

Abstract
Cancer stem cells (CSCs) generate and sustain tumors due to tumor initiating potential, resulting in recurrence or metastasis. We showed that knockout of the cell cycle inhibitor, p21CIP1, in the PyMT mammary tumor model, inhibits metastasis; however the mechanism remained unknown. Here, we show a pivotal role for p21 in potentiating a cancer stem-like phenotype. p21 knockout in PyMT mammary tumor cells caused dramatic suppression of cancer stem cell properties involving tumorsphere formation, ALDH1 activity, and tumor initiating potential, which were in turn rescued by p21 overexpression into PyMT/p21 knockout cells. Interestingly, p21 knockout dramatically suppresses Wnt/β-catenin signaling as shown by striking inhibition of Wnt3a-stimulated expression of LEF1 and TCF1 transcription factors. TCF1 knockdown in PyMT cells suppressed tumorsphere formation due to Cyclin D1 attenuation. These data demonstrate that p21 promotes a cancer stem cell like phenotype via activation of Wnt/TCF1/Cyclin D1 signaling.
Implication: p21 is a strong promoter of mammary cancer stem cells.
Keywords: p21CIP1, cancer initiating cells, Wnt signaling, TCF1, Cyclin D1, metastasis
Introduction
Cancer stem cells (CSCs) are thought to cause tumor relapse or metastasis due to their ability to indefinitely replenish cancer growth at primary and metastatic sites (1, 2). CSCs constitute a sub-population of cells that is endowed with self-renewing capability, promoted by asymmetric cell divisions which preserve the stem cell pool while giving rise to differentiated progenitors (3–5). CSCs are often maintained in a quiescent state that prevents hyperproliferation and consequent cellular exhaustion. One molecule known to drive the quiescence of stem cells is p21CIP1 (p21), a cell cycle inhibitor, that was shown to protect the hematopoietic stem cell pool from depletion (6, 7). Moreover, p21-mediated cell cycle arrest is necessary for DNA repair that prevents onset of mutations that cause genomic instability and elimination of the CSC pool (8). In further support of this idea, others have shown that inhibitor of differentiation (ID) genes that maintain cancer initiating cells in a undifferentiated stem cell state as well as initiate metastatic activity, upregulate p21. p21 is thought to prevent accumulation of excess DNA damage and subsequent exhaustion of the CSC pool that is necessary for replenished tumor growth (9) (8). Hence, cell cycle arrest by p21 is essential for maintaining quiescence and genomic integrity to prevent CSC exhaustion due to excess proliferation or DNA damage, that may be caused by mutations or chemotherapeutic insults.
We previously showed that p21 knockout in the PyMT mammary tumor model suppresses metastasis (10). Interestingly, this effect was associated with mesenchymal to epithelial transition that results in differentiation and inhibition of cell migration. These results suggested that p21 supports an undifferentiated-mesenchymal state that is conducive of stemness (10). While studies have shown that p21 loss results in CSC depletion due to excessive proliferation (6, 7), (11), none have addressed the possibility that p21 may also promote stemness via activation of signaling that regulates CSC proliferation and/or differentiation.
Here we demonstrate a novel mechanism whereby p21 promotes a cancer stem-like phenotype. We show that p21 knockout in the PyMT mammary tumor model inhibits tumorsphere formation, Aldefluor activity, and importantly tumor initiating potential, that were in turn rescued by p21 overexpression in PyMT/p21 knockout cells. Interestingly, p21 ablation led to complete inhibition of Wnt/β-catenin signaling which was due to TCF1 downregulation. TCF1 knockdown in PyMT tumor cells suppressed Wnt signaling, mammosphere formation which was due to Cyclin D1 attenuation. Thus, p21 acts as a central regulator of the mammary CSC like phenotype via activation of Wnt/TCF1/Cyclin D1 signaling.
Materials and Methods
Animals
Female FVB mice and athymic nude mice were obtained from Charles River. Animal protocols of this study were approved by Institute for Animal Studies at Albert Einstein College of Medicine (AECOM).
DNA constructs and reagents
p21CIP1 expression vector was obtained from Dr. Stuart Aaronson (Mount Sinai School of Medicine, New York, NY, USA). Mouse p21CIP1 shRNA and control shRNA were obtained from Santa Cruz Biotechnology Inc. (Santa Cruz, CA, USA). Mouse LEF1, TCF1, Cyclin D1 shRNA clones and non-silencing control shRNA in the pLKO.1 lentiviral vector (Open Biosystems) used to knock down these molecules were obtained from the genomic core facility at Einstein. To generate viruses, lentiviral vectors were transfected into 293T cells with Tat, Rev, Gag/Pol, and VSV-G vectors. TOPFlash, FOPFlash and Renilla plasmids were obtained from Millipore.
Lentivirus production
Lentiviral particles were generated by transient co-transfection of 293T cells with lentivirus-based vector expressing either the full length clone of desired gene or an shRNA sequence to target specific RNA of gene. Briefly, a 100 mm dish seeded with 3 × 106 cells were transfected with 0.6 μg of lentiviral packaging gene TAT, RVE, GAG/POL and 1.2 μg of VSV-G and 12 μg of DNA of interest in lentiviral backbone. Fugene was used to transfect the cells. Following 48 hrs of transfection, supernatant was collected, centrifuged at 2000 rpm for 10 min and filter sterilized.
Cell lines
PyMT and PyMT/p21KO primary mammary tumor cell lines were derived from the PyMT mouse or the PyMT/p21KO mouse as described in detail (10). PyMT and PyMT/p21 KO mice were in a (Bl6/129S/FVB background) mixed background (10). Met-1, is a PyMT mammary tumor cell line that was derived from an MMTV-PyMT mouse in a FVB background. This cell line was serially transplanted into the mammary fat pad of FVB female mice to generate a highly metastatic mammary tumor cell line, known as Met-1 (12). All cell lines were routinely tested for Mycoplasma, and the genetic identity of the cell lines was confirmed by qPCR for expression of the mouse PyMT oncogene. The cell lines were maximally used within 10 passages in culture.
Wnt3a conditioned media
L-cells expressing Wnt3a were a kind gift from Dr. Stuart Aaronson (Mount Sinai School of Medicine, NY). L/Wnt3a cells were cultured in DMEM/10% FBS and allowed to grow for 3 days till they reach confluency. Media was collected, filtered and mixed 1:1 with DMEM/10% FBS for stimulation of cells.
Antibodies
The antibodies used are against p21 (Milipore), β-actin (Sigma); Slug, LEF1, (Cell Signaling Technology); and Sox9, active-β-catenin, β-catenin, TCF-4 (Millipore). Antibodies against TCF1, Cyclin D1, p-LRP6 and LRP6 were obtained from Santa Cruz Biotechnology.
Immunoblotting
Cells or tissues were extracted in solubilization buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 0.5mM MgCl2, 0.2mM EGTA, 1% Triton X-100) including protease inhibitors. Cells were extracted in solubilization buffer including 1% SDS and sonicated. Thirty micrograms protein were loaded on 7–12% SDS–polyacrylamide gels and transferred onto Immobilon membranes. Blots were probed overnight at 4 °C with indicated antibodies and developed by chemiluminescence using ECL detection reagents (Pierce, Thermo Scientific, Rockford, IL, USA).
Cell Transduction
Cells were seeded at a density of 9 × 104 cells per well of 12 well plates. Next day 1 μg of polybrene was added to viral solution, mixed well and 250 μl was added dropwise to 4 different wells. After gently mixing, plates were placed in incubator for 1 hr with rocking for every 15 min. After 1 hr, DMEM containing 10 % FBS was added without antibiotics. Incubated at 37° C for 24 hrs, cells were collected following trypsinization, pooled, and placed in a 10 cm dish with selective antibiotics.
Real-time PCR
RNA was isolated using Trizol reagent (Invitrogen) following manufacturer’s protocol. Murine p21CIP1, Snail2 (Slug), Sox9, TCF4, TCF1, LEF1, Axin2, LRP6, and Cyclin D1 Taqman primers and RNA-to-CT 1-Step Kit were obtained from Applied Biosystems (Carlsbad, CA, USA). Real-time PCR experiments were performed according to manufacturer’s protocol using a StepOnePlus real-time PCR instrument from Applied Biosystems. GAPDH was used as the reference gene and relative mRNA levels were determined using the 2(-△△Ct) method. Three independent experiments were performed. Statistical significance was calculated using the two-tailed t-test, p<0.05.
