

Abstract
Atrial Fibrillation (AF) is the most common supraventricular tachyarrhythmia that is typically associated with cardiovascular disease (CVD) and poor cardiovascular health. Paradoxically, endurance athletes are also at risk for AF. While it is well-established that persistent AF is associated with atrial fibrosis, hypertrophy and inflammation, intensely exercised mice showed similar adverse atrial changes and increased AF vulnerability, which required tumor necrosis factor (TNF) signaling, even though ventricular structure and function improved. To identify some of the molecular factors underlying the chamber-specific and TNF-dependent atrial changes induced by exercise, we performed transcriptome analyses of hearts from wild-type and TNF-knockout mice following exercise for 2 days, 2 or 6 weeks of exercise. Consistent with the central role of atrial stretch arising from elevated venous pressure in AF promotion, all 3 time points were associated with differential regulation of genes in atria linked to mechanosensing (focal adhesion kinase, integrins and cell-cell communications), extracellular matrix (ECM) and TNF pathways, with TNF appearing to play a permissive, rather than causal, role in gene changes. Importantly, mechanosensing/ECM genes were only enriched, along with tubulin- and hypertrophy-related genes after 2 days of exercise while being downregulated at 2 and 6 weeks, suggesting that early reactive strain-dependent remodeling with exercise yields to compensatory adjustments. Moreover, at the later time points, there was also downregulation of both collagen genes and genes involved in collagen turnover, a pattern mirroring aging-related fibrosis. By comparison, twofold fewer genes were differentially regulated in ventricles vs. atria, independently of TNF. Our findings reveal that exercise promotes TNF-dependent atrial transcriptome remodeling of ECM/mechanosensing pathways, consistent with increased preload and atrial stretch seen with exercise. We propose that similar preload-dependent mechanisms are responsible for atrial changes and AF in both CVD patients and athletes.
Keywords: atrial fibrillation (AF), RNA sequencing (RNA-seq), tumor necrosis factor, inflammation, collagen, mechanotransduction, heart, exercise
Introduction
Atrial fibrillation (AF) is the most common supraventricular tachyarrhythmia seen in clinical practice (Calkins et al., 2018), with its prevalence predicted to double by 2060 (Krijthe et al., 2013). AF is easily identified in electrocardiographic (ECG) recordings by the presence of rapid rates (typically > 110 beats/min) coupled with regular-irregular QRS complexes and is associated with impaired cardiac output regulation, non-pumping “quivering” atria, and an increased risk of stroke. The etiology of AF is complex, with most patients being elderly and also suffering from cardiovascular diseases (esp., hypertension, heart failure, valve disease) or having increased risk for cardiovascular disease (sleep apnea, hyperthyroidism, obesity, and diabetes) (Odutayo et al., 2016; Staerk et al., 2017). A common physiological feature of all these conditions, including aging, is increased filling pressures (De Jong et al., 2011; Park et al., 2014) which is believed to drive the atrial fibrosis, inflammation and hypertrophy invariably seen in AF patients. The importance of addressing the AF epidemic is highlighted by the > twofold increase in all-cause mortality seen in patients with AF.
Paradoxically, the risk of AF is also increased in veteran endurance athletes (Mont et al., 2002; Redpath and Backx, 2015), despite well-established evidence of beneficial physiological remodeling of the ventricles. Although the underlying basis for exercise-induced AF and its differential effects on the atria and ventricles are unclear, it is well known that intense exercise is associated with marked elevations in filling pressure (Reeves et al., 1990). Moreover, rodent models have established that endurance exercise causes atrial fibrosis, inflammation, and hypertrophy (Guasch et al., 2013; Aschar-Sobbi et al., 2015), as seen in persistent AF patients (Oakes et al., 2009; Qu et al., 2009; Gramley et al., 2010). We previously demonstrated that the exercise-induced atrial changes were prevented by pharmacological and genetic blockade of tumor necrosis factor (TNF), a mechanosensitive and pro-inflammatory cytokine (Aschar-Sobbi et al., 2015; Lakin et al., 2019). However, when TNF blockade was introduced 3 weeks after the beginning of intense exercise, cardioprotection was lost, suggesting that pathways linked to exercise-induced adverse atrial remodeling occur early in the response to exercise.
In this study, we performed bioinformatic analyses of transcriptomic changes in atria and ventricles induced by endurance exercise in wild-type and TNF knockout mice. Our results demonstrate that exercise induces TNF-dependent differential activation (enrichment) of pathways associated with mechanosensitive ECM remodeling that are time-dependent and differ between atria and ventricles, in a manner consistent with preferential stretch of atria in response to exercise-induced elevations in venous pressure. Our findings provide insight into the chamber-specific roles of TNF and mechanical strain in cardiac changes induced by exercise and support the general conclusion that exercise-induced adverse atrial remodeling is preload-dependent as seen in AF associated with aging and poor cardiovascular health.
Materials and Methods
Experimental Animals and Endurance Exercise Training Models
This study was carried out in accordance with the recommendations of the Canadian Council of Animal Care. The protocol was approved by the Division of Comparative Medicine at the University of Toronto and York University Animal Care Committee. Mice swam for 2 days (4-sessions), 2 or 6 weeks against water currents in containers, as described previously (Aschar-Sobbi et al., 2015). For the 6 week group, 6 week old CD1 male mice (body weight = 28–34 g, Charles River Laboratories) were acclimatized by swimming twice daily for 30 min (separated by 4 h) after which the duration of the swims was increased by 10 min per day until the duration reach 90 min per swim. Thereafter, the mice swam 2 times per day for 90 min/session for 6 weeks. The swim protocol was similar for the 2 week group, except mice only swam for 2 weeks after acclimatization. These 2 week mice were bred in-house after backcrossing TNF knockout (TNF-KO) mice (c57b, Taconic model #1921) into a CD1 background a minimum of 8 times. After reaching 10 weeks of age, wild-type (WT) mice and their TNF-KO littermates were acclimatized as described for the 6 week mice and swam for 2 weeks. The breeding and housing for the 2 day group were as described for the 2 week group. At 10–12 weeks of age, these mice were familiarized for 3 days with a 10 min swim per day, followed by two consecutive days of twice daily, 90 min swims. The sedentary mice for all groups consisted of the age-matched animals who were placed in swim containers without a water current for 5 min each session to ensure similar handling.
Tissue Harvesting
Prior to harvesting of atrial and ventricular tissue, 0.2 ml of heparin was injected intraperitoneally to prevent blood clotting. After 5 min, mice were anesthetized using 2.5% isoflurane and sacrificed via cervical dislocation. Hearts were quickly excised and placed into cold phosphate-buffered saline (PBS) to prevent protein or RNA degradation as well as cell apoptosis. In cold PBS, the left atrial appendage (LAA) and left ventricular (LV) free wall were separated and collected for RNA extraction. For our 2 day swim protocol, tissue was harvested 2 h after last swim. For 2 and 6 week studies, tissue was harvested 24 h after the last swim session.
RNA Extraction
Total RNA was extracted for both atria and ventricles using RNeasy Mini Kit (Qiagen), where the silica membrane used for this protocol removes RNA shorter than 200 nucleotides, including 5S/5.8S rRNAs, microRNAs and all tRNAs. All RNA samples were stored at −80°C until use. Initial RNA quantity and quality analyses were done using the Nanodrop2000 spectrophotometer (Thermo Fisher Scientific). The integrity and concentration of the RNA was determined with capillary electrophoresis by Agilent 2100 (Bioanalyzer, Agilent Technologies, Santa Clara, CA, United States) and was performed by the UHN Princess Margaret Genomics Centre (MaRS Centre, TMDT, Toronto, ON, Canada).
RNA Library Preparation and Sequencing (RNAseq)
Both cDNA library generation and RNA sequencing were performed by the Donnelly Sequencing Centre at the University of Toronto (Toronto, ON, Canada) using Illumina’s TruSeq stranded mRNA enrichment for library preparation. cDNA was generated from amplified mRNA, which was purified from total RNA. To purify mRNA, oligoT and 3′ poly A tails were hybridized to mRNA only during transcription in the nucleus. Magnetic beads linked to poly T oligo were used to selectively isolate mRNA. The purified mRNA was then fragmented by chemical shear and size selection was performed to generate mRNA fragments > 100 bps. Fragmented mRNA was reversed transcribed by reverse transcriptase and random primers to generate first strand cDNA. Second strand cDNA generated using DNA polymerase I and RNase H. Adapters were ligated on cDNA fragments, followed by enrichment using PCR to generate the final mRNA-derived cDNA library. For 2 week study samples, HiSeq2500 single-end sequencing was performed at 51 cycles. For 2 day study samples, NextSeq500 single-end sequencing was performed at 75 cycles. Technical replicates were generated by running each sample across 2 lanes to ensure there is no technical variability.
Analysis of RNA-Sequencing and Microarray Data
For RNA sequencing (RNA-seq), raw sequencing data were processed using the UseGalaxy server (Afgan et al., 2016). Quality control assessments were performed using FastQC version 0.11.7 (Wingett and Andrews, 2018). Technical replicates were concatenated, then aligned to the mus musculus 10 (mm10) reference genome and quantified using Salmon 0.9.1 with default options (Kim et al., 2015; Patro et al., 2017; Srivastava et al., 2019). A gene was considered differentially expressed if the p-value was less than 0.05 using Student’s t-test. Principal component analysis (PCA) was performed to identify the variance that lies between samples. R script was used for PCA analysis (Ringner, 2008; Jolliffe and Cadima, 2016). An example analysis is shown in Supplementary Figure 1. For our microarray analysis, normalized and processed microarray datasets from sedentary and 6 week exercise left atrial appendages (LAA) were acquired from ArrayExpress (E-MTAB-3106) (Aschar-Sobbi et al., 2015).
Gene Set Enrichment Analysis (GSEA 4.0.3) was used to identify differentially expressed gene sets (p < 0.05 and FDR < 0.20) (Subramanian et al., 2005). The C2 curated gene sets (c2 Canonical pathways) and C5 GO gene sets were used for the GSEA analysis. Weighted enrichment statistic was used and Signal2Noise metric was used for ranking genes. Nominal P-values of each gene set were given using 10,000 and 1,000 permutations of gene sets for analysis using c2 canonical pathway database and c5 GO database, respectively. Gene sets with fewer than 15 genes or more than 500 genes were excluded. Enrichment maps were generated to visually identify clusters of gene sets on Cytoscape 3.4.0 using the gene sets that were statistically different (p < 0.05 and FDR < 0.20) between two groups. Edge and node cut-off values were set to 0.375 (default) and 0.1, respectively. Wordcloud version 3.1.3 was used to annotate clusters. The “difference-of-the-difference” analysis was conducted to identify TNF-dependent pathways by subtracting the gene sets that were significantly differentially regulated between sedentary and swim in WT samples from those differentially regulated between sedentary and swim in KO samples.
Heat maps and hierarchal clustering of genes were performed with MATLAB (version 2016a), using all genes belonging to ECM, focal adhesion, integrin and cell-cell communication- related gene sets. Comparisons of transcripts per kilobase million (TPM) expression for individual genes from RNA-seq between two groups utilized unpaired (two-tailed) t-test, and genes with P-values of less than 0.05 were considered significant. All genes in the heat maps are significantly different between sedentary and swim WT (p < 0.05).
Telemetric Hemodynamics
Radiofrequency emitting hemodynamic telemetry devices (Data Sciences International, Inc.) were implanted sub-dermally into the interscapularis region. A fluid-filled catheter was inserted into the right common carotid artery and advanced into the left ventricle. After 7 days of recovery, a 30 min baseline recording preceded an acute, 90 min swim exercise bout. Left ventricular end-diastolic pressure (LVEDP) was used as an index of left atrial pressure. Data were analyzed using Ponemah Physiological Platform analysis software (Data Sciences International, Inc.).
Cardiac Electrical Remodeling and Arrhythmia Vulnerability
Electrical properties and arrhythmia inducibility were assessed as previously described (Aschar-Sobbi et al., 2015). For these measurements, mice were anesthetized (1.5% isoflurane and oxygen mixture) followed by isolation of the right jugular vein and insertion of a 2.0F octapolar recording/stimulation EP catheter (CI’BER Mouse, Numed), which was subsequently advanced into the right ventricle. Programmed electrical stimulations were delivered to the right atria or right ventricle to assess arrhythmia inducibility. All stimulations were delivered at a magnitude of 1.5 times capture threshold and 1 ms pulse duration. Effective refractory periods (ERPs) were determined by delivering nine pulses at 20 ms below the R-R interval followed by an extra stimulation. The S2 coupling interval was initially delivered above capture (∼40 ms) and reduced by 5 ms increments and adjusted until capture was achieved. For arrhythmia induction, 27 pulses at 40 ms intervals were applied to each chamber and reduced at 2 ms decrements to 20 ms. In the absence of inducibility, a second protocol of 20 trains (every 1.5 s) of 20 pulses (2 ms duration) at a 20 ms interpulse interval were applied. Only reproducible episodes of rapid, chaotic, and continuous atrial or ventricular activity of more than 10 s were defined as a sustained arrhythmic event.
Histology and Macrophage Infiltration
For histology, hearts were perfused with PBS containing 1% KCl followed by 4% paraformaldehyde (PFA) in 0.01 M PBS and stored overnight in 4% PFA in 0.01 M PBS at 4°C. Hearts were then embedded in paraffin and 5 μm thin sections were stained with Picrosirius red (PSR) for collagen visualization and quantification. Collagen expression was quantified using ImageJ software as the ratio of positively stained tissue area to total tissue area of each section using the threshold method (Hadi et al., 2011). To quantify macrophage infiltration, antibodies against mouse Mac-3 (1:200, BD Pharminogen, Cat.#553322) were used with the streptavidin-biotin diaminobenzidine chromogen detection method (Vector Laboratories). Mac-3-positive cells were counted in at least three different left atrial appendage sections (100 μm apart) in each replicate and normalized to the total tissue area of each slice. Images were acquired using Metamorph software (Molecular Devices) and analyzed using ImageJ software.
In vitro ADAM17 Enzymatic Activity Assay
In vitro ADAM17 enzymatic activity was measured, as previously described (Shen et al., 2018). Briefly, atrial protein was extracted using a lysis buffer with a high yield of membrane-bound proteins (Cacodylic acid 10mM, NaCl 150 nM, ZnCl2 1 μM, CaCl2 20 mM, NaN3 1.5 mM, Triton X-100 1%, pH 5.0). Mca-Pro-Leu-Ala-Gln-Ala-Val-Dpa-Arg-Ser-Ser-Ser-Arg-NH2 fluorogenic peptide substrate III (R&D Systems, ES003) was used as the substrate for ADAM17. Mca-Pro-Leu-OH (Bachem, M-1975) calibration standard was used to calculate the conversion factor, and recombinant mouse ADAM17 (R&D Systems, 2978-AD) served as a positive control. The ADAM17 activity assay was carried out per the R&D systems protocol. A total amount of 5 μg protein was used for ADAM17 enzymatic activity assay, which was run as a kinetic assay mode for 2 h. Each sample was run in triplicates. ADAM17 activity is expressed as pmol/min/μg tissue protein.
