

Abstract
Prior studies suggest that β-nicotinamide adenine dinucleotide (β-NAD) is an important inhibitory motor neurotransmitter in the enteric nervous system. Metabolism of β-NAD at the neuroeffector junction (NEJ) is likely to be necessary for terminating inhibitory neurotransmission and may also produce bioactive metabolites. The enteric NEJ consists of enteric neurons and postjunctional cells of the SIP syncytium, smooth muscle cells (SMCs), interstitial cells of Cajal (ICC), and cells expressing platelet-derived growth factor receptor α (PDGFRα+ cells). We examined possible specialized functions of the NEJ in β-NAD metabolism by determining the degradation of 1,N6-etheno-NAD (eNAD) in colonic tunica muscularis of wild-type, Cd38−/−, Nt5e−/−, Enpp1−/−, and Cd38−/−/Nt5e−/− mice and in SIP cells from mice expressing cell-specific fluorescent reporters purified by FACS. We measured eNAD and its metabolites eADP-ribose, eAMP and e-adenosine (eADO) from tissues and sorted SIP cells using liquid chromatography. eNAD exposed to colonic muscularis of wild-type mice produced eADPR, eAMP, and eADO. CD38 mediated the conversion of eNAD to eADPR, whereas ENPP1 mediated degradation of eNAD and eADPR to eAMP. NT5E (aka CD73) was the primary enzyme forming eADO from eAMP. PDGFRα+ cells and SMCs were involved in production of eADO from eNAD, and ICC were not involved in extracellular metabolism of eNAD. CD38 mediated the eNAD metabolism in whole tissues, but CD38 did not appear to be functionally expressed by SMCs or ICC. NT5E was expressed in SMCs > PDGFRα+ cells. Our data show that extracellular metabolism of β-NAD in the colon is mediated by multiple enzymes with cell-specific expression.
Keywords: colon, enteric nervous system, NAD, interstitial cells, SIP syncytium
INTRODUCTION
Enteric inhibitory neurotransmission (EIN) is an essential element in the regulation of colonic motility. Several purines (Burnstock et al., 1970, Mutafova-Yambolieva et al., 2007, Durnin et al., 2012, Durnin et al., 2014), nitric oxide (NO) (Bult et al., 1990, Brookes, 1993, Sanders & Ward, 2019), and neuropeptides (Goyal et al., 1980, McConalogue et al., 1995, Keef et al., 2013) are primary enteric inhibitory neurotransmitters in gastrointestinal muscles. These neurotransmitters are released from terminals/varicosities of enteric inhibitory motor neurons in the tunica muscularis and act on postjunctional cells to transmit neurogenic signals to smooth muscle cells (SMCs). The actions of non-peptide neurotransmitters are usually terminated by rapid degradation at the neuroeffector junction (NEJ). In the gastrointestinal (GI) tract the NEJ consists of enteric motor neurons, SMCs, interstitial cells of Cajal (ICC), and interstitial cells expressing platelet-derived growth factor receptor α (PDGFRα+ cells) (Sanders et al., 2010, Sanders et al., 2014). SMCs, ICC and PDGFRα+ cells form an integrated postjunctional network, referred to as the SIP syncytium (Sanders et al., 2012). ICC and SMCs are primary postjunctional targets for NO (Burns et al., 1996, Beck et al., 2018), and PDGFRα+ cells are the principal postjunctional target for purine neurotransmitters (Kurahashi et al., 2014, Baker et al., 2015). ICC and PDGFRα+ cells are hyperpolarized by the actions of enteric inhibitory neurotransmitters and these responses conduct to SMCs via gap junctions (Sanders et al., 2014). NO is a molecule with short lifespan, and it is eliminated quickly by oxidation or S-nitrosylation (Gow et al., 1999) from the NEJ. Purines are metabolized by various enzymes (Zimmermann et al., 2012) that are likely located on membranes of cells forming the NEJ. The relative contributions of individual postjunctional cell types (e.g., PDGFRα+ cells, ICC, and SMCs) to the extracellular metabolism of purine neurotransmitters in GI muscles is presently unknown.
β-Nicotinamide adenine dinucleotide (β-NAD) is the first non-adenosine 5’-triphosphate (ATP) purine neurotransmitter that was discovered in the colons of mice, monkeys and humans (Mutafova-Yambolieva et al., 2007, Hwang et al., 2011). The extracellular degradation of β-NAD in different systems is accomplished by multiple enzymes (Fig. 1). CD38, a type II glycoprotein, functions as a NAD-hydrolase and converts β-NAD to adenosine 5’-diphospho ribose (ADPR) and nicotinamide (Lee, 2001, Graeff et al., 1998, De Flora et al., 2004, Graeff et al., 2009). The catalytic site of CD38 faces the ecto-cellular space (Lee et al., 1993, Munshi et al., 2000) making this enzyme a suitable regulator of extracellular β-NAD levels. We have previously reported that β-NAD can produce ADPR near the sites of neurotransmitter release in mouse and monkey colon muscularis and that CD38 is likely involved in this conversion (Durnin et al., 2012). ADPR is then degraded to adenosine 5’-monophosphate (AMP) by ecto-nucleotide pyrophosphatases (ENPPs) and AMP in turn is degraded to adenosine (ADO) by 5’-nucleotidase (NT5E, aka CD73) (Zimmermann et al., 2012, Linden et al., 2019). Both ENPPs and NT5E are membrane-bound enzymes capable of degrading extracellular adenine nucleotides (Zimmermann et al., 2012). The roles of β-NAD-degrading enzymes and the types of cells in the NEJ that are responsible for the extracellular degradation of β-NAD in the colon remain to be determined. Therefore, this study was designed to investigate extracellular β-NAD metabolism in the murine colon at tissue and cellular levels.
Figure 1.
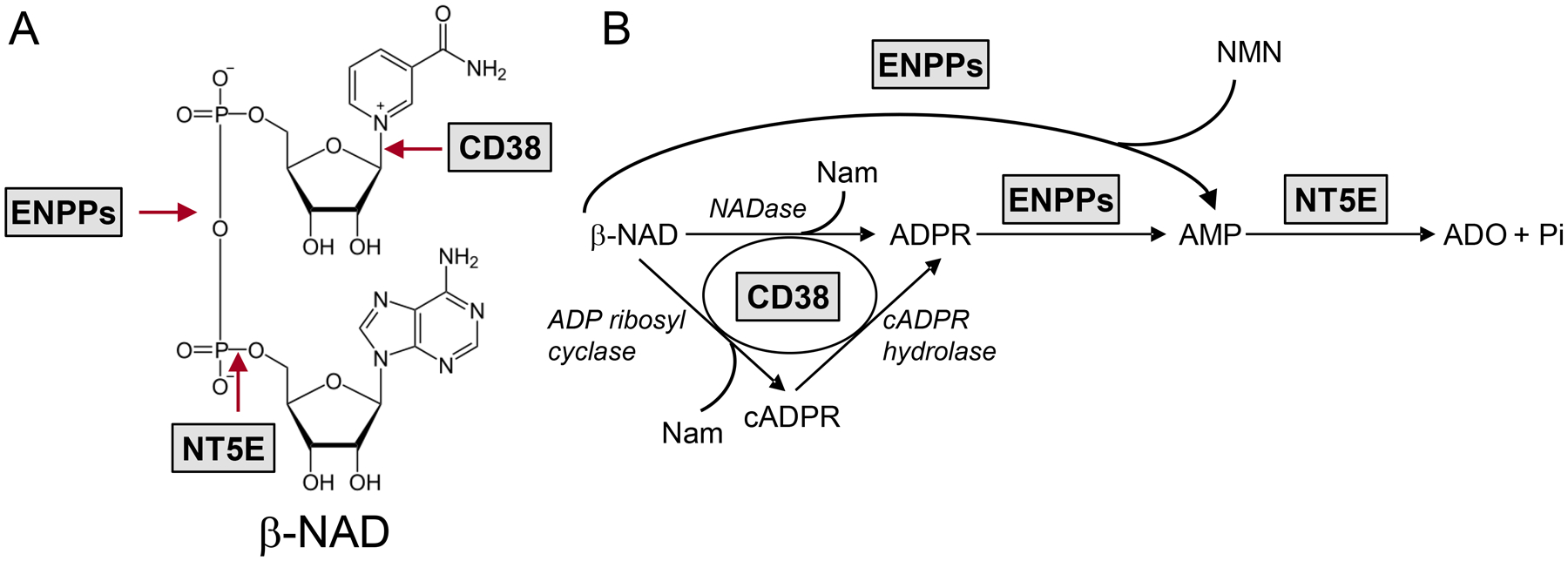
A. Chemical structure of β-NAD and sites of cleavage by ecto-enzymes. B. Extracellular biotransformation pathways for β-NAD demonstrating the sequence of product formation. CD38 serves as NAD glycohydrolase (NADase) (principal activity) as well as ADP ribosyl cyclase and cADPR hydrolase. ENPPs metabolize β-NAD and ADPR to AMP. NT5E degrades AMP to ADO. ENPPs, ecto-nucleotide pyrophosphatases; Nam, nicotinamide; NMN, nicotinamide mononucleotide.
METHODS
Ethical approval
Animals were maintained and experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals and the Institutional Animal Use and Care Committee at the University of Nevada.