TOP/FOP Flash assay
Cells plated in 24-well plates in duplicates. Reporter plasmid TOP-Flash or FOP-Flash, which contain three optimal copies of the TCF/LEF binding site (TOP-FLASH), or mutated copies of the TCF/LEF-binding site (FOP-FLASH) upstream of a minimal thymidine kinase (TK) promoter directing transcription of a luciferase gene. (TOP-FLASH) or (FOP-FLASH) (0.5 μg) together with a Renilla luciferase plasmid (0.1 μg) were transfected using Lipofectamine LTX (Invitrogen) according to the manufacturer’s protocol. To activate the Wnt signaling following 24 hrs of transfection, Wnt3a conditioned media with or without 10μM ICG001 (SelleckChem) was added. Cell lysates were obtained using lysis buffer provided in Dual Luciferase Assay Kit (Promega, Madison, WI, USA). Firefly and Renilla luciferase readings were recorded 48 hr post-transfection using luminometer (Promega). The firefly luciferase activity is then normalized to the Renilla luciferase activity, and fold increase in TOP-Flash activity compared to FOP-Flash will be plotted as Mean± SEM of triplicate tests and validated by t-test, p<0.05
Aldefluor assay
The ALDEFLUOR kit (StemCell Technologies, Durham, NC, USA) was used to measure the cell population with high ALDH enzymatic activity. Dissociated single cells were suspended in Aldefluor assay buffer containing the ALDH substrate, Bodipy-aminoacetaldehyde (BAAA) at 1.5 mM and incubated for 45 min at 37 °C. To distinguish between ALDH-positive and ALDH-negative cells, for each sample of cells an aliquot was incubated under identical condition in the presence of a 10-fold molar excess of the ALDH inhibitor, diethylaminobenzaldehyde (DEAB). This results in a significant decrease in the fluorescence intensity of ALDH-positive cells and was used to calibrate the flow cytometer. After incubation, cells are resuspended in 0.5 mL of ice-cold Aldefluor assay buffer for flow cytometry analysis using the BD LSR II Flow Cytometer in the 488/Green fluorescent channel.
Cell labeling and flow cytometry
Cells were digested by 0.25% Trypsin-EDTA, re-suspended in HBSS plus 2% FBS at 2×105 cells/100 ml and incubated with antibodies against mouse CD49 (PE-Cy7-conjugated, 1:100 from BD) and CD24 (FITC-conjugated, 1:200, from BD) in cold room for 30 minutes. Flow cytometry analysis was performed using the BD LSR II Flow Cytometer at the Albert Einstein Flow Cytometry Core facility.
Mammosphere formation
Single-cell suspensions were plated on a 60-mm ultra-low attachment tissue culture dish (Corning, Lowell, MA, USA) at a density of 1×105/dish in Dulbecco’s modified Eagle’s medium/F12 containing 10 ng/ml FGF-2, 4 μg/ml heparin, 20 ng/ml epidermal growth factor, 5 μg/ml insulin, and 5 μg/ml hydrocortisone for 7 days. Inhibition of mammospheres was tested with ICG001 (Tocris Bioscience), which was added at 5 μM or 10 μM or with 20 μM of Tankyrase inhibitor XAV 939 (Tocris Bioscience) in the medium for 7 days. For secondary sphere formation, primary spheres were dissociated in 1:1 trypsin/Dulbecco’s modified Eagle’s medium at 37 °C, and mechanically dispersed by passing through a 23 gauge needle. Single cells were replated at 5×103/dish, and incubated in 37 °C 5% CO2 for 7 days. At the end of the treatment, cells were transferred to a 35 mm MatTek dish and spheres as well as total cells (including mammospheres, single cells and clusters) per microscopic field over five fields were counted. Mammospheres were expressed as percentage using the average number of spheres per 100 cells.
Limiting dilution assay in vivo
PyMT, PyMT/p21KO (p21KO), p21KO+p21 (p21 rescue) cells or Met1 control and Met1p21shRNA cells were each resuspended in 25% Matrigel PBS at indicated cell numbers and injected into two bilateral sites of mammary fat pads from female athymic nude mice or FVB immunocompetent mice. Tumor onset was determined by palpation and aggregate mammary tumor growth from two sites per each mouse, was measured by calipers to determine tumor volume as an aggregate of two tumors per mouse (10).
Statistical Analysis
All of the data are presented as the mean ± standard errors of mean (SEM). A Student’s t test was used, unless otherwise specified, to calculate the p values; p < 0.05 is considered significant.
Results
p21CIP1 regulates a mammary cancer stem cell-like phenotype
We showed that p21CIP1 gene knockout in the mouse MMTV-PyMT mammary tumor model inhibits lung metastasis and that p21overexpression into PyMT/p21 knockout cells restores metastatic colonization (10). To determine whether metastasis promotion by p21 was associated with cancer stem cell activity, we tested the effect of p21 knockout on cancer stem cell properties, including tumorsphere formation, aldehyde dehydrogenase 1 activity, and importantly, tumor initiating potential (Fig. 1). We used primary tumor cell lines derived from PyMT and PyMT/p21KO mouse mammary tumors (10). Compared to ~60% of PyMT mammary tumor cell lines (PyMT1 and PyMT2) which formed robust tumorpsheres, PyMT/p21KO cell lines (p21KO1 and p21KO2) cell lines were devoid of sphere forming ability (Fig. 1A). Replating single cells from first generation PyMT1 and p21KO1 primary tumorspheres, showed consistent inhibition of sphere formation by p21 loss, shown by the reduced levels of secondary spheroids (Fig. 1B), thus suggesting a role of p21 in CSC/progenitor cell proliferation. Moreover, the percentage of tumor cells with aldehyde dehydrogenase 1 (ALDH1) activity, inherent to tumor stem/progenitor cells, was reduced from 73% in PyMT1 to 19% in p21KO1 cells (Fig. 1C). In addition, FACS sorting for CD49/CD24 expression, known to be medium-high in basal1 mammary stem cells (11), showed that the fraction of CD49/CD24 highly positive cells was reduced from 99% and 98% in PyMT (PyMT1 and PyMT2) cell lines to 27% and 3% in p21KO (p21KO1 and p21KO2) cell lines (Fig. 1D).
Figure 1. p21 knockout in PyMT cells inhibits mammary cancer stem cell properties.
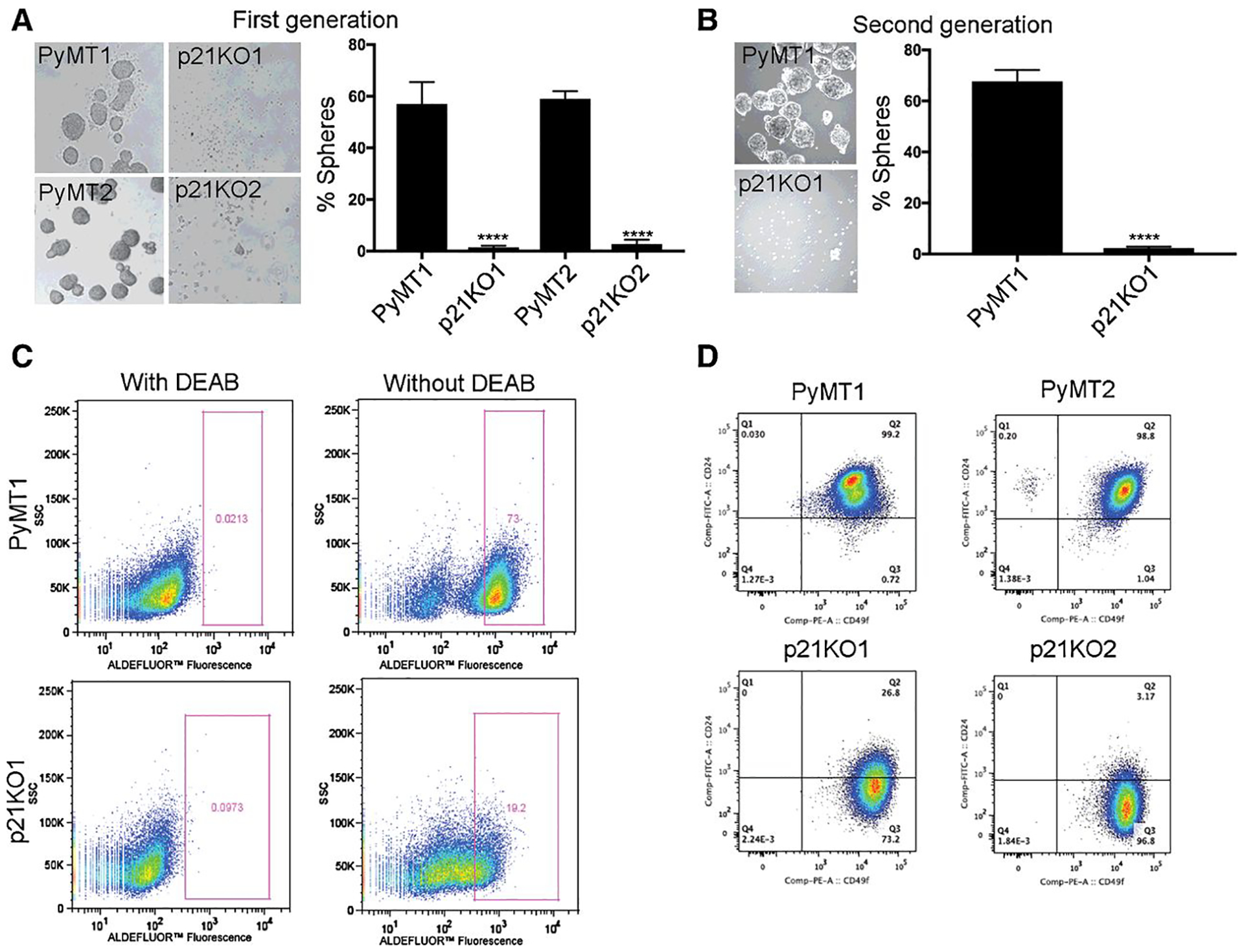
(A) Two primary PyMT tumor cell lines (PyMT1 and PyMT2) were compared to two PyMT/p21KO cell lines (p21KO1 and p21KO2) in first generation tumorsphere formation. (B) PyMT1 were compared to p21KO1 cells in secondary tumorsphere formation. The percent of mammospheres (spheres) in A and B is shown as mean ± SEM, p<0.05. (C) PyMT1 and p21KO1 cell lines were assessed for ALDH1 activity utilizing the Aldefluor assay. p21KO1 cells (bottom panels) showed 3.8 fold decrease in the Aldefluor-positive cell population compared to PyMT1 cells (top panels). Left panels: Negative controls of cells treated with DEAB, an irreversible inhibitor of ALDH1. (D) PyMT (PyMT1 and PyMT2) were compared to p21KO (p21KO1 and p21KO2) cell lines for CD49/CD24 expression using CD49-PE-Cy7 and CD24-FITC flow cytometry, indicating decreased stem marker expression in p21KO cell lines.
