

SUMMARY
Phagocytes reallocate metabolic resources to kill engulfed pathogens, but the intracellular signals that rapidly switch the immunometabolic program necessary to fuel microbial killing are not understood. We report that macrophages use a fast two-step Ca2+ relay to meet the bioenergetic demands of phagosomal killing. Upon detection of a fungal pathogen, macrophages rapidly elevate cytosolic Ca2+ (phase 1), and by concurrently activating the mitochondrial Ca2+ (mCa2+) uniporter (MCU), they trigger a rapid influx of Ca2+ into the mitochondria (phase 2). mCa2+ signaling reprograms mitochondrial metabolism, at least in part, through the activation of pyruvate dehydrogenase (PDH). Deprived of mCa2+ signaling, Mcu−/− macrophages are deficient in phagosomal reactive oxygen species (ROS) production and defective at killing fungi. Mice lacking MCU in their myeloid cells are highly susceptible to disseminated candidiasis. In essence, this study reveals an elegant design principle that MCU-dependent Ca2+ signaling is an electrometabolic switch to fuel phagosome killing.
Graphical Abstract

In Brief
The signaling mechanisms that rapidly reallocate metabolic resources to meet the bioenergetic demands of microbial killing are not understood. Seegren et al. show that mitochondrial Ca2+ signaling serves as a fast electrometabolic switch to fuel microbial killing by phagocytes. This study identifies the mitochondrial Ca2+ channel MCU as a critical component of cell-intrinsic antimicrobial responses.
INTRODUCTION
Sentinel cells such as macrophages and dendritic cells detect, engulf, and digest invading pathogens. A precisely choreographed cell-intrinsic immune response marshals a befitting adaptive immune response through the secretion of inflammatory mediators and presentation of antigenic peptides to T cells (Wang et al., 2019). Efficient killing of the engulfed pathogen by macrophages is therefore a transformative cell-intrinsic defense process in host defense. During phagosome maturation, endosomal fusion events recruit the vacuolar ATPase (V-ATPase) and NADPH oxidase (NOX) complex to the nascent phagosome (Kinchen and Ravichandran, 2008). The V-ATPase complex hydrolyzes ATP to pump protons into the phagosomal lumen, acidifying it and promoting the digestion of pathogens by acid-optimized proteases, lipases, and endonucleases. The NOX complexes oxidize NADPH to inject super oxide (O2·) into the phagosome (Nauseef, 2019), and this oxidative pressure is crucial for the destruction of fungal pathogens. Sustained NOX activity is essential for fungal defense, and patients with mutations in NOX present with frequent fungal infections (Davies et al., 2019; Gazendam et al., 2014). The overall process of phagosomal killing therefore requires a bioenergetic burst, and macrophages are adept at rapidly reprogramming their metabolism to fuel phagosomal killing (Buck et al., 2017). At the same time, within the deadly environment of the phagosome, pathogens fight for survival by adapting their own transcriptional and metabolic programs that subvert the cell-intrinsic defenses and evade destruction (Brown, 2011; Jiménez-López and Lorenz, 2013). Our understanding of the metabolic changes that fuel the antimicrobial machineries in sentinel cells such as macrophages and dendritic is mostly limited to transcriptional changes occurring 24 h after pathogen recognition (O’Neill et al., 2016; Palsson-McDermott et al., 2015). While these responses are critical for inflammation and resolution (Mills et al., 2018), they do not account for the immediate and early bioenergetic demands of pathogen destruction. Our study identifies a fast two-step Ca2+ relay culminating in mitochondrial Ca2+ (mCa2+) signaling that is required for pathogen killing in macrophages.
In screening for ion channels that regulate the cell-intrinsic immunity, we made a serendipitous discovery that the mCa2+ uniporter (MCU) plays a crucial role in the killing of C. albicans. MCU is a Ca2+-selective ion channel located in the inner membrane of mitochondria (Chaudhuri et al., 2013; De Stefani et al., 2011; Kirichok et al., 2004). In conjunction with its regulatory subunits MICU1 and MICU2, the MCU complex can mediate a rapid influx of cytosolic Ca2+ into the mitochondrial matrix (Kamer and Mootha, 2014; Mallilankaraman et al., 2012; Patron et al., 2014). The inner mitochondrial membrane has a resting membrane potential between −160 mV and −200 mV relative to the cytosol, and the enormous driving force for cationic influx into the mitochondrial matrix can be leveraged to convert cytosolic Ca2+ signaling into rapid metabolic responses. We report that in response to a fungal pathogen, macrophages rapidly elevate cytosolic Ca2+, and by concurrently gating open the MCU (Kamer and Mootha, 2015), they trigger a near instantaneous influx of Ca2+ into the electrically charged mitochondria. This fast two-step Ca2+ relay reprograms the mitochondrial metabolism, at least in part, by activating pyruvate dehydrogenase (PDH), a key enzyme in the tricarboxylic acid (TCA) cycle.
Pyruvate is the end product of glycolysis that lies at the vital intersection of cellular metabolic network. Since its catalytic fate can govern the flux of multiple pathways, it is a keystone metabolite for metabolic reprogramming. When transported into the mitochondrial matrix, pyruvate can be directed either toward gluconeogenesis by pyruvate decarboxylase or toward the TCA cycle by the PDH complex (PDC) (Gray et al., 2014). We show that the rapid mCa2+ signaling promotes metabolic reprogramming by activating the PDC in macrophages. In response to fungal pathogens, mCa2+ signaling activates the PDH and increases intracellular citrate, an intermediate of the TCA cycle. Citrate is known to be exported from the mitochondrial matrix to the cytosol for generation of NADPH through the activity of malic enzyme (Williams and O’Neill, 2018). Thus, mCa2+ signaling drives the production of NADPH necessary for phagosomal ROS production. The MCU−/− macrophages are deficient in the generation of phagosomal ROS and this impairs the killing of phagocytosed C. albicans. The higher-order immunological significance is that mice lacking MCU in their myeloid cells and thus deficient in mCa2+ signaling are highly susceptible to disseminated candidiasis. In essence, the study reveals a design principle that the MCU-dependent Ca2+-signaling relay is a fast electrometabolic switch to fuel phagosomal killing. This design principle is likely significant for many other processes of cell-intrinsic immunity, but this study focuses exclusively on phagosomal killing of C. albicans.
RESULTS
MCU Has a Crucial Function in Cell-Autonomous Killing of Fungal Pathogens
To identify the Ca2+ channels necessary for macrophage fungal killing, we developed a small interfering RNA (siRNA) screen in RAW264.7 cells, a murine macrophage cell line. The siRNAs used in the screen were validated to knock down the Ca2+-conducting channels expressed in macrophages (Figure 1A). After siRNA transfection, RAW264.7 macrophages were fed C. albicans (MOI =1) and assessed as described for pathogen killing (Figure 1B). The Z scores, which reflect a change in the capacity to kill relative to macrophages transfected with control siRNA (scrambled siRNA), are plotted against the normalized expression of the targeted ion channels in macrophages (Figure 1C). In this screen, we identified MCU as a regulator of C. albicans killing in macrophages (Figure 1D). A significant defect in C. albicans killing at MOI1 was observed with an ~50% reduction in Mcu transcript levels (Figure S1A). We substantiated this screen with additional tests using siRNA-mediated knockdown of MCU in bone-marrow-derived macrophages (BMDMs) and again observed a significant defect in killing C. albicans (Figure S1B). To further define the function of MCU in pathogen killing by macrophages, we generated a mouse line with a CX3CR1-Cre-driven deletion of Mcu in myeloid cells (herein Mcu(M)−/− mice) (Figure 1E). The bone marrow of Mcu(M)−/− mice differentiated normally into macrophages (data not shown), and both mRNA (Figure 1F) and protein levels (Figures 1G and S1I) of MCU were depleted in Mcu−/− macrophages. In an in vitro assay of mCa2+ uptake, the mitochondria isolated from Mcu−/− macrophages were deficient in their ability to rapidly take up Ca2+ added to the in vitro preparation (Figures 1H and 1I). Additionally, using a permeabilized cell assay, Mcu−/− macrophages showed a striking inability to take up Ca2+ when compared to wild-type (WT) macrophages (Figure 1J). Surprisingly, Mcu−/− macrophages showed no significant defect in basal ATP levels or oxygen consumption rate (OCR) (Figures S1C and S1D). Analysis of mitochondrial membrane potential using JC-1 dye showed that Mcu−/− macrophages show normal mitochondrial membrane potential (Figures S1E and S1F). This was further substantiated by measuring the membrane potential using the dye tetramethylrhodamine, methyl ester (TMRM). Both baseline membrane potential and carbonyl cyanide 4-(trifluoromethoxy)phenylhydrazone (FCCP)-induced depolarization were nearly identical in both WT and Mcu−/− macrophages (Figures S1G and S1H). These analyses show that in absence of a pathogenic challenge, Mcu−/− macrophages do not show any obvious mitochondrial dysfunction. However, when exposed to C. albicans in killing assays (MOI1), the Mcu−/− macrophages exhibited a clear defect in the killing of C. albicans (Figure 1K) and Saccharomyces cerevisiae (Figure 1L). Since macrophages have been reported to undergo pyroptosis during C. albicans phagocytosis (Uwamahoro et al., 2014), we tested whether Mcu−/− macrophages are more susceptible to C. albicans induced pyroptosis. We found no significant release of lactate dehydrogenase (LDH) at 6 h of exposure to C. albicans and no difference between WT and Mcu−/− macrophages (Figures 1M and 1N). Together, these results reveal a role for MCU and mCa2+ signaling in the cell-autonomous killing of C. albicans.
Figure 1. MCU Has a Crucial Function in Phagosomal Killing of Fungal Pathogens.

(A) Schematic depicting the selection of siRNA targets for Ca2+ channel screen in myeloid cells.
(B) Schematic depicting the C. albicans killing experiment in RAW264.7 cells.
(C) Scatterplot showing Z scores of C. albicans killing in relationship to relative gene expression. Ion channel genes that were screened are shown as individual points. Z score calculations are described in STAR Methods. Normalized expression value is the average expression level across four macrophage populations (ImmGen database). Knockdown of Mcu (marked) resulted in a significant deficit in C. albicans killing relative control.
(D) Killing of C. albicans by macrophages transfected with control siRNA and Mcu-targeting siRNA. Individual replicates from the siRNA screen are shown (n = 4). Error bars represent SEM; p < 0.0001 according to Welch’s t test, two tailed.
(E) Schematic showing the breeding strategy used to generate Mcu(M)−/− mice.
(F) Gene expression analysis (qPCR) of Mcu mRNA in WT (n = 6, mice) and Mcu−/− (n = 6, mice) bone-marrow-derived macrophages (BMDMs). Error bars represent SEM; p < 0.0001 according to Welch’s t test, two tailed.
(G) Protein quantification from immunoblots of whole-cell lysates from WT (n = 7, mice) and Mcu−/− (n = 7, mice) BMDMs. Error bars represent SEM; p = 0.0014 according to Welch’s t test, two tailed. See Figure S1I for immunoblots.
(H) Mitochondrial Ca2+ (mCa2+) uptake assay using mitochondria isolated from WT and Mcu−/− BMDMs as reported by the reduction in extramitochondrial Ca2+ signal, after addition of 45 μM Ca2+ to the isolated mitochondria.
(I) Quantification of Ca2+ uptake in mitochondria isolated from WT (n = 4) and Mcu−/− (n = 4) BMDMs. mCa2+ uptake is calculated as percentage of maximum. Error bars represent SEM; p < 0.0001 according to Welch’s t test, two tailed.
(J) mCa2+ uptake assay using digitonin-permeabilized BMDMs. 15 μM Ca2+ was added to 500,000 digitonin-permeabilized BMDMs in a 96-well plate, and Ca2+ Green-5N fluorescence was monitored (Excitation/Emission 505/535 every 2 s for 6 min). 5 μM FCCP was added as positive control to uncouple mitochondria and release Ca2+ at the end of each run.
(K) C. albicans killing by WT (n = 10) and Mcu−/− (n = 10) BMDMs. Error bars represent SEM; p = 0.0015 according to Welch’s t test, two tailed.
(L) Killing of S. cerevisiae by WT (n = 10) and Mcu−/− (n = 10) BMDMs. Error bars represent SEM; p < 0.0001 according to Welch’s t test, two tailed.
(M) LDH release by WT and Mcu−/− BMDMs in response to C. albicans MOI1. LDH release is reported as a percentage of maximum release (black). Error bars represent SEM; no significant difference was detected between WT and Mcu−/− at baseline (n = 4, each group) or 6 h (n = 6, each group) according to ordinary one-way ANOVA with multiple comparisons.
(N) LDH release by WT and Mcu−/− BMDMs in response to zymosan. LDH release is reported as a percent of maximum release (black). Error bars represent SEM; no significant difference was detected between WT and Mcu−/− at baseline (n = 4, each group) or 6 h (n = 6, each group) according to ordinary one-way ANOVA with multiple comparisons.
