dehydrogenase (DHODH) (Figure 5B)21. In accordance with this shared chemical scaffold, GSK983 also demonstrated inhibition of mVP24-induced ARE activity with an IC50 of 0.010 μM (Figure 5B). DHODH is required for de novo pyrimidine synthesis and inhibition leads to depletion of pyrimidines and loss of cellular proliferation21. Inhibitors of DHODH are immunosuppressive and are reported to have broad spectrum antiviral activity through the inhibition of viral RNA synthesis and induction of innate immune gene expression independent of viral infection22–24. Supplementation with exogenous pyrimidine ribonucleosides uridine or cytidine can reverse inhibition of RNA virus replication, while supplementation with deoxycytidine can reverse the cytostatic effects of DHODH inhibitors, relieving repression on DNA replication but not RNA synthesis. However, the connection between inhibition of DHODH and suppression of ARE responses is less clear. Therefore, we assessed the ability of uridine or deoxycytidine to reverse inhibition of mVP24-induced ARE activity in the presence of the DHODH-like inhibitor compounds, J107–0140 and J107–0307, and GSK983. Strikingly, uridine, but not deoxycytidine, was able to restore ARE luciferase activity in the presence of the three compounds, suggesting DHODH de novo synthesis of pyrimidines is required for mVP24-induced ARE responses (Figure 5B).
Figure 5. DHODH-like inhibitors of mVP24-induced ARE promoter activity.
(A) ARE/mVP24 HEK293T cells were distributed in a 384-well plate and treated with increasing concentrations of the indicated compounds in triplicate. Twenty-four hours post-treatment firefly luciferase activity was assessed (left axis, red circles). In parallel, HEK293T cells were plated in a 384-well plate and treated in triplicate with increasing concentrations of compounds. Twenty-four hours post-treatment, ATP content was assessed to determine viability (right axis, black squares). Error bars represent the standard deviation. Structures of compounds are indicated. (B) ARE/mVP24 HEK293T cells were plated in a 384-well plate and treated with increasing concentrations of compound and 1 mM of uridine or deoxycytidine as indicated. Twenty-four hours post-treatment, firefly luciferase activity was assessed. Error bars represent the standard deviation. (C) Expression of Flag-tagged mVP24 (red circles) and endogenous Nrf2 (teal squares) expression determined relative to DMSO control for ARE/mVP24 HEK293T cells treated with the indicated compounds at 5 and 1 μM for 24h. The dotted line represents the DMSO control, error bars represent the standard deviation and individual values for each of the triplicate are indicated. (D) Immunoprecipitation of Flag-tagged mVP24 in cells also expressing HA-tagged Keap1, 24 hours post-treatment with 5 μM of the indicated compounds. Western blots were performed for Flag and HA. (E) HEK293T cells were transfected with plasmids encoding an ARE firefly luciferase reporter, a constitutively expressed Renilla luciferase and the indicated mVP24 and p53 expression plasmids. Twenty-four hours post-transfection, luciferase activity was assessed. Expression of Flag-tagged mVP24 and p53 was determined by western blot. Error bars represent the standard deviation. (F) ARE/mVP24 HEK293T cells were transfected with a scramble siRNA (scr) or one of two p53 targeted siRNAs (p53#1, p53#2) and were treated with compounds at indicated concentrations. Luciferase activity was assessed for the samples in triplicate twenty-four hours post treatment and siRNA knockdown was confirmed by western blot for p53. Statistical significance was assessed using unpaired t test; *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Error bars represent the standard deviation.
To assess if the DHODH-like inhibitor compounds reduce mVP24 or Nrf2 expression, we determined the protein levels of endogenous Nrf2 and Flag-tagged mVP24 following 24h treatment of mVP24/ARE HEK293T cells (Figure 5C). Relative to the DMSO control, mVP24 expression levels were either unaffected or increased in the presence of the five compounds (Figure 5C and Figure S3). Minimal effects on Nrf2 expression levels were detected for the DHODH inhibitor-like compounds, while GSK983 decreased Nrf2 expression (Figure 5C and Figure S3). These data suggest that all DHODH inhibitor-like compounds function independently of effects on mVP24 and Nrf2 expression levels. In contrast, modest reduction of Nrf2 protein levels may contribute to the inhibitory activity of GSK983.
Notably, J107–0137 and J107–0181 exhibited inhibition specific to mVP24-induced Nrf2 activity (Table 1). To determine if this specificity was mediated by loss of the mVP24:Keap1 interaction, a co-immunoprecipitation assay in the presence of 5 μM of indicated compound was used (Figure 5D). Disruption of the mVP24:Keap1 interaction was not demonstrated by the DHODH-like or known inhibitors, suggesting that the inhibition of ARE activity by these compounds is not dependent upon loss of mVP24 interaction with Keap1.
Interestingly, DHODH inhibitors have been shown to increase p53 synthesis, which has separately been demonstrated to suppress Nrf2-dependent ARE responses25–27. To test if p53 can inhibit mVP24-induced ARE activity we co-expressed the ARE luciferase reporter, mVP24 and increasing concentrations of p53 in HEK293T cells. A significant decrease in reporter activity was detected even at the lowest concentration of p53, indicating that p53 does inhibit mVP24-induced ARE activity (Figure 5E). To assess the role of p53 expression on the capacity of DHODH and DHODH-like inhibitors to suppress mVP24 activated ARE responses we knocked down p53 expression using two different siRNAs. Both siRNAs efficiently reduced p53 levels in the mVP24/ARE HEK293T cells with no effect on mVP24 expression (Figure 5F). Loss of p53 led to an increase in mVP24-induced ARE activity over that of the scramble control siRNA, in both the mock treated sample and those treated with J107–0307 or GSK983. However, loss of p53 did not result in a reversal of inhibition by the compounds, suggesting that although p53 has a role in inhibiting ARE responses induced by mVP24, it does not have a role in the suppression mediated by DHODH inhibitors.
