

Cryo-EM studies of the yeast HDAC complex with and without nucleosome reveal mechanistic insight into chromatin modification.
Abstract
The chromatin-modifying histone deacetylases (HDACs) remove acetyl groups from acetyl-lysine residues in histone amino-terminal tails, thereby mediating transcriptional repression. Structural makeup and mechanisms by which multisubunit HDAC complexes recognize nucleosomes remain elusive. Our cryo–electron microscopy structures of the yeast class II HDAC ensembles show that the HDAC protomer comprises a triangle-shaped assembly of stoichiometry Hda12-Hda2-Hda3, in which the active sites of the Hda1 dimer are freely accessible. We also observe a tetramer of protomers, where the nucleosome binding modules are inaccessible. Structural analysis of the nucleosome-bound complexes indicates how positioning of Hda1 adjacent to histone H2B affords HDAC catalysis. Moreover, it reveals how an intricate network of multiple contacts between a dimer of protomers and the nucleosome creates a platform for expansion of the HDAC activities. Our study provides comprehensive insight into the structural plasticity of the HDAC complex and its functional mechanism of chromatin modification.
INTRODUCTION
Histone deacetylases (HDACs) exist in cells as part of multiprotein assemblies that contain chromatin-modifying deacetylase activities, nucleosome recognition modules, and scaffold proteins that mediate structural organization (1, 2). Two major HDAC-containing transcriptional repression complexes in budding yeast are class I Rpd3 and class II Hda1 (3, 4). The Saccharomyces cerevisiae class II HDAC complex is composed of three stably associated subunits Hda1, Hda2, and Hda3 (5). The catalytic component Hda1 self-associates as dimer and interacts with two noncatalytic proteins Hda2 and Hda3, essential for Hda1 deacetylase activity both in vivo and in vitro (5, 6). The Hda1 polypeptide consists of an N-terminal HDAC domain (HDACD), a zinc metalloenzyme, followed by a C-terminal Argonaute-binding protein 2 domain (ARB2D). Hda2 and Hda3 are paralogs and share an overall domain organization with an N-terminal globular domain (NTD) and a C-terminal coiled-coil domain (CCD) (Fig. 1A). Intriguingly, the N-terminal halves of Hda2 and Hda3 resemble the second helicase domain of an SF2 adenosine triphosphatase motor that lacks the adenosine 5′-triphosphate binding motif, along with the SWI2/SNF2 (SWItch2/Sucrose Non-Fermentable2) chromatin remodeler-specific subdomain, and are capable of binding DNA (6). The CCDs of Hda2 and Hda3 act as a scaffold for the assembly of the HDAC complex. Previous studies demonstrated that the S. cerevisiae class II HDAC complex preferentially deacetylates acetyl-lysine residues not only of histones H2B/H3 at specific protomers or inactive genes in a Tup1-dependent manner to delay gene induction but also of histone H4 at actively transcribed genes (7, 8). HAST (Hda1-affected subtelomeric) domains, which are located near the ends of most chromosomes adjacent to the telomeric heterochromatin, are also deacetylated by the class II HDAC complex (9). Despite essential roles of the HDAC complex in gene regulation, we lack a molecular understanding of how the catalytic component is incorporated into the functional transcriptional repression complex to catalyze histone modification of its nucleosome substrate, which role the noncatalytic subunits play to modulate HDAC activity, and how the class II HDAC complex recognizes the nucleosome using a Tup1 repressor–independent mechanism.
Fig. 1. A triangle-shaped HDAC complex and its size heterogeneity were captured by cryo-EM.
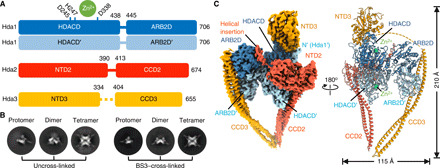
(A) Domain organization of HDAC protomer complex (HDAC-PC) subunits. (B) Representative 2D class averages of uncross-linked and BS3–cross-linked HDAC complexes. (C) A 3.11-Å-resolution cryo-EM map (left) and cartoon representation (right) of HDAC-PC. The subunit color code is used throughout.
RESULTS
The HDAC complex captured by cryo–electron microscopy reveals heterogeneity in the size of particles
To determine the structure of S. cerevisiae full-length class II HDAC complex, we coexpressed constructs encoding the entire Hda1, Hda2, and Hda3 gene in Escherichia coli and copurified them to homogeneity (fig. S1A). Multiangle light scattering coupled with size exclusion chromatography (SEC-MALS) confirmed that the reconstituted complex is consistent with a calculated molecular mass of 313.4 kDa, corresponding to an Hda12-Hda2-Hda3 heterotetrameric complex (hereafter called HDAC protomer complex or HDAC-PC) (fig. S1B). We also noticed that SEC often showed fractions larger than the heterotetramer. When characterizing purified HDAC-PC, we observed a gradual oligomerization of the complex over time, indicating that the HDAC-PC tends to form high–molecular weight oligomers (fig. S1C; see below). The catalytic activities of the purified proteins were assessed by an in vitro deacetylation assay using an acetylated peptide substrate and trichostatin A (TSA). On their own, purified Hda1 and its truncated HDACD are catalytically inactive but become active when bound to the Hda2-Hda3 heterodimer (fig. S1D). Initially, HDAC-PC was applied to a glow-discharged Quantifoil R2/1 holey carbon grid, and its cryo–electron microscopy (cryo-EM) images were recorded on a 300-kV FEG (field emission gun) Titan Halo using a Falcon 3 detector. However, the vast majority of particles were observed to fall apart. To overcome the instability of cryo-EM specimens, we used continuous graphene oxide (GO)–coated EM grids. Cryo-EM micrographs of vitrified specimens on GO grids revealed a heterogeneous mixture of particles. The predominant portion of particles consisted of HDAC-PC. Unexpectedly, other particles had larger sizes with clearly distinguishable two-dimensional (2D) classes featuring multiple copies of HDAC-PC (Fig. 1B).
The consensus reconstruction of the HDAC-PC with 466,972 particles extended to an overall resolution of 3.11 Å, as determined by gold-standard Fourier shell correlation (FSC; see fig. S2). The final map enabled us to unambiguously assign Hda1, Hda2, and Hda3 to their corresponding electron densities (Fig. 1C and fig. S3). For model building, crystal structures of the yeast Hda1 ARB2D monomer [Protein Data Bank (PDB) 5J8J] (10) and Hda3 NTD3 (PDB 3HGT) (6) were rigidly fitted into the cryo-EM map and then manually rebuilt according to the electron densities. The Hda1 HDACD dimer, Hda2 NTD2, and antiparallel CCDs of Hda2 and Hda3 were built de novo (Fig. 1C and fig. S3). The middle segment of Hda3 between NTD3 and CCD3 (residues 335 to 403) is not defined in the cryo-EM map, presumably because of conformational flexibility.
HDAC-PC exhibited higher-order assemblies larger than a 313-kDa heterotetramer, suggesting yet unknown functional roles of the class II HDAC complexes. However, the ratio of these large oligomeric particles was very low on GO-coated grids (fig. S4A). To boost the percentage of higher-order assemblies, we cross-linked cryo-EM specimens of HDAC-PC with 0.5 mM bisulfosuccinimidyl suberate (BS3) and then subjected them to further SEC before application on the grid. Mild cross-linking of HDAC-PC not only proved essential to increase the number of higher-order HDAC assemblies but also helped to enhance the orientational diversity of particles in vitreous ice (fig. S4B). Intriguingly, 2D classification and 3D reconstructions of larger particles revealed self-assemblies of two or four copies of HDAC-PC. The 3.11-Å cryo-EM map of HDAC-PC was used to interpret two distinct oligomeric arrangements, a (Hda12-Hda2-Hda3)2 octameric complex at 8.55 Å displaying C2 symmetry and (Hda12-Hda2-Hda3)4 higher-order assembly at 3.80 Å with D2 symmetry (table S1).