Mcu(M)−/− Mice Are Highly Susceptible to Disseminated Candidiasis
To determine if the deletion of MCU in myeloid cells increased susceptibility to fungal infections, we tested Mcu(M)−/− mice in a model of disseminated candidiasis. Mice were tail vein injected with 1 × 106 viable blastospores of C. albicans and monitored for disease progression for 4 days post-infection. The clinical scores for the infected mice were recorded over time in a blinded protocol. Mcu(M)−/− mice showed increased morbidity and lethargy compared to WT mice (Figure 2A). Mcu(M)−/− mice also had a strikingly lower survival rate, with only 9% of mice surviving until day 4 compared to 46% in the WT control group (Figure 2B). The Mcu(M)−/− mice succumbed to disease at a faster rate (median survival, 48 h) than WT (median survival 72 h) (Figure 2B). We excised the livers and kidneys of the infected mice to analyze them for fungal burden. The organ homogenates were plated on YPD agar plates to derive colony-forming units (CFUs)/g tissue. Mcu(M)−/− mice had increased fungal burden in both kidney and liver (Figures 2C and 2D). To assess fungal burden using tissue histology, we stained kidney sections with periodic acid-Schiff (PAS), which stains mucopolysaccharides and can thereby detect fungal burden in the stained tissue. Histology of the kidney sections 2 days post-infection (dpi) showed severe fungal burden in Mcu(M)−/− mice (Figure 2E). The H&E tissue sections (Figure 2F) of WT and Mcu(M)−/− kidneys showed comparable number of immune cells (Figure 2G), indicating that the Mcu−/− macrophages are not deficient in the recruitment of immune cells. In fact, in comparison to WT mice, the Mcu(M)−/− mice exhibited substantially increased pro-inflammatory cytokines in their serum (Figure 2H). Vehicle controls had no detectable cytokine levels or any discernible changes in H&E and PAS staining (data not shown). These results indicate that the striking susceptibility of Mcu(M)−/− mice to C. albicans is not a result of defective detection of C. albicans or reduced output of inflammatory cytokines. It is predominantly due to defective cell-autonomous killing mechanisms. Since MCU mediates mitochondrial Ca2+ influx, these results suggest that mCa2+ signaling is a crucial component of fungal killing.
Figure 2. Mcu(M)−/− Mice Are Highly Susceptible to Disseminated Candidiasis.
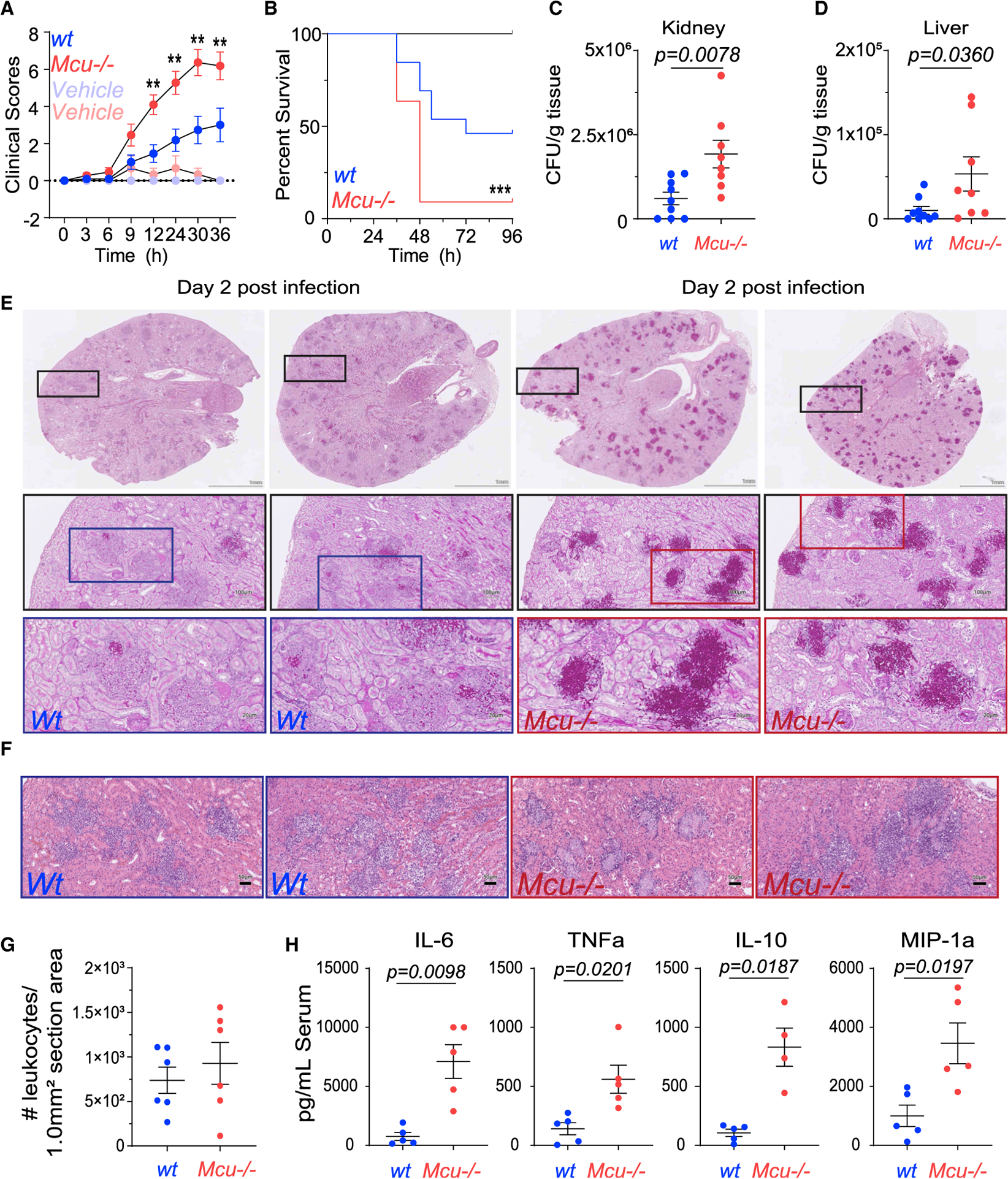
(A) Clinical scores in WT and Mcu(M)−/− mice injected in the tail vein with 100 μL 1 × 106 C. albicans or vehicle control (100 μL 0.9% saline). Infected Mcu(M)−/− mice (n = 11, mice) trace is shown in red and WT (n = 13, mice) in blue. Vehicle controls for Mcu(M)−/− (n = 3, mice) are shown in light red and WT (n = 6, mice) in light blue. Error bars represent SEM for individual time points across three individual experiments; significance (*p < 0.0500 and **p < 0.005) is reported for each time point according to two-way ANOVA with multiple comparisons.
(B) Kaplan-Meier survival curves for WT and Mcu(M) −/− mice infected with tail vein injections of 100 μL 1 × 106 C. albicans or vehicle control (100 μL 0.9% saline). Infected Mcu(M)−/− (n = 11, mice) trace is shown in red and WT (n = 13, mice) in blue. Vehicle controls for Mcu(M)−/− (n = 3, mice) and WT (n = 6, mice) are shown in black (no mice died). At the 48-h time point, 69.2% of WT mice are alive compared to 9.1% of Mcu(M)−/−. Curve comparison was made using a log-rank (Mantel-Cox) test. Data were collected from three individual experiments; p = 0.0008(***).
(C) Colony-forming units (CFUs) of C. albicans counted per gram of kidney at time of sacrifice in wt (n = 9, mice) and Mcu(M)−/− (n = 8, mice) mice infected with tail vein injection of 100 μL 1 × 106 C. albicans. Vehicle control (100 μL 0.9% saline) for both groups had no CFUs. Error bars represent SEM from three individual experiments; p = 0.0078 according to Mann-Whitney test, two tailed.
(D) CFUs of C. albicans counted per gram of liver at time of sacrifice in WT (n = 9, mice) and Mcu(M)−/− (n = 8, mice) mice infected with tail vein injection of C. albicans (100 μL 1 × 106). Vehicle control (100 μL 0.9% saline) for both groups had no CFUs. Error bars represent SEM from three individual experiments; p = 0.0360 according to Mann-Whitney test, two tailed.
(E) Representative periodic acid-Schiff (PAS) staining on kidney sections from WT and Mcu(M) −/− mice, 2 days after tail vein injection of 100 μL (1 × 106) C. albicans. Sections were mounted on slides and entire slides were imaged at 203. Boxes represent the region of each subsequent magnification. Scale is 1 mm for whole kidney section. Scale is 100 μM and 20 μM for subsequent zooms.
(F) Representative H&E sections from WT and Mcu−/− kidneys. Scale bar, 50 μM.
(G) H&E stained kidneys were blindly scored for recruitment of leukocytes at time of sacrifice in infected mice (tail vein injection of 100 μL 1 × 106 C. albicans). Control mice (100 μL of 0.9% saline) did not show any immune cell recruitment and are not shown. Error bars represent SEM; no significant difference was detected between WT (n = 6, mice) and Mcu(M)−/− (n = 6, mice) according to Mann-Whitney test, two tailed.
(H) Measurement of serum cytokines using Luminex Multiplex assay. Serum samples were collected from WT (n = 5, mice) and Mcu(M)−/− mice (n = 4–5, mice) at the time of sacrifice and the indicated cytokines were measured. Vehicle controls had no detectable levels of serum cytokines. Error bars represent SEM; indicated p values calculated according to Welch’s t test, two tailed.
Fungal Pathogens Trigger SOCE, Phase 1 of mCa2+ Signaling
The first phase of the two-step Ca2+ relay in response to fungal pathogens is the elevation of cytosolic Ca2+. The crucial requirement of cytosolic Ca2+ elevation is revealed by the drastically reduced killing of fungi by macrophages loaded with a fast Ca2+ chelator BAPTA-AM (Figure 3A). To record cytosolic Ca2, we loaded BMDMs with the ratiometric Ca2+ indicator Fura-2 AM. Aqueous extracts of heat-killed S. cerevisiae were obtained in 0 mM and 2 mM Ca2+ solutions. Macrophages stimulated with both fungal extracts elicited a robust cytosolic Ca2+ response (Figure 3B). The elevation of cytosolic Ca2+ in macrophages stimulated in 0 mM extracellular Ca2+ indicates that the Ca2+ is released from an intracellular store such as the endoplasmic reticulum (ER). To measure store-operated Ca2+ entry (SOCE), 10 mM Ca2+ was added to the bath once the intracellular release of Ca2+ returned to baseline. This resulted in a robust uptake of extracellular Ca2+, presumably through the store-operated Orai channels. Similar results were observed when macrophages were stimulated with the fungal cell wall component, β-glucan, a potent agonist of Dectin-1 (Figure 3C). Fungal stimulation of Dectin-1 has been proposed to facilitate Ca2+ elevations through Syk-mediated activation of phospholipase C (PLC) (Goodridge et al., 2007; Strijbis et al., 2013). Indeed, the Ca2+ elevations were completely arrested in the presence of GSK143 and U73122, potent inhibitors of Syk and PLC, respectively (Figure 3D). We measured cytosolic Ca2+ in WT and Mcu−/− macrophages in response to zymosan (Figures 3E and 3F) and C. albicans extracts (Figures 3G and 3H). These results show that cytosolic Ca2+ was modestly higher in Mcu−/− macrophages, as the rapid MCU-dependent mCa2+ uptake was absent. Overall, these results show that the fungal pathogen-associated molecular patterns (PAMPs) trigger an immediate SOCE (phase 1) as a prelude to mCa2+ signaling (phase 2; see below).
Figure 3. Fungal Pathogens Trigger SOCE, Phase 1 of MCa2+ Signaling.
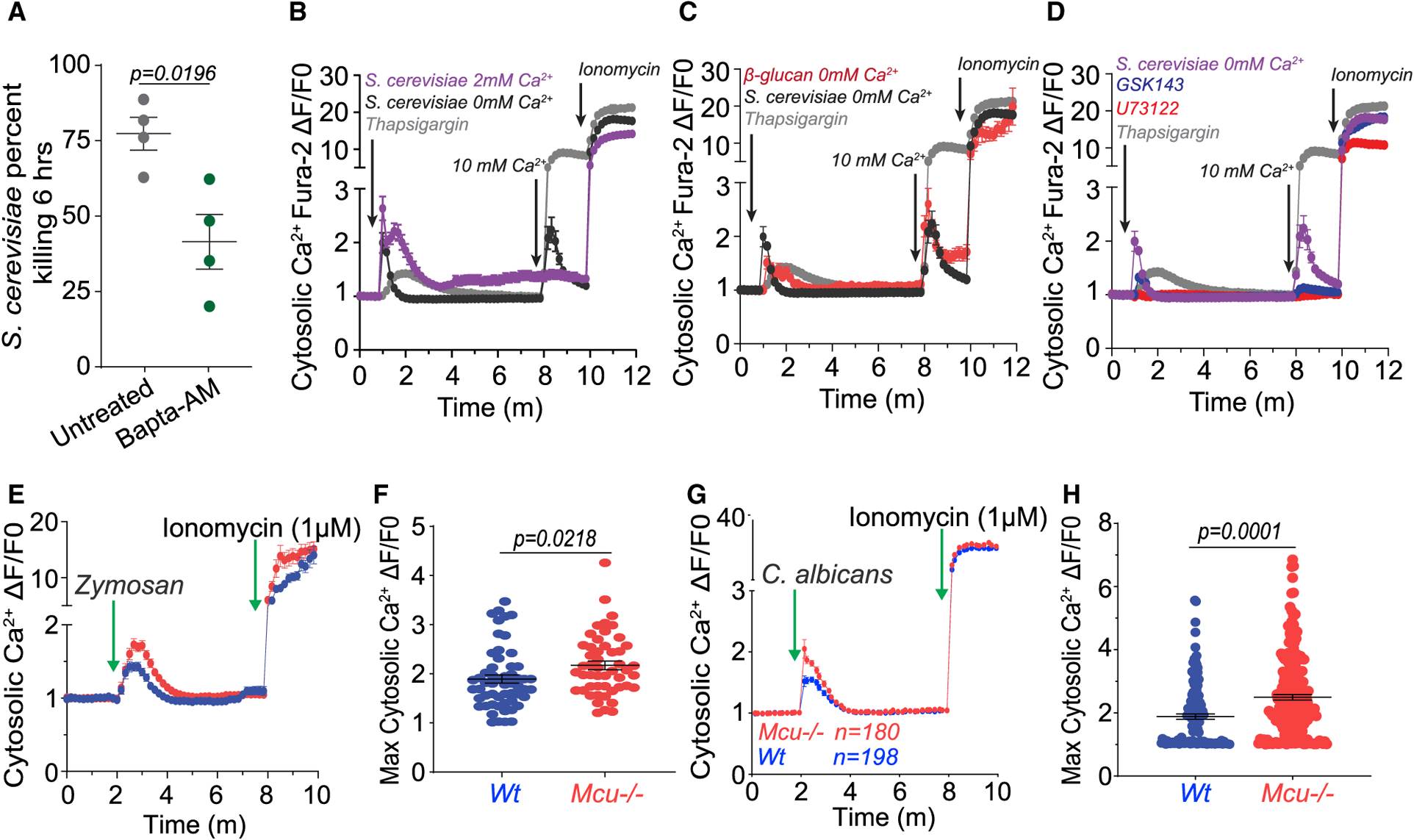
(A) Killing of S. cerevisiae by BMDMs preloaded with vehicle (n = 4) or 5 μM BAPTA-AM (n = 4). Error bars represent SEM; p = 0.0196 according to Welch’s t test, two tailed.
