

A genome-wide screening revealed that TRIM26 is a host factor for HCV replication and a new determinant for host tropism.
Abstract
Hepatitis C virus (HCV) remains a major human pathogen that requires better understanding of virus-host interactions. In this study, we performed a genome-wide CRISPR-Cas9 screening and identified TRIM26, an E3 ligase, as a critical HCV host factor. Deficiency of TRIM26 specifically impairs HCV genome replication. Mechanistic studies showed that TRIM26 interacts with HCV-encoded NS5B protein and mediates its K27-linked ubiquitination at residue K51, and thus promotes the NS5B-NS5A interaction. Moreover, mouse TRIM26 does not support HCV replication because of its unique six–amino acid insert that prevents its interaction with NS5B. Ectopic expression of human TRIM26 in a mouse hepatoma cell line that has been reconstituted with other essential HCV host factors promotes HCV infection. In conclusion, we identified TRIM26 as a host factor for HCV replication and a new determinant of host tropism. These results shed light on HCV-host interactions and may facilitate the development of an HCV animal model.
INTRODUCTION
Hepatitis C virus (HCV) is an enveloped, single-stranded RNA virus belonging to the family Flaviviridae. The HCV RNA genome is 9.6 kb in length and consists of a single open reading frame (ORF) flanked by highly conserved 5′ and 3′ untranslated regions (UTRs). The ORF encodes a single polyprotein of over 3000 amino acids, which is cleaved by cellular and viral proteases into structural proteins (core, E1, and E2) and nonstructural proteins (p7, NS2, NS3, NS4A, NS4B, NS5A, and NS5B). NS5B, an RNA-dependent RNA polymerase (RdRp), together with other nonstructural proteins NS3, NS4A, NS4B, and NS5A, forms intracellular membrane–associated replication complex and catalyzes viral genomic RNA replication. HCV infects over 71 million people worldwide (1). Among this infected population, about 80% develop persistent infection, leading to severe liver diseases, such as liver cirrhosis and hepatocellular carcinoma (HCC). Recently developed direct-acting antiviral agents (DAAs) targeting viral NS3 protease, NS5A and NS5B polymerases are highly effective in curing patients with HCV. However, global eradication of HCV remains challenging due to lack of an HCV vaccine, potential drug-resistant mutations, severe liver disease progression in DAA-cured patients, and other newly emerging problems (2). Better understanding of viral life cycle and virus-host interactions is still imperative for the prevention and control of HCV infection.
The tripartite motif (TRIM) family that consists of more than 70 members in human plays roles in multiple cellular processes including intracellular signaling, development, apoptosis, protein quality control, innate immunity, autophagy, and carcinogenesis (3). An increasing number of studies have focused on the roles of TRIM family proteins in host responses to virus infection. TRIM5α recognizes retroviral capsids to induce premature core disassembly and inhibits reverse transcription of the viral genome (4, 5). TRIM69 restricts dengue virus (DENV) replication by ubiquitinating viral NS3 (6). TRIM22 and TRIM41 inhibit influenza A virus infection by targeting nucleoprotein for degradation (7, 8). TRIM56 suppresses Zika virus (ZIKV) replication through sequestration of its genomic RNA (9).
As an E3 ligase, TRIM26 contains an N-terminal RING domain, B-box domain, coiled-coil domain, and a C-terminal SPRY domain. One study showed that TRIM26 promotes interferon regulatory factor 3 (IRF3) degradation and thus suppresses interferon-β (IFN-β) signaling (10), while another showed that TRIM26 promotes the interaction between the kinases TBK1 and NEMO, leading to activation of IFN signaling (11). In this study, we identified TRIM26 as a critical host factor of HCV by genome-wide CRISPR screening. A mechanistic study demonstrated that TRIM26 mediates NS5B ubiquitination and enhances its interaction with NS5A, which is crucial for HCV genome replication. Furthermore, we showed that mouse TRIM26 does not support HCV replication because of its unique six–amino acid insert that prevents its interaction with NS5B, providing new evidence for understanding the genetic basis underlying the exceptionally narrow host tropism of HCV infection. Ectopic expression of human TRIM26 in a mouse hepatoma cell line that has been reconstituted with other essential HCV host factors promotes HCV infection, providing clues for the development of an HCV animal model.
RESULTS
Identification of host factors essential for HCV infection
Taking advantage of the NIrD (NS3-4A inducible rtTA-mediated dual-reporter) reporter system to monitor HCV infection in real-time and live-cell fashion (12), we performed a genome-wide CRISPR-Cas9 screening to identify host factors essential for HCV infection (13) (Fig. 1A). Huh7.5 cells harboring the NIrD reporter showed red fluorescence (mCherry) upon cell culture-derived HCV (HCVcc) infection in the presence of doxycycline. The reporter cells transduced with a CRISPR single guide RNA (sgRNA) library targeting human protein–coding genes were infected with HCVcc at multiplicity of infection (MOI) of 0.1, and mCherry-negative cells were enriched by cell sorting. The abundance of each sgRNA in the enriched mCherry-negative cells was measured through deep sequencing and analyzed with the RIGER (RNAi Gene Enrichment Ranking) algorithm (tables S1 and S2). Many host factors were identified from this screening (Fig. 1B). Among the top candidates, CD81, occludin (OCLN), and claudin 1 (CLDN1) are the well-defined HCV entry factors (14–16). Peptidylproly isomerase A (PPIA), also known as cyclophilin A (CypA), has been shown critical for HCV replication (17). ELAV-like RNA binding protein 1 (ELAVL1) interacted with HCV 3′UTR and enhanced HCV replication (18). These positive results validated our CRISPR screening. Except for these previously described hits, TRIM26 is a top hit from our screening and was also identified in a previous screening (19). To investigate the role of TRIM26 in HCV infection, we silenced TRIM26 expression in Huh7 cells through CRISPR interference (CRISPRi) (20). Two TRIM26 sgRNAs (sg1 and sg2) that efficiently reduced the TRIM26 expression (Fig. 1C), which had no effect on cell viability (Fig. 1D), were selected for the following experiments. TRIM26 knockdown cells were infected with HCVcc at MOI of 0.1, and the intracellular HCV RNA, NS3 protein levels, and extracellular HCV titer were measured at the indicated time points after HCVcc infection. As shown in Fig. 1 (E to G), TRIM26 knockdown reduced the HCV RNA level, NS3 protein expression, and extracellular HCV titer. We further reconstituted wild-type TRIM26 and the RING domain–deleted mutant (TRIM26ΔR) in the TRIM26 knockdown and control cells. The expression of TRIM26 and TRIM26ΔR in these cells was verified by Western blot (fig. S1A). As shown in fig. S1 (B to D), exogenous expression of wild-type TRIM26, but not TRIM26ΔR, restored HCV infection in the TRIM26 knockdown cells.
Fig. 1. Identification of host factors essential for HCV infection.
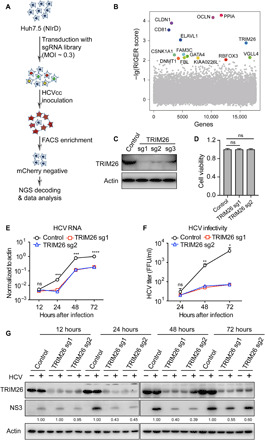
(A) Schematic of whole-genome scale CRISPR screening. (B) The hits identified in CRISPR screening were shown after RIGER analysis. Top hits in the screening were marked by the gene symbols with different colors. (C) Western blot analysis of TRIM26 expression in three TRIM26 knockdown Huh7 cells. (D) Effect of TRIM26 knockdown on cell viability. (E to G) Control and TRIM26 knockdown Huh7 cells were infected with HCVcc at MOI of 0.1 for the indicated time points, and intracellular HCV RNA (E), extracellular HCV titer (F), and NS3 protein (G) were determined. HCV RNA was expressed as values relative to the actin mRNA level. The error bars represent SDs from two independent experiments. FFU, focus-forming units. One-way ANOVA was used for statistical analysis. Not significant (ns), P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. The protein levels were quantified by ImageJ, normalized against internal actin, and expressed as values relative to control cells.