Colon tunica muscularis tissue preparation
C57BL/6 mice, B6.129P2-Cd38tm1Lnd/J (Cd38−/−) mice, C57BL/6J-Enpp1asj/GrsrJ (Enpp1−/−) mice (Jackson Laboratory, Bar Harbor, MN), Nt5e−/− mice (donated by Linda Thompson, Oklahoma Medical Research Center, Oklahoma City), and Cd38−/−/Nt5e−/− knockout mice (generated in house) were euthanized by sedation with isoflurane followed by cervical dislocation and exsanguination. The GI tract was removed and placed in oxygenated Krebs solution for further dissection. Krebs solution had the following composition (mM): 118.5 NaCl, 4.2 KCl, 1.2 MgCl2, 23.8 NaHCO3, 1.2 KH2PO4, 11.0 dextrose, 1.8 CaCl2 (pH 7.4). Colon preparations were dissected free from the remaining GI tract and opened along the mesenteric border. Mouse tunica muscularis preparations were prepared by peeling away the mucosa and submucosa as previously (Mutafova-Yambolieva et al., 2007, Durnin et al., 2014).
Tissue dispersion and SMCs, ICC and PDGFRα+ cell purification
Transgenic mice with green fluorescent proteins (eGFP or copGFP) tagged to cell-specific promoters (smMHC for SMCs, Kit for ICC, and PDGFRα for PDGFRα+ cells) were used to isolate each cell type. smMHC(Myh11)/Cre/eGFP mice and Pdgfratm11(EGFP)Sor/J heterozygote mice (Jackson Laboratory, Bar Harbor, ME, USA), and Kit+/copGFP (developed in our department; Zhu et al., 2009) were anaesthetized with isoflurane and euthanized by cervical dislocation. Colons were removed and placed in Ca2+-free Hanks’ solution with the following composition (mM): 125 NaCl, 5.36 KCl, 15.5 NaHCO3, 0.336 Na2HPO4, 0.44 KH2PO4, 10.0 glucose, 2.9 sucrose, 11.0 N-2-hydroxyethylpiperazine-N′−2-ethanesulfonic acid (HEPES). Colon preparations were opened along the mesenteric border and the mucosa and submucosa were removed. Cells were isolated from tunica muscularis of the entire colon as described previously (Peri et al., 2013, Koh et al., 1997, Zhu et al., 2009). Briefly, dissected colons were incubated at 37°C with Ca2+-free Hanks’ solution containing 4 mg/mL collagenase type II (Worthington, Lakewood, NJ, USA), 8 mg/mL bovine serum albumin (Sigma, St. Louis, MO, USA) and 8 mg/mL trypsin inhibitor (Sigma) for 30 min. Tissues were washed three times with Ca2+-free Hanks’ solution, resuspended in 2 mL Ca2+-free Hanks’ solution and tissues were gently triturated to create a cell suspension. eGFP-SMCs, copGFP-ICC and eGFP-PDGFRα+ cells and were purified by fluorescence-activated cell sorting (FACS; Becton Dickinson FACSAriaII) using a blue laser (488 nm) and the GFP/FITC emission detector (530/30 nm).
β-NAD metabolism in tunica muscularis
Tissues were loaded in small (200 μl)-superfusion chambers (BRANDEL, Gaithersburg, MD, USA) and superfused with oxygenated Krebs solution at 37 °C (Mutafova-Yambolieva et al., 2007, Durnin et al., 2012, Durnin et al., 2014). Tissue preparations isolated from two animals were loaded in one perfusion chamber. After 45 minutes equilibration, the tissue preparations were superfused for 30 s with 1,N6-etheno-NAD (eNAD) at a maximum saturation concentration of 0.2 μM that was determined in pilot experiments. The content of each chamber was collected in ice-cold Eppendorf tubes that were subsequently immersed in liquid nitrogen to stop the enzymatic reactions. HPLC-FLD was used to measure eNAD and its products eADPR, eAMP and eADO. Samples collected after contact of eNAD with tissue [(+ tissue), S1] were compared with eNAD samples collected before contact with tissue [(−) tissue), S0]. The degradation of eNAD was evaluated by assessing the decrease in eNAD and/or the increase in eADPR, eAMP, and eADO in (+) tissue samples.
AMP metabolism in tunica muscularis
Tissues were processed as described in β-NAD metabolism in tunica muscularis. After 45 minutes equilibration, the tissue preparations were superfused for 30 s with 1,N6-etheno-AMP (eAMP) at a maximum saturation concentration of 0.05 μM as previously established for adenine nucleotides (Durnin et al., 2016). The content of each chamber was collected in ice-cold Eppendorf tubes that were subsequently immersed in liquid nitrogen to stop the enzymatic reactions. HPLC-FLD was used to measure eAMP and its product eADO. Samples collected after contact of eAMP with tissue [(+ tissue)] were compared with eAMP samples collected before contact with tissue [(−) tissue), S0]. The degradation of eAMP was evaluated by assessing the decrease in eAMP and the increase in eADO in (+) tissue samples (S1).
β-NAD metabolism in FACS-sorted SIP cells
Thirty five to sixty two thousand SMCs, ICC or PDGFRα+ cells were obtained from colons of 4 or 5 animals and applied under gentle suction onto Whatman GF/B filter paper which was placed in a 0.45 μm Cameo 3N syringe filter serving as a perfusion chamber (Yamboliev et al., 2009). The chamber was perfused with Krebs-bicarbonate-HEPES (KBH) buffer of the following composition (mM): 58.44 NaCl, 74.55 KCl, 246.5 MgSO4, 136.09 KH2PO4, 84.01 NaHCO3, 147 CaCl2, 180 glucose, 238.3 HEPES. After equilibration, chambers were superfused with eNAD (0.2 μM) in KBH solution (Jamal et al., 1988, Todorov et al., 1997, Durnin et al., 2012, Durnin et al., 2016). Four hundred μl-samples from substrate solution (no cells present, S0) and from superfusate after 1-, 2- or 3-minute contact of cells with substrate (S1) were collected in ice-cold Eppendorf tubes and immersed in liquid N2 to stop the enzymatic reactions. Purines in samples were assayed by HPLC-FLD and enzymatic activities were determined by the appearance of 1,N6-etheno-products.
HPLC-FLD assay of 1,N6-etheno-nucleotide and nucleoside formation from eNAD
A reverse phased gradient Agilent Technologies 1200 liquid chromatography system equipped with a fluorescence detector (FLD) was used to detect 1,N6-etheno-products as described previously (Mutafova-Yambolieva et al., 2007, Durnin et al., 2012, Durnin et al., 2016). The mobile phase consisted of 0.1 M KH2PO4 (pH 6.0) as eluent A. Eluent B consisted of 65% eluent A and 35% methanol. Gradient elution was employed according to the following linear program: time 0, 0% eluent B; 18 min, 100% eluent B. The flow rate was 1 mL/min and the run time was 20 min. Column and autosampler temperatures were maintained at 25 °C and 4 °C, respectively. The fluorescence detector was set to record 1,N6-etheno-derivatized nucleotide and nucleoside signals at an excitation wavelength of 230 nm and emission wavelength of 420 nm (Bobalova et al., 2002).
Preparation of eNAD and eAMP
eNAD was purchased from BioLog Life Sciences (Bremen, Germany) and diluted in the superfusion solution to the final concentration needed. eAMP was prepared by 1,N6-etheno-derivatization as described previously (Bobalova et al., 2002, Durnin et al., 2016). Briefly, 0.2 mM AMP (dissolved in double distilled water) was acidified to pH 4.0 with citrate phosphate buffer. Chloroacetaldehyde (1 M) was added and the solution was heated to 80°C for 40 min to form eAMP. Substrates were diluted in superfusion solution to the final concentration needed.
Statistics
Data are presented as means ± SD. ‘n’ is the number of observations in each group. One observation was made per perfusion chamber. Each perfusion chamber contained tissue preparations from two animals or 35,000–62,000 cells isolated from 4–5 animals. Groups were analyzed by One-way ANOVA with Dunnet’s multiple comparison test or Paired, two-tailed t-test (GraphPadPrism, v. 8.3.0, GraphPad Software, Inc, San Diego, CA). Differences between means were considered statistically significant when P < 0.05.
RESULTS
Degradation of eNAD in C57BL/6 murine colon
The eNAD substrate solutions contained eNAD, a small amount of eADPR and no eAMP or eADO (Fig. 2A, S0, no tissue). Thus, in S0 (n = 22) eADPR was 6.64 ± 2.14 fmol/mg tissue whereas both eAMP and eADO were 0 ± 0 fmol/mg tissue. Superfusion of colonic tissues from C57BL/6 (WT) mice (n = 12) with 0.2 μM eNAD for 30 s (Fig. 2A, (+) tissue WT) resulted in 8.706 ± 3.928 fmol/mg tissue eADPR (P = 0.1645 vs. S0) (Fig. 2Ba), 15.09 ± 13.06 fmol/mg tissue eAMP (P = 0.0126 vs. S0) (Fig. 2Bb), and 33.68 ± 10.96 fmol/mg tissue eADO, (P < 0.0001 vs. S0) (Fig. 2Bc). The combined etheno-product eADPR + eAMP + eADO was 5.622 ± 3.135 fmol/mg tissue in S0 and increased to 52.44 ± 19.56 fmol/mg tissue after contact of eNAD with tissue; (P < 0.0001 vs. S0) (Fig. 2Bd).
Figure 2.