A highly elongated Hda2-Hda3 heterodimer is the Hda1 HDAC clamp for catalysis
HDAC-PC is triangular shaped and measures 210 Å in its longest dimension and approximately 120 Å in its two other dimensions in the high-resolution cryo-EM map (Fig. 1C). The Hda1 catalytic component bound to Hda2-Hda3 is a pseudo-twofold symmetric Hda1 homodimer composed of 706 residues per chain with a butterfly-shaped N-terminal HDACD dimer linked to a C-terminal ARB2D dimer, an overall architecture resembling that of Schizosaccharomyces pombe ortholog Clr3 (fig. S5, A and B). The fission yeast apo-Clr3 crystal structure (PDB 5IKK) (11) exhibits a symmetric conformation with a common dyad axis for the HDACD and ARB2D dimers. By contrast, Hda1 is bent likely induced by interaction with Hda2 and Hda3, such that the ARB2D dimer rotates by ~45° toward Hda3 to tightly embrace the CCD3. The Hda1 HDACD observed in the cryo-EM structure closely resembles the previously determined crystal structures of mammalian HDAC6 CD1 (PDB 6UO2) (12) [1.4-Å root mean square deviation (RMSD) for 356 Cα atoms in each, Dali Z = 51.8] and CD2 (PDB 5WGL) (13) (1.3-Å RMSD for 356 Cα atoms in each, Dali Z = 54.0) and consists of a β sheet formed by eight parallel strands surrounded by 14 α helices per monomer, the characteristic core α/β structure of a canonical HDAC fold (fig. S5C) (14). Hda1 ARB2D forms an intertwined, protrusion-exchanged dimer. The antiparallel β strands protruding from one ARB2D lobe and the β-bulge protruding from the other interact with each other across a dyad axis (fig. S5A). Two protrusions containing residues 511 to 531 are in close proximity to two linkers and are sandwiched between HDACD and ARB2D, thus possibly helping to stabilize the altered orientation of ARB2D upon binding to CCD3.
The structures of Hda2 and Hda3 have overall similar shapes and share local resemblances essentially throughout their length. The N-terminal domains of Hda2 and Hda3 consist of a RecA-like α/β domain and chromatin remodeler SWI2/SNF2-specific helical domain (fig. S5D) (15, 16). Hda2 NTD2 contains an additional tyrosine-rich helical insertion in a kinked lasso-like arrangement (residues 211 to 229) contacting an amphiphilic cradle on one ARB2D lobe, which forms a concave surface that binds Hda2 (fig. S6B). The electron density of NTD2 attributed to a serine-rich region (residues 183 to 210) is not visible, likely because of a structural flexibility of this region. The C-terminal domains of Hda2 and Hda3 fold back on themselves at the hinge regions, forming antiparallel coiled coils that extend approximately 147 and 173 Å, respectively, for CCD2 and CCD3 (figs. S5E and S6, A and C). Hinge-mediated dimerization of Hda2 and Hda3, producing a V-shaped heterodimer, represents highly elongated structures of the two noncatalytic subunits clamping onto the Hda1 dimer. Hda2 spans a single lobe of Hda1 HDACD and ARB2D dimers in a reciprocal fashion, while Hda3 NTD3 and CCD3 lie along the other HDAC lobe and the entire diameter of ARB2D dimer, respectively, making extensive intermolecular interactions (fig. S6, A and D). The Hda1 homodimer and the Hda2-Hda3 subcomplex interfaces bury a total solvent-accessible surface area of ~5760 Å2, 49% of which is contributed by Hda1-Hda2 and 51% by Hda1-Hda3. Ordered N-terminal segments of the two Hda1 chains are embedded into Hda1-Hda2 and Hda1-Hda3 interfaces, cross each other, and thus possibly contribute to the structural integrity of the HDAC-PC (fig. S6, E and F).
Multiple contacts with DNA and histone H2B orient HDAC-PC on the nucleosome
We had previously demonstrated that the Hda2 and Hda3 N-terminal halves, which adopt an SF2-type helicase fold, were essential for the interaction between class II HDAC complex and DNA substrates, generating deacetylase activity for relaxed chromatin (6). To assess whether the previously identified DNA binding domains of the HDAC complex contribute to the interaction with the nucleosome, we first measured in vitro nucleosome binding and kinetics of wild-type HDAC complex (WT HDAC-PC) and its mutant (Mut HDAC-PC) (Fig. 2A). Electrophoretic mobility shift assay (EMSA) and microscale thermophoresis (MST) measurement revealed that WT HDAC-PC efficiently bound mononucleosome but the designed variant, which behaves identically to the WT HDAC-PC in protein purification, abolished interaction with the nucleosome substrate, supporting our interpretation of the importance of key residues in nucleosome recognition (see below).
Fig. 2. Nucleosome recognition by the class II HDAC complex.
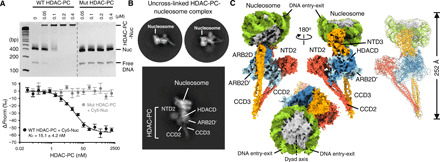
(A) EMSA (top) and MST analysis (bottom) of HDAC-PC and its variant carrying Hda1 K664A/R666A/K667A/K668A, Hda2 K216A, and Hda3 K98A/R103A/K166A/K168A/Q169A/K170A binding to unmodified nucleosome. A dissociation constant (KD) was measured from three independent replicates (shown as means ± SD). bp, base pairs. (B) Representative 2D classes of HDAC-PC bound to nucleosome. (C) Three views of cryo-EM reconstruction of HDAC-PC in complex with nucleosome at a resolution of 4.43 Å (left) and ribbon diagram of the HDAC-nucleosome complex with cryo-EM map (far-right).
We then determined the structure of the nucleosome-bound HDAC complex by cryo-EM. We calculated a 3D reconstruction on the basis of an extensive 2D classification of cryo-EM data, interpreted as one HDAC-PC in complex with mononucleosome (Fig. 2, B and C, and fig. S7). The electron density map for HDAC-PC and canonical nucleosome containing a histone octamer and a 147–base pair (bp) 601 DNA enabled building of a model of the nucleosome-bound HDAC complex, guided by the existing high-resolution structure of HDAC-PC. The HDAC-nucleosome complex was assembled by rigid-body docking of apo-HDAC-PC and nucleosome (PDB 3LZ0) (17), whose orientation is uniquely defined by the DNA entry-exit and the dyad symmetry axis of the nucleosome, followed by subsequent real-space refinement into the cryo-EM reconstruction at 4.43 Å (Fig. 2C). In the complex, HDAC-PC embraces the nucleosome by multiple DNA contacts at superhelical locations 4.5 and 5 (SHL+4.5 and +5) with NTD3 (residues K166, K168, Q169, K170, and N172) and at SHL−2.5 and −3 with one ARB2D lobe (residues K664, R666, and K667) and NTD2 helical insertion (residue K216), exhibiting substantial domain movements of ARB2Ds and NTD3-CCD3 by as much as ~7 Å upon nucleosome binding (Fig. 3A and fig. S7, G and H). These DNA contacts not only enable the Hda1 ARB2D (residues 633 to 636) interactions with the C-terminal helix of one histone H2B but also position a single lobe of the HDACD dimer in proximity to the other H2B N-terminal tail, thus representing a specific configuration of HDAC-PC assembled onto the nucleosome for histone H2B deacetylation characterized by a distance of ~52 Å between K28 and the zinc ion (Fig. 3B). The histone H3 tails protruding between two gyres of nucleosomal DNA near the DNA entry-exit sites and the histone H4 tails located on the lateral surface of the nucleosome are freely accessible in this specific complex, and they are located ~115 and ~ 79 Å, respectively, away from the zinc ion, raising the possibility that additional HDAC-PCs may be engaged in further interactions with the nucleosome to modify the acetylated histone H3 and/or H4 (Fig. 3B; see below).