To further confirm the role of TRIM26 in HCV infection, we generated Huh7.5.1 TRIM26 knockout cells (fig. S2, A and B). We then infected Huh7.5.1 TRIM26 knockout and control cells with HCVcc at MOI of 0.1. Consistently, TRIM26 knockout reduced the HCV RNA level, NS3 expression, and extracellular HCV titer (fig. S2, C to E). Together, these results demonstrated that TRIM26 plays an important role in HCV infection.
Deficiency of TRIM26 impairs HCV genome replication
Previous studies showed that TRIM26 is involved in IFN signaling (10, 11). To examine the potential effect of IFN signaling in TRIM26 knockdown cells on HCV infection, Huh7-TRIM26 knockdown and control cells were infected with HCVcc at MOI of 0.1. IFN-β and IFN-stimulated gene (ISG) mRNA levels were determined by reverse transcription quantitative polymerase chain reaction (RT-qPCR). As shown in fig. S3, no difference was observed between the TRIM26 knockdown and control cells after HCV infection, suggesting that involvement of TRIM26 in HCV infection is not mediated by its potential action on host IFN signaling.
To investigate in which step of the HCV life cycle TRIM26 is involved, we used HCVΔE1 that lacks the E1 region in viral genome and only undergoes single-round infection in Huh7 cells (21). Huh7-TRIM26 knockdown and control cells were infected with the HCVΔE1 virus at MOI of 0.1, and HCV RNA level was determined. As shown in Fig. 2A, TRIM26 knockdown reduced HCVΔE1 RNA level for about sevenfold at 72 hours after infection, suggesting that TRIM26 may contribute to HCV entry or genome replication. Next, we used the pseudotyped HCV particles (HCVpp) that harbor HCV envelope glycoproteins and serve as a surrogate model for HCV entry (22). Huh7-TRIM26 knockdown cells and control cells were infected with HCVpp. As shown in Fig. 2B, no substantial difference was observed between the TRIM26 knockdown and control cells, suggesting that TRIM26 has no effect on HCV entry. To assess the potential effect of TRIM26 on HCV polyprotein cleavage and translation, the TRIM26 knockdown and control cells were transfected with plasmids expressing nonstructural proteins NS3-5B or an RdRp-deficient HCV genome (JFH1-GND-Rz) that recapitulates internal ribosomal entry site (IRES)–dependent viral protein translation and polyprotein cleavage. As shown in fig. S4, TRIM26 knockdown had no effect on HCV polyprotein cleavage and translation. Last, we examined the impact of TRIM26 on HCV genome replication using HCV subgenomic replicon that serves as a surrogate model for viral genome replication (23). The JFH1 subgenomic replicon cells were transduced with lentiviruses expressing TRIM26-sgRNA or control EGFP-sgRNA. HCV RNA and NS3 protein levels were analyzed by RT-qPCR and Western blot, respectively. As shown in Fig. 2 (C and D), TRIM26 knockdown reduced both HCV RNA and NS3 protein levels. Together, these results demonstrated that TRIM26 is likely involved in HCV genome replication.
Fig. 2. Deficiency of TRIM26 impairs HCV genome replication.
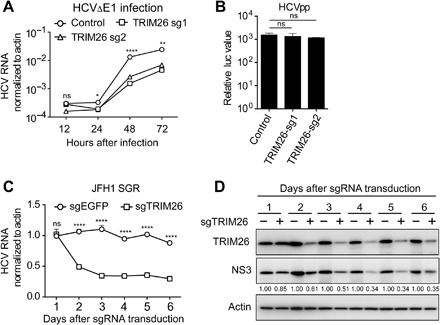
(A) Control and TRIM26 knockdown Huh7 cells were infected with the single-round infectious HCVΔE1 for the indicated time points. The HCV RNA level was detected by RT-qPCR. (B) Control and TRIM26 knockdown Huh7 cells were infected with HCVpp. The infectivity was quantified by luciferase assay. (C and D) JFH1-SGR cells were transduced with sgEGFP or sgTRIM26 for the indicated time points. HCV RNA (C) and NS3 protein levels (D) were determined by RT-qPCR and Western blot, respectively. The error bars represent SDs from two independent experiments. One-way ANOVA (A and B) and t test (C) were used for statistical analysis. ns, P > 0.05; *P < 0.05; **P < 0.01; ****P < 0.0001. The protein levels were quantified by ImageJ, normalized against internal actin, and expressed as values relative to control.
Next, we determined whether TRIM26 is required for the replication of other HCV genotypes. Con1 (genotype 1b) (23) and PR87 (genotype 3a) (24) subgenomic replicon-harboring cells were transduced with lentiviruses expressing TRIM26-sgRNA or control EGFP-sgRNA. As shown in fig. S5 (A to D), TRIM26 knockdown reduced Con1 and PR87 viral RNA and NS5A protein expression. Furthermore, we showed that TRIM26 knockdown inhibited the infection of PR63cc, another genotype 2a HCVcc strain (fig. S5E) (25). Collectively, these results demonstrated that TRIM26 contributes to replication of HCV from multiple genotypes.
Next, we examined whether TRIM26 functions in infection of other flaviviruses, such as DENV and ZIKV. Huh7-TRIM26 knockdown and control cells were infected with DENV or ZIKV at MOI of 0.1. Intracellular viral RNA (fig. S6, A and D), extracellular viral titer (fig. S6, B and E), and DENV E protein level (fig. S6C) were measured. The results showed that TRIM26 knockdown had no effect on DENV and ZIKV infection.
TRIM26 interacts with HCV NS5B protein
To elucidate the underlying mechanism by which TRIM26 promotes HCV replication, we determined the interactions of TRIM26 with HCV-encoded proteins. Plasmids expressing TRIM26 and FLAG-tagged HCV proteins were cotransfected into human embryonic kidney (HEK) 293T cells to perform coimmunoprecipitation (co-IP) assays. As shown in fig. S7 (A to H), TRIM26 was coimmunoprecipitated with NS5B, but not with core, E1, E2, NS2, NS3, NS4B, or NS5A. Conversely, NS5B was coimmunoprecipitated with hemagglutinin (HA)–tagged TRIM26 (Fig. 3A). We further confirmed the colocalization of NS5B and TRIM26 by confocal microscopy (fig. S7I). To verify the TRIM26-NS5B interaction in the context of HCV infection, Huh7 cells ectopically expressing FLAG-tagged TRIM26 were infected with HCVcc at MOI of 1 for 48 hours, and co-IP assay was performed. Consistently, NS5B, but not NS3, was coimmunoprecipitated with TRIM26 (Fig. 3B). This interaction was also confirmed in JFH1 subgenomic replicon cells (Fig. 3C).
Fig. 3. TRIM26 interacts with HCV NS5B and promotes its ubiquitination.
