

A cryo-EM structure reveals unique traits of a human adenovirus that has acquired gastrointestinal tropism.
Abstract
Human adenovirus (HAdV) types F40 and F41 are a prominent cause of diarrhea and diarrhea-associated mortality in young children worldwide. These enteric HAdVs differ notably in tissue tropism and pathogenicity from respiratory and ocular adenoviruses, but the structural basis for this divergence has been unknown. Here, we present the first structure of an enteric HAdV—HAdV-F41—determined by cryo–electron microscopy to a resolution of 3.8 Å. The structure reveals extensive alterations to the virion exterior as compared to nonenteric HAdVs, including a unique arrangement of capsid protein IX. The structure also provides new insights into conserved aspects of HAdV architecture such as a proposed location of core protein V, which links the viral DNA to the capsid, and assembly-induced conformational changes in the penton base protein. Our findings provide the structural basis for adaptation of enteric HAdVs to a fundamentally different tissue tropism.
INTRODUCTION
Adenoviruses (AdVs) are common pathogens in human, causing diseases not only in airways, eyes, and intestine but also in the liver, urinary tract, and/or adenoids (1). To date, more than 100 human AdV (HAdV) types have been isolated, characterized, and classified into seven species (A to G) (2). In addition to causing serious diseases in humans, several AdVs are being explored as vaccine vehicles against infectious diseases such as coronavirus disease 2019, Middle East respiratory syndrome (MERS), Ebola disease, AIDS, Lassa fever, and Zika disease (3). These efforts include a vaccine candidate based on HAdV-F41 that elicits neutralizing antibodies against MERS coronavirus in vivo (4). The two sole members of HAdV species F, HAdV-F40 and HAdV-F41, stand out as the only HAdVs with a pronounced gastrointestinal tropism. These so-called enteric AdVs are a leading cause of diarrhea and diarrhea-associated mortality (5) in young children, inferior only to Shigella and rotavirus (6). Diarrhea is estimated to cause ~530,000 deaths/year in children younger than 5 years worldwide (7). Thus, there are strong incentives to understand the structural and molecular basis of enteric HAdVs.
AdVs are double-stranded DNA viruses with an ~35–kilo–base pair genome sheltered in a large (~950 Å in diameter), nonenveloped capsid with icosahedral symmetry (8–13). At each of the 12 capsid vertices, penton base (PB) subunits organize as homopentamers (14), anchoring the N-terminal tails of the protruding, trimeric fibers (15, 16) to the capsid. Another so-called major capsid protein is the hexon protein (17), present in 240 trimers per virion. Hexons are the main structural component of the virion facets and are organized to give the virion a pseudo T = 25 symmetry (Fig. 1A). Hexon assemblies are stabilized by minor capsid protein IIIa (IIIa), VI, and VIII (located in the capsid interior) and by IX (exposed on the capsid exterior) (9, 10, 18). To date, two HAdVs, HAdV-C5 (11, 12, 19, 20) and HAdV-D26 (21), as well as individual capsid proteins or their subdomains (14, 17, 22) of multiple AdV types have been structurally determined at high resolution.
Fig. 1. The overall structure of HAdV-F41.
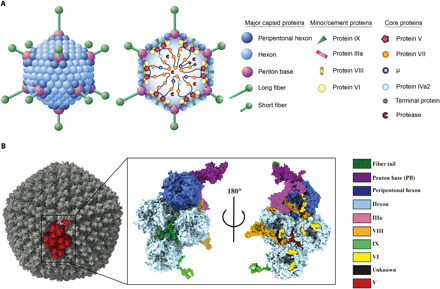
(A) Schematic representation of the capsid and core structure of HAdV-F41. (B) Surface representation of the HAdV-F41 electron density with one asymmetric unit (ASU) highlighted in red (left) and a surface representation of the ASU of the HAdV-F41 atomic model viewed from the virion exterior (middle) and interior (right).
The enteric HAdVs have adapted to a distinct tissue tropism from other AdVs, which is presumably reflected in their capsid structure. One major known difference is that enteric HAdVs contain two different types of fiber proteins, long and short (23, 24), whereas other AdVs contain only one type of fiber. Virions dock on to cells through fiber interactions with cellular receptors, followed by internalization mediated by PB interactions with cellular integrins (25). HAdV-F41 long fibers bind to the Coxsackievirus and AdV receptor (26), but no binding partners have been identified for the short fiber. The short fibers—structural hallmarks of enteric HAdVs—contribute to resistance to low pH of enteric HAdVs (27). All other HAdVs contain a conserved, integrin-interacting Arg-Gly-Asp (RGD) motif in the PB (28). Notably, the enteric HAdV-F40 and HAdV-F41 lack this conserved RGD motif and thus use different integrins for entry (29), which may explain their different and much slower entry mechanism (30, 31). Knowledge about the capsid proteins, their structural organization, and host molecule interactions is important for design and development of AdVs as vectors and vaccine vehicles.
Despite the medical importance of enteric AdVs as a major cause of childhood mortality through diarrhea, the structural basis for their infection is not known. We used cryo–electron microscopy (cryo-EM) to determine near-atomic structures of the HAdV-F41 virion at pH 7.4 and at pH 4.0, the latter sets as an average of the diurnal pH in the stomach of young children (32). These structures reveal a capsid that is structurally unchanged by stomach pH and has an extensively remodeled surface as compared to nonenteric HAdVs. We further propose a conserved location of core protein V, which links the AdV genome to the capsid. Last, we describe the assembly-induced structural changes to the PB protein. We believe that these findings will lay the foundation for a detailed molecular understanding of enteric AdVs, how to prevent their infection, and how to further explore AdVs as vehicles for vaccine development.
RESULTS
The structure of HAdV-F41 reveals a capsid with an altered surface charge distribution, structurally unaffected by low pH
To elucidate the structural basis of enteric AdV infection, we determined the structure of HAdV-F41 using cryo-EM. In parallel, the genome of the purified virus (strain “Tak”) was sequenced, revealing one nonsynonymous mutation (Val77Ala in VIII) compared to the deposited sequence for the same strain (GenBank: DQ315364.2). Further, its proteome was determined using the high-recovery filter-aided sample preparation (FASP) mass spectrometry (MS) (33), revealing a total of 22 viral proteins present in the purified virus (table S1). At an average resolution of 3.8 Å, the three-dimensional (3D) reconstruction of HAdV-F41 had continuous electron density with well-defined secondary structure elements and side-chain density (fig. S1 and movie S1). Local resolution estimates revealed a resolution of better than 3 Å for large parts of the icosahedral capsid (fig. S1). This allowed us to build and refine an atomic model of the asymmetric unit (ASU), which describes the icosahedral part of the virus (Fig. 1B and movie S2). The final ASU model contained four hexon homotrimers, single chains of the PB protein and IIIa (fig. S2), 10 chains of VI, two chains of VIII (fig. S2), four chains of the triskelion protein IX, and five chains of unknown identity, confirming known and revealing unknown protein-protein interaction surfaces (table S2). Electron density for the fibers was present at the interface with the icosahedral capsid but was of insufficient quality for extensive model building due to increasing flexibility in more distal parts. Compared to the two other reported structures of HAdVs, HAdV-C5 [Protein Data Bank (PDB): 6B1T (20)] and HAdV-D26 [PDB: 5TX1 (21)], the sequence identity of the capsid proteins ranges from 30 to 80% (table S3). This generally correlated with the average structural similarity between the three structures, with more divergent proteins showing higher structural difference in terms of Cα root mean square deviation (RMSD) (table S3).