Gelatin Zymography
MMP2 and MMP9 activity levels were assessed by in vitro gelatin zymography, as previously described (Jana et al., 2020). In brief, equal amounts (20 μg) of non-reduced atrial tissue lysate were run on 8% SDS-PAGE gel containing 1 mg/ml gelatin. Following electrophoresis, gels were renatured with 2.5% Triton X-100 buffer for 60 min (room temperature). The gels were then put in calcium assay buffer (50 mM Tris-Cl, pH 7.5, 5 mM CaCl2, 150 mM NaCl) and incubated overnight (37°C). Gels were then stained with 0.05% Coomassie Blue G-250, and grayscale images were scanned and inverted for densitometric quantification. Band intensity was quantified using the inbuilt ImageQuant TL software (Version 7.0 GE Healthcare) and normalized to a loading control.
Immunofluorescent Analyses
Left atrial tissue from each heart was flash-frozen in mounting compound (OCT). Immunohistochemical staining was performed on 5 μm sections (all other staining). Immunostaining for OPN (ab8448; Abcam), SPARC (MAB941, R&D Systems), and GAPDH (#2118. Cell Signaling Technology) were performed on frozen OCT sections, as previously described (Sakamuri et al., 2016).
Protein Extraction and Western Blot Analyses
Flash-frozen atria were freeze-crushed in tissue lysis buffer containing EDTA-free protease inhibitor cocktails and processed for protein extraction and immunoblotting as previously described (Sakamuri et al., 2016). Antibodies used for immunoblotting were as follows: OPN (ab8448; Abcam), SPARC (MAB941, R&D Systems), and GAPDH (#2118, Cell Signaling Technology). Band intensities were quantified using densitometry analysis software (ImageQuant TL 7.0; GE) and values were normalized to GAPDH expressions for each sample.
Statistics
Statistical analyses of transcriptional changes are described above. Unpaired (two-tailed) t-test was used to assess differences in ADAM17 activity, gel zymography, and western blot analyses. P-values of less than 0.05 were considered significant.
Results
Consistent with previous work (Aschar-Sobbi et al., 2015), 6 weeks of swimming increased (P < 0.001) AF inducibility (Figure 1A), which was associated with increased (P < 0.003) left atrial (LA) inflammatory (Mac-3+) cell infiltrations (Figures 1B,C) and fibrotic remodeling (Figures 1D,E), as well as hypertrophy (not shown) in left atrial appendages (LAA). By contrast, the left ventricle (LV) showed enhanced function without fibrosis (Figures 1D,E), inflammation (data not shown) nor increased arrhythmia vulnerability in response to exercise, consistent with a chamber-specific effect of intense exercise.
FIGURE 1.
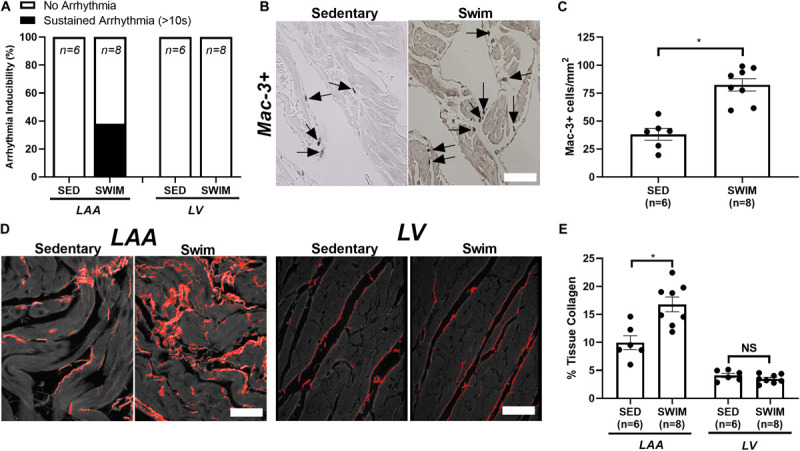
Intense exercise-induced adverse atrial remodeling and atrial fibrillation (AF) vulnerability. (A) 6 week swim exercise training is associated with increased atrial fibrillation (AF) inducibility, with no evidence of left ventricular (LV) arrhythmia vulnerability, compared to sedentary mice. (B,C) Swim exercise was associated with increased inflammatory cell infiltration (macrophage, Mac-3+) compared to sedentary mice. (D,E) Increased fibrosis (%tissue collagen) was observed in the left atrial appendage (LAA) with 6 week swim exercise compared to sedentary mice, with no elevations in fibrosis observed in the LV. Data presented as mean ± SEM. *P < 0.05.
Our previous microarray data from atria of mice after 6 weeks (Aschar-Sobbi et al., 2015) revealed exercise-induced transcriptional changes consistent with increases in inflammatory genes. As shown in Supplementary Figures 2,3, additional bioinformatics analyses verified enrichment in exercised atria of gene sets/clusters associated with inflammation (P < 0.05, FDR < 0.2) along with pathways involved in cell cycle regulation, mitochondrial fatty acid, and biological oxidation as well as metabolism/processing of DNA, RNA, and amino acids. Additionally, gene sets linked to mechanosensitive pathways (i.e., focal adhesion kinases (FAK), integrins, cell-cell communication) as well as extracellular matrix (ECM) remodeling (i.e., collagen formation, degradation, biosynthesis, assembly, cross-linking, and matrisome enzymes) were also differentially regulated between WT exercised and sedentary atria. However, despite the elevations in atrial fibrosis after 6 week of exercise, transcriptional levels of individual collagen genes (i.e., Col1a1, Col3a1, and Col4a1) were paradoxically reduced in WT exercised atria.
The unexpected lack of transcriptional elevations in collagen genes, combined with the inability of TNF blockade to prevent atrial fibrosis when started 3 weeks after exercise initiation (Aschar-Sobbi et al., 2015), suggests that pathways driving fibrosis are engaged early following exercise initiation. Therefore, we investigated the transcriptome responses after 2 weeks of exercise. In order to get greater gene coverage, we used deep RNA sequencing (RNA-seq) for these studies. Since TNF gene disruption prevented adverse atrial changes and exercise-induced AF in a chamber-dependent manner, RNA-seq was performed on atria and ventricles from both WT and TNF-KO mice. We first present the transcriptome changes with exercise in atria and discuss the ventricular results thereafter. For clarity, we illustrate our bioinformatic results by displaying each differentially regulated gene set as an individual dot (blue for enriched in swim and red enriched in sedentary). All closely related gene sets were represented by lines using Cytoscape 3.4.0 which allowed gene sets to be grouped into “gene clusters” with common and overlapping function and/or genes. To help focus our discussion, our graphic representations did not include clusters of gene sets with 2 or fewer related gene sets. The total number of differentially regulated gene sets and the number of gene sets in each cluster are presented in each figure.
Transcriptional Responses in Atria After 2 Weeks of Exercise
Principal component analysis (PCA) showed (an expected) distinct separation between atria and ventricles (Supplementary Figure 1). Surprisingly, there was little separation between WT and TNF-KO, regardless of chamber or exercise status. A remarkable feature of PCA results is the much larger effect of exercise on atrial vs. ventricular transcriptomes, for either genotype. These findings establish that exercise has a far greater impact on atrial vs. ventricular transcriptomes while TNF ablation has a relatively minor impact on exercise-induced changes in either chamber.
The specific gene sets that were differentially affected in WT atria by 2 weeks of exercise are represented in Figure 2 and Supplementary Table 1. The 2 week results in the TNF-KO mice are presented later. These analyses identified 112 differentially regulated (P < 0.05 and FDR < 0.2) gene sets, with 64 of these falling into clusters with common function (Figure 2A). Of these, gene sets linked to ATP synthesis and oxidative phosphorylation were enriched with swimming, which is not unexpected given the known cardiac bioenergetic adaptations to exercise (Vega et al., 2017). More interesting perhaps, was the finding that ∼28% of the differentially regulated gene sets were linked to mechanosensing (i.e., integrin/focal adhesion signaling), cell-cell communication, and ECM (i.e., collagen turnover, cross-linking, and matrisome remodeling), compared to only 15% after 6 weeks of exercise. As discussed later, differential regulation of these gene sets seems highly relevant because atrial stretch is central to AF pathogenesis (Vranka et al., 2007; Remes et al., 2008). Nevertheless, these gene sets were enriched in sedentary atria of WT mice (see Supplementary Table 2 for separation of these gene sets into different functional categories). Importantly, the expression levels of the major cardiac collagen types did not vary between the groups, suggesting that fibrotic responses to exercise after 2 weeks are limited to collagen turnover and stability (discussed below). Consistent with this notion, notch as well as the closely related Ephrin-related pathways were also enriched in the atria of WT sedentary compared to swim mice, which both play central roles in early embryogenesis (Sanz-Ezquerro et al., 2017) and are associated with hypertrophy and fibrosis in the heart as well as other tissues (Su et al., 2017).
FIGURE 2.
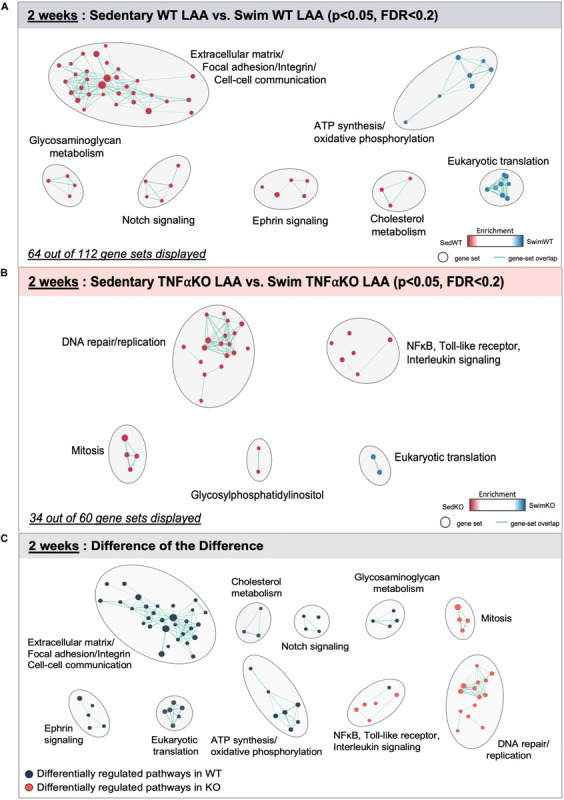
Differentially regulated pathways in the atria of 2 week swim exercised wild-type (WT) and tumor necrosis factor (TNF) knockout (KO) mice. (A) Gene set enrichment analysis (GSEA) and enrichment mapping showing clusters of differentially regulated pathways in the left atrial appendage (LAA) between WT 2 week swim (blue dots) and sedentary (red dots) mice. (B) Enrichment map showing clusters of differentially regulated pathways in the left atrial appendage (LAA) of TNF-KO 2 week swim and sedentary mice. (C) Enrichment map of the difference of the difference analysis revealing clusters of exercise-induced differentially regulated pathways in WT (blue dots) vs. TNF-KO (orange dots) mice. Only gene sets that form clusters are shown for clarity, with connecting lines indicating gene set overlap. Nominal P-value < 0.05, false discovery rate (FDR) < 0.20.
Despite the protective effects of TNF blockade on the atrial changes induced by exercise, the 2 week WT exercised atria did not show clear evidence of differential regulation of genes related to inflammation or TNF signaling, although several TNF-related pathways were just beyond our cut off criteria [NFκB-IKK (P = 0.04, FDR = 0.327), RelA (P = 0.06, FDR = 0.329), TNFR1 (P = 0.08, FDR = 0.384), and IL1R (P = 0.10, FDR = 0.384)]. By contrast, TNF-related gene sets [e.g., Toll-like receptor, interleukin, and TNF-related pathways (NF-κB and p38 MAPK)] were differentially regulated between swim and sedentary atria from TNF-KO mice, with enrichment in the sedentary group (Figure 2B and Supplementary Table 1). It is important to note that the number of gene sets related to ECM/mechanosensing was far less (i.e., 2 vs. 30) and did not form clusters in TNF-KO compared to WT mice, which aligns with the absence of exercised-induced atrial fibrosis when TNF is inhibited. Although at first glance this pattern of differential regulation with exercise in TNF-KO atria seems unexpected, we provide additional data below supporting the conclusion that TNF plays a permissive, rather than a primary role, in exercise-mediated atrial remodeling.
The TNF-dependence of the gene sets that are differentially regulated with exercise are summarized in Figure 2C as the “difference-of-the-difference” results (see section “Materials and Methods”). These analyses reveal, not unexpectedly, that exercise induces TNF-dependent changes in gene sets involved with ECM/mechanosensing, collagen production/turnover, fatty acid metabolism, oxidative phosphorylation as well as notch and ephrin signaling, with all these gene sets being enriched in the atria of WT compared to TNF-KO mice. To better understand the involvement of TNF in exercise-induced atrial remodeling, we further assessed the TNF-dependence of specific differentially regulated genes by generating heat maps of genes related to mechanotransduction and ECM (Figure 3). For these purposes, genes were separated into TNF-dependent (cluster 1 genes, whose expression differed in WT only) vs. TNF-independent genes (cluster 2 genes whose expression differ in both genotypes). As shown in Figure 3, far more genes were regulated in a TNF-dependent than a TNF-independent manner. Since TNF-KO abrogates atrial fibrosis as well as AF inducibility, we focused our attention initially on TNF-dependent collagen/ECM/mechanosensing genes (Supplementary Table 2). Of these, Mmp2 and Mrc2 are reduced in atria after 2 weeks of exercise which is of particular interest because these genes are also reduced in the age-related fibrosis of multiple tissues (Podolsky et al., 2020) and AF is strongly linked to aging (Heeringa et al., 2006).
FIGURE 3.

Heat map and clustering analysis of the tumor necrosis factor (TNF)-dependence of extracellular matrix (ECM)-, integrin-, and cell-cell communication-associated genes in 2 week swim exercised mice. Heat map and cluster analysis of individual genes belonging to ECM-, integrin-, and/or cell-cell communication-associated gene sets that are TNF-dependent (cluster 1) or TNF-independent (cluster 2). Note that all individual genes were enriched in sedentary (Sed) vs. swim mice. TNF-dependent (cluster 1) is: SedWT vs. SwimWT (P < 0.05) and SedKO vs. Swim KO (not P < 0.05). TNF-independent (cluster 2) is: SedWT vs. SwimWT (P < 0.05) and SedKO vs. SwimKO (P < 0.05).
It is also worth stating that many specific ECM/mechanosensing-related genes whose expression was downregulated in TNF-dependent manner with exercise have been linked previously to AF and ECM remodeling, including Comp (Zou et al., 2018; Thomas et al., 2019), Thbs2 (Yang et al., 2000), Ltbp1 and Fbn1 (Zhang et al., 2020; Figure 3 and Table 1). On the other hand, most genes linked to collagen production (i.e., Col1a1, Col1a2, Col3a1) were TNF-independent. Since TNF-KO abrogates fibrosis and AF inducibility, these results suggest (see section “Discussion”) that the cluster 2 genes are not central to the adverse atrial changes induced by exercise. In light of the impact of TNF on atria induced by exercise, it is worth pointing out that only two genes, Comp (TNF-dependent) and PIK3R2 (TNF-independent) are upregulated with exercise in swim WT vs. swim TNF-KO atria (see Discussion). Additional gene sets and individual genes linked to ECM/mechanosensing and/or AF are listed in Table 1 and Supplementary Tables 1–3, respectively.