Inhibitors specific to mVP24-induced ARE activity
Six compounds demonstrated inhibition that was specific to mVP24-induced ARE activity (Table 1). Two of these were the DHODH inhibitor-like compounds J107–0137 and J107–0181 (Figure 5A). The remaining four compounds, C798–0517, E975–1448, F2468–0184 and F2468–0196, demonstrated IC50 values between 1.6 and 3.8 μM (Table 1 and Figure 6A). Using a co-immunoprecipitation assay we determined that treatment of HEK293T cells with each of these compounds at 5 μM does not result in disruption of mVP24 interaction with Keap1 (Figure 6B).
Figure 6. Inhibitors specific to mVP24-induced ARE promoter activity.
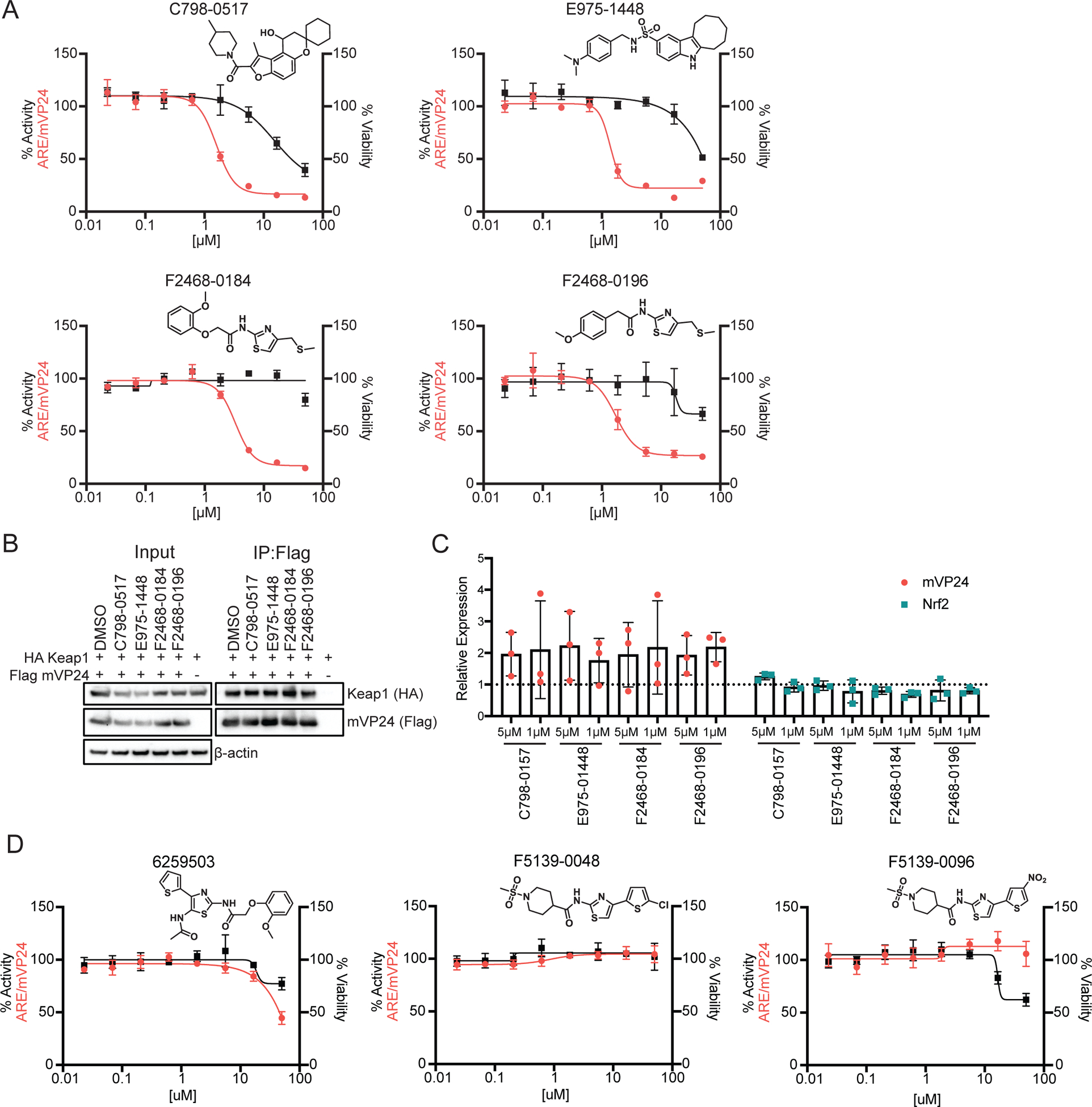
(A) ARE/mVP24 HEK293T cells were plated in a 384-well plate and treated with increasing concentrations of the indicated compounds in triplicate. Twenty-four hours post-treatment firefly luciferase activity was assessed (left axis, red circles). In parallel, HEK293T cells were plated in a 384-well plate and treated in triplicate with increasing concentrations of compounds. Twenty-four hours post-treatment, ATP content was assessed to determine viability (right axis, black squares). Error bars represent the standard deviation. Structures of compounds are indicated. (B) Immunoprecipitation of Flag-tagged mVP24 in cells also expressing HA-tagged Keap1, 24 hours post-treatment with 5 μM of the indicated compounds. Western blots were performed for Flag and HA. (C) Expression of Flag-tagged mVP24 (red circles) and endogenous Nrf2 (teal squares) expression determined relative to DMSO control for ARE/mVP24 HEK293T cells treated with the indicated compounds at 5 and 1 μM for 24h. The dotted line represents the DMSO control, error bars represent the standard deviation and individual values for each of the triplicate are indicated. (D) ARE/mVP24 HEK293T cells were plated and analyzed as in (A) with the indicated compounds.