In their adaptation to gastrointestinal tropism, a major obstacle for enteric AdVs was likely the passage through the low pH of the stomach to their intestinal site of infection. To investigate the adaptation to the hostile environment of the stomach, we solved the structure of HAdV-F41 at pH 4.0, which resembles the diurnal average gastric pH of young children (Fig. 2A) (32). At the achieved resolution of 5.0 Å, the overall structure of the capsid at pH 4.0 was largely unchanged (overall Cα RMSD of 3.0 Å for proteins listed in Fig. 2B), and no marked local movements were observed (Fig. 2B), showing that the icosahedral part of the HAdV-F41 capsid (which does not include the fibers) does not undergo any large conformational changes at gastric pH. We reasoned that the gastrointestinal adaptation might have altered the distribution of acidic and basic residues exposed on the outer surface of the capsid. To investigate this, the surface charge distribution of HAdV-F41 along with the other two existing HAdV structures (HAdV-C5 and HAdV-D26) was calculated for pH 7.4. A visual comparison of the charge distribution revealed substantial differences between the three HAdVs (Fig. 2C). The exposed surface of HAdV-D26 is almost entirely covered by negative charge at pH 7.4, and HAdV-C5 has mostly negatively charged surfaces on the top of the hexons [in a calculation underestimating the amount of negative charge due to exclusion of the flexible, and highly negatively charged, hypervariable region 1 (HVR1; Fig. 2D), which was not built in any of the HAdV-C5 structures (19, 20, 34–37)]. By comparison, the capsid of HAdV-F41 is predominantly uncharged at pH 7.4, especially at the top of the hexon towers and PB protein (Fig. 2C). The surface charge distribution of HAdV-F41 at pH 4.0 revealed two distinct regions as still being relatively uncharged at this extreme pH: the N-terminal part of IX, largely occluded between hexons, and the solvent-exposed loops at the top of the hexons (Fig. 2C).
Fig. 2. Structure and surface charge distribution of HAdV-F41 at pH 7.4 and pH 4.0.
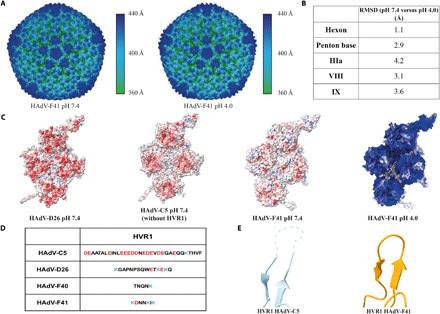
(A) Surface representation of the HAdV-F41 electron density at pH 7.4 and pH 4.0, colored by distance from the virion center. (B) pH-dependent structural changes in selected HAdV-F41 capsid proteins as measured by RMSD at Cα level. (C) Surface charge distribution for the atomic models of HAdV-C5 (PDB: 6CGV), HAdV-D26 (PDB: 5TX1), and HAdV-F41 at pH 7.4 as well as HAdV-F41 at pH 4.0. Red represents a local net negative charge, blue represents positive, and white represents uncharged. (D) Comparison of the HVR1 sequences between HAdV-C5, HAdV-D26, HAdV-F40, and HAdV-F41. (E) Cartoon representation of the HVR1-containing loop for HAdV-C5 and HAdV-F41. The unbuilt loop for HAdV-C5 is shown as a dashed line.
Whereas the overall structural fold of hexon chains is conserved among AdVs, they differ in the seven HVRs. Comparing HAdV-F41 (at pH 7.4) to HAdV-C5 and HAdV-D26, substantial differences were found in all seven HVRs (fig. S3 and table S4). In particular, HVR1 stands out in the comparison because it is much shorter in enteric HAdVs (Fig. 2D). The highly negatively charged HVR1 loop has not been built in any reported HAdV-C5 structure (11, 20), indicating its flexibility. On the other hand, the much shorter HVR1 in HAdV-F41 forms a loop with a rigid conformation that allowed tracing the entire length of the polypeptide (Fig. 2E).
Together, these findings show that the icosahedral part of the HAdV-F41 capsid is structurally unperturbed by exposure to stomach-like pH and has evolved to expose fewer charged residues on its exterior as compared to nonenteric HAdVs, most prominently exemplified by a near-complete deletion of the HVR1.
Protein IX arranges in a unique manner in HAdV-F41
Among the so-called minor capsid proteins, IX forms the most extended and complex arrangements. In previously reported structures of HAdV-C5 and HAdV-D26, this amounts to a tight mesh of ordered protein density that stretches through the canyons between hexons across the virion surface (Fig. 3A) (11, 21, 38). In HAdV-F41, only the N-terminal residues 1 to 58 (henceforth “IX-N”) were sufficiently ordered to trace the protein chain, thus ruling out the same sort of ordered, virus-spanning IX cage seen in other HAdVs (Fig. 3A). To further investigate whether there was any ordered protein density where the C terminus of IX resides in HAdV-C5 and HAdV-D26, we performed a localized asymmetric 3D classification at this position (39). Neither of the 3D classes showed any ordered protein density corresponding to the four-helical bundle that IX forms in the other HAdVs at this position (fig. S4). Analogously to other HAdVs, IX-N trimerizes to form a triskelion (Fig. 3B) located between three nonperipentonal hexon units (Fig. 1B). Each facet of the virion harbors four copies of the IX-N triskelion in two distinct structural surroundings: three copies at the local threefold symmetry axes of each ASU and one at the icosahedral threefold symmetry axis at the center of the facet (fig. S5). The conformations of IX-N in these two surroundings are virtually identical (Fig. 3B). The three IX chains come together at the center of this triskelion, where interactions between residues Phe12, Phe17, and Tyr20 from each chain form a hydrophobic core (Fig. 3C). This hydrophobic core is differently arranged compared to the nonenteric HAdV-C5 and HAdV-D26 (fig. S5) and has more large hydrophobic residues at its center.
Fig. 3. The triskelion-forming protein IX assembles in a unique way in HAdV-F41.
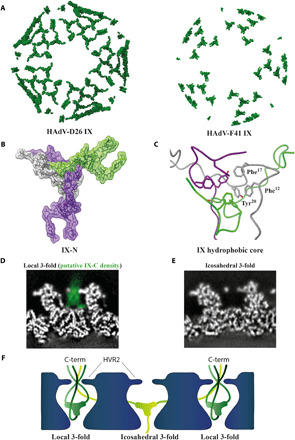
(A) Surface representation of IX assembly in HAdV-D26 and HAdV-F41 including all ordered protein density. (B) Graphical representation of HAdV-F41 IX-N triskelion assembly. The three IX chain atoms (green, gray, and purple) are shown in stick representation covered by a semitransparent surface. (C) Close-up view of the hydrophobic core at the center of the IX triskelion assembly, formed by residues Phe12, Phe17, and Tyr20 of each IX chain. (D) Computational slice through the electron density of HAdV-F41 at pH 7.4, at the position of IX at the local threefold axis. Electron density is white except the proposed density of IX-C, which is highlighted in green. (E) As (D), but at the icosahedral threefold axis. (F) Schematic representation of one possible arrangement of IX-C.
Sequence comparisons of HAdV-C5, HAdV-D26, and HAdV-F41 protein IX revealed that their respective IX-N have a higher sequence homology than the C-terminal parts of IX (residues 59 to 133; “IX-C”) (fig. S5). However, the sequences of IX-C are near identical in HAdV-F41 and the related HAdV-F40, indicating conservation between enteric AdVs. MS analysis detected the entire IX sequence in the purified HAdV-F41, confirming the presence of IX-C in the purified virions (table S1). We reasoned that the substantial protein mass corresponding to IX-C, emanating in the constricted space between the hexons, should be visible in the electron density map at a lower threshold even if it is flexible. Indeed, the interhexonal space above IX-N harbors electron density corresponding to a flexible protein at the local threefold axes (Fig. 3D and figs. S5 and S6). This density appears to pass to the outside of the capsid between the HVR2 loops of the three surrounding hexons. In a localized asymmetric reconstruction, the three HVR2 could be resolved in their entirety, showing that they are present in a single conformation and form a constriction of defined size (fig. S6) from which this density protrudes. In contrast, there is clearly no electron density above IX-N at the icosahedral threefold position (Fig. 3E and fig. S4), suggesting that the spatial organization of IX-C differs between these two positions (Fig. 3F). Adjacent to the position where the C terminus of IX resides in HAdV-C5 and HAdV-D26, one minor class representing 15% of the positions had a weak density somewhat resembling the protruding density at the local threefold axis (fig. S4).