Fig. 3. Multiple contacts with DNA and histone H2B orient HDAC-PC on the nucleosome.
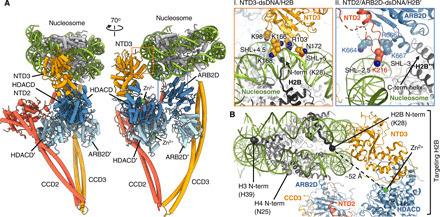
(A) Two views of a cartoon representation of HDAC-PC bound to nucleosome (left) and close-up views of the interfaces between HDAC-PC and nucleosome. Two histone H2Bs are indicated by arrows. (B) Overview of recognition of histone H2B N-terminal tail by HDAC-PC.
The class II HDAC complex is enriched at the nucleosome by dimerization
To explore the possibility of multiple engagement of HDAC-PCs on the nucleosome substrate, we first investigated a fraction of cryo-EM specimens in the absence of nucleosome that formed substantially larger particles in vitreous ice. Successive rounds of 2D classification of an uncross-linked HDAC-PC revealed an ensemble of distinct dimeric states (HDAC dimeric complex or HDAC-DC) (fig. S8A). Three different HDAC-DC conformers characterized by markedly distinguishable symmetry were sufficiently populated to generate the well-resolved 2D class averages, whereas 3D reconstruction failed to visualize individual dimeric states because of insufficient particle images and their conformational heterogeneity. Nevertheless, comparing the three different conformers observed in 2D classes of uncross-linked HDAC-DC particles clearly shows the scale of conformational variability in the dimeric assembly, which may represent dimeric intermediates of HDAC-PC before assembly with the nucleosome. We then determined an 8.55-Å cryo-EM structure of an oligomeric assembly at a lower abundance, revealing a cross-linked HDAC-DC in which two identical HDAC-PCs are arranged in C2 symmetry with a stoichiometry of (Hda12-Hda2-Hda3)2 (fig. S8, B and C). This association is identical to “conformer 1” of the uncross-linked dimer and occurs through a small interface of ~306 Å2 surface area by a single lobe of ARB2D in one HDAC-PC and its symmetry mate along with NTD3 in the other complex. Electrostatic interactions made by E636, R666, K667, and K668 between two adjacent ARB2Ds form a complementary dimer interface with a smaller contribution from the interactions between ARB2D (residues E498, P585, and E700) and C2 symmetry–related NTD3 (residues K98, R103, and K166) (fig. S8D). The small interface between two HDAC-PCs and a low abundance of particles attributed to HDAC-DC suggest that HDAC-PCs dimerize with low intrinsic affinity.
Intriguingly, extensive 2D classification of cryo-EM particle images for the yeast class II HDAC complex bound to nucleosome identified 2D classes of HDAC-DC assembled onto the mononucleosome at a lower abundance (Fig. 4A and fig. S9, B and C). The HDAC-DC observed in the nucleosome-bound particles highly resembles “conformer 3” of the uncross-linked HDAC-DC alone (Fig. 4A and fig. S8A). Its resulting class averages correspond closely to the calculated reprojection images of the HDAC-DC-nucleosome model (fig. S9H; see below). To visualize how multiple HDAC complexes are engaged in interactions with the nucleosome substrate, we determined the cryo-EM reconstruction of the HDAC-DC-nucleosome complex at 10.60 Å (Fig. 4B and fig. S9). A model was built by rigid-body docking of high-resolution structures of two individual HDAC-PCs and mononucleosome (PDB 3LZ0) in the cryo-EM map. The assembly has a good agreement with the low-resolution map when fitted as three rigid bodies and clearly shows one additional HDAC-PC that contacts the nucleosomal DNA at near the entry-exit side where the histones H3 N-terminal tail protrudes. In the cryo-EM model, the histones H3 and H4 N-terminal tails are located ~55 and ~54 Å, respectively, away from a zinc ion within the active site of one HDAC lobe (Fig. 4E). Intriguingly, when the nucleosomes observed in the nucleosome-bound structures of the HDAC-PC and HDAC-DC were superimposed, we recognized in one HDAC-PC large domain movements of its CCDs by as much as ~63 Å along with relatively smaller movements of its two NTDs and HDACD, such that one Hda1 HDACD is positioned closer to the histone H2B N-terminal tail (Fig. 4, C to E). The HDAC-PC pair is held together by nucleosome binding and by intermolecular interaction between ARB2D and NTD2 (fig. S9, F and G). The structure resembles an imperfect pinwheel with only two blades and suggests how the intricate HDAC-DC-nucleosome assembly may enable deacetylation of multiple histone tails simultaneously (Fig. 4B).
Fig. 4. The class II HDAC complex is enriched at the nucleosome by dimerization.
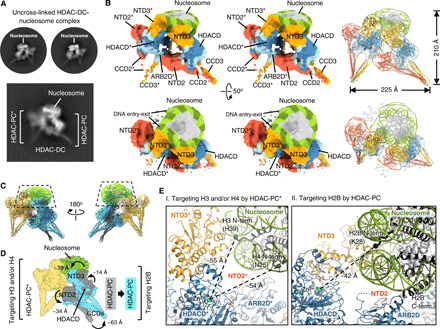
(A) Representative 2D class averages of the uncross-linked HDAC-DC-nucleosome complex. (B) Stereo views of cryo-EM reconstruction of HDAC-DC in complex with nucleosome at a resolution of 10.60 Å (left) and ribbon diagrams of the HDAC-DC-nucleosome complex with cryo-EM map (far right). (C) Overlay of HDAC-DC with HDAC-PC observed in the nucleosome-bound complexes. Two nucleosomes (dashed trapezoid) are superimposed. (D) Relative domain movements within one HDAC-PC upon asymmetric dimerization with the other HDAC-PC in the nucleosome-bound state. A surface representation of the constructed model is shown. (E) Overview of recognition of histone H2B, H3, and/or H4 N-terminal tails by two individual HDAC-PCs in the nucleosome-bound HDAC-DC.