We further assessed these compounds for effects on mVP24 or Nrf2 expression, following treatment of the ARE/mVP24 HEK293T cell line at 5 and 1 μM for 24h (Figure 6C). Interestingly, all compounds appeared to increase mVP24 expression levels relative to the DMSO control, while minimal effects on Nrf2 levels were detected (Figure 6C and Figure S4). This suggests that although these compounds demonstrate inhibitory specificity towards mVP24-induced responses, this effect is not mediated through inhibition of mVP24 expression.
Interestingly, F2468–0184 and F2468–0196 share a core structure (Figure 6A). Three additional compounds with similar structures were present in the initial screen but did not demonstrate any suppression of mVP24-induced ARE responses (Figure 6D). We completed dose response assays and cytotoxicity curves with compounds 6259503, F5139–0048 and F5139–0096, confirming the lack of inhibition (Figure 6D). These results suggest that modifications and relocations of the methoxy phenyl are tolerated and that large substituents on the thiazole, such as a thiophene, render the compounds inactive.
Discussion
Dysregulation of the Nrf2-dependent antioxidant response plays a major role in several diseases, making it an attractive target for therapeutics. In this study, using both canonical and non-canonical activators of the Nrf2 pathway, we identified several novel small molecule inhibitors of antioxidant gene expression. These distinct patterns of inhibition suggest additional levels of regulation of the antioxidant response that are dependent upon the inducer.
Our HTS assays identified the adenosine kinase inhibitor 5-iodotubercidin as having a broad spectrum of inhibitory activity reaching across both canonical and non-canonical activators of the antioxidant response. Identification of 5-iodotubercidin provides proof-of-concept for the ability of our screening assays to identify inhibitors of antioxidant activity as this kinase inhibitor has previously been used to block Nrf2 activity28. Inhibition of the NF-κB reporter was also demonstrated by 5-iodotubercidin, although the IC50 for inhibition of NF-κB was 5–100-fold greater than those for suppressing ARE activation, suggesting greater specificity for inhibition of antioxidant response gene expression than NF-κB in these assays.
Three microtubule destabilizing compounds, podophyllotoxin, colchicine and nocodazole, were identified as inhibitors of mVP24-induced antioxidant activity. Podophyllotoxin was further shown to inhibit antioxidant activity induced by p62 expression and tBHQ treatment, but not over-expression of Nrf2. Of note, podophyllotoxin is considered to be a cytotoxic compound29. However, this was not detected in our cytotoxicity assays, nor did we see inhibition of luciferase activity across all secondary screens, indicating a lack of general toxicity by podophyllotoxin under the conditions of our assays. The small step down in the podophyllotoxin cytotoxicity curve may suggest possible cytostatic activity and it is possible that treatment for longer than 24 hours would increase the detected cytotoxicity. While further studies are required to determine the mechanism of inhibition, these results suggest that podophyllotoxin exerts its inhibitory activity at a point in the activation process that occurs prior to accumulation of free Nrf2. This suggests that microtubule dynamics may play a role in regulating activation of the Nrf2 pathway. Additionally, a reduction in mVP24 and Nrf2 expression levels were detected, indicating that regulation of inducer expression may contribute to inhibition.
Activation of the antioxidant response by mVP24 is through a non-canonical mechanism, while activation by tBHQ can be considered canonical, as it leads to modifications of cysteine residues in Keap1 that prevent Nrf2 degradation16. Therefore, it is interesting that four compounds were identified that inhibit antioxidant responses induced by both mVP24 and tBHQ, but not p62, another non-canonical activator. A decrease in mVP24 expression following treatment by F2336–0041 and F5001–2941 could contribute to the inhibition detected with these compounds following mVP24-induced Nrf2 activity. Additionally, these two compounds and F0015–0327 decrease endogenous Nrf2 expression, suggesting this could contribute to the inhibition of both mVP24 and tBHQ-induced responses. F5540–0057 had minimal effects on mVP24 and Nrf2 expression levels, suggesting a mechanism of action unrelated to their expression levels. However, it is important to note that while F5540–0057 reached an IC50 value, inhibitory activity against mVP24-induced responses plateaued soon after, indicating incomplete inhibition of the antioxidant response. The reason for the lack of corresponding inhibition of p62-induced antioxidant responses by these compounds is not clear. If the decrease in Nrf2 expression following treatment by three of these compounds contributes to inhibition of mVP24 and tBHQ-induced responses, it could be expected to also reduce responses induced by p62. As it does not, this may suggest that the inhibitory activity of these compounds is mediated through a regulatory step in the antioxidant pathway that affects mVP24 and tBHQ-induced responses but does not affect p62-induced responses.
Interestingly, our screen identified compounds with structural similarity to the DHODH inhibitor GSK983, two compounds that have activity against antioxidant responses induced by mVP24 and p62, both non-canonical activators of the Nrf2 response, and two compounds with specificity for inhibition of mVP24-induced activity. We further showed that this ARE inhibitory activity extended to GSK983. Notably, supplementation of the mVP24-induced Nrf2 activity assay with the pyrimidine analog uridine, but not the deoxyribonucleoside deoxycytidine, reversed inhibition of the antioxidant response. This suggests that de novo pyrimidine synthesis is required for antioxidant gene expression. Previous studies have shown that p53 synthesis is upregulated following inhibition of DHODH and, separately, that p53 can inhibit Nrf2 responses25–27. We found that over-expression of p53 can also inhibit mVP24-induced antioxidant responses, however, loss of p53 did not reverse inhibition by GSK983 and the DHODH-like inhibitor compounds. Therefore, while p53 can have a role in modulating Nrf2 activity induced by mVP24, this does not extend to regulating the inhibitory activity of these compounds. Together, these data indicate that DHODH and DHODH-like inhibitor compounds block mVP24-induced activation of antioxidant responses through a mechanism that requires de novo pyrimidine synthesis, although further study is required to understand this role.