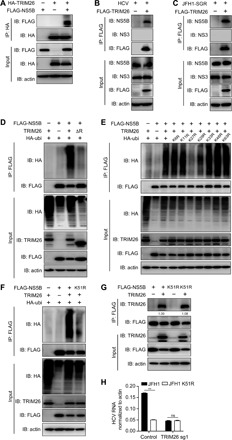
(A) HEK293T cells were transfected with plasmids expressing HA-tagged TRIM26 and FLAG-tagged NS5B. The co-IP assay was performed with an anti-HA antibody. IB, immunoblot; IP: immunoprecipitation. (B) Huh7 cells transfected with FLAG-tagged TRIM26 for 24 hours were infected with HCVcc for another 48 hours, and the cell lysates were immunoprecipitated with anti-FLAG antibody. (C) JFH1 subgenomic replicon cells were transfected with FLAG-tagged TRIM26 for 48 hours, and the cell lysates were immunoprecipitated with anti-FLAG antibody. (D) HEK293T cells were transfected with plasmids expressing FLAG-tagged NS5B, TRIM26, or TRIM26ΔR together with HA-tagged ubiquitin for 48 hours. The cell lysates were immunoprecipitated with anti-FLAG antibody and analyzed by indicated antibodies. (E) HEK293T cells were transfected with plasmids expressing FLAG-tagged NS5B and TRIM26 along with HA-tagged ubiquitin or ubiquitin mutants. The cell lysates were immunoprecipitated with anti-FLAG antibody and analyzed by indicated antibodies. (F) HEK293T cells were transfected with plasmids expressing FLAG-tagged NS5B or NS5B K51R and TRIM26 along with HA-tagged ubiquitin. The cell lysates were immunoprecipitated with anti-FLAG antibody and analyzed by indicated antibodies. (G) HEK293T cells were transfected with plasmids expressing FLAG-tagged NS5B or NS5B K51R and TRIM26. The cell lysates were immunoprecipitated with anti-FLAG antibody and analyzed by indicated antibodies. (H) Control and TRIM26 knockdown Huh7 cells were electroporated with in vitro transcribed JFH1 or JFH1-K51R subgenomic RNA. After G418 selection, the subgenomic replicon cells were harvested for detecting HCV RNA level by RT-qPCR. The error bars represent SDs from two independent experiments. t test was used for statistical analysis. ns, P > 0.05; **P > 0.01. The protein levels were quantified by ImageJ, normalized against TRIM26 input protein level, and expressed as values relative to FLAG IP protein level.
Next, we sought to map the key domains that are indispensable for TRIM26-NS5B interaction. We constructed TRIM26 mutants with a deletion in the N-terminal RING domain (TRIM26ΔR) or the C-terminal SPRY domain (TRIM26ΔSPRY) (fig. S8A), as well as NS5B mutants with a deletion in the N-terminal finger domain (NS5BΔN), the central palm domain (NS5BΔ188-371), or the C-terminal thumb domain (NS5BΔC) (fig. S8B). The co-IP assays showed that the SPRY domain of TRIM26 and the C-terminal region of NS5B were required for the interaction (fig. S8, A and B).
TRIM26 promotes ubiquitination of NS5B
As the function of TRIM26 in HCV replication requires its RING domain that is known to be responsible for ubiquitinating its substrates (fig. S1, B to D), we next examined whether TRIM26 mediates NS5B ubiquitination. HEK293T cells were cotransfected with plasmids expressing wild-type or RING-deleted TRIM26, FLAG-tagged NS5B, and HA-tagged ubiquitin, and then, NS5B was immunoprecipitated and its ubiquitination was analyzed by Western blot. As shown in Fig. 3D, wild-type, but not the RING-deleted TRIM26, promoted the ubiquitination of NS5B. Ubiquitin is known to link to substrate protein through its internal lysine residues at position 6, 11, 27, 29, 33, 48, or 63 (26); therefore, we constructed a series of ubiquitin mutants with the lysine (K)–to–arginine (R) change at each of these positions. We found that ubiquitin with the K27R mutation significantly reduced the TRIM26-mediated NS5B ubiquitination (Fig. 3E), suggesting that K27 is likely the ubiquitin lysine residue linked to NS5B.
Next, we sought to identify critical lysine residues of NS5B targeted by TRIM26. There are 30 lysine residues in NS5B of JFH1. We analyzed the conservation for these individual lysine residues among different HCV genotypes and their positions in the three-dimensional structure of NS5B (Protein Data Bank ID: 2XYM) (fig. S9A) (27). Given that TRIM26 is involved in the replication of HCV genotypes 1, 2, and 3, we selected 11 lysine residues that are highly conserved among the three genotypes (conservation, >90%) and located on the surface of NS5B structure (highlighted red in fig. S9A). As shown in Fig. 3F and fig. S9C, the K51R mutation in NS5B significantly reduced TRIM26-mediated NS5B ubiquitination, suggesting that TRIM26 promotes K27-linked ubiquitination of NS5B at the residue of K51.
Next, we investigated whether the K51R mutation in NS5B affects its interaction with TRIM26. Plasmids expressing TRIM26 and either wild-type or K51R-mutated NS5B were cotransfected into HEK293T cells to perform a co-IP experiment. As shown in Fig. 3G, the K51R mutation did not impair the interaction between NS5B and TRIM26.
Last, we assessed the effect of K51R mutation on HCV replication. The wild-type or NS5B-K51R mutant JFH1 subgenomic replicon was established in wild-type and TRIM26 knockdown Huh7 cells. As shown in Fig. 3H, NS5B K51R mutation reduced HCV replication in wild-type cells but not in the TRIM26 knockdown cells. Together, these results demonstrated that the TRIM26-mediated ubiquitination of NS5B at the K51 residue is critical for HCV replication.
TRIM26 enhances the interaction between NS5B and NS5A
To investigate how TRIM26-mediated NS5B ubiquitination enhances HCV replication, we examined the interaction of NS5B with other nonstructural proteins involved in the viral replication complex. Huh7-TRIM26 knockdown and control cells were transfected with the plasmid expressing the NS3-5B polyprotein. As shown in Fig. 4A, TRIM26 knockdown reduced the interaction between NS5B and NS5A but had little effect on the interaction between NS5B and NS3. Consistently, TRIM26 overexpression specifically enhanced the interaction between NS5B and NS5A (Fig. 4, B and C).
Fig. 4. TRIM26 promotes the interaction between NS5B and NS5A.
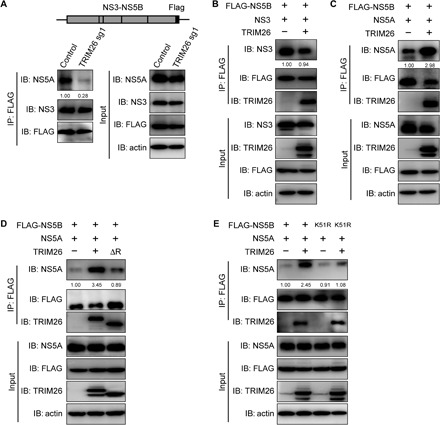
(A) Control and TRIM26- knockdown Huh7 cells were transfected with plasmids expressing NS3-5B-3 × FLAG. The cell lysates were immunoprecipitated with anti-FLAG antibody and then immunoblotted by indicated antibodies. (B and C) HEK293T cells were transfected with plasmids expressing FLAG-tagged NS5B, TRIM26, and NS3 (B) or NS5A (C). The cell lysates were immunoprecipitated with anti-FLAG antibody and then immunoblotted by indicated antibodies. (D) HEK293T cells were transfected with plasmids expressing FLAG-tagged NS5B and NS5A together with TRIM26 or TRIM26ΔR. The cell lysates were immunoprecipitated with anti-FLAG antibody and then immunoblotted by indicated antibodies. (E) HEK293T cells were transfected with plasmids expressing FLAG-tagged NS5B or NS5B K51R and NS5A together with TRIM26. The cell lysates were immunoprecipitated with anti-FLAG antibody and then immunoblotted by indicated antibodies. The NS3 and NS5A protein levels were quantified by ImageJ, normalized against their input protein levels, respectively, and expressed as values relative to FLAG IP protein levels.
To assess whether TRIM26-mediated NS5B ubiquitination affects NS5B-NS5A interaction, FLAG-tagged NS5B, NS5A, and TRIM26 or TRIM26ΔR expression plasmids were cotransfected into HEK293T cells to perform a co-IP assay. As shown in Fig. 4D, deletion of RING domain in TRIM26 reduced the interaction of NS5B and NS5A. To further confirm it, HEK293T cells were cotransfected with plasmids expressing wild-type or K51R-mutated NS5B, together with NS5A and TRIM26. As shown in Fig. 4E, NS5B K51R mutation reduced the NS5B-NS5A interaction. Together, these results demonstrated that TRIM26 mediates ubiquitination of NS5B and promotes its interaction with NS5A.