Furthermore, tumor initiating potential, a gold standard test for stemness activity, was measured by mammary implantation of limiting dilutions of tumor cells into two sites at the mammary fat pads of female athymic nude mice. Compared to PyMT1 cells which formed exponentially growing tumors at high (1×106) and low (5×104) cell densities, p21KO1 cells were unable of forming sizeable tumors at both dilutions (Fig. 2A). Interestingly, rescue of p21 expression in p21 KO1 cells (Fig. 2B), induced tumor forming ability as compared to p21KO1 cells, at both high (1×106) and low (5×104) cell densities (Fig. 2 B–C). Of note, p21 rescued cells (p21KO1+p21) grew tumors with a significant delay in tumor onset as compared to PyMT cells (24 days vs 4 days) (Fig 2A–C). This may be due to either a partial rescue by exogenous p21 expression, noted in only 60% of p21KO1 cells or to delayed cell growth caused by that p21 re-expression in p21 KO cells. Despite a gap in tumor onset, p21KO1+p21 cells were able of forming similar tumor size as PyMT controls, when compared 15 days post respective tumor onset times (Fig. 2A–C). By contrast, p21KO1 tumors remained consistently reduced in size throughout the whole time course (Fig. 2B–C). In support of these data, p21 overexpression in p21KO1 cells was also able of restoring sphere formation to a similar level as PyMT1 cells (Fig. 2D), and caused a 4.5 fold increase in the fraction of ALDH1 positive cells (Fig. 2E). Consistent with a partial rescue by p21, ALDH1 activity was noted in 30% of p21KO+p21 cells as compared to 73% of PyMT1 control cells (73%) (see Fig. 1C). Thus, these data demonstrate that p21 promotes a powerful cancer stem-like phenotype that is consistent with its pro-metastatic activity (10).
Figure 2. p21 knockout in PyMT cells inhibits tumor initiating potential that can be rescued by p21 overexpression in p21KO cells.
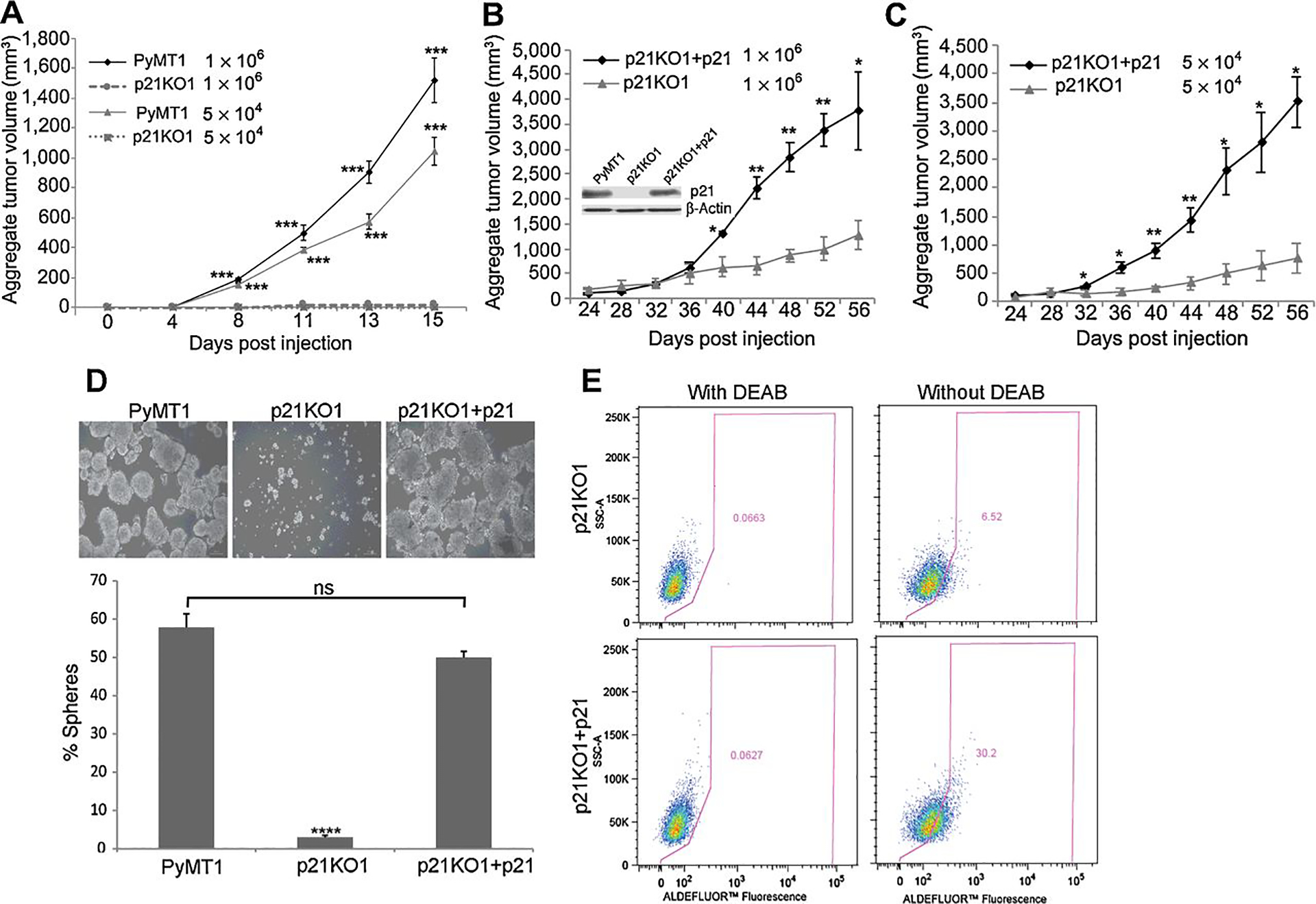
(A) PyMT1 tumor cells were compared to p21KO1 cells for ability to form primary tumors following fat pad implantation of 1×106 and 5×104 cells in two mammary sites of 6 athymic nude mice. (B-C) p21KO1 cells in which p21 was overexpressed (p21KO1+p21) were compared to p21KO1 cells expressing control vector (immunoblot in B shows p21 rescue). Each of these cell lines were implanted in two sites of the mammary fat pads of 6 athymic nude mice at 1×106 and (B) 5×104 (C) cells per site. Mammary tumor growth was measured as aggregate tumor volume (sum of two tumors per mouse) over a period of 15 days for (A) and 56 days for (B-C). Results are shown as mean aggregate tumor volume ± SEM. Differences in aggregate tumor volume at each cell dilution and time point between PyMT1 and p21KO1 (A) and p21KO1 and p21KO1+p21 (B-C) were determined by two-tail non-parametric t-test, and were statistically significant (p<0.05). (D) PyMT1, p21KO1, p21KO+p21 cells were each tested for mammosphere formation in triplicate cultures. The percent of spheres is shown as mean ± SEM. Significant differences (p=0.0001) were observed between PyMT1 and p21KO1, but not between PyMT1 and p21KO1+p21 cells (ns). (D) p21KO1 and p21KO1+p21 cell lines were assessed for ALDH1 activity; p21KO1+p21 cells (bottom panels) showed 5 fold increase in the Aldelfuor-positive cell population as compared to p21KO1 cells (top panels). Left panels: Negative controls include cells treated with DEAB, an irreversible inhibitor of ALDH1.
p21CIP1 knockdown in the metastatic PyMT/Met-1 cell line, suppresses cancer stem like properties.