(B) Cytosolic Ca2+ elevations in BMDMs stimulated with S. cerevisiae extracts in 0 mM (n = 78 cells) or 2 mM Ca2+ (n = 44 cells) or in response to thapsigargin (w/extracellular 0 mM Ca2+) (n = 44 cells) (positive control). [Ca2+] in the bath was raised to 2 mM to record the store-operated Ca2+ entry (SOCE). Ionomycin (1 μM) added last revealed maximum Ca2+ responsiveness of all the Fura-2-AM-loaded cells. The plotted values were calculated as a change in fluorescence/initial fluorescence (ΔF/F0). Error bars represent SEM.
(C) Cytosolic Ca2+ elevations in BMDMs stimulated with S. cerevisiae extracts in 0 mM Ca2+ (n = 78 cells), β-glucan in 0 mM Ca2+ (n = 68 cells), and thapsigargin (positive control). Error bars represent SEM.
(D) Cytosolic Ca2+ elevations in BMDMs stimulated with S. cerevisiae extracts in 0 mM Ca2+ and thapsigargin (positive control). BMDMs were stimulated in the presence of either GSK143 (2 μM, n = 92 cells) or U73122 (1 μM, n = 22 cells). Error bars represent SEM.
(E) Cytosolic Ca2+ elevations in response to zymosan in WT (n = 56 cells) and Mcu−/− (n = 50 cells) loaded with Fura-2 AM. These values are calculated as a change in fluorescence/ initial fluorescence (ΔF/F0). Error bars represent SEM.
(F) Peak cytosolic Ca2+ elevations in response to zymosan in WT (n = 56 cells) and Mcu−/− (n = 50 cells) loaded with Fura-2 AM. Error bars represent SD; p values were calculated according to Welch’s t test, two tailed.
(G) Cytosolic Ca2+ elevations in response to C. albicans extracts in WT (n = 198 cells) and Mcu−/− (n = 180 cells) loaded with Fura-2 AM. These values are calculated as a change in fluorescence/initial fluorescence (ΔF/F0). Error bars represent SEM.
(H) Peak cytosolic Ca2+ elevations in response to C. albicans supernatant in WT (n = 180 cells) and Mcu−/− (n = 198 cells) loaded with Fura-2 AM. Error bars represent SEM; p values were calculated according to Welch’s t test, two tailed.
Rapid Elevations in mCa2+ (Phase 2) Are Detected Immediately after the Recognition of C. albicans
To license the activation of MCU, cytosolic Ca2+ must be sufficiently elevated to allow the gatekeeper MICU1 to promote mCa2+ uptake (Mallilankaraman et al., 2012). In response to fungal pathogens, cytosolic Ca2+ is elevated through SOCE. The close juxtaposition of mitochondria to the ER then allows the second phase of the Ca2+ relay into the mitochondria. To measure mCa2+ elevations in macrophages, we established a RAW264.7 stable cell line expressing the genetically encoded mCa2+ sensor CEPIA3mt (Suzuki et al., 2014) (herein RAW-3mt). In these cells, CEPIA3mt colocalized with the mitochondrial marker dye MitoTracker red (Figure S2A) and reported sharp mCa2+ transients when the cells were treated with the Ca2+ ionophore ionomycin (Figures S2B and S2C). Recognition and phagocytosis of fungal pathogens is commonly simulated by zymosan particles, a sterile cell wall preparation from S. cerevisiae that stimulates cell surface receptors Toll-like receptor 2 (TLR2) and Dectin-1. When RAW-3mt cells are exposed to fluorescent zymosan, they readily phagocytose the particles and trigger the elevation of mCa2+. As reported previously (West et al., 2011), a concurrent movement of the mitochondria to the phagosome is also seen (Video S1). mCa2+ elevations were recorded by drawing regions of interest (ROIs) around the mitochondrial network in macrophages actively engulfing zymosan particles (Figure 4A). Zymosan-triggered mCa2+ elevations were significantly reduced in RAW-3mt cells wherein MCU was knocked down with siRNA (Figure 4B). Similarly, C. albicans supernatant elicited robust mCa2+ elevations (Figure 4C) but with faster kinetics. The mCa2+ elevations in response to C. albicans are also MCU dependent (Figure 4D). The movement of the mitochondria to the C.-albicans-containing phagosome led us to investigate the membrane proximity through transmission electron microscopy (TEM). We observed intimate mitochondrial juxtaposition to C.-albicans-containing phagosomes with a distance of ~50 nm separating the phagosomal membrane from the mitochondrial outer membrane (Figure 4E). Similar association was observed in response to zymosan (Figure 4F). The close juxtaposition is therefore similar to that observed in ER-mitochondrial contact sites and such repositioning can facilitate mCa2+ uptake from the Ca2+ released from the phagosome or from the adjoining ER (Csordas et al., 2008, 2010). To analyze the mCa2+ elevations in the mitochondria juxtaposed with the phagosome, we analyzed the ROIs around the phagosome for changes in periphagosomal mCa2+ (PPMiCa). The PPMiCa elevations were then normalized to the mCa2+ across the mitochondrial network in that cell (Figure S2D) to reveal the PPMiCa dynamics during phagocytosis. Despite the heterogeneity of mCa2+ elevations in terms of amplitude and kinetics, the analysis showed that PPMiCa was significantly higher relative to the mCa2+ across the mitochondrial network, and in comparison to control cells, the PPMiCa elevations were suppressed in cells wherein MCU was knocked down using siRNA (Figures 4G and 4H; Video S2). Time courses of PPMiCa responses are shown in (Figure S2E; Video S3). These analyses reveal that detection and phagocytosis of fungal particles elicits the activation of MCU and robust mCa2+ elevations throughout the mitochondrial network. The mitochondria that are in close proximity to the phagosome show especially increased mCa2+ signaling. The precise regulatory mechanism underlying the gating of MCU channel during phagosomal killing is not a focus of this study. However, since it was recently reported that Mcu is activated by AMP-activated protein kinase (AMPK) during cell division (Zhao et al., 2019), we tested the role of AMPK inhibition during fungal killing. Macrophages treated with the AMPK inhibitor dorsomorphin dihydrochloride did not show an impairment in fungal killing (Figure 4I), indicating that MCU is not regulated by AMPK during phagosomal killing.
Figure 4. Rapid Elevations in MCa2+ (Phase 2) Are Detected after Fungal Recognition.
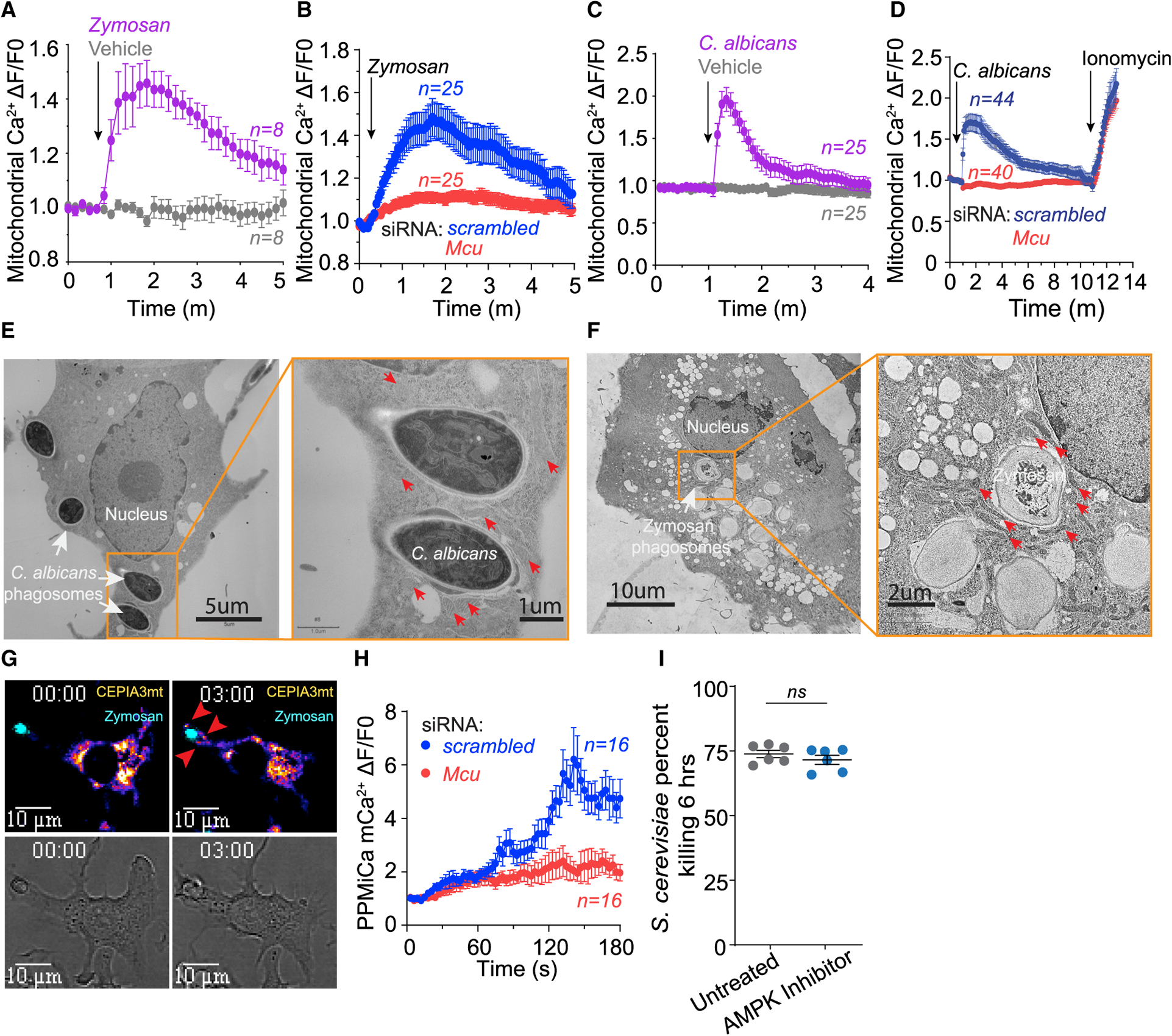
(A) Whole-cell mCa2+ elevations in zymosan-stimulated (n = 8 cells) and untreated (n = 8 cells) RAW3mt macrophages. Whole-cell mCa2+ is taken from the average fluorescence intensity of ROIs drawn over the entire mitochondrial network. Cells were sampled every 10 s. Error bars represent SEM.
(B) Whole-cell mCa2+ elevations in zymosan-stimulated RAW3mt macrophages. RAW3mt cells treated with scrambled siRNA (n = 25 cells) or Mcu siRNA (n = 25 cells) were imaged every 3 s. Error bars represent SEM.
(C) Whole-cell mCa2+ elevations in RAW3mt macrophages stimulated with C. albicans extract (n = 25 cells) and vehicle (n = 25 cells). Cells were sampled every 5 s. Error bars represent SEM.
(D) Whole-cell mCa2+ elevations in RAW3mt macrophages stimulated with C. albicans extract. RAW3mt cells treated with scrambled siRNA (n = 40 cells) or Mcu siRNA (n = 44 cells) were imaged every 3 s. 1 μM ionomycin was added at the end of the experiment to reveal maximum mCa2+ uptake. Error bars represent SEM.
(E) Representative TEM image of BMDMs infected with C. albicans for 6 h. Magnification of C. albicans phagosomes shows peri-phagosomal juxtaposition of mitochondria indicated by red arrows.
(F) Representative TEM image of BMDMs exposed to zymosan for 6 h. Magnification of zymosan phagosomes shows peri-phagosomal juxtaposition of mitochondria indicated by red arrows.