TRIM26 contributes to the HCV species tropism
TRIM26 is expressed in a wide range of animals and highly conserved among different species (fig. S10 and Fig. 5A). Next, we determined whether TRIM26 from different species supports HCV replication. We cloned TRIM26 from mouse that does not efficiently support HCV replication and from tupaia (tree shrew) that has been reported to moderately support HCV replication (28). Human, mouse, and tupaia TRIM26, designated hTRIM26, mTRIM26, and tTRIM26, respectively, were stably expressed in control and TRIM26 knockdown Huh7 cells (Fig. 5B). The resulting cells were infected with HCVcc at MOI of 0.1, and HCV RNA level and NS3 protein expression at 48 hours after infection were measured. As shown in Fig. 5 (C and D), ectopic expression of hTRIM26 and tTRIM26 in the TRIM26 knockdown cells restored HCV replication, whereas ectopic expression of mTRIM26 did not restore HCV replication in the TRIM26 knockdown cells or exert a dominant-negative effect on HCV infection in Huh7 cells.
Fig. 5. TRIM26 determines the HCV species tropism.
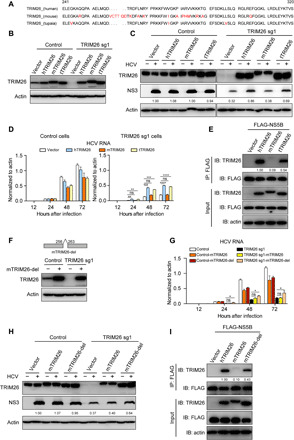
(A) Alignment of TRIM26 protein of different species. (B) Western blot analysis of reconstituted TRIM26 of different species in control and Huh7-TRIM26 knockdown cells. (C and D) The reconstituted TRIM26 cells were infected with HCVcc at MOI of 0.1 for the indicated time points. HCV NS3 protein expression (C) and RNA level (D) were analyzed at 48 hours after infection. (E) Huh7-TRIM26 knockdown cells reconstituted with hTRIM26, mTRIM26, and tTRIM26 were transfected with plasmids expressing FLAG-tagged NS5B. The cell lysates were immunoprecipitated with anti-FLAG antibody and then immunoblotted by indicated antibodies. (F) Western blot analysis of reconstituted mTRIM26-del expression in control cells and Huh7-TRIM26 knockdown cells. (G and H) The reconstituted TRIM26 cells were infected with HCVcc at MOI of 0.1 for the indicated time points. HCV RNA level (G) and NS3 protein expression (H) at 48 hours after infection were analyzed. (I) Huh7-TRIM26 knockdown cells reconstituted with hTRIM26, mTRIM26, or mTRIM26-del were transfected with plasmids expressing FLAG-tagged NS5B. The cell lysates were immunoprecipitated with anti-FLAG antibody and then immunoblotted by indicated antibodies. The error bars represent SDs from two independent experiments. One-way ANOVA was used for statistical analysis. ns, P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. The protein levels were quantified by ImageJ, normalized against internal actin, and expressed as relative to control (C and H) or normalized against TRIM26 input protein level and expressed as values relative to immunoprecipitated FLAG-tagged protein level (E and I).
Next, we performed a co-IP assay to determine the interaction of NS5B and TRIM26 of different species. Huh7-TRIM26 knockdown cells reconstituted with hTRIM26, mTRIM26, and tTRIM26 were transfected with FLAG-tagged NS5B. As shown in Fig. 5E, NS5B interacted with hTRIM26 and tTRIM26, while it had very weak interaction with mTRIM26. The amino acid sequence alignment showed that there is a unique insert of six amino acids in mTRIM26 (located at 257–262) but not in TRIM26 of human, chimpanzees, rhesus monkey, or tupaia (Fig. 5A and fig. S10). To determine whether this six–amino acid insert within mTRIM26 influences its function in HCV infection, we deleted it in mTRIM26 (designated mTRIM26-del) and stably expressed it in control and TRIM26 knockdown Huh7 cells (Fig. 5F). The resulting cells were infected with HCVcc at MOI of 0.1, and HCV RNA level and NS3 protein expression at 48 hours after infection were measured. As shown in Fig. 5 (G to H), in contrast to the full-length mTRIM26, mTRIM26-del reconstitution in TRIM26 knockdown cells partially restored HCV replication. Consistently, mTRIM26-del acquired the ability to interact with NS5B (Fig. 5I). Collectively, these results demonstrated that TRIM26 not only plays a role in HCV replication but also contributes to viral host tropism.
Human TRIM26 enhances HCV infection in murine hepatoma cells
Last, we examined whether human TRIM26 can enhance HCV replication in murine hepatocytes that normally do not support HCV infection. For this purpose, we used a previously reported murine hepatoma cell line Hep56·1D·7A7 that had been reconstituted with CD81, SRBI, CLDN1, OCLN, SEC14L2, and miR122, which are host factors essential for HCV infection (29). Hep56·1D·7A7 and its parental control Hep56·1D cells were first transfected with hTRIM26 or mTRIM26 for 24 hours and then infected with Gluc-labeled HCVcc (JC1-Gluc) that can secret Gluc into culture supernatants upon infection. The expression of hTRIM26 and mTRIM26 was verified by Western blot (Fig. 6A). The culture supernatants at days 0, 1, 2, 3 after infection were harvested for the luciferase assay. As shown in Fig. 6B, hTRIM26 expression enhanced about eightfold HCV infection in Hep56·1D·7A7 cells but not in Hep56·1D cells, while mTRIM26 had no effect in the both cells. To confirm this, we established Hep56·1D·7A7 cells that stably express hTRIM26 by lentiviral transduction (designated Hep56·1D·7A7-hTRIM26). The expression of hTRIM26 was identified by Western blot (Fig. 6C). Next, Hep56·1D·7A7 and Hep56·1D·7A7-hTRIM26 cells were infected with HCV and then analyzed by NS5A immunofluorescence staining. As shown in Fig. 6D, although the infection in the both cells was not very efficient, there were more HCV-positive cells in Hep56·1D·7A7-hTRIM26 cells. Consistently, flow cytometry analysis showed that HCV infection was more efficient in hTRIM26-transduced Hep56·1D·7A7 cells, and this increased HCV infection was more evident in the hTRIM26–high expressing Hep56·1D·7A7 cells (Fig. 6E). Together, these data suggested that hTRIM26 reconstitution enhances HCV infection in murine hepatocytes.
Fig. 6. hTRIM26 enhances HCV infection in murine hepatoma cells.
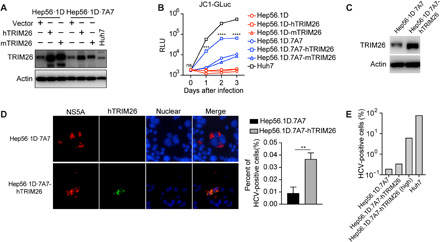
(A) Western blot analysis of hTRIM26 and mTRIM26 expression in Hep56·1D and Hep56·1D·7A7 cells. (B) Hep56·1D and Hep56·1D·7A7 cells transfected with hTRIM26 or mTRIM26 as well as Huh7 cells were infected with JC1-Gluc for the indicated time points. The culture supernatants were harvested for the luciferase assay. The error bars represent SDs from two independent experiments. RLU, relative light units. (C) Western blot analysis of hTRIM26 expression in Hep56·1D·7A7 and Hep56·1D·7A7-hTRIM26 cells. (D) Parental and hTRIM26-transuced Hep56·1D·7A7 cells were infected with HCVcc for 72 hours and then stained with anti-NS5A antibody (red) for immunofluorescence microscopy. ZsGreen coexpressed in the same lentiviral vector with hTRIM26 was labeled green. The error bars represent SDs from the number of NS5A-positive cells in three wells from one representative experiment. t test was used for statistical analysis. ns, P > 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. (E) Parental, hTRIM26-transuced Hep56·1D·7A7 cells, or Huh7 cells were infected with HCVcc for 72 hours and then stained with anti-NS5A antibody for flow cytometry analysis of HCV-positive cells. Meanwhile, ZsGreen-positive hTRIM26-transuced Hep56·1D·7A7 cells (2.8% of total) were also analyzed.