TABLE 1.
Differentially regulated genes with 2 weeks exercise of the ECM-receptor, integrin, and cell-cell communication pathways associated with hypertrophic remodeling and/or atrial fibrillation (AF).
| Gene | Gene product | Directional change with swim exercise | Tumor necrosis factor (TNF) dependent? | Link(s) to fibrotic and/or hypertrophic remodeling and AF vulnerability |
| Adam10 | A disintegrin and metalloprotease (ADAM) 10 | ↓ | No | Processes membrane bound TNF to a soluble form, which in turn can induce MMPs and drive ECM remodeling (Manso et al., 2006; Murphy, 2008) |
| Adam12 | A disintegrin and metalloprotease (ADAM) 12 | ↓ | Yes | Regulator of MMPs and linked to prevention of cardiac hypertrophic and fibrotic remodeling through reductions in TGF-β- and integrinβ1-mediated FAK/Akt, ERK, and Smad signaling (Frangogiannis, 2012; Nakamura et al., 2020) |
| Adam17 | A disintegrin and metalloprotease (ADAM) 17 | ↓ | No | A metalloprotease and disintegrin, also known as TNF-α converting enzyme (TACE), that cleaves TNF to its soluble form and has been implicated in pressure overload-induced hypertrophic and fibrotic remodeling (Xu et al., 2016) and AF vulnerability (Weeke et al., 2014) |
| Adamts2 | Adamts2 | ↓ | Yes | Extracellular enzyme that activates pro-collagens I, II, III, and V (Wang et al., 2017b), and is a regulator of MMPs linked to cardiac hypertrophic and fibrotic remodeling in pressure-overload (Dong et al., 2013) |
| Bcl2 | B-cell lymphoma 2 | ↓ | No | Increased expression of BCL-2/BCL-2-associated X protein (BAX) linked to fibrosis and apoptosis in AF (Xu et al., 2013; Diao et al., 2016) |
| Bmp1 | Bone morphogenetic protein 1 | ↓ | Yes | A peptidase that cleaves the C-terminal pro-peptides of procollagen I, II, and III and mediates the proteolytic activation of lysyl oxidase LOX (Rodriguez and Martinez-Gonzalez, 2019) |
| Col4a4, Col5a1, and Col5a3 | Collagen type IV, alpha 4 chain; collagen type V, alpha 1 chain; and collagen type V, alpha 3 chain | ↓ | No | Collagen transcripts linked to AF and rhythm outcome following ablation (Husser et al., 2016) |
| Col6a6 | Collagen type VI Alpha 6 Chain | ↓ | No | Collagen protein encoding gene containing multiple von Willebrand factor (vWF) domains. Linked to extracellular matrix (ECM)-receptor interactions and is upregulated in AF (Zou et al., 2018) |
| Ctsb | Cathepsin B | ↓ | Yes | Lysosomal cysteine proteases localized in lysosomes and endosomes that function to degrade cellular substrates (Turk et al., 2000). Stimulated by TNF and linked to hypertrophic and fibrotic cardiac remodeling (Cheng et al., 2012b). Upregulated in AF (Thomas et al., 2019) |
| Erbb2 | HER-2; receptor tyrosine kinase | ↓ | No | A receptor involved in physiological cardiac adaptations and hypertrophic remodeling through the activation of MAPK, PI3K/Akt and Src/FAK signaling pathways (Vermeulen et al., 2016) |
| Fbln2 | Fibulin-2 | ↓ | Yes | ECM glycoprotein involved in angiotensin-II-mediated TGF-β signaling and cardiac hypertrophy (Zhang et al., 2014) |
| Fbn1 | Fibrillin 1 | ↓ | Yes | Large ECM glycoprotein and constituent of the myocardial ECM that is linked to reactive and reparative fibrotic remodeling (Bouzeghrane et al., 2005) and is upregulated in AF (Zhang et al., 2020) |
| Igf1 | Insulin-like growth factor-1 | ↓ | Yes | A hormone that has pleiotropic actions in the heart and mediated eccentric hypertrophy through PI3K- and MAPK-dependent mechanisms (Lavandero et al., 1998) |
| Itga4, Itgb1, Itgb6 | Integrin alpha-4, beta-1, and beta-6 precursors | ↓ | No | ECM proteins involved in mechanotransduction linked to AF incidence and rhythm outcome following ablation (Husser et al., 2016) |
| Ltbp1, Ltbp3, and Ltbp4 | Latent transforming growth factor (TGF)- β binding proteins | ↓ | Yes | Family of secreted multidomain proteins that bind to and regulate TGFβ-dependent activation and pro-fibrotic remodeling (Goumans and Ten Dijke, 2018) as well as AF (Thomas et al., 2019; Zhang et al., 2020) |
| Mfap4 | Microfibril-associated protein 4 | ↓ | Yes | A matricellular protein associated with AF and atrial fibrotic remodeling (Zhang et al., 2019) |
| Pgf | Placental growth factor | ↓ | Yes | A member of the VEGF (vascular endothelial growth factor) family known to induce cardiac fibroblasts to secrete TNF and other pro-hypertrophic factors (Accornero and Molkentin, 2011) |
| Pdgfb | Platelet-derived growth factor subunit B | ↓ | No | Increased PDGF-B expression linked to focal cardiac fibrosis and moderate cardiac hypertrophy (Gallini et al., 2016) |
| Pdgfra | Platelet-derived growth factor α receptor | ↓ | Yes | Cardiac mast cells synthesize and release PDGF-A and mediates both fibrosis and AF in pressure-overloaded hearts (Liao et al., 2010; Wang et al., 2017a) |
| Plod2 | Procollagen-lysine, 2-oxoglutarate 5-dioxygenase 2 (PLOD2) | ↓ | No | Lysyl hydroxylase linked to both TNF- and TGFβ1-mediated pyridinoline cross-linking inherent to fibrotic cardiac remodeling (van der Slot et al., 2005) |
| Pten | Phosphatase and tensin homolog (PTEN) | ↓ | No | A protein linked to increased pathological hypertrophy and progression to heart failure in response to biomechanical stress (Oudit et al., 2008) |
| Reln | Reelin | ↓ | No | A large secreted ECM glycoprotein that regulates pathways associated with ECM-receptor interaction and focal adhesion and its expression is linked to AF (Husser et al., 2016; Zhao et al., 2018; Zou et al., 2018) |
| Sdc4 | Syndecan-4 | ↓ | Yes | A transmembrane (type I) heparan sulfate proteoglycan that interacts with the ECM and is a primary determinant in collagen cross-linking and LOX induction in pressure-overload hearts (Herum et al., 2015) |
| Serpinh1 | Heat shock protein 47 (HSP47) | ↓ | Yes | A chaperone protein for collagen involved in cardiac injury-induced fibrosis and reductions in hypertrophy in cardiac pressure-overload (Khalil et al., 2019) |
| Sparc | Secreted protein acidic and cysteine rich (Sparc) | ↓ | Yes | A matricellular protein that is activated by several MMPs (i.e., MMP-2) and is elevated in cardiac hypertrophy and fibrosis in pressure-overload (Bradshaw et al., 2009) and aging (Bradshaw et al., 2010) |
| Src | Proto-ongogene tyrosine-protein kinase Src | ↓ | No | A tyrosine kinase that phosphorylates FAK (Frame et al., 2010) and is activated in response to integrin clustering and activation as well as LOX-mediated ECM remodeling (Schneider et al., 2008) |
| Thbs2 | Thrombospondin-2 | ↓ | Yes | A matricellular protein involved in myocardial matrix integrity (Schroen et al., 2004) and linked to protection against inflammatory-induced cardiac injury and dysfunction (Papageorgiou et al., 2012) |
| Tln1 | Talin-1 | ↓ | Yes | A ubiquitously expressed protein localized to costameres that become more prominent during mechanical stress-induced cardiac hypertrophy and fibrosis (Manso et al., 2013) and is upregulated in AF (Weeke et al., 2014) |
| Vwf | von Willebrand factor | ↓ | Yes | A prothrombotic plasma marker and index of endothelial damage and dysfunction that is prominently linked to AF incidence, rhythm outcome following ablation (Husser et al., 2016) and stroke risk (Zhong et al., 2018; Ye et al., 2020) |
Transcriptional Responses in Atria After 2 Days of Exercise
The observation that ECM/FAK/integrin gene sets pathways were generally enriched in sedentary atria at 2 and 6 weeks, despite atrial fibrosis at 6 weeks in exercised mice, prompted us to perform RNAseq measurements in hearts after only 2 days of exercise (i.e., 4-sessions of 90 min swims). Consistent with 2 week data, PCA showed the expected separation between atria and ventricles (Supplementary Figure 1). Surprisingly, while there were distinct separations between exercise and sedentary atrial samples, this was not true in ventricles, suggesting a much smaller effect of exercise on ventricular transcriptomic remodeling. Moreover, there was overlap between WT and TNF-KO, regardless of chamber or exercise status, suggesting TNF ablation may have a minor impact on acute exercise-induced changes.
The results of our GSEA analyses for mice after 2 days of exercise are summarized in Figure 4 and Supplementary Table 4. The data reveals that 101 gene sets were differentially regulated (P < 0.05, FDR < 0.2) between swim (total of 61 differentially regulated gene sets) vs. sedentary (40 gene sets) atria in WT mice, with 60 sets clustering into common pathways (Figure 4A). Importantly, unlike what was seen after 2 and 6 weeks of exercise, ECM/mechanosensing gene sets were now more enriched in exercise vs. sedentary atria (Supplementary Table 4). Acute exercise also induced enrichment in gene sets related to actin/tubulin folding, which cross-talks to many hypertrophic signaling pathways (i.e., MAPK/FGFR1/2) linked to dilated and hypertrophic cardiomyopathy (Caporizzo et al., 2019), as well as IQGAPs, PAKs, and AMPK activation (see section “Discussion”) (Hedman et al., 2015; Daskalopoulos et al., 2016). On the other hand, TNF-related gene sets in acute exercise were only differentially regulated in TNF-KO atria (Figure 4B) with reductions in many specific pro-inflammatory genes in exercised mice [i.e., toll-like receptor 10 (p < 0.008, FDR = 0.178), IL-2 (p = 0.018, FDR = 0.187), IL-2-STAT5 (p = 0.018, FDR = 0.240), TAK1 (P < 0.037, FDR = 0.278), and NFκB (P < 0.005, FDR = 0.124)].
FIGURE 4.
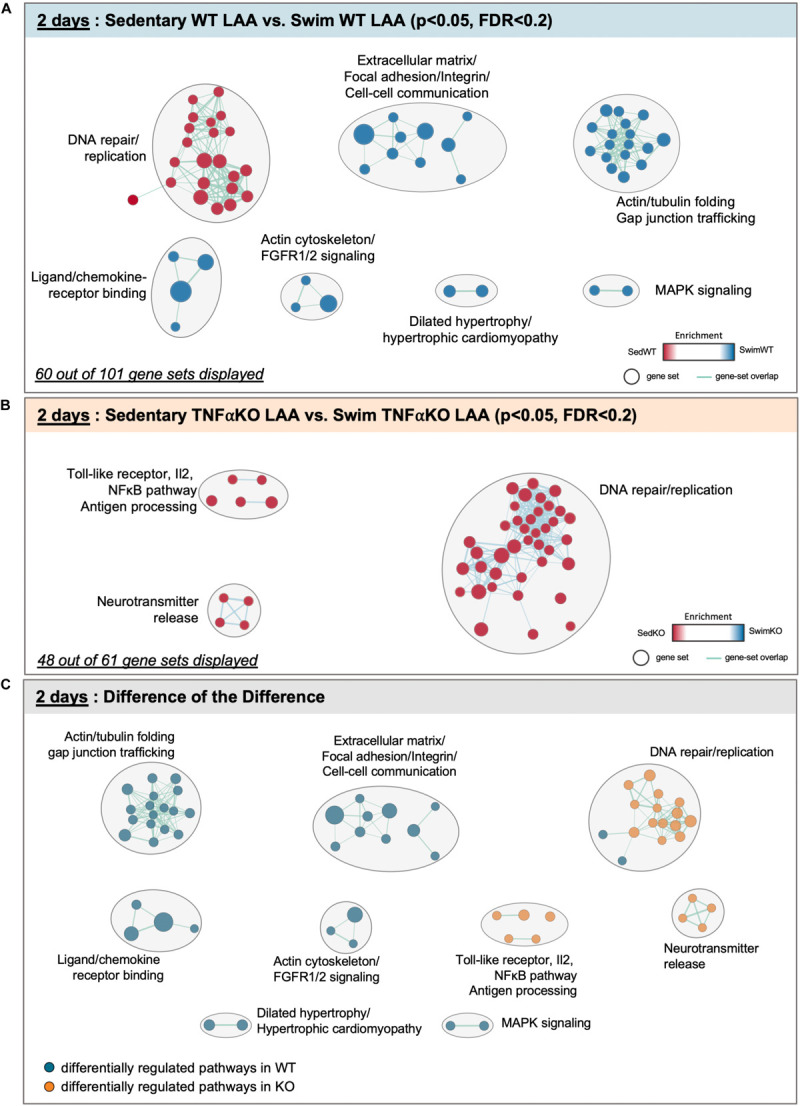
Differentially regulated pathways in the atria of 2 day swim exercised wild-type (WT) and tumor necrosis factor (TNF) knockout (KO) mice. (A) Gene set enrichment analysis (GSEA) and enrichment mapping showing clusters of differentially regulated pathways in the left atrial appendage (LAA) between WT 2 day swim (blue dots) and sedentary (red dots) mice. (B) Enrichment map showing clusters of differentially regulated pathways in the LAA of TNF-KO 2 day swim (blue dots) and sedentary (red dots) mice. (C) Enrichment map of the difference of the difference analysis revealing clusters of exercise-induced differentially regulated pathways in WT (blue dots) vs. TNF-KO (orange dots) mice. Only gene sets that form clusters are shown for clarity, with connecting lines indicating gene set overlap. Nominal P-value < 0.05, false discovery rate (FDR) < 0.20.
The difference-of-the-difference analyses after 2 day exercise clearly establish that gene sets involved in mechanosensitive pathways are uniquely differentially enriched in exercised WT atria, while TNF-related signaling and DNA replication/repair pathways are uniquely enriched in sedentary KO mice (Figure 4C). Heat maps of differentially (P < 0.05) regulated genes revealed (Figure 5) a distinct pattern after 2 days of exercise compared to 2 weeks. Now, many TNF-dependent genes linked to ECM remodeling as well as AF are upregulated with exercise in WT atria compared to sedentary, including Mmp14 (Simmers et al., 2016), Fgf2 (i.e., fibroblast growth factor 2, which activates p38 and induces cardiac hypertrophy as well as fibrosis) (Itoh and Ohta, 2013), Gsk3β (Sugden et al., 2008), and Tln1 (Manso et al., 2013). On the other hand, only Fgf2 was upregulated (P = 0.0142) in WT vs. KO exercised atria, suggesting TNF-dependent activation of FGF signaling pathways may be important early responses that regulate exercise-induced atrial remodeling. Nonetheless, these results suggest that acute exercise leads to TNF-dependent and TNF-independent transcriptome changes affecting ECM remodeling. Additional genes and their links to ECM/mechanosensing and/or AF are listed in Table 2.