(G) Representative PPMiCa responses in RAW3mt cells. Shown are merged images of CEPIA3mt (Fire LUT) and zymosan (cyan LUT) (top) and bright-field images (bottom). Red arrows indicate PPMiCa signals.
(H) PPMiCa traces from zymosan-stimulated RAW3mt macrophages. The values are calculated as a change in fluorescence/ initial fluorescence (ΔF/F0). RAW3mt cells treated with scrambled siRNA (n = 16 cells) or Mcu siRNA (n = 16 cells) were imaged every 3 s. Error bars represent SEM.
(I) Killing of S. cerevisiae by macrophages treated with vehicle (n = 5) or 1 μM AMPK inhibitor dorsomorphin dihydrochloride (n = 5). Error bars represent SEM; p < 0.0001 according to Welch’s t test, two tailed.
mCa2+ Signaling Controls Phagosomal ROS Production
The acidification of the nascent phagosome is carried out by V-ATPase complex, and we reasoned that the process may put an unusually high demand on ATP. We hypothesized that mCa2+ signaling during phagocytosis drives the acidification of phagosome through a burst of ATP generation in the proximity of the phagosome. To assess the engulfment of C. albicans and the subsequent acidification of the phagosome, we labeled C. albicans with two dyes, CellTrace violet (CTV), which is relatively insensitive to acidic pH, and CypHer-5E, which fluoresces brightly with low pH. The dually labeled C. albicans was then offered to WT and Mcu−/− macrophages and the cells were analyzed by flow cytometry to assess engulfment (CTV fluorescence) and phagosome acidification (CypHer-5E fluorescence). Surprisingly, these experiments revealed that Mcu−/− macrophages were not defective in engulfment (Figures 5A and 5B) or phagosome acidification (Figures 5C–5E). The gating strategy and representative flow cytometry plots are shown (Figures S3A–S3C). The ATP levels during phagocytosis of C. albicans were also normal in Mcu−/− macrophages (Figure 5F). In both WT and Mcu−/− macrophages, cellular ATP levels decreased only modestly at 1 h and returned to baseline at 6 h post-stimulation. Recent reports have shown a role for mitochondrial ROS (mROS) in pathogen destruction (Geng et al., 2015; West et al., 2011). Using MitoSox, a fluorescent sensor of mROS, we did not see an impairment in the acute production of mROS in Mcu−/− macrophages during their response to the pathogenic stimuli (Figures S4A and S4B). Only after 18 h of zymosan stimulation did Mcu−/− macrophages begin to show a modest defect in mROS production (Figures S4C and S4D). The phagosomal destruction of pathogens is also mediated by injection of ROS in the phagosome by NADPH oxidase complex (NOX). The activity of NOX requires substantial bioenergetic expenditure in the form of NADPH. To assess phagosomal ROS generation by the NOX complex, we developed a flow cytometry-based assay. We labeled C. albicans with CellRox (a dye that fluoresces when oxidized by ROS) and CTV. This assay reports the NOX-mediated generation of ROS with high fidelity, and this is demonstrated by the validation control wherein the macrophages are pretreated with diphenyleneiodonium (DPI), a potent inhibitor of NOX (Altenhöfer et al., 2015), prior to pathogen exposure. Macrophages treated with DPI displayed a significant decrease in CellRox fluorescence (Figures S4E–S4H). We found that production of phagosomal ROS in Mcu−/− macrophages engulfing C. albicans was also significantly reduced (to ~50% relative to WT cells) 3 h post-engulfment (Figures 5G–5I). These results indicate that during phagocytosis, V-ATPase activity is not influenced by mCa2+ signaling, but phagosomal ROS production is highly dependent on MCU-dependent mCa2+ signaling.
Figure 5. mCa2+ Signaling Controls Phagosomal ROS Production.
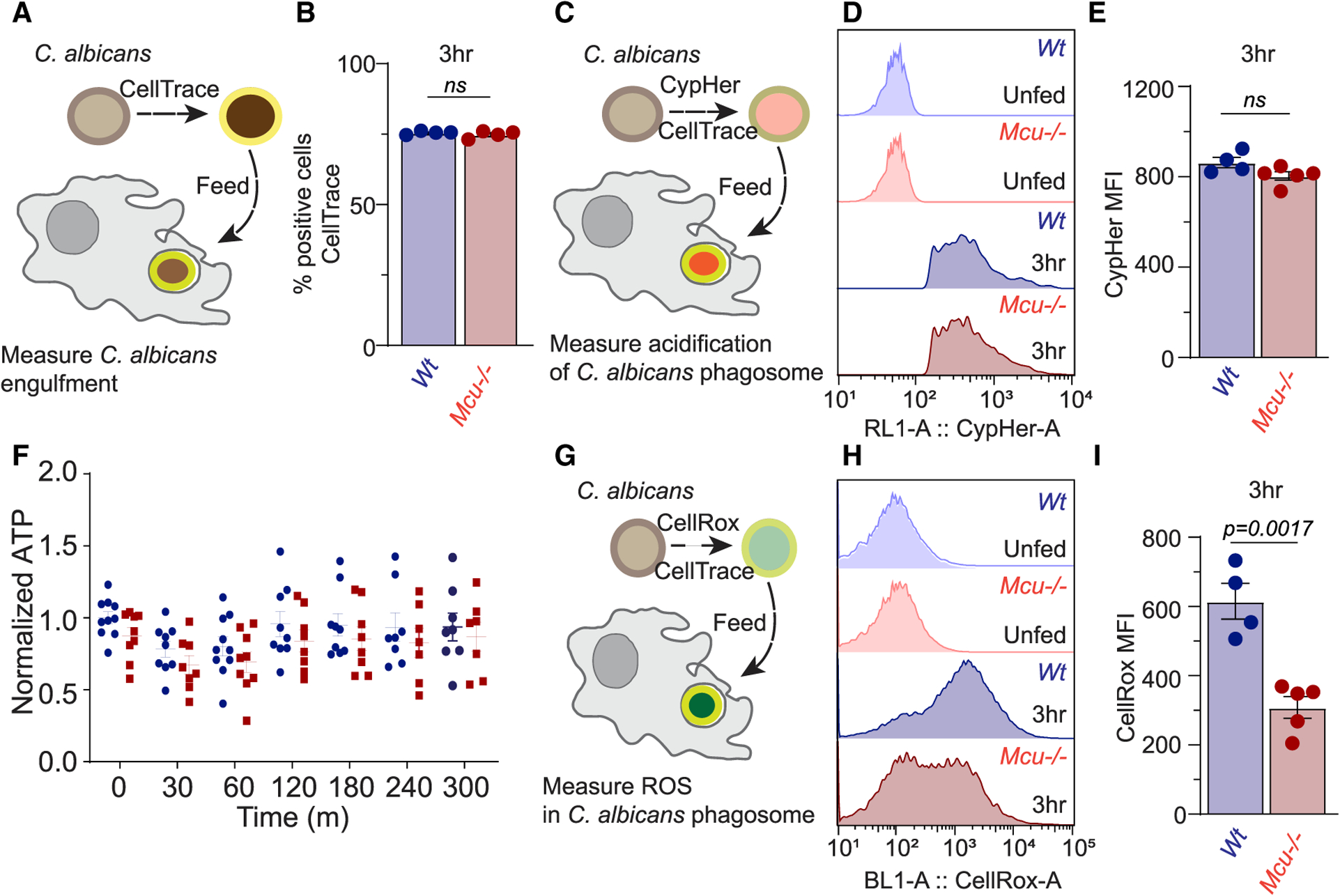
(A) Schematized experimental design to measure C. albicans engulfment in macrophages using flow cytometry. C. albicans were labeled with CellTrace Violet (CTV) prior to phagocytosis.
(B) Quantification of CTV-positive cells at 3 h post-phagocytosis for WT (n = 4) and Mcu−/− (n = 4) BMDMs. Error bars represent SEM; no significance was detected according to Welch’s t test, two tailed.
(C) Schematized experimental design to measure phagosome acidification in macrophages using flow cytometry. C. albicans were labeled with CypHer5E (fluoresces with acidic pH) and CTV (to confirm engulfment) prior to phagocytosis.
(D) Flow cytometry histograms of CypHer5E fluorescence intensity in WT and Mcu−/− BMDMs after 3 h of phagocytosis.
(E) Quantification CypHer5E MFI in WT (n = 4) and Mcu−/− (n = 5) BMDMs after 3 h of phagocytosis. Error bars represent SEM; no significance was detected according to Welch’s t test, two tailed.
(F) Quantification of relative ATP levels in WT (n = 8–10 for each time point) and Mcu−/− (n = 8–10 for each time point) BMDMs. Cells were stimulated with zymosan, and ATP levels were measured at each time point. Error bars represent SEM; no significance was detected according to two-way ANOVA, two tailed.
(G) Schematized experimental design to measure phagosomal ROS in BMDMs. C. albicans were labeled with CellRox (reports oxidative stress) and CTV (to confirm engulfment) prior to phagocytosis.
(H) Flow cytometry histograms of CellRox fluorescence intensity in WT and Mcu−/− BMDMs after 3 h of phagocytosis.
(I) Quantification CellRox MFI in WT (n = 4) and Mcu−/− (n = 5) BMDMs after 3 h of phagocytosis. Error bars represent SEM; p = 0.0017 according to Welch’s t test, two tailed.
Mcu−/− Macrophages Are Unable to Meet the NADPH Demand of Phagosomal Killing
Since NOX activity is NADPH dependent, we reasoned that mCa2+ signaling controls the metabolic shift needed to increase cellular NADPH metabolism required for phagocytosis. To measure this metabolic shift, we took advantage of the native fluorescent properties of NAD(P)H and used fluorescence lifetime imaging microscopy (FLIM) for a quantitative analysis of cellular NAD(P)H levels. The NAD+/NADH and NADP+/NADPH redox couples are critical determinants of the cellular redox (Blacker et al., 2014; Wallrabe et al., 2018). FLIM enables the study of NAD(P)H photochemistry in living cells by measuring the rates of fluorescence decay (fluorescence lifetime). The lifetime of NAD(P)H fluorescence is sensitive to its microenvironment, and metabolic bursts that increase NAD(P)H levels register a sharp increase in enzyme-bound NAD(P)H with increased fluorescence lifetimes. Since the enzyme-bound fraction of NAD(P)H (fraction a2%) can be calculated based on FLIM, we explored the technique in measuring the metabolic shifts that alter NAD(P)H synthesis and utilization in macrophages engulfing and killing C. albicans. The WT macrophages engulfing C. albicans exhibit a steady increase in bound NAD(P)H fraction reflecting the increased a2% fraction (Figure 6A). Strikingly, the Mcu−/− macrophages show a severe deficit in a2% fraction during phagocytosis (Figures 6A and 6B). These results indicate that mCa2+ signaling controls the NAD(P)H metabolism during response to fungal pathogens. To substantiate these findings further, we measured NADP+ and NADPH levels using biochemical assays (Figures S5A–S5C). We found no significant change in the NADPH/NADP+ ratio at baseline, but the ratio was significantly reduced in Mcu−/− macrophages after 3 h of exposure to zymosan. This difference is largely accounted by a significantly reduced levels of NADPH (~30% reduction) in Mcu−/− macrophages (Figure S5B). The difference in NADP+ is relatively modest (Figure S5C).
Figure 6. Mcu−/− Macrophages Exhibit an Abnormal Immunometabolic Shift in Response to Fungal Pathogens.
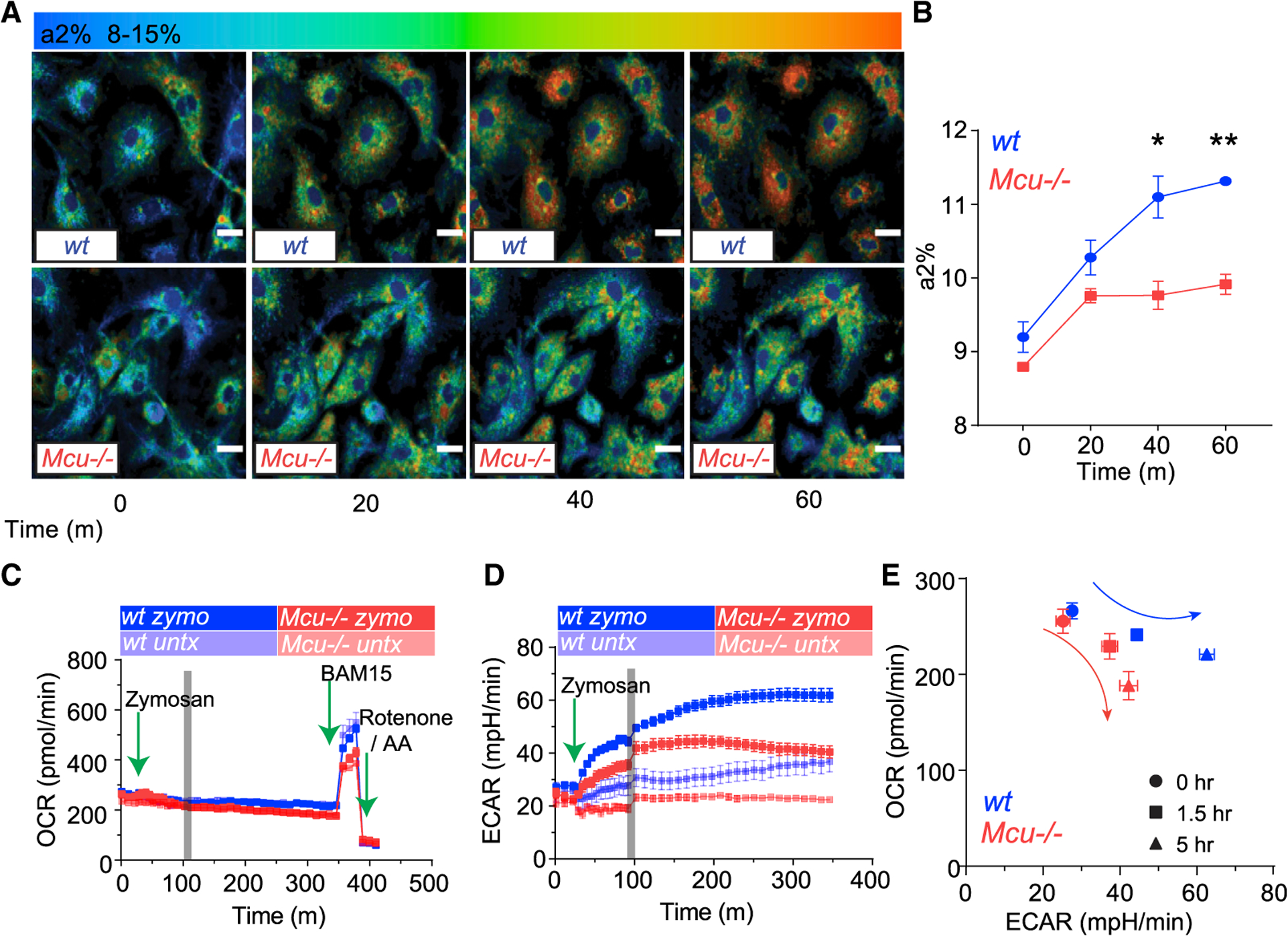
(A) Representative FLIM images from before and after C. albicans engulfment. a2% is shown as a shift from blue to red. Scale bar is 10 μm.