Together, these data suggest a very different arrangement of IX in enteric AdVs as compared to respiratory (C5) and ocular (D26) HAdVs. In HAdV-F41, the C-terminal half of IX is flexible and appears to expose its C terminus to the capsid exterior at three of the four IX positions in each virion facet.
The HAdV-F41 PB undergoes assembly-induced conformational changes
Located on the fivefold symmetry axes of AdV capsids, the PB protein forms a homopentamer that contains integrin-binding motifs and serves as an assembly hub connecting the icosahedral capsid to the fibers (Fig. 1A). In our reconstruction of the entire HAdV-F41, the electron density for the PB was less well resolved than other parts of the capsid. To improve the map of the PB, we performed a localized asymmetric reconstruction of the PB monomer (fig. S7). The improved map allowed the building of an atomic model for the PB, which was placed into a composite atomic model of the entire ASU. Overall, the HAdV-F41 PB is very similar to the PB in HAdV-C5 (11, 12, 20) and HAdV-D26 (21), with a β sheet–rich fold that can roughly be divided into four domains: crown, head, body, and tail, with the body and head as the main domains, separated by loop regions (Fig. 4A).
Fig. 4. The HAdV-F41 PB undergoes assembly-induced conformational changes.
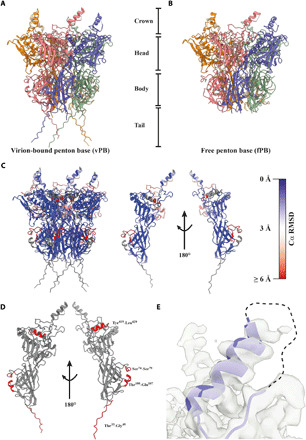
(A) Cartoon representation of the virion-bound PB (vPB), which can be divided into a crown, head, body, and tail. (B) Cartoon representation of the free PB (fPB). (C) Cartoon representation of the vPB and a single vPB monomer chain, each colored by Cα RMSD indicating the local degree of difference in Cα positioning between the vPB and fPB structures. (D) Cartoon representation of a single vPB monomer chain (gray). Missing residues in the fPB structure are highlighted in red. (E) Cartoon representation of the helix and disordered loop region containing the integrin-binding IGDD motif (dashed line) located in the crown. The electron density is shown as a transparent surface.
During assembly of the virus particle, the PB forms a plethora of interactions with peripentonal hexons, the fiber, and IIIa (table S2). To investigate conformational changes induced during the PB assembly process, we solved the structure of a recombinantly expressed HAdV-F41 PB in solution [free PB (fPB)] by cryo-EM. The map had an average resolution of 3.7 Å (fig. S8), allowing for the placement of an atomic model (Fig. 4B). Comparing the atomic models of the fPB and the virion-bound PB (vPB), the overall Cα shift (RMSD) was very small (~0.9 Å). However, color-coding vPB by its structural deviations from fPB revealed regions with higher RMSD, indicating localized assembly-induced conformational changes (Fig. 4C and movie S3). Moreover, four sequence segments that were built in the vPB model could not be built in the fPB model (Fig. 4A), indicating that these regions are disordered in solution and only become stabilized in a defined conformation upon assembly into the virion. One such region is the tail, a 17-residue (Thr33-Gly49) random coil region (Fig. 4A), which is disordered in the fPB (Fig. 4B). It is stabilized through interactions with two loop regions from IIIa (table S2). The sequence of the tail domain is largely conserved between HAdVs (fig. S9), suggesting a conserved role as an assembly motif. The second motif (Tyr419-Leu429) becoming ordered upon assembly is an α helix consisting of residues Gln416-Thr427, located close to the fivefold axis of the PB (Fig. 4D) and close to where the fiber binds. Although the HAdV-F41 capsid map shows only weak density for the proximal fiber in connection with the PB, the vPB structure did allow tracing of a fragment of the conserved fiber tail (fig. S9 and table S2). Thus, the folding of Gln416-Thr427 may be dependent on interactions within the capsid and/or binding of the fiber to the PB. Additional assembly-dependent interactions take place in two loop regions located between residues Val70-Asn110 in the body domain (Fig. 4D). These loops are disordered in the fPB but well resolved in the vPB where their conformation is stabilized by interactions with the peripentonal hexon. The first loop (Ser74-Ser79), located at the top of the body, is stabilized as an extended coil structure upon interaction with hexon chain Ser663-Tyr671 loop. The second region (Thr100-Gln107), located at the bottom of body, is stabilized as a short α helix upon binding to an uncharged pocket formed by peripentonal hexon residues Ala623-Ile640.
Peculiarly for enteric HAdVs, the otherwise conserved integrin-binding RGD motif has been replaced by Ile-Gly-Asp-Asp (IGDD) in HAdV-F41 [Arg-Gly-Ala-Asp(RGAD) for HAdV-F40)]. In the HAdV-F41 structure, the IGDD-containing loop is the only surface-exposed part of the PB for which we find no continuous electron density (Fig. 4E), despite being shorter than in most other HAdVs (fig. S9) (28). This parallels the observed flexibility of the RGD motifs in the two previously reported structures of HAdVs (19–21), indicating that the function of the IGDD sequence may be dependent on it being flexible until interacting with a target molecule.
In summary, a comparison of the PB in solution and in the virus capsid revealed several distinct motifs that become folded only upon assembly of the PB into the capsid and further revealed that the noncanonical integrin-binding motif IGDD is disordered also in the context of the assembled virus.
The DNA binding protein V is located at a conserved position at the inner face of the capsid
After initial model building of the virion at pH 7.4, the ASU contained five peptide chains that still had not been assigned an identity. Four of the five chains were deemed too short to assign an identity to. Three of those four chains interlace with different copies of VI at positions where VI interacts with VIII or IIIa at the inner face of the capsid (fig. S10 and movie S2). The fifth unidentified peptide chain is markedly longer and is located at the inner face of the capsid, in a pocket formed by three nonperipentonal hexons (Fig. 5, A and B, and table S5). Its electron density is resolved well enough to identify large side-chain residues, and we thus reasoned that a structural bioinformatics workflow might be devised to reveal its identity. With the initial constraint being only that residues number 4 and 21 in the identified 24-mer peptide must have large side chains, we used a combination of the proteomics data, exclusion of proteins with known locations, real-space refinement scores, and other considerations to elucidate its identity (see Supplementary Methods and fig. S11 in the Supplementary Materials). After sequential exclusion of candidates based on these criteria, a single most likely candidate remained, a sequence from the center of protein V: Gln170-Asp194. V has been reported to bind directly to DNA and to VI, thereby bridging the core and the surrounding capsid, but it has not been localized in any AdV structure. The built sequence of V fits the density without any clashes or unlikely interactions with surrounding proteins and, furthermore, shows a high degree of sequence conservation with V in HAdV-C5 and HAdV-D26 (Fig. 5, C and D, and movie S4). In both hitherto published HAdV structures, HAdV-C5 (20) and HAdV-D26 (21), there is similarly shaped electron density at the corresponding position of the virus capsid (Fig. 5E). In HAdV-C5, no atomic model was built into it (20). Similarly, at the corresponding position in the HAdV-D26 structure, two shorter peptide chains of unknown identity were placed (21). The location of the 24-mer chain of V is such that both the N and C termini of V, which are not resolved in our structure, may protrude toward the interior of the virion in agreement with the proposed role of V to link the viral genome to the capsid. In summary, we used a systematic structural bioinformatics approach to proposing a likely conserved position of core protein V in HAdVs.
Fig. 5. Location of DNA binding protein V at the interface of the three nonperipentonal hexon subunits.