The higher-order tetrameric assembly is a nucleosome binding–deficient HDAC complex
During the examination of the HDAC complex 2D class averages, we realized that the predominant particles of higher-order assemblies are represented by two copies of HDAC-DC, showing three distinct twofold axes (here called HDAC tetrameric complex or HDAC-TC). The symmetry of the oligomer is most apparent in the position of four CCD2-CCD3 legs, which adopt V-shaped peripheral extensions. We calculated the 3D reconstruction of HDAC-TC with imposed D2 symmetry at 3.80-Å resolution (Fig. 5A and figs. S10, D to H, and S11). The HDAC-TC structure was initially modeled by rigid-body docking of four HDAC-PCs into the electron density map, manual rebuilding, and then refining of one of the four HDAC-PC copies that fits best into the map (Fig. 5B). A 1.3-MDa HDAC-TC adopts a four-legged starfish-like architecture of ~310 Å in the longest dimension. In this structure, four individual NTD3 are sandwiched between HDACDs and C2 symmetry–related copies of NTD2, resulting in a tetrapartite interface of ~2,500-Å2 buried surface area (Fig. 5, B and C). The tetrameric structure assembles essentially as two dimers of HDAC-DC, albeit with a staggered swinging movement of two HDAC-PCs on the dyad axis, which pulls each of four HDAC-PCs inward by ~8 Å, forming an interwoven set of four HDAC-PCs (Fig. 5D). Two HDAC-DCs are inverted with respect to one another so that each convex NTD side faces inward, resulting in D2 symmetry of the protein components. In this tetrameric assembly of HDAC-PCs, nucleosome substrate binding as observed in HDAC-PC-nucleosome structure is hindered. Intriguingly, when deacetylase activity was tested against an acetylated peptide substrate, BS3–cross-linked HDAC-TC represented an approximately fourfold higher catalytic activity compared with HDAC-PC, indicating that the catalytic potential of HDAC-TC is unchanged upon oligomerization (Fig. 5E). Thus, it raises the possibility that the higher-order assembly of HDAC-PC, which is unable to recognize the nucleosome substrate, may play a role in regulation of chromatin modification or deacetylation against nonhistone proteins (see Discussion).
Fig. 5. The higher-order assembly is a nucleosome binding–deficient HDAC complex.
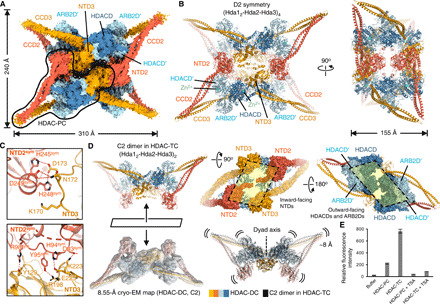
(A) Cryo-EM map of the 1.3-MDa HDAC-TC at a resolution of 3.80 Å. (B) Two orthogonal views of a cartoon representation of HDAC-TC. (C) Magnified views showing tetramerization interfaces between NTD3 and C2 symmetry–related NTD2 (NTD2sym). The HDAC-TC structure was built by rigid-body fitting of four high-resolution structures of the HDAC-PC followed by PHENIX real-space refinement. (D) Quaternary structure (left) and subunit distribution (top right and middle) of HDAC-TC and overlay of HDAC-DC with C2 dimer observed in the halves of HDAC-TC (bottom center). (E) In vitro deacetylase activities of purified HDAC complexes with and without TSA. The assay was performed in triplicate, and the error bars are the SD of the data (n = 3).
Multisubunit HDAC complexes share a general regulatory mechanism in HDAC activity
A correlation between the flexibility of loops flanking the HDAC active site and deacetylase activity has been suggested previously (18). X-ray crystallography and molecular dynamic simulation studies of human HDAC3 also revealed conformational mobility for the catalytic tyrosine Y298 in the active site, which adopts the “out” conformation in the absence of corepressor components and the catalytically required “in” conformation upon corepressor binding (19, 20). Likewise, structural studies of human HDAC8 and its mutants showed that a conformational shift of the catalytic tyrosine Y306 by 1.7 Å from the “in” conformation causes severely compromised deacetylase activity and a 500-fold loss of catalytic efficiency compared with that of WT HDAC8 (21).
Intriguingly, a comparison of the yeast class II HDAC complex structure with two class I HDAC complexes, HDAC3–SMRT–DAD–inositol tetraphosphate (19) and HDAC1-MTA1-sulfates (22), reveals that the noncatalytic components interact with similar regions of the catalytic subunits (residues 68 to 87 of Hda1, residues 16 to 26 of HDAC3, and residues 22 to 32 of HDAC1), which may induce the conformational change of the HDAC to facilitate substrate access to the active site, suggesting a general mechanism for the regulation of deacetylase activity in multiprotein class I and class II HDAC complexes (Fig. 6). On forming a functional HDAC-PC with three subunits, Hda2 and Hda3 buttress loop 2, helix 1, and loop 3 (hereafter L2-H1-L3, residues 68 to 87) of Hda1 flanking the deacetylase active site, which reduces HDACD surface dynamics. Superposition of the two individual HDAC lobes reveals that L2-H1-L3 exhibits a substantial conformational shift upon Hda2 and Hda3 binding. L2-H1-L3 observed in the NTD3-bound HDAC lobe moves by ~8 Å toward Hda2, which is farther away, to interact with CCD2. The zinc atoms and several key residues (catalytic tyrosine Y378, tandem histidine dyad H205 and H206, as well as zinc-coordinating D245, H247, and D338) in the active sites of the Hda1 dimer are universally conserved, whereas the conformation of Y378, which is vital for efficient HDAC catalysis, differs in the individual chains of the Hda1 dimer (Fig. 6B and fig. S3C). The catalytic Y378 in one HDACD lobe bound to NTD2 and CCD2 adopts the “in” conformation required for substrate binding and catalysis, while Y378 in the other HDACD lobe interacting with NTD3 has its phenolic hydroxyl group shifted 3.7 Å outward, suggesting that the subunits of the dimeric Hda1 HDACD may have distinct deacetylase activities in the HDAC-PC (Fig. 6C).
Fig. 6. Multisubunit class I and II HDAC complexes share a general regulatory mechanism in HDAC activity.
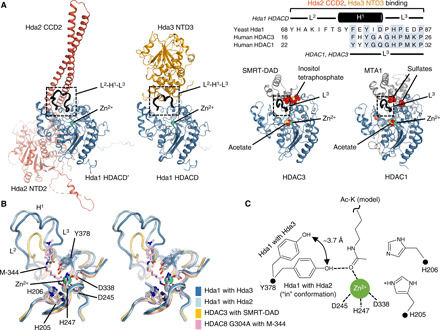
(A) Ribbon diagrams of the yeast Hda1 HDACD-Hda2 NTD2/CCD2, Hda1 HDACD-Hda3 NTD3, human class I HDAC3 bound to SMRT-DAD (PDB 4A69), and human class I HDAC1 in complex with MTA1 (PDB 4BKX) are illustrated on the basis of superposition. The multiple-sequence alignment of HDAC from the yeast and human is shown. (B) Superposition in stereo of one Hda1 HDACD with the other Hda1 HDACD and class I HDACs (PDB 4A69 and 5THU for HDAC3-SMRT-DAD and HDAC8 G304A-M-344, respectively). The cryo-EM density for Y378 in the active site of Hda1 in complex with Hda3 NTD3 is shown as blue mesh. (C) Schematic representation of the catalytic sites of S. cerevisiae Hda1 HDACD dimer with an acetyl-lysine model.