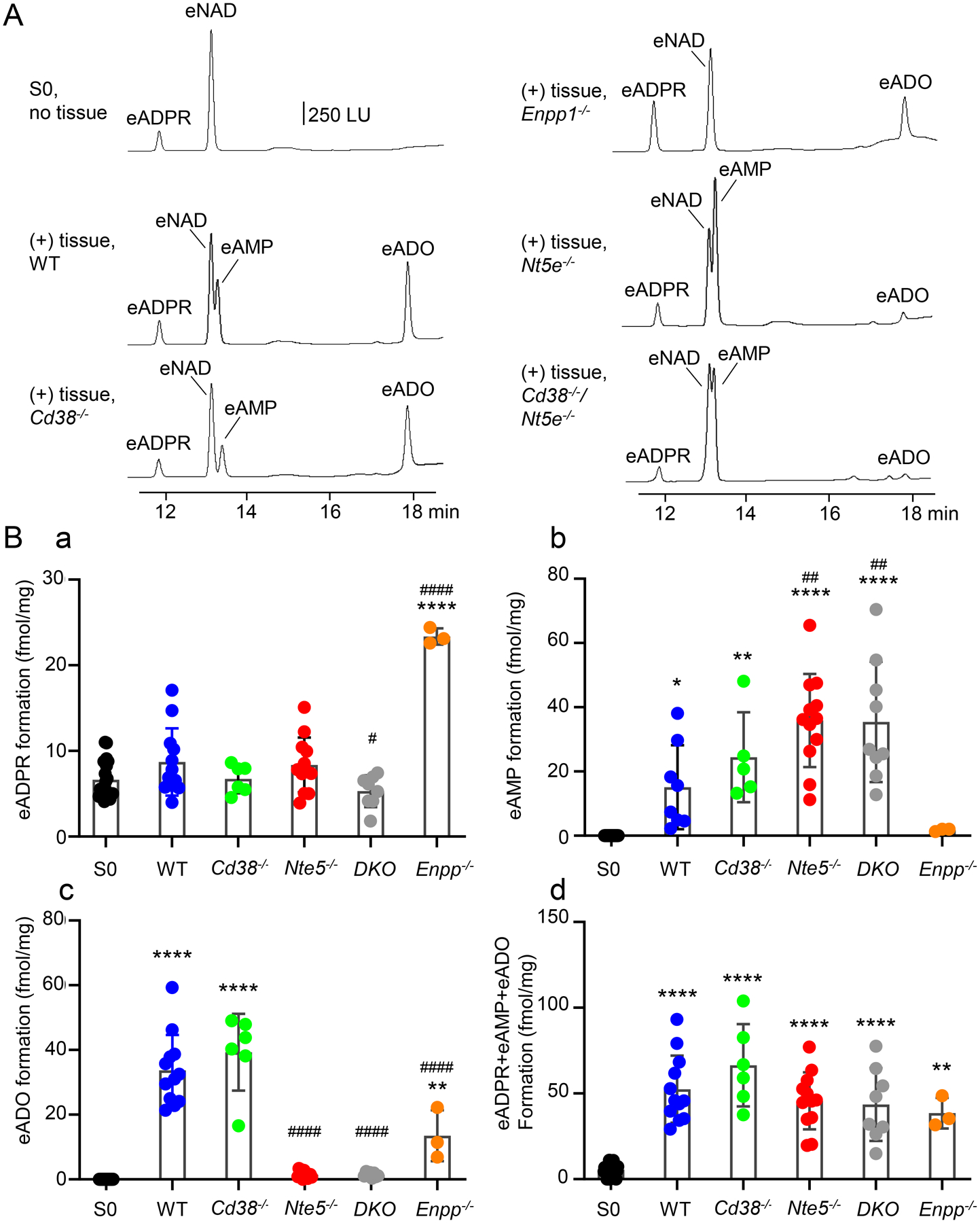
A. Original chromatograms representing the degradation of eNAD in tunica muscularis preparations of colons of wild-type (WT) and knockout mice. LU, luminescence units. B. Scattered plots (means, SD) of eNAD metabolites produced after contact of eNAD substrate with colonic muscularis. Each data point (closed circles) represents a result from one experiment. Each experiment is performed in one perfusion chamber containing tissue preparations from two mice. Asterisks demonstrate significant differences from S0 (no tissue) (* P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001). Number signs demonstrate statistically significant differences from WT controls (# P < 0.05; ###P < 0.001; ####P < 0.0001). Data analyzed with One-way ANOVA with Dunnett’s multiple comparison test (GraphPad Prism, v. 8.3.0, GraphPad Software, Inc, San Diego, CA). DKO, double knockout mice = Cd38−/−/Nt5e−/− mice.
Degradation of eNAD in Cd38−/− mouse colon
We next examined the eNAD degradation in Cd38−/− mouse colons (n = 6) that lacked the main β-NAD metabolizing enzyme CD38. Superfusion of colons from Cd38−/− mice with eNAD resulted in no significant change of eADPR (P = 0.4822 vs. WT); however, significant formation of eAMP and eADO was observed (Fig. 2). Thus, after contact of eNAD with tissue, the amounts of eADPR, eAMP, and eADO were 6.73 ± 1.657 fmol/mg tissue (P > 0.9999 vs. S0), 24.44 ± 14.01 fmol/mg tissue (P = 0.004 vs S0), and 39.30 ± 11.89 fmol/mg tissue (P < 0.0001 vs. S0), respectively. The amounts of both eAMP and eADO formed from eNAD in Cd38−/− mouse colons were similar to the amounts of eAMP and eADO in WT colons (P = 0.4834 and P = 0.2701, respectively). The combined etheno-product eADPR + eAMP + eADO after contact of eNAD with tissue was 66.39 ± 24.01 fmol/mg tissue (P < 0.0001 vs. S0 and P = 0.2500 vs. WT). These results suggest that in the absence of CD38, β-NAD cannot form ADPR, but it forms AMP and ADO by an alternative NAD-degradation pathway that is not associated with CD38.
Degradation of eNAD in Enpp1−/− mouse colon
In colon muscularis of Enpp1−/− mice, amounts of eADPR were 5.33 ± 1.07 and 23.37 ± 0.9426 fmol/mg tissue before and after contact of tissue with eNAD (n = 3), which is significantly higher than eADPR produced from eNAD in WT colons (P < 0.0001) (Fig. 2). The amounts of eAMP in samples collected from Enpp1−/− colons were small (i.e., 1.77 ± 0.4272 fmol/mg tissue) and could be resolved under magnification (P = 0.9997 vs. S0 and P = 0.3133 vs. WT). eADO was increased from 0 ± 0 to 13.52 ± 7.878 fmol/mg tissue before and after contact with eNAD (n = 3, P = 0.0037 vs. S0), which represents about 50% of eADO formed in tissues from WT mice (P < 0.0001). The combined etheno-product eADPR + eAMP + eADO after contact of eNAD with tissue was 38.54 ± 8.95 fmol/mg tissue (P = 0.0032 vs. S0 and P = 0.4959 vs. WT).
Degradation of eNAD in Nt5e−/− mouse colon
Superfusion of Nt5e−/− mouse colons with eNAD (n = 12) resulted in accumulation of eAMP to 35.873 ± 14.48 fmol/mg tissue (P = 0.0011 vs. WT colon) and amounts of eADO reduced to 1.44 ± 1.104 fmol/mg tissue (P < 0.0001 vs. WT colon) (Fig. 2). eADPR formed from eNAD in colons of Nt5e−/− mice was 8.365 ± 3.192 fmol/mg tissue and was not significantly different from eADPR in S0 (P = 0.3221) or in WT colons (P = 0.9983). Based on these data, NT5E appears to be the primary enzyme for ADO formation from β-NAD in the colon. We validated this observation by demonstrating that eAMP did not produce any eADO in colonic muscularis preparations of Nt5e−/− mice, suggesting that NT5E is the only detectable enzyme that degrades AMP to ADO in this preparation (Fig. 3). The combined etheno-product eADPR + eAMP + eADO after contact of eNAD with tissue was 45.68 ± 16.64 fmol/mg tissue (P < 0.0001 vs. S0 and P = 0.7282 vs. WT).
Figure 3.
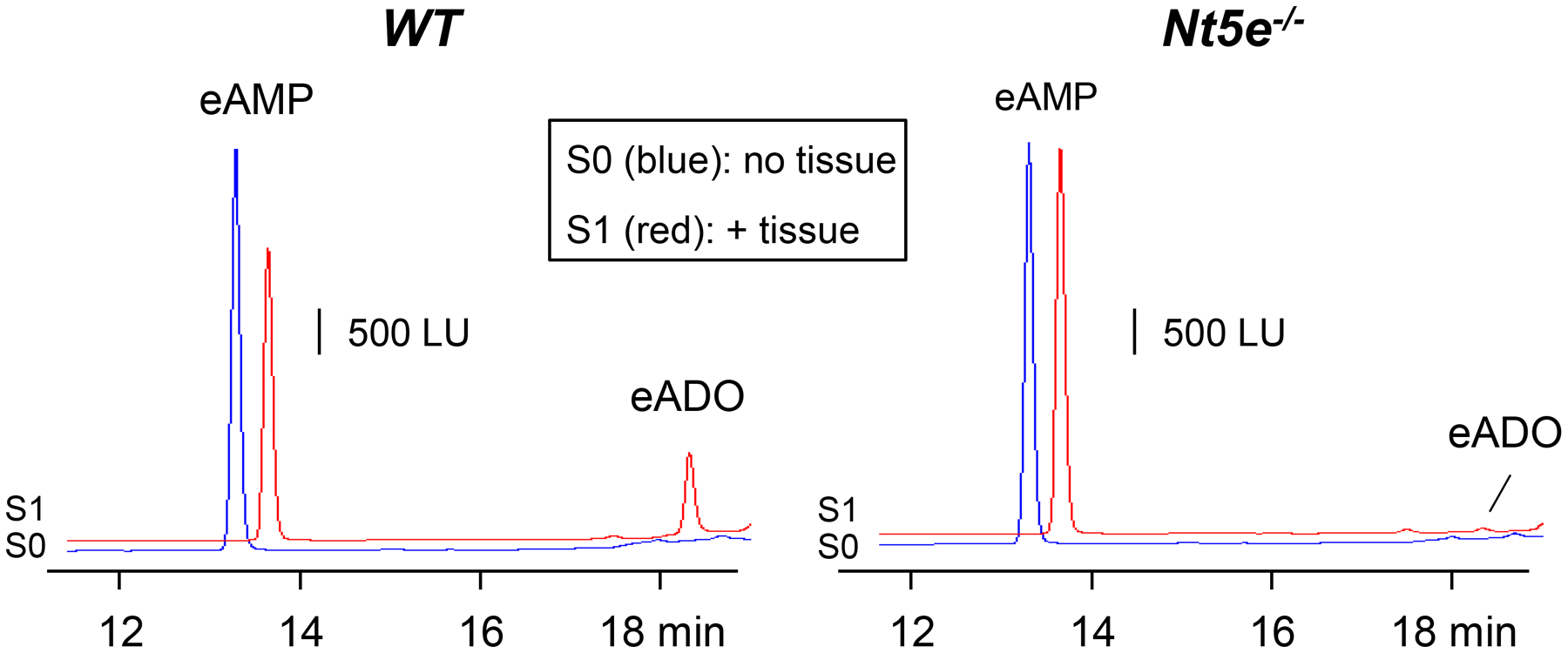
Degradation of eAMP in colon muscularis from WT and Nt5e−/− mice. Original chromatograms demonstrate that the formation of eADO from eAMP is abolished in preparations lacking Nt5e. S0 (blue): eAMP substrate without tissue; S1 (red): eAMP substrate + tissue. WT, wildtype; LU, luminescence units.