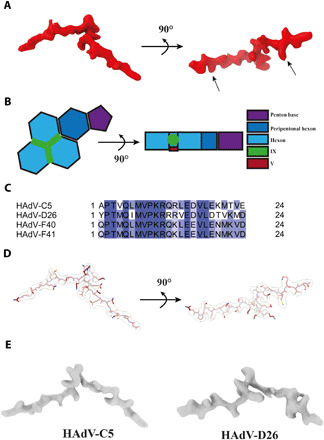
(A) Surface representation of V electron density. Arrows indicate the positions fixed during bioinformatics analysis. (B) Schematic representation of V and its location in the HAdV-F41 ASU. (C) Alignment of the identified V amino acid sequence from HAdV-C5, HAdV-D26, HAdV-F40, and HAdV-F41. Coloring represents percent sequence identity, with dark blue illustrating 100% homology. (D) Graphical representation of the modeled HAdV-F41 V peptide, shown in maroon, and stick representation covered by the corresponding electron density, shown as transparent surface. (E) Surface representation of the HAdV-C5 [EMD-7034 (20)] and HAdV-D26 [EMD-8471 (21)] electron densities located at the same position in their respective ASUs.
DISCUSSION
Here, we present the structure of a major cause of diarrhea and diarrhea-associated mortality in young children: the enteric adenovirus HAdVF41. As the first structure of an AdV with pronounced gastrointestinal tropism, it reveals a capsid whose structure is virtually unaltered by stomach pH and has substantial changes to the virion surface compared to respiratory and ocular HAdVs. Overall, HAdV-F41 has fewer charged, i.e., pH-dependent, residues exposed on the surface of its capsid. This is especially prominent at the top of the hexons where HVR1 is long and rich in negatively charged residues in HAdV-C5. This allows interaction of HAdV-C5 with lactoferricin through a charge-dependent mechanism (40), which contributes to an extended tissue tropism (41, 42). Evolution of HAdV-F41 has resulted in a largely truncated and less charged HVR1 (Fig. 2D), seemingly to adapt to the specific conditions in the gastrointestinal tract. Note that our study could not address one major distinguishing feature of enteric AdVs: the presence of two different fibers (Fig. 1A). The fibers were too flexible to be resolved to a larger extent in the current structure, and it is thus, e.g., possible that the fibers change their conformation at stomach pH more than the icosahedral part of the virus capsid discussed here.
Another major change to the capsid exterior of HAdV-F41 is the starkly different conformation of protein IX. Instead of forming a virus-covering, rigid mesh, the C-terminal half of IX (IX-C) is flexible. An unaccounted density protrudes to the outside of the capsid right above the IX-N triskelia, kept in place by hexon HVR2-containing loops. We favor the interpretation that this density is IX-C due to its proximity to the IX-N triskelia, the presence of IX-C in the purified virus as determined by MS and because IX-C is not found at any other position in the virus structure. Notably, this putative IX-C density is observed above all IX-N trimers except at the icosahedral threefold axis (Fig. 3, D and E, and figs. S4 and S5). In the model, in which this density belongs to IX-C, each IX-C chain could either emerge in cis (i.e., above the same IX-N trimer to which it belongs) or stretch across the virion surface to emerge above another IX-N trimer (in a trans arrangement). The length of IX-C in HAdV-F41 is compatible with both of these arrangements. A cis arrangement of all IX-C would be reminiscent of IX in some non-HAdVs, in which the conformation of IX-C is also more defined (38, 43, 44). Whereas our data do not allow tracing of individual chains of IX-C, the clear lack of any IX-C density above the IX-N trimer at the icosahedral threefold rules out such a pure cis arrangement of IX-C. One possible, parsimonious interpretation would be that the central IX trimer adopts a trans arrangement, donating one IX-C chain to each of the three IX trimers at local threefold positions that, in turn, have their IX-Cs in cis (Fig. 3F). Although other models for the IX-C arrangement may be consistent with our data, it is clear from the data that IX arranges in a unique manner in enteric AdVs compared to other HAdVs studied to date. All these modifications to the virion surface of HAdV-F41 are likely related to the different set of interactors and different pH range that this virus encounters throughout the gastrointestinal tract. Other gastrointestinal viruses interact with components such as bile (calicivirus) (45) and lipopolysaccharides [poliovirus (46), mouse mammary tumor virus (47)], which is crucial for infection of these viruses. Besides low-pH resistant interactions of HAdV-F41 with gastrointestinal phospholipids (48), little is known about the HAdV-F41:gastrointestinal interactome. Finding these interaction partners, e.g., of the disordered and exposed IX-C region, will yield further insights into the infection cycle and tropism of enteric AdVs.
Our study further unveiled how several motifs in the HAdV-F41 PB are disordered in solution and only adopt a defined conformation upon assembly into the virus capsid, laying out another piece of the still largely unfinished puzzle of AdV assembly (10). The observation that the modified integrin-interacting motif of the HAdV-F41 PB is still disordered in the assembled virus particle highlights the need for structural studies of the interactions with its proposed binding partner, laminin-binding integrins (29).
Biochemical data have defined core protein V as a key protein linking the AdV genome to the capsid (49), but, despite its conserved function in AdVs, it had not been located in the AdV capsid. Here, we propose the point of interaction of V to the interior of the capsid and provide data suggesting that this position, at the junction between three nonperipentonal hexons, is conserved between HAdVs. Previous biochemical data have not suggested V to interact with the hexons but have instead suggested interactions between V and the minor protein VI (49, 50). These data are not mutually exclusive with our identification of the V anchoring point to the hexons, because most of the V sequence is still unaccounted for in the structure and several copies of VI are found in the vicinity of the anchoring point where they may form additional interactions.
Together, the structure of the enteric AdV HAdV-F41 revealed key conserved aspects of AdV architecture and highly divergent features of enteric AdVs, thus laying the foundation for structure-based approaches to developing effective antivirals, for preventing this prominent cause of diarrhea-associated mortality in young children, and for further development of these structurally divergent AdV types as vaccine vehicles.
MATERIALS AND METHODS
Virus propagation and purification
Human A549 cells (gift from A. Kidd) were maintained in Dulbecco’s modified Eagle medium (DMEM; Sigma-Aldrich) supplemented with 5% fetal bovine serum (FBS; HyClone, GE Healthcare), 20 mM Hepes (Sigma-Aldrich), and penicillin (20 U/ml) and streptomycin (20 μg/ml) (Gibco).
For HAdV-F41 (strain Tak) propagation, 30 bottles of A549 cells (175 cm2, at a 90% confluency) were infected with HAdV-F41 inoculation material (produced in A549 cells) in 5 ml of growth media (1% FBS) for 90 min on a rocking table at 37°C. Thereafter, additional 25 ml of growth media (1% FBS) were added to each flask, and the cells were further incubated at 37°C. Infected cells were harvested after approximately 1 week or when cells displayed clear signs of cytopathic effect. Cells were collected by centrifugation, resuspended in DMEM, and disrupted to release virions by freeze-thawing and by addition of equal volume of Vertrel XF (Sigma-Aldrich). After vigorous resuspension, the cell extract was centrifuged at 3000 rpm for 10 min. The upper phase was transferred onto a discontinuous CsCl gradient [densities: 1.27, 1.32, and 1.37 g/ml, in 20 mM tris-HCl (pH 8.0); Sigma-Aldrich] and centrifuged at 25,000 rpm in a Beckman SW40 rotor for 2.5 hours at 4°C. The virion band was collected and desalted on a NAP column (GE Healthcare) into sterile phosphate-buffered saline (PBS).