DISCUSSION
The budding yeast Hda1 HDAC, which is the founding member of the class II HDACs, is best known as an “epigenetic eraser” that has a strong specificity for the acetyl marks on histones H2B (K11 and K16) and H3 (K9, K14, K18, K23, and K27) to repress gene activities (7). In addition, a recent study showed that the yeast class II HDAC complex preferentially deacetylases histone H4 within hyperactive genes to partially inhibit transcription elongation (8). Until now, despite the underlined important functional roles of HDAC, molecular structures and mechanisms by which multiprotein HDAC complexes recognize the nucleosome to target histone N-terminal tails remain unknown. Furthermore, it was unclear which molecular mechanisms promote nuclear enrichment of HDACs in relaxed chromatin, as oligomeric states in large HDAC complexes, which may underlie mechanism regulating substrate deacetylation and expansion of HDAC activities under different cellular conditions, were structurally unknown, although it has been suggested that several mammalian HDACs exist as high–molecular weight oligomers in vivo and in vitro (19, 23, 24). Here, we provide the first 3D views of the multisubunit HDAC assemblies and their nucleosome-bound complexes, illuminating detailed architectures and mechanistic features. The ensemble of cryo-EM structures presented in this study outlines a model for how the class II HDAC complex regulates chromatin modification and histone deacetylation.
Previous studies demonstrated that Hda2 and Hda3 are required for HDAC catalysis by Hda1 in vivo and in vitro (5, 6). A high-resolution cryo-EM structure of HDAC-PC provides the mechanistic basis for how the noncatalytic subunits (Hda2 and Hda3) enable the catalytic subunit (Hda1) to afford HDAC activity: Hda2 and Hda3 bind to the Hda1 homodimer by an intricate network of intermolecular interactions. This causes a conformational change within the active sites of Hda1 HDACDs to form the heterotetrameric Hda12-Hda2-Hda3 assembly as the catalytically active HDAC complex (Figs. 1 and 6). In this complex, the Hda1 ARB2Ds rotate by almost 45° and tightly embrace Hda3 CCD3, allowing access of the nucleosome substrate. We propose that Hda2 and Hda3 contribute to nucleosome recognition by making critical contacts with the superhelical DNA and by positioning the Hda1 catalytic component for productive interactions with histones of the nucleosome. The cryo-EM structure of the HDAC-PC bound to nucleosome presented here supports a model in which a network of multiple intermolecular interactions between HDAC-PC and the nucleosome substrate enables the class II HDAC complex to orient the nucleosome for histone H2B deacetylation (Fig. 3). The Hda3 NTD3 serves as the nucleosomal anchor to position the Hda1 HDACD in close proximity to the histone H2B N-terminal tail. Intriguingly, only one of the two Hda1 HDACDs appears to support the deacetylation of histone H2B in this specific configuration, where the active site of the other HDAC lobe interacting primarily with Hda2 is located ~87, ~132, and ~ 94 Å away from histones H2B, H3, and H4 N-terminal tails, respectively (Fig. 3). This arrangement for deacetylation of a particular histone tail within the nucleosome may increase specificity by decreasing the likelihood of deacetylating other histone tails. In addition, given the different substrate specificity between two individual HDACDs in tandem of zebrafish and human class II HDAC6s (25), we speculate that the two active sites of the yeast class II HDAC complex may play distinct roles in deacetylation by regulating histones within the nucleosome substrate and/or by targeting nonhistones, respectively.
The specific arrangement of HDAC-PC assembled onto the nucleosome for the deacetylation of histone H2B allows Hda2 NTD2 to buttress an additional HDAC complex by interacting with the Hda1 ARB2D of the adjacent HDAC-PC (Fig. 4), offering an explanation of the strong enrichment of HDAC activities for multiple histone deacetylation in relaxed chromatin as described in recent work identifying enrichment of the class II HDAC complex at highly transcribed genes throughout all yeast chromosomes (8). The asymmetric HDAC-DC bound to mononucleosome described here may underlie the mechanism by which multiple engagement of the HDAC complexes at both near the DNA entry-exit site and the nucleosome periphery, respectively, can expand the catalytic activities for regulating gene expression. In addition, given the dyad symmetry of the mononucleosome, one more HDAC-DC may bind at the nucleosome, enabling further deacetylation of multiple histone tails.
Unexpectedly, we have found that the purified class II HDAC complex tends to oligomerize over time. Previous studies suggested that several mammalian class I and II HDACs form oligomers in vivo and in vitro, although structural and mechanistic insights of the oligomerization were not elucidated (19, 23, 24). Here, we show that the tetrameric higher-order assembly of HDAC-PC adopts a closed conformation that is unable to engage the nucleosome substrate. The nucleosome recognition sites of four individual HDAC-PCs are completely occluded, while the active sites of the Hda1 HDACDs are on the convex side of the complex and are exposed to enable substrate binding (Fig. 5). Upon incubation with an acetylated peptide, the cross-linked HDAC-TC readily deacetylases the substrate in vitro and shows an approximately fourfold increase of HDAC activity compared with HDAC-PC, suggesting unchanged catalysis, while our structural analysis suggests impaired nucleosome binding. It may represent an inactive storage form to allow fast activation when needed, and it may have different substrate binding activity capable of deacetylation of nonhistone proteins. Previous studies demonstrated that the mammalian class II HDAC6, which contains two HDACDs in tandem, not only is enriched in active genes through the interaction with phosphorylated RNA polymerase II but also regulates the acetylation levels of the α-tubulin and the molecular chaperone heat shock protein 90 (Hsp90) (26–28). Regulation of the yeast Hsp90 function by the class I Rdp3 and class II Hda1 was also reported (29), and further studies are required to understand the precise roles of the class II HDAC complex and its oligomeric forms toward nonhistone substrates.
S. cerevisiae is a nonpathogenic fungus but phylogenetically related to human fungal pathogens such as Candida glabrata and Candida albicans that develop life-threatening diseases (30, 31). Previous studies demonstrated that the fungal-specific Hda2 and Hda3 are key to regulate morphological switches in C. albicans, which is responsible for millions of infections annually (32). The amino acid conservation in the interfaces between the catalytic domain and noncatalytic subunits among fungi (fig. S6G) and the molecular structures described here may be the key for strategies to design and develop novel fungicides. The structural diversity between human and fungal HDACs may assist in this endeavor lessen cross-reactivity and toxicity issues.
In summary, our structural and biochemical analyses of class II HDAC complex provide key observations for understanding the architectures of the multisubunit chromatin-modifying HDAC complex and its supramolecular assembly as well as mechanisms by which Hda2 and Hda3 activate Hda1 HDACD. We demonstrate that the HDAC complex embraces one DNA gyre of the nucleosome through DNA binding sites of NTD3 and binds to the other DNA gyre through ARB2D and NTD2, representing Tup1-independent processing of histone H2B by HDAC at the relaxed chromatin. Furthermore, we suggest structural and mechanistic insights into multiple engagements of the HDAC complex with the nucleosome to enable catalytic activities for regulating gene expression. The molecular structures also provide a basis for further definition of the precise roles of the higher-order HDAC oligomers.