Degradation of eNAD in Cd38−/−/Nt5e−/− mouse colon
We also investigated the degradation of eNAD in the absence of both Cd38 and Nt5e. As shown in Fig. 2, eADPR was 5.307 ± 1.866 fmol/mg tissue (n = 8) after contact with eNAD (P = 0.7058 vs. S0 and P = 0.035 vs. WT colons), suggesting that no ADPR was formed from β-NAD in tissues lacking CD38. eAMP was increased to 35.42 ± 18.74 fmol/mg tissue after contact with eNAD (P < 0.0001 vs. S0 and P = 0.0031 vs. WT colons) whereas negligible amounts of eADO (i.e., 1.456 ± 0.7349 fmol/mg tissue) were detected in samples collected from preparations of double knockout mice (P < 0.0001 vs. WT colons). Therefore, in these preparations, AMP was likely formed by ENPP(s) but could not get degraded further due to the absence of NT5E. The combined etheno-product eADPR + eAMP + eADO after contact of eNAD with tissue was 43.57 ± 21.28 fmol/mg tissue (P < 0.0001 vs. S0 and P = 0.5935 vs. WT).
Degradation of eNAD in unsorted cells of tunica muscularis
As in colonic tissue preparations of WT mice (Fig. 2), superfusion of freshly dispersed unsorted cells (Fig. 4) isolated from tunica muscularis of mice with GFP-tagged cells resulted in the formation of eADPR and eADO after a 1-, 2- or 3-minute contact of cells with eNAD. The degradation after a 2-minute contact with eNAD was greater than the eNAD degradation after a 1-minute contact of substrate with cells (not shown). However, increasing the time of eNAD contact with cells to 3 minutes resulted in no significant additional degradation than the degradation after a 2-minute contact between eNAD and cells. In particular, a 2-minute contact of eNAD with freshly dispersed unsorted cells (n = 12) resulted in an increase of eADPR from 20.958 ± 3.513 fmol/1000 cells in S0 (i.e., substrate in absence of cells) to 26.46 ± 4.294 fmol/1000 cells after contact with cells (P < 0.0001). eADO was increased from 0 ± 0 fmol/1000 cells in S0 to 25.54 ± 8.236 fmol/1000 cells (P < 0.0001) after contact of eNAD with cells (Fig. 4). Therefore, eNAD degradation can be detected in isolated cells from murine colonic preparations using the small chamber superfusion assay.
Figure 4.
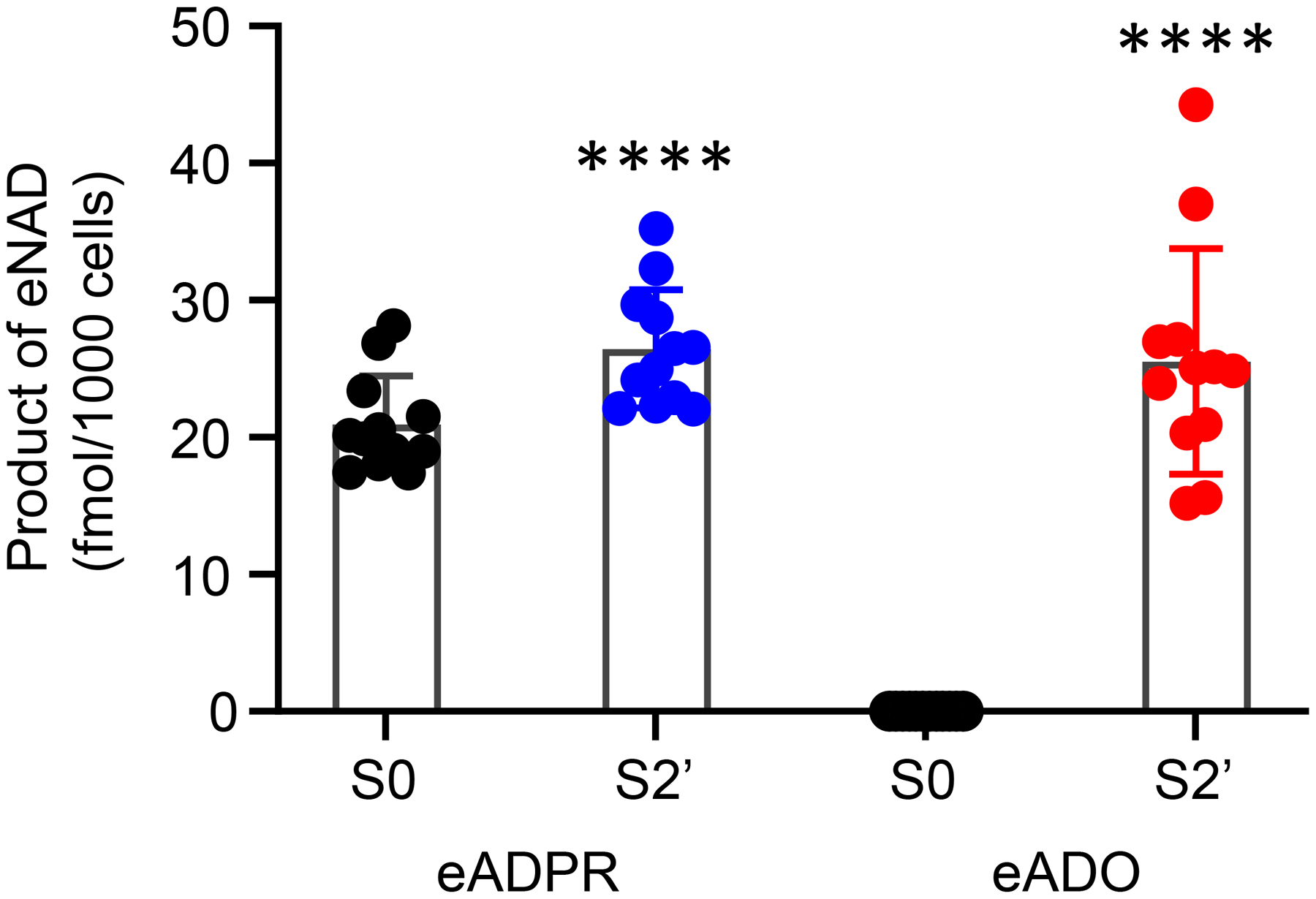
Degradation of eNAD after a 2-minute contact with freshly dispersed unsorted cells of murine colon tunica muscularis. Scattered plots (mean, SD) of 12 experiments demonstrating the formation of eADPR and eADO from eNAD. Each experiment is performed in one perfusion chamber containing 35,000–62,000 cells collected from 4 mouse colons. S0 is eNAD substrate in the absence of cells. S2’ is sample collected after contact of eNAD with cells. Asterisks denote significant difference of S2’ vs. S0. ****P < 0.0001. Paired, two-tailed t-test (GraphPadPrism v. 8.3.0., GraphPad Software, Inc, San Diego, CA).
Degradation of eNAD in purified SMCs
Experiments were also performed to determine contributions from individual cell types of the SIP syncytium to the degradation of eNAD (Fig. 5). We measured the formation of eADPR and eADO as the major indicators for eNAD degradation by SIP cells. Superfusion of FACS-sorted eGFP-positive cells isolated from tunica muscularis of eGFP-SMC mouse colons with 0.2 μM eNAD for 1, 2 or 3 minutes resulted in no significant changes in the eADPR amounts. Fig. 5B shows results of the 2-minute contact between eNAD and cells. Thus, eADPR was 21.243 ± 1.614 fmol/1000 cells (n = 4) and 21.36 ± 1.576 fmol/1000 cells (n = 4) before and after superfusion of SMCs with eNAD (P > 0.0.9999) (Fig. 5Ba). In contrast to eADPR, the eADO amounts in the cell superfusate increased from 0.0 ± 0.0 fmol/1000 cells to 11.662 ± 2.688 fmol/1000 cells (P < 0.0001, n = 4) (Fig. 5Bb). No additional degradation occurred after a 3-minute contact of cells with substrate.
Figure 5.