DISCUSSION
Host factors participate in each step of the HCV life cycle. In this study, we performed a genome-wide CRISPR-Cas9 screen to uncover host factors crucial for HCV replication. A variety of host factors were identified from this screen, including the well-defined HCV entry factors CD81, OCLN, and CLDN1 (14–16). PPIA has been shown to be critical for HCV replication and as a druggable target for HCV (17). ELAVL1 binds the 3′ ends of the HCV RNAs and protects viral RNAs from degradation (18). The aforementioned candidates were also identified from a previous CRISPR-based screen, which was also performed in a hepatoma cell line (19). Besides, CSNK1A1 is responsible for NS5A hyperphosphorylation and crucial for viral production (30). DNA methyltransferase 1 (DNMT1), a key factor involved in establishing and maintaining DNA methylation, is also required for HCV propagation (31). Collectively, our CRISPR screening results are highly consistent with previous findings.
We found that TRIM26 is a critical host factor for efficient HCV replication. This conclusion is supported by multiple lines of evidence. First, deficiency of TRIM26 reduces replication of HCV from multiple genotypes (Fig. 2 and fig. S5). Second, TRIM26 specifically interacts with viral polymerase NS5B (Fig. 3 and fig. S7). Third, TRIM26 promotes K27-linked ubiquitination of NS5B at residue K51, which enhances its binding to NS5A, a critical interaction for assembly of viral replication complex (Figs. 3 and 4). The role of TRIM26 in HCV replication seems virus specific, as it is not involved in the life cycle of other closely related flaviviruses such as DENV and ZIKV (fig. S6). It is important to point out that TRIM26 knockout greatly diminishes HCV replication but does not completely abolish it. This implies that a possible redundant host function may compensate the TRIM26 deficiency.
Previous studies showed that TRIM26 plays a role in the regulation of antiviral IFN response (10, 11). Our results showed that neither IFNs nor ISGs were induced significantly in HCV-infected Huh7 cells (fig. S3). This observation was consistent with many previous studies demonstrating that HCV has multiple strategies to antagonize host innate immune responses (32–34). There was no difference in the levels of IFNs or ISGs between the TRIM26 knockdown and control cells upon HCV infection, which further rules out the possibility that TRIM26 contributes to HCV replication via regulation of IFN signaling. In addition, TRIM26 was reported to function as a tumor suppressor of HCC, as TRIM26 knockdown promotes cell proliferation and metastasis (35). We found that TRIM26 knockdown had no effect on the cell viability of Huh7, a human HCC cell line (Fig. 1D).
Together with host factors, HCV nonstructural proteins form a membrane-associated replication complex, which is required for HCV genome replication. In this study, we found that TRIM26 binds NS5B, which requires both the SPRY domain of TRIM26 and the C-terminal region of NS5B (fig. S8). TRIM26-mediated ubiquitination of NS5B at residue K51 enhances the interaction between NS5B and NS5A and, in turn, enhances HCV genome replication (Figs. 3 and 4). As shown in fig. S9A, K51 is highly conserved among all HCV genotypes. In addition, we found K51 is 99.8% conserved among 2045 HCV sequences in the ViPR-HCV database, highlighting a critical role of this residue in the biological function of NS5B. NS5B functions as an RdRp, which that contains finger, palm, and thumb subdomains (36–38). While the C-terminal thumb domain is required for the NS5B-TRIM26 interaction, residue K51 is located at the base of a finger loop, which is not essential for the interaction. However, there is an extensive interaction between the finger and thumb subdomains, leading to an encircled catalytic active site located in the central palm subdomain (36–38). Therefore, we speculate that the finger and thumb interaction also serves as a platform for TRIM26-mediated ubiquitination of NS5B at residue K51. A cocrystal structure of NS5B and uridine 5′-triphosphate (UTP) showed that K51 is adjacent to the triphosphate moiety of UTP and makes an electrostatic interaction with UTP (39). In addition, NS5B K51 has been shown to be a contacting residue with nascent RNA during RNA synthesis (40). Our result demonstrated that ubiquitination of NS5B at residue K51 is required for its interaction with NS5A (Fig. 4E). Future studies will be needed to investigate whether the NS5B K51 ubiquitination affects its contact with RNA substrates.
Humans are the sole known natural host for HCV infection. Although chimpanzees can be experimentally infected by HCV, it has become ethically difficult to use it as an in vivo model to study the virus and evaluate HCV vaccine candidates. In recent years, much progress has been made to develop small-animal models supporting HCV infection. Tupaia (also called tree shrew), a nonrodent small mammal, moderately supports HCV infection. However, its application for HCV animal model has been impeded by its outbred genetic background. Mice are excellent animal model for its inbred genetic background and related research tools but are not naturally permissive for HCV infection because of lack of critical HCV host factors (41). Reconstitution of human HCV entry factors in mice renders limited HCV infection (16, 42), raising a possibility that mice may still lack other HCV host factors. In our study, we found that human and tupaia TRIM26 are capable of supporting HCV replication, while mouse TRIM26 is not (Fig. 5B). Consistently, NS5B interacts with human and tupaia TRIM26, but not with mouse TRIM26 that contains a mouse specific six–amino acid insert. Deletion of this insert restores at least partially the ability of mouse TRIM26 to bind NS5B and to support HCV replication (Fig. 5, G and H). Although this six–amino acid insert (257–262) is not located within the SPRY domain (294–539) of TRIM26 that is required for its interaction with NS5B, its relative close proximity to the SPRY domain suggests that this extra insert may interfere with access of NS5B to the binding site of TRIM26. A more detailed structural analysis of the NS5B-TRIM26 complex is needed.
Expression of hTRIM26 can further boost HCV infection in Hep56·1D·7A7 cells that have already been reconstituted with six critical human factors for HCV entry and replication (Fig. 6). This raises a possibility that introduction of hTRIM26 into a previously developed transgenic mouse model that expresses human HCV entry factors CD81, SR-B1, CLDN1, and OCLN (42) may further increase its permissiveness to HCV infection, an important step to develop a fully permissive small-animal model for HCV infection.
MATERIALS AND METHODS
Experimental design
This study aimed to identify host factors essential for HCV infection by genome-wide CRISPR-Cas9 screening. Among the top candidates, TRIM26 was identified as a novel host factor for HCV infection. We then analyzed at which step of the HCV life cycle TRIM26 is involved and deciphered the underlying mechanism by which TRIM26 promotes HCV replication. Last, we compared the role of TRIM26 from different species in HCV infection and explored its potential roles in contribution to host tropism.
Cell culture
HEK293T and Huh7 cells were obtained from the Cell Bank of Shanghai Institute of Biological Sciences, Chinese Academy of Sciences. Huh7.5.1 cells were obtained from F. Chisari at Scripps Research Institute. Hep56·1D and Hep56·1D·7A7 cells were obtained from A. Ploss at Princeton University. The cells were maintained in complete Dulbecco’s modified Eagle’s medium (DMEM) (Invitrogen) supplemented with 10% fetal bovine serum, 10 mM Hepes, 2 mM l-glutamine, 100 U of penicillin/ml, and 100 mg of streptomycin/ml. All cells were cultured in humidified air containing 5% CO2 at 37°C.