MATERIALS AND METHODS
Purification of S. cerevisiae class II HDAC complex
All constructs were generated by using a standard polymerase chain reaction. For coexpression of HDAC-PC components, three expression plasmids (pET21b, pET30a, and pCDFDuet-1) (Novagen) encoding individual full-length Hda1, Hda2, and Hda3 genes were cotransformed into E. coli Rosetta(DE3) (Novagen). HDAC-PC and its variant were purified by using a His-affinity chromatography with a 5-ml HisTrap FF crude column (GE Healthcare) followed by anion-exchange chromatography with Resource Q (GE Healthcare) and SEC with Superdex 200 or Superose 6 (GE Healthcare). Peak fractions were concentrated to 1 mg ml−1. For chemical cross-linking, HDAC-PC (0.14 mg ml−1) in a protein storage buffer containing 25 mM Hepes (pH 8.0), 100 mM NaCl, and 1 mM dithiothreitol (DTT) was cross-linked with 0.5 mM BS3 (Thermo Fisher Scientific) for 45 min on ice. The reaction was quenched with 100 mM tris-HCl (pH 7.5), and then the mixture was further purified by SEC (Superose 6 10/600) in buffer supplemented with 25 mM Hepes (pH 8.0), 100 mM NaCl, and 1 mM DTT. Peak fractions were concentrated by ultracentrifugation to 0.72 mg ml−1.
Purification of mononucleosome
Purification of unmodified Xenopus laevis histone proteins and nucleosome reconstruction using a 147-bp Widom 601 positioning sequence were performed as described previously (33). Recombinant X. laevis histones (H2A, H2B, H3, and H4) and 601 DNA were mixed in buffer supplemented with 20 mM tris-HCl (pH 7.5), 2 M NaCl, and 1 mM EDTA. The mixture was then transferred to a D-Tube Dialyzer (Millipore) and dialyzed against buffer containing 20 mM tris-HCl (pH 7.5), 2 M NaCl, and 1 mM EDTA [RB (Reconstitution Buffer) high buffer] for 5 hours at 4°C. A peristaltic pump was used for replacing RB high buffer with low-salt buffer [20 mM tris-HCl (pH 7.5), 200 mM NaCl, and 1 mM EDTA]. The same was lastly dialyzed against the protein storage buffer containing 20 mM tris-HCl (pH 7.5), 20 mM NaCl, and 1 mM EDTA at 4°C.
SEC-MALS analysis
The molecular mass of HDAC-PC was determined by the quantitative analysis using MALS. The protein sample (1 mg ml−1) was injected into a WTC-030S5 column (Wyatt Technology) coupled to an 18-angle light-scattering detector (DAWN HELEOS II) and a refractive index detector (Optilab T-rEX) (Wyatt Technology). All data were collected at 25°C (0.5 ml min−1 of flow rate) and analyzed using ASTRA 6 software.
HDAC activity assay
All protein samples (0.25 μM) were mixed with fluoro-substrate peptide (20 μM) in assay buffer supplemented with 25 mM tris-HCl (pH 8.0), 125 mM NaCl, and 1% glycerol. Reaction mixtures with and without TSA (1 μM) were incubated at room temperature for 1 hour and then quenched by adding 50 μl of buffer containing lysyl endopeptidase (1 mAU ml−1). After further incubation for 15 min at room temperature, fluorescence intensities were measured by a fluoro-spectrophotometer with excitation at 350 nm and emission at 440 nm. Each reaction was performed in triplicate with the same preparation of protein, and the SD of the raw data is reported.
Electrophoretic mobility shift assay
For the EMSA, 0.1 μM mononucleosome containing an X. laevis histone octamer and 147-bp Widom 601 double-stranded DNA was mixed with HDAC-PC or its variant at the indicated concentrations and incubated on ice for 1 hour in EMSA buffer [20 mM tris-HCl (pH 8.0), 50 mM NaCl, 1 mM MgCl2, and 1 mM DTT]. The samples were then resolved by electrophoresis onto a 6% native TBE (Tris-borate-EDTA) gel at 4°C. The gel was stained with SYBR Safe DNA gel stain (Thermo Fisher Scientific) to visualize DNA bands.
Microscale thermophoresis
For the MST experiments, a concentration series of WT HDAC-PC or its variant was prepared using a 1:1 serial dilution of HDAC complexes in buffer supplemented with 20 mM Hepes (pH 8.0), 50 mM NaCl, 1 mM MgCl2, 1 mM DTT, and 0.05% Tween 20. The range of HDAC complexes was from 3.3 μM to a final 0.1 nM, over 16 serial diluted Monolith NT.115 premium capillaries (NanoTemper) with 10-μl samples. The interaction was initiated by the addition of 10 μl of 3.74 nM Cy5-labeled mononucleosome to each reaction mixture, resulting in a 1.87 nM final concentration of nucleosome substrate. The measurements were performed on a Monolith NT.115 pico (NanoTemper). Data were analyzed by MO.Control and MO.Affinity software (NanoTemper).
Cryo-EM sample preparation
Three microliters of uncross-linked HDAC-PC at a concentration of 1 mg ml−1 was applied onto GO-coated R2/1 copper 200 mesh (Agar Scientific Ltd.) followed by a 60-s wait time and plunge-frozen in liquid ethane using a Vitrobot Mark IV (Thermo Fisher Scientific) with settings at 4°C, 95% humidity, and 4-s blot time. For BS3–cross-linked HDAC-TC, 4 μl of the cross-linked specimen at a concentration of 0.72 mg ml−1 (fig. S10A) was applied onto a glow-discharged R2/1 copper 200-mesh holey carbon grid (Quantifoil) and immediately frozen in liquid ethane. The HDAC-nucleosome complex was obtained by mixing 2.0 μM of the uncross-linked HDAC-PC with 0.3 μM nucleosome. The nucleosome-bound complex was then applied onto GO-coated R2/1 copper 200 mesh (Agar Scientific Ltd.) and plunge-frozen in liquid ethane with settings at 4°C, 95% humidity, and 6-s blot time of Vitrobot Mark IV (Thermo Fisher Scientific).
Cryo-EM data collection and image processing
All cryo-EM data were acquired at the cryo-EM facility at the Max Planck Institute of Biochemistry (table S1). Cryo-EM data of HDAC-PC, BS3–cross-linked HDAC-DC, and the nucleosome-bound HDAC-DC were acquired on a Glacios transmission electron microscope (Thermo Fisher Scientific) operated at 200 keV, equipped with a K2 summit direct detector and a GIF quantum energy filter (Gatan). For HDAC-PC dataset, cryo-EM data were collected at a nominal dose of 62.1 e−/Å2 with 75 frames per movie and a pixel size of 1.181 Å. For the cross-linked HDAC-DC dataset, data were collected at a nominal dose of 62.0 e−/Å2 and the target defocus range between 1.5 and 3.0 μm. Cryo-EM data of HDAC-TC and the nucleosome-bound HDAC-PC were acquired on a Titan Krios (Thermo Fisher Scientific) at 300 keV using a K3 direct detector (Gatan) in electron counting mode at a nominal magnification of 105,000 and a pixel size of 0.8512 Å. A total of 7290, 1873, 20,594, 15,049, and 2388 movies were collected for HDAC-PC, HDAC-DC, HDAC-TC, HDAC-PC-nucleosome, and HDAC-DC-nucleosome, respectively. All cryo-EM data were recorded using a SerialEM (34) multirecord mode.
HDAC-PC dataset was imported into cryoSPARC (35), and movie frames were aligned using patch motion. CTF (Contrast Transfer Function) was estimated in a patch manner, and micrographs that were not suitable for image analysis (for example, because of a large drift or heavy contamination with crystalline ice) were removed by manual inspection. Particles were picked with cryoSPARC reference-free blob picker. An initial set of 1,721,559 particles was then extracted, and four rounds of 2D classification were performed to clean up the dataset. Ab initio model generation using cryoSPARC with three classes resulted in one highly populated class and two “junk” classes. A total of 520,040 particles attributed to the best class were iteratively refined using heterogeneous refinement, homogeneous refinement, and nonuniform refinement to produce a map of 3.11-Å resolution (a final set of 466,972 particles).
