

Graphical Abstract:
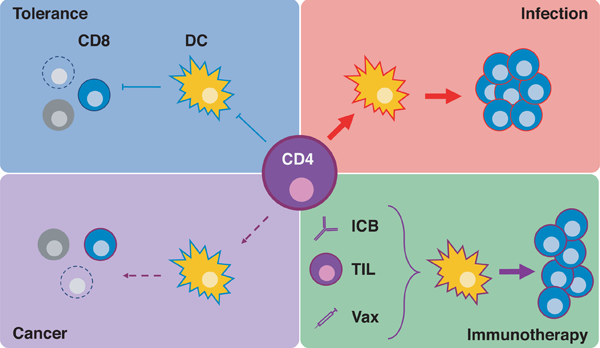
CD4+ T cells can either bolster or constrain CD8+ T cell responses by conditioning dendritic cells. In the context of anti-tumor immunity, the capacity of CD4+ T cells to do so is greatly diminished. Thus, in this review we will discuss how augmenting neoantigen-specific CD4+ T cells via immune checkpoint blockade (ICB), adoptive cellular therapy (ACT), and tumor-specific vaccines (Vax) could lead to robust CD8+ T cell responses.
Introduction
The idea that cells of the adaptive immune system—specifically of the T lineage—surveil, recognize, and eliminate cells expressing mutated-self antigens (neoantigens, NeoAg) lies at the heart of the current immunotherapy revolution in oncology. While these ideas are hardly new (1), our ability to clinically manipulate the immune system to elicit such potent antitumor response has only recently matured. Somatic mutations in the functional domains of key genes not only set in motion the transformation of normal cells into cancer but also serve as potential targets for T cells (2). The major avenues of cancer immunotherapy–immune checkpoint blockade (ICB), cancer vaccines, and adoptive cell therapy (ACT)—each focus on directing a cytotoxic T lymphocyte (CTL) response against the tumor; however, the role of helper CD4+ T cells in enhancing CTL function is often overlooked. In this review, we aim to highlight our current understanding and therapeutic value of CD4+ T cell help in cancer immunotherapy.
T helper immunity in the context of cancer immunotherapy
Helper T cells shape and orchestrate immune responses through direct cellular interactions and soluble factors. For example, direct TCR:MHCII interactions result in the selection of high affinity B cell clones in germinal centers via CD40-CD40L interactions (3). As antigen-presenting cells (APC), B cells can engage in this direct communication with CD4+ T cells. Similarly, CD4+ T cells help CTLs but through an APC intermediate. Older models suggested that CD4+ T cell cytokine production (particularly IL-2) in proximity to CD8+ T cells interacting with the same dendritic cell (DC) imparts the coveted help signal (4,5). However, numerous subsequent studies have upheld a dynamic, stepwise model involving coordinated cellular interactions. Following initial TCR:MHCII interactions, CD4+ T cells condition an APC via CD40-CD40L to provide proper costimulation to cognate CD8+ T cells reacting to a cross-presented antigen on the same APC (6–9). Recently, key studies have refined previous models and identified cellular interactions between different DC subsets and T cells that are spatiotemporally distinct (10). Specifically, incoming, antigen-loaded migratory DCs prime CD4+ T cells and transfer antigen to lymph node (LN)-resident DCs capable of efficient cross-presentation and CTL priming (11–13). While these studies primarily used viral infection models, the question of whether these same rules apply, differ, or are rendered defunct in the context of cancer immunity is an active area of research (14). Importantly, this inter-DC antigen transfer phenomenon was shown to be highly efficient and maintains peripheral tolerance (15–17). Thus, we propose that the paucity of presented NeoAgs relative to autoantigens and lack of pattern recognition receptor (PRR) and, thereby, innate immune system engagement, both contribute to the impaired initiation of a proper CTL response (Figure 1).
Figure 1. Context dependent CTL activation.

Migratory DCs capture antigen and traffic to the LN where they can present to CD4+ T cells and transfer antigen to LN-resident DCs. (A) In the case of self-antigens (blue), CD4+ T cells are not activated and thus LN-resident DCs capable of cross presentation are not licensed or conditioned to provide proper costimulation to potentially autoreactive CTLs, leading to no activation or AICD. (B) In the context of an acute pathogenic insult, abundant foreign antigen (red) and PRR engagement leads to CD4+ activation and proper conditioning of LN-resident DCs via CD40:CD40L interactions. Ultimately, cognate CD8+ T cells undergo robust expansion and memory formation due to optimal costimulation. (C) Rare tumor antigens (purple) relative to autoantigens (blue) and lack of PRR activation leads to incomplete costimulation. The resulting helpless or exhausted CTLs may be insufficient to control the tumor. Some CTLs might receive all necessary cues and form proper memory; however, the clonal diversity of the effective CTL response is dramatically decreased and may lead to tumor escape.
Without this highly choreographed dance between T cell and DC subsets, the consequences of a “helpless” CTL response include poor memory formation, secondary expansion, effector function, and survival (18–22). There exist a number of virulent infections which apparently do not require T help to generate a sufficient cytotoxic response, and in these cases, overzealous PRR activation has been thought to circumvent the requirements for help (9,23). However, in the context of a relatively non-inflammatory tumor, helpless cytotoxic responses are likely to be inadequate to control or eradicate the malignancy (Figure 1C). Strong evidence for the help requirement was demonstrated by seminal experiments in Rag2−/− mice: a significantly higher frequency of these mice developed sarcomas when exposed to the mutagen 3’-methylcholanthrene (MCA) than did their wildtype counterparts. Importantly, when tumors from Rag2−/− mice were transplanted into wildtype mice, many were spontaneously rejected in a CD4+ or CD8+ T cell dependent manner (24). This line of investigation established two key observations: 1) Both CD4+ and CD8+ T cells react and respond to cancer antigens and 2) the cytotoxic response alone is insufficient to control tumor progression. Thus, ICB, cancer vaccines, and ACT should aim to exploit the rules of proper T cell activation.
Which antigens do T cells recognize in cancer?
Tumor antigens can be broadly divided into two classes: self-antigens derive from proteins selectively expressed or overexpressed by tumor cells, and non-self-antigens deriving from mutated proteins, or NeoAg. Self-antigens include the tissue-specific cancer-testis antigens, such as MAGE-A, NY-ESO-1, and SSX-2, and lineage-specific proteins, such as MART-1, gp100, and tyrosinase (25). Given that these antigens arise from wildtype proteins, they are more likely to be shared between patients than NeoAg. Several clinical trials have been conducted investigating vaccines and cellular therapies targeting these shared tumor antigens. In one such trial, melanoma patients immunized with NY-ESO-1 peptides and CpG adjuvant mounted CD8+ and CD4+ T cell responses (26). Despite apparent antigen-specific T cell responses, clinical efficacy has been limited in patients with established tumors receiving these and similar vaccines. This lack of efficacy may be due to either the immunosuppressive tumor microenvironment or an inherent defect in the T cell repertoire recognizing these antigens. One preclinical study has demonstrated that the avidity of TCRs recognizing tumor self-antigens TRP-2 and gp100 are lower than those of TCRs recognizing a non-self, viral tumor antigen, resulting in worse tumor control after vaccination (27). This may be due to elimination of highly avid T cells recognizing tumor self-antigens during thymic selection. Indeed, another group has demonstrated that the tumor self-antigen carcinoembryonic antigen (CEA) is expressed by medullary thymic epithelial cells in CEA-transgenic mice, thus limiting the CD4+ T cell repertoire responding to CEA vaccination (28). In addition to concerns regarding efficacy, safety concerns abound with immunotherapies targeting wildtype antigens (29,30).