To further confirm the effect of p21 gene knockout on CSC properties, we used shRNA to knock down p21 expression in a mammary PyMT tumor cell line, known as Met-1, which was derived from an independent PyMT model in the FVB background (12). This cell line was serially transplanted into the mammary fat pad of FVB female mice to generate a highly metastatic cell line, that can be tested in a immunocompetent FVB mouse (12). p21 shRNA mediated knockdown in Met-1 cells attenuated sphere forming efficiency from 30% to 12% (Fig. 3A) and reduced the aldefluor positive fraction of Met-1 cells from 20% to 7% (Fig. 3B). Moreover, p21 knockdown in Met-1 cells reduced tumor incidence following mammary fat pad inoculation of 5×105 and 5×104 tumor cells into syngeneic FVB female mice (Fig. 3C). The number of mice developing tumors at 14 weeks (end points) post fat pad injection of 5×105 cells was higher for Met-1 control cells (5 out 6 mice) than for Met-1/p21shRNA cells (2 out of 6 mice) (Fig. 3C). At the lower cell density (5×104), Met1 cells produced tumors in 3 out of 6 mice whereas Met-1/p21shRNA cells in 2 out 6 mice. Overall, Met-1 control cells produced substantially larger tumors than Met-1/ p21shRNA cells at both cell densities. These data confirmed that ablation of p21 expression by shRNA recapitulates the effects of p21 gene knockout in PyMT cells by attenuating CSC-like properties.
Figure 3. p21 shRNA knockdown in PyMT/Met-1 cells suppresses cancer stem cell like properties.
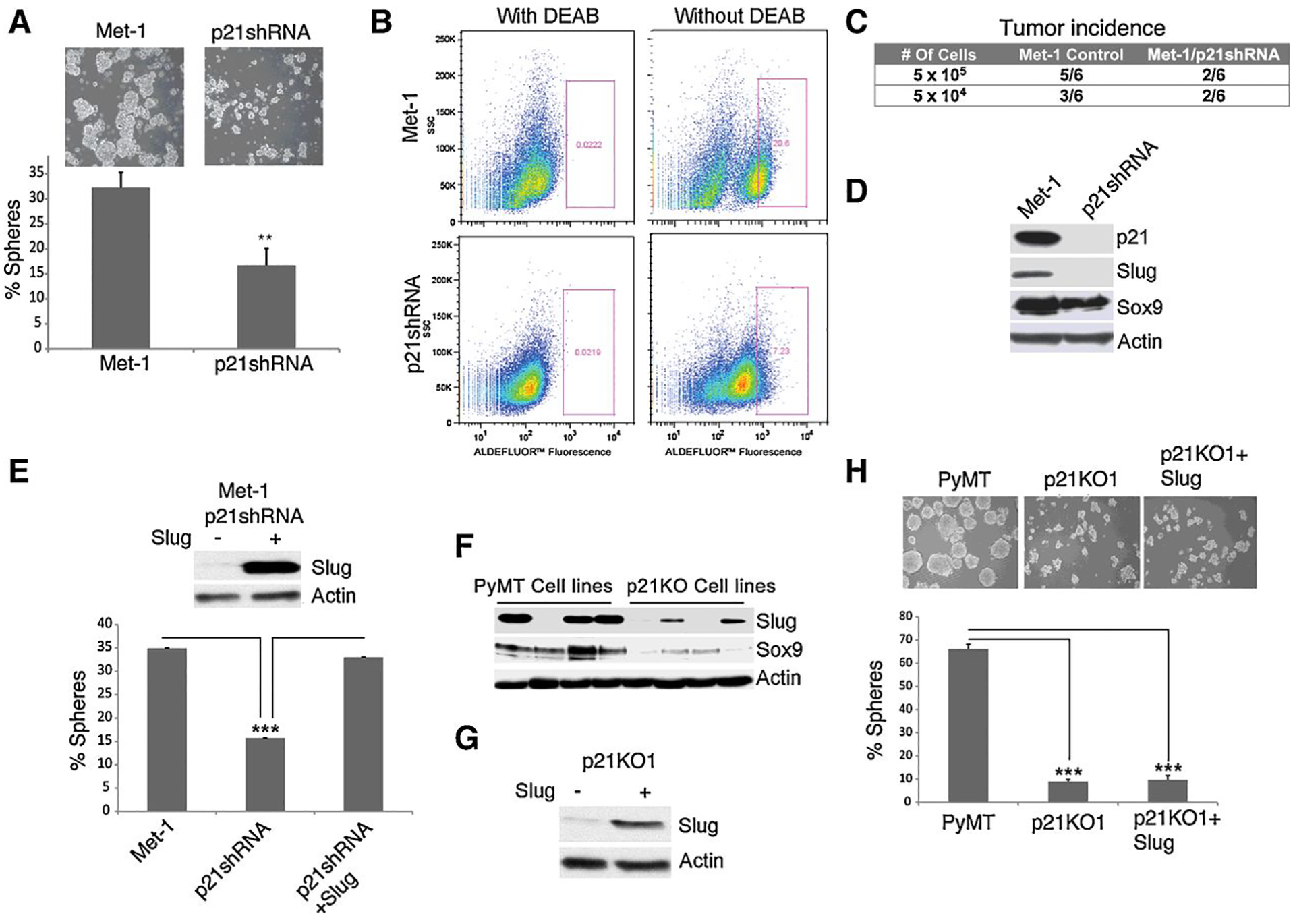
(A) PyMT/Met-1 cells expressing scrambled vector or p21shRNA, were tested for mammosphere forming ability. The percent of spheres in triplicate cultures is shown as mean ± SEM; p<0.05. (B) Met-1 control and Met-1/p21shRNA (p21shRNA) cells were each tested for ALDH1 activity. Met-1/p21shRNA cells (bottom panels) showed 3 fold reduction in the Aldelfluor-positive cell population relative to Met-1 control cells (top panels). Left panels: Negative controls include cells treated with DEAB. (C) Met-1 control and Met-1/p21shRNA cells were each implanted into two sites at the mammary fat pads of 6 athymic nude mice at 5×105 and 5×104 cells per site. Tumor growth was monitored over a period of 14 weeks post tumor onset (end points); the number of mice developing tumors at the two cell densities is indicated. (D) Met-1 control and Met-1/p21shRNA (p21shRNA) cells were immunoblotted for expression of p21, Slug, Sox9 and Actin. (E) Met-1/p21shRNA cells were infected with control vector and mouse Slug lentiviral vector; cell lysates were immunoblotted for Slug and Actin; sphere formation was determined for each condition; the percent of spheres in triplicate cultures comparing Met-1 control to Met-1/p21shRNA cells before and after Slug re-expression is shown as mean ± SEM, p<0.05. (F) Four PyMT (PyMT1–4) were compared to four p21KO (p21KO1–4) cell lines by immunoblotting for Slug, Sox 9, and Actin expression. (G) Control or Slug lentiviral construct was each infected in p21KO1 cells as shown by Slug immunoblotting. (H) PyMT cells were compared to p21KO1 and (p21KO1+Slug) cells for sphere formation. The percentage of spheres in triplicate samples is shown; no significant differences in sphere formation between p21KO1 and (p21KO1+Slug) cells.
It has been reported that Slug and Sox9 are two transcription factors that cooperatively drive the mammary stem cell state as well as tumorigenic and metastasis-seeding abilities (13) (14). Indeed, p21 knockdown in Met-1 cells (Fig. 3D), inhibits Slug, and to a lesser extent, also Sox9 expression (Fig. 3D). Interestingly, Slug rescue into Met-1/p21shRNA cells was able of restoring sphere formation (Fig. 3E). By comparison, Slug and Sox9 were both substantially reduced in four of the p21KO cell lines as compared to PyMT control cell lines (Fig. 3F); however, re-expression of Slug in p21KO1 cells (Fig. 3G) did not rescue sphere formation (Fig. 3H). This might be due to the dramatically lower levels of Sox9 in p21 KO1 cell lines relative to Met-1/p21 shRNA cells. However, re-expression of Slug and Sox9 into p21KO1 cells was unable of rescuing sphere formation (not shown), which could be due to insufficient expression. Alternatively, PyMT cells may be deficient in other stemness factors that may be present in Met-1 cells such as Sox10 (15, 16). Nevertheless, these data are consistent with that Slug and Sox9 can cooperate in creating a mammary stem cell phenotype.
p21 deletion results in inhibition of canonical Wnt/β-catenin signaling pathway
The Wnt signaling pathway is known to promote stemness by increasing levels of transcriptionally active β-catenin induced by Wnt ligands, resulting in gene transcription leading to stem cell renewal (17). Wnt/β-catenin is therefore thought to promote self-renewal and suppress differentiation of stem cells (17). Comparison of four p21KO to four PyMT mammary tumor cell lines, revealed that p21KO cell lines expressed significantly reduced levels of unphosphorylated or stabilized β-catenin (active-β-catenin) as compared to PyMT control cell lines, whereas total β-catenin expression was unchanged (Fig. 4A).