FIGURE 5.
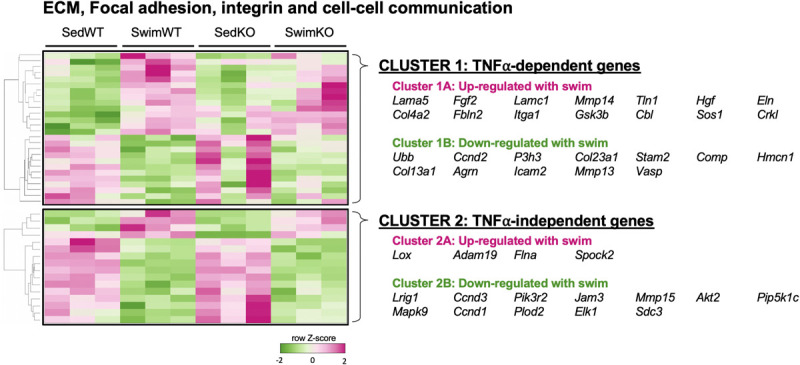
Heat map and clustering analysis of the tumor necrosis factor (TNF)-dependence of extracellular matrix (ECM)-, integrin-, and cell-cell communication-associated genes in 2 day swim exercised mice. Heat map and cluster analysis of up- or down-regulation of individual genes belonging to ECM-, integrin-, and/or cell-cell communication-associated gene sets that are TNF-dependent (cluster 1) or TNF-independent (cluster 2) in 2 day swim mice. TNF-dependent (cluster 1) is: SedWT vs. SwimWT (P < 0.05) and SedKO vs. Swim KO (not P < 0.05). TNF-independent (cluster 2) is: SedWT vs. SwimWT (P < 0.05) and SedKO vs. SwimKO (P < 0.05).
TABLE 2.
Differentially regulated genes with acute (2 day) exercise of the ECM-receptor, integrin, and cell-cell communication pathways associated with hypertrophic remodeling and/or atrial fibrillation (AF).
| Gene | Gene product | Directional change with swim exercise | Tumor necrosis factor (TNF) dependent? | Link(s) to fibrotic and/or hypertrophic remodeling and atrial fibrillation |
| Ccnd3 | Cyclin D3 | ↓ | No | A protein shown to be upregulated during hypertrophic growth (Busk et al., 2002) and linked to the pathogenesis of AF (Li et al., 2019) |
| Elk1 | Elk-1 | ↓ | No | A transcription factor that is phosphorylated via MEK/ERK kinases and plays a role in cardiac hypertrophy (Babu et al., 2000) as well as regulating stretch-mediated atrial natriuretic factor (ANF) expression (Mahida, 2013) |
| Fgf2 | Fibroblast growth factor 2 | ↑ | Yes | A multifunctional polypeptide that is upregulated by stress (Kardami et al., 2004) and involved in p38 MAPK-mediated cardiac hypertrophy (Tanaka et al., 1999) as well as fibrosis (Itoh and Ohta, 2013) |
| Gsk3β | Glycogen synthase kinase 3 beta | ↑ | Yes | An enzyme that serves as a hub for the regulation of both physiological and pathological hypertrophic and fibrotic remodeling (Lal et al., 2015) via TNF-related pathways (Sugden et al., 2008) |
| Itga1 | Integrin, alpha 1 | ↑ | Yes | Cell-cell and cell-matrix adhesion (ECM-receptor interactions), upregulated in AF and linked to rhythm outcome of AF catheter ablation (Husser et al., 2016) |
| Lama5 | Laminin subunit alpha-5 | ↑ | Yes | A component of the ECM, specifically basement membranes, that is upregulated in AF (Husser et al., 2016) |
| Mmp13 | Matrix metalloproteinase 13 | ↓ | Yes | A matrix metalloproteinase and collagenase that targets collagens I and III and is linked to cardiac hypertrophy and fibrosis in pressure overload hearts (Spinale, 2002) |
| Plod2 | Procollagen-lysine, 2-oxoglutarate 5-dioxygenase 2 (PLOD2) | ↓ | No | Lysyl hydroxylase linked to both TNF- and TGFβ1-mediated pyridinoline cross-linking inherent to fibrotic cardiac remodeling (van der Slot et al., 2005) |
| Tln1 | Talin-1 | ↑ | Yes | A protein mediating cell-cell adhesion linked to AF (Weeke et al., 2014) and becomes more prominent at costameres during mechanical stress and modulates hypertrophic and fibrotic remodeling (Manso et al., 2013) |
Transcriptional Changes in Ventricles With Exercise
As mentioned, after 2 weeks of exercise, PCA showed relatively small effects of exercise on ventricles (compared to atria) at all-time points. Before directly comparing LV and LA transcriptomic remodeling, we present the effects of exercise on LV genetic plasticity. After 2 weeks, genes sets associated with oxidative phosphorylation and ribosome translation were enriched in exercised WT mice while the genes sets related to ECM/mechanosensing (as in atria) and cardiomyopathy (i.e., HCM, DCM, ARVC), as well as notch, ephrin/Rho GTPases, and MAPK signaling were enriched in sedentary atria (Supplementary Figure 4A). By comparison, TNF-KO mice showed enrichment of gene sets linked to amino acid metabolism and TCA cycle with exercise while gene sets associated with ECM/mechanosensing, chemokine, interleukin, and T-cell/B-cell receptor pathways were enriched in the sedentary group (Supplementary Figure 4B). Interestingly, after performing the difference-of-the-difference analyses (Supplementary Figure 4C) the majority of gene sets that remained were related to TNF-mediated signaling with differential regulation in TNF-KO mice, suggesting that TNF also serves a role in LV remodeling with exercise, albeit less than in LA.
The results above establish that the gene sets inked to ECM/mechanosensitive pathways are quite similar after 2 weeks of exercise between the LA and LV (i.e., Figure 2C and Supplementary Figure 4C), even though the exercise-induced remodeling is different between the chambers. To more directly assess the differential effects of exercise and TNF on chamber-specific transcriptomic remodeling at 2 weeks, a modified difference-of-the-difference analysis was performed in which TNF-dependent pathways were determined by subtracting the gene sets that were significantly differentially regulated between swim in WT and KO samples within each chamber from those differentially regulated between LA and LV samples. As shown in Figure 6A, the number of TNF-dependent differentially regulated gene sets with exercise is far smaller for LVs (i.e., 4) vs. LAs (i.e., 43) at 2 weeks, with little overlap between the gene sets between chambers (i.e., 12). Indeed, while ECM/mechanosensitive pathways were enriched in both the LAs and LVs of sedentary mice at 2 weeks, direct comparisons between chambers highlight the predominance of TNF-dependent differentially gene sets (Figure 6B), including ECM/integrin signaling, dilated/hypertrophic cardiomyopathy, and actin/tubulin folding, in the atria, which reinforces the chamber-specific effects of both exercise and TNF on transcriptome remodeling.
FIGURE 6.
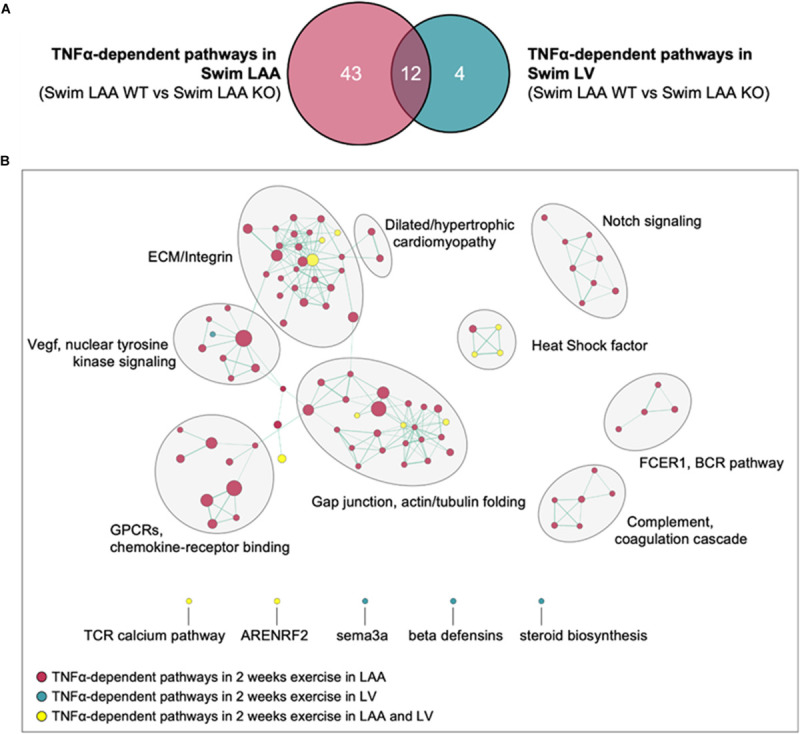
Differential roles of TNF in exercise-induced transcriptomic remodeling with 2 week swim exercise in the left atrial appendage (LAA) vs. left ventricle (LV). (A) Venn diagram of exercise- and TNF-dependent (i.e., Swim WT vs. swim KO) differentially regulated gene sets in the LAA vs. LV. (B) Gene set enrichment analysis (GSEA) and enrichment mapping showing clusters of TNF-dependent differentially regulated gene sets in the LAA (red dots) and LV (green dots). Gene sets that were TNF-dependent and differentially regulated in both LAA and LV are indicated by yellow dots. Only gene sets that form clusters are shown for clarity. Nominal P-value < 0.05, false discovery rate (FDR) < 0.20.
The differential impact of exercise on the LA and LV is also apparent in 2 day acutely exercised mice. Indeed, while we found clear evidence of ECM/mechanosensitive pathway enrichment in WT mice at 2 days in the atria (discussed above), when we assessed ventricular changes after 2 day acute exercise, only 16 gene sets were differentially regulated in WT LVs with most of the pathways linked to cell cycle and DNA replication processes (Supplementary Figure 5A). By contrast, exercised LV from TNF-KO mice had far greater numbers (211) of differentially regulated gene sets, including ECM/integrin, gap junction and actin/tubulin folding, as well as cell cycle and DNA repair/replication (Supplementary Figure 5B), all of which were enriched in sedentary mice. The difference-of-the-difference analysis (Supplementary Figure 5C) confirmed enrichment of the above pathways in TNF-KO compared to WT mice. Indeed, heat map and cluster analysis (Supplementary Figure 6) identified only three genes linked to ECM/mechanosensing, Mmp16, Dst, and Reln, that were upregulated in a TNF-dependent manner with acute swim, compared to the 14 genes identified in the LA. The absence of enrichment of gene sets linked to ECM/mechanosensitive pathways in the LV and their upregulation in the LA with 2 day acute exercise supports our contention that strain-dependent signaling mediates exercise-induced atrial remodeling.
Acute Effects of Exercise on Atrial Pressures and Collagen Metabolism/Remodeling
Since AF is primarily observed in cardiovascular conditions associated with elevated diastolic filling pressures (De Jong et al., 2011), we previously postulated (Aschar-Sobbi et al., 2015) that elevated filling pressures seen with exercise may explain the increased incidence of AF in endurance athletes. Consistent with this conjecture, we found that diastolic filling pressures increase from 10 mmHg to ∼45 mmHg within the first 10 min after mice begin swimming exercise (Figure 7). Thereafter the pressure falls to ∼20 mmHg over the next 30–50 min after which the filling pressure steadily rise to 40–45 mmHg after 90 min. Such increases in venous filling pressures would be expected to preferentially stretch the thin-walled and compliant atria, which may explain our observation of mechanosensitive and compensatory hypertrophic pathways being disproportionately activated in LAs compared to LVs.
FIGURE 7.
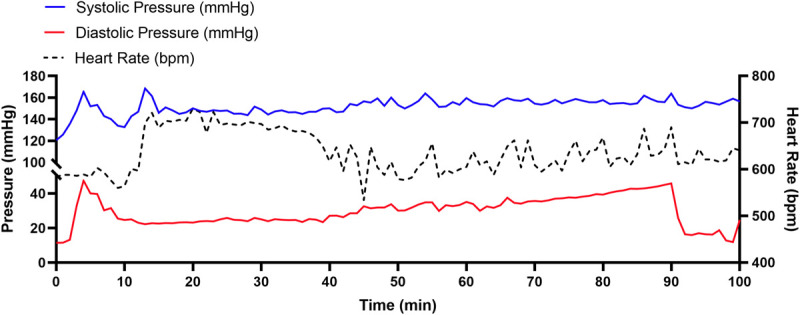
Representative left ventricular (LV) hemodynamic changes measured by implantable pressure-telemetry during an acute swim bout in mice. During a 90 min swim bout, LV diastolic filling pressures (red) increase rapidly from a baseline of ∼10 mmHg to ∼45 mmHg within the first 10 min. Thereafter, the pressure falls to ∼20 mmHg, only to rise steadily to ∼40–45 mmHg by the end of the 90 min swim.
Taken together, our findings demonstrate prominent time-dependent transcriptional changes in genes related to strain-dependent pathways in response to exercise. However, despite the induction of fibrosis by exercise, the absence of increased collagen expression at all-time points following exercise led us to explore the potential contribution of factors that have previously been shown to mediate post-translational changes in collagen maturation and deposition in the heart. For these studies, we used the 2 day mice and made the measurements 2 h following the final 90 min exercise bout, consistent with our RNA-seq measurements. Given that soluble TNF is required for exercise-induced atrial changes (Lakin et al., 2019), we first measured the activity of TNF-converting enzyme (TACE, or ADAM17), which is upregulated with mechanical stretch and releases active (soluble) TNF (Zhan et al., 2007). Indeed, TACE activity tended to be increased (P = 0.211) with swim (142 ± 4 pmol/min/μg, n = 5) compared to sedentary mice (129 ± 8 pmol/min/μg, n = 6), suggesting activation. It is conceivable that earlier assessment would have displayed even greater TACE activity since we previously found upregulation of TNF-dependent p38 MAPK signaling within 10 min post-exercise (Aschar-Sobbi et al., 2015). We also measured MMP2 and MMP9 activity since these are increased with atrial stretch as well as in CV disease (Yabluchanskiy et al., 2013). We found that pro-MMP2 activity was increased (P = 0.0003) while pro-MMP9 activity tended (P = 0.098) to be increased in atria after swim completion in 2 day swim compared to sedentary mice (Supplementary Figure 7A), establishing increased collagen turnover with acute exercise.
Since previous studies reported increases in matricellular proteins that mediate post-synthetic collagen turnover in several models (Frangogiannis, 2012; McDonald et al., 2018), we also measured osteopontin (OPN) and SPARC expression levels. Although SPARC expression in atria was unaffected by acute exercise (P = 0.562), OPN was decreased (P = 0.006), suggesting these matricellular proteins contribute minimally to adverse atrial remodeling in the early response to exercise.