Protein identification by MS
Protein digestion
The samples were split for tryptic and chymotryptic digestion and processed using a modified protocol of FASP (33). Briefly, triethylammonium bicarbonate (TEAB) was added to a final concentration of 50 mM TEAB before reduction using 100 mM dithiothreitol at 56°C for 30 min. The reduced samples were loaded onto 10-kDa molecular weight cutoff of Pall Nanosep centrifugal filters (Sigma-Aldrich), washed with 8 M urea and 1% sodium deoxycholate (SDC), and alkylated with 10 mM methyl methane thiosulfonate. Two-step digestion was performed on filters using trypsin and chymotrypsin as digestive enzymes in 50 mM TEAB and 0.5% SDC buffer. The first step was performed overnight, and the second step, with an additional portion of proteases, was performed for 4 hours the next day. Tryptic digestion was performed at 37°C using Pierce MS Grade Trypsin Protease (Thermo Fisher Scientific). Chymotryptic digestion was performed at room temperature using Pierce MS Grade Chymotrypsin Protease (Thermo Fisher Scientific). The peptides were collected by centrifugation, and SDC was precipitated by acidifying the sample with trifluoroacetic acid (final concentration, 1%). The digested sample was desalted using Pierce Peptide Desalting Spin Columns (Thermo Fisher Scientific) according to the manufacturer’s protocol.
Liquid chromatography–tandem mass spectrometry
The digested and desalted samples were analyzed using a QExactive HF mass spectrometer interfaced with an Easy-nLC 1200 liquid chromatography system (both Thermo Fisher Scientific). Peptides were trapped on an Acclaim PepMap 100 C18 trap column (100 μm by 2 cm; particle size, 5 μm; Thermo Fischer Scientific) and separated on an in-house packed analytical column (75 μm by 300 mm; particle size, 3 μm; Reprosil-Pur C18, Dr. Maisch). A stepped gradient used was from 7 to 35% solvent B in 97 min, followed by an increase to 48% in 8 min and to 100% solvent B in 5 min at a flowrate of 300 nl/min. Solvent A was 0.2% formic acid, and solvent B was 80% acetonitrile in 0.2% formic acid. The mass spectrometer was operated in data-dependent acquisition (DDA) mode where the MS1 scans were acquired at a resolution of 60,000 and a scan range from 400 to 1600 mass/charge ratio (m/z). The 10 most intense ions with a charge state of 2 to 4 were isolated with an isolation window of 1.2 m/z and fragmented using normalized collision energy of 28. The MS2 scans were acquired at a resolution of 30,000, and the dynamic exclusion time was set to 20 s.
Database search
Data analysis was performed using Proteome Discoverer (version 1.4, Thermo Fisher Scientific). The data were searched against an in-house database containing the amino acid sequences of HAdV-F41. Mascot (version 2.5.1, Matrix Science) was used as search engine with a precursor mass tolerance of 5 parts per million for MS1 and 30 milli mass unit (mmu) for MS2 spectra. Tryptic peptides were accepted with a maximum of one missed cleavage, and chymotryptic peptides were accepted with maximum three missed cleavages. Variable modification of methionine oxidation and fixed methylthio of cysteines were selected. The Mascot significance threshold for peptides was set to 0.01.
Cryo-EM sample preparation
Purified HAdV-F41 was used at 4.2 mg/ml (pH 7.4) and 1.6 mg/ml (pH 4.0). The recombinant HAdV-F41 PB (fPB) was purified as described before (29) and used at 1 mg/ml in PBS buffer, supplemented with 5% glycerol. A HAdV-F41 sample at pH 4.0 was prepared by adding 2 μl of a 0.5 M citric acid/1 M Na2HPO4 (pH 3.4) solution to 25 μl of HAdV-F41 (pH 7.4), followed by incubating on ice for 15 min. Samples were vitrified on QUANTIFOIL Cu R200 2/2 (Electron Microscopy Sciences, catalog no. Q2100CR2) and QUANTIFOIL Cu R200 1.2/1.3 (Electron Microscopy Sciences, catalog no. Q3100CR1.3) grids for the virus particles and the recombinant protein, respectively. Before sample application, the grids were glow discharged using a PELCO easiGlow device (Ted Pella Inc.) at 15 mA for 30 s. Sample was applied by transferring 3 μl of sample onto the glow-discharged side of the grid, blotted, and plunge-frozen in liquid ethane, using a Vitrobot plunge freezer (Thermo Fisher Scientific), with the following settings: 22°C, 80% humidity, a blot force of −20, and a blotting time of 3 s. For HAdV-F41 at pH 7.4, sample was applied twice with a blotting step, using the same settings as above, between applications (51).
Data collection
All data were collected on an FEI Titan Krios transmission electron microscope (Thermo Fisher Scientific) operated at 300 keV and equipped with a Gatan BioQuantum energy filter and a K2 direct electron detector. A condenser aperture of 70 μm (HAdV-F41 at pH 7.4 and pH 4.0) and an objective aperture of 100 μm were chosen for data collection. A C2 aperture of 100 μm was selected for the PB41 data collection. Coma-free alignment was performed with AutoCtf/Sherpa. Data were acquired in parallel illumination mode using EPU (Thermo Fisher Scientific) software at a nominal magnification of 130,000× (1.041-Å pixel size). Both datasets for the HAdV-F41 structure at pH 7.4 were collected in superresolution mode. Because of a preferred orientation of PB41, a second dataset was collected at a 30° tilted stage. Data collection parameters are listed in table S6.
Data processing and structure determination HAdV-F41 at pH 7.4
Two datasets were collected on HAdV-F41 at pH 7.4 and initially processed independently. Data were initially processed using RELION 3.0 (52) and continued in RELION 3.1 (beta) (53). Beam-induced motion was corrected using RELION’s MotionCor2 (54) implementation, at which step the superresolution movies were binned once, and the per-micrograph contrast transfer function (CTF) estimated using GCTF software (55) for all datasets. Particles were manually picked and subjected to reference-free 2D classification, and well-resolved classes were combined and subjected to 3D classification, applying icosahedral symmetry [I3 according to Crowther (56)] and a mask of the capsid structures. A low-pass filtered (50 Å) volume of HAdV-C5 [EMD-7034 (20)] was used as a reference volume. Particles were classified into two classes, resulting in 99% of particles allocated to one well-resolved class that was used for downstream processing. 3D refinement was performed using the output of the 3D classification as a reference model, low-pass filtered to 50 Å, with no additional Fourier padding. Following refinement, data were postprocessed, and the particles were subjected to per-particle CTF refinement, Bayesian polishing, and another round of per-particle CTF refinement. The particles were subjected to an additional round of 3D refinement before combining both datasets and performing a final 3D refinement, with no additional Fourier padding. The resolution was calculated using the gold standard Fourier shell correlation [FSC threshold, 0.143)] to 3.84 Å after postprocessing. Last, the data were corrected for the Ewald’s sphere curvature using RELION, which led to a local improvement of the electron density map with a new average resolution of 3.77 Å. Local resolution estimates were calculated using ResMap (57).
A homology model was generated using the SWISS-MODEL server (58), the HAdV-F41 capsid protein sequences, for which homologs have been structurally determined. The resulting homology model was based on the reported HAdV-D26 structure [PDB: 5TX1 (21)]. The model was manually docked into the HAdV-F41 electron density in ChimeraX (59) and the map corresponding to the ASU extracted. The ASU map was locally sharpened using Phenix’s (60) autosharpen tool. Subsequently, the HAdV-F41 homology model was docked and subjected to an initial round of real-space refinement using Phenix. The structure was fully refined using iterative cycles of Phenix’s real-space refinement and model building in Coot (61).
Localized asymmetric reconstruction
To improve the map quality surrounding the PB monomer, the HVR2-loop containing region (local threefold axis), the icosahedral threefold axis, and the region analogous to the region where the four-helical IX-C bundles in HAdV-C5 and HAdV-D26 are located, the map was improved using the localized asymmetric reconstruction workflow reported by Ilca et al. (39) and implemented in Scipion v2.0 (62). Coordinates for the subparticles were determined in ChimeraX and subsequently located by applying icosahedral symmetry and extracted in Scipion v2.0. Subparticles were subsequently filtered to exclude particles not present within a [−20°, 20°] range from the image plane. The resulting subparticles were then subjected to an asymmetric 3D classification. To increase the probability of convergence during classification, changes in the origins and orientations were not allowed. A subsequent 3D refinement yielded a 3D reconstruction of the PB monomer and the HVR2-loop containing region to a resolution of 3.0 and 3.35 Å, respectively. Average resolutions were calculated according to the gold standard FSC calculations (threshold, 0.143). Data processing statistics for the HVR2-containing loop region and the PB monomer are given in table 7. 3DFSC curves were calculated using the Remote 3DFSC Processing Server (63).