Plasmid construction
The coding sequences of human, mouse, and tupaia TRIM26 were amplified by PCR/RT-PCR from an hTRIM26-containing plasmid (provided by J. Han at Xiamen University), murine L929 cells, and tupaia liver tissue, respectively, using the following primers: hTRIM26 (forward: CGAAAATATCGAACGCCTCAAGGTGGACAAGGGCAGGC; reverse: GAGGCGTTCGATATTTTCGACCAGGCTGGCCAGTTGC), mTRIM26 (forward: GCGCTCGAGATGGCAGTGTCAGCCCCCTTGAGGAG; reverse: GCGTCTAGATCAGGGTCTCAGCAGAAGGCGTGCTC), and tTRIM26 (forward: GCGCTCGAGATGGCCACGTCAGCCCCTCTGCG; reverse: GCGTCTAGATCAGGGTCTCAGCAGGAGGCGTG). The amplified PCR products were cloned into pLVX-IRES-Puro vector or pLVX-IRES-zsGreen vector (Fig. 6, C to E). The plasmids expressing core, NS2, NS3, NS4B, NS5A, and NS5B contain a FLAG tag at the N terminus. The E1- and E2-expressing plasmids contain a signal peptide at the N terminus and a FLAG tag at the C terminus. The mutant TRIM26 and NS5B plasmids were made by homologous recombination using a Gibson assembly cloning kit [New England Biolabs (NEB), USA]. All the constructs were verified by DNA sequencing.
Analysis of NS5B lysine residues of different HCV genotypes
The amino acids of NS5B of different HCV genotypes were downloaded from the HCV database site map, and the number of the sequences from each genotype is listed in fig. S9. The conservation and location of NS5B lysine residues among different HCV genotypes were analyzed by Vector NTI and PyMOL, respectively.
sgRNA library construction and CRISPR screening
The human whole-genome sgRNA library that consists of approximately 180,000 sgRNAs that target 19,271 genes was designed. sgRNA oligos were synthesized (CustomArray), and the sgRNA-coding DNA fragments were amplified with PCR from the synthesized oligos with primers flanking the sgRNA target sequences. The amplified sgRNA-coding DNA fragments were purified (DNA Clean & Concentrator TM-5 Kit, Zymo Research) and ligated into the lentiviral vector expressing green fluorescent protein (GFP) by Golden Gate Assembly. The plasmids were transformed into trans1-T1 competent cells. The plasmid library was packaged into pseudotyped lentiviral particles by cotransfection with pCMVR8.74 and VSV-G (glycoprotein of vesicular stomatitis virus) plasmids. Huh7.5-NIrD, a reporter cell line described in (12), was transduced with lentiviral vectors at MOI of 0.3, and GFP-expressing cells were enriched by fluorescence-activated cell sorting (FACS) (BD FACSAria III), which were ready for the following screening experiments after cell culture for 2 weeks.
The cell library was equally divided into two parts: the reference and experimental groups. The genomic DNA from the reference group was extracted, and the sgRNA-coding sequences integrated into chromosomes were amplified by PCR, followed by next-generation sequencing (Illumina HiSeq 2500). The cells of the experimental group were infected with HCVcc at MOI of 0.1 for 30 days, during which most cells died due to HCV replication. After that, mCherry-negative cells were enriched again by FACS in the presence of doxycycline (1 μg/ml). The sgRNA sequences from the mCherry-negative cells were decoded by deep sequencing. Comparison of sgRNA abundance between the experimental group and the reference group was analyzed by the RIGER algorithm. The low count reads (less than 10) were filtered out.
Generation of the TRIM26 knockdown Huh7 cell line via CRISPRi
The following three sgRNA sequences were used for the TRIM26 knockdown via CRISPRi (43) (sg1: GCGGCACCCCTCCTCTCTCA; sg2: GGAATAGCCGGGAGATTACG; sg3: GCTCGTGCAGGAGCGGGACC). The sgRNA targeting AAVS1 transcription start site (TSS) region was used as control (AAVS1 sg: CGGAACCTGAAGGAGGCGGC). The sgRNAs were cloned into a lentiviral vector expressing GFP and later packaged into VSV-G pseudotyped lentiviral particles by cotransfection with pCMVR8.74 and pVSV-G plasmids into HEK293T cells. Meanwhile, the KRAB-dCas9-P2A-mCherry (Addgene, #60954) vector was also packaged by cotransfection with pCMVR8.74 and pVSV-G plasmids. Huh7 cells were then transduced with both pseudotyped lentiviral vectors that express sgRNA and KRAB-dCas9-P2A-mCherry. Three days after transduction, the GFP and mCherry double-positive cells were sorted by FACS and further cultured. The knockdown efficiency of TRIM26 was measured by Western blot.
Generation of TRIM26 knockout Huh7.5.1 single-cell clone
Oligonucleotides (TRIM26 oligo F: ACCGTGTGGCAACTGGCCAGCCTGG and TRIM26 oligo R: AAACCCAGGCTGGCCAGTTGCCACA) of TRIM26 sgRNA were synthesized (Ruibiotech) and annealed in 50 μl of TransTaq HiFi Buffer II at a final concentration of 9 μM. The annealed oligos were ligated into a lentiviral vector bearing a puromycin selection marker with Golden Gate Assembly (NEB). The ligation products were then transformed into Trans1-T1 competent cells (Transgen, CD501). Pseudotyped lentiviral vectors expressing sgRNA were generated by cotransfection of a vector expressing the VSV-G, pCMVR8.74 (containing lentiviral gal/pol), and the lentiviral vector expressing sgRNAs. Huh7.5.1 cells were transduced with the pseudotyped lentiviral vectors expressing sgRNA and selected with puromycin (1 μg/ml). The single-cell clones resistant to puromycin were selected. Genomic DNA were extracted from the single-cell clones, and the insertions and deletions (indels) caused by sgRNA/Cas9 in each cell clone were confirmed by Sanger sequencing after PCR amplification.
RNA isolation and RT-qPCR
The protocols and sequences of primers for quantifying HCV RNA, human IFN-β, MxA, ISG56, and actin were described previously (34). The cells were lysed in TRIzol (Tiangen), and RNA was isolated according to the manufacturer’s protocol. The cDNA was synthesized using the ReverTra Ace qPCR RT kit (Toyobo). RT-qPCR was performed using quantitative PCR SYBR green RT-PCR master mix (Toyobo). The sequences of the primers targeting DENV and ZIKV were as follows: ZIKV, CAACTACTGCAAGTGGAAGGGT (forward) and AAGTGGTCCATATGATCGGTTGA (reverse); DENV, ACAAGTCGAACAA CCTGGTCCAT (forward) and GCCGCACCATTGGTCTTCTC (reverse). The expression of target genes was normalized to actin.
Virus preparation and titration
The JC1-GLuc virus was constructed as previously described (44). The GLuc gene and an autocleaving peptide 2A were inserted between p7 and NS2 of the JC1 cDNA clone. HCVcc, ZIKV, and DENV preparation and titration were as previously described. Briefly, 1 × 105 Vero cells were seeded in a 24-well plate for 24 hours, then washed with phosphate-buffered saline (PBS), and infected with the serially diluted ZIKV for 1 hour. Viral inoculations were replaced with 1.2 ml of DMEM containing 1.5% fetal bovine serum and 1% carboxymethyl cellulose sodium salt. Viral plaques were developed at day 4 after infection. Huh7.5.1 (1 × 104) cells were seeded in a 96-well plate and infected with serially diluted DENV for 72 hours. The cells were fixed with 4% paraformaldehyde and incubated with a monoclonal antibody against DENV envelope protein (clone D1-4G2-4-15; Millipore) followed by incubation with Alexa Fluor 488–conjugated secondary antibody and Hoechst 33258. The stained cells were analyzed by fluorescence microscopy.
HCVpp infection
HCVpp were generated as previously described (22). HEK293T cells were cotransfected with plasmids expressing HCV envelope glycoproteins, retroviral core packaging component, and luciferase. Supernatants were collected 72 hours later and filtered through 0.45-μM pore size membranes. For infection, targeted cells were seeded in 96-well plates and infected with HCVpp. At 72 hours after infection, the firefly luciferase activity was measured by luciferase assay according to the manufacturer’s instructions (Promega).