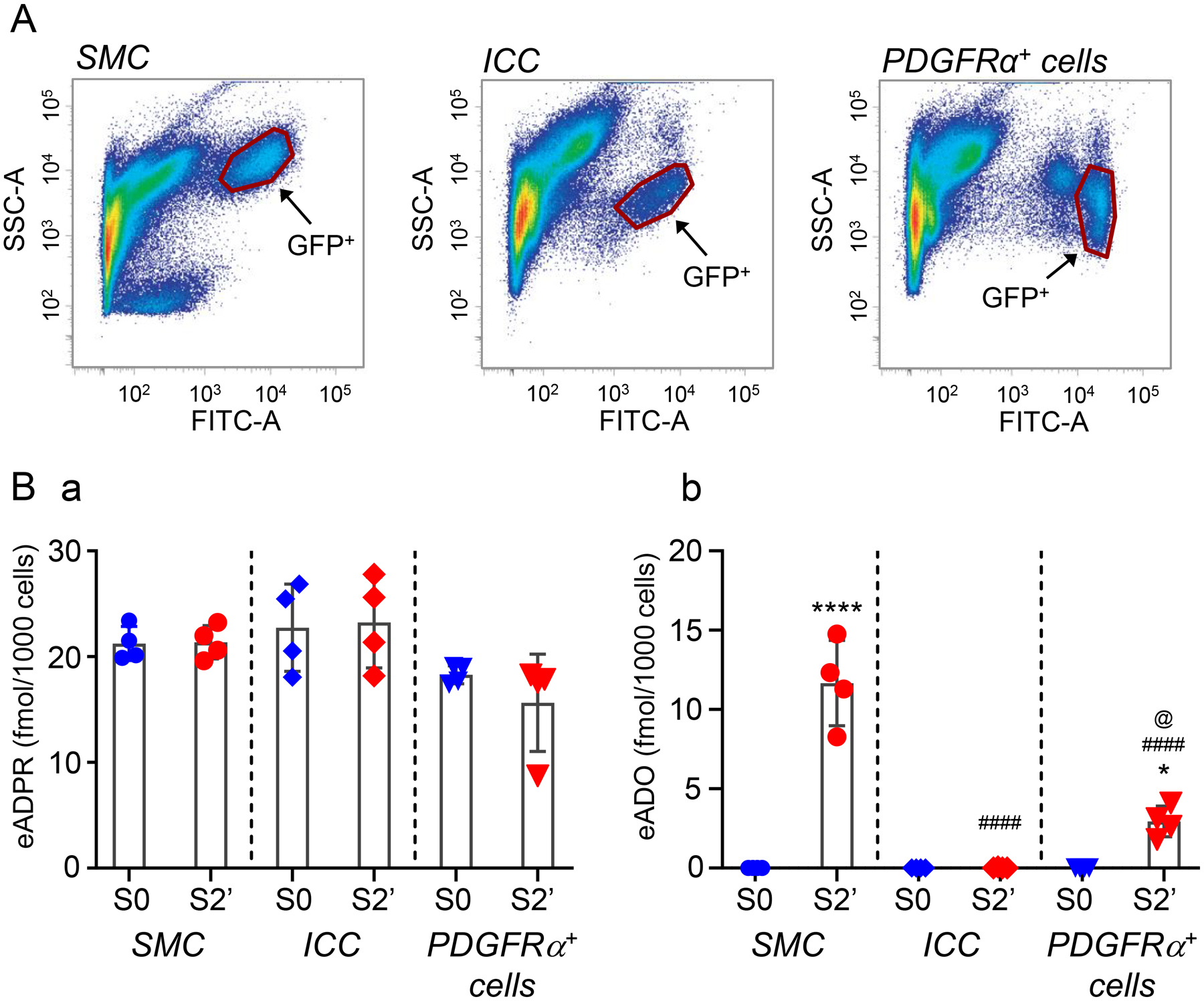
A. SMCs, ICC and PDGFRα+ cells of colonic tunica muscularis purified by fluorescence-activated cells sorting. B. Scattered plots (mean, SD) demonstrating product formation after exposure of cells to eNAD substrate for 2 minutes. Each data point represents a result from a single experiment performed with 35,000–62,000 cells collected from 4–5 mouse colons. Asterisks denote significant differences from S0 (no tissue) (* P < 0.05; ****P < 0.0001). Number signs denote significant differences from S2’ of SMC (####P < 0.0001). The at symbol denotes significant difference from S2’ of ICC (@ < 0.05). One-way ANOVA with Dunnett’s multiple comparison test (GraphPad Prism v. 8.3.0, GraphPad Software, Inc, San Diego, CA).
Degradation of eNAD in purified ICC
Superfusion of cells with 0.2 μM eNAD for 1 – 3 minutes resulted in no formation of eADPR or eADO (Fig. 5Ba,b), suggesting that ICC cells are not the predominant cell type expressing CD38 or NT5E. In particular, eADPR was 22.729 ± 4.128 fmol/1000 cells before exposure of ICC to eNAD and 23.24 ± 4.296 fmol/1000 cells after a 2-minute contact of cells with substrate (P > 0.9999, n = 4) (Fig. 5). eADO was 0.0 ± 0.0 and 0.018 ± 0.0355 fmol/1000 cells before and after eNAD, respectively (P > 0.9999, n = 4).
Degradation of eNAD in purified PDGFRα+ cells
Superfusion of PDGFRα+ cells with 0.2 μM eNAD for 1–3 min resulted in no change of eADPR amounts (usually mediated by CD38) and in production of smaller amounts of eADO than SMC (usually mediated by NT5E) (Fig. 5), suggesting that PDGFRα+ cells are not the predominant cell types expressing CD38 or NT5E. In particular, eADPR was 18.29 ± 0.8583 and 15.643 ± 4.594 fmol/1000 cells before and after 2-minute contact of eNAD with cells, respectively (P = 0.8494, n = 4) (Fig. 5Ba). eADO was 0.0 ± 0.0 fmol/1000 cells before and 2.941 ± 0.9627 fmol/1000 cells after contact of cells with eNAD (P = 0.0227). eADO formation in PDGFRα+ cells was significantly lower than the eADO formation in SMC (P < 0.0001) but significantly higher than eADO formation in ICC (P = 0.0237, n = 4) (Fig. 5Bb).
DISCUSSION
The present study provides functional evidence for the complex extracellular metabolism of β-NAD in the large intestine and the first characterization of the roles of SMCs, ICC and PDGFRα+ cells in the β-NAD degradation. β-NAD is a key inhibitory motor neurotransmitter in the enteric nervous system in the colon (Mutafova-Yambolieva et al., 2007, Hwang et al., 2011, Durnin et al., 2013, Mutafova-Yambolieva & Durnin, 2014). β-NAD released from motor neurons activates purinergic P2Y1 receptors on PDGFRα+ cells to trigger activation of small-conductance Ca2+-activated K+ channels, SK channels (Kurahashi et al., 2014, Peri et al., 2013, Baker et al., 2015). The immediate metabolite of β-NAD, ADPR, also meets key criteria for an enteric inhibitory neurotransmitter as it activates the P2Y1 receptor-SK3 channel pathway causing membrane hyperpolarization of PDGFRα+ cells, conducted hyperpolarization to SMC, and relaxation of SMCs (Durnin et al., 2012, Kurahashi et al., 2014). It is currently unknown whether ADPR is exclusively a result of extracellular degradation of β-NAD or if it is also released from nerve varicosities during firing of action potentials. Regardless of the underlying mechanism, the presence of ADPR at the NEJ would cause activation of the P2Y1 receptor-SK3 channel pathway in PDGFRα+ cells resulting in smooth muscle relaxation. The more distant metabolite of β-NAD, ADO, also has biological activity by acting on G-protein coupled adenosine receptors (Fredholm et al., 2011). Previous reports suggest that ADO acts on adenosine receptors localized on neuronal terminals and possibly SMCs to modulate neurotransmitter release and gut motility (Giaroni et al., 2002, Burnstock, 2014). However, recent expression data suggest that ADO receptors (Adora1) may be localized to PDGFRα+ cells, and therefore a portion of responses to ADO may be contributed by these cells (Lee et al., 2015, Lee et al., 2017, Ha et al., 2017). Enzymatic transformation of extracellular β-NAD to ADPR and ADO can affect the activation of specific purinergic receptors, including the Gq-coupled P2Y1 receptor and the Gs- or Gi-coupled adenosine receptors. Little was known previously about the extracellular metabolism of β-NAD in the large intestine. Knowledge of the β-NAD metabolism in the gut might suggest strategies for terminating or facilitating purinergic inhibitory motor neurotransmission and for altering extra-junctional effects of β-NAD and its metabolites in the large intestine. Clarifying β-NAD metabolism may also provide pharmacological depth in understanding concentration-response information, as these responses undoubtedly include activation of multiple receptors in colonic tissues. Therefore, in this study, we investigated how extracellular β-NAD is degraded in the murine colon at tissue and cellular levels.
As illustrated in Fig. 1, the extracellular degradation of β-NAD is complex. It is performed by multiple enzymes that likely have differential distribution in different organs and cells. We first examined the degradation of β-NAD in colonic tunica muscularis preparations that contain the motor NEJs. Sufficiently selective and specific inhibitors of the enzymes involved in β-NAD degradation are currently lacking. Therefore, to understand the involvement of individual enzymes in the degradation of β-NAD, we utilized preparations from mice in which the genes for CD38, NT5E, and ENPP1 were inactivated genetically. To determine the ability of SIP cell types to degrade β-NAD, we used FACS-purified cells isolated from colons of transgenic mice with fluorescent reporters tagged to cell-specific promoters for SMCs, ICC, and PDGFRα+ cells.