A schematic overview of the time-dependent and atrial-specific exercise-induced TNF-dependent transcriptomic changes mediating adverse atrial remodeling and AF vulnerability in response to increased filling pressures and atrial stretch are shown in Figure 8.
FIGURE 8.
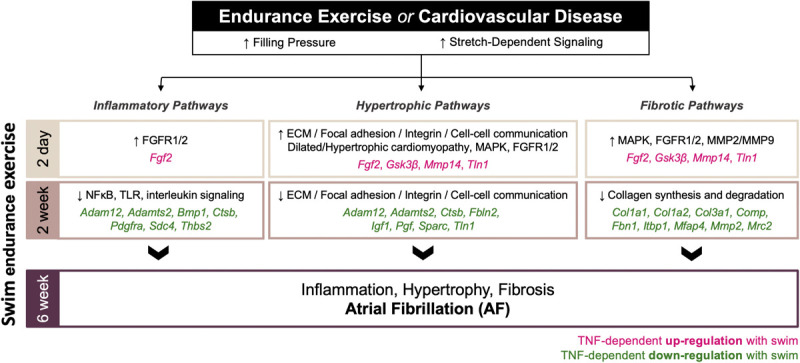
Schematic overview of the time-dependent and atrial-specific exercise-induced tumor necrosis factor (TNF)-dependent transcriptional responses to elevated venous filling pressures and atrial stretch mediating adverse atrial remodeling (inflammation, fibrosis, hypertrophy) and atrial fibrillation (AF) vulnerability.
Discussion
The transcriptional changes in WT atria were generally similar after 2 vs. 6 weeks of exercise, with about 50% of the differentially regulated gene sets related to ECM and mechanosensing enriched in sedentary mice. We believe that changes in gene sets associated with ECM/mechanosensitive pathways at these time points are relevant because elevated venous filling pressures (i.e., preloads) are seen invariably in virtually all AF conditions (Vranka et al., 2007; Remes et al., 2008), including intense exercise (Reeves et al., 1990; Aschar-Sobbi et al., 2015). Such elevations in filling pressure are expected to favor atrial vs. ventricular stretch and thereby preferentially driving stretch-dependent atrial remodeling (De Jong et al., 2011). This would help explain the appearance of fibrosis in atria but not ventricles. Moreover, the number of differentially regulated genes/gene sets related to ECM/mechanosensing were ∼twofold greater after 2 weeks vs. 6 weeks of exercise suggesting that the atrial responses to exercise adapt and diminish with time, as expected with the appearance of fibrosis. A puzzling finding, however, is the absence of increased collagen mRNA levels in exercised atria at either time point, despite the fibrosis after 6 weeks of exercise. This was surprising because fibrosis seen in most cardiac conditions (i.e., heart failure, hypertension, and cardiomyopathies) is associated with elevated collagen mRNA (Frangogiannis, 2019). The absence of elevated collagen mRNA in fibrotic exercised atria is similar to the discordant pattern seen with age- (Horn and Trafford, 2016; Podolsky et al., 2020) and stress-related (Kandalam et al., 2011) fibrosis in several tissues. As discussed below, we believe these findings have important implications on the mechanisms underlying adverse atrial changes and AF, regardless of the inciting factors.
Consistent with the TNF-dependence of adverse atrial changes with exercise, TNF- and inflammation-related gene sets were differentially regulated after 2 weeks of exercise (similar to 6 weeks of exercise) with the number of differentially regulated gene sets being ∼twofold less in TNF-KO vs. WT atria. On the other hand, after 2 weeks of exercise atria from sedentary TNF-KO mice, but not WT mice, showed enrichment in TNF-related inflammatory gene sets. Since adverse atrial changes do not occur in TNF-KO mice, it appears that TNF plays a permissive role in exercise-induced inflammation rather than being a primary factor. By contrast, the primary exercise-induced gene changes in TNF-KO atria are related to DNA replication and repair, whose significance will require further studies. Collectively, the differences in atrial transcriptome remodeling after 2 weeks of exercise are consistent with the pleiotropic actions of TNF (Tracey and Cerami, 1994).
To gain insight into possible mechanisms underlying the TNF-dependent atrial changes induced by 2 weeks of exercise, we examined transcript levels of individual genes between the 4 groups. Consistent with our enrichment maps, the bulk of the differentially regulated genes related to ECM/mechanotransduction had lower expression levels in exercised atria of both WT and KO mice. Moreover, most of these genes have been linked to increased tissue fibrosis, despite the absence of atrial fibrosis and AF inducibility in exercised TNF-KO mice, which suggests that many of these gene changes are likely of limited relevance in driving exercise-induced fibrosis (Chou et al., 1996; Sivasubramanian et al., 2001). However, it is noted in previous studies that reductions in the metalloproteinase, Mmp2, and the canonical collagen endocytic receptor (Engelholm et al., 2003), Mrc2 (which are both TNF-dependent genes that are reduced in WT exercised atria), are linked to age-dependent fibrosis (Podolsky et al., 2020). These TNF-dependent changes are particularly interesting since atrial fibrosis and AF are generally seen with aging (Gramley et al., 2009; Ravassa et al., 2019), and AF is especially prevalent in veteran endurance athletes (Karjalainen et al., 1998; Mont et al., 2002). Also, these Mmp2 reductions paralleled reductions in Mmp14, albeit independently of TNF, which has been associated with AF (Simmers et al., 2016) and shown to activate MMP-2 (Jr and Nagase, 2000). On the other hand, when we directly compared WT and TNF-KO swim atria, the majority of genes were increased in exercised atria from TNF-KO relative to WT, with the only gene that was increased in a TNF-dependent manner after 2 weeks of exercise in WT atria was Comp, which is a biomarker for cardiac fibrosis and hypertrophic remodeling (Huang et al., 2013; Zhao et al., 2018). Comp is involved in non-collagen ECM-receptor interaction (Rosenberg et al., 1998) and appears from multiple studies to contribute to the pathogenesis of AF (Zou et al., 2018; Thomas et al., 2019).
As the ECM/mechanosensitive pathways did not show enrichment at 2 and 6 weeks in our exercised mice, we also performed RNAseq measurements after only 2 days of exercise. Importantly, at this time point gene sets associated with ECM/mechanosensitive as well as hypertrophic signaling pathways were enriched in exercised atria from WT mice, which would appear to align with the elevated filling pressures seen in exercise and AF-related conditions (Reeves et al., 1990; De Jong et al., 2011; Aschar-Sobbi et al., 2015). Of particular note is the enrichment of tubulin folding and MAPK pathways in exercised WT (but not TNK-KO) atria. Tubulin assembly/disassembly in microtubules is involved in mechanosensing (White, 2011) in a number of cell types and is interdependent on MAPKs (Samaj et al., 2004), particularly p38 kinases (Ramkumar et al., 2018). These results suggest that strain-dependent signaling via microtubule assembly/disassembly may play a role in driving early atrial responses to atrial stretch occurring during exercise, possibly in concert with the recruitment of TNF-dependent transduction, consistent with TNF’s mechanosensing properties (Kroetsch et al., 2017). Microtubule involvement is consistent with the pioneering studies by George Cooper (4th) who showed that p21-activated kinase-1 (Pak1)-dependent microtubule assembly plays a central role in the early response to pressure overload and mechanical stretch in right ventricular cardiomyocytes (Cheng et al., 2012a). Indeed, Pak1 regulates exercise-induced cardiac hypertrophy (Davis et al., 2015), which aligns nicely with our 2 day atrial analyses showing exercise-induced upregulation of Flna (filamen A), a cytoprotective protein that is upregulated with mechanical stress (D’Addario et al., 2003) and is essential for actin/cytoskeletal dynamics (Vadlamudi et al., 2002) through interdependent p38- (D’Addario et al., 2002) and Pak1-mediated signaling (Zhang et al., 1995; Shifrin et al., 2012). We also found enrichment of other pathways, including IQGAPs and AMPKs, in acutely exercised WT atria which are involved in the early compensatory responses to pressure overload stimuli that may be harbingers of fibrotic remodeling in the long-term (Hermida et al., 2013; Hedman et al., 2015; Daskalopoulos et al., 2016). Taken together, these observations suggest that the loss of TNF leads to an inhibitory modulation of mechanosensitive signaling pathways which is consistent with the stretch-dependence of TNF activation (Kroetsch et al., 2017) and the regulation of FAK by TNF (Funakoshi-Tago et al., 2003; Murphy et al., 2019) via MAPK signaling and IL-6 expression (Schlaepfer et al., 2007). Given the absence of exercise-induced adverse atrial remodeling and AF with TNF inhibition, our results suggest stretch-activation of TNF may tip the scales toward maladaptive compensatory remodeling that is unique to the atria.
As in the 2 week group, far fewer gene sets (61 vs. 101) were differentially regulated with 2 day exercise in TNF-KO vs. WT mice. In particular, TNF-KO mice again showed enrichment of NF-κB, toll-like receptor, and interleukin pathways in the sedentary group, further supporting a permissive role for TNF in regulating exercise-induced atrial changes. With regards to individual genes, our analyses showed that fewer atrial genes were differentially regulated between swim WT and KO mice. Of these, the differentially regulated genes related to ECM/mechanosensitive include Mmp14, Tln1, Lox, and Gsk3β as well as Fgf2, the latter being the only gene upregulated in a TNF-dependent manner exclusively in exercised WT mice. The presence of only one TNF-dependent differentially regulated gene in the WT compared to TNF-KO exercise group is unexpected given the increased filling pressures we observed with swim. However, exercise is an intermittent hemodynamic overload stimulus (Moreira-Goncalves et al., 2015), and the nature and time course of cardiac transcriptomic remodeling in response to mechanical stretch is highly dependent on stretch duration (Rysa et al., 2018). Therefore, as we only looked at atrial transcriptomic remodeling 2 h following the last acute swim bout, and focused our analysis on genes linked to mechanotransduction and ECM remodeling, a larger window is likely necessary to capture the full impact of swim exercise on stretch-induced transcriptomic remodeling and enzyme activity (i.e., TACE/ADAM17, pro-MMP2).
Relative to atria, the number of genes sets in ventricles that were differentially regulated in response to exercise was much smaller. This is not unexpected because ventricles are far less compliant than atria due to differences in ECM as well as wall thickness (La Gerche et al., 2011) in agreement with our previous studies showing that exercise activates p38 in atria but not ventricles (Aschar-Sobbi et al., 2015). Presumably, preferential atrial stretch underlies prominent bi-atrial enlargement in athletes (D’Andrea et al., 2010; D’Ascenzi et al., 2016, 2018) as well as pronounced atrial hypertrophy and fibrosis in our exercised mice. These responses would be expected to normalize atrial wall stress which explains nicely the evolving pattern of time-dependent exercised-induced atrial changes in stretch-dependent signaling pathways in the current study.
Given the link between TNF and mechanical stress, it is tempting to speculate that the degree of TNF elevation with elevated filling pressures and its effects on mechanosensitive signaling cascades may determine the threshold between compensatory (i.e., physiological) or maladaptive transcriptomic activation early in the response to exercise training. This would be consistent with the pleiotropic functions of TNF (Tracey and Cerami, 1994). Indeed, TNF and its downstream factors such as NFκB and p38 can promote protective and pathophysiological responses (Schumacher and Naga Prasad, 2018). Moreover, time-dependent adaptations (i.e., hypertrophy and fibrosis) may serve to blunt or normalize acute elevations in wall stress with exercise, which would explain enrichment in ECM/mechanosensitive genes in sedentary mice over time and compensatory deactivation of stretch-mediated remodeling. This would further promote an early transition to reduced collagen transcription, mimicking fibrotic processes seen in aging wherein collagen expression is also not increased (Podolsky et al., 2020).
Implications
AF increases strongly with age, CVD and conditions associated with poor cardiovascular health (i.e., diabetes, obesity, and metabolic syndrome). Even though physical activity reduces AF risk (Drca et al., 2014; Malmo et al., 2016), endurance athletes, especially elite veteran athletes, have AF risks rivaling that seen with hypertension and other CVD conditions (Mont et al., 2002; Redpath and Backx, 2015; Goodman et al., 2018). In CVD patients, persistent AF is invariably associated with atrial fibrosis, inflammation and hypertrophy, along with variable electrical changes (Daoud et al., 1996; Xu et al., 2004; Nattel and Harada, 2014). Although historically AF in athletes has often been referred to as “lone AF” (Calvo et al., 2016) because of the absence of CVD, the term is no longer considered appropriate since AF is associated with a multitude of conditions (Wyse et al., 2014). In this regard, animal studies have established, and some human studies suggest, that intense exercise leads to adverse atrial changes resembling those seen in persistent AF patients. In this regard, our studies reveal that exercise induces dynamic transcriptional adaptations involving, in particular, pronounced changes in strain-dependent pathways related to ECM/integrin/focal adhesion. These observations seem particularly relevant since elevated filling pressures and atrial stretch are both prominent features of aging, CVD and exercise. A novel and remarkable finding of our analyses was the link between genes associated with collagen turnover, rather than collagen transcripts, and fibrosis in exercised atria, a pattern that mirrors aging-related fibrosis (Podolsky et al., 2020). This is especially interesting because the strongest predictor of AF is age (Staerk et al., 2018). Moreover, our findings revealed TNF plays a permissive rather than primary role in exercise-mediated atrial structural and transcriptomic remodeling.
TNF involvement in exercise-induced structural and transcriptional adaptations are of particular interest because TNF has been implicated in the pathogenesis of AF (Ren et al., 2015) and persistent AF is associated with elevated atrial TNF levels and inflammatory infiltrates (Li et al., 2010; Guo et al., 2012). Collectively, the many common atrial features between persistent AF patients and exercised mice suggests to us that AF and adverse remodeling seen with intense exercise and CVD share common mechanisms. Thus, while our findings of an arrhythmogenic substrate requires confirmation in athletes presenting with AF, the genetic changes seen in our studies may have broader implications for the general AF population. By comparison, ventricular responses to exercise were relatively muted, although the differentially regulated gene sets were similar to those in the atria, consistent with clinical and epidemiological evidence for exercise-induced arrhythmogenic remodeling being chamber-specific (Guasch et al., 2018).
Limitations
Obviously the use of whole tissue samples prevents us from determining the individual contributions of cardiomyocytes, endothelial cells, and fibroblasts to the transcriptomic remodeling induced by exercise which would be highly desirable since TNF is a mechanosensitive cytokine expressed in cardiomyocytes (Kapadia et al., 1997; Sun et al., 2007), fibroblasts (Yokoyama et al., 1999), and endothelial cells (Yin et al., 2017).
Although the studies presented here were limited to male mice, we have plan to examine the effects of exercise on female mice. It is worth noting that even though female athletes remain underrepresented generally in previous studies examining exercise-induced AF (Andersen et al., 2013), several studies have found a reduced incidence of AF in females (Mohanty et al., 2016) making our future studies potentially highly relevant.