NeoAg are ideal targets for immunotherapy because they are exclusively expressed by malignant tissue, and because NeoAg-specific clones are likely to survive thymic selection or peripheral tolerance. Indeed, ACT with tumor infiltrating lymphocytes (TIL) harboring NeoAg specificities has induced tumor regression in patients with metastatic breast cancer and melanoma (31,32). Furthermore, patients with higher mutational burden malignancies (and therefore a greater diversity of NeoAg targets) experience a greater response rate to both TIL ACT and ICB (33–35). NeoAg-specific T cells have also been identified as effectors of antitumor immunity in mouse models (36–38). Specifically, NeoAg-specific T cell populations increased in frequency and a greater percentage produced effector cytokines in mice bearing MCA-induced sarcomas receiving ICB (37). This phenomenon has also been observed in patient responders to ICB (34).
Methods of NeoAg identification
In order to test immune recognition of putative NeoAg, tumor mutations within the protein coding region of the genome must first be identified. The advent of next generation sequencing (NGS) technology permits high-throughput sequencing of multiple tumor specimens and provides a comprehensive map of somatic mutations across a range of human malignancies (39). These data reveal that 10–1000s of nonsynonymous mutations exist in tumor tissue and could serve as targets for immune therapies. Accordingly, numerous strategies have been developed to identify NeoAg and test them for immunogenicity. To date, in silico prediction algorithms have been utilized to identify putative NeoAg peptides based on their calculated ability to bind MHC-I and II (40,41). These prediction algorithms have been a necessary first step for NeoAg identification for high mutation rate malignancies; however, this approach carries the risk that de facto NeoAg will be missed and therefore remain untested (42,43). Indeed, many predicted NeoAg are unable to generate detectable T cell responses in vivo after therapeutic vaccination (44,45). While additional bioinformatics packages exist that may be used to augment the performance of MHC prediction algorithms, a recent study from our colleagues in the Peters lab found that the addition of predictions for proteasomal cleavage, TAP transport, and MHC-peptide complex stability did not improve the predictive power of NetMHCpan—a widely used prediction algorithm for MHC I binding (46). Lastly, in a study from the Riddell group, mutations were selected for screening in an HLA agnostic manner (47). Instead, mutations were ranked based on their mean expression levels in The Cancer Genome Atlas for the given cancer type or expression as determined by RNA sequencing of a patient derived xenograft. The expression of mutated genes within a tumor is an important metric to consider given the influence of the transcriptome on antigen presentation (48). In a retrospective analysis they found that only one of three validated CD4+ T cell epitopes was predicted to bind its restricting MHC allele using NetMHCIIpan.
Alongside the development of algorithms for the prediction of NeoAg, experimental approaches have been investigated to verify presentation of putative NeoAg. Some groups have been able to identify NeoAg by eluting peptides from MHC molecules on the cell surface and subsequently using mass spectrometry to verify the presentation of mutant epitopes (43). However, such an approach will likely not be feasible for widespread clinical applications, at least not on a per-patient basis. Moreover, given that most tumors do not express MHC class II, NeoAg identified in this manner would be highly if not entirely biased towards class I epitopes. Rosenberg and colleagues have employed traditional functional assays such as ELISpot to identify NeoAg-specific responses among TIL and circulating T cells. In these assays, autologous APCs are either pulsed with 8–11mer peptides corresponding to mutant epitopes or transfected with tandem minigene thus allowing to screen both MHC class I and II, NeoAg-specific T cell responses (49). Given that CD4+ T cells predominantly recognize antigen derived from endocytosed dying cells or cell debris in vivo, the degree to which either peptide pulsing or transfection with tandem minigenes accurately reflects this process remains in question. So called “type B” CD4+ T cells have been identified in the context of autoimmune disease and recognize APCs directly pulsed with soluble peptides but are unable to recognize APCs incubated with the larger parent protein (50,51). To rule out such responses, which may be unproductive in vivo, surrogate assays for class II presentation should be employed. This can be accomplished by either feeding HLA-matched DCs irradiated or otherwise killed cell lines expressing the mutant proteins or employing in vitro transcribed RNA constructs linking the target antigen to a membrane trafficking sequence and transmembrane domain (52). In toto, future approaches should couple bioinformatic identification of putative NeoAgs in an HLA-agnostic manner followed by rigorous testing using standard immunoassays.
Help in Therapeutic Context
Immune Checkpoint Blockade
Currently, the most widely used immunotherapeutic strategy is ICB. Blockade of the inhibitory molecules CTLA4 and/or PD1/PDL1 results in the activation of a preexisting pool of NeoAg or tumor-specific T cells within the patient (53). Tumor regression in the context of PD1:PDL1 blockade presumably works via the transient reversal of CD8+ T cell exhaustion (54); however, the role of help in PD1 blockade efficacy is yet to be ascertained. Importantly, a new report reveals a de novo tumor-specific cytotoxic response following PD1 blockade in humans with basal or squamous cell carcinoma rather than the expansion of preexisting clonotypes (55). This phenomenon supports other recent findings indicating that T cells within the tumor microenvironment enter a state of epigenetically programmed, irreversible exhaustion (56,57). Thus, whether the recruitment of these novel CD8+ NeoAg specificities is dependent on help remains an open question.
In contrast to PD1, CTLA4 outcompetes CD28 for B7–1 and 2 (58,59), and thus imparts a strong inhibitory effect on T cell activation. Consequently, CTLA4 deficient mice succumb to a lymphoproliferative disease similar to FOXP3-deficient mice, indicating that CTLA4 is a key mediator of peripheral tolerance (60,61). In Nobel-worthy mouse studies, CTLA4 blockade lead to potent anti-tumor responses (62). Importantly, the antitumor response in certain tumor models sensitive to CTLA4 blockade was found to be CD4 dependent (63–65). Whether this dependence was due to help provided or Treg depletion remains controversial (66,67); however, recent studies have highlighted that in humans, CTLA4 therapy does not deplete Tregs but results in T cell activation (68,69). Despite the lack of clarity surrounding its effects on the natural course of a bona fide cytotoxic response, ICB’s clinical results are impressive especially in highly mutated tumors such as smoking induced lung cancer and melanoma (70,71).
The moniker of “exhaustion” was originally applied to CTLs, but the dysfunction that ICB aims to reverse also applies to helper T cells. Indeed, chronic antigen exposure drives CD4+ T cells into a dysfunctional state similar to the exhaustion phenotype seen in CD8+ T cells (72). Exhausted CD4+ and CD8+ T cells express many of the same coinhibitory receptors, but there is evidence in chronic viral infection models of biased expression (72). Specifically, dysfunctional CD4+ T cells appear to express higher levels of CTLA4 at late time points in their activation cycle than their CD8+ counterparts. This may partially explain why, in many murine cancer models, ICB with anti-CTLA4 alone or in combination with anti-PD1 induces the expansion of a TH1-like subset of CD4+ T cells (73,74). Whether this expansion is the result of a “rescue” of dysfunctional CD4+ T cells or the priming of novel clonotypes requires further investigation. Combination treatments employing NeoAg vaccines and ICB will likely yield optimal responses by overcoming exhaustion of newly primed clones. Such combinations may also lead to responses in patients with a lower mutational burden that would have previously been unresponsive to ICB alone.
Cancer Vaccines
Studies of NeoAg vaccines to date have identified a peculiar phenomenon: peptides selected in silico for their ability to bind MHC class I largely yield CD4+ T cell responses in vivo (45). Indeed, NeoAg-specific CD4+ T cells arise spontaneously in various human malignancies and are readily identifiable (32,75). The role of these NeoAg-specific CD4+ T cells, however, is incompletely understood. We propose that these cells have a dominant function as “helpers” for cytotoxic CD8+ T cells through the licensing of dendritic cells via CD40/CD40L interactions (7,76). CD8+ T cells primed in the absence of CD4+ T cell help are unable to undergo secondary expansion and instead are subject to activation-induced cell death (AICD) (18). It therefore stands to reason that CD4+ T cell epitopes are a crucial component of effective NeoAg vaccination (Figure 2A). It has been demonstrated that the inclusion of “helper” epitopes in therapeutic cancer vaccines improves the antitumor response by increasing the expansion of CD8+ T cells, as well as reducing their expression of coinhibitory receptors and increasing their migratory potential (77).