Tissues or sorted cells were exposed to the highly-fluorescent analog of β-NAD, 1,N6-etheno-NAD (eNAD), used as a substrate for β-NAD metabolizing enzymes (Durnin et al., 2012). The process of etheno-derivatization involves insertion of acyl group between nitrogen 1 and 6 of the adenine moiety and renders the molecule fluorescent but leaves unaltered the phosphate chain. Therefore, 1,N6-etheno-nucleotides and non-derivatized nucleotides share identical sites for hydrolysis 1,N6-etheno-nucleotides (Todorov et al., 1997, Bobalova et al., 2002, Durnin et al., 2012). Use of eNAD as substrate in place of non-derivatized β-NAD has clear advantages: (i) 1,N6-etheno-derivatization of adenine nucleotides and nucleosides increases the sensitivity of detection about 1,000,000 fold (Bobalova et al., 2002), which allows small changes in product concentrations to be detected in small tissue or cell samples, and (ii) use of eNAD avoids the need of 1,N6-etheno-derivatization of collected samples to detect its metabolites; this leaves undetected the endogenous purines that might have been released in the extracellular space during tissue or cell perfusion. As discussed above, the degradation of eNAD was evaluated by the appearance or increase in its metabolites in tissue or cell superfusates. Since the elution times of eNAD and eAMP were very close (Fig. 2A), the two signals were not always well separated, which made it challenging to accurately measure the AMP amounts. In tissue preparations, eNAD and eAMP were still separated allowing a split-peak analysis (see Fig. 2A) whereas eAMP was not resolved in the cell superfusates. Therefore, the degradation of eNAD in colon muscularis was determined by the formation of eADPR, eAMP and eADO whereas the degradation of eNAD in cells was determined by the formation of eADPR and eADO only.
In wild-type tissue preparations, eADPR was elevated after contact of tissues with eNAD, whereas no changes in the amounts of eADPR were observed in the CD38−/− preparations. Therefore, CD38 appears to be involved in the degradation of eNAD to ADPR in the colon. Although eADPR was not produced in colons from CD38−/− mice, eAMP and eADO were still formed after contact of tissue with eNAD. These are intriguing observations that suggest that in addition to CD38, other enzymes are also involved in the degradation of eNAD in the colon. Moreover, the amounts of eAMP and eADO formed from eNAD in the CD38−/− preparations exceeded the amounts of eAMP and eADO in WT preparations (Durnin et al., 2012; the present study), suggesting that the alternative pathways for eAMP and eADO formation are functionally upregulated in the absence of CD38. ENPP1 appears to provide a pathway for eNAD degradation as the formation of eAMP from eNAD was significantly diminished in colonic preparations from Enpp1−/− mice. The appearance of eADO in these studies appeared to be entirely dependent on the presence of NT5E. Thus, in colonic muscularis isolated from Nt5e−/− mice, the eAMP was enhanced whereas the eADO was significantly diminished, suggesting that the degradation of eAMP to eADO was inhibited. As anticipated, in the absence of both Cd38 and Nt5e, large amounts of eAMP were accumulated after contact of tissue with eNAD, because eAMP that was formed by ENPP1 could not be degraded to eADO. Therefore, the studies in tunica muscularis preparations isolated from the murine colon demonstrated that β-NAD is degraded to ADPR by CD38, to AMP by ENPP1, and to ADO by NT5E.
We next sought to determine whether the three types of cells that form the postjunctional domain of the NEJ demonstrate cell-specific enzymatic activities that degrade eNAD. Cells were treated with substrates for longer periods of time than tissues to ensure a reliable detection of changes in substrate and product amounts in cell suspensions containing lower number of cells (hence lower amounts of enzymes) than intact colon muscularis. Purified SMCs failed to produce eADPR from eNAD, but were able to produce significant amounts of eADO, suggesting that SMCs likely express high levels of NT5E and possibly some ENPP1, but lack CD38. These functional data are in agreement with previous studies demonstrating that the expression of Nt5e is highest in purified SMCs, lower in PDGFRα+ cells and not resolved in ICC (Peri et al., 2013, Lee et al., 2015, Breland et al., 2019). These studies also demonstrated that SMCs express Enpp1, but the amounts of Cd38 were low in FACS-purified SMCs.
Previous studies using quantitative reverse transcription-polymerase chain reaction (qRT-PCR) (Peri et al., 2013) and deep-sequencing analyses (Lee et al., 2015, Breland et al., 2019) have demonstrated very low expression levels of Cd38, Nt5e and Enpp1 in ICC purified by FACS. The present study also found that purified ICC are not involved in a significant way in the extracellular metabolism of β-NAD. ICC may be involved in modulation of purinergic neurotransmission in the colon (Durnin et al., 2017). However, this effect appears to be due to nitrergic modulation of purine neurotransmitter release and not an effect of ICC on purine metabolism.
Superfusion of PDGFRα+ cells with eNAD resulted in insignificant changes in eADPR in the absence or presence of cells, suggesting that either eADPR was not formed from eNAD or that eADPR was formed, but was quickly degraded to eAMP and eADO. The latter possibility is likely, as purified PDGFRα+ cells are enriched in Enpp1 and show moderate expression of Nt5e and little Cd38 (Peri et al., 2013, Lee et al., 2015, Breland et al., 2019). Since PDGFRα+ cells generated eADO from eNAD, it is likely that these cells contribute to termination of the neurotransmitter action of β-NAD. These cells are also enriched in Adora1, the gene for the adenosine A1 receptor (Peri et al., 2013, Lee et al., 2015, Breland et al., 2019). Thus, ADO formed from the neurotransmitter β-NAD in the NEJ would tend to activate adenosine receptors on PDGFRα+ cells. Further studies are warranted to determine the role of adenosine A1 receptors in PDGFRα+ cells in colon muscularis.
CD38 activity appears to be present in whole tissue and in unsorted cells, but not in SMCs, ICC or PDGFRα+ cells, suggesting that cell types that are not included in the SIP syncytium are the primary location of CD38 in the colon. We have previously demonstrated that CD38 is present in perivascular nerve terminals (Smyth et al., 2006). In analogy with such observations, enteric neurons or enteric glia might be principal locations of CD38 in the colon muscularis. Pilot studies that attempted to determine the localization of CD38 in the mouse colon, however, failed to produce reliable information due to the poor quality of several anti-CD38 antibodies that were tested. Further studies are warranted to determine the exact localization of CD38 in the colon that contains a variety of cell types.
The main findings of the present study suggest differential prevalence of β-NAD-metabolizing enzymatic activities in the cell types of the SIP syncytium. These findings are in remarkable congruence with previous molecular studies reporting quantitative comparisons of specific gene expression in the SIP cells (e.g., Peri et al., 2013, Lee et al., 2015, Breland et al., 2019).
In summary (Fig. 6), β-NAD is degraded in tunica muscularis of the murine colon to ADPR, AMP and ADO by CD38, ENPP1, and NT5E, respectively. CD38 metabolizes β-NAD to ADPR and appears to be expressed primarily in cells that are not involved in the SIP syncytium. ENPP1 metabolizes β-NAD and ADPR to AMP in colonic tunica muscularis, but the level of its contribution to the β-NAD metabolism in SIP cells remains elusive. NT5E metabolizes AMP to ADO and it is functionally expressed in SMCs and to a lesser extent in PDGFRα+ cells. ICC are not equipped for significant β-NAD metabolism in the colon whereas PDGFRα+ cells and SMCs contribute to the junctional and extrajunctional metabolism of β-NAD in colonic tunica muscularis. Further studies are needed to understand the role of other cell types, such as enteric neurons, enteric glia or resident macrophages in the extracellular breakdown of β-NAD.
Figure 6.