(B) Assessment of a2% (reflects NADPH levels) in WT (n = 4) and Mcu−/− (n = 3) BMDMs. Error bars represent SEM; p = 0.0123 at 40 min and p = 0.0038 at 60 min according to Welch’s, two-tailed test.
(C) Mitochondrial oxygen consumption rate (OCR) in WT (n = 4) and Mcu−/− (n = 4) macrophages in response to zymosan. Cells were measured at baseline for 30 min followed by the injection of zymosan. BAM15 (mitochondrial uncoupler) and rotenone and antimycin A injected at indicated times (green arrows).
(D) Extracellular acidification rate (ECAR) of WT (n = 4) and Mcu−/− (n = 4) macrophages in response to zymosan. Cells were measured at baseline for 30 min followed by the injection of zymosan (green arrow). The gray bar denotes a change in sampling rate from a measurement every 5 min to a measurement every 10 min.
(E) Seahorse bioenergetics profile, shown as relationship of OCR and ECAR, in WT (n = 4) and Mcu−/− (n = 4) BMDMs responding to zymosan.
Mcu−/− Macrophages Exhibit an Abnormal Immunometabolic Shift in Response to Fungal Pathogens
To further characterize the defects in metabolic reprogramming, we used the Seahorse analyzer, an instrument measuring key cellular bioenergetic parameters such as OCR, which reflects oxidative phosphorylation in the mitochondria and extracellular acidification rate (ECAR), which reflects the glycolytic activity. After 30 min of baseline measurement, we injected zymosan particles directly onto the macrophages and monitored the change in OCR and ECAR over 6 h. At baseline, WT and Mcu−/− macrophages showed no significant differences in OCR, but after zymosan stimulation, the Mcu−/− macrophages were unable to maintain the OCR (Figure 6C). Maximal OCR that the mitochondria are capable of is revealed by addition of BAM15, a mitochondrial uncoupler. Interestingly, absence of MCU modestly reduced the maximal OCR that the mitochondria are capable of during phagosomal killing (Figure S5E). Exposure to zymosan significantly increased the ECAR in both WT and Mcu−/− macrophages, but when compared to WT macrophages, the increase in ECAR was significantly lower in the Mcu−/− macrophages (Figure 6D) The difference was especially evident at 5 h post-engulfment (Figure S5F). To observe the combined bioenergetics profile revealed by seahorse analysis, we plotted OCR by ECAR during the course of zymosan stimulation (Figure 6E). This reveals that Mcu−/− macrophages had an impaired metabolic response to fungal stimulation. Wild-type macrophages increase their bioenergetic output as measured by OCR and ECAR, while Mcu−/− macrophages do not.
Metabolomic Analysis of Mcu−/− Macrophages Responding to Zymosan
Metabolomic analysis of Mcu−/− and WT macrophages before and after zymosan stimulation revealed considerable differences in the levels of glycolytic metabolites, but the levels of pyruvate and lactate, the final products of glycolysis, were not significantly different at 6 h (Figure S6B). There was no significant difference in the glycolytic capacity of Mcu−/− macrophages, as measured by the glycolyticstress test(GST) using the Seahorse analyzer (Figures S6C and S6D). The intermediates of the pentose phosphate pathway (PPP) were also altered in Mcu−/− macrophages (Figure S6E). We observed decreased glucose-6-phosphate and increased phosphoribosyl pyrophosphate (PRPP) in Mcu−/− macrophages at both 3 h and 6 h of zymosan exposure. Interestingly, we saw no significant difference in G6PGDH activity following zymosan stimulation in Mcu−/− macrophages (Figure S6F). Nevertheless, we tested the possibility that the PPP is a key contributor of NADPH necessary for efficient fungal killing in macrophages. To test the role of the PPP in fungal killing, we inhibited G6PGDH with 6-aminonicotinamide (6-AN) but did not observe significant defects infungalkilling (FigureS6G). These findings therefore indicate thatalthough mCa2+dynamics influence the PPP,NADPH production by the PPP is dispensable for fungal killing.
Dysregulated TCA Cycle Underlies Defective Killing by Mcu−/− Macrophages
Small Molecule Pathway Database (SMPDB) analysis of metabolites revealed significant dysregulation of TCA cycle metabolites (Figures 7A and 7B). Citrate and aconitate, the TCA metabolites immediately downstream of PDH were significantly reduced in zymosan-stimulated Mcu−/− macrophages at 6 h (Figures 7C and 7D). We observed similar decreases in citrate at 3 h (Figure S7B). Previously, it has been suggested that the TCA cycle intermediate citrate is essential for metabolic reprogramming of macrophages during inflammatory stimulations (Williams and O’Neill, 2018). Our analysis supports these findings as the relative abundance of citrate and aconitate increased in response to zymosan stimulation (Figure S7B). However, Mcu−/− macrophages do not increase the relative abundance of citrate and aconitate after zymosan stimulation, indicating that mCa2+ signaling is essential for this metabolic response. These data indicate that mCa2+ signaling has a direct impact on TCA cycle in general and more specifically on the accumulation of citrate and aconitate during phagosomal killing. We hypothesized that mCa2+-dependent accumulation of citrate is necessary for the cytosolic NADPH generation, because citrate can be exported into the cytosol and then utilized by malic enzymes to generate NADPH. To test this hypothesis, we tested the prediction that supplementation of citrate will rescue the defective killing in Mcu−/− macrophages. We ascertained the ability of WT and Mcu−/− macrophages to kill C. albicans in media supplemented with 2 mM citrate. The citrate supplementation completely rescued the killing defect of Mcu−/− macrophages (Figures 7E and 7F). Together, these data reveal a vital role for mCa2+ signaling in the regulation of TCA-cycle- and citrate-dependent NADPH generation necessary for fungal killing.
Figure 7. Dysregulated TCA Cycle Underlies Defective Killing by Mcu−/− Macrophages.
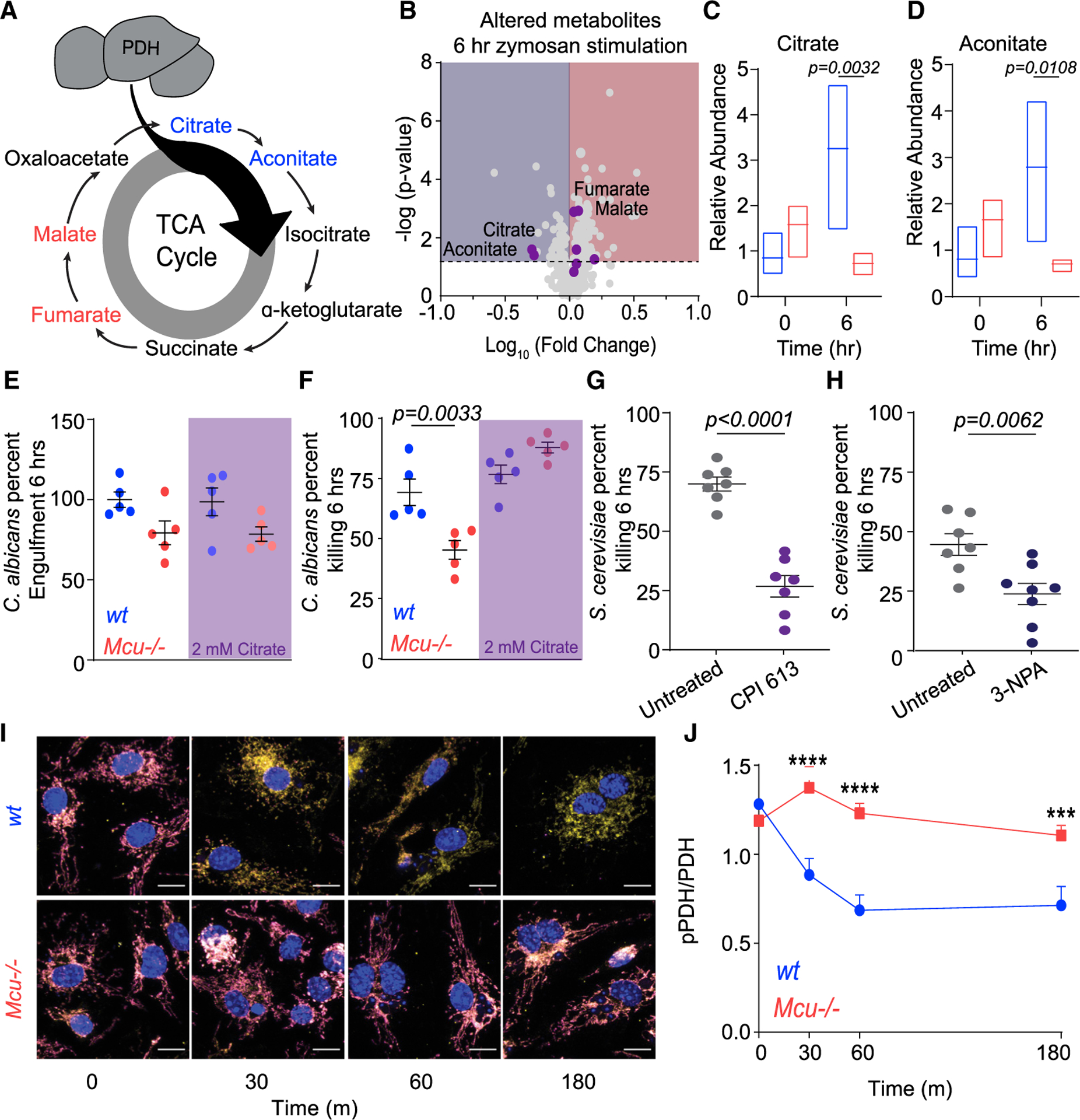
(A) Schematic of TCA cycle. Metabolites reduced in Mcu−/− BMDMs responding to zymosan (6 h) are blue. Metabolites that increased in Mcu−/− BMDMs are red.
(B) Volcano plot of significantly altered metabolites in Mcu−/− macrophages compared to WT following 6 h of zymosan stimulation. The dotted horizontal line represents the 75th percentile for −log (p values). TCA cycle metabolites are shown as purple dots.
(C) Relative abundance of citrate measured in WT (blue) and Mcu−/− (red) BMDMs at baseline and 6 h following zymosan stimulation. Floating bars represent the minimum to maximum, and the line is at the mean for each dataset; p = 0.0032 was detected according to two-way ANOVA, two tailed.
(D) Relative abundance of aconitate measured in WT (blue) and Mcu−/− (red) BMDMs at baseline and 6 h following zymosan stimulation. Floating bars represent the min to max and the line is at the mean for each dataset; p = 0.0108 was detected according to two-way ANOVA, two tailed.
(E) C. albicans engulfment in WT (n = 5) and Mcu−/− (n = 5) BMDMs in the absence and presence of 2 mM citrate. Error bars represent SEM; p = 0.0033 according to one-way ANOVA.
(F) C. albicans killing by WT (n = 5) and Mcu−/− (n = 5) BMDMs in the absence and presence of 2 mM citrate. Error bars represent SEM; p = 0.0033 according to one-way ANOVA.
(G) Killing of S. cerevisiae by BMDMs treated with vehicle (n = 7) or 100 μM CPI 613 (n = 7). Error bars represent SEM; p < 0.0001 according to Welch’s t test, two tailed.
(H) Killing of S. cerevisiae by BMDMs treated with vehicle (n = 7) or 100 μM 3-nitropropionic acid (n = 8). Error bars represent SEM; p < 0.0001 according to Welch’s t test, two tailed.