Our studies were limited to only three time points leaving many uncertainties regarding the evolving effects of intense exercise. Nevertheless, our results showed time-dependent adaptations to exercise with generally comparable responses at the 2 and 6 week time points, suggestive of compensatory adaptations in fibrotic, hypertrophic and inflammatory pathways. In this regard, we did not extend our swim training beyond 6 weeks given the clear evidence of adverse atrial remodeling and increased AF vulnerability at this time point. However, given the presence of inflammatory infiltrates and enrichment of TNF-mediated inflammatory signaling pathways at 6 weeks of exercise, it is conceivable that the arrhythmogenic substrate and the degree of AF vulnerability may be more pronounced if we were to extend our exercise protocol. We cannot rule out increased collagen synthesis or mechanisms involved in post-translational modifications/deposition (i.e., SPARC/OPN, LOXs) or degradation (i.e., MMPs) as contributing to exercise-induced atrial remodeling, which might not have been fully captured at 2 day, 2 or 6 week exercise.
While we used microarray analyses for 6 week data and RNAseq for the other time points to assess exercise-induced transcriptional changes, our analyses focused on RNAseq data at the earlier time points. In this regard, it was reassuring to find that the 6 weeks microarray results align well with the 2 week RNAseq results, thus robustly supporting the compensatory nature of the atrial responses to exercise at the later time points.
Conclusion
Our results demonstrate clear exercise-induced TNF-dependent differential activation (enrichment) of pathways associated with mechanosensitive ECM remodeling, which are both time-dependent and differ between atria and ventricles in a manner consistent with preferential stretch of atria in response to exercise-induced elevations in venous pressure. Our findings provide insight into the chamber-specific roles of TNF and mechanical strain in cardiac changes induced by exercise, which supports the general conclusion that exercise-induced adverse atrial remodeling and AF vulnerability is linked to elevated filling pressures, consistent with AF associated with aging and poor cardiovascular health. The common atrial features between persistent AF patients and exercised mice suggests that AF and adverse remodeling seen with intense exercise may share common mechanisms, which may have broader implications for the general AF population.
Data Availability Statement
The data generated in this study can be found in the BioProject database (accession: PRJNA663094).
Ethics Statement
The animal study was reviewed and approved by the Division of Comparative Medicine at the University of Toronto and York University Animal Care Committee (ACC).
Author Contributions
Experiments were performed at York University Department of Biology, the University of Toronto Department of Physiology, and the University of Alberta Department of Physiology. YO, SY, RL, and PB were responsible for the conception and design of the work, the acquisition, analysis, and interpretation for the work, drafting the work, and revising it critically for important intellectual content. XL, SJ, FI, XG, RD, and D-KK were involved in acquisition, analysis, and interpretation of data for the work. All authors approved the final version of this manuscript and agreed to be accountable for all aspects of the work.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
Funding. This work was supported by the Queen Elizabeth II/Heart and Stroke Foundation of Ontario Graduate Scholarships in Science and Technology Award, Queen Elizabeth II Scholarship in Science and Technology (QEII-GSST), and University of Ottawa Heart Institute Endowed Scholarship to YO, the Natural Science Funding from the Science and Technology Department of Jilin Province, China (No. 20200201562JC) to SY, the Canadian Institutes of Health Research (CIHR) Fellowship to RL, the Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant to K-HK, the Canadian Institutes of Health Research, Operating grant (MOP125950 and MOP119339) and a Canada Research Chair in Cardiovascular Biology to PB, and the Canadian Foundation for Innovation, John Evans Leader Award to PB.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2020.605671/full#supplementary-material
References
- Accornero F., Molkentin J. D. (2011). Placental growth factor as a protective paracrine effector in the heart. Trends Cardiovasc. Med. 21 220–224. 10.1016/j.tcm.2012.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afgan E., Baker D., Van Den Beek M., Blankenberg D., Bouvier D., Cech M., et al. (2016). The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Res. 44 W3–W10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen K., Farahmand B., Ahlbom A., Held C., Ljunghall S., Michaelsson K., et al. (2013). Risk of arrhythmias in 52 755 long-distance cross-country skiers: a cohort study. Eur. Heart J. 34 3624–3631. 10.1093/eurheartj/eht188 [DOI] [PubMed] [Google Scholar]
- Aschar-Sobbi R., Izaddoustdar F., Korogyi A. S., Wang Q., Farman G. P., Yang F., et al. (2015). Increased atrial arrhythmia susceptibility induced by intense endurance exercise in mice requires TNFalpha. Nat. Commun. 6:6018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babu G. J., Lalli M. J., Sussman M. A., Sadoshima J., Periasamy M. (2000). Phosphorylation of elk-1 by MEK/ERK pathway is necessary for c-fos gene activation during cardiac myocyte hypertrophy. J. Mol. Cell Cardiol. 32 1447–1457. 10.1006/jmcc.2000.1185 [DOI] [PubMed] [Google Scholar]
- Bouzeghrane F., Reinhardt D. P., Reudelhuber T. L., Thibault G. (2005). Enhanced expression of fibrillin-1, a constituent of the myocardial extracellular matrix in fibrosis. Am. J. Physiol. Heart Circ. Physiol. 289 H982–H991. [DOI] [PubMed] [Google Scholar]
- Bradshaw A. D., Baicu C. F., Rentz T. J., Van Laer A. O., Boggs J., Lacy J. M., et al. (2009). Pressure overload-induced alterations in fibrillar collagen content and myocardial diastolic function: role of secreted protein acidic and rich in cysteine (SPARC) in post-synthetic procollagen processing. Circulation 119 269–280. 10.1161/circulationaha.108.773424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradshaw A. D., Baicu C. F., Rentz T. J., Van Laer A. O., Bonnema D. D., Zile M. R. (2010). Age-dependent alterations in fibrillar collagen content and myocardial diastolic function: role of SPARC in post-synthetic procollagen processing. Am. J. Physiol. Heart Circ. Physiol. 298 H614–H622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busk P. K., Bartkova J., Strom C. C., Wulf-Andersen L., Hinrichsen R., Christoffersen T. E., et al. (2002). Involvement of cyclin D activity in left ventricle hypertrophy in vivo and in vitro. Cardiovasc. Res. 56 64–75. 10.1016/s0008-6363(02)00510-2 [DOI] [PubMed] [Google Scholar]
- Calkins H., Hindricks G., Cappato R., Kim Y. H., Saad E. B., Aguinaga L., et al. (2018). 2017 HRS/EHRA/ECAS/APHRS/SOLAECE expert consensus statement on catheter and surgical ablation of atrial fibrillation: executive summary. Europace 20 157–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo N., Ramos P., Montserrat S., Guasch E., Coll-Vinent B., Domenech M., et al. (2016). Emerging risk factors and the dose-response relationship between physical activity and lone atrial fibrillation: a prospective case-control study. Europace 18 57–63. 10.1093/europace/euv216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporizzo M. A., Chen C. Y. X., Prosser B. L. (2019). Cardiac microtubules in health and heart disease. Exp. Biol. Med. 244 1255–1272. 10.1177/1535370219868960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng G., Kasiganesan H., Baicu C. F., Wallenborn J. G., Kuppuswamy D., Cooper G. T. (2012a). Cytoskeletal role in protection of the failing heart by beta-adrenergic blockade. Am. J. Physiol. Heart Circ. Physiol. 302 H675–H687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X. W., Shi G. P., Kuzuya M., Sasaki T., Okumura K., Murohara T. (2012b). Role for cysteine protease cathepsins in heart disease: focus on biology and mechanisms with clinical implication. Circulation 125 1551–1562. 10.1161/circulationaha.111.066712 [DOI] [PubMed] [Google Scholar]
- Chou D. H., Lee W., Mcculloch C. A. (1996). TNF-alpha inactivation of collagen receptors: implications for fibroblast function and fibrosis. J. Immunol. 156 4354–4362. [PubMed] [Google Scholar]
- D’Addario M., Arora P. D., Ellen R. P., Mcculloch C. A. G. (2002). Interaction of p38 and Sp1 in a mechanical force-induced, beta(1), integrin-mediated transcriptional circuit that regulates the actin-binding protein filamin-A. J. Biol. Chem. 277 47541–47550. 10.1074/jbc.m207681200 [DOI] [PubMed] [Google Scholar]
- D’Addario M., Arora P. D., Ellen R. P., Mcculloch C. A. G. (2003). Regulation of tension-induced mechanotranscriptional signals by the microtubule network in fibroblasts. J. Biol. Chem. 278 53090–53097. 10.1074/jbc.m309027200 [DOI] [PubMed] [Google Scholar]
- D’Andrea A., Riegler L., Cocchia R., Scarafile R., Salerno G., Gravino R., et al. (2010). Left atrial volume index in highly trained athletes. Am. Heart J. 159 1155–1161. 10.1016/j.ahj.2010.03.036 [DOI] [PubMed] [Google Scholar]
- Daoud E. G., Bogun F., Goyal R., Harvey M., Man K. C., Strickberger S. A., et al. (1996). Effect of atrial fibrillation on atrial refractoriness in humans. Circulation 94 1600–1606. 10.1161/01.cir.94.7.1600 [DOI] [PubMed] [Google Scholar]
- D’Ascenzi F., Anselmi F., Focardi M., Mondillo S. (2018). Atrial enlargement in the Athlete’s heart: assessment of atrial function may help distinguish adaptive from pathologic remodeling. J. Am. Soc. Echocardiogr. 31 148–157. 10.1016/j.echo.2017.11.009 [DOI] [PubMed] [Google Scholar]
- D’Ascenzi F., Solari M., Anselmi F., Maffei S., Focardi M., Bonifazi M., et al. (2016). Atrial chamber remodelling in healthy pre-adolescent athletes engaged in endurance sports: a study with a longitudinal design. The CHILD study. Int. J. Cardiol. 223 325–330. 10.1016/j.ijcard.2016.08.231 [DOI] [PubMed] [Google Scholar]
- Daskalopoulos E. P., Dufeys C., Bertrand L., Beauloye C., Horman S. (2016). AMPK in cardiac fibrosis and repair: actions beyond metabolic regulation. J. Mol. Cell Cardiol. 91 188–200. 10.1016/j.yjmcc.2016.01.001 [DOI] [PubMed] [Google Scholar]
- Davis R. T., Simon J. N., Utter M., Mungai P., Alvarez M. G., Chowdhury S. A. K., et al. (2015). Knockout of p21-activated kinase-1 attenuates exercise-induced cardiac remodelling through altered calcineurin signalling. Cardiovasc. Res. 108 335–347. 10.1093/cvr/cvv234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jong A. M., Maass A. H., Oberdorf-Maass S. U., Van Veldhuisen D. J., Van Gilst W. H., Van Gelder I. C. (2011). Mechanisms of atrial structural changes caused by stretch occurring before and during early atrial fibrillation. Cardiovasc. Res. 89 754–765. 10.1093/cvr/cvq357 [DOI] [PubMed] [Google Scholar]
- Diao S. L., Xu H. P., Zhang B., Ma B. X., Liu X. L. (2016). Associations of MMP-2, BAX, and Bcl-2 mRNA and protein expressions with development of atrial fibrillation. Med. Sci. Monit. 22 1497–1507. 10.12659/msm.895715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C., Li H. J., Chang S., Liao H. J., Zhang Z. P., Huang P., et al. (2013). A disintegrin and metalloprotease with thrombospondin motif 2 may contribute to cirrhosis in humans through the transforming growth factor-beta/SMAD pathway. Gut Liver 7 213–220. 10.5009/gnl.2013.7.2.213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drca N., Wolk A., Jensen-Urstad M., Larsson S. C. (2014). Atrial fibrillation is associated with different levels of physical activity levels at different ages in men. Heart 100 1037–1042. 10.1136/heartjnl-2013-305304 [DOI] [PubMed] [Google Scholar]
- Engelholm L. H., List K., Netzel-Arnett S., Cukierman E., Mitola D. J., Aaronson H., et al. (2003). uPARAP/Endo180 is essential for cellular uptake of collagen and promotes fibroblast collagen adhesion. J. Cell Biol. 160 1009–1015. 10.1083/jcb.200211091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frame M. C., Patel H., Serrels B., Lietha D., Eck M. J. (2010). The FERM domain: organizing the structure and function of FAK. Nat. Rev. Mol. Cell Biol. 11 802–814. 10.1038/nrm2996 [DOI] [PubMed] [Google Scholar]
- Frangogiannis N. G. (2012). Matricellular proteins in cardiac adaptation and disease. Physiol. Rev. 92 635–688. 10.1152/physrev.00008.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frangogiannis N. G. (2019). Cardiac fibrosis: cell biological mechanisms, molecular pathways and therapeutic opportunities. Mol. Aspects Med. 65 70–99. 10.1016/j.mam.2018.07.001 [DOI] [PubMed] [Google Scholar]
- Funakoshi-Tago M., Sonoda Y., Tanaka S., Hashimoto K., Tago K., Tominaga S., et al. (2003). Tumor necrosis factor-induced nuclear factor kappaB activation is impaired in focal adhesion kinase-deficient fibroblasts. J. Biol. Chem. 278 29359–29365. 10.1074/jbc.m213115200 [DOI] [PubMed] [Google Scholar]
- Gallini R., Lindblom P., Bondjers C., Betsholtz C., Andrae J. (2016). PDGF-A and PDGF-B induces cardiac fibrosis in transgenic mice. Exp. Cell Res. 349 282–290. 10.1016/j.yexcr.2016.10.022 [DOI] [PubMed] [Google Scholar]
- Goodman J. M., Banks L., Connelly K. A., Yan A. T., Backx P. H., Dorian P. (2018). Excessive exercise in endurance athletes: Is atrial fibrillation a possible consequence? Appl. Physiol. Nutr. Metab. 43 973–976. 10.1139/apnm-2017-0764 [DOI] [PubMed] [Google Scholar]
- Goumans M. J., Ten Dijke P. (2018). TGF-β signaling in control of cardiovascular function. Cold Spring Harb. Perspect. Biol. 10:a022210. 10.1101/cshperspect.a022210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gramley F., Lorenzen J., Knackstedt C., Rana O. R., Saygili E., Frechen D., et al. (2009). Age-related atrial fibrosis. Age 31 27–38. 10.1007/s11357-008-9077-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gramley F., Lorenzen J., Koellensperger E., Kettering K., Weiss C., Munzel T. (2010). Atrial fibrosis and atrial fibrillation: the role of the TGF-beta1 signaling pathway. Int. J. Cardiol. 143 405–413. 10.1016/j.ijcard.2009.03.110 [DOI] [PubMed] [Google Scholar]