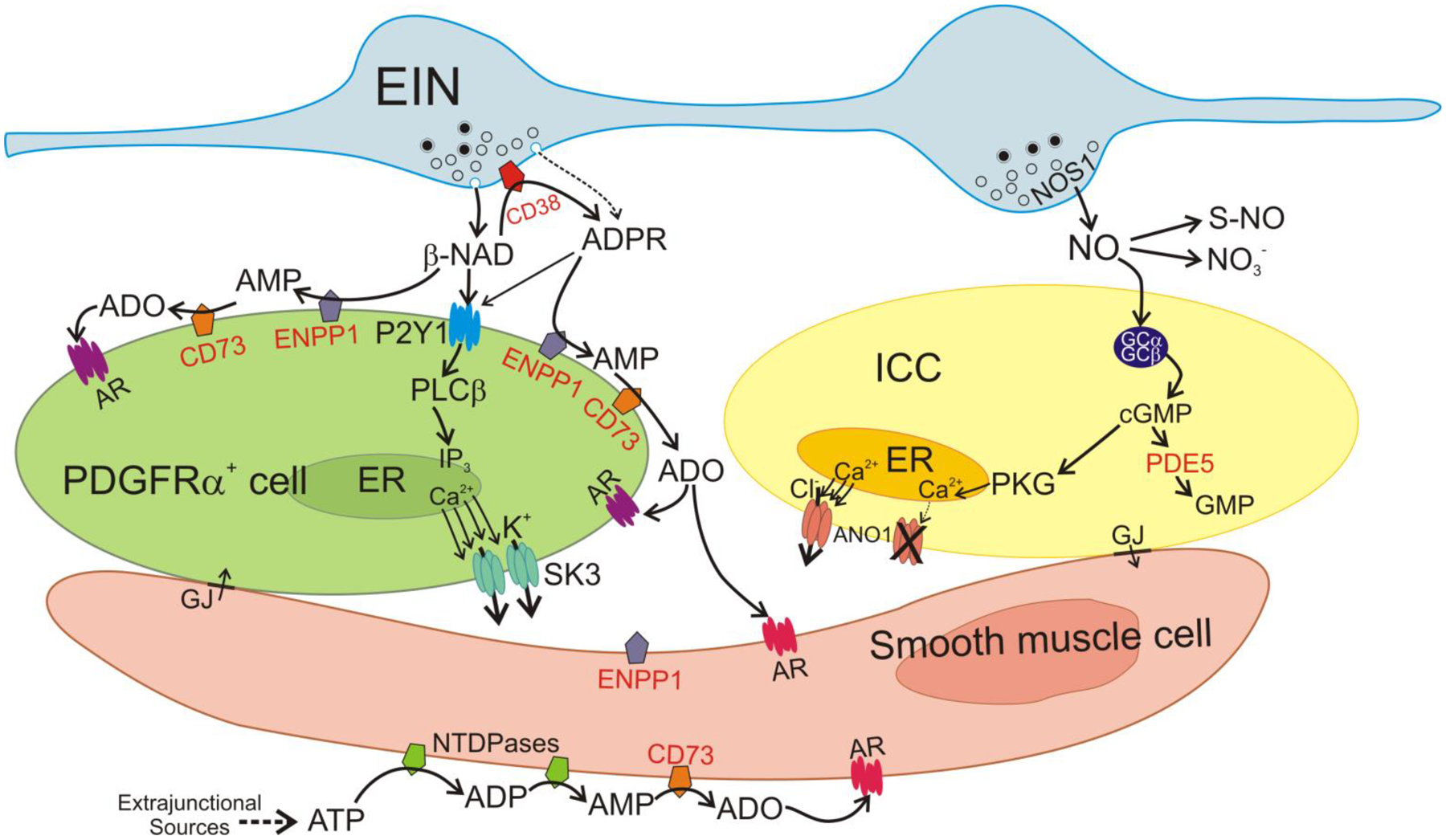
Mechanisms of non-peptide enteric inhibitory neurotransmitters and their metabolism. The SIP syncytium is composed of SMCs, ICC and PDGFRα+ cells, and it is innervated by enteric excitatory (not shown) and inhibitory motor neurons (EIN). Firing of EIN causes release of NO and β-NAD into the neuroeffector junction (NEJ). ADPR is also possibly released as a primary neurotransmitter (Durnin et al., 2012). The primary receptors mediating purinergic responses in the GI tract are P2Y1 receptors (Hwang et al., 2012, gallego et al., 2012, Gallego et al., 2006). P2Y1 receptors are dominantly expressed by PDGFRα+ cells (Peri et al., 2013). β-NAD binds to P2Y1 in the membrane of PDGFRα+ cells. P2Y1 receptors couple through Gq/11 to activate phospholipase Cβ (PLCβ) and generate IP3. IP3 binds to IP3 receptors in the ER membrane, increases the release of Ca2+ from stores and activates small-conductance K+ channels (SK3). SK3 channels generate outward currents that conduct to SMCs through gap junctions (GJ), producing a net inhibitory input of membrane hyperpolarization and reduced excitability and contractions of SMCs. β-NAD comes in contact with membrane-bound enzymes in the NEJ and is metabolized. CD38, likely present on EIN membranes, hydrolyses β-NAD to ADPR. ADPR also binds to P2Y1 and activates the same pathway activated by β-NAD. ADPR is also degraded to AMP by ENPP1expressed in PDGFRα+ cells. AMP is degraded to adenosine (ADO) by NT5E on membranes of PDGFRα+ cells and SMCs. ADO binds to adenosine receptors (AR) in PDGFRα+ cells and SMCs. Adora1 is the dominant AR expressed by PDGFRα+ cells (Ha et al., 2017). β-NAD can be directly degraded to AMP (bypassing ADPR formation) by ENPP1 on PDGFRα+ cells. Purine neurotransmitters and metabolites may also reach SMCs. SMCs express NT5E and hence high amounts of ADO can be formed. ADO can also bind to ARs on SMCs. The primary source for ATP in the colon tunica muscularis is extraneuronal (Durnin et al., 2013). ATP released from other sources that reaches SMCs is degraded by membrane-bound nucleotide tri- and diphosphatases (ENTDPases) to ADP and AMP. AMP is degraded to adenosine by NT5E in SMCs. NO released by EIN diffuses to ICC and SMCs (pathway in SMCs not shown) and binds to soluble guanylyl cyclase (sGS) that is made up of 2 subunits, GCα and GCβ. Binding of NO activates sGC and enhances production of cGMP which activates protein kinase G (PKG). Ca2+ is released in a stochastic manner spontaneously from the ER in ICC and activates Ca2+-activated Cl− channels (ANO1) (Drumm et al., 2019, Zhu et al., 2015). This generates an inward current that is conducted to SMCs via gap junctions and provides excitatory input to the SIP syncytium and increases contraction. PKG has multiple effects in ICC, but a major effect is to block Ca2+ release from the ER. This blocks activation of ANO1 and serves as a potent inhibitory signal in the SIP syncytium. NO is metabolized by oxidation to NO3− (intermediates not shown) or bound to activated thiol groups of proteins, forming S-nitrosothiols (S-NO). cGMP is metabolized by phosphodiesterases, mainly PDE5, which is strongly expressed in ICC and SMCs.
Supplementary Material
A Key Point List:
β-NAD is a key inhibitory neurotransmitter in the colon.
The neuroeffector junction in the gut consists of enteric motor neurons and SIP syncytium, including smooth muscle cells (SMCs), interstitial cells of Cajal (ICC), and cells expressing platelet-derived growth factor receptor α (PDGFRα+ cells).
Measuring metabolism of 1,N6-etheno-NAD (eNAD) in colonic tunica muscularis and in SMCs, ICC, and PDGFRα+ cells with HPLC-FLD, we report that 1) in tissues, eNAD is degraded to eADP-ribose, eAMP, and e-adenosine (eADO) by CD38, ENPP1, and NT5E, 2) with SMCs and PDGFRα+ cells, eNAD is metabolized to eADO by ENPP1 and NT5E, 3) eNAD is not metabolized by ICC, 4) NT5E is expressed chiefly by SMCs and moderately by PDGFRα+ cells, 5) SIP cells are not the primary location of CD38.
These data argue that the duration and strength of purinergic neurotransmission can be modulated by targeting multiple enzymes with specialized cellular distribution in the colon.
Acknowledgments
We are thankful for the excellent technical assistance of Nancy Horowitz, Byoung Koh and Robyn Highison.
Funding
This study was supported by NIH grants DK41315 and DK120759.
Funding: HHS | NIH | National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK): Violeta N Mutafova-Yambolieva, DK041315; HHS | NIH | National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK): Kenton M Sanders, DK041315; HHS | NIH | National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK): Kenton M Sanders, DK120759
Footnotes
Data Availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Competing interests
None
Reference List
- Baker SA, Hennig GW, Ward SM, & Sanders KM (2015). Temporal sequence of activation of cells involved in purinergic neurotransmission in the colon. J Physiol. 593(8); 1945–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck K, Friebe A, & Voussen B (2018). Nitrergic signaling via interstitial cells of Cajal and smooth muscle cells influences circular smooth muscle contractility in murine colon. Neurogastroenterol Motil 30, e13300. [DOI] [PubMed] [Google Scholar]
- Bobalova J, Bobal P, & Mutafova-Yambolieva VN (2002). High-Performance Liquid Chromatographic Technique for Detection of a Fluorescent Analogue of ADP-Ribose in Isolated Blood Vessel Preparations. Analytical Biochemistry 305, 269–276. [DOI] [PubMed] [Google Scholar]
- Breland A, Ha SE, Jorgensen BG, Jin B, Gardner TA, Sanders KM, & Ro S (2019). Smooth Muscle Transcriptome Browser: offering genome-wide references and expression profiles of transcripts expressed in intestinal SMC, ICC, and PDGFRalpha(+) cells. Sci Rep 9, 387–36607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brookes SJ (1993). Neuronal nitric oxide in the gut. J Gastroenterol Hepatol 8, 590–603. [DOI] [PubMed] [Google Scholar]
- Bult H, Boeckxstaens GE, Pelckmans PA, Jordaens FH, Van Maercke YM, & Herman AG (1990). Nitric oxide as an inhibitory non-adrenergic non-cholinergic neurotransmitter. Nature 345, 346–347. [DOI] [PubMed] [Google Scholar]
- Burns AJ, Lomax AE, Torihashi S, Sanders KM, & Ward SM (1996). Interstitial cells of Cajal mediate inhibitory neurotransmission in the stomach. Proc Natl Acad Sci U S A 93, 12008–12013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnstock G (2014). Purinergic signalling in the gastrointestinal tract and related organs in health and disease. Purinergic Signal 10, 3–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnstock G, Campbell G, Satchell D, & Smythe A (1970). Evidence that adenosine triphosphate or a related nucleotide is the transmitter substance released by non-adrenergic inhibitory nerves in the gut. Br J Pharmacol 40, 668–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Flora A, Zocchi E, Guidal L, Franco L, & Bruzzone S (2004). Autocrine and Paracrine Calcium Signaling by the CD38/NAD+/Cyclic ADP-Ribose System. Ann NY Acad Sci 1028, 176–191. [DOI] [PubMed] [Google Scholar]
- Drumm BT, Hwang SJ, Baker SA, Ward SM, & Sanders KM (2019). Ca(2+) signalling behaviours of intramuscular interstitial cells of Cajal in the murine colon. J Physiol 597, 3587–3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durnin L, Hwang SJ, Kurahashi M, Drumm BT, Ward SM, Sasse KC, Sanders KM, & Mutafova-Yambolieva VN (2014). Uridine adenosine tetraphosphate is a novel neurogenic P2Y1 receptor activator in the gut. Proc Natl Acad Sci U S A 111, 15821–15826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durnin L, Hwang SJ, Ward SM, Sanders KM, & Mutafova-Yambolieva VN (2012). Adenosine 5-diphosphate-ribose is a neural regulator in primate and murine large intestine along with beta-NAD(+). J Physiol 590, 1921–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durnin L, Lees A, Manzoor S, Sasse KC, Sanders KM, & Mutafova-Yambolieva VN (2017). Loss of nitric oxide-mediated inhibition of purine neurotransmitter release in the colon in the absence of interstitial cells of Cajal. Am J Physiol Gastrointest Liver Physiol 313, G419–G433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durnin L, Moreland N, Lees A, & Mutafova-Yambolieva VN (2016). A commonly used ecto-ATPase inhibitor, ARL-67156, blocks degradation of ADP more than the degradation of ATP in murine colon. Neurogastroenterol Motil 28, 1370–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durnin L, Sanders KM, & Mutafova-Yambolieva VN (2013). Differential release of beta-NAD(+) and ATP upon activation of enteric motor neurons in primate and murine colons. Neurogastroenterol Motil 25(3), e194–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fredholm BB, IJzerman AP, Jacobson KA, Linden J, & Muller CE (2011). International Union of Basic and Clinical Pharmacology. LXXXI. Nomenclature and classification of adenosine receptors--an update. Pharmacol Rev 63, 1–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallego D, Hernandez P, Clave P, & Jimenez M (2006). P2Y1 receptors mediate inhibitory purinergic neuromuscular transmission in the human colon. Am J Physiol Gastrointest Liver Physiol 291, G584–G594. [DOI] [PubMed] [Google Scholar]