For HDAC-TC dataset, the 60 frames of each image stack were aligned using cryoSPARC patch motion. CTF values of the summed micrographs were then determined using cryoSPARC patch CTF estimation. Particle picking was carried out using the reference-free blob picker, and 472,885 particles were cleaned up by removing outlier particles using three rounds of reference-free 2D classification. After removing false positives and noisy particles, the remaining 94,838 particles were used for ab initio 3D reconstruction into three classes using cryoSPARC. A final set of 53,757 particles were then imported into the cisTEM (36) to perform a final 3D refinement with D2 symmetry and yielded the final reconstruction at 3.80-Å overall resolution.
For HDAC-PC in complex with nucleosome dataset, beam-induced motion correction and CTF estimation were performed as described for the HDAC-PC and HDAC-TC data collections. In total, 5286 micrographs were selected after removing poor-quality ones. Particle picking was carried out as described for the HDAC-PC and HDAC-TC data collections and 1,141,091 particles were then subjected to four rounds of reference-free 2D classification in cryoSPARC. A total of 50,531 particles were used to generate ab initio models with three classes in cryoSPARC. 3D classifications were carried out, and 41,279 particles belonging to homogeneous well-defined classes were subjected to homogeneous refinement and nonuniform refinement in cryoSPARC. The final reconstruction was refined to an overall resolution of 4.43 Å.
All resolutions reported here were determined by an FSC criterion of 0.143, and maps were sharpened by applying an automated local resolution–weighted negative B factor. Local map resolutions were estimated by cryoSPARC.
Model building and refinement
To build an atomic structure of HDAC-PC, crystal structures of S. cerevisiae Hda1 ARB2D monomer (PDB 5J8J) (10) and Hda3 NTD3 (PDB 3HGT) (6) were fitted into the refined 3D reconstruction map using UCSF (University of California at San Francisco) Chimera (37) and then manually rebuilt in Coot (38) to fit the densities. Hda1 HDACD, Hda2 NTD2, and CCD2/3 were built de novo from a polyalanine model. The entire model of HDAC-PC was manually improved in Coot. Sequence assignment was guided mainly by bulky side-chain residues. The final atomic structure was refined in real space using PHENIX (39) and validated using MolProbity (40). For HDAC-DC and HDAC-TC model building, the coordinates of HDAC-PC determined in this work were rigid body–fitted into the refined cryo-EM map with imposed C2 and D2 symmetry, respectively, for HDAC-DC and HDAC-TC using UCSF Chimera and then subjected to PHENIX real-space refinement to generate final model statistics.
To build cryo-EM models of the nucleosome-bound HDAC complexes, the published crystal structure of X. laevis nucleosome (PDB 3LZ0) (17) and HDAC-PC solved in this study were rigid body–fitted into the cryo-EM map using UCSF Chimera. The final model for the HDAC-PC-nucleosome complex was further refined in real space using PHENIX and validated using MolProbity. Structural analysis was performed in Coot, and figures were prepared using PyMOL (The PyMOL Molecular Graphics System, v.2.3.4) and ChimeraX (41).
Supplementary Material
Acknowledgments
We thank T. Ziegler, A. Rechenberg, and L. Lercher for assistance with nucleosome preparation; D. Witte and E. Schneider for assistance with MST; M. Zobawa for assistance with SEC-MALS; P. Liebhart for preparing laboratory consumables; and T. Neuefeind for support. Funding: This work was supported by a Nobel Laureate Fellowship of the Max Planck Society (J.-H.L.). Author contributions: J.-H.L. and R.H. conceived the study. J.-H.L. carried out all experiments and data analysis. D.B. assisted with cryo-EM sample preparation and data acquisition of HDAC-PC, HDAC-TC, and HDAC-PC in complex with nucleosome. T.S. assisted with cryo-EM sample preparation and data acquisition of HDAC-DC. J.-H.L. and R.H. interpreted the data and wrote the manuscript, with contributions from all authors. Competing interests: The authors declare that they have no competing interests. Data and materials availability: Cryo-EM maps have been deposited in the Electron Microscopy Data Bank (EMDB) under accession numbers EMD-11092 (HDAC-PC), EMD-11094 (HDAC-DC), EMD-11101 (HDAC-TC), EMD-11102 (HDAC-PC-nucleosome complex), and EMD-11712 (HDAC-DC-nucleosome complex). Model coordinates have been deposited in the PDB under accession codes 6Z6F (HDAC-PC), 6Z6H (HDAC-DC), 6Z6O (HDAC-TC), and 6Z6P (HDAC-PC-nucleosome complex). All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the corresponding authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/2/eabd4413/DC1
REFERENCES AND NOTES
- 1.Inoue A., Fujimoto D., Enzymatic deacetylation of histone. Biochem. Biophys. Res. Commun. 36, 146–150 (1969). [DOI] [PubMed] [Google Scholar]
- 2.Ayer D. E., Histone deacetylases: Transcriptional repression with SINers and NuRDs. Trends Cell Biol. 9, 193–198 (1999). [DOI] [PubMed] [Google Scholar]
- 3.Rundlett S. E., Carmen A. A., Kobayashi R., Bavykin S., Turner B. M., Grunstein M., Hda1 and Rpd3 are members of distinct yeast histone deacetylase complexes that regulate silencing and transcription. Proc. Natl. Acad. Sci. U.S.A. 93, 14503–14508 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kasten M. M., Dorland S., Stillman D. J., A large protein complex containing the yeast Sin3p and Rpd3p transcriptional regulators. Mol. Cell. Biol. 17, 4852–4858 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu J., Carmen A. A., Kobayashi R., Suka N., Grunstein M., Hda2 and Hda3 are related proteins that interact with and are essential for the activity of the yeast histone deacetylase Hda1. Proc. Natl. Acad. Sci. U.S.A. 98, 4391–4396 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee J.-H., Maskos K., Huber R., Structural and functional studies of the yeast class II Hda1 histone deacetylase complex. J. Mol. Biol. 391, 744–757 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Wu J., Suka N., Carlson M., Grunstein M., Tup1 utilizes histone H3/H2B-specific Hda1 deacetylase to repress gene activity in yeast. Mol. Cell 7, 117–126 (2001). [DOI] [PubMed] [Google Scholar]
- 8.Ha S. D., Ham S., Kim M. Y., Kim J. H., Jang I., Lee B. B., Lee M. K., Hwang J.-T., Roh T.-Y., Kim T., Transcription-dependent targeting of Hda1C to hyperactive genes mediates H4-specific deacetylation in yeast. Nat. Commun. 10, 4270 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robyr D., Suka Y., Xenarios I., Kurdistani S. K., Wang A., Suka N., Grunstein M., Microarray deacetylation maps determine genome-wide functions for yeast histone deacetylases. Cell 109, 437–446 (2002). [DOI] [PubMed] [Google Scholar]