- Guasch E., Benito B., Qi X., Cifelli C., Naud P., Shi Y., et al. (2013). Atrial fibrillation promotion by endurance exercise: demonstration and mechanistic exploration in an animal model. J. Am. Coll. Cardiol. 62 68–77. 10.1016/j.jacc.2013.01.091 [DOI] [PubMed] [Google Scholar]
- Guasch E., Mont L., Sitges M. (2018). Mechanisms of atrial fibrillation in athletes: what we know and what we do not know. Neth. Heart J. 26 133–145. 10.1007/s12471-018-1080-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y., Lip G. Y., Apostolakis S. (2012). Inflammation in atrial fibrillation. J. Am. Coll. Cardiol. 60 2263–2270. [DOI] [PubMed] [Google Scholar]
- Hadi A. M., Mouchaers K. T., Schalij I., Grunberg K., Meijer G. A., Vonk-Noordegraaf A., et al. (2011). Rapid quantification of myocardial fibrosis: a new macro-based automated analysis. Cell Oncol. 34 343–354. 10.1007/s13402-011-0035-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedman A. C., Smith J. M., Sacks D. B. (2015). The biology of IQGAP proteins: beyond the cytoskeleton. EMBO Rep. 16 427–446. 10.15252/embr.201439834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heeringa J., Van Der Kuip D. A., Hofman A., Kors J. A., Van Herpen G., Stricker B. H., et al. (2006). Prevalence, incidence and lifetime risk of atrial fibrillation: the Rotterdam study. Eur. Heart J. 27 949–953. 10.1093/eurheartj/ehi825 [DOI] [PubMed] [Google Scholar]
- Hermida N., Markl A., Hamelet J., Van Assche T., Vanderper A., Herijgers P., et al. (2013). HMGCoA reductase inhibition reverses myocardial fibrosis and diastolic dysfunction through AMP-activated protein kinase activation in a mouse model of metabolic syndrome. Cardiovasc. Res. 99 44–54. 10.1093/cvr/cvt070 [DOI] [PubMed] [Google Scholar]
- Herum K. M., Lunde I. G., Skrbic B., Louch W. E., Hasic A., Boye S., et al. (2015). Syndecan-4 is a key determinant of collagen cross-linking and passive myocardial stiffness in the pressure-overloaded heart. Cardiovasc. Res. 106 217–226. 10.1093/cvr/cvv002 [DOI] [PubMed] [Google Scholar]
- Horn M. A., Trafford A. W. (2016). Aging and the cardiac collagen matrix: novel mediators of fibrotic remodelling. J. Mol. Cell Cardiol. 93 175–185. 10.1016/j.yjmcc.2015.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Xia J., Zheng J., Geng B., Liu P., Yu F., et al. (2013). Deficiency of cartilage oligomeric matrix protein causes dilated cardiomyopathy. Basic Res. Cardiol. 108:374. [DOI] [PubMed] [Google Scholar]
- Husser D., Büttner P., Ueberham L., Dinov B., Sommer P., Arya A., et al. (2016). Genomic contributors to rhythm outcome of atrial fibrillation catheter ablation – pathway enrichment analysis of GWAS data. PLoS One 11:e0167008. 10.1371/journal.pone.0167008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh N., Ohta H. (2013). Pathophysiological roles of FGF signaling in the heart. Front Physiol 4:247. 10.3389/fphys.2013.00247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jana S., Chute M., Hu M., Winkelaar G., Owen C. A., Oudit G. Y., et al. (2020). ADAM (a disintegrin and metalloproteinase) 15 deficiency exacerbates Ang II (Angiotensin II)-induced aortic remodeling leading to abdominal aortic aneurysm. Arterioscler. Thromb. Vasc. Biol. 40 1918–1934. 10.1161/ATVBAHA.120.314600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolliffe I. T., Cadima J. (2016). Principal component analysis: a review and recent developments. Philos. Trans. A Math. Phys. Eng. Sci. 374:20150202. 10.1098/rsta.2015.0202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jr J. W., Nagase H. (2000). Activation of the Zymogen Forms of MMPs. New York, NY: Oxford University Press. [Google Scholar]
- Kandalam V., Basu R., Moore L., Fan D., Wang X., Jaworski D. M., et al. (2011). Lack of tissue inhibitor of metalloproteinases 2 leads to exacerbated left ventricular dysfunction and adverse extracellular matrix remodeling in response to biomechanical stress. Circulation 124 2094–2105. 10.1161/circulationaha.111.030338 [DOI] [PubMed] [Google Scholar]
- Kapadia S. R., Oral H., Lee J., Nakano M., Taffet G. E., Mann D. L. (1997). Hemodynamic regulation of tumor necrosis factor-alpha gene and protein expression in adult feline myocardium. Circ. Res. 81 187–195. 10.1161/01.res.81.2.187 [DOI] [PubMed] [Google Scholar]
- Kardami E., Jiang Z. S., Jimenez S. K., Hirst C. J., Sheikh F., Zahradka P., et al. (2004). Fibroblast growth factor 2 isoforms and cardiac hypertrophy. Cardiovasc. Res. 63 458–466. 10.1016/j.cardiores.2004.04.024 [DOI] [PubMed] [Google Scholar]
- Karjalainen J., Kujala U. M., Kaprio J., Sarna S., Viitasalo M. (1998). Lone atrial fibrillation in vigorously exercising middle aged men: case-control study. BMJ 316 1784–1785. 10.1136/bmj.316.7147.1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khalil H., Kanisicak O., Vagnozzi R. J., Johansen A. K., Maliken B. D., Prasad V., et al. (2019). Cell-specific ablation of Hsp47 defines the collagen-producing cells in the injured heart. JCI Insight 4:e128722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D., Langmead B., Salzberg S. L. (2015). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 12 357–360. 10.1038/nmeth.3317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krijthe B. P., Kunst A., Benjamin E. J., Lip G. Y., Franco O. H., Hofman A., et al. (2013). Projections on the number of individuals with atrial fibrillation in the European Union, from 2000 to 2060. Eur. Heart J. 34 2746–2751. 10.1093/eurheartj/eht280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroetsch J. T., Levy A. S., Zhang H., Aschar-Sobbi R., Lidington D., Offermanns S., et al. (2017). Constitutive smooth muscle tumour necrosis factor regulates microvascular myogenic responsiveness and systemic blood pressure. Nat. Commun. 8:14805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Gerche A., Heidbuchel H., Burns A. T., Mooney D. J., Taylor A. J., Pfluger H. B., et al. (2011). Disproportionate exercise load and remodeling of the athlete’s right ventricle. Med. Sci. Sports Exerc. 43 974–981. 10.1249/mss.0b013e31820607a3 [DOI] [PubMed] [Google Scholar]
- Lakin R., Polidovitch N., Yang S., Guzman C., Gao X., Wauchop M., et al. (2019). Inhibition of soluble TNFalpha prevents adverse atrial remodeling and atrial arrhythmia susceptibility induced in mice by endurance exercise. J. Mol. Cell Cardiol. 129 165–173. 10.1016/j.yjmcc.2019.01.012 [DOI] [PubMed] [Google Scholar]
- Lal H., Ahmad F., Woodgett J., Force T. (2015). The GSK-3 family as therapeutic target for myocardial diseases. Circ. Res. 116 138–149. 10.1161/circresaha.116.303613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavandero S., Foncea R., Perez V., Sapag-Hagar M. (1998). Effect of inhibitors of signal transduction on IGF-1-induced protein synthesis associated with hypertrophy in cultured neonatal rat ventricular myocytes. FEBS Lett. 422 193–196. 10.1016/s0014-5793(98)00008-8 [DOI] [PubMed] [Google Scholar]
- Li J., Solus J., Chen Q., Rho Y. H., Milne G., Stein C. M., et al. (2010). Role of inflammation and oxidative stress in atrial fibrillation. Heart Rhythm 7 438–444. 10.1016/j.hrthm.2009.12.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y., Tan W., Ye F., Xue F., Gao S., Huang W., et al. (2019). Identification of microRNAs and genes as biomarkers of atrial fibrillation using a bioinformatics approach. J. Int. Med. Res. 47 3580–3589. 10.1177/0300060519852235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao C. H., Akazawa H., Tamagawa M., Ito K., Yasuda N., Kudo Y., et al. (2010). Cardiac mast cells cause atrial fibrillation through PDGF-A-mediated fibrosis in pressure-overloaded mouse hearts. J. Clin. Invest. 120 242–253. 10.1172/jci39942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahida S. (2013). Transcription factors and atrial fibrillation. Cardiovasc. Res. 101 194–202. 10.1093/cvr/cvt261 [DOI] [PubMed] [Google Scholar]
- Malmo V., Nes B. M., Amundsen B. H., Tjonna A. E., Stoylen A., Rossvoll O., et al. (2016). Aerobic interval training reduces the burden of atrial fibrillation in the short term: a randomized trial. Circulation 133 466–473. 10.1161/circulationaha.115.018220 [DOI] [PubMed] [Google Scholar]
- Manso A. M., Elsherif L., Kang S. M., Ross R. S. (2006). Integrins, membrane-type matrix metalloproteinases and ADAMs: potential implications for cardiac remodeling. Cardiovasc. Res. 69 574–584. 10.1016/j.cardiores.2005.09.004 [DOI] [PubMed] [Google Scholar]
- Manso A. M., Li R., Monkley S. J., Cruz N. M., Ong S., Lao D. H., et al. (2013). Talin1 has unique expression versus talin 2 in the heart and modifies the hypertrophic response to pressure overload. J. Biol. Chem. 288 4252–4264. 10.1074/jbc.m112.427484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald L. T., Zile M. R., Zhang Y., Van Laer A. O., Baicu C. F., Stroud R. E., et al. (2018). Increased macrophage-derived SPARC precedes collagen deposition in myocardial fibrosis. Am. J. Physiol. Heart Circ. Physiol. 315 H92–H100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohanty S., Mohanty P., Tamaki M., Natale V., Gianni C., Trivedi C., et al. (2016). Differential association of exercise intensity with risk of atrial fibrillation in men and women: evidence from a meta-analysis. J. Cardiovasc. Electrophysiol. 27 1021–1029. 10.1111/jce.13023 [DOI] [PubMed] [Google Scholar]
- Mont L., Sambola A., Brugada J., Vacca M., Marrugat J., Elosua R., et al. (2002). Long-lasting sport practice and lone atrial fibrillation. Eur. Heart J. 23 477–482. 10.1053/euhj.2001.2802 [DOI] [PubMed] [Google Scholar]
- Moreira-Goncalves D., Henriques-Coelho T., Fonseca H., Ferreira R., Padrao A. I., Santa C., et al. (2015). Intermittent cardiac overload results in adaptive hypertrophy and provides protection against left ventricular acute pressure overload insult. J. Physiol. 593 3885–3897. 10.1113/jp270685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy G. (2008). The ADAMs: signalling scissors in the tumour microenvironment. Nat. Rev. Cancer. 8 929–941. [DOI] [PubMed] [Google Scholar]
- Murphy J. M., Jeong K., Rodriguez Y. A. R., Kim J. H., Ahn E. E., Lim S. S. (2019). FAK and Pyk2 activity promote TNF-alpha and IL-1beta-mediated pro-inflammatory gene expression and vascular inflammation. Sci. Rep. 9:7617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y., Kita S., Tanaka Y., Fukuda S., Obata Y., Okita T., et al. (2020). A disintegrin and metalloproteinase 12 prevents heart failure by regulating cardiac hypertrophy and fibrosis. Am. J. Physiol. Heart Circ. Physiol. 318 H238–H251. [DOI] [PubMed] [Google Scholar]
- Nattel S., Harada M. (2014). Atrial remodeling and atrial fibrillation: recent advances and translational perspectives. J. Am. Coll. Cardiol. 63 2335–2345. [DOI] [PubMed] [Google Scholar]
- Oakes R. S., Badger T. J., Kholmovski E. G., Akoum N., Burgon N. S., Fish E. N., et al. (2009). Detection and quantification of left atrial structural remodeling with delayed-enhancement magnetic resonance imaging in patients with atrial fibrillation. Circulation 119 1758–1767. 10.1161/circulationaha.108.811877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odutayo A., Wong C. X., Hsiao A. J., Hopewell S., Altman D. G., Emdin C. A. (2016). Atrial fibrillation and risks of cardiovascular disease, renal disease, and death: systematic review and meta-analysis. BMJ 354:i4482. 10.1136/bmj.i4482 [DOI] [PubMed] [Google Scholar]
- Oudit G. Y., Kassiri Z., Zhou J., Liu Q. C., Liu P. P., Backx P. H., et al. (2008). Loss of PTEN attenuates the development of pathological hypertrophy and heart failure in response to biomechanical stress. Cardiovasc. Res. 78 505–514. 10.1093/cvr/cvn041 [DOI] [PubMed] [Google Scholar]
- Papageorgiou A. P., Swinnen M., Vanhoutte D., Vandendriessche T., Chuah M., Lindner D., et al. (2012). Thrombospondin-2 prevents cardiac injury and dysfunction in viral myocarditis through the activation of regulatory T-cells. Cardiovasc. Res. 94 115–124. 10.1093/cvr/cvs077 [DOI] [PubMed] [Google Scholar]
- Park J., Joung B., Uhm J. S., Young Shim C., Hwang C., Hyoung Lee M., et al. (2014). High left atrial pressures are associated with advanced electroanatomical remodeling of left atrium and independent predictors for clinical recurrence of atrial fibrillation after catheter ablation. Heart Rhythm 11 953–960. 10.1016/j.hrthm.2014.03.009 [DOI] [PubMed] [Google Scholar]
- Patro R., Duggal G., Love M. I., Irizarry R. A., Kingsford C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14 417–419. 10.1038/nmeth.4197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podolsky M. J., Yang C. D., Valenzuela C. L., Datta R., Huang S. K., Nishimura S. L., et al. (2020). Age-dependent regulation of cell-mediated collagen turnover. JCI Insight 5:e137519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu Y. C., Du Y. M., Wu S. L., Chen Q. X., Wu H. L., Zhou S. F. (2009). Activated nuclear factor-kappaB and increased tumor necrosis factor-alpha in atrial tissue of atrial fibrillation. Scand. Cardiovasc. J. 43 292–297. 10.1080/14017430802651803 [DOI] [PubMed] [Google Scholar]
- Ramkumar A., Jong B. Y., Ori-Mckenney K. M. (2018). ReMAPping the microtubule landscape: how phosphorylation dictates the activities of microtubule-associated proteins. Dev. Dyn. 247 138–155. 10.1002/dvdy.24599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravassa S., Ballesteros G., Diez J. (2019). Aging and atrial fibrillation: a matter of fibrosis. Aging (Albany NY) 11 9965–9966. 10.18632/aging.102501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redpath C. J., Backx P. H. (2015). Atrial fibrillation and the athletic heart. Curr. Opin. Cardiol. 30 17–23. 10.1097/hco.0000000000000130 [DOI] [PubMed] [Google Scholar]
- Reeves J. T., Groves B. M., Cymerman A., Sutton J. R., Wagner P. D., Turkevich D., et al. (1990). Operation Everest II: cardiac filling pressures during cycle exercise at sea level. Respir. Physiol. 80 147–154. 10.1016/0034-5687(90)90078-d [DOI] [PubMed] [Google Scholar]
- Remes J., Van Brakel T. J., Bolotin G., Garber C., De Jong M. M., Van Der Veen F. H., et al. (2008). Persistent atrial fibrillation in a goat model of chronic left atrial overload. J. Thorac. Cardiovasc. Surg. 136 1005–1011. 10.1016/j.jtcvs.2008.05.015 [DOI] [PubMed] [Google Scholar]