Figure 2. Help-centric therapeutic avenues.
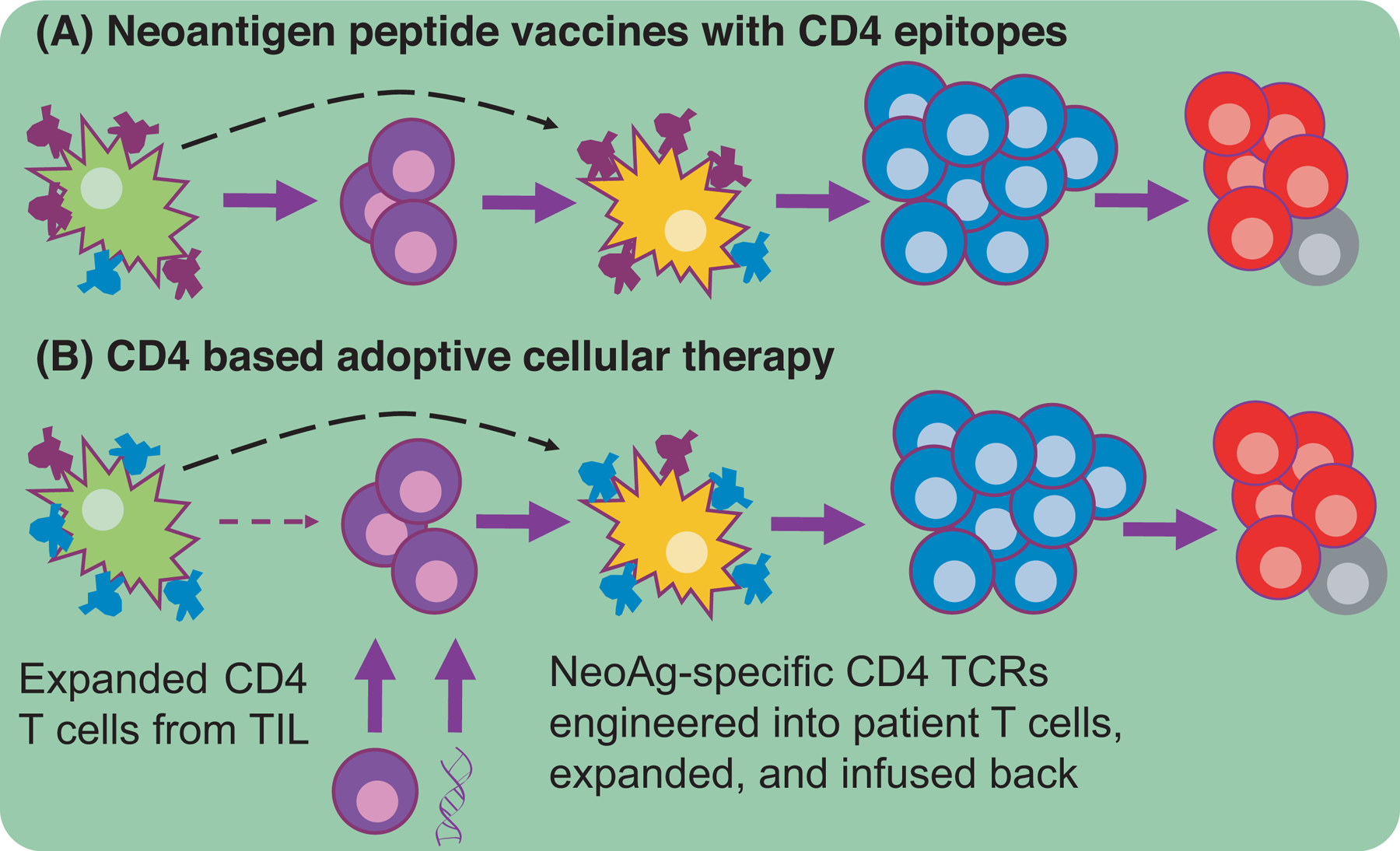
A major issue with anti-tumor immunity is the paucity of NeoAgs available for cross-presentation. (A) Peptide vaccines containing CD4-NeoAg epitopes (purple) increase the amount of NeoAg migratory DCs carry to LNs where activated CD4+ T cells will subsequently result in LN-resident DC conditioning, proper CTL priming, robust CTL expansion, and memory formation. (B) An alternative therapeutic modality functions via the expansion of CD4+ T cells found in the TIL of a patient or the genetic introduction of NeoAg-specificity. Both ACT strategies circumscribe the need for migratory DC to carry NeoAgs to the LN. The expanded helper cells will presumably condition the resident DC and ultimately lead to polyfunctional CTLs.
In addition to aiding in the productive priming of CD8+ T cells in secondary lymphoid organs, effector roles of CD4+ T cells in the tumor microenvironment have been proposed. These include the activation of local NK cells via secretion of effector cytokines, recruitment of CD8+ T cells by IFNγ-inducible chemokines such as CXCL-10, and even class-II dependent killing of tumor cells (78–80). In a recent publication from the Schreiber laboratory, KPC (LSL-KrasG12D/+; LSL-Trp53R172H/+; Pdx-1-Cre) tumors lacking natural NeoAg were transduced to express a single MHC-I-restricted NeoAg with or without an additional MHC-II-restricted NeoAg (81). Contralateral injection resulted in a significant increase in the numbers of antigen-specific CD8+ T cells, total CD8+ and CD4+ T cells, and iNOS+ macrophages infiltrating the MHC-II NeoAg-expressing tumor. Importantly, KPC tumors not expressing MHC-II NeoAg continued to grow indicating that CD4+ T cell effectors must recognize antigen locally to mediate their antitumor effects. Furthermore, another recent study has demonstrated that across a range of solid tumor types MHC-II expression is rare among tumor cells, which implies that the local effects of CD4+ T cells previously mentioned are likely dependent on antigen recognition in the context of APCs and stromal cells (82). Overall, these and other effector roles of CD4+ T cells require further exploration. If the dominant role of CD4+ T cells in the antitumor immune response is the provision of help, NeoAg vaccines can simply include promiscuous helper epitopes such as the pan-DR binding epitope (83). If, however, there is an additional role for tumor-specific CD4+ within the tumor, class II NeoAg must be identified and targeted specifically.
Adoptive Cellular Therapy
The promise of ACT is that it provides anti-tumor specificities that were never present or are exceeding rare to be beneficial. Broadly, ACT can be divided into 3 categories based on the cellular product delivered: Chimeric antigen receptor (CAR) T cells, TIL, and TCR engineered T cells. While TIL and TCR engineered T cells recognize antigen by conventional peptide-MHC interactions, CAR T cells target cell lineage determining antigens such as CD19 via the introduction of antibody specificity linked to intracellular T cell activating signal motifs into autologous T cells (84–86).
CART-19 therapy has vastly improved overall survival in certain B cell lymphomas relative to past standard of care (87,88), but both normal and malignant B cells are eliminated. While CAR T cells to date target cell surface proteins, TCR “mimic” (TCRm) antibodies have been described that bind tumor associated antigens in the context of HLA (89). Thus, NeoAg-reactive TCRm antibodies could be used with current CAR technology to provide specificity against NeoAgs in the near future. Surprisingly, recent mouse studies have highlighted that CD4+ CAR T cells alone mediate superior antitumor activity relative to their CD8+ counterparts (90–93). While the cytotoxic capacity of CD4+ T cells in tumor models is hardly novel (80,94–96), in the context of CAR T therapy, CD4+ T cells also mediate tumor cytotoxicity directly, resist AICD, do not help CTLs resist exhaustion, and are in fact impeded by their CD8+ compatriots (90). Future CAR trials should test whether enriched CD4+ T cells are similarly superior in humans as they are in mice.