Figure 4. p21 knockout inhibits Wnt/β-catenin signaling.
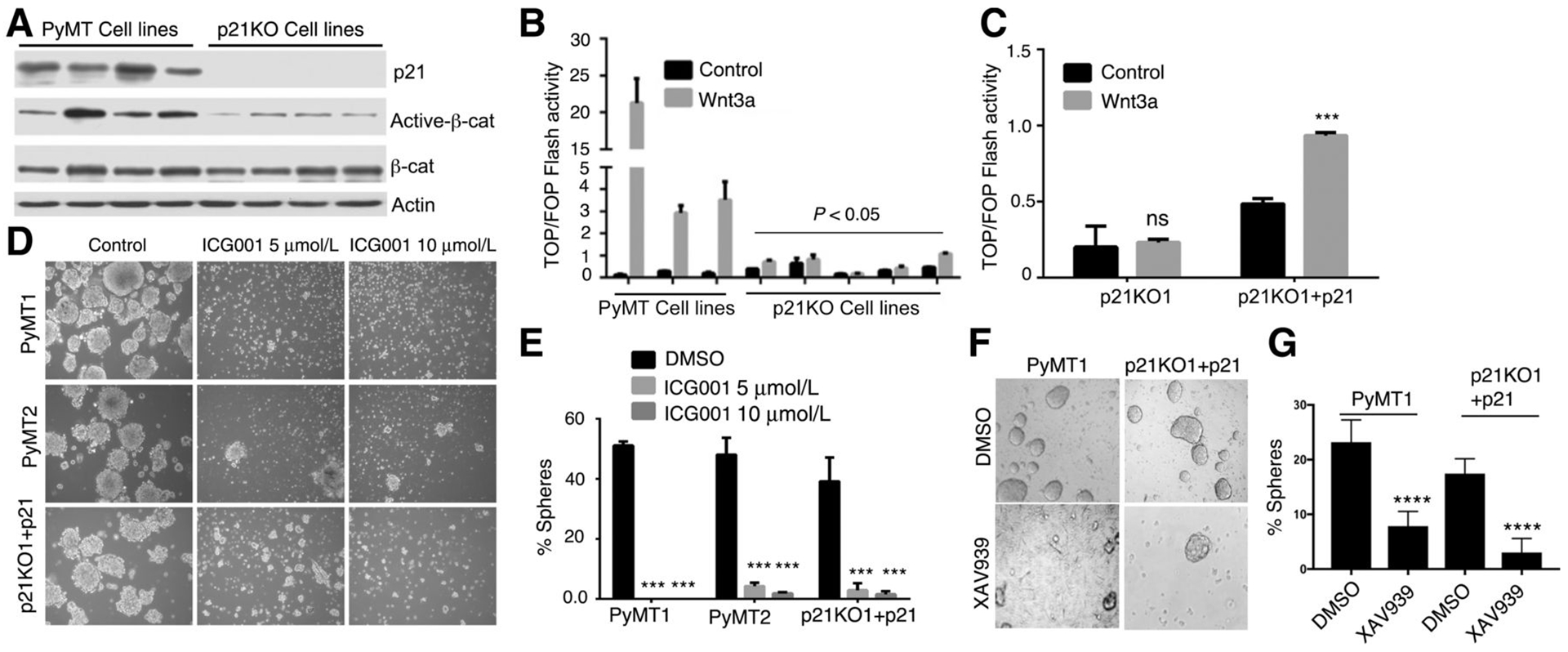
(A) Four primary PyMT cell lines were compared to four p21KO cell lines for expression of p21, active-β-catenin, total β-catenin, and Actin expression by western blotting. (B) Three primary PyMT cell lines were compared to five p21KO cell lines before (black) and after Wnt3a stimulation (grey), for TOP/FOP Flash activity. Results are shown as mean ± SEM; TOP/FOP Flash units were not different between ligand untreated PyMT and p21KO cell lines (black bars); however there was a significant difference (p<0.05) between Wnt3a-treated PyMT and p21KO cell lines (grey bars). (C) p21KO1 and (p21KO1+p21) cells that were untreated (black bars) or treated (grey bars) with Wnt3a for 18 hrs, were assayed for TOP/FOP Flash activity. Results are shown as Mean± SEM; significant differences (p<0.05) were observed between (p21KO1+p21) and p21KO1 cells before and after Wnt3a treatment. (D) PyMT1, PyMT2, and (p21KO1+p21) cells were assayed for mammosphere formation in the absence and presence of 5 and 10 μM Wnt antagonist ICG001; the effect of ICG001 on TOPFlash activation is shown in Supplemental Fig. 1A. (E) The percent of spheres before and after ICG001 treatment is shown as mean ± SEM; p<0.05. (F) PyMT1 and (p21KO1+p21) cells were plated under sphere forming conditions for 5 days in the absence (DMSO) or presence of tankyrase inhibitor, XAV 939, at 20 μM; sphere formation was determined as above. Results are shown as Mean± SEM; p<0.05.
To test whether p21 promotes canonical Wnt signaling, we assayed for β-catenin activation of the T cell factor (TCF)/lymphoid enhancer factor (LEF) transcription factors, using a TCF/LEF reporter plasmid, which consists of 3 TCF binding sites fused to luciferase (13, 18). β-catenin-mediated TCF/LEF transcriptional activation was measured as fold induction of a consensus TCF-β-catenin reporter (TOP-FLASH) with respect to FOP-FLASH (a scrambled consensus reporter), normalized for transfection efficiency. Comparison of PyMT to p21KO cell lines, showed an overall significant decrease in Wnt3a-stimulated TOP/FOP FLASH reporter activation in p21KO (p21KO1–5) cell lines relative to control PyMT (PyMT 1–3) cell lines, which overall expressed higher, yet variable levels of TOP/FOP FLASH, likely due to tumor cell heterogeneity (Fig. 4B). Moreover, p21 rescue in p21KO1 cells enhanced TOP/FOP FLASH reporter activity by 2 fold in ligand-untreated cells, and by 4 fold in Wnt3a-stimulated cells (Fig. 4C), implying a stimulatory effect of p21 on Wnt signaling which occurs even in the absence of Wnt ligand. Collectively, these data confirm an active role for p21 in potentiating Wnt/β-catenin/TCF signaling.
Inhibition of Wnt/β-catenin signaling suppresses mammosphere formation in a p21-dependent fashion
To confirm that Wnt/β-catenin activity regulates CSC-like behavior in PyMT cells, we used a selective Wnt/β-catenin signaling inhibitor ICG001, which antagonizes β-catenin/TCF/CBP mediated transcription (19). Treatment of PyMT cell lines with ICG001, which abrogates TOPFlash reporter activation by Wnt3a (Supplemental Fig. 1A), dramatically suppressed sphere forming activity (Fig. 4D–E). Importantly, ICG001 blocked rescue of sphere formation by p21 overexpression in p21KO1 cells (Fig. 4D–E). Similarly, a tankyrase inhibitor, XAV939, which directly inhibits Wnt/β-catenin signaling by causing Axin stabilization (20), was also efficient in blocking sphere formation by PyMT1 cells as well as by p21-rescued p21KO1 cells (p21KO1+p21) (Fig. 4F–G). These data underscore an active role for p21 in promoting CSC activity via Wnt/β-catenin/TCF transactivation.
p21CIP1 stimulates LEF1 upregulation
We tested whether p21 stimulation of TCF/LEF transcriptional activity was due to increases in expression of Wnt signal transducers including LEF1 and TCF4 (21, 22). There was no difference in TCF4 mRNA expression between PyMT1 and p21KO1 cells before or after Wnt3a treatment (Fig. 5A–B). LEF1 expression was also unchanged between PyMT1 and p21KO1 cells that were untreated with Wnt3a; however, Wnt3a stimulated a dramatic increase in LEF1 mRNA levels in PyMT1 cells, but not in p21KO1 cells (Fig. 5 A–B). At the contrary, Wnt3a reduced LEF1 expression in p21KO1 cells below levels present in Wnt-untreated p21KO1 cells, implying a synergy between p21 and Wnt3a in regulating LEF1 expression (Fig. 5A–B).