- Ren M., Li X., Hao L., Zhong J. (2015). Role of tumor necrosis factor alpha in the pathogenesis of atrial fibrillation: a novel potential therapeutic target? Ann. Med. 47 316–324. 10.3109/07853890.2015.1042030 [DOI] [PubMed] [Google Scholar]
- Ringner M. (2008). What is principal component analysis? Nat. Biotechnol. 26 303–304. [DOI] [PubMed] [Google Scholar]
- Rodriguez C., Martinez-Gonzalez J. (2019). The role of Lysyl oxidase enzymes in cardiac function and remodeling. Cells 8:1483. 10.3390/cells8121483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg K., Olsson H., Morgelin M., Heinegard D. (1998). Cartilage oligomeric matrix protein shows high affinity zinc-dependent interaction with triple helical collagen. J. Biol. Chem. 273 20397–20403. 10.1074/jbc.273.32.20397 [DOI] [PubMed] [Google Scholar]
- Rysa J., Tokola H., Ruskoaho H. (2018). Mechanical stretch induced transcriptomic profiles in cardiac myocytes. Sci. Rep. 8:4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakamuri S. S., Takawale A., Basu R., Fedak P. W., Freed D., Sergi C., et al. (2016). Differential impact of mechanical unloading on structural and nonstructural components of the extracellular matrix in advanced human heart failure. Transl. Res. 172 30–44. 10.1016/j.trsl.2016.02.006 [DOI] [PubMed] [Google Scholar]
- Samaj J., Baluska F., Hirt H. (2004). From signal to cell polarity: mitogen-activated protein kinases as sensors and effectors of cytoskeleton dynamicity. J. Exp. Bot. 55 189–198. 10.1093/jxb/erh012 [DOI] [PubMed] [Google Scholar]
- Sanz-Ezquerro J. J., Munsterberg A. E., Stricker S. (2017). Editorial: signaling pathways in embryonic development. Front. Cell Dev. Biol. 5:76. 10.3389/fcell.2017.00076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlaepfer D. D., Hou S., Lim S. T., Tomar A., Yu H., Lim Y., et al. (2007). Tumor necrosis factor-alpha stimulates focal adhesion kinase activity required for mitogen-activated kinase-associated interleukin 6 expression. J. Biol. Chem. 282 17450–17459. 10.1074/jbc.m610672200 [DOI] [PubMed] [Google Scholar]
- Schneider S., Weydig C., Wessler S. (2008). Targeting focal adhesions:Helicobacter pylori-host communication in cell migration. Cell Commun. Signal 6:2. 10.1186/1478-811x-6-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroen B., Heymans S., Sharma U., Blankesteijn W. M., Pokharel S., Cleutjens J. P., et al. (2004). Thrombospondin-2 is essential for myocardial matrix integrity: increased expression identifies failure-prone cardiac hypertrophy. Circ Res 95 515–522. 10.1161/01.res.0000141019.20332.3e [DOI] [PubMed] [Google Scholar]
- Schumacher S. M., Naga Prasad S. V. (2018). Tumor necrosis factor-alpha in heart failure: an updated review. Curr. Cardiol. Rep. 20:117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen M., Hu M., Fedak P. W. M., Oudit G. Y., Kassiri Z. (2018). Cell-specific functions of ADAM17 regulate the progression of thoracic aortic aneurysm. Circ. Res. 123 372–388. 10.1161/circresaha.118.313181 [DOI] [PubMed] [Google Scholar]
- Shifrin Y., Pinto V. I., Hassanali A., Arora P. D., Mcculloch C. A. (2012). Force-induced apoptosis mediated by the Rac/Pak/38 signalling pathway is regulated by filamin A. Biochem. J. 445 57–67. 10.1042/bj20112119 [DOI] [PubMed] [Google Scholar]
- Simmers M. B., Cole B. K., Ogletree M. L., Chen Z., Xu Y., Kong L. J., et al. (2016). Hemodynamics associated with atrial fibrillation directly alters thrombotic potential of endothelial cells. Thromb. Res. 143 34–39. 10.1016/j.thromres.2016.04.022 [DOI] [PubMed] [Google Scholar]
- Sivasubramanian N., Coker M. L., Kurrelmeyer K. M., Maclellan W. R., Demayo F. J., Spinale F. G., et al. (2001). Left ventricular remodeling in transgenic mice with cardiac restricted overexpression of tumor necrosis factor. Circulation 104 826–831. 10.1161/hc3401.093154 [DOI] [PubMed] [Google Scholar]
- Spinale F. G. (2002). Matrix metalloproteinases: regulation and dysregulation in the failing heart. Circ. Res. 90 520–530. 10.1161/01.res.0000013290.12884.a3 [DOI] [PubMed] [Google Scholar]
- Srivastava A., Malik L., Smith T., Sudbery I., Patro R. (2019). Alevin efficiently estimates accurate gene abundances from dscRNA-seq data. Genome Biol. 20:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staerk L., Sherer J. A., Ko D., Benjamin E. J., Helm R. H. (2017). Atrial fibrillation: epidemiology, pathophysiology, and clinical outcomes. Circ. Res. 120 1501–1517. 10.1161/circresaha.117.309732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staerk L., Wang B., Preis S. R., Larson M. G., Lubitz S. A., Ellinor P. T., et al. (2018). Lifetime risk of atrial fibrillation according to optimal, borderline, or elevated levels of risk factors: cohort study based on longitudinal data from the Framingham Heart Study. BMJ 361:k1453. 10.1136/bmj.k1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su S. A., Yang D., Wu Y., Xie Y., Zhu W., Cai Z., et al. (2017). EphrinB2 regulates cardiac fibrosis through modulating the interaction of Stat3 and TGF-beta/Smad3 signaling. Circ. Res. 121 617–627. 10.1161/circresaha.117.311045 [DOI] [PubMed] [Google Scholar]
- Subramanian A., Tamayo P., Mootha V. K., Mukherjee S., Ebert B. L., Gillette M. A., et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A. 102 15545–15550. 10.1073/pnas.0506580102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugden P. H., Fuller S. J., Weiss S. C., Clerk A. (2008). Glycogen synthase kinase 3 (GSK3) in the heart: a point of integration in hypertrophic signalling and a therapeutic target? A critical analysis. Br. J. Pharmacol. 153(Suppl. 1) S137–S153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M., Chen M., Dawood F., Zurawska U., Li J. Y., Parker T., et al. (2007). Tumor necrosis factor-alpha mediates cardiac remodeling and ventricular dysfunction after pressure overload state. Circulation 115 1398–1407. 10.1161/circulationaha.106.643585 [DOI] [PubMed] [Google Scholar]
- Tanaka K., Abe M., Sato Y. (1999). Roles of extracellular signal-regulated kinase 1/2 and p38 mitogen-activated protein kinase in the signal transduction of basic fibroblast growth factor in endothelial cells during angiogenesis. Jpn. J. Cancer Res. 90 647–654. 10.1111/j.1349-7006.1999.tb00796.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas A. M., Cabrera C. P., Finlay M., Lall K., Nobles M., Schilling R. J., et al. (2019). Differentially expressed genes for atrial fibrillation identified by RNA sequencing from paired human left and right atrial appendages. Physiol. Genomics 51 323–332. 10.1152/physiolgenomics.00012.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracey K. J., Cerami A. (1994). Tumor necrosis factor: a pleiotropic cytokine and therapeutic target. Annu. Rev. Med. 45 491–503. 10.1146/annurev.med.45.1.491 [DOI] [PubMed] [Google Scholar]
- Turk B., Turk D., Turk V. (2000). Lysosomal cysteine proteases: more than scavengers. Biochim. Biophys. Acta 1477 98–111. 10.1016/s0167-4838(99)00263-0 [DOI] [PubMed] [Google Scholar]
- Vadlamudi R. K., Li F., Adam L., Nguyen D., Ohta Y., Stossel T. P., et al. (2002). Filamin is essential in actin cytoskeletal assembly mediated by p21-activated kinase 1. Nat. Cell Biol. 4 681–690. 10.1038/ncb838 [DOI] [PubMed] [Google Scholar]
- van der Slot A. J., Van Dura E. A., De Wit E. C., De Groot J., Huizinga T. W., Bank R. A., et al. (2005). Elevated formation of pyridinoline cross-links by profibrotic cytokines is associated with enhanced lysyl hydroxylase 2b levels. Biochim. Biophys. Acta 1741 95–102. 10.1016/j.bbadis.2004.09.009 [DOI] [PubMed] [Google Scholar]
- Vega R. B., Konhilas J. P., Kelly D. P., Leinwand L. A. (2017). Molecular mechanisms underlying cardiac adaptation to exercise. Cell Metab. 25 1012–1026. 10.1016/j.cmet.2017.04.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermeulen Z., Segers V. F., De Keulenaer G. W. (2016). ErbB2 signaling at the crossing between heart failure and cancer. Basic Res. Cardiol. 111:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vranka I., Penz P., Dukat A. (2007). Atrial conduction delay and its association with left atrial dimension, left atrial pressure and left ventricular diastolic dysfunction in patients at risk of atrial fibrillation. Exp. Clin. Cardiol. 12 197–201. [PMC free article] [PubMed] [Google Scholar]
- Wang L., Yue Y., Yang X., Fan T., Mei B., Hou J., et al. (2017a). Platelet derived growth factor alpha (PDGFRalpha) induces the activation of cardiac fibroblasts by activating c-Kit. Med. Sci. Monit. 23 3808–3816. 10.12659/msm.906038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Chen W., Zhang J., Khan A., Li L., Huang F., et al. (2017b). Critical role of ADAMTS2 (a disintegrin and metalloproteinase with thrombospondin Motifs 2) in cardiac hypertrophy induced by pressure overload. Hypertension 69 1060–1069. 10.1161/hypertensionaha.116.08581 [DOI] [PubMed] [Google Scholar]
- Weeke P., Muhammad R., Delaney J. T., Shaffer C., Mosley J. D., Blair M., et al. (2014). Whole-exome sequencing in familial atrial fibrillation. Eur. Heart J. 35 2477–2483. 10.1093/eurheartj/ehu156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- White E. (2011). Mechanical modulation of cardiac microtubules. Pflugers Arch. 462 177–184. 10.1007/s00424-011-0963-0 [DOI] [PubMed] [Google Scholar]
- Wingett S. W., Andrews S. (2018). FastQ Screen: a tool for multi-genome mapping and quality control. F1000Res. 7:1338. 10.12688/f1000research.15931.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyse D. G., Van Gelder I. C., Ellinor P. T., Go A. S., Kalman J. M., Narayan S. M., et al. (2014). Lone atrial fibrillation: does it exist? J. Am. Coll. Cardiol. 63 1715–1723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G. J., Gan T. Y., Tang B. P., Chen Z. H., Mahemuti A., Jiang T., et al. (2013). Accelerated fibrosis and apoptosis with ageing and in atrial fibrillation: adaptive responses with maladaptive consequences. Exp. Ther. Med. 5 723–729. 10.3892/etm.2013.899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J., Cui G., Esmailian F., Plunkett M., Marelli D., Ardehali A., et al. (2004). Atrial extracellular matrix remodeling and the maintenance of atrial fibrillation. Circulation 109 363–368. 10.1161/01.cir.0000109495.02213.52 [DOI] [PubMed] [Google Scholar]
- Xu J., Mukerjee S., Silva-Alves C. R., Carvalho-Galvao A., Cruz J. C., Balarini C. M., et al. (2016). A disintegrin and metalloprotease 17 in the cardiovascular and central nervous systems. Front. Physiol. 7:469. 10.3389/fphys.2016.00469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yabluchanskiy A., Ma Y., Iyer R. P., Hall M. E., Lindsey M. L. (2013). Matrix metalloproteinase-9: Many shades of function in cardiovascular disease. Physiology (Bethesda) 28 391–403. 10.1152/physiol.00029.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z., Kyriakides T. R., Bornstein P. (2000). Matricellular proteins as modulators of cell-matrix interactions: adhesive defect in thrombospondin 2-null fibroblasts is a consequence of increased levels of matrix metalloproteinase-2. Mol. Biol. Cell 11 3353–3364. 10.1091/mbc.11.10.3353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y. Z., Chang Y. F., Wang B. Z., Ma Y. T., Ma X. (2020). Prognostic value of von Willebrand factor for patients with atrial fibrillation: a meta-analysis of prospective cohort studies. Postgrad. Med. J. 96 267–276. 10.1136/postgradmedj-2019-136842 [DOI] [PubMed] [Google Scholar]
- Yin J. C., Platt M. J., Tian X., Wu X., Backx P. H., Simpson J. A., et al. (2017). Cellular interplay via cytokine hierarchy causes pathological cardiac hypertrophy in RAF1-mutant Noonan syndrome. Nat. Commun. 8:15518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama T., Sekiguchi K., Tanaka T., Tomaru K., Arai M., Suzuki T., et al. (1999). Angiotensin II and mechanical stretch induce production of tumor necrosis factor in cardiac fibroblasts. Am. J. Physiol. 276 H1968–H1976. [DOI] [PubMed] [Google Scholar]
- Zhan M., Jin B., Chen S. E., Reecy J. M., Li Y. P. (2007). TACE release of TNF-alpha mediates mechanotransduction-induced activation of p38 MAPK and myogenesis. J. Cell Sci. 120 692–701. 10.1242/jcs.03372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H., Wu J., Dong H., Khan S. A., Chu M. L., Tsuda T. (2014). Fibulin-2 deficiency attenuates angiotensin II-induced cardiac hypertrophy by reducing transforming growth factor-β signalling. Clin. Sci. (Lond.) 126 275–288. 10.1042/cs20120636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang P. P., Sun J., Li W. (2020). Genome-wide profiling reveals atrial fibrillation-related circular RNAs in atrial appendages. Gene 728:144286. 10.1016/j.gene.2019.144286 [DOI] [PubMed] [Google Scholar]
- Zhang S. J., Han J. H., Sells M. A., Chernoff J., Knaus U. G., Ulevitch R. J., et al. (1995). Rho-family gtpases regulate P38 mitogen-activated protein-kinase through the downstream mediator Pak1. J. Biol. Chem. 270 23934–23936. 10.1074/jbc.270.41.23934 [DOI] [PubMed] [Google Scholar]
- Zhang X., Li H., Kou W., Tang K., Zhao D., Zhang J., et al. (2019). Increased plasma microfibrillar-associated protein 4 is associated with atrial fibrillation and more advanced left atrial remodelling. Arch. Med. Sci. 15 632–640. 10.5114/aoms.2018.74953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J., Lv T., Quan J., Zhao W., Song J., Li Z., et al. (2018). Identification of target genes in cardiomyopathy with fibrosis and cardiac remodeling. J. Biomed. Sci. 25:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong C., Xin M., He L., Sun G., Shen F. (2018). Prognostic value of von Willebrand factor in patients with atrial fibrillation: a meta-analysis. Medicine (Baltimore) 97:e11269. 10.1097/md.0000000000011269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou R., Yang M., Shi W., Zheng C., Zeng H., Lin X., et al. (2018). Analysis of genes involved in persistent atrial fibrillation: comparisons of ‘Trigger’ and ‘Substrate’ differences. Cell Physiol. Biochem. 47 1299–1309. 10.1159/000490225 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study can be found in the BioProject database (accession: PRJNA663094).