TIL therapy expands a preexisting pool of T cells isolated from the patient’s tumor. Tumor tissue is cultured with high concentrations of IL-2 to allow for TIL extravasation and growth and expanded TIL cultures are then infused back into the patient (97). While NeoAg-specific T cells are found within TIL, so too are T cells with irrelevant specificities (98). We propose that selection and expansion of TIL cultures with a high frequency of NeoAg-specific T cells as assessed by the methods previously described holds the most promise for effective cancer therapy (99) (Figure 2B). Apart from tumor specificity, several studies have correlated higher objective responses with a higher proportion of CD8+ T cells within TIL products in melanoma patients (100,101); however, a randomized control trial failed to prove this hypothesis with a CD8+ T cell enrichment step (102). Thus, understanding the relative contributions of CD4+ or CD8+ T cells with TIL studies is incomplete.
Specific anticancer responses may be achieved by engineering patient autologous T cells from blood with NeoAg-specific TCRs. Indeed, T cells have been engineered to express TCRs targeting tumor self-antigens using retroviral and more recently CRISPR based methodologies capable of simultaneously disrupting the endogenous TCR (103,104). This approach may require lower total numbers of T cells for ACT given that the frequency of engineered tumor-specific cells will be much higher than in TIL. In addition, T cell clones within TIL may be dysfunctional as a consequence of the immunosuppressive tumor microenvironment and chronic antigen stimulation (56,57). Therefore, engineered “fresh” T cells may have enhanced antitumor function on a per cell basis. Moreover, some tumor-specific TCRs are of such high affinity that they can recognize antigen in a co-receptor independent manner (105). Since CD4+ T cells can now recognize NeoAg directly on tumors via MHC class I, the consequences of these interactions will be an important avenue of investigation. A clinical trial investigating TCR engineering with NeoAg-specific TCRs targeting metastatic disease in a range of malignancies is currently underway at the National Cancer Institute (NCT03412877).
Although efficacy endpoints for CD4+ T cells in ACT thus far focus on direct tumor cytotoxicity, assessing how ACT functions through the help paradigm will be informative (Figure 2B). The value of tumor-specific CD4+ T cells in the context of ACT has been demonstrated in numerous mouse models implicating a downstream role for macrophages, NK cells, and CTLs (95,96). Compelling clinical evidence exists in which a patient receiving TIL therapy containing a BRAFV600E-specific CD4+ clone experienced complete remission (32). In this case, the researchers identified the subsequent expansion of CD8+ T cells recognizing several tumor-associated antigens, which may not have been primed in the absence of this expanded and activated CD4+ clone (Figure 2B). Whether similar results can be obtained using NeoAg-specific CD4+ TCR engineering remains unknown.
Lastly, many studies have indicated that a TH1 phenotype is preferable for ACT; however, recent work indicates that TH17 polarized CD4+ T cells are able to exert superior antitumor immunity over TH1 cells by resisting apoptosis and senescence (106). Importantly, tumor destruction still depends on the TH1 cytokine IFNγ and CD8+ T cells. Adoptively transferred TH17 cells also may have a greater capacity to recruit DC subsets, promote CD8+ T cell differentiation directly via IL-17, and ultimately control large tumors (107,108). Given that other studies have identified IL-17 and related gene signatures as negative prognostic markers in non-small cell lung and colorectal cancers, therapeutic engagement of TH17 CD4+ T cells requires further investigation (109,110).
Considerations for NeoAg-specific therapies
Personalized NeoAg vaccines tested thus far have demonstrated cautiously encouraging clinical responses that can be improved to provide a less costly alternative to ACT with either TIL or engineered T cells. RNA- and peptide-based NeoAg vaccines in melanoma and glioblastoma have induced the expansion of T cells recognizing targeted epitopes (44,111,112). Patients receiving RNA-based multiepitope vaccines experienced significantly reduced incidence of metastatic events compared to pretreatment (112). We echo what many have already shown and proposed: in order to achieve optimal CD8+ T cell priming, NeoAg vaccines must include CD4+ helper epitopes (111,113,114) (Figure 2A). We also speculate that these epitopes should be physically linked to ensure their delivery to the same APC, given that cross presentation is dependent on CD4+ T cell help (115). In the case of multimer nucleic acid vaccines, a single vector encoding both CD8+ and CD4+ epitopes would ensure their expression within the same cell. For peptide vaccines, a flexible linking moiety joining these epitopes or nanoparticles should be considered for efficient codelivery. This “linker” strategy ensures that both helper and CTL neoepitopes will be deposited and transferred to appropriate DC subsets within the draining LNs thus maximizing the cytotoxic response (10–13). Lastly, attention must be given to proper adjuvant selection in order to drive a potent CTL response. Failure to induce a TH1 and/or TH17 response can result not only in poor anti-tumor cytotoxicity but also tolerize the immune response to the malignancy (116–118). The nuances of proper cancer adjuvant selection are beyond the scope of this review but are well summarized elsewhere (119).
In addition to including epitopes recognized by both CD8+ and CD4+ T cells, epitopes across multiple HLA alleles should be targeted to reduce the chance of immune escape by loss of heterozygosity (LOH), which is now a well-documented phenomenon in patients experiencing relapse after immunotherapy (111,120,121). We speculate that this may be an inherent benefit of CD4+ T cell-directed therapies because help functions through antigen presented by APCs and not the tumor cells themselves (122). Moreover, CD4+ T cells can direct a diverse CTL response through a variety of MHCI alleles rather than one in addition to recruiting NK cells and macrophages, thus trapping the tumor from mutating further. Indeed, the same considerations apply for TCR engineering. Whereas the majority of early studies have prioritized single TCRs recognizing targets in the context of the common HLA-A*02:01 allele (of North American and European Caucasian populations), only recently have investigators searched for TCRs restricted to other common alleles (123). Priority targeting of known driver mutations, which are more likely to be clonal “trunk” mutations, will be advantageous so as to limit immune escape from antigen loss or incomplete clearance due to preexisting intratumor heterogeneity (124–126). A final consideration for effective NeoAg-specific therapies is that of optimal TCR avidity. Studies employing minimally altered tumor epitopes to model varying TCR-MHC interaction strengths have demonstrated that vaccination with intermediate affinity epitopes provides optimal tumor control, while the highest affinity epitope vaccinations result in a dysfunctional T cell response (127). Few published studies exist defining the rules of CD4+ TCR affinity in the context of cancer. We would speculate that, similar to CD8+ T cells, higher affinity TCR-MHC interactions in CD4+ T cells may lead to dysfunctional responses, but this hypothesis must be experimentally tested.
Concluding Remarks
NeoAg-specific therapies represent perhaps the most advanced foray into personalized medicine for cancer. While NeoAg prediction methods require further investigation in a broader range of patient cohorts, we are beginning to learn which approaches are optimal for which contexts. Additionally, we are uncovering roles for CD4+ T cells that may inform the next wave of therapies specifically designed to target this cell type. By employing appropriate methods for detection and validation along with rational design of both vaccination and cellular therapies, we can improve upon the already impressive recent track record of immunotherapies against cancer.
Acknowledgments:
Joseph Dolina for lively discussion on all matters related to help.