Figure 5. p21 knockout inhibits LEF1, but not TCF4, expression.
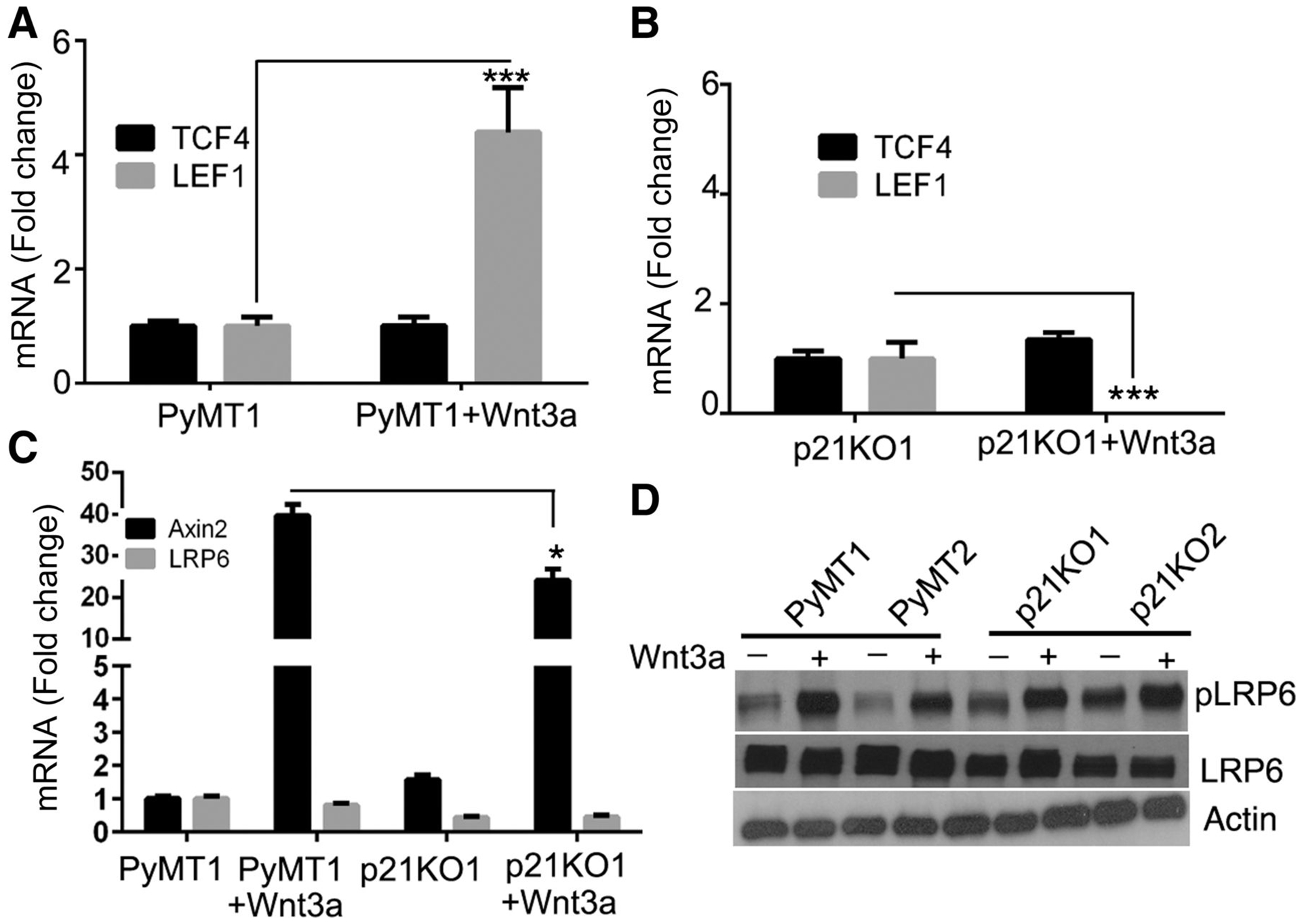
(A, B) PyMT1 and p21KO1 cell lines that were untreated or treated with Wnt3a for 18 hrs, were tested by Taqman qRT-PCR for expression of LEF1 and TCF4 mRNA, or for (C) Axin 2 or LRP6 mRNA expression. (D) Two PyMT (PyMT 1–2) and p21KO (p21KO1–2) cell lines that were untreated or treated with Wnt3a for 18hrs were immunoblotted for levels of p-LRP6, LRP6, and Actin. Densitometry of LRP6 and p-LRP6 immunoblots did not reveal any significant differences between PyMT and p21KO cells (Supplemental Fig. 1B–C).
To determine whether p21 ablation in PyMT cells affects the expression of Wnt transducing effectors that are upstream of β-cat/TCF/LEF transactivation, we measured the expression of Axin2, a central node in the Wnt transduction cascade, as well as LRP6, a Frizzled co-receptor, which regulates Wnt ligand activation (22) (23). Wnt3a stimulated Axin2 mRNA expression in PyMT1 cells and to a lesser degree also in p21KO1 cells, (Fig. 5C). LRP6 mRNA (Fig. 5C) or protein (Fig. 5D) expression was not significantly affected by p21 knockout. LRP6 phosphorylation which regulates binding of Wnt to Frizzled (23) was not significantly changed in p21KO relative to PyMT cell lines (Fig. 5D). Levels of p-LRP6 and LRP6 were not significantly changed as shown by densitometric analysis of immunoblots (Supplemental Fig. 1B–C). Frizzled 7 expression could not be assessed by western blot due to variability in detection levels by different antibodies. In sum, our data demonstrate that p21 ablation in PyMT cells readily suppresses the expression of LEF1, a main Wnt signal effector and target gene (21) (22).
LEF1 knockdown in PyMT cells does not inhibit Wnt signaling or mammosphere formation
To further examine the role of p21-mediated LEF1 upregulation in Wnt signal activation, we knocked down LEF1 in PyMT cells by 2 independent shRNAs. This led to dramatic suppression of LEF1 mRNA expression, in Wnt3a-treated and untreated cells (Supplemental Fig. 2A). LEF1 knockdown, however, did not inhibit Wnt3a-mediated TOP/FOP Flash reporter activation as compared to marked inhibition by the Wnt signal antagonist ICG001 (Supplemental Fig. 2B). In support of this finding, LEF1 shRNA knockdown by two targeting sequences in PyMT cells did not affect sphere formation (Supplemental Fig. 2C–D).
TCF1 knockdown in PyMT cells inhibits Wnt signaling and sphere formation via Cyclin D1 attenuation.
Since TCF1 might be redundant to LEF1 in recruiting β-catenin to de-repress Wnt target gene transactivation (21, 24, 25), we tested whether p21 knockout inhibits TCF1 expression. Of note, TCF1 mRNA was significantly attenuated in p21KO1 relative to PyMT1 cells under basal or Wnt3a-treated conditions (Fig. 6A), suggesting p21 supports TCF1 expression. Knockdown of TCF1 in PyMT1 cells by two independent shRNAs led to ~60% reduction in TCF1 mRNA expression before or after Wnt3a treatment (Fig. 6B). Consistently, both TCF1shRNAs reduced Wnt3a stimulated TOPFlash activation by 50% as compared to control and ICG001 treated cells (Fig. 6C), resulting in marked inhibition of spheroids (Fig. 6D). Thus, p21 knockout attenuates TCF1 expression, thereby attenuating canonical Wnt signaling and sphere formation.
Figure 6. TCF1 knockdown in PyMT cells attenuates sphere formation through Cyclin D1 downregulation.

(A) PyMT1 and p21KO1 cell lines that were untreated or treated with Wnt3a for 18 hrs were assayed for TCF1 mRNA expression by Taqman qRT-PCR. Significant differences in TCF1 expression (p<0.05) were noted between untreated or Wnt3a-treated PyMT1 and p21KO1 cell lines. (B) PyMT1 cells expressing control shRNA, TCF1shRNA1 or TCF1shRNA2, were untreated or treated with Wnt3a for 18 hrs, and tested for TCF1 mRNA expression, or (C) assayed for TOP/FOP Flash activity in the absence or presence of 10μM ICG001. Significant differences were seen between PyMT1 and PyMT/TCF1shRNA1 and PyMT/TCF1shRNA2; p<0.05. (D) PyMT1 were compared to PyMT1/TCF1shRNA1 (sh1) and PyMT1/TCF1shRNA2 (sh2) for mammosphere formation. The percent of spheres in triplicate experiments is shown as mean ± SEM, p<0.05. (E) PyMT1, PyMT1/TCF1shRNA1 and PyMT1/TCF1shRNA2 cells were assayed for levels of Cyclin D1 using qRT-PCR. Significant differences in Cyclin D1 mRNA level were noted between PyMT1 and TCF1 knockdown cells; p<0.05. (F) PyMT1 cells were treated with control shRNA (lane 1) and 4 Cyclin D1 shRNAs; levels of Cyclin D1 assessed by western blotting. Note shRNA 3 and 4 were most efficient in knocking down Cyclin D1 expression. PyMT1 expressing control and CyclinD1 shRNA3 and shRNA4 were assayed for mammosphere formation. The percentage of spheres was measured in triplicates, showing significant differences between PyMT1 and Cyclin D1 knockdown cultures (p<0.05).