Image processing and model building for HAdV-F41 at pH 4.0
Data were processed as described for the HAdV-F41 at pH 7.4 structure up until the first 3D refinement. The volume HAdV-F41 at pH 7.4 was low-pass filtered to 10 Å and used as a reference. The resolution was estimated to 5.0 Å using the gold standard FSC (threshold, 0.143) after postprocessing. Local resolution estimates were calculated using ResMap. The HAdV-F41 (pH 7.4) model was fitted into the reconstructed HAdV-F41 (pH 4.0) density using ChimeraX, an ASU was extracted, and the resulting map was locally sharpened using Phenix. The model was then further fitted and energy minimized using Namdinator (64).
Data processing and structure determination recombinant HAdV-F41 PB
The HAdV-F41 PB (PB41) data (untilted and tilted at 30°) were processed using RELION 3.1 (beta), with beam-induced motion correction and CTF estimation performed as for the HAdV-F41 structure. Particles were picked using the automated particle picker crYOLO (65) using the available Phosaurus generalized model. Reference-free 2D classification of particles was performed in RELION and revealed a substantial proportion of particles with the same view, suggesting a preferred orientation of the specimen. From the 0° data, an initial model was generated in cryoSPARC (66). Well-resolved 2D classes were combined and subjected to 3D refinement using the same reference model as, during 3D classification, low pass filtered to 10 Å. Following refinement, data were postprocessed, and the particles were subjected to per-particle CTF refinement, Bayesian polishing, and another round of per-particle CTF refinement before performing a final round of 3D refinement. Inspection of the final volume revealed poor resolution along one of the axes (fig. S8). We therefore collected data on a tilted specimen stage. As data collection at a tilted stage leads to a defocus gradient along the image path, per-particle CTF refinement was performed after particle extraction and before reference-free 2D classification, using GCTF. Subsequent processing steps were performed as described for the data collected on an untilted specimen stage. The average resolutions were estimated to 3.4 Å (untilted) and 3.7 Å (30° tilt), using the gold standard FSC (threshold, 0.143) after postprocessing. Local resolution estimates were calculated using ResMap. 3DFSC curves were calculated using the Remote 3DFSC Processing Server (63).
The PB41 volume generated from the data collected on the tilted stage was used for downstream model building and model refinement. The PB monomer chain from the HAdV-F41 pH 7.4 model was initially fitted into PB41 volume using Namdinator (64) and outlying residues pruned in Coot. Subsequently, the model was fully built and refined using iterative cycles of real-space refinement in Phenix and model building in Coot.
Calculation of surface charge distribution
Surface charges for HAdV-C5 [PDB: 6CGV (19)], HAdV-D26 [PDB: 5TX1 (21)], and HAdV-F41 were calculated using the PDB2PQR-APBS software package (67) at pH 7.4 and pH 4.0.
Bioinformatics workflow to determine the protein identity of the unknown chain
For each direction of the unknown chain, a 24-mer poly-alanine chain was manually placed into the respective density and initially real-space refined using Coot. A list of sequences was screened using a job pipeline including a mutation step in Coot and real-space refinement in Phenix. Custom bash scripts written for this purpose are available upon request. An extended description is given in Supplementary Methods.
Molecular graphics and visualization
Figures of protein structures and electron densities were generated using ChimeraX.
Supplementary Material
Acknowledgments
We are grateful to P. Emsley for help with Coot scripting, M. Hall for help with cryoSPARC, and T. Terwilliger for help with Phenix. We thank S. Nord, J. Näslund, and A. Sjödin (FOI, Swedish Defence Research Institute, Umeå, Sweden) for help with DNA sequencing. Protein identification was performed at the Proteomics Core Facility of Sahlgrenska Academy, University of Gothenburg. Funding: We are thankful for funding from the Human Frontier Science Program (Career Development Award CDA00047/2017-C to L.-A.C.), Stiftelsen Olle Engkvist Byggmästare (postdoctoral fellowship to K.R.), the Knut and Alice Wallenberg Foundation (through the Wallenberg Centre for Molecular Medicine Umeå), and the Swedish Research Council (Dnr 2019-01472 and 2017-00859). Cryo-EM data were collected at the Umeå Core Facility for Electron Microscopy (SciLifeLab National Cryo-EM facility and part of National Microscopy Infrastructure, NMI VR-RFI 2016-00968), supported by instrumentation grants from the Knut and Alice Wallenberg Foundation and the Kempe Foundations. We thank the High Performance Computing Center North (HPC2N) at Umeå University for providing computational resources and valuable support during test and performance runs (SNIC projects 2018/5-158, 2019/3-668, and 2019/5-76). Author contributions: K.R., A.L., N.A., and L.-A.C. conceived and designed the study. A.L. produced and purified virus particles. A.R. purified recombinant PB protein. K.R. collected cryo-EM data and performed image processing, model building, and validation. K.R., A.L., N.A., and L.-A.C. interpreted structural data. J.F. performed proteomics analysis. K.R., A.L., N.A., and L.-A.C wrote the original manuscript with input from J.F. All authors reviewed and approved the final manuscript. Competing interests: The authors declare that they have no competing interests. Data and materials availability: The scripts used for the bioinformatics analysis are available upon request. Coordinates reported in this study have been deposited with the PDB with accession codes 6Z7N (HAdV-F41 ASU) and 6Z7Q [HAdV-F41 (free) PB]. Electron microscopy maps and half maps have been deposited in the Electron Microscopy Data Bank with the accession codes EMD-11108 (HAdV-F41 at pH 7.4), EMD-11111 (HAdV-F41 at pH 4.0), EMD-11112 [HAdV-F41 (free) PB], EMD-11109 (localized asymmetric reconstruction of the HAdV-F41 PB), and EMD-11110 (localized asymmetric reconstruction of the HAdV-F41 HVR2-containing loop).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/2/eabe0974/DC1
REFERENCES AND NOTES
- 1.W. S. M. Wold, M. G. Ison, Adenoviruses, in Fields Virology, D. M. Knipe, P. M. Howley, Eds. (Lippincott Williams & Wilkins, 2016), vol. 2, chap. 56, pp. 1732–1767. [Google Scholar]
- 2.HAdV Working Group, (2019); http://hadvwg.gmu.edu/.