Non-canonical activation of the antioxidant pathway occurs through the competitive interaction of proteins with Keap1 that results in loss of interaction with Nrf2. These non-canonical activating proteins include the host protein p62 and the viral protein mVP245, 9, 14, 15. Despite Nrf2 and p62 binding to overlapping sites in the Kelch domain of Keap1, the compound K67 was demonstrated to specifically disrupt the interaction of phosphorylated p62 with Keap1, while the interaction of Nrf2:Keap1 remained intact30. This facilitated Nrf2 degradation and inhibition of the pathway. mVP24 and Nrf2 also bind to an overlapping site in the Kelch domain31. The specificity of K67 for disruption of the p62 interaction suggests the potential for identification of specific inhibitors of mVP24 interaction with Keap1, while maintaining the Nrf2:Keap1 interaction. Given this possibility, we used co-immunoprecipitation assays to assess the capacity for mVP24 to interact with Keap1 in the presence of those inhibitors specific to mVP24-induced antioxidant activity. However, no disruption of interaction was detected. Furthermore, mVP24 expression was enhanced in the presence of compounds specific for inhibition of mVP24-induced antioxidant responses. These data indicate that the mechanism of action of the inhibitors specific to mVP24-induced antioxidant responses does not involve disrupting the interaction of mVP24 with Keap1 or decreasing mVP24 expression.
Mice lacking Nrf2 demonstrate better control of the virus, suggesting the possibility that activation of Nrf2 by mVP24 may promote virulence9. This effect implies that therapeutic inhibition of Nrf2 activity could be used to control MARV infection. However, it is not yet fully understood how Nrf2 contributes to MARV replication either in vitro or in vivo. For these reasons, small molecule inhibitors of mVP24 activity that can serve as molecular probes would be useful to clarify the role of mVP24 activation of Nrf2. Initial studies in Huh7 and THP-1 cells with infectious MARV demonstrated minimal effects on viral titer with several of the identified compounds (data not shown). Further studies will be required to fully determine the contribution of Nrf2 activation by mVP24 to MARV infection.
In summary, we screened more than 55,000 compounds for inhibition of mVP24-induced antioxidant activity. These compounds were further tested for activity against antioxidant gene expression induced by the canonical activator, tBHQ, the non-canonical activator, p62, and over-expression of Nrf2. The identified compounds exhibited inducer-specific inhibitory activity, suggesting the presence of distinct mechanisms of regulation of the Nrf2-dependent antioxidant response. Together, these assays will allow for further screening and the potential identification of additional inducer-specific inhibitors. Such compounds should be useful to clarify not only the role of mVP24 activation of Nrf2 but also mechanisms that regulate the Keap1-Nrf2-antioxidant pathway.
Material and Methods
Plasmids and lentiviruses
pCAGGS Flag-tagged Nrf2, pcDNA/TO HA-tagged p62 and the NF-κB firefly luciferase reporter were previously described5, 17. The pGL4.37[luc2P/ARE/Hygro] (ARE) reporter plasmid was purchased from Promega. The constitutively expressed firefly (ce-FF) plasmid used contains the firefly luciferase open reading frame cloned into the plasmid pCAGGS. pcDNA Flag-tagged p53 was purchased from Addgene (#10838). Replication-defective lentiviruses were generated as previously described32. Lentiviruses expressing GFP alone were generated from a lentiviral vector derived from pHR-SIN-CSIGW, which encodes for GFP under an internal ribosome entry site (IRES). Flag-tagged mVP24 was cloned into this lentiviral vector and was used to generate lentiviruses expressing both GFP and mVP24.
Cells
HEK293T cells were purchased from ATCC (CRL-3216). To generate stable HEK293T ARE/mVP24 and ARE/GFP cell lines, HEK293T cells were first transfected with a plasmid encoding the antioxidant response element (ARE) firefly luciferase reporter (pGL4.37[luc2P/ARE/Hygro] (Promega)) using Lipofectamine 2000 (Invitrogen). Two days post-transfection, hygromycin was used to select for cells that stably integrated the plasmid. Clonal cell populations were screened for luciferase activity in the presence of 10 μM tert-butyl hydroquinone (tBHQ) and a single ARE clone was selected for optimal signal to background ratio. This clone was then transduced with either control or mVP24-expressing lentiviruses. As the lentiviruses express GFP under the control of an IRES, GFP was used as a selection marker for cell sorting to generate clonal populations. ARE/GFP control cell lines were screened for optimal ARE luciferase activity following tBHQ treatment; ARE/mVP24 cell lines were screened for mVP24 expression and optimal mVP24-induced ARE luciferase activity. A single clonal population of each cell line was selected for further use. HEK293T cells and stable ARE/mVP24 and ARE/GFP HEK293T cells were maintained in Dulbecco’s minimal essential medium supplemented with 10% fetal bovine serum (FBS) and cultured at 37°C and 5% CO2.