Abbreviations:
- NeoAg
Neoantigen
- TCR
T Cell Receptor
- CTL
Cytotoxic T Lymphocyte
- MHC
Major Histocompatability Complex
- HLA
Human Leukocyte Antigen
- APC
Antigen Presenting Cell
- DC
Dendritic Cell
- ICB
Immune Checkpoint Blockade
- ACT
Adoptive Cellular Therapy
- AICD
Activation-Induced Cell Death
- PRR
Pattern Recognition Receptor
Footnotes
Conflict of Interest Disclosure: None
REFERENCES:
- 1.Burnet FM. The concept of immunological surveillance. Prog Exp Tumor Res. 1970;13:1–27. [DOI] [PubMed] [Google Scholar]
- 2.Tyzzer EE. Tumor Immunity. The Journal of Cancer Research. American Association for Cancer Research Journals; 1916. April 1;1(2):125–56. [Google Scholar]
- 3.Bannard O, Cyster JG. Germinal centers: programmed for affinity maturation and antibody diversification. Current Opinion in Immunology. 2017. April;45:21–30. [DOI] [PubMed] [Google Scholar]
- 4.Cassell D, Forman J. Linked recognition of helper and cytotoxic antigenic determinants for the generation of cytotoxic T lymphocytes Ann N Y Acad Sci. John Wiley & Sons, Ltd (10.1111); 1988;532(1 Cytotoxic T C):51–60. [DOI] [PubMed] [Google Scholar]
- 5.Keene JA, Forman J. Helper activity is required for the in vivo generation of cytotoxic T lymphocytes. J Exp Med. 1982. March 1;155(3):768–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Toes RE, Schoenberger SP, van der Voort EI, Offringa R, Melief CJ. CD40-CD40Ligand interactions and their role in cytotoxic T lymphocyte priming and anti-tumor immunity. Semin Immunol. 1998. December;10(6):443–8. [DOI] [PubMed] [Google Scholar]
- 7.Schoenberger SP, Toes RE, van der Voort EI, Offringa R, Melief CJ. T-cell help for cytotoxic T lymphocytes is mediated by CD40-CD40L interactions Nature. Nature Publishing Group; 1998. June 4;393(6684):480–3. [DOI] [PubMed] [Google Scholar]
- 8.Bennett SR, Carbone FR, Karamalis F, Flavell RA, Miller JF, Heath WR. Help for cytotoxic-T-cell responses is mediated by CD40 signalling Nature. Nature Publishing Group; 1998. June 4;393(6684):478–80. [DOI] [PubMed] [Google Scholar]
- 9.Ridge JP, Di Rosa F, Matzinger P. A conditioned dendritic cell can be a temporal bridge between a CD4+ T-helper and a T-killer cell Nature. Nature Publishing Group; 1998. June 4;393(6684):474–8. [DOI] [PubMed] [Google Scholar]
- 10.Borst J, Ahrends T, Bąbała N, Melief CJM, Kastenmüller W. CD4+ T cell help in cancer immunology and immunotherapy Nature Reviews Immunology. Nature Publishing Group; 2018. October;18(10):635–47. [DOI] [PubMed] [Google Scholar]
- 11.Allan RS, Waithman J, Bedoui S, Jones CM, Villadangos JA, Zhan Y, et al. Migratory dendritic cells transfer antigen to a lymph node-resident dendritic cell population for efficient CTL priming. Immunity. 2006. July;25(1):153–62. [DOI] [PubMed] [Google Scholar]
- 12.Hor JL, Whitney PG, Zaid A, Brooks AG, Heath WR, Mueller SN. Spatiotemporally Distinct Interactions with Dendritic Cell Subsets Facilitates CD4+ and CD8+ T Cell Activation to Localized Viral Infection. Immunity. 2015. September 15;43(3):554–65. [DOI] [PubMed] [Google Scholar]
- 13.Eickhoff S, Brewitz A, Gerner MY, Klauschen F, Komander K, Hemmi H, et al. Robust Anti-viral Immunity Requires Multiple Distinct T Cell-Dendritic Cell Interactions. Cell. 2015. September 10;162(6):1322–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Böttcher JP, Reis E Sousa C. The Role of Type 1 Conventional Dendritic Cells in Cancer Immunity. Trends Cancer. 2018. November;4(11):784–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Inaba K, Turley S, Yamaide F, Iyoda T, Mahnke K, Inaba M, et al. Efficient presentation of phagocytosed cellular fragments on the major histocompatibility complex class II products of dendritic cells. J Exp Med. 1998. December 7;188(11):2163–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scheinecker C, McHugh R, Shevach EM, Germain RN. Constitutive presentation of a natural tissue autoantigen exclusively by dendritic cells in the draining lymph node. J Exp Med. 2002. October 21;196(8):1079–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Turley S, Poirot L, Hattori M, Benoist C, Mathis D. Physiological beta cell death triggers priming of self-reactive T cells by dendritic cells in a type-1 diabetes model. J Exp Med. 2003. November 17;198(10):1527–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Janssen EM, Lemmens EE, Wolfe T, Christen U, Herrath von MG, Schoenberger SP. CD4+ T cells are required for secondary expansion and memory in CD8+ T lymphocytes Nature. Nature Publishing Group; 2003. February 20;421(6925):852–6. [DOI] [PubMed] [Google Scholar]
- 19.Sun JC, Bevan MJ. Defective CD8 T cell memory following acute infection without CD4 T cell help. Science. 2003. April 11;300(5617):339–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun JC, Williams MA, Bevan MJ. CD4+ T cells are required for the maintenance, not programming, of memory CD8+ T cells after acute infection Nat Immunol. Nature Publishing Group; 2004. September;5(9):927–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shedlock DJ, Shen H. Requirement for CD4 T cell help in generating functional CD8 T cell memory. Science. 2003. April 11;300(5617):337–9. [DOI] [PubMed] [Google Scholar]
- 22.Novy P, Quigley M, Huang X, Yang Y. CD4 T cells are required for CD8 T cell survival during both primary and memory recall responses. JI. 2007. December 15;179(12):8243–51. [DOI] [PubMed] [Google Scholar]
- 23.Bevan MJ. Helping the CD8(+) T-cell response Nature Reviews Immunology. Nature Publishing Group; 2004. August;4(8):595–602. [DOI] [PubMed] [Google Scholar]
- 24.Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. IFNgamma and lymphocytes prevent primary tumour development and shape tumour immunogenicity Nature. Nature Publishing Group; 2001. April 26;410(6832):1107–11. [DOI] [PubMed] [Google Scholar]
- 25.Lee C-H, Yelensky R, Jooss K, Chan TA. Update on Tumor Neoantigens and Their Utility: Why It Is Good to Be Different. Trends Immunol. 2018. July;39(7):536–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Baumgaertner P, Costa Nunes C, Cachot A, Maby-El Hajjami H, Cagnon L, Braun M, et al. Vaccination of stage III/IV melanoma patients with long NY-ESO-1 peptide and CpG-B elicits robust CD8+ and CD4+ T-cell responses with multiple specificities including a novel DR7-restricted epitope Oncoimmunology. Taylor & Francis; 2016;5(10):e1216290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pedersen SR, Sørensen MR, Buus S, Christensen JP, Thomsen AR. Comparison of vaccine-induced effector CD8 T cell responses directed against self- and non-self-tumor antigens: implications for cancer immunotherapy. J Immunol. 2013. October 1;191(7):3955–67. [DOI] [PubMed] [Google Scholar]
- 28.Bos R, van Duikeren S, van Hall T, Kaaijk P, Taubert R, Kyewski B, et al. Expression of a natural tumor antigen by thymic epithelial cells impairs the tumor-protective CD4+ T-cell repertoire. Cancer Res. 2005. July 15;65(14):6443–9. [DOI] [PubMed] [Google Scholar]