- 3.Garofalo M., Staniszewska M., Salmaso S., Caliceti P., Pancer K. W., Wieczorek M., Kuryk L., Prospects of replication-deficient adenovirus based vaccine development against SARS-CoV-2. Vaccines 8, 293 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guo X., Deng Y., Chen H., Lan J., Wang W., Zou X., Hung T., Lu Z., Tan W., Systemic and mucosal immunity in mice elicited by a single immunization with human adenovirus type 5 or 41 vector-based vaccines carrying the spike protein of Middle East respiratory syndrome coronavirus. Immunology 145, 476–484 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Collaborators G. B. D. D. D., Estimates of the global, regional, and national morbidity, mortality, and aetiologies of diarrhoea in 195 countries: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Infect. Dis. 18, 1211–1228 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu J., Platts-Mills J. A., Juma J., Kabir F., Nkeze J., Okoi C., Operario D. J., Uddin J., Ahmed S., Alonso P. L., Antonio M., Becker S. M., Blackwelder W. C., Breiman R. F., Faruque A. S., Fields B., Gratz J., Haque R., Hossain A., Hossain M. J., Jarju S., Qamar F., Iqbal N. T., Kwambana B., Mandomando I., McMurry T. L., Ochieng C., Ochieng J. B., Ochieng M., Onyango C., Panchalingam S., Kalam A., Aziz F., Qureshi S., Ramamurthy T., Roberts J. H., Saha D., Sow S. O., Stroup S. E., Sur D., Tamboura B., Taniuchi M., Tennant S. M., Toema D., Wu Y., Zaidi A., Nataro J. P., Kotloff K. L., Levine M. M., Houpt E. R., Use of quantitative molecular diagnostic methods to identify causes of diarrhoea in children: A reanalysis of the GEMS case-control study. Lancet 388, 1291–1301 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.GBD 2017 Diarrhoeal Disease Collaborators , Quantifying risks and interventions that have affected the burden of diarrhoea among children younger than 5 years: An analysis of the Global Burden of Disease Study 2017. Lancet Infect. Dis. 20, 37–59 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.S. J. Flint, G. R. Nemerow, Adenovirus composition, structure, and biophysical properties, in Human Adenoviruses: From Villains to Vectors (World Scientific, 2017), chap. 2, pp. 15–41. [Google Scholar]
- 9.Nemerow G. R., Stewart P. L., Reddy V. S., Structure of human adenovirus. Curr. Opin. Virol. 2, 115–121 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.San Martin C., Latest insights on adenovirus structure and assembly. Viruses 4, 847–877 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu H., Jin L., Koh S. B., Atanasov I., Schein S., Wu L., Zhou Z. H., Atomic structure of human adenovirus by cryo-EM reveals interactions among protein networks. Science 329, 1038–1043 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reddy V. S., Natchiar S. K., Stewart P. L., Nemerow G. R., Crystal structure of human adenovirus at 3.5 A resolution. Science 329, 1071–1075 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stewart P. L., Burnett R. M., Cyrklaff M., Fuller S. D., Image reconstruction reveals the complex molecular organization of adenovirus. Cell 67, 145–154 (1991). [DOI] [PubMed] [Google Scholar]
- 14.Zubieta C., Schoehn G., Chroboczek J., Cusack S., The structure of the human adenovirus 2 penton. Mol. Cell 17, 121–135 (2005). [DOI] [PubMed] [Google Scholar]
- 15.van Raaij M. J., Mitraki A., Lavigne G., Cusack S., A triple beta-spiral in the adenovirus fibre shaft reveals a new structural motif for a fibrous protein. Nature 401, 935–938 (1999). [DOI] [PubMed] [Google Scholar]
- 16.Xia D., Henry L. J., Gerard R. D., Deisenhofer J., Crystal structure of the receptor-binding domain of adenovirus type 5 fiber protein at 1.7 A resolution. Structure 2, 1259–1270 (1994). [DOI] [PubMed] [Google Scholar]
- 17.Roberts M. M., White J. L., Grütter M. G., Burnett R. M., Three-dimensional structure of the adenovirus major coat protein hexon. Science 232, 1148–1151 (1986). [DOI] [PubMed] [Google Scholar]
- 18.Benevento M., Di Palma S., Snijder J., Moyer C. L., Reddy V. S., Nemerow G. R., Heck A. J., Adenovirus composition, proteolysis, and disassembly studied by in-depth qualitative and quantitative proteomics. J. Biol. Chem. 289, 11421–11430 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kundhavai Natchiar S., Venkataraman S., Mullen T. M., Nemerow G. R., Reddy V. S., Revised crystal structure of human adenovirus reveals the limits on protein IX quasi-equivalence and on analyzing large macromolecular complexes. J. Mol. Biol. 430, 4132–4141 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Dai X., Wu L., Sun R., Zhou Z. H., Atomic structures of minor proteins VI and VII in human adenovirus. J. Virol. 91, e00850-17 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu X., Veesler D., Campbell M. G., Barry M. E., Asturias F. J., Barry M. A., Reddy V. S., Cryo-EM structure of human adenovirus D26 reveals the conservation of structural organization among human adenoviruses. Sci. Adv. 3, e1602670 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Seiradake E., Cusack S., Crystal structure of enteric adenovirus serotype 41 short fiber head. J. Virol. 79, 14088–14094 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kidd A. H., Chroboczek J., Cusack S., Ruigrok R. W., Adenovirus type 40 virions contain two distinct fibers. Virology 192, 73–84 (1993). [DOI] [PubMed] [Google Scholar]
- 24.Yeh H. Y., Pieniazek N., Pieniazek D., Gelderblom H., Luftig R. B., Human adenovirus type 41 contains two fibers. Virus Res. 33, 179–198 (1994). [DOI] [PubMed] [Google Scholar]
- 25.Wickham T. J., Mathias P., Cheresh D. A., Nemerow G. R., Integrins αvβ3 and αvβ5 promote adenovirus internalization but not virus attachment. Cell 73, 309–319 (1993). [DOI] [PubMed] [Google Scholar]
- 26.Roelvink P. W., Lizonova A., Lee J. G., Li Y., Bergelson J. M., Finberg R. W., Brough D. E., Kovesdi I., Wickham T. J., The coxsackievirus-adenovirus receptor protein can function as a cellular attachment protein for adenovirus serotypes from subgroups A, C, D, E, and F. J. Virol. 72, 7909–7915 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rodriguez E., Romero C., Rio A., Miralles M., Raventos A., Planells L., Burgueno J. F., Hamada H., Perales J. C., Bosch A., Gassull M. A., Fernandez E., Chillon M., Short-fiber protein of ad40 confers enteric tropism and protection against acidic gastrointestinal conditions. Hum. Gene Ther. Methods 24, 195–204 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Madisch I., Hofmayer S., Moritz C., Grintzalis A., Hainmueller J., Pring-Akerblom P., Heim A., Phylogenetic analysis and structural predictions of human adenovirus penton proteins as a basis for tissue-specific adenovirus vector design. J. Virol. 81, 8270–8281 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rajan A., Persson B. D., Frangsmyr L., Olofsson A., Sandblad L., Heino J., Takada Y., Mould A. P., Schnapp L. M., Gall J., Arnberg N., Enteric species F human adenoviruses use laminin-binding integrins as co-receptors for infection of Ht-29 cells. Sci. Rep. 8, 10019 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Albinsson B., Kidd A. H., Adenovirus type 41 lacks an RGD α(v)-integrin binding motif on the penton base and undergoes delayed uptake in A549 cells. Virus Res. 64, 125–136 (1999). [DOI] [PubMed] [Google Scholar]
- 31.Leung T. K., Brown M., Block in entry of enteric adenovirus type 41 in HEK293 cells. Virus Res. 156, 54–63 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Nagita A., Amemoto K., Yoden A., Aoki S., Sakaguchi M., Ashida K., Mino M., Diurnal variation in intragastric pH in children with and without peptic ulcers. Pediatr. Res. 40, 528–532 (1996). [DOI] [PubMed] [Google Scholar]
- 33.Wisniewski J. R., Zougman A., Nagaraj N., Mann M., Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362 (2009). [DOI] [PubMed] [Google Scholar]