Chemicals
The chemical libraries used for the screens were the SCREEN-WELL Kinase Inhibitor library (Enzo Life Sciences), the Prestwick Chemical library (Prestwick Chemical), and a subset of Microbiotix libraries of >200,000 discrete compounds previously used in high-throughput screening for inhibitors of anti-infective targets33–39. GSK983, podophyllotoxin, colchicine, nocodazole, 5-iodotubercidin, tert-butylhydroquinone (tBHQ), uridine, deoxycytidine and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich. F0015–0327, F2336–0041, F5540–0057, F5001–2941, C798–0517, E975–1448, F2468–0184, F2468–0196, 6259503, F5139–0048, F5139–0096, J107–0137, J107–0140, J107–0181 and J107–0307 were purchased from Life Chemicals Inc. All compounds were diluted to a concentration of 20 mM in DMSO before use. tBHQ was diluted in ethanol to a concentration of 50 mM. Uridine and deoxycytidine were diluted to 10 mM in distilled water. TNFα was purchased from PeproTech.
mVP24-ARE Screen
Kinase Inhibitor Library:
ARE/mVP24 and ARE/GFP (negative control) HEK293T cells (2 × 104 cells/40 μL/well) were distributed in 384-well plates using a MultiDrop Combi reagent dispenser (Thermo Scientific). Plates were centrifuged for five minutes at 1000 rpm, after which cells were allowed to rest for one hour before compounds were transferred in triplicate onto ARE/mVP24 cells (final concentration: 10 μM). Twenty-four hours post-compound addition, neolite (PerkinElmer) substrate was added and luciferase activity was read using an EnVision plate reader (PerkinElmer). Z-factor values were calculated using the equation Z-factor = 1 − [(3σc+ + 3σc−)/(|μc+ − μc−|)]; the positive control was ARE/mVP24 cells, and the negative control was ARE/GFP cells.
Prestwick Chemical Library:
Cells were plated as in the Kinase Inhibitor Library screen. Compounds were transferred in duplicate using a high-density pin tool (V&P Scientific) attached to the pipetting head of a Nimbus liquid handler (20 nL/well, final concentration: 5 μM). Twenty-four hours post-compound addition, luciferase activity was read using neolite and a H1 synergy plate reader (BioTek). Z-factor values were calculated as in the Kinase Inhibitor Library.
Microbiotix Libraries:
ARE/mVP24 and ARE/GFP HEK293T cells were plated as described above. Compounds were transferred in quadruplicate using a pintool (SciClone) (100 nL/well, final concentration: 5 μM). Plates were developed after a 24 hour-incubation with compounds and analyzed as in the Kinase Inhibitor Library.
For all libraries, the half-maximal inhibitory concentrations (IC50) of selected compounds were determined. ARE/mVP24 HEK293T cells (2 × 104 cells/40 μL/well) were distributed in 384-well plates using a MultiDrop Combi reagent dispenser (Thermo Scientific) and compounds were added in triplicate (0–50 μM, 3-fold dilution series). Twenty-four hours post-treatment, luciferase activity was determined as above. IC50 values were calculated with Prism 8 (GraphPad) using a four-parameter, nonlinear regression analysis.
Secondary Screens
Constitutively expressed firefly luciferase (ce-FF):
HEK293T cells (7.5 × 106) were transfected in a T75 flask with 3 μg of a plasmid encoding constitutively expressed firefly luciferase (ce-FF) or pCAGGS empty vector. Twenty-four hours post-transfection cells were trypsinized and resuspended in DMEM with 10% FBS and were distributed in 384-well plates (2 × 104 cells/40 μL/well). Twenty-four hours post-plating firefly luciferase activity was assessed as above. Z-factor values were calculated as above; the positive control was ce-FF transfected cells, and the negative control was pCAGGS empty vector transfected cells.
ARE/p62 and ARE/Nrf2:
HEK293T cells (7.5 × 106) were transfected in a T75 flask with 3 μg of a plasmid encoding ARE firefly luciferase reporter alone or in combination with 3 μg of HA-tagged p62, a non-canonical activator of the Nrf2 antioxidant response, or Flag-tagged Nrf2 expression plasmids, to induce ARE firefly luciferase gene expression. The assay was carried out as above for ce-FF. Z-factor values were calculated using ARE/p62 or ARE/Nrf2 transfected cells as the positive control and ARE firefly luciferase reporter transfected cells as the negative control.
ARE/GFP/tBHQ:
ARE/GFP HEK293T cells were plated in 384-well format (2 × 104 cells/40 μL/well) and one-hour post-plating cells were treated with 10 μM tBHQ. Twenty-four hours post treatment luciferase activity was assessed as above. Z-factor values were calculated using ARE/GFP cells treated with tBHQ as the positive control and untreated ARE/GFP cells as the negative control.
NF-κB:
HEK293T cells (7.5 × 106) were transfected in a T75 flask with 3 μg of a plasmid encoding NF-κB firefly luciferase reporter. Twenty-four hours post transfection, cells were distributed in a 384-well plate (2 × 104/40 uL/well) and the assay was carried out as above for ce-FF with the exception that at six hours prior to assessing luciferase activity, cells were treated with TNFα (10 ng/ml).
For all secondary screens, the half-maximal inhibitory concentrations (IC50) of selected compounds were determined. Cells were manipulated as above for each screen and were plated in 384-well plates using a MultiDrop Combi reagent dispenser (Thermo Scientific) and compounds were added in triplicate (0–50 μM, 3-fold dilution series). Twenty-four hours post-treatment, luciferase activity was assessed as above and IC50 values were calculated with Prism 8 using a four-parameter, nonlinear regression analysis.
Determining Half-Maximal Cell Cytotoxicity Concentration (CC50)
HEK293T cells (1 × 103 cells/40 μL/well) were plated in 384-well plates and were centrifuged at 1000 rpm for five minutes, after which they were allowed to rest for one hour. Compounds were then added as indicated (0–50 μM, 3-fold dilution series) in triplicate. Twenty-four hours post-treatment, CellTiter-Glo (Promega) was added, and ATP content was determined by reading luminescence using an EnVision plate reader. CC50 values were calculated with Prism 8 using a four-parameter, nonlinear regression analysis.