Co-IP assay
HEK293T cells were cotransfected with indicated plasmids and lysed in NP-40 buffer containing 50 mM tris-HCl (pH 7.5), 150 mM NaCl, and 0.5% NP-40, with protease inhibitor (Sigma-Aldrich) at 48 hours after transfection. The cell lysates were centrifuged at 10,000g at 4°C for 10 min, and supernatants were transferred to new tubes and incubated with normal immunoglobulin G (Santa Cruz Biotechnology) as well as protein A/G agarose beads at 4°C for 30 min to eliminate nonspecific binding proteins. After centrifugation at 1000g at 4°C for 5 min to remove the protein A/G agarose beads, the supernatants were incubated overnight with specific primary antibody at 4°C and then with protein A/G agarose beads for an additional 2 hours. The samples were collected by centrifugation at 1000 g at 4°C for 5 min, washed with NP-40 buffer for four times, and then resuspended in 40 μl of loading buffer for Western blot.
Western blot
The protocol was as described previously (34). The following antibodies were used: anti-FLAG (Sigma-Aldrich); anti-HA and anti-actin (Abmart); anti-TRIM26 antibody, goat–anti-mouse horseradish peroxidase (HRP) antibody, and goat–anti-rabbit HRP antibody (Santa Cruz Biotechnology); monoclonal antibodies against HCV NS3 and NS5A (generated by J. Zhong’s laboratory); and anti-DENV E protein (D1-11, Abcam).
Luciferase assay
The protocol was as described previously (45). A BioLux Gaussia luciferase assay kit (NEB) was used to measure the GLuc activity.
Immunofluorescence microcopy
Hep56·1D·7A7 cells (1 × 104) were seeded in a 96-well plate and infected with HCV for 72 hours. The cells were fixed with 4% paraformaldehyde and incubated with a monoclonal antibody against HCV NS5A followed by incubation with Alexa Fluor 555–conjugated secondary antibody and Hoechst 33258. The stained cells were analyzed by fluorescence microscopy.
Confocal microscopy
HEK293T cells transfected with the indicated plasmids were seeded on 14-mm-diameter glass coverslips for 48 hours. The cells were washed with PBS and fixed with paraformaldehyde for 1 hour. Then, the cells were incubated with the primary antibody for 1 hour followed by incubation with the secondary antibody conjugated with Alexa Fluor 488 (Invitrogen) or Alexa Fluor 555 (Invitrogen). Images were acquired with an Olympus FV1200 laser scanning confocal microscope (Olympus, Tokyo, Japan) and analyzed using ImageJ software. Pearson’s coefficiency functioned as the indicator of colocalization.
Cell viability
Huh7 and Huh7-TRIM26 knockdown cells were seeded in a 96-well plate for 72 hours. Then, luminescent signal was acquired for cell viability analysis using the CellTiter-Glo 2.0 assay.
Flow cytometry
Hep56·1D·7A7, Hep56·1D·7A7-hTRIM26, and Huh7 cells were infected with HCV for 72 hours. The cells were fixed with 4% paraformaldehyde and incubated with permeabilization buffer. Next, the cells were incubated with a monoclonal antibody against HCV NS5A followed by incubation with Alexa Fluor 555–conjugated secondary antibody. The cells were analyzed by flow cytometry. For each staining, at least 5000 events were collected for analysis. The FlowJo software was used for HCV-positive cell analysis.
Statistical analysis
Statistical analysis was performed using GraphPad Prism 8 software. Student’s t test was used for analyzing the difference between two groups, and one-way analysis of variance (ANOVA) followed by Tukey post hoc test was used for analyzing the differences among groups of more than three. Not significant (ns), P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
Supplementary Material
Acknowledgments
Funding: This study was supported by the grants from the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB29010205, to J.Z.); the National Natural Science Foundation of China (31670172, to J.Z.; 31930016, to W.W.; 31670169, to Z.Z.; and 31770189, to Y.T.); the support from Beijing Municipal Science and Technology Commission (Z181100001318009), the Beijing Advanced Innovation Center for Genomics at Peking University, and the Peking-Tsinghua Center for Life Sciences to W.W.; the West Light Foundation of the Chinese Academy of Sciences (xbzg-zdsys-201909) to J.Z.; and the Natural Science Foundation of Guangdong Province (2019A1515110668) to G.Z. Author contributions: J.Z., W.W., and G.Z. conceived and designed the study. Y.L., G.Z., and Q.L. designed, performed, and analyzed the experiments. L.H. and Y.X. contributed to the DENV and ZIKV infection experiments. Y.G. and G.Z. performed the bioinformatics analysis. X.H., X.Z., and Q.D. contributed to the establishment of murine hepatoma cells. W.T., M.G., T.G., Y.T., and Z.Z. participated in data analysis. Y.L. and G.Z. drafted the manuscript. All authors contributed to and revised the final version of the manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/2/eabd9732/DC1
REFERENCE AND NOTES
- 1.W. H. Organization, Global Hepatitis Report 2017 (World Health Organization, 2017).
- 2.Li D., Huang Z., Zhong J., Hepatitis C virus vaccine development: Old challenges and new opportunities. Natl. Sci. Rev. 2, 285–295 (2015). [Google Scholar]
- 3.Hatakeyama S., TRIM family proteins: Roles in autophagy, immunity, and carcinogenesis. Trends Biochem. Sci. 42, 297–311 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Stremlau M., Perron M., Lee M., Li Y., Song B., Javanbakht H., Diaz-Griffero F., Anderson D. J., Sundquist W. I., Sodroski J., Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5α restriction factor. Proc. Natl. Acad. Sci. 103, 5514–5519 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grütter M. G., Luban J., TRIM5 structure, HIV-1 capsid recognition, and innate immune signaling. Curr. Opin. Virol. 2, 142–150 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang K., Zou C., Wang X., Huang C., Feng T., Pan W., Wu Q., Wang P., Dai J., Interferon-stimulated TRIM69 interrupts dengue virus replication by ubiquitinating viral nonstructural protein 3. PLOS Pathog. 14, e1007287 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Di Pietro A., Kajaste-Rudnitski A., Oteiza A., Nicora L., Towers G. J., Mechti N., Vicenzi E., TRIM22 inhibits influenza A virus infection by targeting the viral nucleoprotein for degradation. J. Virol. 87, 4523–4533 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Patil G., Zhao M., Song K., Hao W., Bouchereau D., Wang L., Li S., TRIM41-mediated ubiquitination of nucleoprotein limits influenza A virus infection. J. Virol. 92, e00905–e00918 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang D., Li N. L., Wei D., Liu B., Guo F., Elbahesh H., Zhang Y., Zhou Z., Chen G.-Y., Li K., The E3 ligase TRIM56 is a host restriction factor of Zika virus and depends on its RNA-binding activity but not miRNA regulation, for antiviral function. PLOS Negl. Trop. Dis. 13, e0007537 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang P., Zhao W., Zhao K., Zhang L., Gao C., TRIM26 negatively regulates interferon-β production and antiviral response through polyubiquitination and degradation of nuclear IRF3. PLOS Pathog. 11, e1004726 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ran Y., Zhang J., Liu L.-L., Pan Z.-Y., Nie Y., Zhang H.-Y., Wang Y.-Y., Autoubiquitination of TRIM26 links TBK1 to NEMO in RLR-mediated innate antiviral immune response. J. Mol. Cell Biol. 8, 31–43 (2016). [DOI] [PubMed] [Google Scholar]
- 12.Ren Q., Li C., Yuan P., Cai C., Zhang L., Luo G. G., Wei W., A Dual-reporter system for real-time monitoring and high-throughput CRISPR/Cas9 library screening of the hepatitis C virus. Sci. Rep. 5, 8865 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhou Y., Zhu S., Cai C., Yuan P., Li C., Huang Y., Wei W., High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature 509, 487–491 (2014). [DOI] [PubMed] [Google Scholar]
- 14.Pileri P., Uematsu Y., Campagnoli S., Galli G., Falugi F., Petracca R., Weiner A. J., Houghton M., Rosa D., Grandi G., Abrignani S., Binding of hepatitis C virus to CD81. Science 282, 938–941 (1998). [DOI] [PubMed] [Google Scholar]