- 34.Rux J. J., Kuser P. R., Burnett R. M., Structural and phylogenetic analysis of adenovirus hexons by use of high-resolution x-ray crystallographic, molecular modeling, and sequence-based methods. J. Virol. 77, 9553–9566 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fabry C. M., Rosa-Calatrava M., Conway J. F., Zubieta C., Cusack S., Ruigrok R. W., Schoehn G., A quasi-atomic model of human adenovirus type 5 capsid. EMBO J. 24, 1645–1654 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schmid M., Ernst P., Honegger A., Suomalainen M., Zimmermann M., Braun L., Stauffer S., Thom C., Dreier B., Eibauer M., Kipar A., Vogel V., Greber U. F., Medalia O., Pluckthun A., Adenoviral vector with shield and adapter increases tumor specificity and escapes liver and immune control. Nat. Commun. 9, 450 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bottermann M., Lode H. E., Watkinson R. E., Foss S., Sandlie I., Andersen J. T., James L. C., Antibody-antigen kinetics constrain intracellular humoral immunity. Sci. Rep. 6, 37457 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reddy V. S., The role of hexon protein as a molecular mold in patterning the protein IX organization in human adenoviruses. J. Mol. Biol. 429, 2747–2751 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ilca S. L., Kotecha A., Sun X., Poranen M. M., Stuart D. I., Huiskonen J. T., Localized reconstruction of subunits from electron cryomicroscopy images of macromolecular complexes. Nat. Commun. 6, 8843 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Persson B. D., Lenman A., Frangsmyr L., Schmidt M., Ahlm C., Pluckthun A., Jenssen H., Arnberg N., Lactoferrin-hexon interactions mediate CAR-independent adenovirus infection of human respiratory cells. J. Virol. 94, e00542-20 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Garnett C. T., Talekar G., Mahr J. A., Huang W., Zhang Y., Ornelles D. A., Gooding L. R., Latent species C adenoviruses in human tonsil tissues. J. Virol. 83, 2417–2428 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Johansson C., Jonsson M., Marttila M., Persson D., Fan X. L., Skog J., Frangsmyr L., Wadell G., Arnberg N., Adenoviruses use lactoferrin as a bridge for CAR-independent binding to and infection of epithelial cells. J. Virol. 81, 954–963 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cheng L., Huang X., Li X., Xiong W., Sun W., Yang C., Zhang K., Wang Y., Liu H., Huang X., Ji G., Sun F., Zheng C., Zhu P., Cryo-EM structures of two bovine adenovirus type 3 intermediates. Virology 450-451, 174–181 (2014). [DOI] [PubMed] [Google Scholar]
- 44.Schoehn G., El Bakkouri M., Fabry C. M., Billet O., Estrozi L. F., Le L., Curiel D. T., Kajava A. V., Ruigrok R. W., Kremer E. J., Three-dimensional structure of canine adenovirus serotype 2 capsid. J. Virol. 82, 3192–3203 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kilic T., Koromyslova A., Hansman G. S., Structural basis for human norovirus capsid binding to bile acids. J. Virol. 93, e01581-18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuss S. K., Best G. T., Etheredge C. A., Pruijssers A. J., Frierson J. M., Hooper L. V., Dermody T. S., Pfeiffer J. K., Intestinal microbiota promote enteric virus replication and systemic pathogenesis. Science 334, 249–252 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kane M., Case L. K., Kopaskie K., Kozlova A., MacDearmid C., Chervonsky A. V., Golovkina T. V., Successful transmission of a retrovirus depends on the commensal microbiota. Science 334, 245–249 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Favier A. L., Burmeister W. P., Chroboczek J., Unique physicochemical properties of human enteric Ad41 responsible for its survival and replication in the gastrointestinal tract. Virology 322, 93–104 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Perez-Vargas J., Vaughan R. C., Houser C., Hastie K. M., Kao C. C., Nemerow G. R., Isolation and characterization of the DNA and protein binding activities of adenovirus core protein V. J. Virol. 88, 9287–9296 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chatterjee P. K., Vayda M. E., Flint S. J., Interactions among the three adenovirus core proteins. J. Virol. 55, 379–386 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Snijder J., Borst A. J., Dosey A., Walls A. C., Burrell A., Reddy V. S., Kollman J. M., Veesler D., Vitrification after multiple rounds of sample application and blotting improves particle density on cryo-electron microscopy grids. J. Struct. Biol. 198, 38–42 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zivanov J., Nakane T., Forsberg B. O., Kimanius D., Hagen W. J., Lindahl E., Scheres S. H., New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7, e42166 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zivanov J., Nakane T., Scheres S. H. W., Estimation of high-order aberrations and anisotropic magnification from cryo-EM data sets in RELION-3.1. IUCrJ 7, 253–267 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li X., Mooney P., Zheng S., Booth C. R., Braunfeld M. B., Gubbens S., Agard D. A., Cheng Y., Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods 10, 584–590 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zhang K., Gctf: Real-time CTF determination and correction. J. Struct. Biol. 193, 1–12 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Crowther R. A., Procedures for three-dimensional reconstruction of spherical viruses by Fourier synthesis from electron micrographs. Philos. Trans. R. Soc. Lond. Ser. B Biol. Sci. 261, 221–230 (1971). [DOI] [PubMed] [Google Scholar]
- 57.Kucukelbir A., Sigworth F. J., Tagare H. D., Quantifying the local resolution of cryo-EM density maps. Nat. Methods 11, 63–65 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Waterhouse A., Bertoni M., Bienert S., Studer G., Tauriello G., Gumienny R., Heer F. T., de Beer T. A. P., Rempfer C., Bordoli L., Lepore R., Schwede T., SWISS-MODEL: Homology modelling of protein structures and complexes. Nucleic Acids Res. 46, W296–W303 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Goddard T. D., Huang C. C., Meng E. C., Pettersen E. F., Couch G. S., Morris J. H., Ferrin T. E., UCSF ChimeraX: Meeting modern challenges in visualization and analysis. Protein Sci. 27, 14–25 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liebschner D., Afonine P. V., Baker M. L., Bunkoczi G., Chen V. B., Croll T. I., Hintze B., Hung L. W., Jain S., McCoy A. J., Moriarty N. W., Oeffner R. D., Poon B. K., Prisant M. G., Read R. J., Richardson J. S., Richardson D. C., Sammito M. D., Sobolev O. V., Stockwell D. H., Terwilliger T. C., Urzhumtsev A. G., Videau L. L., Williams C. J., Adams P. D., Macromolecular structure determination using x-rays, neutrons and electrons: Recent developments in Phenix. Acta Crystallogr. D Struct. Biol. 75, 861–877 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Emsley P., Lohkamp B., Scott W. G., Cowtan K., Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.de la Rosa-Trevin J. M., Quintana A., Del Cano L., Zaldivar A., Foche I., Gutierrez J., Gomez-Blanco J., Burguet-Castell J., Cuenca-Alba J., Abrishami V., Vargas J., Oton J., Sharov G., Vilas J. L., Navas J., Conesa P., Kazemi M., Marabini R., Sorzano C. O., Carazo J. M., Scipion: A software framework toward integration, reproducibility and validation in 3D electron microscopy. J. Struct. Biol. 195, 93–99 (2016). [DOI] [PubMed] [Google Scholar]
- 63.Tan Y. Z., Baldwin P. R., Davis J. H., Williamson J. R., Potter C. S., Carragher B., Lyumkis D., Addressing preferred specimen orientation in single-particle cryo-EM through tilting. Nat. Methods 14, 793–796 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kidmose R. T., Juhl J., Nissen P., Boesen T., Karlsen J. L., Pedersen B. P., Namdinator—Automatic molecular dynamics flexible fitting of structural models into cryo-EM and crystallography experimental maps. IUCrJ 6, 526–531 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wagner T., Merino F., Stabrin M., Moriya T., Antoni C., Apelbaum A., Hagel P., Sitsel O., Raisch T., Prumbaum D., Quentin D., Roderer D., Tacke S., Siebolds B., Schubert E., Shaikh T. R., Lill P., Gatsogiannis C., Raunser S., SPHIRE-crYOLO is a fast and accurate fully automated particle picker for cryo-EM. Commun. Biol. 2, 218 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Punjani A., Rubinstein J. L., Fleet D. J., Brubaker M. A., cryoSPARC: Algorithms for rapid unsupervised cryo-EM structure determination. Nat. Methods 14, 290–296 (2017). [DOI] [PubMed] [Google Scholar]
- 67.Jurrus E., Engel D., Star K., Monson K., Brandi J., Felberg L. E., Brookes D. H., Wilson L., Chen J., Liles K., Chun M., Li P., Gohara D. W., Dolinsky T., Konecny R., Koes D. R., Nielsen J. E., Head-Gordon T., Geng W., Krasny R., Wei G. W., Holst M. J., McCammon J. A., Baker N. A., Improvements to the APBS biomolecular solvation software suite. Protein Sci. 27, 112–128 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/2/eabe0974/DC1