Pyrimidine Supplementation Assay
ARE/mVP24 HEK293T cells (2 × 104 cells/40 μL/well) were plated in 384-well plates using a MultiDrop Combi reagent dispenser (Thermo Scientific). Indicated compounds were added in triplicate (0–50 μM, 3-fold dilution series) in the presence of 1 mM of uridine or deoxycytidine, as indicated. Luciferase activity was read at twenty-four hours post-treatment as above.
p53 ARE Luciferase Assay
HEK293T cells (1 × 105/well) were transfected with 30 ng of a plasmid encoding ARE firefly luciferase reporter, 30ng of a plasmid encoding constitutively expressed Renilla luciferase reporter (pRLTK), 200 ng of pCAGGS encoding Flag-tagged mVP24 and increasing concentrations of pcDNA encoding Flag-tagged p53 (25–200 ng), using Lipofectamine 2000 (Thermo Fisher Scientific). Twenty-four hours post-transfection, luciferase activity was assessed using a dual luciferase assay (Promega) and read on an EnVision plate reader. Firefly luciferase values were normalized to Renilla luciferase values. The assay was performed in triplicate; error bars indicate the standard deviation for the triplicate.
siRNA ARE Luciferase Assay
mVP24/ARE HEK293T cells (7 × 105) were transfected with 1uM of siRNA using Lipofectamine RNAiMAX (Thermo Fisher Scientific); either one of two against p53 (#1 A-003329–22 and #2 A-003329–23 (Dharmacon)) or a scrambled siRNA control (D-001810–01 (Dharmacon)). Twenty-four hours post-transfection cells were trypsinized and replated in a 96-well plate (1 × 105/well). Cells were allowed to rest for one hour after which they were treated with DMSO, J107–0307 or GSK983 at the indicated concentrations. Twenty-four hours post-treatment, luciferase activity was assessed and analyzed as under the ARE Luciferase Assay. Cell lysates were subjected to western blot analysis for Flag-tagged mVP24 and endogenous p53 expression.
Co-immunoprecipitation Assay
HEK293T cells (1 × 106) were transfected with 2 μg of pCAGGS encoding HA-tagged Keap1 and Flag-tagged Nrf2, as indicated, using Lipofectamine 2000 (Thermo Scientific). At 24 hours post-transfection, cells were treated with 5 μM of the indicated compounds for an additional 24 hours. Following treatment, cells were lysed in NP-40 lysis buffer (50 mM Tris [pH 7.5], 280 mM NaCl, 0.5% Nonidet P-40, 0.2 mM EDTA, 2 mM EGTA, 10% glycerol, protease inhibitor (cOmplete; Roche)). Anti-FLAG M2 magnetic beads (Sigma-Aldrich) were incubated with lysates for one hour at 4°C, washed five times in NP-40 lysis buffer, and eluted using 3X FLAG peptide (Sigma-Aldrich) at 4°C for 30 minutes. Input and co-precipitation samples were analyzed by western blot.
Western Blots
Lysates were run on 10% Bis-Tris Plus polyacrylamide gels (Thermo Fisher) and transferred to PVDF membrane (Bio-Rad). Membranes were probed with the indicated antibodies and were developed by Western Lightning Plus ECL (Perkin Elmer) and imaged on a ChemiDoc MP Imaging System (Bio-Rad).
Relative Expression Determination
ARE/mVP24 HEK293T cells (3 × 105) were treated with the indicated compounds at 5 μM and 1 μM for 24 hours. Cells were lysed in NP-40 lysis buffer and samples were analyzed by western blot. Relative expression was determined by normalizing expression of the indicated protein to its β-actin loading control and expressing the value relative to the expression level of the DMSO treated control. The assay was repeated in triplicate.
Antibodies
Rabbit and mouse anti-Flag, rabbit anti-HA and mouse anti-β-tubulin antibodies were purchased from Sigma-Aldrich. Rabbit anti-β-actin and mouse anti-p53 antibodies were purchased from Cell Signaling. Rabbit anti-Nrf2 antibody was purchased from Abcam.
Quantification and Statistical Analysis
Statistical analysis was performed using GraphPad Prism 8 with significance determined by an unpaired t test. Statistical details can be found in the figure legends. Data points were considered significantly different if the p-value was < 0.05. Quantification of protein expression on western blots was performed using Image Lab Software V5.2.1 to determine mean intensity of each protein band (Bio-Rad).
Supplementary Material
Acknowledgements.
Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases of the National Institutes of Health under Award Numbers R01AI114654, R56AI127835 and R01AI123926 to CFB and GKA; U19109664 to AB, CFB, GKA, TLB and DTM; and R43AI138739 to TLB, DTM and CFB.