- 29.Morgan RA, Chinnasamy N, Abate-Daga D, Gros A, Robbins PF, Zheng Z, et al. Cancer regression and neurological toxicity following anti-MAGE-A3 TCR gene therapy. J Immunother. 2013. February;36(2):133–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Parkhurst MR, Yang JC, Langan RC, Dudley ME, Nathan D-AN, Feldman SA, et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol Ther. 2011. March;19(3):620–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zacharakis N, Chinnasamy H, Black M, Xu H, Lu Y-C, Zheng Z, et al. Immune recognition of somatic mutations leading to complete durable regression in metastatic breast cancer Nature Medicine. Nature Publishing Group; 2018. June;24(6):724–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Veatch JR, Lee SM, Fitzgibbon M, Chow I-T, Jesernig B, Schmitt T, et al. Tumor-infiltrating BRAFV600E-specific CD4+ T cells correlated with complete clinical response in melanoma J Clin Invest. American Society for Clinical Investigation; 2018. April 2;128(4):1563–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lauss M, Donia M, Harbst K, Andersen R, Mitra S, Rosengren F, et al. Mutational and putative neoantigen load predict clinical benefit of adoptive T cell therapy in melanoma Nat Commun. Nature Publishing Group; 2017. November 23;8(1):1738–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic basis for clinical response to CTLA-4 blockade in melanoma N Engl J Med. Massachusetts Medical Society; 2014. December 4;371(23):2189–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Rooij N, van Buuren MM, Philips D, Velds A, Toebes M, Heemskerk B, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. J Clin Oncol. 2013. November 10;31(32):e439–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature Publishing Group. Nature Publishing Group; 2012. February 8;482(7385):400–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature Publishing Group. Nature Publishing Group; 2014. November 27;515(7528):577–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fehlings M, Simoni Y, Penny HL, Becht E, Loh CY, Gubin MM, et al. Checkpoint blockade immunotherapy reshapes the high-dimensional phenotypic heterogeneity of murine intratumoural neoantigen-specific CD8+ T cells Nat Commun. Nature Publishing Group; 2017. September 15;8(1):562–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature Publishing Group. Nature Publishing Group; 2013. August 22;500(7463):415–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lundegaard C, Lamberth K, Harndahl M, Buus S, Lund O, Nielsen M. NetMHC-3.0: accurate web accessible predictions of human, mouse and monkey MHC class I affinities for peptides of length 8–11. Nucleic Acids Res. 2008. July 1;36(Web Server issue):W509–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vita R, Overton JA, Greenbaum JA, Ponomarenko J, Clark JD, Cantrell JR, et al. The immune epitope database (IEDB) 3.0. Nucleic Acids Res. 2015. January;43(Database issue):D405–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bjerregaard A-M, Nielsen M, Jurtz V, Barra CM, Hadrup SR, Szallasi Z, et al. An Analysis of Natural T Cell Responses to Predicted Tumor Neoepitopes Front Immunol. Frontiers; 2017;8:1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yadav M, Jhunjhunwala S, Phung QT, Lupardus P, Tanguay J, Bumbaca S, et al. Predicting immunogenic tumour mutations by combining mass spectrometry and exome sequencing. Nature Publishing Group. Nature Publishing Group; 2014. November 27;515(7528):572–6. [DOI] [PubMed] [Google Scholar]
- 44.Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature Publishing Group. Nature Publishing Group; 2017. July 5;:1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kreiter S, Vormehr M, van de Roemer N, Diken M, Löwer M, Diekmann J, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature Publishing Group. Nature Publishing Group; 2015. April 30;520(7549):692–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koşaloğlu-Yalçın Z, Lanka M, Frentzen A, Logandha Ramamoorthy Premlal A, Sidney J, Vaughan K, et al. Predicting T cell recognition of MHC class I restricted neoepitopes Oncoimmunology. Taylor & Francis; 2018;7(11):e1492508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Veatch JR, Jesernig BL, Kargl J, Fitzgibbon M, Lee SM, Baik C, et al. Endogenous CD4+ T Cells Recognize Neoantigens in Lung Cancer Patients, Including Recurrent Oncogenic KRAS and ERBB2 (Her2) Driver Mutations. Cancer Immunol Res. 2019. June;7(6):910–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fortier M-H, Caron E, Hardy M-P, Voisin G, Lemieux S, Perreault C, et al. The MHC class I peptide repertoire is molded by the transcriptome. J Exp Med. 2008. March 17;205(3):595–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lu Y-C, Yao X, Crystal JS, Li YF, El-Gamil M, Gross C, et al. Efficient identification of mutated cancer antigens recognized by T cells associated with durable tumor regressions. Clin Cancer Res. 2014. July 1;20(13):3401–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mohan JF, Unanue ER. Unconventional recognition of peptides by T cells and the implications for autoimmunity. Nature Reviews Immunology. Nature Publishing Group; 2012. October;12(10):721–8. [DOI] [PubMed] [Google Scholar]
- 51.Mohan JF, Levisetti MG, Calderon B, Herzog JW, Petzold SJ, Unanue ER. Unique autoreactive T cells recognize insulin peptides generated within the islets of Langerhans in autoimmune diabetes Nat Immunol. Nature Publishing Group; 2010. April;11(4):350–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kreiter S, Selmi A, Diken M, Sebastian M, Osterloh P, Schild H, et al. Increased Antigen Presentation Efficiency by Coupling Antigens to MHC Class I Trafficking Signals. JI. 2007. December 20;180(1):309–18. [DOI] [PubMed] [Google Scholar]
- 53.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy Nat Rev Cancer. Nature Publishing Group; 2012. March 22;12(4):252–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sharpe AH, Pauken KE. The diverse functions of the PD1 inhibitory pathway Nature Reviews Immunology. Nature Publishing Group; 2018. March;18(3):153–67. [DOI] [PubMed] [Google Scholar]
- 55.Yost KE, Satpathy AT, Wells DK, Qi Y, Wang C, Kageyama R, et al. Clonal replacement of tumor-specific T cells following PD-1 blockade Nature Medicine. Nature Publishing Group; 2019. August;25(8):1251–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schietinger A, Philip M, Krisnawan VE, Chiu EY, Delrow JJ, Basom RS, et al. Tumor-Specific T Cell Dysfunction Is a Dynamic Antigen-Driven Differentiation Program Initiated Early during Tumorigenesis. Immunity. 2016. August 16;45(2):389–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature Publishing Group. Nature Publishing Group; 2017. May 25;545(7655):452–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Collins AV, Brodie DW, Gilbert RJC, Iaboni A, Manso-Sancho R, Walse B, et al. The interaction properties of costimulatory molecules revisited. Immunity. 2002. August;17(2):201–10. [DOI] [PubMed] [Google Scholar]
- 59.van der Merwe PA, Bodian DL, Daenke S, Linsley P, Davis SJ. CD80 (B7–1) binds both CD28 and CTLA-4 with a low affinity and very fast kinetics. J Exp Med. 1997. February 3;185(3):393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995. November 10;270(5238):985–8. [DOI] [PubMed] [Google Scholar]
- 61.Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995. November;3(5):541–7. [DOI] [PubMed] [Google Scholar]
- 62.Leach DR, Krummel MF, Allison JP. Enhancement of antitumor immunity by CTLA-4 blockade. Science. 1996. March 22;271(5256):1734–6. [DOI] [PubMed] [Google Scholar]
- 63.Demaria S, Kawashima N, Yang AM, Devitt ML, Babb JS, Allison JP, et al. Immune-mediated inhibition of metastases after treatment with local radiation and CTLA-4 blockade in a mouse model of breast cancer. Clin Cancer Res. 2005. January 15;11(2 Pt 1):728–34. [PubMed] [Google Scholar]
- 64.Paradis TJ, Floyd E, Burkwit J, Cole SH, Brunson B, Elliott E, et al. The anti-tumor activity of anti-CTLA-4 is mediated through its induction of IFN gamma. Cancer Immunol Immunother. 2001. May;50(3):125–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shrikant P, Khoruts A, Mescher MF. CTLA-4 blockade reverses CD8+ T cell tolerance to tumor by a CD4+ T cell- and IL-2-dependent mechanism. Immunity. 1999. October;11(4):483–93. [DOI] [PubMed] [Google Scholar]
- 66.Simpson TR, Li F, Montalvo-Ortiz W, Sepulveda MA, Bergerhoff K, Arce F, et al. Fc-dependent depletion of tumor-infiltrating regulatory T cells co-defines the efficacy of anti-CTLA-4 therapy against melanoma. J Exp Med. 2013. August 26;210(9):1695–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, et al. Anti-CTLA-4 antibodies of IgG2a isotype enhance antitumor activity through reduction of intratumoral regulatory T cells. Cancer Immunol Res. 2013. July;1(1):32–42. [DOI] [PubMed] [Google Scholar]