- 15.Beard M. R., Warner F. J., Claudin-1, a new junction in the hepatitis C virus entry pathway. Hepatology 46, 277–279 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Ploss A., Evans M. J., Gaysinskaya V. A., Panis M., You H., de Jong Y. P., Rice C. M., Human occludin is a hepatitis C virus entry factor required for infection of mouse cells. Nature 457, 882 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Madan V., Paul D., Lohmann V., Bartenschlager R., Inhibition of HCV replication by cyclophilin antagonists is linked to replication fitness and occurs by inhibition of membranous web formation. Gastroenterology 146, 1361–1372.e9 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Shwetha S., Kumar A., Mullick R., Vasudevan D., Mukherjee N., Das S., HuR displaces polypyrimidine tract binding protein to facilitate La binding to the 3’ untranslated region and enhances hepatitis C virus replication. J. Virol. 89, 11356–11371 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marceau C. D., Puschnik A. S., Majzoub K., Ooi Y. S., Brewer S. M., Fuchs G., Swaminathan K., Mata M. A., Elias J. E., Sarnow P., Carette J. E., Genetic dissection of Flaviviridae host factors through genome-scale CRISPR screens. Nature 535, 159–163 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qi L. S., Larson M. H., Gilbert L. A., Doudna J. A., Weissman J. S., Arkin A. P., Lim W. A., Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell 152, 1173–1183 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tong Y., Chi X., Yang W., Zhong J., Functional analysis of hepatitis C virus (HCV) envelope protein E1 using a trans-complementation system reveals a dual role of a putative fusion peptide of E1 in both HCV entry and morphogenesis. J. Virol. 91, e02468-16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bartosch B., Dubuisson J., Cosset F.-L., Infectious hepatitis C virus pseudo-particles containing functional E1-E2 envelope protein complexes. J. Exp. Med. 197, 633–642 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lohmann V., Körner F., Koch J., Herian U., Theilmann L., Bartenschlager R., Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285, 110–113 (1999). [DOI] [PubMed] [Google Scholar]
- 24.Guo M., Lu J., Gan T., Xiang X., Xu Y., Xie Q., Zhong J., Construction and characterization of Genotype-3 hepatitis C virus replicon revealed critical genotype-3-specific polymorphism for drug resistance and viral fitness. Antiviral Res. 171, 104612 (2019). [DOI] [PubMed] [Google Scholar]
- 25.Lu J., Xiang Y., Tao W., Li Q., Wang N., Gao Y., Xiang X., Xie Q., Zhong J., A novel strategy to develop a robust infectious hepatitis C virus cell culture system directly from a clinical isolate. J. Virol. 88, 1484–1491 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hershko A., Ciechanover A., The ubiquitin system. Annu. Rev. Biochem. 67, 425–479 (1998). [DOI] [PubMed] [Google Scholar]
- 27.Schmitt M., Scrima N., Radujkovic D., Caillet-Saguy C., Simister P. C., Friebe P., Wicht O., Klein R., Bartenschlager R., Lohmann V., Bressanelli S., A comprehensive structure-function comparison of hepatitis C virus strain JFH1 and J6 polymerases reveals a key residue stimulating replication in cell culture across genotypes. J. Virol. 85, 2565–2581 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xie Z.-C., Riezu-Boj J. I., Lasarte J. J., Guillen J., Su J. H., Civeira M. P., Prieto J., Transmission of hepatitis C virus infection to tree shrews. Virology 244, 513–520 (1998). [DOI] [PubMed] [Google Scholar]
- 29.Gaska J. M., Balev M., Ding Q., Heller B., Ploss A., Differences across cyclophilin A orthologs contribute to the host range restriction of hepatitis C virus. eLife 8, e44436 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Masaki T., Matsunaga S., Takahashi H., Nakashima K., Kimura Y., Ito M., Matsuda M., Murayama A., Kato T., Hirano H., Endo Y., Lemon S. M., Wakita T., Sawasaki T., Suzuki T., Involvement of hepatitis C virus NS5A hyperphosphorylation mediated by casein kinase I-α in infectious virus production. J. Virol. 88, 7541–7555 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen C., Pan D., Deng A.-M., Huang F., Sun B.-L., Yang R.-G., DNA methyltransferases 1 and 3B are required for hepatitis C virus infection in cell culture. Virology 441, 57–65 (2013). [DOI] [PubMed] [Google Scholar]
- 32.Meylan E., Curran J., Hofmann K., Moradpour D., Binder M., Bartenschlager R., Tschopp J., Cardif is an adaptor protein in the RIG-I antiviral pathway and is targeted by hepatitis C virus. Nature 437, 1167–1172 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Ding Q., Cao X., Lu J., Huang B., Liu Y.-J., Kato N., Shu H.-B., Zhong J., Hepatitis C virus NS4B blocks the interaction of STING and TBK1 to evade host innate immunity. J. Hepatol. 59, 52–58 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Liang Y., Cao X., Ding Q., Zhao Y., He Z., Zhong J., Hepatitis C virus NS4B induces the degradation of TRIF to inhibit TLR3-mediated interferon signaling pathway. PLOS Pathog. 14, e1007075 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y., He D., Yang L., Wen B., Dai J., Zhang Q., Kang J., He W., Ding Q., He D., TRIM26 functions as a novel tumor suppressor of hepatocellular carcinoma and its downregulation contributes to worse prognosis. Biochem. Biophys. Res. Commun. 463, 458–465 (2015). [DOI] [PubMed] [Google Scholar]
- 36.Bressanelli S., Tomei L., Roussel A., Incitti I., Vitale R. L., Mathieu M., De Francesco R., Rey F. A., Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Proc. Natl. Acad. Sci. U.S.A. 96, 13034–13039 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ago H., Adachi T., Yoshida A., Yamamoto M., Habuka N., Yatsunami K., Miyano M., Crystal structure of the RNA-dependent RNA polymerase of hepatitis C virus. Structure 7, 1417–1426 (1999). [DOI] [PubMed] [Google Scholar]
- 38.Lesburg C. A., Cable M. B., Ferrari E., Hong Z., Weber P. C., Crystal structure of the RNA-dependent RNA polymerase from hepatitis C virus reveals a fully encircled active site. Nat. Struct. Biol. 6, 937–943 (1999). [DOI] [PubMed] [Google Scholar]
- 39.Rigat K., Wang Y., Hudyma T. W., Ding M., Zheng X., Gentles R. G., Beno B. R., Gao M., Roberts S. B., Ligand-induced changes in hepatitis C virus NS5B polymerase structure. Antiviral Res. 88, 197–206 (2010). [DOI] [PubMed] [Google Scholar]
- 40.Vaughan R., Fan B., You J.-S., Kao C. C., Identification and functional characterization of the nascent RNA contacting residues of the hepatitis C virus RNA-dependent RNA polymerase. RNA 18, 1541–1552 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berggren K. A., Suzuki S., Ploss A., Animal models used in hepatitis C virus research. Int. J. Mol. Sci. 21, 3869 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dorner M., Horwitz J. A., Robbins J. B., Barry W. T., Feng Q., Mu K., Jones C. T., Schoggins J. W., Catanese M. T., Burton D. R., Law M., Rice C. M., Ploss A., A genetically humanized mouse model for hepatitis C virus infection. Nature 474, 208–211 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Horlbeck M. A., Gilbert L. A., Villalta J. E., Adamson B., Pak R. A., Chen Y., Fields A. P., Park C. Y., Corn J. E., Kampmann M., Weissman J. S., Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. eLife 5, e19760 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Phan T., Beran R. K., Peters C., Lorenz I. C., Lindenbach B. D., Hepatitis C virus NS2 protein contributes to virus particle assembly via opposing epistatic interactions with the E1-E2 glycoprotein and NS3-NS4A enzyme complexes. J. Virol. 83, 8379–8395 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tao W., Gan T., Guo M., Xu Y., Zhong J., Novel Stable Ebola Virus Minigenome Replicon Reveals Remarkable Stability of the Viral Genome. J. Virol. 91, e01316-17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/2/eabd9732/DC1