References
- 1.Baird L; Dinkova-Kostova AT, The cytoprotective role of the Keap1-Nrf2 pathway. Arch Toxicol 2011, 85 (4), 241–72. [DOI] [PubMed] [Google Scholar]
- 2.Ahmed SM; Luo L; Namani A; Wang XJ; Tang X, Nrf2 signaling pathway: Pivotal roles in inflammation. Biochim Biophys Acta Mol Basis Dis 2017, 1863 (2), 585–597. [DOI] [PubMed] [Google Scholar]
- 3.Burdette D; Olivarez M; Waris G, Activation of transcription factor Nrf2 by hepatitis C virus induces the cell-survival pathway. J Gen Virol 2010, 91 (Pt 3), 681–90. [DOI] [PubMed] [Google Scholar]
- 4.Cho HY; Imani F; Miller-DeGraff L; Walters D; Melendi GA; Yamamoto M; Polack FP; Kleeberger SR, Antiviral activity of Nrf2 in a murine model of respiratory syncytial virus disease. Am J Respir Crit Care Med 2009, 179 (2), 138–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Edwards MR; Johnson B; Mire CE; Xu W; Shabman RS; Speller LN; Leung DW; Geisbert TW; Amarasinghe GK; Basler CF, The Marburg virus VP24 protein interacts with Keap1 to activate the cytoprotective antioxidant response pathway. Cell Rep 2014, 6 (6), 1017–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ivanov AV; Smirnova OA; Ivanova ON; Masalova OV; Kochetkov SN; Isaguliants MG, Hepatitis C virus proteins activate NRF2/ARE pathway by distinct ROS-dependent and independent mechanisms in HUH7 cells. PLoS One 2011, 6 (9), e24957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kesic MJ; Simmons SO; Bauer R; Jaspers I, Nrf2 expression modifies influenza A entry and replication in nasal epithelial cells. Free Radic Biol Med 2011, 51 (2), 444–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee J; Koh K; Kim YE; Ahn JH; Kim S, Upregulation of Nrf2 expression by human cytomegalovirus infection protects host cells from oxidative stress. J Gen Virol 2013, 94 (Pt 7), 1658–68. [DOI] [PubMed] [Google Scholar]
- 9.Page A; Volchkova VA; Reid SP; Mateo M; Bagnaud-Baule A; Nemirov K; Shurtleff AC; Lawrence P; Reynard O; Ottmann M; Lotteau V; Biswal SS; Thimmulappa RK; Bavari S; Volchkov VE, Marburgvirus hijacks nrf2-dependent pathway by targeting nrf2-negative regulator keap1. Cell Rep 2014, 6 (6), 1026–1036. [DOI] [PubMed] [Google Scholar]
- 10.Rojo de la Vega M; Chapman E; Zhang DD, NRF2 and the Hallmarks of Cancer. Cancer Cell 2018, 34 (1), 21–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schaedler S; Krause J; Himmelsbach K; Carvajal-Yepes M; Lieder F; Klingel K; Nassal M; Weiss TS; Werner S; Hildt E, Hepatitis B virus induces expression of antioxidant response element-regulated genes by activation of Nrf2. J Biol Chem 2010, 285 (52), 41074–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wu S; Lu H; Bai Y, Nrf2 in cancers: A double-edged sword. Cancer Med 2019, 8 (5), 2252–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kerins MJ; Ooi A, A catalogue of somatic NRF2 gain-of-function mutations in cancer. Sci Rep 2018, 8 (1), 12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Komatsu M; Kurokawa H; Waguri S; Taguchi K; Kobayashi A; Ichimura Y; Sou YS; Ueno I; Sakamoto A; Tong KI; Kim M; Nishito Y; Iemura S; Natsume T; Ueno T; Kominami E; Motohashi H; Tanaka K; Yamamoto M, The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 2010, 12 (3), 213–23. [DOI] [PubMed] [Google Scholar]
- 15.Lau A; Wang XJ; Zhao F; Villeneuve NF; Wu T; Jiang T; Sun Z; White E; Zhang DD, A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol 2010, 30 (13), 3275–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Abiko Y; Miura T; Phuc BH; Shinkai Y; Kumagai Y, Participation of covalent modification of Keap1 in the activation of Nrf2 by tert-butylbenzoquinone, an electrophilic metabolite of butylated hydroxyanisole. Toxicol Appl Pharmacol 2011, 255 (1), 32–9. [DOI] [PubMed] [Google Scholar]
- 17.Edwards MR; Basler CF, Marburg Virus VP24 Protein Relieves Suppression of the NF-kappaB Pathway Through Interaction With Kelch-like ECH-Associated Protein 1. J Infect Dis 2015, 212 Suppl 2, S154–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schilstra MJ; Martin SR; Bayley PM, The effect of podophyllotoxin on microtubule dynamics. J Biol Chem 1989, 264 (15), 8827–34. [PubMed] [Google Scholar]
- 19.Skoufias DA; Wilson L, Mechanism of inhibition of microtubule polymerization by colchicine: inhibitory potencies of unliganded colchicine and tubulin-colchicine complexes. Biochemistry 1992, 31 (3), 738–46. [DOI] [PubMed] [Google Scholar]
- 20.Vasquez RJ; Howell B; Yvon AM; Wadsworth P; Cassimeris L, Nanomolar concentrations of nocodazole alter microtubule dynamic instability in vivo and in vitro. Mol Biol Cell 1997, 8 (6), 973–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deans RM; Morgens DW; Okesli A; Pillay S; Horlbeck MA; Kampmann M; Gilbert LA; Li A; Mateo R; Smith M; Glenn JS; Carette JE; Khosla C; Bassik MC, Parallel shRNA and CRISPR-Cas9 screens enable antiviral drug target identification. Nat Chem Biol 2016, 12 (5), 361–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hoffmann HH; Kunz A; Simon VA; Palese P; Shaw ML, Broad-spectrum antiviral that interferes with de novo pyrimidine biosynthesis. Proc Natl Acad Sci U S A 2011, 108 (14), 5777–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Luthra P; Naidoo J; Pietzsch CA; De S; Khadka S; Anantpadma M; Williams CG; Edwards MR; Davey RA; Bukreyev A; Ready JM; Basler CF, Inhibiting pyrimidine biosynthesis impairs Ebola virus replication through depletion of nucleoside pools and activation of innate immune responses. Antiviral Res 2018, 158, 288–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cramer DV; Chapman FA; Jaffee BD; Jones EA; Knoop M; Hreha-Eiras G; Makowka L, The effect of a new immunosuppressive drug, brequinar sodium, on heart, liver, and kidney allograft rejection in the rat. Transplantation 1992, 53 (2), 303–8. [DOI] [PubMed] [Google Scholar]