(I) Representative IF images from WT and Mcu−/− BMDMs in response to C. albicans. pPDH (magenta LUT), PDH (yellow LUT), and nucleus (DAPI; blue LUT) were acquired using LSM880 confocal microscope. Scale bar, 10 μm.
(J) Quantification of pPDH/PDH from WT and Mcu−/− macrophages in response to C. albicans. Cells quantified for each time point are 0 min (n = 31), 30 min (n = 23), 60 min (n = 31), and 180 min (n = 22) for wt. For Mcu−/−, n = 40 cells were quantified (all time points). Error bars represent SEM. ***p < 0.001 and ****p < 0.0001; p values were calculated according to two-way ANOVA, two tailed.
mCa2+ Triggers Dephosphorylation and Activation of PDH
As shown above, a major abnormality of the TCA cycle in Mcu−/− macrophages is that citrate and aconitate, the TCA metabolites immediately downstream of PDH, are significantly reduced during phagocytosis (Figures 7C and 7D).
To further substantiate the metabolic salience of the TCA cycle in phagosomal killing, we tested the inhibitors of PDH and succinate dehydrogenase (SDH), two key enzymes in the TCA cycle. Inhibition of PDH and α-ketoglutarate dehydrogenase using CPI 613 and SDH with 3-nitropropionic acid (3-NPA) reduced the killing of S. cerevisiae (Figures 7G and 7H). These results show that the TCA cycle is critical for efficient fungal killing in macrophages. PDC is a tightly regulated multi-component enzymatic complex that catalyzes the conversion of pyruvate into acetyl-coenzyme A (acetyl-CoA). PDH, the catalytic subunit of the PDC, is phospho-regulated by PDH kinases (PDKs) and PDH phosphatases (PDPs) (Gray et al., 2014). Phosphorylation of PDH is inhibitory, whereas dephosphorylation at S293 activates the enzyme, increasing the flux of pyruvate into the TCA cycle. The PDPs are Ca2+-activated phosphatases, and we hypothesized that mCa2+ signaling during phagocytosis can activate the PDC through PDH dephosphorylation. Using antibodies specific for phosphor-PDH, we monitored the activation state of PDH during phagocytosis of C. albicans. Macrophages engulfing C. albicans were stained with antibodies against PDH and phospho-PDH (pPDH). Confocal immunofluorescence microscopy was used to visualize and quantify total PDH and pPDH at multiple time points. These results show striking dephosphorylation of PDH in WT macrophages, but not in Mcu−/− macrophages (Figure 7I). Representative images are shown for WT and Mcu−/− macrophages at 30, 60, and 180 min after exposure to C. albicans (Figures S7C–S7F). Quantification of images enabled the derivation of pPDH/PDH ratio (Figure 7J), and the results clearly demonstrate that dephosphorylation of PDH, a pivotal regulatory event in cellular metabolism, is highly dependent on MCU-mediated mCa2+ signaling initiated for the cell-intrinsic response to C. albicans.
DISCUSSION
Our current understanding of immunometabolic reprogramming relies heavily on transcriptional changes initiated during macrophage activation. Such reprogramming undoubtedly plays an important role during an immune response, but transcriptional changes are too slow to mediate the metabolic burst required by the sentinel phagocytes when they first encounter and engulf a pathogen. How do the phagocytes rapidly switch the primary gears of their cellular metabolism during phagocytosis? Our study provides an important insight into this fundamental question. Although our study focuses on the killing of C. albicans by macrophages, the underlying design principle may be of relevance to fast immunometabolic reprogramming in a variety of scenarios. In essence, we propose that the MCU-dependent mCa2+ signaling is an electrometabolic switch to fuel cell-intrinsic immune responses.
Macrophages recognize C. albicans through the pattern-recognition receptors TLR2 and Dectin-1 (Goodridge et al., 2007). We show that recognition of C. albicans elicits a sharp elevation in cytosolic Ca2+ through SOCE. The phagocytes use this sharp but transient increase in cytosolic Ca2+ to mediate a MCU-dependent influx of Ca2+ into the mitochondrial matrix. The outer membrane of the mitochondria is not a barrier to this process, because it is freely permeable to cations, and once MCU is gated open, the membrane potential (−180 mV) across the inner membrane serves as a strong driving force for Ca2+ entry into the matrix (Kamer and Mootha, 2014). The mechanisms that activate MCU are not fully understood yet, but it is clear that MICU1 and MICU2, the two Ca2+-sensing subunits of the MCU complex, play a key role in gating MCU. A recent study reported the direct gating of the MCU pore by AMPK-mediated phosphorylation (Zhao et al., 2019), but we observed that AMPK inhibition has no impact on mCa2+ signaling triggered by fungal pathogens. Whether the MCU complex is regulated through rapid post-translational modifications during cell-intrinsic immune responses remains an outstanding question that we plan to address in an independent line of investigation. In any case, when macrophages encounter C. albicans, the sharp increases in cytosolic Ca2+ and concurrent gating of MCU mediate a rapid influx of Ca2+ into the matrix of the charged mitochondria.
Inside the mitochondrial matrix, Ca2+ can activate multiple enzyme complexes to stimulate the TCA cycle. These include the PDH, isocitrate dehydrogenase (IDH) and oxoglutarate dehydrogenase (OGDH) (Denton, 2009). As a major determinant of the metabolic fate of pyruvate, the PDC is central node in metabolism, and it is especially sensitive to Ca2+-mediated phosphoregulation. We have shown that in macrophages engulfing C. albicans, the dephosphorylation (activation) of PDH is highly dependent on mCa2+ signaling. These findings are also supported by metabolomics analysis of the TCA cycle. Concomitant regulation of IDH and OGDH is also likely, but this was not investigated in our study. Together, the effect of mCa2+ signaling is rapid and profound. Interestingly, Mcu−/− macrophages can sustain the ATP levels necessary for the immune response, but mCa2+ signaling is critical for increasing cellular NAD(P)H levels. In macrophages phagocytosing C. albicans, there is increased NADPH production to drive NOX2-dependent phagosomal ROS production. The generation of NADPH is largely dependent on the PPP and TCA cycle. During phagosomal killing, we show that inhibition of the PPP has little or no effect on killing, but inhibition of TCA enzymes drastically reduces killing. This suggests that mCa2+ signaling initiated during phagosomal killing drives the production of NADPH through the TCA cycle. Indeed, the defective killing by Mcu−/− macrophages can be rescued by supplementation of citrate, the key TCA intermediate that accumulates in an Mcu-dependent manner. Citrate is known to be exported out of the mitochondria and serve as a substrate for malic-enzyme-mediated NADPH production. The major sources of NADPH in the cell are the PPP, folate metabolism, and malic enzyme. We show that the PPP is dispensable for early pathogen killing, but instead, the TCA cycle, and specifically citrate, are necessary for fungal killing. We have shown that mCa2+ signaling is a key regulator of the TCA cycle, and in the absence of mCa2+ signaling, there is a reduction in citrate levels. Citrate is also of long-term importance to immunometabolism, because it is a necessary intermediate for fatty acid synthesis (Williams and O’Neill, 2018). It is therefore highly likely that over a more extended period, mCa2+ signaling also controls the citrate shuttle necessary for fatty acid synthesis and membrane biogenesis. In addition to the direct regulation of PDH and NADPH levels, mCa2+ signaling may also govern other important anaplerotic demands of phagosomal killing. However, since this study relied on snapshots of metabolite levels and did not evaluate metabolic flux using radioisotope labeling studies, a comprehensive understanding of the metabolic deficits in Mcu−/− macrophages is still pending.
Previous studies have proposed a crucial role for mROS in phagosomal killing of bacteria (West et al., 2011). This study reported that ECSIT-depleted macrophages were deficient in bacterial killing over a 12- to 24-h period because of a deficit in the production of mROS. A more recent study from the same lab (Carneiro et al., 2018) indicates that ECSIT−/− macrophages constitutively generate high levels of mROS but remain deficient in additional production of mROS during TLR stimulation. In our study, both WT and Mcu−/− macrophages increased mROS production comparably in response to zymosan. Mcu−/− macrophages showed no significant difference in the immediate production of mROS production, but when we measured mROS production 18 h after zymosan stimulation, we detected a modest deficit in Mcu−/− macrophages. While it is possible that these modest deficits in mROS production contribute to phagosomal ROS in case of bacterial killing, our findings support a model wherein mCa2+ signaling is a key determinant of NOX-mediated phagosomal ROS production. We propose a simple and elegant design principle that mCa2+ signaling is an electrometabolic switch to fuel phagosomal killing and other cell-intrinsic defense mechanisms.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Bimal N. Desai: bdesai@virginia.edu
Materials Availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
Requests for data should be directed to Lead Contact, Bimal Desai. Data will be made available upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse Strains
Male and female mice aged 7 to 14 weeks were used for all experiments. Mcu(M)fl/fl Cx3cr1 cre mice were generated by crossing B6;129S-Mcutm1.1Jmol/J (Jackson Laboratories; 029817) mice to B6J.B6N(Cg)-Cx3cr1tm1.1(cre)Jung/J (Jackson Laboratories; 025524). Genotyping of Mcu(M)fl/fl was performed using Jax genotyping protocols for B6J.B6N(Cg)-Cx3cr1tm1.1(cre)Jung/J and B6;129S-Mcutm1.1Jmol/J strains. Genetic deletion was confirmed using quantitative real-time PCR analysis, western blot analysis, and mitochondrial Ca2+ uptake assays on isolated mitochondria. Mice were housed and bred in accordance with policies and procedures of the University of Virginia Institutional Animal Care and Use Committee (IACUC).