- 68.Maker AV, Attia P, Rosenberg SA. Analysis of the cellular mechanism of antitumor responses and autoimmunity in patients treated with CTLA-4 blockade. JI. 2005. December 1;175(11):7746–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sharma A, Subudhi SK, Blando J, Scutti J, Vence L, Wargo J, et al. Anti-CTLA-4 Immunotherapy Does Not Deplete FOXP3+ Regulatory T Cells (Tregs) in Human Cancers. Clin Cancer Res. 2019. February 15;25(4):1233–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob J-J, Cowey CL, et al. Overall Survival with Combined Nivolumab and Ipilimumab in Advanced Melanoma N Engl J Med. Massachusetts Medical Society; 2017. October 5;377(14):1345–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015. April 3;348(6230):124–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Crawford A, Angelosanto JM, Kao C, Doering TA, Odorizzi PM, Barnett BE, et al. Molecular and Transcriptional Basis of CD4+ T Cell Dysfunction during Chronic Infection Immunity. Elsevier Inc; 2014. February 20;40(2):289–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.PPG AR. Dual PD-1 and CTLA-4 Checkpoint Blockade Promotes Antitumor Immune Responses through CD4. 2018. August 16;:1–14. [DOI] [PubMed]
- 74.Wei SC, Levine JH, Cogdill AP, Zhao Y, Anang N-AAS, Andrews MC, et al. Distinct Cellular Mechanisms Underlie Anti-CTLA-4 and Anti-PD-1 Checkpoint Blockade Cell. Elsevier Inc; 2017. September 7;170(6):1120–1125.e17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Linnemann C, van Buuren MM, Bies L, Verdegaal EME, Schotte R, Calis JJA, et al. High-throughput epitope discovery reveals frequent recognition of neoantigens by CD4+ T cells in human melanoma Nature Medicine. Nature Publishing Group; 2015. January;21(1):81–5. [DOI] [PubMed] [Google Scholar]
- 76.Feau S, Garcia Z, Arens R, Yagita H, Borst J, Schoenberger SP. The CD4+ T-cell help signal is transmitted from APC to CD8+ T-cells via CD27-CD70 interactions Nat Commun. Nature Publishing Group; 2012. July 10;3(1):948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ahrends T, Spanjaard A, Pilzecker B, Bąbała N, Bovens A, Xiao Y, et al. CD4+ T Cell Help Confers a Cytotoxic T Cell Effector Program Including Coinhibitory Receptor Downregulation and Increased Tissue Invasiveness. Immunity. 2017. November 21;47(5):848–861.e5. [DOI] [PubMed] [Google Scholar]
- 78.Doorduijn EM, Sluijter M, Salvatori DC, Silvestri S, Maas S, Arens R, et al. CD4+ T Cell and NK Cell Interplay Key to Regression of MHC Class Ilow Tumors upon TLR7/8 Agonist Therapy Cancer Immunol Res. American Association for Cancer Research; 2017. August;5(8):642–53. [DOI] [PubMed] [Google Scholar]
- 79.Sato Y, Bolzenius JK, Eteleeb AM, Su X, Maher CA, Sehn JK, et al. CD4+ T cells induce rejection of urothelial tumors after immune checkpoint blockade JCI Insight. American Society for Clinical Investigation; 2018. December 6;3(23):1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Quezada SA, Simpson TR, Peggs KS, Merghoub T, Vider J, Fan X, et al. Tumor-reactive CD4(+) T cells develop cytotoxic activity and eradicate large established melanoma after transfer into lymphopenic hosts. J Exp Med. 2010. March 15;207(3):637–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Alspach E, Lussier DM, Miceli AP, Kizhvatov I, DuPage M, Luoma AM, et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature Publishing Group; Springer US; 2019. October 22;:1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Abelin JG, Harjanto D, Malloy M, Suri P, Colson T, Goulding SP, et al. Defining HLA-II Ligand Processing and Binding Rules with Mass Spectrometry Enhances Cancer Epitope Prediction Immunity. Elsevier Inc; 2019. August 30;:1–32. [DOI] [PubMed] [Google Scholar]
- 83.Alexander J, Sidney J, Southwood S, Ruppert J, Oseroff C, Maewal A, et al. Development of high potency universal DR-restricted helper epitopes by modification of high affinity DR-blocking peptides. Immunity. 1994. December;1(9):751–61. [DOI] [PubMed] [Google Scholar]
- 84.Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015. April 3;348(6230):62–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.June CH, Sadelain M. Chimeric Antigen Receptor Therapy N Engl J Med. Massachusetts Medical Society; 2018. July 5;379(1):64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hinrichs CS, Rosenberg SA. Exploiting the curative potential of adoptive T-cell therapy for cancer Immunological Reviews. Wiley/Blackwell (10.1111); 2014. January;257(1):56–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Neelapu SS, Locke FL, Bartlett NL, Lekakis LJ, Miklos DB, Jacobson CA, et al. Axicabtagene Ciloleucel CAR T-Cell Therapy in Refractory Large B-Cell Lymphoma N Engl J Med. Massachusetts Medical Society; 2017. December 28;377(26):2531–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Crump M, Neelapu SS, Farooq U, Van Den Neste E, Kuruvilla J, Westin J, et al. Outcomes in refractory diffuse large B-cell lymphoma: results from the international SCHOLAR-1 study. Blood. 2017. October 19;130(16):1800–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Trenevska I, Li D, Banham AH. Therapeutic Antibodies against Intracellular Tumor Antigens. Front Immunol. 2017;8:1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Wang D, Aguilar B, Starr R, Alizadeh D, Brito A, Sarkissian A, et al. Glioblastoma-targeted CD4+ CAR T cells mediate superior antitumor activity JCI Insight. American Society for Clinical Investigation; 2018. May 17;3(10):985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sommermeyer D, Hudecek M, Kosasih PL, Gogishvili T, Maloney DG, Turtle CJ, et al. Chimeric antigen receptor-modified T cells derived from defined CD8+ and CD4+ subsets confer superior antitumor reactivity in vivo Leukemia. Nature Publishing Group; 2016. February;30(2):492–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Turtle CJ, Hanafi L-A, Berger C, Hudecek M, Pender B, Robinson E, et al. Immunotherapy of non-Hodgkin’s lymphoma with a defined ratio of CD8+ and CD4+ CD19-specific chimeric antigen receptor-modified T cells. Sci Transl Med. 2016. September 7;8(355):355ra116–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Turtle CJ, Hanafi L-A, Berger C, Gooley TA, Cherian S, Hudecek M, et al. CD19 CAR-T cells of defined CD4+:CD8+ composition in adult B cell ALL patients J Clin Invest. American Society for Clinical Investigation; 2016. June 1;126(6):2123–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Xie Y, Akpinarli A, Maris C, Hipkiss EL, Lane M, Kwon E-KM, et al. Naive tumor-specific CD4(+) T cells differentiated in vivo eradicate established melanoma. J Exp Med. 2010. March 15;207(3):651–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Corthay A, Skovseth DK, Lundin KU, Røsjø E, Omholt H, Hofgaard PO, et al. Primary antitumor immune response mediated by CD4+ T cells. Immunity. 2005. March;22(3):371–83. [DOI] [PubMed] [Google Scholar]
- 96.Perez-Diez A, Joncker NT, Choi K, Chan WFN, Anderson CC, Lantz O, et al. CD4 cells can be more efficient at tumor rejection than CD8 cells Blood. American Society of Hematology; 2007. June 15;109(12):5346–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Rohaan MW, van den Berg JH, Kvistborg P, Haanen JBAG. Adoptive transfer of tumor-infiltrating lymphocytes in melanoma: a viable treatment option. Journal for ImmunoTherapy of Cancer. 2018. October 3;6(1):102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Simoni Y, Becht E, Fehlings M, Loh CY, Koo S-L, Teng KWW, et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature Publishing Group. Nature Publishing Group; 2018. May;557(7706):575–9. [DOI] [PubMed] [Google Scholar]