- 25.Faraonio R; Vergara P; Di Marzo D; Pierantoni MG; Napolitano M; Russo T; Cimino F, p53 suppresses the Nrf2-dependent transcription of antioxidant response genes. J Biol Chem 2006, 281 (52), 39776–84. [DOI] [PubMed] [Google Scholar]
- 26.Chen W; Jiang T; Wang H; Tao S; Lau A; Fang D; Zhang DD, Does Nrf2 contribute to p53-mediated control of cell survival and death? Antioxid Redox Signal 2012, 17 (12), 1670–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ladds M; van Leeuwen IMM; Drummond CJ; Chu S; Healy AR; Popova G; Pastor Fernandez A; Mollick T; Darekar S; Sedimbi SK; Nekulova M; Sachweh MCC; Campbell J; Higgins M; Tuck C; Popa M; Safont MM; Gelebart P; Fandalyuk Z; Thompson AM; Svensson R; Gustavsson AL; Johansson L; Farnegardh K; Yngve U; Saleh A; Haraldsson M; D’Hollander ACA; Franco M; Zhao Y; Hakansson M; Walse B; Larsson K; Peat EM; Pelechano V; Lunec J; Vojtesek B; Carmena M; Earnshaw WC; McCarthy AR; Westwood NJ; Arsenian-Henriksson M; Lane DP; Bhatia R; McCormack E; Lain S, A DHODH inhibitor increases p53 synthesis and enhances tumor cell killing by p53 degradation blockage. Nat Commun 2018, 9 (1), 1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sid B; Glorieux C; Valenzuela M; Rommelaere G; Najimi M; Dejeans N; Renard P; Verrax J; Calderon PB, AICAR induces Nrf2 activation by an AMPK-independent mechanism in hepatocarcinoma cells. Biochem Pharmacol 2014, 91 (2), 168–80. [DOI] [PubMed] [Google Scholar]
- 29.Magedov IV; Manpadi M; Rozhkova E; Przheval’skii NM; Rogelj S; Shors ST; Steelant WF; Van slambrouck S; Kornienko A, Structural simplification of bioactive natural products with multicomponent synthesis: dihydropyridopyrazole analogues of podophyllotoxin. Bioorg Med Chem Lett 2007, 17 (5), 1381–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Saito T; Ichimura Y; Taguchi K; Suzuki T; Mizushima T; Takagi K; Hirose Y; Nagahashi M; Iso T; Fukutomi T; Ohishi M; Endo K; Uemura T; Nishito Y; Okuda S; Obata M; Kouno T; Imamura R; Tada Y; Obata R; Yasuda D; Takahashi K; Fujimura T; Pi J; Lee MS; Ueno T; Ohe T; Mashino T; Wakai T; Kojima H; Okabe T; Nagano T; Motohashi H; Waguri S; Soga T; Yamamoto M; Tanaka K; Komatsu M, p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming. Nat Commun 2016, 7, 12030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Johnson B; Li J; Adhikari J; Edwards MR; Zhang H; Schwarz T; Leung DW; Basler CF; Gross ML; Amarasinghe GK, Dimerization Controls Marburg Virus VP24-dependent Modulation of Host Antioxidative Stress Responses. J Mol Biol 2016, 428 (17), 3483–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yen B; Mulder LC; Martinez O; Basler CF, Molecular basis for ebolavirus VP35 suppression of human dendritic cell maturation. J Virol 2014, 88 (21), 12500–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aiello D; Barnes MH; Biswas EE; Biswas SB; Gu S; Williams JD; Bowlin TL; Moir DT, Discovery, characterization and comparison of inhibitors of Bacillus anthracis and Staphylococcus aureus replicative DNA helicases. Bioorg Med Chem 2009, 17 (13), 4466–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Aiello D; Williams JD; Majgier-Baranowska H; Patel I; Peet NP; Huang J; Lory S; Bowlin TL; Moir DT, Discovery and characterization of inhibitors of Pseudomonas aeruginosa type III secretion. Antimicrob Agents Chemother 2010, 54 (5), 1988–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Basu A; Antanasijevic A; Wang M; Li B; Mills DM; Ames JA; Nash PJ; Williams JD; Peet NP; Moir DT; Prichard MN; Keith KA; Barnard DL; Caffrey M; Rong L; Bowlin TL, New small molecule entry inhibitors targeting hemagglutinin-mediated influenza a virus fusion. J Virol 2014, 88 (3), 1447–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Basu A; Li B; Mills DM; Panchal RG; Cardinale SC; Butler MM; Peet NP; Majgier-Baranowska H; Williams JD; Patel I; Moir DT; Bavari S; Ray R; Farzan MR; Rong L; Bowlin TL, Identification of a small-molecule entry inhibitor for filoviruses. J Virol 2011, 85 (7), 3106–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Moir DT; Di M; Wong E; Moore RA; Schweizer HP; Woods DE; Bowlin TL, Development and application of a cellular, gain-of-signal, bioluminescent reporter screen for inhibitors of type II secretion in Pseudomonas aeruginosa and Burkholderia pseudomallei. J Biomol Screen 2011, 16 (7), 694–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Opperman TJ; Kwasny SM; Williams JD; Khan AR; Peet NP; Moir DT; Bowlin TL, Aryl rhodanines specifically inhibit staphylococcal and enterococcal biofilm formation. Antimicrob Agents Chemother 2009, 53 (10), 4357–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wallace J; Bowlin NO; Mills DM; Saenkham P; Kwasny SM; Opperman TJ; Williams JD; Rock CO; Bowlin TL; Moir DT, Discovery of bacterial fatty acid synthase type II inhibitors using a novel cellular bioluminescent reporter assay. Antimicrob Agents Chemother 2015, 59 (9), 5775–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.