Cell lines and Cell Culture
The complete list of cell lines used in this study are given in the Key Resources Table. All cell lines were grown at 37°C 5% CO2 and maintained according to ATCC guidelines. Bone marrow-derived macrophages (BMDMs) were isolated and cultured as previously described (Schappe et al., 2018). In brief, bone marrow was extracted from mouse femur and tibia via centrifugation. The RBCs were lysed with ACK lysis buffer and the remaining cells were counted and plated on Petri dishes at a density of 2–4×106 cells/ plates in BMDM Media (RPMI 1640 + 10% FBS + 20% L929-conditioned media). Cells were differentiated for 7 days and media was replaced every 3 days. For experiments BMDMs were used between days 9 – 14 post-harvest. RAW264.7 cell line (ATCC TIB-71) was maintained according to ATCC guidelines.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Pyruvate Dehydrogenase E1-alpha subunit antibody [9H9AF5] | Abcam | Abcam Cat# ab110330, RRID:AB_10858459 |
| Recombinant Anti-Pyruvate Dehydrogenase E1-alpha subunit (phospho S293) antibody [EPR12200] | Abcam | Abcam Cat# ab177461, RRID:AB_2756339 |
| Anti-MCU antibody | Abcam | Cat#ab121499 |
| Tom20 (D8T4N) Rabbit mAb | Cell Signaling Technology | Cell Signaling Technology Cat# 42406, RRID:AB_2687663 |
| Pyruvate Dehydrogenase (C54G1) Rabbit mAb | Cell Signaling Technology | Cell Signaling Technology Cat# 3205, RRID:AB_2162926 |
| Bacterial and Virus Strains | ||
| pCMV-CEPIA3mt | Suzuki et al. | Addgene Cat#58219, RRID: addgene_58219 |
| Lenti-X Packaging Single Shots (VSV-G) | Takara | Cat#631275 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Carbonyl cyanide 4-(trifluoromethoxy) phenylhydrazone (FCCP) | Sigma | Cat# C2920-10MG |
| JC-1 Dye (Mitochondrial Membrane Potential Probe) | ThermoFisher Scientific | Cat#T3168 |
| Calcium Green-5N, Hexapotassium Salt, cell impermeant | ThermoFisher Scientific | Cat#C3737 |
| Fura-2, AM, cell permeant | ThermoFisher Scientific | Cat#F1221 |
| Probenecid | Enzo | Cat# ALX-430-113-G005 |
| Pluronic F-127 (20% solution in DMSO) | ThermoFisher Scientific | Cat#P3000MP |
| Ionomycin | Cayman Chemical | Cat#10004974 |
| MitoTracker Red | ThermoFisher Scientific | Cat#M22425 |
| CypHer5E NHS Ester | GE healthcare | Cat#PA15401 |
| BAM15 | Cayman Chemical | Cat#17811 |
| Antimycin A | Sigma | Cat# A8674-25MG |
| Rotenone | Sigma | Cat#R8875-1G |
| Zymosan A S. cerevisiae BioParticles, Texas Red conjugate | ThermoFisher Scientific | Cat#Z2843 |
| Zymosan A S. cerevisiae BioParticles, unlabeled | ThermoFisher Scientific | Cat#Z2849 |
| Dorsomorphin dihydrochloride | Tocris | Cat#3093 |
| 6-Aminonicotinamide | Cayman Chemical | Cat#10009315 |
| 6,8-Bid(benzylthio)octanoic acid | Tocris | Cat#5348 |
| BAPTA-AM, cell permeant chelator | ThermoFisher Scientific | Cat#B6769 |
| Beta-glucan peptide | Invivogen | Cat#tlrl-bgp |
| Thapsigargin | ThermoFisher Scientific | Cat#T7458 |
| Critical Commercial Assays | ||
| CellTiter-Glo® 2.0 Cell Viability Assay | Promega | Cat#G9241 |
| CellTrace Violet Cell Proliferation Kit, for flow cytometry | ThermoFisher Scientific | Cat#C34557 |
| CellTrace Yellow Cell Proliferation Kit, for flow cytometry | ThermoFisher Scientific | Cat#C34567 |
| CellROX Green Reagent, for oxidative stress detection | ThermoFisher Scientific | Cat#C10444 |
| Pierce LDH Cytotoxicity Assay Kit | ThermoFisher Scientific | Cat#88953 |
| Experimental Models: Cell Lines | ||
| RAW264.7 ATCC TIB-71 | ATCC | ATCC Cat# TIB-71, RRID:CVCL_0493 |
| Experimental Models: Organisms/Strains | ||
| S. cerevisiae sy1022 fy5 | This paper | Gift: Jeff Smith (UVA) |
| Culti-Loops Candida albicans ATCC 10231 | ThermoFisher Scientific | Cat#R4601503 |
| Mouse: B6;129S-Mcutm1.1Jmol/J | The Jackson Laboratory | IMSR Cat# JAX:029817, RRID:IMSR_JAX:029817 |
| Mouse: B6J.B6N(Cg)-Cx3cr1tm1.1(cre)/Jung/J | The Jackson Laboratory | IMSR Cat# JAX:025524, RRID:IMSR_JAX:025524 |
| Oligonucleotides | ||
| MCU Exon 5 Forward Primer | This Paper | N/A |
| 5’-TGAACGACGTGAAGACCCT G-3’ | ||
| MCU Exon 5 Reverse Primer | This Paper | N/A |
| 5’-TTCGTACCTTCTCCAGGGGG-3’ | ||
| Software and Algorithms | ||
| ImageJ | https://imagej.nih.gov/ij/ | |
| SPCM | Wallrabe et al., 2018 | https://www.becker-hickl.com/products/spcm/ |
| Graphpad Prism | GraphPad Prism | https://www.graphpad.com/scientific-software/prism/ |
| Seahorse Wave | Agilent | https://www.agilent.com/en/products/cell-analysis/software-download-for-wave-desktop |
| Other | ||
| CFX Connect Real-Time system (Bio-Rad) | qPCR | N/A |
| XF24 Extracellular Flux Analyzer (Agilent Technologies) | Seahorse | N/A |
| FlexStation 3 | In vitro mitochondrial Ca2+ uptake, LDH assay, ATP assay | N/A |
| Leica SP5 × Confocal/Spectral Imaging Microscopy System with White Light Laser | Live mCa2+ imaging for RAW-3mt | N/A |
| Zeiss Axio Observer with DG4 Illuminator and ORCA-Flash 4.0 V2 CMOS camera | Fura-2 Ca2+ imaging | N/A |
| LSM880 confocal and a Chameleon multiphoton light source | Immunofluorescence | N/A |
| Attune NxT equipped with four lasers (488nm, 640nm, 405nm, and 561nm) | Flow Cytometry | N/A |
| JEOL 1230 Transmission Electron Microscope | TEM | N/A |
| FUIJ Film LAS-4000 | Western Blotting | N/A |
METHOD DETAILS
Ca2+ Channel siRNA screen
RAW264.7 cells were collected and aliquoted into individual cell suspension for each siRNA target. RAW264.7 cells were resuspended in OptiMem (ThermoFisher; 31985062) with 10 nM siRNA (Dharmacon) and Lipofectamine 3000 (ThermoFisher; L3000015), according to manufacturer’s instructions. Cells were plated at 0.5×106 cells/mL in 6-well tissue culture treated plates for 48 h. After 48 h, cells were washed 3X with HBSS and detached by gentle scraping. Cells were then counted and 0.5×106 cells were aliquoted into individual tubes for each siRNA target and resuspended in siRNA mix as described above. Cells were plated at 0.1×106 cells/well in 24-well tissue culture treated plate for another 48h. A single colony of C. albicans was grown overnight at 30°C in 5 mL YPD broth. On the day of the experiment, C. albicans were washed 3X in complete media and counted using OD600. C. albicans were resuspended in complete media at desired multiplicity of infection (MOI = 1). RAW264.7 cells were washed 3X with HBSS and C. albicans were added to each well at MOI1 in 100uL complete media. RAW cells were incubated with C. albicans for 30 minutes at 37°C 5% CO2. After 30 minutes, cells were washed 3X with HBSS to remove non-engulfed C. albicans and resuspended in complete media. One plate of cells was harvested at this 30-minute time point and lysed in sterile diH2O for 30 minutes before being serial diluted and plated on YPD agar plates for T0 colony forming units (CFUs). Remaining cells were grown for 8h (T2). Cells were harvested as described above. Percent killing was calculated by the formula (T0-T2)/(T0)*100. Gene knockdown was confirmed via quantitative real-time PCR. Z-scores were calculated as the difference in percent killing between siRNA and siScramble-treated cells for a given siRNA target, divided by the standard deviation of the dataset; z-scores across the experiment were then averaged and plotted as shown.
In vivo (mouse) disseminated candidiasis model
In vivo candidiasis was performed as described (Conti et al., 2014). In brief, WT and Mcu(M)−/− mice (ages between 7–12 weeks) were tail vein injected with 100 μL of 1×106 viable C. albicans. Mice were monitored every 3 h for first 24 h and every 6 h for remainder of the study for disease symptoms using a blinded clinical score system (Supplemental Methods S1). Mice were euthanized at humane endpoints: > 25% loss in body weight or > 7 clinical score for two consecutive time points. Mice kidneys and livers were harvested for CFU analysis and histology. Serum was analyzed by Flow Cytometry for luminex analysis.
Isolation of mitochondria
Bone-marrow derived macrophages from WT and Mcu−/− mice were lysed in IB++ buffer (100 mM KCl, 50 mM Tris-HCl pH 7.4, 2 mM EDTA-KOH pH 7.4, 0.5% fatty acid-free BSA, 1X protease inhibitors (Roche)). Lysates were homogenized at 4°C using a dounce homogenizer. Whole cell fraction was spun at 600xg for 10 min at 4°C. Supernatant was transferred to prechilled Eppendorf tubes and centrifuged at 8000xg for 10 min at 4°C. Mitochondria were resuspended in IB++ w/o BSA for BCA.
Killing assays
RAW264.7 and BMDM cells were incubated with C. albicans or S. cerevisiae for 30 min (37°C, 5% CO2). After 30 min, cells were washed 3X with HBSS to remove non-engulfed fungus and resuspended in complete media. One plate of cells was harvested at this 30 min time point and lysed in sterile distilled H2O for 30 min before being serially diluted and plated on YPD agar plates for T0 colony forming units (CFUs). Remaining cells were co-incubated with C. albicans for indicated times (T2). C. albicans percent survival was calculated by the formula (T2/T0)*100.
In vitro Mitochondrial Ca2+ uptake assay
Crude mitochondria from WT and Mcu−/− macrophages were diluted to 1 mg/mL in RB++ (137 mM KCl, 10 mM HEPES-KOH, 2.5 mM MgCl2, 3 mM KH2PO4−, 25 μM EDTA, 5 mM Succinate, 5 mM glutamate, 5 mM malate and 0.4 nM Ca2+ Green-5N (pH = 7.4) and added to a 96-well glass bottom black wall plate (CellVis, Cat#P96–1-N). Mitochondrial Ca2+ uptake was recorded on a FlexStation 3 with excitation/emission 515/530nm. 45 μM CaCl2 was injected at indicated time point. Fluorescence was recorded every 2 s for the duration of experiment.
Preparation of fungal aqueous extracts
Single colonies of C. albicans or S. cerevisiae were grown overnight in 5mL of YPD at 30°C. Cells were washed 3x in BMDM Media (RPMI 1640 + 10% FBS + 20% L929-conditioned media). Cells were counted using OD600 and normalized to 4×107 cells/mL and incubated at 4°C overnight. The following day cells were heat-killed at 95°C for 30 min and centrifuged at 10000xg for 10 min to pellet cell debris. The resulting supernatants (C. albicans extracts) was at 5X concentration for experiments and used to stimulated macrophages for indicated experiments.
LDH Release Assay
BMDM cells were incubated with C. albicans for 30 min at 37°C, 5% CO2. After 30 min, cells were washed 3X with HBSS to remove un-engulfed C. albicans and resuspended in complete media. After 6 h, LDH was measured in the supernatants and Triton-X-treated positive controls. For zymosan treatment, cells were stimulated with zymosan (2:1, particles:cell) for 6 h before measuring LDH in the resulting supernatant. LDH was measured with Pierce™ LDH Cytotoxicity Assay Kit (ThermoScientific) according to manufacturer instructions using a Flexstation 3 plate reader.
Generation of RAW3mt stable cell line
RAW264.7 macrophages were transduced with CEPIA3mt lentivirus and seeded on 6-well tissue culture treated plates. Transduced cells were replenished with Selection Media (DMEM + 10% FBS + 1mg/mL puromycin) every 24h for 10 days. Surviving cells were counted and single cell cloned on 96 well plates. Single colonies were selected for validation of CEPIA3mt protein and Ca2+ sensitivity.
Live cell mitochondrial Ca2+ imaging during zymosan phagocytosis
RAW3mt cells were plated overnight on coverslips prior to imaging. Coverslips were washed 1x in Ringer Solution (155 mM NaCl, 4.5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 5 mM HEPES, 10 mM D-glucose) and placed in the imaging chamber. Coverslips were imaged at 37°C ± 1°C. Microscopy was performed with an open pinhole on the Leica SP5 microscope with excitation from ‘white light’ and 488 nm argon lasers using Leica Applicate Suite Software (Leica). The images were processed using ImageJ software. Texas Red-conjugated zymosan A S. cerevisiae Bioparticles (ThermoFisher; Z2843) were fed to macrophages on coverslips and imaged for phagocytosis. “Whole cell mCa2+” was analyzed by drawing ROIs around individual cells during zymosan phagocytosis. For measurements of PPMiCa response, data analysis was performed as described in Extended Figure 5B.
Transmission Electron Microscopy (TEM)
BMDMs were seeded onto 13 mm diameter Thermanox plastic coverslips (EMS; #72280) at 0.1×106 cell per well in a 24-well plate. C. albicans and Zymosan were fed for 30 min (37°C, 5% CO2). After 30 min, cells were washed 3X with HBSS to remove un-engulfed C. albicans and resuspended in complete media. Cells were then cultured for indicated times before fixation (4% PFA and 2.5% EM-grade glutaraldehyde). Samples were submitted to the Advanced Microscopy Facility Core (UVA), where they were stained and cut for imaging on JOEL 1230 TEM.
Cytosolic Ca2+ imaging using ratiometric Fura-2
For ratiometric Ca2+ imaging, macrophages were incubated for 30 min with gentle agitation at RT with 5 μM Fura-2-AM, 0.02% of pluronic acid and 500 μM probenecid in Ringer solution ([in mM] 155 NaCl, 4.5 KCl, 2 CaCl2, 1 MgCl2, 5 HEPES, 10 glucose, pH 7.4). Fura-2 emissions were collected at 510 nm and with 340/380 nm excitation. Excitation was performed using a DG4 Illuminator (Sutter Instruments) and fluorescence was detected using an ORCA-Flash 4.0 V2 CMOS camera (Hamamatsu) using SlideBook 6 software. Cells were imaged every 10 s for the duration of the experiment. C. albicans extracts and ionomycin were perfused at indicated times.
ATP Measurements
BMDM cells were plated at 2×105 cells per well in a 96-well plate overnight (~16 h). On the day of the experiment, the wells were treated in triplicated for indicated times with zymosan (2:1, particles:cell). Cells were lysed and ATP was measured using the Cell-Titer-Glo® Luminescent cell viability assay (Promega) according to manufacturer’s instruction.
Flow cytometry
BMDMs were plated at 4×105 cells per well in a 12-well non-tissue culture treated plate overnight (~16 h). A single colony of C. albicans was grown overnight at 30°C in 5mL YPD broth. On the day of the experiment, C. albicans were washed 3X in complete media and counted using OD600. C. albicans were resuspended in complete media at desired multiplicity of infection (MOI = 1). C. albicans were washed in 3X in HBSS (w/ Mg2+ and Ca2+) and stained with 10 μM CellTrace dye and/or 10 μM CellRox dye, 2 μM CypHer5E dye for 45 min at 37°C and 5% CO2. Labeled C. albicans were washed 3X in complete media and added to macrophages. Cells with centrifuged for 5 min at 500 xg to facilitate macrophage phagocytosis. Macrophages were incubated with labeled C. albicans for 30 min (37°C, 5% CO2). After 30 min, cells were washed 3X with HBSS to remove un-engulfed C. albicans and resuspended in complete media. Macrophages were then incubated for indicated times. Prior to flow cytometry, cells were detached from wells using 0.25% trypsin + EDTA, washed 2X with HBSS and analyzed using Attune NxT flow cytometer. CellTrace violet was detected using the 405nm laser with bandpass filter 440/50. CellRox was detected using 488nm laser with bandpass filter 530/30. CypHer was detected using 640nm laser with bandpass filter 670/14. Macrophages exposed to unlabeled yeast were used for gating controls. Gating strategies are shown in the extended figures.
Mitochondrial ROS
BMDMs were plated into a 96-well plate at a 200,000 cells per well. Cells were stained with MitoSox according to manufacturer’s protocol and imaged using the FlexStation 3 (Molecular Devices). Zymosan or vehicle control was added to the wells prior to imaging. MitoSox fluorescence intensity was measured every 5 min (Ex/ Em 510/580) for 1 hour.