- 10.Shen H., Zhu Y., Wang C., Yan H., Teng M., Li X., Structural and histone binding ability characterization of the ARB2 domain of a histone deacetylase Hda1 from Saccharomyces cerevisiae. Sci. Rep. 6, 33905 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Job G., Brugger C., Xu T., Lowe B. R., Pfister Y., Qu C., Shanker S., Baños Sanz J. I., Partridge J. F., Schalch T., SHREC silences heterochromatin via distinct remodeling and deacetylation modules. Mol. Cell 62, 207–221 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Osko J. D., Christianson D. W., Structural basis of catalysis and inhibition of HDAC6 CD1, the enigmatic catalytic domain of histone deacetylase 6. Biochemistry 58, 4912–4924 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Porter N. J., Mahendran A., Breslow R., Christianson D. W., Unusual zinc-binding mode of HDAC6-selective hydroxamate inhibitors. Proc. Natl. Acad. Sci. U.S.A. 114, 13459–13464 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marmorstein R., Structure of histone deacetylases: Insights into substrate recognition and catalysis. Structure 9, 1127–1133 (2001). [DOI] [PubMed] [Google Scholar]
- 15.Hauk G., Bowman G. D., Structural insights into regulation and action of SWI2/SNF2 ATPases. Curr. Opin. Struct. Biol. 21, 719–727 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yan L., Chen Z., A unifying mechanism of DNA translocation underlying chromatin remodeling. Trends Biochem. Sci. 45, 217–227 (2020). [DOI] [PubMed] [Google Scholar]
- 17.Vasudevan D., Chua E. Y. D., Davey C. A., Crystal structures of nucleosome core particles containing the ’601′ strong positioning sequence. J. Mol. Biol. 403, 1–10 (2010). [DOI] [PubMed] [Google Scholar]
- 18.Lombardi R. M., Cole K. E., Dowling D. P., Christianson D. W., Structure, mechanism, and inhibition of histone deacetylases and related metalloenzymes. Curr. Opin. Struct. Biol. 21, 735–743 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Watson P. J., Fairall L., Santos G. M., Schwabe J. W. R., Structure of HDAC3 bound to co-repressor and inositol tetraphosphate. Nature 481, 335–340 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Arrar M., Turnham R., Pierce L., de Oliviera C. A. F., McCammon J. A., Structural insight into the separate roles of inositol tetraphosphate and deacetylase-activating domain in activation of histone deacetylase 3. Protein Sci. 22, 83–92 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Porter N. J., Christianson N. H., Decroos C., Christianson D. W., Structural and functional influence of the glycine-rich loop G302GGGY on the catalytic tyrosine of histone deacetylase 8. Biochemistry 55, 6718–6729 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Millard C. J., Watson P. J., Celardo I., Gordiyenko Y., Cowley S. M., Robinson C. V., Fairall L., Schwabe J. W. R., Class I HDACs share a common mechanism of regulation by inositol phosphates. Mol. Cell 51, 57–67 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang W.-M., Tsai S.-C., Wen Y.-D., Fejer G., Seto E., Functional domains of histone deacetylase-3. J. Biol. Chem. 277, 9447–9454 (2002). [DOI] [PubMed] [Google Scholar]
- 24.Guo L., Han A., Bates D. L., Cao J., Chen L., Crystal structure of a conserved N-terminal domain of histone deacetylase 4 reveals functional insights into glutamine-rich domains. Proc. Natl. Acad. Sci. U.S.A. 104, 4297–4302 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Miyake Y., Keusch J. J., Wang L., Saito M., Hess D., Wang X., Melancon B. J., Helquist P., Gut H., Matthias P., Structural insights into HDAC6 tubulin deacetylation and its selective inhibition. Nat. Chem. Biol. 12, 748–754 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Hubbert C., Guardiola A., Shao R., Kawaguchi Y., Ito A., Nixon A., Yoshida M., Wang X.-F., Yao T.-P., HDAC6 is a microtubule-associated deacetylase. Nature 417, 455–458 (2002). [DOI] [PubMed] [Google Scholar]
- 27.Kovacs J. J., Murphy P. J. M., Gaillard S., Zhao X., Wu J.-T., Nicchitta C. V., Yoshida M., Toft D. O., Pratt W. B., Yao T.-P., HDAC6 regulates Hsp90 acetylation and chaperone-dependent activation of glucocorticoid receptor. Mol. Cell 18, 601–607 (2005). [DOI] [PubMed] [Google Scholar]
- 28.Wang Z., Zang C., Cui K., Schones D. E., Barski A., Peng W., Zhao K., Genome-wide mapping of HATs and HDACs reveals distinct functions in active and inactive genes. Cell 138, 1019–1031 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robbins N., Leach M. D., Cowen L. E., Lysine deacetylases Hda1 and Rpd3 regulate Hsp90 function thereby governing fungal drug resistance. Cell Rep. 2, 878–888 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fidel P. L. Jr., Vazquez J. A., Sobel J. D., Candida glabrata: Review of epidemiology, pathogenesis, and clinical disease with comparison to C. albicans. Clin. Microbiol. Rev. 12, 80–96 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gow N. A. R., van de Veerdonk F. L., Brown A. J. P., Netea M. G., Candida albicans morphogenesis and host defence: Discriminating invasion from colonization. Nat. Rev. Microbiol. 10, 112–122 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.M. R. Peterson, R. J. Price, S. Gourlay, A. May, J. Tullet, A. Buscaino, The fungal-specific Hda2 and Hda3 proteins regulate morphological switches in the human fungal pathogen Candida albicans bioRxiv 340364 [Preprint]. 7 June, 2018. 10.1101/340364. [DOI]
- 33.Luger K., Rechsteiner T. J., Richmond T. J., Expression and purification of recombinant histones and nucleosome reconstitution. Methods Mol. Biol. 119, 1–16 (1999). [DOI] [PubMed] [Google Scholar]
- 34.Mastronarde D. N., Automated electron microscope tomography using robust prediction of specimen movements. J. Struct. Biol. 152, 36–51 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Punjani A., Rubinstein J. L., Fleet D. J., Brubaker M. A., cryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 36.Grant T., Rohou A., Grigorieff N., cisTEM, user-friendly software for single-particle image processing. eLife 7, e35383 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E., UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Emsley P., Cowtan K., Coot: Model-building tools for molecular graphics. Acta. Crystallogr. D Biol. Crystallogr. 60, 2126–2132 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Afonine P. V., Poon B. K., Read R. J., Sobolev O. V., Terwilliger T. C., Urzhumtsev A., Adams P. D., Real-space refinement in PHENIX for cryo-EM and crystallography. Acta. Crystallogr. D Biol. Crystallogr. 74, 531–544 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen V. B., Arendall W. B. III, Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., Richardson D. C., MolProbity: All-atom structure validation for macromolecular crystallography. Acta. Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Goddard T. D., Huang C. C., Meng E. C., Pettersen E. F., Couch G. S., Morris J. H., Ferrin T. E., UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hai Y., Shinsky S. A., Porter N. J., Christianson D. W., Histone deacetylase 10 structure and molecular function as a polyamine deacetylase. Nat. Commun. 8, 15368 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Farnung L., Vos S. M., Wigge C., Cramer P., Nucleosome-Chd1 structure and implications for chromatin remodelling. Nature 550, 539–542 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li M., Xia X., Tian Y., Jia Q., Liu X., Lu Y., Li M., Li X., Chen Z., Mechanism of DNA translocation underlying chromatin remodelling by Snf2. Nature 567, 409–413 (2019). [DOI] [PubMed] [Google Scholar]
- 45.Yan L., Wu H., Li X., Gao N., Chen Z., Structures of the ISWI-nucleosome complex reveal a conserved mechanism of chromatin remodeling. Nat. Struct. Mol. Biol. 26, 258–266 (2019). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/2/eabd4413/DC1