- 99.Tran E, Turcotte S, Gros A, Robbins PF, Lu Y-C, Dudley ME, et al. Cancer immunotherapy based on mutation-specific CD4+ T cells in a patient with epithelial cancer Science. American Association for the Advancement of Science; 2014. May 9;344(6184):641–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Radvanyi LG, Bernatchez C, Zhang M, Fox PS, Miller P, Chacon J, et al. Specific lymphocyte subsets predict response to adoptive cell therapy using expanded autologous tumor-infiltrating lymphocytes in metastatic melanoma patients. Clin Cancer Res. 2012. December 15;18(24):6758–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Itzhaki O, Hovav E, Ziporen Y, Levy D, Kubi A, Zikich D, et al. Establishment and large-scale expansion of minimally cultured “young” tumor infiltrating lymphocytes for adoptive transfer therapy. J Immunother. 2011. March;34(2):212–20. [DOI] [PubMed] [Google Scholar]
- 102.Dudley ME, Gross CA, Somerville RPT, Hong Y, Schaub NP, Rosati SF, et al. Randomized selection design trial evaluating CD8+-enriched versus unselected tumor-infiltrating lymphocytes for adoptive cell therapy for patients with melanoma J Clin Oncol. American Society of Clinical Oncology; 2013. June 10;31(17):2152–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Roth TL, Puig-Saus C, Yu R, Shifrut E, Carnevale J, Li PJ, et al. Reprogramming human T cell function and specificity with non-viral genome targeting. Nature Publishing Group. Nature Publishing Group; 2018. July;559(7714):405–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Robbins PF, Kassim SH, Tran TLN, Crystal JS, Morgan RA, Feldman SA, et al. A pilot trial using lymphocytes genetically engineered with an NY-ESO-1-reactive T-cell receptor: long-term follow-up and correlates with response. Clin Cancer Res. 2015. March 1;21(5):1019–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhao Y, Bennett AD, Zheng Z, Wang QJ, Robbins PF, Yu LYL, et al. High-affinity TCRs generated by phage display provide CD4+ T cells with the ability to recognize and kill tumor cell lines. JI. 2007. November 1;179(9):5845–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Bowers JS, Nelson MH, Majchrzak K, Bailey SR, Rohrer B, Kaiser ADM, et al. Th17 cells are refractory to senescence and retain robust antitumor activity after long-term ex vivo expansion. JCI Insight. 2017. March 9;2(5):289–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Muranski P, Boni A, Antony PA, Cassard L, Irvine KR, Kaiser A, et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood. 2008. July 15;112(2):362–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Martin-Orozco N, Muranski P, Chung Y, Yang XO, Yamazaki T, Lu S, et al. T Helper 17 Cells Promote Cytotoxic T Cell Activation in Tumor Immunity Immunity. Elsevier Ltd; 2009. November 20;31(5):787–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tosolini M, Kirilovsky A, Mlecnik B, Fredriksen T, Mauger S, Bindea G, et al. Clinical Impact of Different Classes of Infiltrating T Cytotoxic and Helper Cells (Th1, Th2, Treg, Th17) in Patients with Colorectal Cancer. Cancer Res [Internet]. 2011. February 14;71(4):1263–71. Available from: http://cancerres.aacrjournals.org/lookup/doi/10.1158/0008-5472.CAN-10-2907 [DOI] [PubMed] [Google Scholar]
- 110.Xu C, Hao K, Yu L, Zhang X. Serum interleukin-17 as a diagnostic and prognostic marker for non-small cell lung cancer. Biomarkers. 2014. March 29;19(4):287–90. [DOI] [PubMed] [Google Scholar]
- 111.Sahin U, Derhovanessian E, Miller M, Kloke B-P, Simon P, Löwer M, et al. Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature Publishing Group. Nature Publishing Group; 2017. July 13;547(7662):222–6. [DOI] [PubMed] [Google Scholar]
- 112.Keskin DB, Anandappa AJ, Sun J, Tirosh I, Mathewson ND, Li S, et al. Neoantigen vaccine generates intratumoral T cell responses in phase Ib glioblastoma trial Nature Publishing Group. Nature Publishing Group; 2019. January;565(7738):234–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kenter GG, Welters MJP, Valentijn ARPM, Lowik MJG, Berends-van der Meer DMA, Vloon APG, et al. Vaccination against HPV-16 oncoproteins for vulvar intraepithelial neoplasia. N Engl J Med. 2009. November 5;361(19):1838–47. [DOI] [PubMed] [Google Scholar]
- 114.Ott PA, Hu Z, Keskin DB, Shukla SA, Sun J, Bozym DJ, et al. An immunogenic personal neoantigen vaccine for patients with melanoma. Nature Publishing Group. Nature Publishing Group; 2017. July 13;547(7662):217–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bennett SR, Carbone FR, Karamalis F, Miller JF, Heath WR. Induction of a CD8+ cytotoxic T lymphocyte response by cross-priming requires cognate CD4+ T cell help. J Exp Med. 1997. July 7;186(1):65–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Toes RE, Blom RJ, Offringa R, Kast WM, Melief CJ. Enhanced tumor outgrowth after peptide vaccination. Functional deletion of tumor-specific CTL induced by peptide vaccination can lead to the inability to reject tumors. JI. 1996. May 15;156(10):3911–8. [PubMed] [Google Scholar]
- 117.Toes RE, Offringa R, Blom RJ, Melief CJ, Kast WM. Peptide vaccination can lead to enhanced tumor growth through specific T-cell tolerance induction. Proc Natl Acad Sci USA. 1996. July 23;93(15):7855–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Hailemichael Y, Dai Z, Jaffarzad N, Ye Y, Medina MA, Huang X-F, et al. Persistent antigen at vaccination sites induces tumor-specific CD8+ T cell sequestration, dysfunction and deletion. Nature Medicine. 2013. April;19(4):465–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Melief CJM, van Hall T, Arens R, Ossendorp F, van der Burg SH. Therapeutic cancer vaccines. J Clin Invest. 2015. September;125(9):3401–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Tran E, Robbins PF, Lu Y-C, Prickett TD, Gartner JJ, Jia L, et al. T-Cell Transfer Therapy Targeting Mutant KRAS in Cancer N Engl J Med. Massachusetts Medical Society; 2016. December 8;375(23):2255–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zaretsky JM, Garcia-Diaz A, Shin DS, Escuin-Ordinas H, Hugo W, Hu-Lieskovan S, et al. Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma N Engl J Med. Massachusetts Medical Society; 2016. September 1;375(9):819–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ossendorp F, Mengedé E, Camps M, Filius R, Melief CJ. Specific T helper cell requirement for optimal induction of cytotoxic T lymphocytes against major histocompatibility complex class II negative tumors J Exp Med. Rockefeller University Press; 1998. March 2;187(5):693–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Bethune MT, Li X-H, Yu J, McLaughlin J, Cheng D, Mathis C, et al. Isolation and characterization of NY-ESO-1-specific T cell receptors restricted on various MHC molecules Proc Natl Acad Sci USA. National Academy of Sciences; 2018. November 6;115(45):E10702–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.McGranahan N, Furness AJS, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, et al. Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade Science. American Association for the Advancement of Science; 2016. March 25;351(6280):1463–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Chen F, Zou Z, Du J, Su S, Shao J, Meng F, et al. Neoantigen identification strategies enable personalized immunotherapy in refractory solid tumors J Clin Invest. American Society for Clinical Investigation; 2019. May 1;129(5):2056–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Rosenthal R, Cadieux EL, Salgado R, Bakir MA, Moore DA, Hiley CT, et al. Neoantigen-directed immune escape in lung cancer evolution Nature Publishing Group. Nature Publishing Group; 2019. March;567(7749):479–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.McMahan RH. Relating TCR-peptide-MHC affinity to immunogenicity for the design of tumor vaccines. Journal of Clinical Investigation. 2006. August 24;:1–9. [DOI] [PMC free article] [PubMed]