Fluorescence Lifetime Imaging (FLIM) Microscopy
For imaging BMDMs, cells were plated on 25 mm round #1.5 glass coverslips (overnight) to obtain 70%–90% confluence. Cells were washed 1X in Ringer Solution (155 mM NaCl, 4.5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 5 mM HEPES, 10 mM D-glucose) and placed in imaging chamber at 37°C ± 1°C for imaging. Baseline NAD(P)H was monitored followed by the addition of C. albicans extract. NAD(P) H lifetime was monitored for indicated times. Cells were imaged on a Zeiss LSM-780 NLO confocal/multiphoton microscopy system with Chameleon Vission-II (Coherent Inc.) ultrafast Ti:sapphire laser with dispersion compensation (W.M. Keck Center UVA). Channel 2 460–500nm was used to collect NAD(P)H signal. Time-resolved fluorescence is recorded in Time correlated single photon counting (TCSPC) mode (256 × 256) using a Becker-Hickl TCSPC module and SPCM (9.74) acquisition software. Note that TCSPC is a highly sensitive technique for recording low-level light signals with high time resolution and high precision but the spatial resolution is low (256 × 256).
Seahorse Assays
Extracellular flux assays were performed on the XF24 Extracellular Flux Analyzer (Agilent Technologies). Cells were plated at a density of 1×105 cells per well overnight (~16h, 37°C, 5% CO2). At beginning of the experiment, assay media was changed to DMEM containing pyruvate (Thermo-Fisher Cat#12800017; pH = 7.35 at 37°C) and cells were equilibrated for 30 min. OCR (pmol 02/min) was measured every 5 min for the first hour of recording and then changed to 15 min intervals for the remainder of experiment (indicated by gray bar on figure). Zymosan (2:1 particles:cell) particles were injected following 3 baseline measurements of OCR. To measure maximal OCR, 2 μM BAM15 was injected at the indicated time point. Antimycin A (1 μM) and Rotenone (100 nM) were injected at the end of experiment. Respiration was calculated by subtracting the average of the post-Antimycin A and Rotenone measurements from the recorded measurements.
Immunofluorescence
Cells were plated overnight on coverslips prior to experiments. Following treatments, coverslips were washed 3x in PBS to remove media and loose/non-adherent cells. Coverslips were fixed in 4% PFA (30 min, RT). Coverslips were washed 3X in wash buffer (PBS with 0.05% Tween-20), blocked and permeabilized at RT for 1 h in B/P buffer (2.5% donkey serum, 2.5% goat serum, 1% BSA, 0.1% fish gelatin, 0.1% Triton X-100, and 0.05% Tween-20 in PBS), and then incubated with primary antibody diluted in B/P buffer overnight at 4°C. Coverslips were washed 3X in wash buffer and incubated at RT with the appropriate secondary antibody in B/P buffer for 2 h, followed by 3X washes in wash buffer. Coverslips were mounted on glass slides (ProLong Gold Antifade; ThermoFisher #P36930), stored in a desiccated box at 4°C and imaged within 48h. Confocal microscopy was performed on Zeiss LSM880. Data were acquired with Zen Black and analyzed using ImageJ.
Metabolomics
Confluent 12-cm plates of bone-marrow derived macrophages were used for each condition with 3–5 replicates per condition. Cells were plated overnight prior to experiment. On the day of the experiment, plates were stimulated with zymosan (2:1 particles:cell) or vehicle control for indicated time points (3 hours or 6 hours). Following stimulation, cells were washed 3X in HBSS and packed into 50–100uL pellets by centrifugation at 1000 xg for 5 min. Cells were resuspended in HBSS and transferred to pre-labeled 2.0 mL polypropylene tubes. Cells were centrifuged at 1000 xg for 5 min. Supernatant was removed and pellets were flash frozen in liquid nitrogen and stored at −80°C until shipped to Metabolon Inc. for analysis. MetaboAnalyst was used for data processing and visualization. Volcano plots were generated in Graphpad Prism by plotting the −log(p value) relative to the Log10 (fold change) for individual metabolites.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics
All data were analyzed using Excel (Microsoft) and GraphPad Prism 8 (GraphPad) software. Data are presented as means with error bars which reflect standard error of the mean (SEM) as indicated in figure legends. Statistical significance (p < 0.05) was computed using one-way ANOVA, 2-way ANOVA, and welch’s t test (two-tailed) as indicated in figure legends. Survival curves were analyzed using Kaplan-Meier method and statistical significance (p < 0.05) was computed using Log-rank (Mantel-Cox) test. The sample size and representation of ‘n’ (mice, experimental repeats, or cells) is indicated in figure legends.
Supplementary Material
Highlights.
Identification of MCU as a major regulator of phagosomal killing by macrophages
Mice lacking Mcu in myeloid cells are highly susceptible to in vivo candidiasis
Fungal pathogens elicit mCa2+ elevations using a fast two-phase Ca2+ relay
mCa2+ signaling activates pyruvate dehydrogenase during phagocytosis
ACKNOWLEDGMENTS
We thank the members of the Desai and Leitinger laboratories for scientific insights and technical help. We also thank Drs. Hervé Agaisse (UVA), Doug Bayliss (UVA), Michelle Bland (UVA), Sarah Ewald (UVA), and Kevin Lynch (UVA) for helpful and timely advice. We thank the lab of Dr. Sarah Ewald (UVA) for the use of an LSM880 microscope and the W.M. Keck Center for Cellular Imaging (UVA) for the use of Zeiss 780 multiphoton FLIM microscope and Leica SP5X confocal; these instruments are supported by NIH instrumentation grants OD016446 and RR025616, respectively (A.P.). We also thank the following core facilities for their technical resources and support: UVA Flow Cytometry Core, Carter Immunology Center Flow Cytometry Core, Advanced Microscopy Facility Core (UVA), and the UVA Research Histology Core. The work was predominantly funded by NIH research grants GM108989 (B.N.D), T32 GM007055-46 (P.V.S), and AG067048 (A.P.).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2020.108411.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Altenhöfer S, Radermacher KA, Kleikers PW, Wingler K, and Schmidt HH (2015). Evolution of NADPH oxidase inhibitors: selectivity and mechanisms for target engagement. Antioxid. Redox Signal 23, 406–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blacker TS, Mann ZF, Gale JE, Ziegler M, Bain AJ, Szabadkai G, and Duchen MR (2014). Separating NADH and NADPH fluorescence in live cells and tissues using FLIM. Nat. Commun 5, 3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown GD (2011). Innate antifungal immunity: the key role of phagocytes. Annu. Rev. Immunol 29, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck MD, Sowell RT, Kaech SM, and Pearce EL (2017). Metabolic instruction of immunity. Cell 169, 570–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carneiro FRG, Lepelley A, Seeley JJ, Hayden MS, and Ghosh S (2018). An essential role for ECSIT in mitochondrial complex I assembly and mitophagy in macrophages. Cell Rep. 22, 2654–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri D, Sancak Y, Mootha VK, and Clapham DE (2013). MCU encodes the pore conducting mitochondrial calcium currents. eLife 2, e00704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conti HR, Huppler AR, Whibley N, and Gaffen SL (2014). Animal models for candidiasis. Curr. Protoc. Immunol 105, 19.16.11–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordas G, Renken C, Varnai P, Walter L, Weaver D, Buttle KF, Balla T, Mannella CA, and Hajnoczky G (2008). Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Neurochem 104, 6–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordás G, Várnai P, Golenár T, Roy S, Purkins G, Schneider TG, Balla T, and Hajnóczky G (2010). Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol. Cell 39, 121–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies LC, Rice CM, McVicar DW, and Weiss JM (2019). Diversity and environmental adaptation of phagocytic cell metabolism. J. Leukoc. Biol 105, 37–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefani D, Raffaello A, Teardo E, Szabò I, and Rizzuto R (2011). A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton RM (2009). Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 1787, 1309–1316. [DOI] [PubMed] [Google Scholar]
- Gazendam RP, van Hamme JL, Tool AT, van Houdt M, Verkuijlen PJ, Herbst M, Liese JG, van de Veerdonk FL, Roos D, van den Berg TK, and Kuijpers TW (2014). Two independent killing mechanisms of Candida albicans by human neutrophils: evidence from innate immunity defects. Blood 124, 590–597. [DOI] [PubMed] [Google Scholar]
- Geng J, Sun X, Wang P, Zhang S, Wang X, Wu H, Hong L, Xie C, Li X, Zhao H, et al. (2015). Kinases Mst1 and Mst2 positively regulate phagocytic induction of reactive oxygen species and bactericidal activity. Nat. Immunol 16, 1142–1152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodridge SH, Simmons RM, and Underhill DM (2007). Dectin-1 stimulation by Candida albicans yeast or zymosan triggers NFAT activation in macrophages and dendritic cells. J. Immunol 178, 3107–3115. [DOI] [PubMed] [Google Scholar]
- Gray LR, Tompkins SC, and Taylor EB (2014). Regulation of pyruvate metabolism and human disease. Cell. Mol. Life Sci 71, 2577–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez-López C, and Lorenz MC (2013). Fungal immune evasion in a model host-pathogen interaction: Candida albicans versus macrophages. PLoS Pathog. 9, e1003741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamer KJ, and Mootha VK (2014). MICU1 and MICU2 play nonredundant roles in the regulation of the mitochondrial calcium uniporter. EMBO Rep. 15, 299–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamer KJ, and Mootha VK (2015). The molecular era of the mitochondrial calcium uniporter. Nat. Rev. Mol. Cell Biol 16, 545–553. [DOI] [PubMed] [Google Scholar]
- Kinchen JM, and Ravichandran KS (2008). Phagosome maturation: going through the acid test. Nat. Rev. Mol. Cell Biol 9, 781–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirichok Y, Krapivinsky G, and Clapham DE (2004). The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427, 360–364. [DOI] [PubMed] [Google Scholar]
- Mallilankaraman K, Doonan P, Cárdenas C, Chandramoorthy HC, Müller M, Miller R, Hoffman NE, Gandhirajan RK, Molgó J, Birnbaum MJ, et al. (2012). MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca(2+) uptake that regulates cell survival. Cell 151, 630–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, Jedrychowski MP, Costa ASH, Higgins M, Hams E, et al. (2018). Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 556, 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nauseef WM (2019). The phagocyte NOX2 NADPH oxidase in microbial killing and cell signaling. Curr. Opin. Immunol 60, 130–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Neill LA, Kishton RJ, and Rathmell J (2016). A guide to immunometabolism for immunologists. Nat. Rev. Immunol 16, 553–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MAR, Sheedy FJ, Gleeson LE, van den Bosch MWM, Quinn SR, Domingo-Fernandez R, Johnston DGW, et al. (2015). Pyruvate kinase M2 regulates Hif-1α activity and IL-1β induction and is a critical determinant of the warburg effect in LPS-activated macrophages. Cell Metab. 21, 65–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patron M, Checchetto V, Raffaello A, Teardo E, Vecellio Reane D, Mantoan M, Granatiero V, Szabò I, De Stefani D, and Rizzuto R (2014). MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol. Cell 53, 726–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schappe MS, Szteyn K, Stremska ME, Mendu SK, Downs TK, Seegren PV, Mahoney MA, Dixit S, Krupa JK, Stipes EJ, et al. (2018). Chanzyme TRPM7 mediates the Ca(2+) influx essential for lipopolysaccharide-induced Toll-like receptor 4 endocytosis and macrophage activation. Immunity 48, 59–74.e55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strijbis K, Tafesse FG, Fairn GD, Witte MD, Dougan SK, Watson N, Spooner E, Esteban A, Vyas VK, Fink GR, et al. (2013). Bruton’s tyrosine kinase (BTK) and Vav1 contribute to Dectin1-dependent phagocytosis of Candida albicans in macrophages. PLoS Pathog. 9, e1003446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki J, Kanemaru K, Ishii K, Ohkura M, Okubo Y, and Iino M (2014). Imaging intraorganellar Ca2+ at subcellular resolution using CEPIA. Nat. Commun 5, 4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uwamahoro N, Verma-Gaur J, Shen HH, Qu Y, Lewis R, Lu J, Bambery K, Masters SL, Vince JE, Naderer T, and Traven A (2014). The pathogen Candida albicans hijacks pyroptosis for escape from macrophages. MBio 5, e00003–e00014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallrabe H, Svindrych Z, Alam SR, Siller KH, Wang T, Kashatus D, Hu S, and Periasamy A (2018). Segmented cell analyses to measure redox states of autofluorescent NAD(P)H, FAD & Trp in cancer cells by FLIM. Sci. Rep 8, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang A, Luan HH, and Medzhitov R (2019). An evolutionary perspective on immunometabolism. Science 363, 363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West AP, Brodsky IE, Rahner C, Woo DK, Erdjument-Bromage H, Tempst P, Walsh MC, Choi Y, Shadel GS, and Ghosh S (2011). TLR signalling augments macrophage bactericidal activity through mitochondrial ROS. Nature 472, 476–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams NC, and O’Neill LAJ (2018). A role for the Krebs cycle intermediate citrate in metabolic reprogramming in innate immunity and inflammation. Front. Immunol 9, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Li T, Wang K, Zhao F, Chen J, Xu G, Zhao J, Li T, Chen L, Li L, et al. (2019). AMPK-mediated activation of MCU stimulates mitochondrial Ca2+ entry to promote mitotic progression. Nat. Cell Biol 21, 476–486. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Requests for data should be directed to Lead Contact, Bimal Desai. Data will be made available upon request.