- Gallego d, Gil V, Martinez-Cutillas M, Mane N, Martin MT, & jimenez m (2012). Purinergic neuromuscular transmission is absent in the colon of P2Y1 knocked out mice. J Physiol 590, 1943–1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaroni C, Knight GE, Ruan HZ, Glass R, Bardini M, Lecchini S, Frigo G, & Burnstock G (2002). P2 receptors in the murine gastrointestinal tract. Neuropharmacology 43, 1313–1323. [DOI] [PubMed] [Google Scholar]
- Gow AJ, Luchsinger BP, Pawloski JR, Singel DJ, & Stamler JS (1999). The oxyhemoglobin reaction of nitric oxide. Proc Natl Acad Sci U S A 96, 9027–9032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal RK, Rattan S, & Said SI (1980). VIP as a possible neurotransmitter of non-cholinergic non-adrenergic inhibitory neurones. Nature 288, 378–380. [DOI] [PubMed] [Google Scholar]
- Graeff R, Liu Q, Kriksunov IA, Kotaka M, Oppenheimer N, Hao Q, & Lee HC (2009). Mechanism of cyclizing NAD to cyclic ADP-ribose by ADP-ribosyl cyclase and CD38. J Biol Chem 284, 27629–27636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graeff RM, Franco L, De Flora A, & Lee HC (1998). Cyclic GMP-dependent and -independent effects on the synthesis of the calcium messengers cyclic ADP-ribose and nicotinic acid adenine dinucleotide phosphate. J Biol Chem 273, 118–125. [DOI] [PubMed] [Google Scholar]
- Ha SE, Lee MY, Kurahashi M, Wei L, Jorgensen BG, Park C, Park PJ, Redelman D, Sasse KC, Becker LS, Sanders KM, & Ro S (2017). Transcriptome analysis of PDGFRalpha+ cells identifies T-type Ca2+ channel CACNA1G as a new pathological marker for PDGFRalpha+ cell hyperplasia. PLoS One 12, e0182265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SJ, Blair PJ, Durnin L, Mutafova-Yambolieva V, Sanders KM, & Ward SM (2012). P2Y1 purinoreceptors are fundamental to inhibitory motor control of murine colonic excitability and transit. J Physiol 590, 1957–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SJ, Durnin L, Dwyer L, Rhee PL, Ward SM, Koh SD, Sanders KM, & Mutafova-Yambolieva VN (2011). beta-Nicotinamide Adenine Dinucleotide Is an Enteric Inhibitory Neurotransmitter in Human and Nonhuman Primate Colons. Gastroenterology 140, 608–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jamal Z, Afkham-Ebrahimi A, & Saggerson ED (1988). A novel assay for 5’-nucleotidase using 1,N6-etheno-AMP as substrate, and comments on the properties of the reaction product, ethenoadenosine. Biochem J 250, 369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keef KD, Saxton SN, McDowall RA, Kaminski RE, Duffy AM, & Cobine CA (2013). Functional role of vasoactive intestinal polypeptide in inhibitory motor innervation in the mouse internal anal sphincter. J Physiol 591, 1489–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh SD, Dick GM, & Sanders KM (1997). Small-conductance Ca2+-dependent K+ channels activated by ATP in murine colonic smooth muscle. Am J Physiol Cell Physiol 273, C2010–C2021. [DOI] [PubMed] [Google Scholar]
- Kurahashi M, Mutafova-Yambolieva V, Koh SD, & Sanders KM (2014). Platelet-derived growth factor receptor-alpha-positive cells and not smooth muscle cells mediate purinergic hyperpolarization in murine colonic muscles. Am J Physiol Cell Physiol 307, C561–C570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HC (2001). Physiological functions of cyclic ADP-ribose and NAADP as calcium messengers. Annu Rev Pharmacol Toxicol 41, 317–345. [DOI] [PubMed] [Google Scholar]
- Lee HC, Zocchi E, Guida L, Franco L, Benatti U, & De Flora A (1993). Production and hydrolysis of cyclic ADP-ribose at the outer surface of human erythrocytes. Biochem Biophys Res Commun 191, 639–645. [DOI] [PubMed] [Google Scholar]
- Lee MY, Ha SE, Park C, Park PJ, Fuchs R, Wei L, Jorgensen BG, Redelman D, Ward SM, Sanders KM, & Ro S (2017). Transcriptome of interstitial cells of Cajal reveals unique and selective gene signatures. PLoS One %20;12, e0176031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MY, Park C, Berent RM, Park PJ, Fuchs R, Syn H, Chin A, Townsend J, Benson CC, Redelman D, Shen TW, Park JK, Miano JM, Sanders KM, & Ro S (2015). Smooth Muscle Cell Genome Browser: Enabling the Identification of Novel Serum Response Factor Target Genes. PLoS One 10, e0133751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linden J, Koch-Nolte F, & Dahl G (2019). Purine Release, Metabolism, and Signaling in the Inflammatory Response. Annu Rev Immunol 37, 325–347. [DOI] [PubMed] [Google Scholar]
- McConalogue K, Furness JB, Vremec MA, Holst JJ, Tornoe K, & Marley PD (1995). Histochemical, pharmacological, biochemical and chromatographic evidence that pituitary adenylyl cyclase activating peptide is involved in inhibitory neurotransmission in the taenia of the guinea-pig caecum. J Auton Nerv Syst 50, 311–322. [DOI] [PubMed] [Google Scholar]
- Munshi C, Aarhus R, Graeff R, Walseth TF, Levitt D, & Lee HC (2000). Identification of the Enzymatic Active Site of CD38 by Site-directed Mutagenesis. J Biol Chem 275, 21566–21571. [DOI] [PubMed] [Google Scholar]
- Mutafova-Yambolieva VN & Durnin L (2014). The purinergic neurotransmitter revisited: A single substance or multiple players? Pharmacol Ther 144(2):162–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutafova-Yambolieva VN, Hwang SJ, Hao X, Chen H, Zhu MX, Wood JD, Ward SM, & Sanders KM (2007). beta-Nicotinamide adenine dinucleotide is an inhibitory neurotransmitter in visceral smooth muscle. PNAS 104, 16359–16364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peri LE, Sanders KM, & Mutafova-Yambolieva VN (2013). Differential expression of genes related to purinergic signaling in smooth muscle cells, PDGFRalpha-positive cells, and interstitial cells of Cajal in the murine colon. Neurogastroenterol Motil 25, e609–e620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders KM, Hwang SJ, & Ward SM (2010). Neuroeffector apparatus in gastrointestinal smooth muscle organs. J Physiol 588, 4621–4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders KM, Koh SD, Ro S, & Ward SM (2012). Regulation of gastrointestinal motility--insights from smooth muscle biology. Nat Rev Gastroenterol Hepatol 9, 633–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders KM & Ward SM (2019). Nitric oxide and its role as a non-adrenergic, non-cholinergic inhibitory neurotransmitter in the gastrointestinal tract. Br J Pharmacol 176, 212–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders KM, Ward SM, & Koh SD (2014). Interstitial cells: regulators of smooth muscle function. Physiol Rev 94, 859–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth LM, Breen LT, Yamboliev IA, & Mutafova-Yambolieva VN (2006). Novel localization of CD38 in perivascular sympathetic nerve terminals. Neuroscience 139, 1467–1477. [DOI] [PubMed] [Google Scholar]
- Todorov LD, Mihaylova-Todorova S, Westfall TD, Sneddon P, Kennedy C, Bjur RA, & Westfall DP (1997). Neuronal release of soluble nucleotidases and their role in neurotransmitter inactivation. Nature 387, 76–79. [DOI] [PubMed] [Google Scholar]
- Yamboliev IA, Smyth LM, Durnin L, Dai Y, & Mutafova-Yambolieva VN (2009). Storage and secretion of beta-NAD, ATP and dopamine in NGF-differentiated rat pheochromocytoma PC12 cells. Eur J Neurosci 30, 756–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu MH, Kim TW, Ro S, Yan W, Ward SM, Koh SD, & Sanders KM (2009). A Ca(2+)-activated Cl(−) conductance in interstitial cells of Cajal linked to slow wave currents and pacemaker activity. J Physiol 587, 4905–4918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu MH, Sung TS, O’Driscoll K, Koh SD, & Sanders KM (2015). Intracellular Ca(2+) release from endoplasmic reticulum regulates slow wave currents and pacemaker activity of interstitial cells of Cajal. Am J Physiol Cell Physiol 308, C608–C620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmermann H, Zebisch M, & Strater N (2012). Cellular function and molecular structure of ecto-nucleotidases. Purinergic Signal 8, 437–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.