Among the TCF/LEF target genes, Cyclin D1 was shown to regulate mammary CSC renewal (25, 26). We tested whether TCF1 knockdown in PyMT cells affects Cyclin D1 expression. Indeed, TCF1 knockdown in PyMT1 cells (Fig. 6E), led to significant attenuation in Cyclin D1 expression. In support of this idea, knockdown of Cyclin D1 by two shRNA sequences in PyMT1 cells markedly reduced sphere formation (Fig. 6F).
Importantly, most of these findings were recapitulated in the metastatic PyMT/Met-1 cell line (Fig. 7). Namely, p21 knockdown by shRNA in Met-1 cells, caused a dramatic reduction in LEF1 expression but had no effect on TCF4 or LRP6 mRNA expression, although it reduced Axin 2 mRNA (Fig. 7A–C). Moreover, Wnt3a stimulation caused a dramatic increase in LEF1 and TCF1 protein expression that was suppressed by p21 shRNA in Met-1 cells (Fig. 7D). Furthermore, TCF1 knockdown in Met-1 cells (Fig. 7E) reduced sphere formation (Fig. 7F). Interestingly, TCF1 knockdown in Met-1 cells also reduced Cyclin D1 expression, similar to the effect of p21 shRNA (Fig. 7G). Consistent with a role of TCF1/Cyclin D1 in promoting stem like properties, Cyclin D1 attenuation by shRNA in Met-1 cells (Fig. 7H), caused a significant decrease in sphere formation (Fig. 7I).
Figure 7. p21 knockdown in Met-1 cells inhibits TCF1 and Cyclin D1 expression, resulting in sphere suppression.
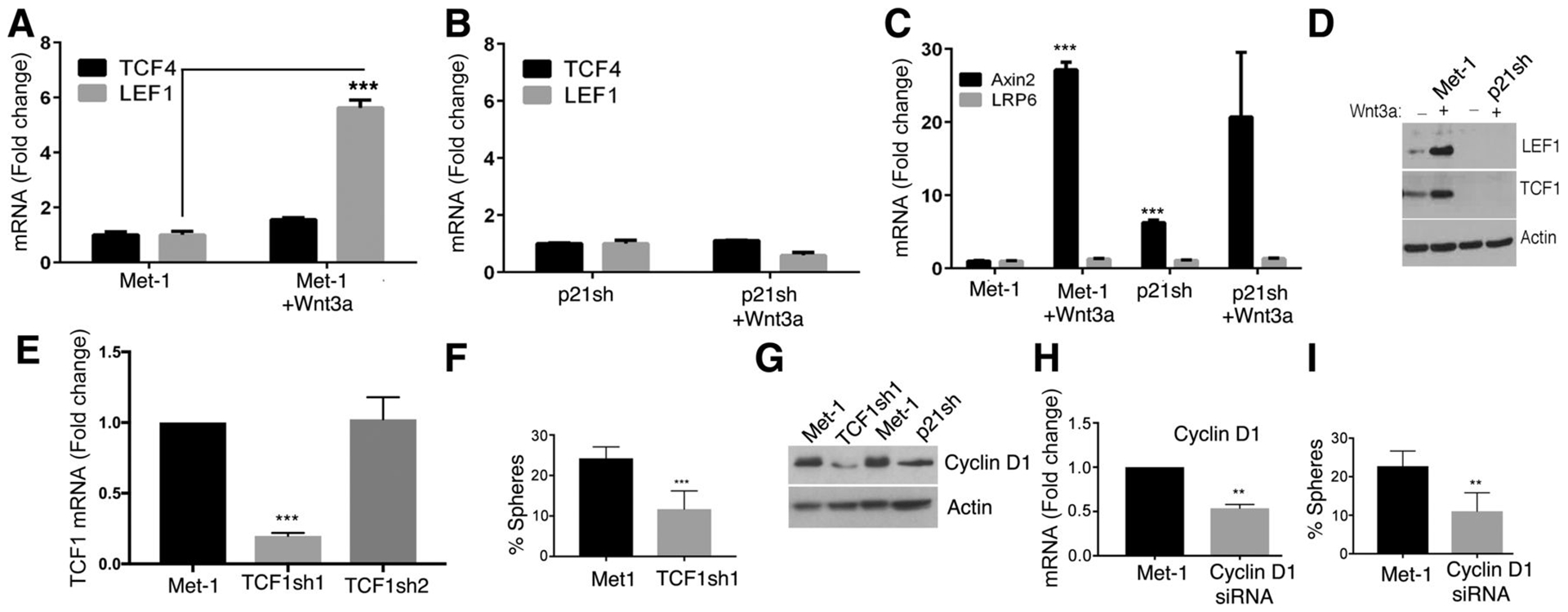
(A, B) Met-1 and Met-1/p21shRNA (sh) cell lines that were untreated or treated with Wnt3a for 18 hrs, were tested by Taqman qRT-PCR for expression of LEF1 and TCF4 mRNA, or for (C) Axin 2 or LRP6 mRNA expression. Differences in mRNA expression between Met-1 and Met-1/p21shRNA were determined by unpaired t-test and significance set at p<0.05. (D) Met-1 and Met-1/p21sh cells were untreated or treated with Wnt3a for 18 hrs and tested by Western blot for expression of LEF-1, TCF-1, or Actin. (E) Met-1 cells were treated with control shRNA, TCF1shRNA1, and TCF1shRNA2 and levels of TCF1 mRNA were tested by qRT-PCR. (F) Control Met-1 and Met- 1/TCF1shRNA1 cells were tested for mammosphere formation; the percent of spheres in triplicate experiments is shown as mean ± SEM, p<0.05. (G) Met-1 control cells were compared to Met-1/TCF1shRNA1 and Met-1/p21shRNA cells by Western blot for expression of Cyclin D1 relative to Actin. (H) Met-1 cells were treated with control siRNA or Cyclin D1 siRNA and the level of mRNA knockdown was tested by qRT-PCR. (I) Met-1 and Met-1/CyclinD1 siRNA cells were tested for sphere formation. Results are shown as mean percent of spheres ± SEM; p<0.05.
Thus, the cumulative data demonstrate that p21 regulates sphere formation or CSC/progenitor proliferation by upregulating TCF1 and consequently Cyclin D1, thereby promoting a CSC like phenotype.
Discussion
p21CIP1 is a cell cycle inhibitor that acts as a tumor suppressor by halting cell proliferation and promoting DNA repair in response to genotoxic stress; however, increasing evidence suggests that p21 may also act as a malignancy promoter (8). Wnt/β-catenin signaling is known to regulate stem cells by promoting dedifferentiation and self-renewal (17). It also promotes quiescence via p21 upregulation to protect cells from excessive proliferation that leads to cancer stem cell exhaustion (27). p21 was also shown to act downstream of genes that regulate cancer initiating cells and metastasis such as “inhibitor of differentiation” (ID) genes, thereby preventing excess DNA damage by stress or chemotherapy which results in CSC exhaustion (9).
We previously showed that p21 knockout in the PyMT mammary tumor model inhibits lung metastasis (10). We speculate that p21 loss may inhibit metastasis by suppressing cancer stem cell behavior. Indeed, we show that p21 knockout in PyMT mammary cancer cells dramatically suppresses CSC properties, involving tumorsphere formation, AlDH1 activity, and tumor initiating potential. The stem promoting effect of p21 was corroborated by genetic rescue of p21 in knockout cells, resulting in re-activation of CSC properties, including tumor regenerative potential.
Interestingly, p21 appears to stimulate stemness properties via activation of canonical Wnt signaling, a key pathway implicated in stem and cancer stem cell self-renewal (17). Consistent with this idea, p21 knockout in PyMT cells suppressed Wnt3a-stimulated TCF/β-catenin transactivation, and importantly, p21 overexpression in p21 knockout cells was able of rescuing Wnt reporter activity. We show that p21 activates canonical Wnt signaling via positive regulation of TCF1 and Cyclin D1, two main effectors as well as target genes of Wnt signaling (21). Our data demonstrate that p21 strongly supports the expression of LEF1 and TCF1; however, only TCF1, and not LEF1, knockdown in PyMT cells was able of attenuating Wnt reporter activation and sphere formation. This implied that TCF1 is the likely protein recruiting β-catenin to the transcriptional complex to de-repress Wnt signaling PyMT cells, which is consistent with the higher abundance of TCF1 relative to LEF1 protein in PyMT cells. We believe that TCF1 regulates Cyclin D1, a bona fide Wnt target gene, which in turn promotes CSC/progenitor proliferation (25, 26). In support of this idea, we found that TCF1 knockdown in PyMT cells attenuates Cyclin D1 expression and Wnt/TCF TOPFLASH reporter activation as well as sphere formation, implying a role for Cyclin D1 upregulation by p21 in driving CSC/progenitor self-renewal (28). In addition, to promoting dedifferentiation and self-renewal, p21 also activates EMT and stemness factors such as Slug and Sox9, which may also be activated by Wnt signaling. These data are partly consistent with our observation that Slug rescue in p21-knockdown mammary tumor cells restores sphere formation.
At last remains the question whether p21 loss inhibits cancer stem cells by enhancing cell proliferation, which might lead to CSC exhaustion (6, 7). We previously showed that p21 loss in PyMT mammary tumor cells causes increased cell growth in vitro, but not in vivo, where it surprisingly reduced mammary tumor growth, an effect which was associated with striking epithelial differentiation (10). We speculate that the effect of p21 loss on proliferation is offset by its effect on differentiation, which in turn stops cell proliferation. This effect was mostly observed in vivo, but not in vitro, likely due to abundance of growth factors in the culture media that tilt the balance towards proliferation over differentiation. These data suggest that p21 loss causes a switch from proliferation to differentiation, which in turn inhibits EMT, migration and ultimately stemness, a matter that warrants further investigation.
Our cumulative data point to an interesting and novel function of p21 in potentiating cancer stem cells via activation of canonical Wnt signaling due to TCF1/Cyclin D1 upregulation, resulting in promotion of self-renewal, leading to proliferation of CSC/progenitor cells that fuels tumor growth and also metastasis. Further investigation on the precise molecular mechanism whereby p21 activates canonical Wnt signaling is warranted.
Supplementary Material
Acknowledgments
This work was supported by grants from the Avon foundation, Breast Cancer Research Foundation, and Cure Breast Cancer Foundation to (R.B Hazan and L. Norton).
Footnotes
Conflict of interest statement: No potential conflict of interest to disclose.
References
- 1.Dontu G, Al-Hajj M, Abdallah WM, Clarke MF, Wicha MS. Stem cells in normal breast development and breast cancer. Cell Prolif. 2003;36 Suppl 1:59–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Korkaya H, Liu S, Wicha MS. Breast cancer stem cells, cytokine networks, and the tumor microenvironment. J Clin Invest. 2011;121(10):3804–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dontu G, Liu S, Wicha MS. Stem cells in mammary development and carcinogenesis: implications for prevention and treatment. Stem Cell Rev. 2005;1(3):207–13. [DOI] [PubMed] [Google Scholar]
- 4.Morrison SJ, Kimble J. Asymmetric and symmetric stem-cell divisions in development and cancer. Nature. 2006;441(7097):1068–74. [DOI] [PubMed] [Google Scholar]
- 5.Blick T, Hugo H, Widodo E, Waltham M, Pinto C, Mani SA, et al. Epithelial mesenchymal transition traits in human breast cancer cell lines parallel the CD44(hi/)CD24 (lo/-) stem cell phenotype in human breast cancer. J Mammary Gland Biol Neoplasia. 15(2):235–52. [DOI] [PubMed] [Google Scholar]
- 6.Cheng T, Rodrigues N, Shen H, Yang Y, Dombkowski D, Sykes M, et al. Hematopoietic stem cell quiescence maintained by p21cip1/waf1. Science. 2000;287(5459):1804–8. [DOI] [PubMed] [Google Scholar]
- 7.Cheng T, Scadden DT. Cell cycle entry of hematopoietic stem and progenitor cells controlled by distinct cyclin-dependent kinase inhibitors. Int J Hematol. 2002;75(5):460–5. [DOI] [PubMed] [Google Scholar]
- 8.Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9(6):400–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Brien CA, Kreso A, Ryan P, Hermans KG, Gibson L, Wang Y, et al. ID1 and ID3 regulate the self-renewal capacity of human colon cancer-initiating cells through p21. Cancer Cell. 2012;21(6):777–92. [DOI] [PubMed] [Google Scholar]
- 10.Qian X, Hulit J, Suyama K, Eugenin EA, Belbin TJ, Loudig O, et al. p21CIP1 mediates reciprocal switching between proliferation and invasion during metastasis. Oncogene. 2013;32(18):2292–303 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cai S, Kalisky T, Sahoo D, Dalerba P, Feng W, Lin Y, et al. A Quiescent Bcl11b High Stem Cell Population Is Required for Maintenance of the Mammary Gland. Cell Stem Cell. 2017;20(2):247–60 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Borowsky AD, Namba R, Young LJ, Hunter KW, Hodgson JG, Tepper CG, et al. Syngeneic mouse mammary carcinoma cell lines: two closely related cell lines with divergent metastatic behavior. Clin Exp Metastasis. 2005;22(1):47–59. [DOI] [PubMed] [Google Scholar]
- 13.Marjanovic ND, Weinberg RA, Chaffer CL. Cell plasticity and heterogeneity in cancer. Clin Chem. 2013;59(1):168–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo W, Keckesova Z, Donaher JL, Shibue T, Tischler V, Reinhardt F, et al. Slug and sox9 cooperatively determine the mammary stem cell state. Cell. 148(5):1015–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dravis C, Spike BT, Harrell JC, Johns C, Trejo CL, Southard-Smith EM, et al. Sox10 Regulates Stem/Progenitor and Mesenchymal Cell States in Mammary Epithelial Cells. Cell Rep. 2015;12(12):2035–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dravis C, Chung CY, Lytle NK, Herrera-Valdez J, Luna G, Trejo CL, et al. Epigenetic and Transcriptomic Profiling of Mammary Gland Development and Tumor Models Disclose Regulators of Cell State Plasticity. Cancer Cell. 2018;34(3):466–82 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434(7035):843–50. [DOI] [PubMed] [Google Scholar]
- 18.Staal FJ, van Noort M, Strous GJ, Clevers HC. Wnt signals are transmitted through N-terminally dephosphorylated beta-catenin. EMBO Rep. 2002;3(1):63–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Emami KH, Nguyen C, Ma H, Kim DH, Jeong KW, Eguchi M, et al. A small molecule inhibitor of beta-catenin/CREB-binding protein transcription [corrected]. Proc Natl Acad Sci U S A. 2004;101(34):12682–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Huang SM, Mishina YM, Liu S, Cheung A, Stegmeier F, Michaud GA, et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461(7264):614–20. [DOI] [PubMed] [Google Scholar]
- 21.Cadigan KM, Waterman ML. TCF/LEFs and Wnt signaling in the nucleus. Cold Spring Harb Perspect Biol. 2012;4(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Logan CY, Nusse R. The Wnt signaling pathway in development and disease. Annu Rev Cell Dev Biol. 2004;20:781–810. [DOI] [PubMed] [Google Scholar]
- 23.Nusse R, Fuerer C, Ching W, Harnish K, Logan C, Zeng A, et al. Wnt signaling and stem cell control. Cold Spring Harb Symp Quant Biol. 2008;73:59–66. [DOI] [PubMed] [Google Scholar]
- 24.MacDonald BT, Tamai K, He X. Wnt/beta-catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009;17(1):9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jeselsohn R, Brown NE, Arendt L, Klebba I, Hu MG, Kuperwasser C, et al. Cyclin D1 kinase activity is required for the self-renewal of mammary stem and progenitor cells that are targets of MMTV-ErbB2 tumorigenesis. Cancer Cell. 17(1):65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shtutman M, Zhurinsky J, Simcha I, Albanese C, D’Amico M, Pestell R, et al. The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proc Natl Acad Sci U S A. 1999;96(10):5522–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fleming HE, Janzen V, Lo Celso C, Guo J, Leahy KM, Kronenberg HM, et al. Wnt signaling in the niche enforces hematopoietic stem cell quiescence and is necessary to preserve self-renewal in vivo. Cell Stem Cell. 2008;2(3):274–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jeselsohn R, Brown NE, Arendt L, Klebba I, Hu MG, Kuperwasser C, et al. Cyclin D1 kinase activity is required for the self-renewal of mammary stem and progenitor cells that are targets of MMTV-ErbB2 tumorigenesis. Cancer Cell. 2010;17(1):65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.